
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Plant Sci., 19 April 2016
Sec. Plant Breeding
Volume 7 - 2016 | https://doi.org/10.3389/fpls.2016.00450
This article is part of the Research TopicAdvances in legume researchView all 45 articles
 Deepak Bajaj1
Deepak Bajaj1 Rishi Srivastava1
Rishi Srivastava1 Manoj Nath2
Manoj Nath2 Shailesh Tripathi3
Shailesh Tripathi3 Chellapilla Bharadwaj3
Chellapilla Bharadwaj3 Hari D. Upadhyaya4
Hari D. Upadhyaya4 Akhilesh K. Tyagi1
Akhilesh K. Tyagi1 Swarup K. Parida1*
Swarup K. Parida1*The large-scale mining and high-throughput genotyping of novel gene-based allelic variants in natural mapping population are essential for association mapping to identify functionally relevant molecular tags governing useful agronomic traits in chickpea. The present study employs an alternative time-saving, non-laborious and economical pool-based EcoTILLING approach coupled with agarose gel detection assay to discover 1133 novel SNP allelic variants from diverse coding and regulatory sequence components of 1133 transcription factor (TF) genes by genotyping in 192 diverse desi and kabuli chickpea accessions constituting a seed weight association panel. Integrating these SNP genotyping data with seed weight field phenotypic information of 192 structured association panel identified eight SNP alleles in the eight TF genes regulating seed weight of chickpea. The associated individual and combination of all SNPs explained 10–15 and 31% phenotypic variation for seed weight, respectively. The EcoTILLING-based large-scale allele mining and genotyping strategy implemented for association mapping is found much effective for a diploid genome crop species like chickpea with narrow genetic base and low genetic polymorphism. This optimized approach thus can be deployed for various genomics-assisted breeding applications with optimal expense of resources in domesticated chickpea. The seed weight-associated natural allelic variants and candidate TF genes delineated have potential to accelerate marker-assisted genetic improvement of chickpea.
Allele mining is an efficient strategy to unlock a wealth of largely untapped natural and functional allelic variation/diversity existing within wild and cultivated genetic resources for crop genetic enhancement, thereby improving the productivity and sustainability of global agriculture. The vast available germplasm (core and mini-core) repositories and different recently developed high-throughput array-based next-generation sequencing (NGS) and sequence-based marker genotyping strategies are found expedient in large-scale mining and genotyping of genome/gene-based SNP (single nucleotide polymorphism) alleles among these germplasm accessions for driving genomics-assisted crop improvement through genetic and association mapping. The allele mining strategies commonly adopted in laboratories equipped with advanced infrastructural facilities (like high-throughput genotyping platforms and modern computational genomics tools), require prior information of SNP alleles (nature/types and flanking sequences) for their discovery, validation and genotyping in the targeted gene/genomic regions of multiple crop accessions. Conversely, EcoTILLING (Ecotype Targeting Induced Local Lesions IN Genomes), a rapid, inexpensive and well-established allele mining approach is found much proficient in large-scale mining and high-throughput genotyping of novel natural and functional allelic variants (without prior knowledge of SNP alleles) of known and candidate genes related to useful agronomic traits in diverse crop germplasm accessions (McCallum et al., 2000; Comai et al., 2004; Till et al., 2006, 2007, 2010; Raghavan et al., 2007; Wang et al., 2010; Xia et al., 2012). The implication of EcoTILLING to identify potential novel functional alleles in the known and candidate genes/transcription factors (TFs) regulating qualitative and quantitative agronomic traits by association/genetic mapping is well-documented for expediting the genetic enhancement of crop plants (Mejlhede et al., 2006; Barkley and Wang, 2008; Ibiza et al., 2010; Negrao et al., 2011; Yu et al., 2012; Frerichmann et al., 2013).
EcoTILLING usually employs a mismatch-specific CEL-I nuclease to cleave the PCR amplified fragments at the site of heteroduplex formation involving nucleotide (SNP-allelic) polymorphism. Most of the EcoTILLING studies utilize the advanced genotyping platforms (LICOR NEN Model 4300 DNA Analyzer, Transgenomic WAVE-HS denaturing high performance liquid chromatography, ABI 377 sequencer and eGene capillary electrophoresis systems) for efficient resolution of fluorescent dye (IRDye 700/800 and SYBR green)-labeled CEL-I cleaved heteroduplex PCR amplified fragments. Consequently, these efforts led to the discovery and genotyping of novel potential alleles specifically derived from the trait-associated known and candidate genes in natural population of diverse crop plants (Perry et al., 2003; Caldwell et al., 2004; Comai et al., 2004; Henikoff et al., 2004; Yang et al., 2004; Suzuki et al., 2005). The added-advantage of agarose gel-based EcoTILLING vis-à-vis the commonly utilized LICOR genotyper for large-scale mining and genotyping of allelic variants in accessions exhibiting low level polymorphism, is well-demonstrated in many crop plants (Raghavan et al., 2007; Negrao et al., 2011; Yu et al., 2012). This is merely because efficacy of an agarose gel-based EcoTILLING approach in precise resolution of unlabeled CEL I-cleaved heteroduplex PCR amplified fragments by a simpler, economical and time-saving agarose gel-based detection assay as compared to a standard EcoTILLING method that requires labeled CEL I-cleaved heteroduplex PCR amplicons for resolution in a LICOR genotyper. The broader utility and deployment of this agarose gel-based EcoTILLING approach in manifold large-scale genotyping applications is well-documented by the research laboratories with minimal resources (Raghavan et al., 2007; Negrao et al., 2011; Yu et al., 2012). This includes understanding the natural allelic diversity, population genetic structure and domestication pattern among accessions, molecular mapping and genetic association analysis for identification of potential molecular tags like alleles and genes/QTLs (quantitative trait loci) governing vital agronomic traits and marker-assisted breeding for selecting desirable accessions for crop genetic improvement.
Chickpea, a member of genus Cicer, is rich in cultivated and wild germplasm resource (core/mini-core collections) with a wealth of trait diversity (Upadhyaya and Ortiz, 2001; Upadhyaya et al., 2001, 2002). More in-depth characterization of these core/mini-core germplasm resources at both genotypic and phenotypic level for diverse important abiotic/biotic stress tolerance and yield/quality component traits is essential to discover and deploy valuable alleles and allelic combinations scanned from these germplasm accessions, more effectively for genetic improvement of chickpea (Upadhyaya et al., 2008, 2011; Varshney et al., 2013a; Saxena et al., 2014a,b; Bajaj et al., 2015). The existing diverse germplasm collections are thus “gold mines” for analysis of functional as well as natural allelic variation/diversity in the known and candidate genes controlling important agronomic traits of chickpea. Considering the importance of allele mining in crop genetic enhancement, EcoTILLING can be employed in multiple cultivated (desi and kabuli) and wild chickpea accessions for identifying novel functional/natural allelic variants in the candidate and known genes associated with multiple traits of agricultural importance in chickpea.
The agarose gel-based EcoTILLING strategy mostly utilizes pooling of genomic DNA isolated from two diverse accessions rather than multiple accessions for robust mining and genotyping of alleles in the view of anticipating more allelic variations between distant accessions of crop plants (Raghavan et al., 2007). However, the level of allelic variation and diversity captured specifically from different sequence components of genes/genomes among germplasm accessions of chickpea is known to be very low due to its narrow genetic base and extensive domestication bottlenecks as compared to other crop plants (Abbo et al., 2003, 2005; Berger et al., 2003, 2005; Singh et al., 2008; Toker, 2009; Jain et al., 2013; Varshney et al., 2013b; Saxena et al., 2014a). Therefore, in a diploid self-pollinated crop species like chickpea with a lower occurrence of SNP-allelic variations, the agarose gel-based EcoTILLING approach can easily be expanded to multiple accessions regardless of selecting only two accessions for DNA pooling in allele mining. Consequently, the efficient resolution and estimation of allelic variants scanned from the pooled DNA of multiple chickpea accessions will be relatively convenient, even in a low-resolution agarose gel than that of other crop species exhibiting higher allelic polymorphism. Such a strategy of multiple accessions pooling-based EcoTILLING coupled with agarose gel detection approach has been found beneficial for various high-throughput allele mining and large-scale genotyping applications, including genetic and association mapping of alleles/genes (TFs) regulating drought and salinity stress tolerance traits in rice (Negrao et al., 2011; Yu et al., 2012). Henceforth, the utilization of this multiple accessions-pooling agarose gel-based EcoTILLING approach can certainly accelerate the process of rapid selection of informative SNP alleles/markers as well as identification of accessions exhibiting higher allelic variations for their robust genotyping at a genome-wide scale. This strategy will be thus useful for various high-throughput genetic analysis in chickpea with sub-optimal use of resources. A large-scale novel as well as functional allelic genotyping information cataloged from diverse germplasm (core/mini-core) accessions and bi-parental mapping populations by use of agarose gel-based EcoTILLING assay can serve as a vital resource for trait association and genetic mapping. This will be helpful to identify favorable natural allelic variants undergoing selection during the course of domestication in desi, kabuli and wild accessions that are adapted to diverse agro-climatic conditions for genomics-assisted crop improvement of chickpea.
In light of the above, the present study employed a simpler non-laborious and rapid yet cost-effective agarose gel-based EcoTILLING assay (Figure 1) for high-throughput mining of natural allelic variants derived from diverse coding and non-coding regulatory sequence components of 1133 TF genes by genotyping in 192 core/mini-core germplasm accessions constituting a seed weight association panel. As a proof of concept, the high-throughput genotyping data of 1133 TF gene-derived SNPs was correlated with seed weight field phenotypic information of the 192 accessions to delineate functionally relevant natural allelic variants in the candidate TF genes regulating 100-seed weight in chickpea.
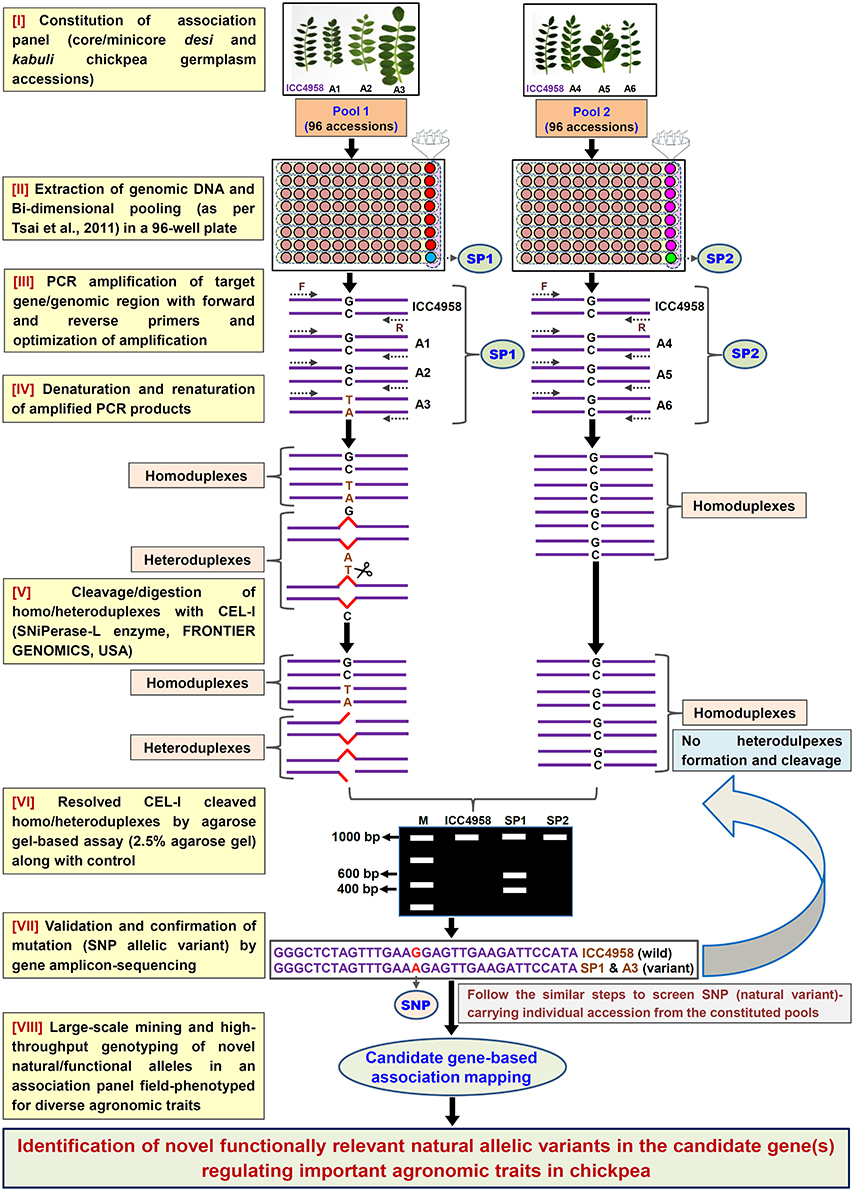
Figure 1. Schematic depicting the major steps followed in an agarose gel-based EcoTILLING assay for efficient identification of functionally relevant molecular tags governing useful agronomic traits in chickpea. This strategy is optimized for successful large-scale mining of novel SNP allelic variants from the target genomic regions (genes) by genotyping in a constituted field-phenotyped association panel (desi and kabuli core/mini-core germplasm lines). A, Accessions; SP, Superpool; F, (Forward); and R, (Reverse) primers.
For large-scale mining and genotyping of gene-based SNP alleles by EcoTILLING in chickpea, a set of 1248 TF genes annotated from desi and kabuli genomes were acquired. The selected TFs include 819 desi and kabuli TF genes and 429 TF-encoding transcripts of desi accession (ICC 4958), which were specifically selected from the previous studies of Jhanwar et al. (2012) and Kujur et al. (2015), respectively based on the presence of at least one SNP in the CDS (coding sequence) and regulatory sequences of these genes. The multiple forward and reverse primer combination (at least two primer-pairs per TF gene) with expected amplification product size of 1000–1500 bp (per primer) targeting the diverse CDS and 2000-bp upstream/downstream regulatory regions (URRs/DRRs) of 1248 TF genes were designed. The amplification of each target gene regions was optimized (specifically the annealing temperature) with different combination of primer-pairs using the genomic DNA of one desi chickpea accession (ICC 4958) as per the detail PCR protocol described by Jhanwar et al. (2012) and Kujur et al. (2013). Based on these analyses, 1890 (75.7% of 2496 primer-pairs designed in total) primers designed from the 1133 TF genes exhibited reproducible single amplicons (by eliminating the non-specific amplified fragments and duplicate loci) in ICC 4958 using 2.5% agarose gel (Figure 2A). The fragments amplified specific to diverse coding (CDS) and non-coding URR/DRR sequence components of TF genes using the optimized primers were further assayed through agarose gel-based EcoTILLING approach for allele mining.
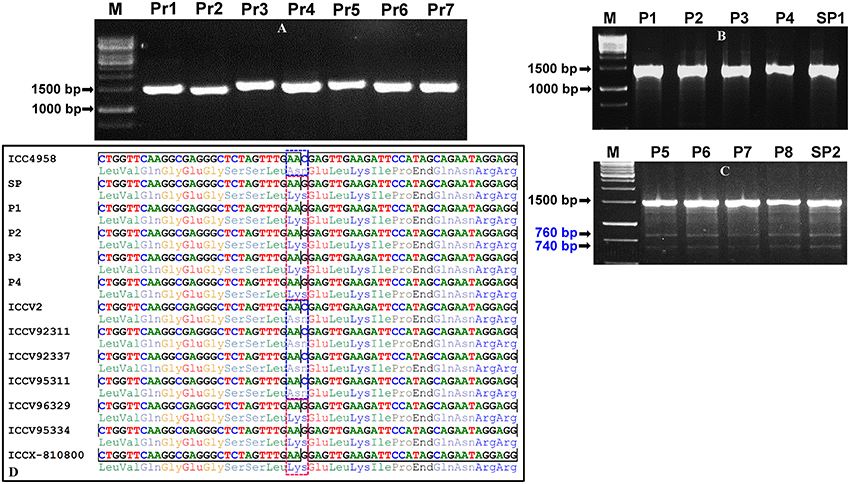
Figure 2. Optimization and validation of pool-based EcoTILLING approach coupled with agarose gel detection assay for large-scale mining of novel allelic variants from diverse coding and regulatory sequence components of TF genes by genotyping in a 192 desi and kabuli germplasm lines belonging to a seed weight association panel. (A) A representative gel illustrating the optimization followed by PCR amplification of seven primer-pairs (Pr1-Pr7) designed targeting various coding and regulatory regions of seven TF genes in the genomic DNA of a desi chickpea accession (ICC 4958) to produce single reproducible amplicons of each primer for EcoTILLING analysis. (B,C) The representative gels depicting the screening of allelic variants from the eight representative micropools (P1–P8) and two superpools (SP1 and SP2) made from the genomic DNA of 192 desi and kabuli germplasm lines (including ICC 4958 as control) employing an agarose gel-based EcoTILLING assay as defined in the Figure 1. The absence (B) and presence (C) of one non-synonymous SNP allelic variant in the pools and superpools based on cleavage/digestion patterns of 1500 bp fragments amplified from the target CDS region of a mTERF TF gene was apparent in 2.5% agarose gel. The occurrence of 1500 bp homoduplex uncut PCR amplicons as well as mismatch-specific CEL I cleavage of 1500 bp heteroduplex PCR amplified fragments into two varied amplicons of 760 and 740 bp fragment sizes due to the effect of single nucleotide polymorphism (SNP-allele) was observed in the four pools (P5–P8) and one superpool (SP2). M: 1 kb DNA ladder size standard. (D) The sequencing of 1500 bp amplified PCR product of an mTERF TF gene followed by multiple alignment of their high-quality sequences ascertained the presence of one coding SNP (C to G) exhibiting missense non-synonymous amino acid substitution [aspargine (AAC) to lysine (AAG)] in the four pools (P5–P8), one superpool (SP2) and seven individual desi and kabuli accessions as per expectation based on agarose gel-based EcoTILLING assay. The sequenced region carrying the non-synonymous SNP is indicated with a dotted box. The detail information of genes used for validation is mentioned in the Table S2.
To access the potential of EcoTILLING in large-scale mining and high-throughput genotyping of TF gene-derived SNP alleles, 192 desi and kabuli chickpea core/minicore germplasm accessions were selected (Table S1) from a 100-seed weight (SW) specific association panel (244 accessions) as constituted previously by Kujur et al. (2014). The high-quality genomic DNA isolated from these 192 accessions was quantified to equal concentration of 1 ng/μl. The bi-dimensional pooling of the uniformly quantified genomic DNA of 192 accessions was performed in two of each 96-well PCR plate to constitute eight micropools and one superpool (per plate) according to Tsai et al. (2011) (Figure 1). The genomic DNA of each of these pools was contrasted with that of ICC 4958 individually with a 1:1 ratio and further PCR amplified with the 1890 optimized primer-pairs designed from the 1133 TF genes (as per aforementioned methods). The amplified PCR product from each pool was denatured and renatured for homoduplex/heteroduplex formation and digested with CEL I-based SNiPerase-L enzyme (FRONTIER GENOMICS, Alaska, USA) following the detail instructions of manufacturer (FRONTIER GENOMICS; Figure 1). The purified CEL I cleaved homo/heteroduplex PCR products of each TF gene amplified from the pools were resolved in 2.5% agarose gel as per the EcoTILLING approach documented by Raghavan et al. (2007) (Figure 1). The individual accession exhibiting putative mutations (SNP allelic variants) was screened from the pools by accessing the digestion pattern of all 1133 TF genes in the row and/or column-wise de-multiplexed genomic DNA following the aforesaid agarose gel-based EcoTILLING method (Figures 2B,C). To ascertain the putative mutations (SNP allelic variants) discovered in the TF genes among accessions constituting the pools, the PCR products of corresponding genes amplified from the pools/accessions were sequenced by an automated 96 capillary ABI 3730xl DNA Analyzer (Applied Biosystems, USA) (Figure 2D). The SNP allelic variants were detected by aligning and comparing the multiple high-quality gene sequences among accessions following Kujur et al. (2013). The above-said analysis of allele mining and genotyping by agarose gel-based EcoTILLING led to discover 1133 SNP allelic variants from the diverse coding and non-coding regulatory sequence components of 1133 TF genes (Table S2). Of these, 406 (35.8%) and 702 (62.0%) SNP alleles exhibited synonymous and missense/nonsense non-synonymous amino acid substitutions, respectively in the CDS regions of 1108 TF genes. The remaining 25 (2.2%) SNP alleles were derived from the regulatory (URR/DRR) sequence components of 25 TF genes (Table S2). To determine the physical localization (bp) of SNPs on the chickpea genome, the 100-bp TF gene sequences flanking the 1133 SNP loci were BLAST searched (≥95% query coverage and percent identity) against the draft genome sequences of desi (Jain et al., 2013) and kabuli (Varshney et al., 2013a) chickpea. Notably, 1042 (92%) and 91 (8%) SNPs of the total discovered 1133 TF gene-derived SNP alleles were physically mapped on the eight chromosomes and unanchored scaffolds of desi and kabuli chickpea genomes, respectively (Table S2). These observations overall infer the efficacy of agarose gel-based EcoTILLING assay in large-scale mining and high-throughput genotyping of natural as well as functional allelic variants among diverse desi and kabuli chickpea germplasm accessions by the optimal expense of time, labor and cost in the research laboratories equipped with limited infrastructural facilities. Notably, this approach seems quite convenient and straightforward for screening the allelic variants more efficiently from the constituted pools containing DNA of numerous germplasm accessions (whole association panel) in a diploid crop species like chickpea with narrow genetic base and low intra-/inter-specific genetic polymorphism. Henceforth, this agarose-based detection assay has potential utility not only for the analysis of EcoTILLING using the pools of natural germplasm accessions but also for TILLING involving the pools of available EMS (ethyl methanesulfonate)-induced mutant lines (~10,000) of desi accession (ICC 4958; Varshney et al., 2009, 2013b) to identify the functionally relevant novel SNP allelic variants (mutations) influencing vital agronomic traits. Therefore, this optimized strategy has utility in accelerating the genomics-assisted crop improvement of chickpea through genetic/association mapping. In the present study, large-scale genotyping data of novel TF gene-based SNP alleles discovered from a seed weight association panel (192 accessions) using an optimized pool-based agarose gel-EcoTILLING strategy were assessed for trait association mapping potential to identify functional and natural allelic variants of the candidate TF genes regulating seed weight in chickpea.
To perform candidate gene-based association analysis, the genotyping information of 1133 TF gene-derived SNP alleles (≥5% minor allele frequency) mined by EcoTILLING was integrated with multi-location replicated SW field phenotyping (100 seed weight: 6–63 g), principal component analysis (P), population genetic structure (Q), and kinship (K) matrix of 192 desi and kabuli accessions (association panel) of chickpea. At the most, we could expect clustering of 192 accessions into two distinct population groups at K = 2, in accordance with our preliminary genetic distance-based phylogenetic tree analysis. Using population genetic structure, the average likelihood value [Ln P(D)] against each K across 20 independent replications was estimated and plotted. The optimal value of K was determined following ad hoc and delta K procedures of Pritchard et al. (2000) and Evanno et al. (2005), respectively. At the optimum value of K = 2, the population structure model representing expected phylogenetic relationships among 192 accessions was constructed. The principal component analysis (PCA) among accessions was performed using GAPIT (Lipka et al., 2012). The kinship matrix (K) was estimated using SPAGeDi 1.2 (Hardy and Vekemans, 2002). For candidate gene-based association analysis, the CMLM (compressed mixed linear model) (P + K, K and Q + K) along with P3D (population parameters previously determined, Kang et al., 2010; Zhang et al., 2010) interfaces of GAPIT were employed following Kujur et al. (2013, 2014), Thudi et al. (2014) and Kumar et al. (2015). To ensure the accuracy and robustness of each SNP marker-trait association, the quantile-quantile plot-based false discovery rate (FDR cut-off ≤ 0.05) corrections (Benjamini and Hochberg, 1995) for multiple comparisons between observed/expected -log10(P)-values and adjusted P-value threshold of significance were performed in accordance with Kujur et al. (2015). The degree of association of SNP loci with SW trait was measured by the R2 (model with the SNP and adjusted P-value following FDR-controlling method). The TF gene-derived SNP loci exhibiting significant association with SW trait at lowest FDR adjusted P-values (threshold P < 10−4) and highest R2 were identified in chickpea.
The CMLM and P3D/EMMAX-based association analysis at a FDR cut-off ≤ 0.05 detected eight TF gene-derived SNPs exhibiting significant association with 100-seed weight at a P ≤ 10−4 (Table 1, Figure S1). Seven and one of these eight SW-associated SNPs were derived from the diverse coding (six non-synonymous and one synonymous SNP loci) and regulatory (URR) sequence components of eight TF genes, respectively. Seven SW-associated TF gene-based SNPs were physically mapped on four desi and kabuli chickpea chromosomes (1, 2, 3, and 4), whereas one SNP mapped on the unanchored scaffold of desi genome (Table 1, Figure S1). The proportion of SW phenotypic variation explained by eight SNP loci derived from eight TF genes [encoding bZIP (Basic-leucine zipper), SBP (Squamosa promoter binding protein) protein, Zinc finger-domain containing protein, NAC (No apical meristem arabidopsis transcription activation factor-cup shaped cotyledon), bHLH (Basic helix-loop-helix) protein, AP2-EREBP (APETALA-2/ethylene response element binding protein), ARF (auxin response factor), and mTERF (mitochondrial transcription termination factor)] among 192 desi and kabuli accessions belonging to an association panel varied from R2: 10 to 15% (Table 1, Figure S1). All significant eight SNP loci in combination explained 31% SW phenotypic variation. Strong association of one non-synonymous SNP in a bZIP TF gene (R2: 15% with P: 1.3 × 10−5) with SW was observed in desi and kabuli chickpea (Table 1, Figure S1). The SW-associated eight TF genes delineated by EcoTILLING-based allele mining, genotyping and trait association mapping in chickpea probably regulate seed growth and development, including determination of seed size/weight in many crop plants (Manning et al., 2006; Agarwal et al., 2007, 2011; Nijhawan et al., 2008; Libault et al., 2009; Wang et al., 2011, 2015; Heang and Sassa, 2012; Martínez-Andújar et al., 2012; Jones and Vodkin, 2013; Ha et al., 2014; Hudson and Hudson, 2015; Liu et al., 2015; Singh and Jain, 2015; Zhang et al., 2015). Especially, the seed weight trait association potential of four TFs (bZIP, SBP, NAC, and bHLH)-derived SNPs mapped on chromosomes 1 and 2, has been ascertained by recent studies in chickpea through identification of similar gene models-containing TFs, integrating seed weight trait-specific association analysis with QTL mapping, differential expression profiling and LD (linkage disequilibrium)-based marker haplotyping (Kujur et al., 2013, 2014). The validation of these TF gene-based SNPs in two of our independent studies suggests the potential significance and robustness of these identified novel functional molecular tags (natural allelic variants and genes) in controlling seed weight, which can essentially be deployed for marker-assisted genetic enhancement of chickpea.
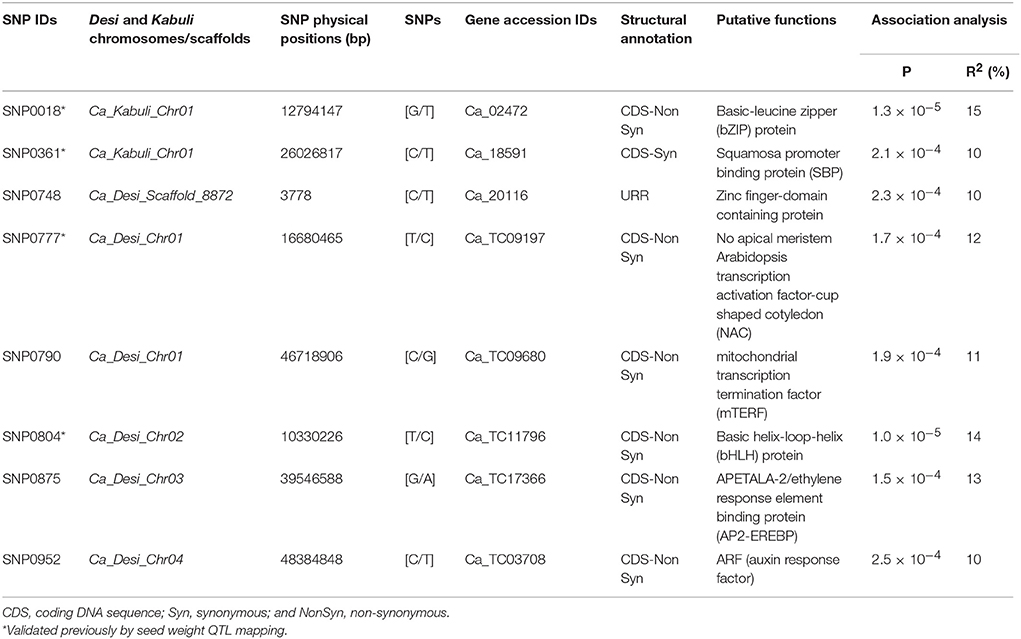
Table 1. Eight seed weight-associated SNP allelic variants of transcription factor genes delineated by EcoTILLING-based trait association mapping.
Collectively, the present study demonstrated the efficacy of an optimized pool-based agarose gel-EcoTILLING strategy (Figure 1) for high-throughput allele mining and genotyping as well as trait association analysis in a natural association panel to delineate novel functional allelic variants of the TF genes governing seed weight in chickpea. Therefore, this approach has potential utility to expedite various genomics-assisted breeding applications, including genetic enhancement targeting diverse qualitative and quantitative stress tolerance and yield component traits by optimal resource expenses in chickpea.
DB, RS, and MN conducted experiments and drafted the manuscript. ST, CB, and HU helped in constitution of association panel and performed phenotyping. SP and AT conceived and designed the study, guided data analysis and interpretation, participated in drafting and correcting the manuscript critically and gave the final approval of the version to be published. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors gratefully acknowledge the financial support by the Department of Biotechnology (DBT), Government of India, through their research grant (102/IFD/SAN/2161/2013-14) for this research work.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00450
Abbo, S., Berger, J., and Turner, N. C. (2003). Evolution of cultivated chickpea: four bottlenecks limit diversity and constrain adaptation. Funct. Plant Biol. 30, 1081–1087. doi: 10.1071/FP03084
Abbo, S., Gopher, A., Rubin, B., and Lev-Yadun, S. (2005). On the origin of Near Eastern founder crops and the ‘dump-heap hypothesis’. Genet. Res. Crop Evol. 52, 491–495. doi: 10.1007/s10722-004-7069-x
Agarwal, P., Arora, R., Ray, S., Singh, A. K., Singh, V. P., Takatsuji, H., et al. (2007). Genome-wide identification of C2H2 zinc-finger gene family in rice and their phylogeny and expression analysis. Plant Mol. Biol. 65, 467–485. doi: 10.1007/s11103-007-9199-y
Agarwal, P., Kapoor, S., and Tyagi, A. K. (2011). Transcription factors regulating the progression of monocot and dicot seed development. Bioessays 33, 189–202. doi: 10.1002/bies.201000107
Bajaj, D., Saxena, M. S., Kujur, A., Das, S., Badoni, S., Tripathi, S., et al. (2015). Genome-wide conserved non-coding microsatellite (CNMS) marker-based integrative genetical genomics for quantitative dissection of seed weight in chickpea. J. Exp. Bot. 66, 1271–1290. doi: 10.1093/jxb/eru478
Barkley, N. A., and Wang, M. L. (2008). Application of TILLING and EcoTILLING as reverse genetic approaches to elucidate the function of genes in plants and animals. Curr. Genomics 9, 212–226. doi: 10.2174/138920208784533656
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300.
Berger, J. D., Abbo, S., and Turner, N. C. (2003). Ecogeography of annual wild Cicer species: the poor state of the world collection. Crop Sci. 43, 1076–1090. doi: 10.2135/cropsci2003.1076
Berger, J. D., Buck, R., Henzell, J. M., and Turner, N. C. (2005). Evolution in the genus Cicer vernalisation response and low temperature pod set in chickpea (C. arietinum L.) and its annual wild relatives. Aust. J. Agric. Res. 56, 1191–1200. doi: 10.1071/AR05089
Caldwell, D. G., McCallum, N., Shaw, P., Muehlbauer, G. J., Marshall, D. F., and Waugh, R. (2004). A structured mutant population for forward and reverse genetics in barley (Hordeum vulgare L.). Plant J. 40, 143–150. doi: 10.1111/j.1365-313X.2004.02190.x
Comai, L., Young, K., Till, B. J., Reynolds, S. H., Greene, E. A., Codomo, C. A., et al. (2004). Efficient discovery of DNA polymorphisms in natural populations by EcoTILLING. Plant J. 37, 778–786. doi: 10.1111/j.0960-7412.2003.01999.x
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Frerichmann, S. L., Kirchhoff, M., Müller, A. E., Scheidig, A. J., Jung, C., and Kopisch-Obuch, F. J. (2013). EcoTILLING in Beta vulgaris reveals polymorphisms in the FLC-like gene BvFL1 that are associated with annuality and winter hardiness. BMC Plant Biol. 13:52. doi: 10.1186/1471-2229-13-52
Ha, C. V., Esfahani, M. N., Watanabe, Y., Tran, U. T., Sulieman, S., Mochida, K., et al. (2014). Genome-wide identification and expression analysis of the CaNAC family members in chickpea during development, dehydration and ABA treatments. PLoS ONE 9:e114107. doi: 10.1371/journal.pone.0114107
Hardy, O. J., and Vekemans, X. (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2, 618–620. doi: 10.1046/j.1471-8286.2002.00305.x
Heang, D., and Sassa, H. (2012). Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in rice. PLoS ONE 7:e31325. doi: 10.1371/journal.pone.0031325
Henikoff, S., Till, B. J., and Comai, L. (2004). TILLING: traditional mutagenesis meets functional genomics. Plant Physiol. 135, 630–636. doi: 10.1104/pp.104.041061
Hudson, K. A., and Hudson, M. E. (2015). A classification of basic helix-loop-helix transcription factors of soybean. Int. J. Genomics 2015:603182. doi: 10.1155/2015/603182
Ibiza, V. P., Cañizares, J., and Nuez, F. (2010). EcoTILLING in Capsicum species: searching for new virus resistances. BMC Genomics 11:631. doi: 10.1186/1471-2164-11-631
Jain, M., Misra, G., Patel, R. K., Priya, P., Jhanwar, S., Khan, A. W., et al. (2013). A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 74, 715–729. doi: 10.1111/tpj.12173
Jhanwar, S., Priya, P., Garg, R., Parida, S. K., Tyagi, A. K., and Jain, M. (2012). Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol. J. 10, 690–702. doi: 10.1111/j.1467-7652.2012.00712.x
Jones, S. I., and Vodkin, L. O. (2013). Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 8:e59270. doi: 10.1371/journal.pone.0059270
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Kujur, A., Bajaj, D., Saxena, M. S., Tripathi, S., Upadhyaya, H. D., Gowda, C. L. L., et al. (2013). Functionally relevant microsatellite markers from chickpea transcription factor genes for efficient genotyping applications and trait association mapping. DNA Res. 20, 355–374. doi: 10.1093/dnares/dst015
Kujur, A., Bajaj, D., Saxena, M. S., Tripathi, S., Upadhyaya, H. D., Gowda, C. L. L., et al. (2014). An efficient and cost-effective approach for genic microsatellite marker-based large-scale trait association mapping: identification of candidate genes for seed weight in chickpea. Mol. Breed. 34, 241–265. doi: 10.1007/s11032-014-0033-3
Kujur, A., Bajaj, D., Upadhyaya, H. D., Das, S., Ranjan, R., Shree, T., et al. (2015). Employing genome-wide SNP discovery and genotyping strategy to extrapolate the natural allelic diversity and domestication patterns in chickpea. Front. Plant Sci. 6:162. doi: 10.3389/fpls.2015.00162
Kumar, V., Singh, A., Amitha Mithra, S. V., Krishnamurthy, S. L., Parida, S. K., Jain, S., et al. (2015). Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res. 22, 133–145. doi: 10.1093/dnares/dsu046
Libault, M., Joshi, T., Benedito, V. A., Xu, D., Udvardi, M. K., and Stacey, G. (2009). Legume transcription factor genes: what makes legumes so special? Plant Physiol. 151, 991–1001. doi: 10.1104/pp.109.144105
Lipka, A. E., Tian, F., Wang, Q., Peiffer, J., Li, M., Bradbury, P. J., et al. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399. doi: 10.1093/bioinformatics/bts444
Liu, K., Yuan, C., Li, H., Lin, W., Yang, Y., Shen, C., et al. (2015). Genome-wide identification and characterization of auxin response factor (ARF) family genes related to flower and fruit development in papaya (Carica papaya L.). BMC Genomics 16:901. doi: 10.1186/s12864-015-2182-0
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Martínez-Andújar, C., Martin, R. C., and Nonogaki, H. (2012). Seed traits and genes important for translational biology-highlights from recent discoveries. Plant Cell Physiol. 53, 5–15. doi: 10.1093/pcp/pcr112
McCallum, C. M., Comai, L., Greene, E. A., and Henikoff, S. (2000). Targeted screening for induced mutations. Nat. Biotechnol. 18, 455–457. doi: 10.1038/74542
Mejlhede, N., Kyjovska, Z., Backes, G., Burhenne, K., Rasmussen, S. K., and Jahoor, A. (2006). EcoTILLING for the identification of allelic variation in the powdery mildew resistance genes mlo and Mla of barley. Plant Breed. 125, 461–467. doi: 10.1111/j.1439-0523.2006.01226.x
Negrao, S., Courtois, B., Ahmadi, N., Abreu, I., Saibo, N., and Oliveira, M. M. (2011). Recent updates on salinity stress in rice: from physiological to molecular responses. Crit. Rev. Plant Sci. 30, 329–377. doi: 10.1080/07352689.2011.587725
Nijhawan, A., Jain, M., Tyagi, A. K., and Khurana, J. P. (2008). Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol. 146, 333–350. doi: 10.1104/pp.107.112821
Perry, J. A., Wang, T. L., Welham, T. J., Gardner, S., Pike, J. M., Yoshida, S., et al. (2003). A TILLING reverse genetics tool and a web-accessible collection of mutants of the legume Lotus japonicus. Plant Physiol. 131, 866–871. doi: 10.1104/pp.102.017384
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Raghavan, C., Naredo, M. E. B., Wang, H., Atienza, G., Liu, B., Qiu, F., et al. (2007). Rapid method for detecting SNPs on agarose gels and its application in candidate gene mapping. Mol. Breed. 19, 87–101. doi: 10.1007/s11032-006-9046-x
Saxena, M. S., Bajaj, D., Das, S., Kujur, A., Kumar, V., Singh, M., et al. (2014b). An integrated genomic approach for rapid delineation of candidate genes regulating agro-morphological traits in chickpea. DNA Res. 21, 695–710. doi: 10.1093/dnares/dsu031
Saxena, M. S., Bajaj, D., Kujur, A., Das, S., Badoni, S., Kumar, V., et al. (2014a). Natural allelic diversity, genetic structure and linkage disequilibrium pattern in wild chickpea. PLoS ONE 9:e107484. doi: 10.1371/journal.pone.0107484
Singh, R., Sharma, P., Varshney, R. K., and Sharma, S. K. (2008). Chickpea improvement: role of wild species and genetic markers. Biotechnol. Genet. Eng. 25, 267–313. doi: 10.5661/bger-25-267
Singh, V. K., and Jain, M. (2015). Genome-wide survey and comprehensive expression profiling of Aux/IAA gene family in chickpea and soybean. Front. Plant Sci. 6:918. doi: 10.3389/fpls.2015.00918
Suzuki, T., Eiguchi, M., Satoh, H., Kumamaru, T., and Kurata, N. (2005). A modified TILLING system for rice mutant screening. Rice Genet. News Lett. 22, 89–91.
Thudi, M., Upadhyaya, H. D., Rathore, A., Gaur, P. M., Krishnamurthy, L., Roorkiwal, M., et al. (2014). Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 9:e96758. doi: 10.1371/journal.pone.0096758
Till, B. J., Cooper, J., Tai, T. H., Colowit, P., Greene, E. A., Henikoff, S., et al. (2007). Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 7:19. doi: 10.1186/1471-2229-7-19
Till, B. J., Jankowicz-Cieslak, J., Sagi, L., Huynh, O. A., Utsushi, H., Swennen, R., et al. (2010). Discovery of nucleotide polymorphisms in the Musa gene pool by EcoTILLING. Theor. Appl. Genet. 121, 1381–1389. doi: 10.1007/s00122-010-1395-5
Till, B. J., Zerr, T., Comai, L., and Henikoff, S. (2006). A protocol for TILLING and EcoTILLING in plants and animals. Nat. Protoc. 1, 2465–2477. doi: 10.1038/nprot.2006.329
Toker, C. (2009). A note on the evolution of kabuli chickpeas as shown by induced mutations in Cicer reticulatum Ladizinsky. Genet. Resour. Crop. Evol. 56, 7–12. doi: 10.1007/s10722-008-9336-8
Tsai, H., Howell, T., Nitcher, R., Missirian, V., Watson, B., Ngo, K. J., et al. (2011). Discovery of rare mutations in populations: TILLING by sequencing. Plant Physiol. 156, 1257–1268. doi: 10.1104/pp.110.169748
Upadhyaya, H. D., Bramel, P. J., and Singh, S. (2001). Development of a chickpea core subset using geographic distribution and quantitative traits. Crop Sci. 41, 206–210. doi: 10.2135/cropsci2001.411206x
Upadhyaya, H. D., Dwivedi, S. L., Baum, M., Varshney, R. K., Udupa, S. M., Gowda, C. L. L., et al. (2008). Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol. 8:106. doi: 10.1186/1471-2229-8-106
Upadhyaya, H. D., and Ortiz, R. (2001). A mini-core subset for capturing diversity and promoting utilization of chickpea genetic resources in crop improvement. Theor. Appl. Genet. 102, 1292–1298. doi: 10.1007/s00122-001-0556-y
Upadhyaya, H. D., Ortiz, R., Bramel, P. J., and Singh, S. (2002). Phenotypic diversity for morphological and agronomic characteristics in chickpea core collection. Euphytica 123, 333–342. doi: 10.1023/A:1015088417487
Upadhyaya, H. D., Thudi, M., Dronavalli, N., Gujaria, N., Singh, S., Sharma, S., et al. (2011). Genomic tools and germplasm diversity for chickpea improvement. Plant Genetic Res. 9, 45–58. doi: 10.1017/S1479262110000468
Varshney, R. K., Close, T. J., Singh, N. K., Hoisington, D. A., and Cook, D. R. (2009). Orphan legume crops enter the genomics era! Curr. Opin. Plant Biol. 12, 202–210. doi: 10.1016/j.pbi.2008.12.004
Varshney, R. K., Mohan, S. M., Gaur, P. M., Gangarao, N. V., Pandey, M. K., and Bohra, A. (2013b). Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol. Adv. 31, 1120–1134. doi: 10.1016/j.biotechadv.2013.01.001
Varshney, R. K., Song, C., Saxena, R. K., Azam, S., Yu, S., Sharpe, A. G., et al. (2013a). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246. doi: 10.1038/nbt.2491
Wang, J. W., Park, M. Y., Wang, L. J., Koo, Y., Chen, X. Y., Weigel, D., et al. (2011). miRNA control of vegetative phase change in trees. PLoS Genet. 7:e1002012. doi: 10.1371/journal.pgen.1002012
Wang, N., Shi, L., Tian, F., Ning, H., Wu, X., Long, Y., et al. (2010). Assessment of FAE1 polymorphisms in three Brassica species using EcoTILLING and their association with differences in seed erucic acid contents. BMC Plant Biol. 10:137. doi: 10.1186/1471-2229-10-137
Wang, Z., Cheng, K., Wan, L., Yan, L., Jiang, H., Liu, S., et al. (2015). Genome-wide analysis of the basic leucine zipper (bZIP) transcription factor gene family in six legume genomes. BMC Genomics 16:1053. doi: 10.1186/s12864-015-2258-x
Xia, Y., Ning, Z., Bai, G., Li, R., Yan, G., Siddique, K. H., et al. (2012). Allelic variations of a light harvesting chlorophyll a/b-binding protein gene (Lhcb1) associated with agronomic traits in barley. PLoS ONE 7:e37573. doi: 10.1371/journal.pone.0037573
Yang, W., Bai, X., Kabelka, E., Eaton, C., Kamoun, S., van der Knaap, E., et al. (2004). Discovery of single nucleotide polymorphisms in Lycopersicon esculentum by computer aided analysis of expressed sequence tags. Mol. Breed. 14, 21–34. doi: 10.1023/B:MOLB.0000037992.03731.a5
Yu, S., Liao, F., Wang, F., Wen, W., Li, J., Mei, H., et al. (2012). Identification of rice transcription factors associated with drought tolerance using the EcoTILLING method. PLoS ONE 7:e30765. doi: 10.1371/journal.pone.0030765
Zhang, S. D., Ling, L. Z., and Yi, T. S. (2015). Evolution and divergence of SBP-box genes in land plants. BMC Genomics 16:787. doi: 10.1186/s12864-015-1998-y
Keywords: allele, association mapping, chickpea, EcoTILLING, seed weight, SNP, transcription factor
Citation: Bajaj D, Srivastava R, Nath M, Tripathi S, Bharadwaj C, Upadhyaya HD, Tyagi AK and Parida SK (2016) EcoTILLING-Based Association Mapping Efficiently Delineates Functionally Relevant Natural Allelic Variants of Candidate Genes Governing Agronomic Traits in Chickpea. Front. Plant Sci. 7:450. doi: 10.3389/fpls.2016.00450
Received: 28 November 2015; Accepted: 22 March 2016;
Published: 19 April 2016.
Edited by:
Nicolas Rispail, Institute for Sustainable Agriculture - Consejo Superior de Investigaciones Científicas, SpainReviewed by:
Milind Ratnaparkhe, Directorate of Soybean Research, IndiaCopyright © 2016 Bajaj, Srivastava, Nath, Tripathi, Bharadwaj, Upadhyaya, Tyagi and Parida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swarup K. Parida, c3dhcnVwQG5pcGdyLmFjLmlu; c3dhcnVwZGJ0QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.