 Elise A. R. Serin
Elise A. R. Serin Harm Nijveen1,2
Harm Nijveen1,2 Henk W. M. Hilhorst
Henk W. M. Hilhorst Wilco Ligterink
Wilco Ligterink- 1Wageningen Seed Lab, Laboratory of Plant Physiology, Wageningen University, Wageningen, Netherlands
- 2Laboratory of Bioinformatics, Wageningen University, Wageningen, Netherlands
Plants are fascinating and complex organisms. A comprehensive understanding of the organization, function and evolution of plant genes is essential to disentangle important biological processes and to advance crop engineering and breeding strategies. The ultimate aim in deciphering complex biological processes is the discovery of causal genes and regulatory mechanisms controlling these processes. The recent surge of omics data has opened the door to a system-wide understanding of the flow of biological information underlying complex traits. However, dealing with the corresponding large data sets represents a challenging endeavor that calls for the development of powerful bioinformatics methods. A popular approach is the construction and analysis of gene networks. Such networks are often used for genome-wide representation of the complex functional organization of biological systems. Network based on similarity in gene expression are called (gene) co-expression networks. One of the major application of gene co-expression networks is the functional annotation of unknown genes. Constructing co-expression networks is generally straightforward. In contrast, the resulting network of connected genes can become very complex, which limits its biological interpretation. Several strategies can be employed to enhance the interpretation of the networks. A strategy in coherence with the biological question addressed needs to be established to infer reliable networks. Additional benefits can be gained from network-based strategies using prior knowledge and data integration to further enhance the elucidation of gene regulatory relationships. As a result, biological networks provide many more applications beyond the simple visualization of co-expressed genes. In this study we review the different approaches for co-expression network inference in plants. We analyse integrative genomics strategies used in recent studies that successfully identified candidate genes taking advantage of gene co-expression networks. Additionally, we discuss promising bioinformatics approaches that predict networks for specific purposes.
Introduction
In plants, the age of systems biology has accelerated the investigation of complex molecular mechanisms underlying intricate developmental and physiological processes. Since plants are anchored to their environment, they cannot escape from stresses by simply moving away. Instead, plants have developed a wide range of mechanisms to cope with environmental fluctuations. This plasticity generally involves changes at the level of DNA, RNA, protein and metabolites, resulting in complex phenotypes governed by multiple genes. Advanced genetic and molecular tools have led to tremendous progress in revealing the genetic architecture but also the regulatory mechanisms of complex traits (Mochida and Shinozaki, 2011). The development of molecular profiling techniques nowadays enables the high-throughput and affordable acquisition of large omics data sets, such as for transcriptomics, proteomics and metabolomics.
While substantial efforts are being made to generate large omics data sets, there is a growing need to develop platforms to integrate these data and derive models describing biological interactions in plants. In this context, networks have rapidly become an attractive approach to manage, display and contextualize these large data sets in order to obtain a system level and molecular understanding of biological key processes (Barabási and Oltvai, 2004; Usadel et al., 2009; Costa et al., 2015; Silva et al., 2016).
Biological networks are generally classified by the nature of the compounds and interactions involved. These networks can be derived from various molecular data resulting in, e.g., gene expression networks (correlation or co-expression networks), protein-protein interaction (PPI) networks, metabolic networks and signaling networks. Graphically, networks are represented as an ensemble of components (nodes or vertices) and interactions depicted by links (edges) connecting pairs of nodes. Such interaction maps provide an attractive framework to study the organizational structure of complex systems and have found many applications in plants (Jiménez-Gómez, 2014).
The fast development of transcriptomic technologies, as compared to other analytical platforms, has supported a range of studies on genetic and environmental perturbations at the transcriptome level in many organisms. Co-expression networks have grown in popularity in the last years as they enable the integration of large transcriptional data sets (Li et al., 2015; Liseron-Monfils and Ware, 2015). Co-expression network analysis allows the simultaneous identification, clustering and exploration of thousands of genes with similar expression patterns across multiple conditions (co-expressed genes). The main procedure for co-expression network inference is explained in Box 1 and illustrated in Figure 1. Briefly, a similarity score (i.e., correlation coefficient) is calculated from the pairwise comparison of the gene expression patterns for each possible pair of genes. Above a certain threshold, genes and gene pairs form a list of nodes and corresponding edges from which the network is constructed. As a rule, the guilt-by-association principle is applied stating that genes sharing the same function or that are involved in the same regulatory pathway will tend to present similar expression profiles and hence form clusters or modules in the network (Wolfe et al., 2005). Thus, within the same module, genes of known function can be used to predict the function of co-expressed unknown genes (Rhee and Mutwil, 2014).
Box 1. Network Inference
Constructing a network of genes from expression data generally consists of the following steps: first a measure of similarity or relatedness is calculated for each of the possible gene pairs. The resulting list of gene pairs is then filtered using a threshold value for the similarity score. The remaining gene pairs form a list of edges from which the network is constructed (Figure 1). As an optional next step, modules of highly related genes can be extracted from the network using gene prioritization approaches.
Similarity Score
Gene expression values are usually log2 transformed before calculating the similarity score in order to scale the values to the same dynamic range.
Several measures are used to determine a similarity score between gene pairs, each with its specific strengths and weaknesses. Simple Pearson or Spearman correlation is often used and performs well compared to more sophisticated methods, both in terms of finding gene relationships and performance on large data sets (Song et al., 2012; Ballouz et al., 2015). Pearson is the most popular correlation measure, although it assumes a linear correlation, normally distributed values and is sensitive to outliers. Spearman's rank correlation is more robust, but also less powerful. Another often used measure that can describe non-linear relations between genes is called Mutual Information (MI) (Meyer et al., 2008). Song et al. (2012) found that in many situations MI does not perform better than correlation. They proposed “bi-weight mid-correlation” (bicor) as an attractive alternative correlation measure that is more robust than Pearson correlation.
Significance Threshold
When the similarity scores between all gene pairs have been determined, a cutoff is applied to select the gene pairs that should be connected in the network. This can be an arbitrary cutoff, but there are several ways to make a more informed choice. Lee et al. (2004) selected only the top 0.5% most positively and the top 0.5% most negatively correlated pairs. Bassel et al. (2011) chose a cutoff that results in a network following a power-law distribution, using the Weighted Gene Co-expression Network Analysis (WGCNA) package (Langfelder Langfelder and Horvath, 2008). Butte and Kohane (2000) used random permutations of the expression data to determine a cutoff for significant interactions. Other approaches calculate a p-value based on the null hypothesis that the correlation between two genes is 0.
Zhang and Horvath (2005) proposed to use soft thresholds instead of hard cutoffs, to produce weighted gene networks and preserve the underlying continuous nature of the correlation. However, visualizing these networks is challenging since the directly linked neighbors of a node are difficult to identify.
Promising Approaches
Correlation networks do not distinguish between direct and indirect interactions. The ARACNE algorithm (Margolin et al., 2006; Meyer et al., 2008) addresses this by pruning edges based on the analysis of gene triplets. If genes A, B, and C are fully connected in the network and the edge between A and C has the lowest weight, this edge could actually be an indirect interaction of A and C through B.
Correlation networks have undirected edges, since no causality can be inferred from two connected genes, although work has been published to address this (Opgen-Rhein and Strimmer, 2007). Regression methods are well-suited to find directed edges, since they try to find the set of genes that best predict the expression of a given target gene. However, because regression methods are generally computational demanding, the set of possible predictor genes is often limited to known transcription factors (Vignes et al., 2011; Marbach et al., 2012). In addition, Bayesian networks also allow the inclusion of prior knowledge, but their application is even more computationally challenging and not feasible for large sets of genes (Tamada et al., 2003; Imoto et al., 2004; Werhli and Husmeier, 2008).
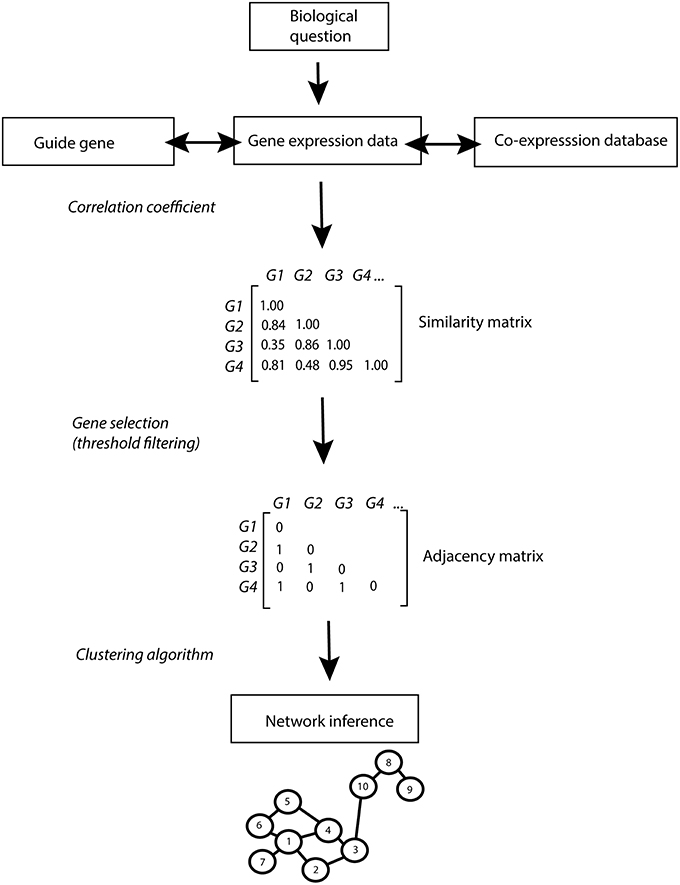
Figure 1. Co-expression network inference pipeline. The biological question addressed drives the strategy for the co-expression network analysis: prior knowledge can be used to identify guide-genes and co-expression databases can be queried to investigate gene co-expression patterns across multiple conditions. Similarity in gene expression patterns is calculated using correlation coefficients (Pearson, Spearman…). A user defined threshold (in this example set at 0.8) enables the selection of genes with high co-expression scores. Significantly co-expressed genes are reported in the binary adjacency matrix as 1. A clustering algorithm is applied on the adjacency matrix to infer networks of significantly co-expressed genes. In the resulting network, significantly co-expressed genes are depicted as numbered nodes (vertices) linked by edges (links). The length of the edges is relative to the expression similarity of the connected genes, with a short edge corresponding to a high co-expression value. A “path” corresponds to the number of edges connecting two nodes (the shortest path from node 9 to 4 is 4 edges). Hubs are identified as highly connected nodes (node 1) and group of connected genes form modules (nodes 1–7). Network properties can be described by different parameters such as:
•The connectivity of a network corresponds to the total number of links in the network.
•The node degree corresponds to the number of connections of a node with other nodes in the network (node 4 has a node degree of 3).
•The betweenness of a node corresponds to the sum of the shortest paths connecting all pair of nodes in the network, passing through that specific node. The betweenness of node 8 corresponds to the sum of the shortest path the connecting node 10–9, 3–9, 4–9 etc…).
The two main applications for co-expression network analysis are to find novel genes involved in the biological process under investigation and to suggest the biological process a gene is involved in. Intuitively, reliable networks are needed to infer meaningful gene function predictions. Such networks heavily depend on a combination of decisions taken throughout the network inference process. From the quality, type and availability of the input data, the correlation coefficient and inference algorithm used, to the prior knowledge, the experimental and computational resources, any negligence can result in unreliable networks and subsequent misleading biological interpretations.
Caveats and opportunities of co-expression network analyses have been discussed previously (Usadel et al., 2009). When handling large data sets, co-expression networks can become very complex which limits their biological interpretation (Usadel et al., 2009). In addition, in contrast to regulatory networks, and because of their static representation, co-expression networks do not provide per se information on the nature of the regulatory relationship of connected genes (Stuart et al., 2003). Careful application of network analysis tools and strategies is thus important to maximize the information extraction, to disentangle reliable network connections and to infer true biological meaning.
In this review, we aim to provide an overview of the different strategies to employ during or after the co-expression network construction with the common aim of exploiting the full predictive potential of co-expression networks. The application of these strategies is illustrated by examples of recent studies. Particular attention is given to available and promising bioinformatics tools. Finally, we will speculate on network aspects worth developing in the near future to strengthen their inference power for a comprehensive understanding of the regulation of important biological processes.
Data Availability for Co-Expression Network Analysis
In the post-genomic era, the reduction of costs for large scale and high-throughput measurement technologies, such as for transcriptomics, has to the extensive collection of gene expression profiles capturing changes in gene expression during development, between different treatments or tissues, etc.
In addition, the sequenced genomes of model plants (e.g., Arabidopsis, medicago, and poplar) and economically important crops (e.g., tomato, potato, tobacco, rice, and soybean) strongly improve our understanding of transcriptional dynamics.
The compendia of generated data led to the development of publicly available gene expression databases (Table 1). These databases still largely contain microarray data and many of them are related to the model plant Arabidopsis. In recent years, RNA-sequencing, using next-generation high-throughput sequencing technologies (RNA-seq) has proven to be a powerful tool for whole transcriptome profiling with enhanced sensitivity for the discovery of new transcripts and enhanced specificity such as for the examination of allele-specific expression. The power of these sequencing technologies has enabled co-expression network analysis in species without a sequenced genome and, as a result, has opened the way for new applications (see Section Comparative Co-expression Network Analysis). RNA-seq based co-expression network construction is still in its infancy (Iancu et al., 2012; Ballouz et al., 2015) but the foreseen predominance of next generation sequencing tools in the coming years will certainly enrich existing databases for the benefit of network studies. Microarrays are still commonly used for transcriptome analysis because they are relatively cheap and their analysis is highly standardized. Comprehensive microarray gene expression sets are available in public repositories such as the Gene Expression Omnibus (GEO, Edgar et al., 2001), Genevestigator (Hruz et al., 2008) or Array Express (Parkinson, 2004). Other tools, such as the online bio-analytical resource for plant biology (BAR, Winter et al., 2007), provide interactive interfaces for the exploratory visualization of gene expression variation.
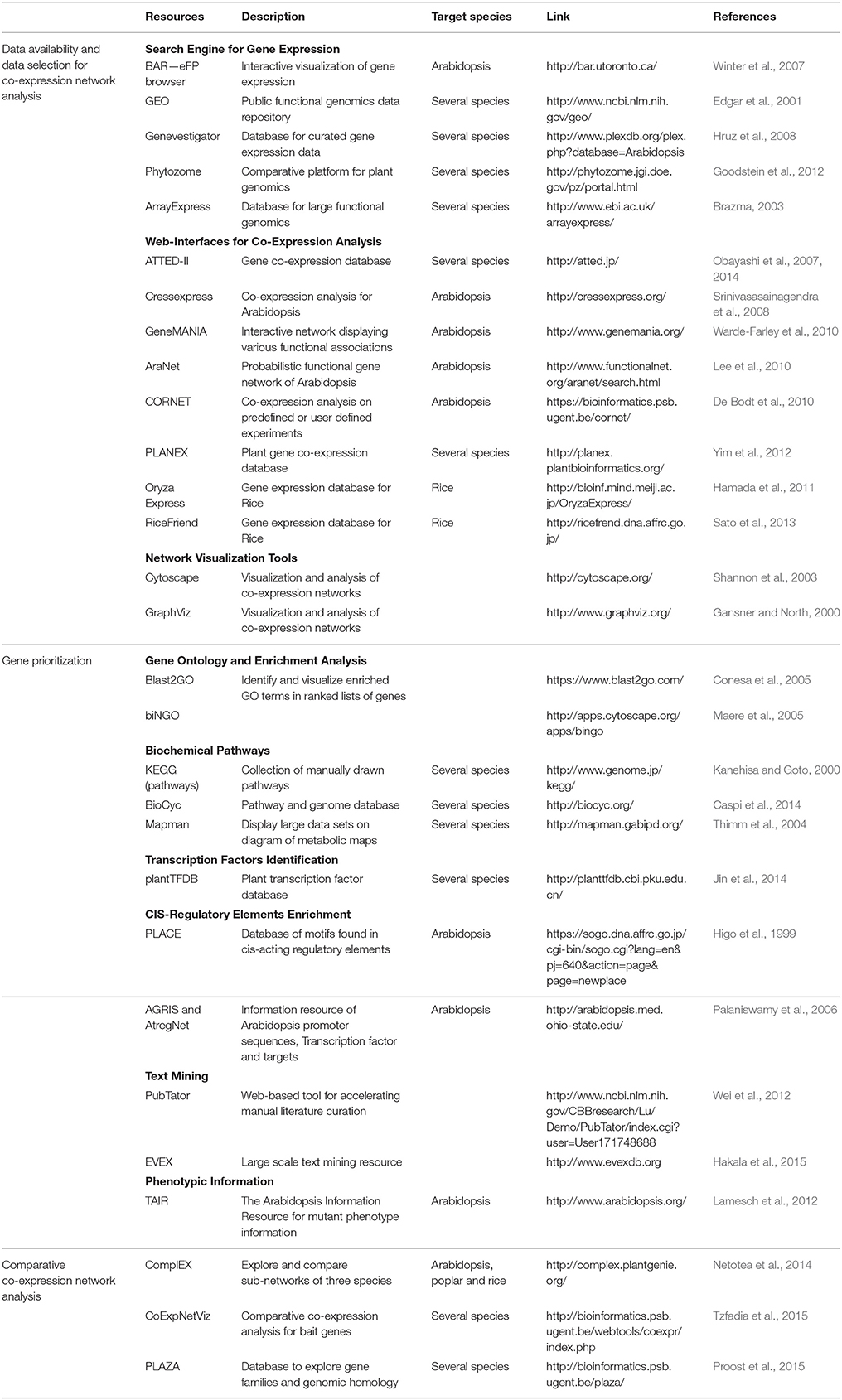
Table 1. Overview of available resources for co-expression network analysis.
Co-expression networks allow the simultaneous investigation of multiple gene co-expression patterns across a wide range of conditions. As a result, publicly available transcriptome data sets represent valuable resources for such analysis. It has been reported that nearly one in four studies uses public data to address a biological problem without generating new raw data (Rung and Brazma, 2013). The reuse of such data strengthens the need for reliable expression studies. A correct experimental design, the proper execution of the wet lab experiments and thorough annotation of the data are essential prerequisites for successful subsequent reuse (Brazma, 2003).
Several gene co-expression databases are available to help researchers in their investigations (reviewed in Brady and Provart, 2009; Usadel et al., 2009; Table 1). These databases provide user-friendly interfaces to facilitate access to the data and most of them also offer integrated data processing tools. ATTED-II (Obayashi et al., 2007, 2014) allows condition specific searches for co-expressed genes in several plant species. For Arabidopsis, CressExpress (Srinivasasainagendra et al., 2008) in addition allows selection of data sets based on a quality score to filter out “bad” microarrays. GeneMANIA (Warde-Farley et al., 2010) uses a large set of functional data of various types (predicted interactions, correlations, physical interactions and shared protein domains) to display all predicted interactions for a query gene list in an interactive network. The probabilistic functional gene network AraNet (Lee et al., 2015b) provides a measure to assess the connectivity of the query genes used in regard to the generated network. Additionally, AraNet integrates enrichment analysis tools for network components for gene ontology terms and biochemical pathways (Mapman, BioCyc and KEGG) (see Section Gene Prioritization). A popular platform for network inference is Cytoscape (Shannon et al., 2003). This open source program with its many plugins and apps allows the integration, visualization and analyses of network data (Saito et al., 2012).
Data Selection for Co-Expression Network Analysis
Publicly available gene expression databases can be queried using two main approaches. These approaches are reported in the literature as “non-targeted” (or “global”) and “targeted” (or “guided-gene”) approaches (Aoki et al., 2007). The use of one or the other approach is largely determined by the biological question addressed and the available knowledge.
The non-targeted approach provides a global overview of co-expression patterns of multiple genes across many conditions. This approach is also termed knowledge-independent or condition-independent, as no a priori information is used to construct the network. As an example, Mao et al. (2009) built an Arabidopsis gene co-expression network using gene expression data from 1094 non-redundant Affymetrix ATH1 arrays from the AtGenExpress consortium. This data set represented nine categories of experimental conditions, such as environmental stresses, hormonal treatments and developmental stages. The resulting network consisted of 6206 nodes and 512,936 edges. These “global” networks are generally used to describe the overall set of connections predicted to occur between gene pairs. Separated modules of functionally related genes can be identified and enable further gene prioritization (see Section Gene Prioritization).
In these global networks, also designated as condition-independent, weak interactions or interactions only occurring under specific conditions are easily missed. This can be circumvented by specifically selecting data from experiments that are relevant to the biological question addressed (Saito et al., 2008; Usadel et al., 2009). The resulting condition-dependent networks provide insights on specific biological processes (Atias et al., 2009). Illustratively, by selecting 138 samples from publicly available gene expression data sets exclusively from mature imbibed Arabidopsis seeds, Bassel et al. (2011) established a seed specific network. This SeedNet enabled the identification of modules associated with seed traits such as germination and dormancy. Childs et al. (2011) reported the improved predictive power for gene functional annotation of such condition-dependent networks. One of the limits of this approach is that the elucidation of system wide properties, such as intersecting biological pathways and genes exhibiting pleiotropic effects, might be overlooked.
An alternative approach allows to mimic condition-dependent data set selection, while using the full potential of gene expression data sets. This approach consists of pre-clustering the samples prior to network construction. In this case, a clustering algorithm is directly applied to the normalized expression matrix (genes × conditions) to partition the input samples into a defined number of groups based on their overall expression similarity. Co-expression networks are then built from each of the clusters obtained. Using this technique, Feltus et al. (2013) have shown that such an unsupervised pre-clustering approach improved capturing of co-expressed genes and the representation of unique biological terms in the derived network modules.
When experimental data have elucidated key components of specific pathways, a guide-gene approach can help to identify novel members of the same pathway in a more targeted manner (Itkin et al., 2013). These known genes, also called bait or seed genes, are used as input genes to build a seeded co-expression network. For example, Yang et al. (2011) used this approach to identify new candidate genes involved in cell-wall biosynthesis. They first established a list of 121 genes known to be involved in cell-wall biosynthesis and by querying available data sets with these seed genes, the initial list was extended to 694 potential candidate genes.
Strategies combining guide-gene queries and condition-dependent approaches may empower the predictive power of co-expression networks. For instance, Li et al. (2009) implemented a pipeline based on QUBIC, a QUalitative BIClustering algorithm, to select the conditions under which seed genes of the plant cell-wall biosynthesis pathway in Arabidopsis were found to be co-expressed among a total set of 351 conditions. These conditions were then used to generate networks of co-expressed gene modules.
Gene Prioritization
Once a co-expression network is obtained, biological relevant information can be mined by gene prioritization. This process consists of integrating diverse data sources to allow the ranking of the nodes in the network and to identify groups of functionally related genes, down to important putative regulatory genes. A panel of databases and tools are available to facilitate the integration of gene information in the network (Table 1).
In nature, a variety of biological networks have displayed evidence of scale-free behavior (Barabási and Oltvai, 2004; Albert, 2005; Atias et al., 2009). Such networks are characterized by a distribution of nodes following a power law distribution. Graphically, this type of network displays a relatively large number of low-connected nodes and a few nodes with a high connectivity, the so called “hubs.” Even though, the assumption of a power law distribution is stated in numerous studies, statistical analyses have also refuted this approach (Khanin and Wit, 2006; Lima-Mendez and Van Helden, 2009).
The network topology encodes preliminary evidences for the understanding of the underpinning biological organization and reveals biological relevant information on the functional importance of individual nodes (Atias et al., 2009). Parameters derived from network local properties such as clustering coefficient, node degree (number of connected nodes), betweenness and centrality are commonly used for node ranking (Pavlopoulos et al., 2011). Nodes with a higher rank, i.e., with a high degree of connection and a high clustering coefficient, are identified as major hubs and are also likely associated to essential genes in the network (Provero, 2002; Carlson et al., 2006). The phenomenon, describing the link between connectivity and essentiality is termed the “lethality-centrality rule” (Jeong et al., 2001). Several studies have associated the non-trivial topological features of scale free networks to an essential buffering system for biological networks robustness and environmental responses (Levy and Siegal, 2008; Fu et al., 2009; Lachowiec et al., 2015).
Groups of highly connected genes in a network tend to form modules. Extracting modules from the network is thus a commonly used approach to generate manageable graph subunits for further study (Aoki et al., 2007; Mao et al., 2009). For this purpose, several clustering algorithms are available. These algorithms can be categorized into hierarchical and non-hierarchical algorithms. Hierarchical clustering algorithms identify clusters by iteratively assigning nodes to clusters. In a first step, weights are assigned to the network vertices, using for instance the calculated correlation coefficient. Clusters are then built from high weight vertices and progressively expanded by including neighboring vertices. The number of final clusters varies, for instance depending on a chosen threshold. A variety of hierarchical clustering methods are available including Weighted Gene Correlation Network Analysis (WGCNA) (Langfelder and Horvath, 2008), Markov Cluster Algorithm (MCL) (Enright et al., 2002; Mao et al., 2009), Normalization Engine for Matching Organizations (NeMo) (Rivera et al., 2010) and Improved Principal Component Analysis (IPCA) (Li M. et al., 2008; Fukushima et al., 2012). Mutwil et al. (2010) suggested a novel Heuristic Cluster Chiseling Algorithm (HCCA). For each node in the network, this algorithm generates node vicinity networks by collecting all nodes within n steps away from the seed node. Non-hierarchical approaches, such as K-mean clustering (Stuart et al., 2003), identify a certain number of modules given the input cluster criteria instead.
The performance of the different clustering algorithms can be assessed by evaluating the functional coherence of the predicted modules and inform, in return, the user on the best clustering algorithm to use (Lysenko et al., 2011). MORPH, an algorithm developed by Tzfadia et al. (2012), combines a guide-gene approach with data set selection and clustering to enable finding the best combination of gene expression data and network clustering to optimally associate candidate genes with a given target pathway.
Modules are often used as the starting point for more detailed studies as they considerably reduce the global network complexity. A panel of tools can be employed to further mine these modules (Table 1). These tools enable the functional annotation of nodes and modules and to unravel the nature of the gene-gene relationships.
Enrichment analysis for the genes within a module is the most widely used technique to associate modules with particular functions. Under the “guilt-by-association” rule, these functional modules provide a powerful framework for the identification of new genes relevant to biological processes and their functional annotation in the absence of strong a priori knowledge. These enrichment analyses mostly rely on annotation databases (Table 1). The most popular ones are the gene ontology (GO) database (Ashburner et al., 2000) and manually curated databases for metabolite pathways such as the Kyoto Encyclopedia for Genes and Genomes (KEGG) (Kanehisa and Goto, 2000), Mapman (Thimm et al., 2004), or BioCyc (Caspi et al., 2014).
Phenotypic data can also be used with the a priori expectation that clustered genes collaborate to control the same phenotypic trait. For example, Mutwil et al. (2010) successfully associated an individual cluster with a specific biological function using phenotypic data and tissue-dependent expression profiles for each gene in the cluster. Similarly, Ficklin et al. (2010) used phenotypic information of rice mutant lines to identify clusters of genes enriched for mutant phenotypic terms such as “sterile” or “dwarf.” In another study, Lee et al. (2010) showed that genes whose disruption is associated with embryonic lethality and pigmentation were significantly more interlinked in the AraNet network than expected by chance, corroborating the aforementioned centrality-essentiality theory.
Other available data can help to unravel the nature of the links connecting genes in the network. Co-expression networks are undirected networks as the edges between two genes do not indicate the direction of the interaction. Additionally, the co-expression link between two connected genes might also indicate an indirect interaction. To further unravel the gene regulatory dynamics in such modules, known gene-gene interactions can be displayed on the network and help to identify gene regulatory relationships (Ulitsky and Shamir, 2009).
One of the common approaches to identify regulatory relationships is to focus on known transcription factors and their known targets in the network. As transcription factors regulate the expression of many genes in the genome, one might also expect to find them as highly connected nodes in the network or connected to hub genes. The range of interactions of a transcription factor is defined by its binding capacity to specific cis-regulatory elements (motifs) identified in the promoter region of its target genes. Consequently, the search for such motifs in the nodes located in the vicinity of identified transcription factors can be a complementary source to functionally annotate genes and infer potential gene regulatory relationships (Vandepoele et al., 2009).
In their approach, Ma et al. (2013) used a bottom-up approach by first creating sub networks of genes based on motif enrichment for specific cis-regulatory elements and then identifying co-expression modules in those sub-networks.
Gene interaction information can also be retrieved from other data sources. The development and application of genome-wide methods for detecting protein-protein interactions, such as yeast two-hybrid (Brückner et al., 2009) or affinity purification methods coupled to mass spectrometry (Morris et al., 2014) have increased available interactome data. The InterProScan (Quevillon et al., 2005) or STRING (Szklarczyk et al., 2014) databases can be investigated to retrieve known physical interactions, both structurally resolved and experimentally validated. Knowledge on genetic interactions enables further inferring of functional relationships between genes and pathways. Besides data storage in databases, information on gene function and interactions can also be found embedded in textual data (Hakala et al., 2015). Text mining methods applied to literature resources, such as PubMed articles, help to extract additional information using manual curation efforts (Szakonyi et al., 2015) or semi-automated tools such as PubTator (Wei et al., 2012).
Previously mentioned data mining approaches essentially rely on available knowledge. Ample knowledge is available for Arabidopsis, but for other less well-studied plant species, the lack of knowledge regarding gene annotation and interactions severely limits network analysis using gene prioritization. Comparing networks from different species can provide an additional source of knowledge for gene functional annotation and gene connectivity using gene orthologs information and network alignment (see Section Comparative Co-Expression Network Analysis). As an example, Lee et al. (2015a) used conserved functional gene associations from networks inferred for Arabidopsis, worm, human and yeast as an additional source of data for the RiceNet, which was initially limited to rice-specific data sets.
The availability of these complementary data has opened the way to integrated approaches for function prediction studies. Multiple independent lines of evidence provide confidence for network functional gene associations. Kourmpetis et al. (2011) employed the Bayesian Markov Random Fields (BMRF) model to integrate protein sequence information, gene expression and protein-protein interaction data in their function prediction approach in Arabidopsis. They demonstrated that the model for network integration had the best performance when all of these data sources were used. One of the best examples of data integration is provided by GeneMANIA. This prediction server relies on a Gaussian Markov Random Fields-based method for protein function prediction combining multiple networks (Warde-Farley et al., 2010).
Together with computational methods, these tools, mobilizing and integrating prior knowledge and network features, have contributed to the establishment of diverse strategies to prioritize candidate genes for further experimentation (Table 2).
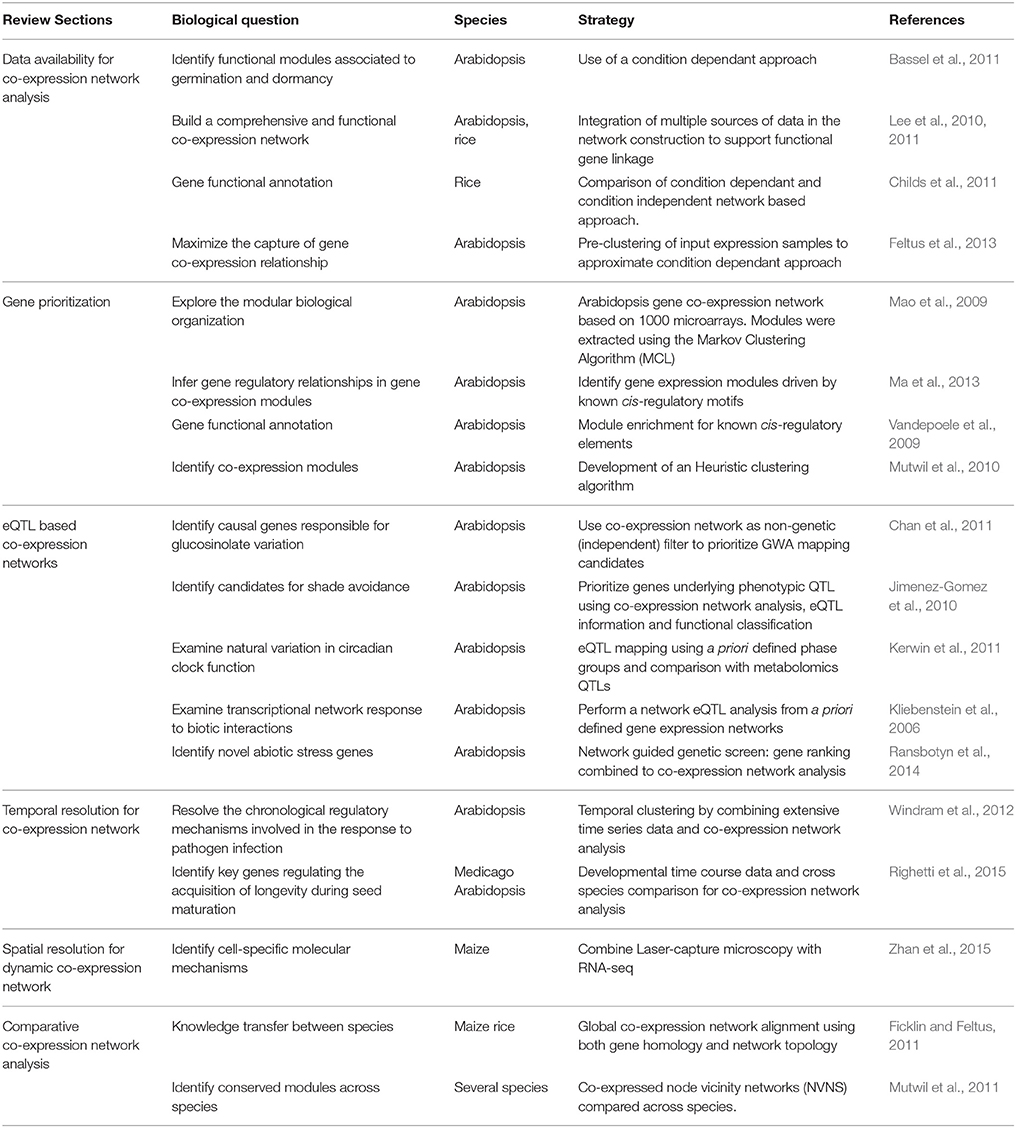
Table 2. Examples of strategies used for co-expression network analysis in regard to the respective biological question addressed.
Co-Expression Network Applications
eQTL Based Co-Expression Networks
Advances in “genetical genomics” have greatly benefited the elucidation of the genetic loci controlling transcription and the inference of regulatory mechanisms underlying complex phenotypic traits. The concept of “genetical genomics” was first introduced by Jansen and Nap in 2001 (Jansen and Nap, 2001), marking a new turn in genetic studies. The basic idea of this approach is to join classical genetic linkage analysis (Quantitative trait Loci (QTL) analysis) with gene expression studies (Keurentjes et al., 2007). The variation in gene expression is regarded as a quantitative trait for which the genetic basis (expression QTL, eQTLs) is investigated in mapping populations, such as recombinant inbred line (RIL) populations. In plants, “genetical genomics” has proven to be a successful strategy to dissect complex traits in a number of studies (for reviews see Joosen et al., 2009; Kliebenstein, 2009; Ligterink et al., 2012).
Detected eQTLs for a specific gene can be classified into “local” or “distant” eQTLs depending on whether they co-localize with the physical position of the studied gene or are located elsewhere in the genome, respectively (Rockman and Kruglyak, 2006). eQTLs can also be classified as cis- or trans-acting based on the location of the associated causal polymorphism in the gene under study or elsewhere in the genome, respectively. Consequently, distant eQTLs are always trans-acting, while local eQTLs can be cis-acting, if the associated causal polymorphism resides in the gene under study, or trans-eQTLs when they are caused by a closely linked allelic variation in a trans-acting factor. Allele specific expression analysis can specifically determine whether a local eQTL is trans or cis-acting (for review see Kliebenstein, 2009).
A common feature of global eQTL studies is the identification of trans-eQTL hotspots (Keurentjes et al., 2007; West et al., 2007). These eQTL hotspots correspond to a high number of co-locating trans-eQTLs in one region of the genome, indicating a hotspot for transcriptional regulation (Kliebenstein, 2009). Due to their analogy to high degree nodes in a network, cis-eQTLs located in these hotspots are sought as candidate master regulators affecting the expression of genes with a trans-eQTL in that same region (West et al., 2007). A regulatory relationship can be inferred by correlating gene expression profiles between the cis-eQTL candidate regulators and their potential downstream trans regulated genes. An iterative group analysis can be used to detect significant associations (Breitling et al., 2004; Keurentjes et al., 2007; Wang et al., 2014). Keurentjes et al. (2007) established a regulatory network for genes involved in the transition of flowering based on eQTL data. The GIGANTEA (GI) protein, known to be involved in the circadian clock controlled flowering time pathway, was identified as a regulator. Phenotypic QTLs associated with flowering and the circadian clock were also identified at the genetic locus of GI. Similarly, Wang et al. (2014) identified eight regulatory groups and their target genes for heading time in rice RILs. One regulatory group centered on Ghd7, an important regulator in heading time and yield potential in rice, was identified with a cis-eQTL connected to nine genes with trans-eQTLs. The network was validated by inspecting the transcript abundance of downstream-regulated targets and supported by co-localizing phenotypic QTLs for yield and heading time. These studies illustrate the usefulness of eQTL based co-expression analysis to guide the identification of candidate genes controlling quantitative traits. Other studies combined eQTL with co-expression analysis to identify regulator candidates underlying eQTLs (Terpstra et al., 2010; Flassig et al., 2013).
Interestingly, eQTL studies have also reported noteworthy properties of eQTLs in regard to their regulatory and evolutionary significance. cis-eQTLs were found to be highly inheritable with a larger genetic effect when compared to trans-eQTLs (Petretto et al., 2006; West et al., 2007; Kloosterman et al., 2012). cis-eQTLs were also found to be more consistent across different genetic backgrounds (Cubillos et al., 2012) and more robust to environmental perturbations (Cubillos et al., 2014), while genes with trans-eQTLs were more frequently reported as tissue or organ specific (Drost et al., 2010; Kloosterman et al., 2012).
QTLs tend to cover large regions of the genome, typically spanning hundreds of genes, and finding the actual gene that causes the observed trait variation is a formidable task. The capacity of gene co-expression networks to handle genome-wide data and filter out genes based on their correlation coefficients offers an attractive approach to prioritize genes. This strategy was successfully applied in the identification of EARLY FLOWERING 3 (ELF3), and its implication in shade avoidance response (Jimenez-Gomez et al., 2010). In this study, a network was built for each of the 363 candidate genes underlying the main phenotypic QTL for shade avoidance, connecting each candidate gene to co-expressed genes across 1.388 (selected) experiments. The eQTLs available for the investigated RIL population allowed pruning of the networks to keep only the co-expressed genes with a cis-eQTL, which is indicative of a regulatory relationship (Hansen et al., 2008). In a similar approach, Chan et al. (2011) used co-expression analysis to prioritize candidate genes resulting from a genome wide association study (GWAS). Alternatively, co-expression networks can be used prior to eQTL analysis (Kliebenstein et al., 2006; Kerwin et al., 2011). Kliebenstein et al. (2006) implemented an a priori network eQTL approach by calculating the mean expression value of the genes within each pre-determined network and using this as a quantitative trait in a subsequent QTL analysis.
One main advantage of eQTL analysis is that regulatory insights can be gained without prior knowledge. Information on the nature of the inferred interaction in such an approach, combined with co-expression network analysis, can substantially accelerate understanding of molecular regulatory interactions (Figure 2). However, the link between phenotype and transcript variation is not always straightforward as changes are also likely to occur at the protein or metabolite levels. The additional integration of other omics data available as QTLs for protein (pQTL) or metabolite (mQTL) variation (Wentzell et al., 2007; Kerwin et al., 2011) can bridge the gap between genotype and phenotype, providing an in-depth understanding of causal mechanisms. As an example, Kerwin et al. (2011) identified overlapping eQTLs and mQTLs for circadian time and glucosinolate variation in Arabidopsis. Specifically, AOP2, a 2-oxoglutarate-dependent dioxygenase, was identified as a potential regulator. Altered AOP2 function resulted in changes in expression of clock output genes, suggesting a causal relationship between changes in clock function and metabolite content.
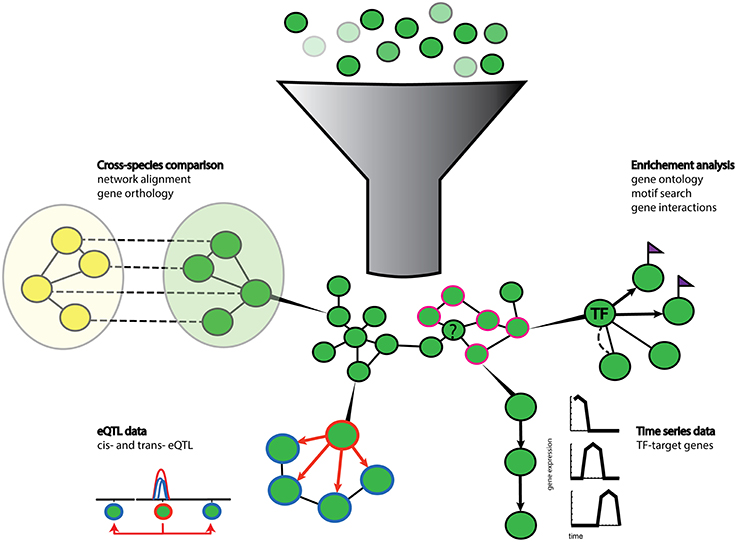
Figure 2. Schematic representation of gene prioritization strategies. Gene sets of different expression values (shades of green) are used for co-expression network inference. Genes with co-expression values above a user defined threshold (dark green nodes) form nodes and edges in the network. Various additional data can then be used to enrich and extract biological relevant information from the network. Enrichment analysis tools such as gene ontology terms (pink contour nodes) can be used to functionally annotate unknown genes (question marked node) clustered in the vicinity. Prior knowledge can also help to highlight known gene-gene interactions (dotted line) and cis-regulatory motif (purple flags) can suggest local regulatory interactions (arrows) between transcription factors (TF node) and their target genes (flagged nodes). Gene regulatory relationships can also be extracted from time series data. Algorithms can extract causal regulatory relationships from shifted gene expression patterns in time series data. Co-localization of trans- and cis-eQTLs (hotspots) can also infer regulatory relationships between genes with a cis-eQTL (orange contour node) and genes with trans-eQTLs (blue contour node). Additional information can be gained from comparisons with networks of other species (yellow nodes) by orthology and network alignment (dotted lines).
High-Resolution Co-Expression Networks
Co-expression networks offer a conceptual framework to study gene interactions. However, their static representation does not capture all possible gene relationships as these do not operate simultaneously due to spatial and temporal variation in gene expression.
Temporal Resolution for Dynamic Co-Expression Networks
In response to developmental or environmental stimuli, plants undergo global transcriptional reprogramming. Monitoring transcriptional changes over time can provide more insight into the cascade of biological processes involved in the signal perception, transduction and final response.
Using time series data sets throughout seed development, Le et al. (2010) identified seed specific transcription factors active in different compartments and tissues of the seed at unique moments of seed development, suggesting a chronology of specific regulatory programs triggering seed development.
Time series experiments are often used to examine the dynamics of gene expression. Wei et al. (2013) used six time points during growth of poplar roots in low nitrogen conditions. GO categories associated with signal transduction were identified for differentially expressed gene sets in the early time points of the response (6 and 24 h), while categories associated with organ morphogenesis were prevalent throughout the later time points (48 and 96 h). By reducing the time scale to minutes, Krouk et al. (2010) observed that within 3 min following nitrate addition in Arabidopsis, functional categories such as ribosomal proteins were over-represented, suggesting the rapid activation of key elements of the translation machinery to synthesize proteins required for nitrogen acquisition. Combining time series and co-expression network analysis can unveil gene interactions associated with the dynamics of transcriptional programs. Global expression patterns can be obtained from the expression similarity calculated across samples collected at different time points. This approach is well suited to find modules of simultaneous expressed genes and gene interactions but is not well suited for time lagged regulations since all genes influencing the expression of downstream target genes are not necessarily captured within a same time point (experiment). This results in complex relationships between co-regulated genes, including co-expression, time shifted and inverted relationships (Zhang et al., 2005): an activated transcription factor gene first has to be transcribed and the resulting mRNA translated before it in turn can activate its downstream targets. The delay further depends on the dynamics of the regulation, and for instance the presence of network motifs like feed forward or negative feedback loops (Alon, 2007).
Windram et al. (2012) dissected the infection response of Arabidopsis to Botrytis cinerea using 48 time points with 2-h intervals. To capture the chronological establishment of the associated transcriptional events and to predict their regulation, the differentially expressed genes were first clustered based on the similarity of gene expression patterns over time or based on the timing of differential expression of each gene. Regulatory predictions were made using a discrete-time causal structure identification algorithm. The expression means of the clusters and Botrytis cinerea growth information were used to build a regulatory network. In this network, a NAC transcription factor identified in one cluster connected to two downstream clusters enriched for the NAC binding motif in their promoter sequence, suggesting a regulatory relationship.
This example shows that causality information of time series on a fine temporal scale can provide valuable information on the directionality of gene interactions. Several algorithms have been proposed to perform time delayed correlation analysis in time series data (De Smet and Marchal, 2010). For instance, Lavenus et al. (2015) proposed a time delay correlation algorithm (TDCor) that includes minimal prior knowledge on the nature of the genes, with transcription factors categorized as repressor, activator, regulator or non-regulator, to build a network of plausible interactions from time series data. Krouk et al. (2010) used a noise reduction state-space modeling algorithm to build a dynamic linear model defining the rate of change in expression between time points t and t + 1. This model was then used to predict the influence of transcription factors on the genes they regulated (influential rate). The authors reasoned that the observed low influential rate of the transcription factors could be due to the functional redundancy that is often observed in biological networks and is consistent with a proposed global buffering system counteracting stresses and evolutionary forces (Fu et al., 2009). Polanski et al. (2014) suggested a module identification procedure based on the Wigwams algorithm capable of mining multiple time series for condition dependent co-expression across a subset of time series. Using such an approach, the reconstruction of co-expression networks can be directed to time specific modules of co-regulated genes.
Together, these studies suggest that new regulatory insights can be gained from integration of co-expression networks with data from time series, for the identification of “subtle” gene clusters, showing condition dependent regulation. Time series are valuable for further disentangling of real co-regulatory gene relationships from co-expression links. For application in more studies, new challenges have to be addressed such as the judicious selection of time points (Vashishtha et al., 2015), the development of performant inference algorithms, the reliable detection of direct and indirect gene interactions and most importantly the connection with their real biological meaning (reviewed by Bar-Joseph et al., 2012). We believe that this approach will offer new venues for deeper insights into the fine-tuned regulation and predictive analysis of gene expression behavior in future studies.
Spatial Resolution for Dynamic Co-Expression Networks
Plants are multicellular organisms whose vegetative and reproductive organs are composed of complex tissues and cell types. Cell differentiation is a fundamental process required to acquire cell identity and consequently ensure the correct execution of essential structural and biological functions. Genome-wide transcriptome and gene network analyses have mostly been conducted on whole plant organs, severely limiting the identification of more specific regulatory interactions occurring at the tissue or single cell level. The development of new highly selective methods has enabled the collection of expression profiles at unprecedented resolution (Nelson et al., 2008; Tang et al., 2011; Belmonte et al., 2013) offering new insights into the various biological levels of transcription regulation. As an example, laser capture microdissection (LCM) enables isolation of specific tissues at cell level while fluorescent activated cell sorting (FACS) allows separation of specific cell types expressing green fluorescent protein (GFP) under control of cell specific promoters.
These techniques were used to get insight into single cell transcriptomic data for well-studied and specialized organs such as roots or pollen (Aya et al., 2011; Becker et al., 2014; Slane et al., 2014; Efroni et al., 2015).
A fluorescent cell sorting technique was used to obtain a high-resolution map of spatiotemporal expression profiles of Arabidopsis roots (Brady et al., 2007). In this study, transcriptome analysis of root transverse sections revealed 51 dominant root radial expression patterns among which 17 showed enrichment in a single cell type, whereas 34 expression patterns were found across 2–5 cell types (Brady et al., 2007). In the same study, the longitudinal root section expression profiling to analyse different developmental stages in root cell-type formation, enabled the identification of specific expression patterns. Transcriptional changes may also occur in response to environmental shifts. Interestingly, a close link was observed between development and stress responses at the cell-type specific level in the Arabidopsis root showing developmental plasticity (Gifford et al., 2008) while adding a layer of complexity, i.e., environment specific effects, to an already intricate system. Together, these results highlight the spatiotemporal transcriptional complexity down to the cellular level and suggest cell-specific transcriptional programs.
Integrating tissue- or cell-type specific high-resolution datasets by co-expression network analysis is a promising approach for the regulatory dissection of specific biological functions. Illustratively, Zhan et al. (2015) combined LCM and RNA-seq to isolate and profile filial and maternal cell types of maize kernels at 8 days after pollination. From the resulting gene co-expression network, 18 endosperm-associated co-expression modules were identified among which 10 were found to be highly compartment- or cell-type-specific. The comparison of these spatial co-expression modules with temporally upregulated gene data sets showed that genes within co-expression modules are regulated both in time and space. Collectively, these results support the effectiveness of co-expression networks analysis to uncover the temporal and spatial organization of specific differentiation processes.
On-going developments to further improve single-cell RNA-seq analysis (Buettner et al., 2015) should strongly benefit the establishment and interpretation of specialized co-expression networks in the coming years. Furthermore, the advancement of computational tools able to manage the increasing amount of data as well as the development of robust and efficient algorithms to analyse large-scale data will be needed to tackle the increasing complexity added to gene regulatory networks.
Comparative Co-Expression Network Analysis
“Nothing in biology makes sense except in the light of evolution” (Dobzhansky, 1973).
Classic research in evolutionary developmental biology (“evo-devo”) has focused on comparative analysis with the help of mutant analysis, heterologous mutant complementation, comparative gene expression studies and phylogenetic analysis. These analyses mostly rely on gene and protein sequence information; however the increasing number of gene expression data in many different species is opening up new perspectives. Cross-species comparison of co-expression networks is a promising approach to understand the interplay between regulatory function and evolution (Movahedi et al., 2012; Hansen et al., 2014).
There are several advantages of cross-species network comparisons. Networks of well-studied plants such as Arabidopsis can enrich sparse networks, such as for crops, reducing the need of extensive functional genomic and phenomic resources. Cross-species comparison can accelerate the functional annotation of genes and the discovery of gene-gene interactions, consequently hastening the gene prioritization process for targeted mutational studies.
There is evidence that networks are shaped by major evolutionary features, such as by neo- or sub-functionalization following whole genome duplications (Conant and Wolfe, 2006; De Smet and Van De Peer, 2012). These adaptive processes may result in an evolutionary functional gene network partitioning associated with a rewiring in the gene regulatory circuitry (Conant and Wolfe, 2006). In this context, co-expression network comparison can be used to identify functionally conserved network patterns and to study their evolution.
Different methods have been proposed to compare co-expression networks. Leal et al. (2014) compared gene co-expression networks obtained for several plant species in reponse to different pathogens using a multivariate analysis. Each network was characterized by eight graph variables which were then summarized in a principal component analysis. Clustered networks identified in the principal component analysis plot suggested similar pathogen specific responses across species.
An obvious method to align networks and to get better insight into the degree of network conservation is to link orthologous genes between different species. The effectiveness of such comparative analysis essentially relies on the consistency of the orthologous information as well as the quality of the underlying co-expression networks. Orthologous gene information can be obtained through various methods (Kuzniar et al., 2008). Simple approaches use best Blast hits or reciprocal hit blast (RHB) for closely related species (Yang et al., 2011). More advanced tools such as the OrthoMCL clustering algorithm (Li et al., 2003) or OrthoFinder (Emms and Kelly, 2015) enable differentiation of true orthologous from paralogous genes. Zarrineh et al. (2011) proposed a cross-species co-clustering approach (COMODO). Network comparisons can be done at the global scale or focused on specific gene modules. In a global approach, Ficklin and Feltus (2011) used an alignment algorithm, IsoRank, that incorporates both gene homology and network topology to compare networks in rice and maize. They identified aligned modules enriched for similar functional terms, suggesting their potential evolutionary conservation.
In another study, Obertello et al. (2015) used orthologous information from OrthoMCL and BlastP, to align genes between Arabidopsis and rice co-expression networks. The authors observed that integrating rice data in an Arabidopsis network did not improve the available interaction knowledge, while Arabidopsis could substantially enrich rice network interactions. This study illustrates the usability of network comparisons to promote translational discoveries. It shows that well-known networks, such as those from model plants like Arabidopsis, can enrich more sparse networks of crops, such as rice, although Lee et al. (2011) demonstrated a higher accuracy for a rice network, RiceNet, derived from data of diverse species (with 15.5% of true positive linkages) than for a rice network derived solely from orthology with AraNet, the Arabidopsis network (with 6.5% true positive linkages).
In a more targeted approach, Yang et al. (2011) investigated conserved co-expression of cell-wall associated genes between Arabidopsis and poplar. An initial list of known cell-wall related genes was used to build a co-expression network with 22 clusters. The orthologous clusters of co-expressed genes identified in poplar did not all correlate in gene expression pattern with the clusters in Arabidopsis (gene expression pattern correlated for 9 of 22 clusters). Additionally, conserved co-expression clusters referred to plant essential biological functions, such as cell-wall formation. More comprehensively, Movahedi et al. (2011) implemented an expression context conservation score (ECC) to quantitatively estimate the degree of conservation of expression similarity between orthologous genes and their co-expression partners. The overall ECC scores revealed that for 4.630 orthologs in rice-Arabidopsis gene pairs, 77% had a conserved expression context. In another study, Netotea et al. (2014) performed an extensive examination of network properties, like node degree distribution and gene centrality, to compare co-expression networks of Arabidopsis, poplar and rice. They analyzed the degree of conservation of gene co-expression links and neighborhood (connected genes) among all orthologs in the three networks and showed that genes with high centrality, typically hubs, were significantly conserved while local regulatory motifs were relatively less well conserved across species.
Additionally, they noted that sequence similarity did not always predict gene regulation conservation. Beyond simple gene sequence comparison, the integration of co-expression networks to cross-species data provides a new dimension in evolutionary studies, revealing conservation and divergence in the regulation of genes.
At the moment, several integrative platforms are available to enquire, display and compare co-expression networks. Examples of these are PLANEX (Yim et al., 2012), ComPLex (Netotea et al., 2014), CoExpNetViz (Tzfadia et al., 2015), PLAZA (Proost et al., 2015) and the “NetworkComparer” pipeline on the PlaNet platform (Mutwil et al., 2011) that integrates genomics, transcriptomics, phenomics and ontology analyses to compare seven plant species.
Conclusion and Perspectives
Co-expression networks are a powerful approach to accelerate the elucidation of molecular mechanisms underlying important biological processes. Importantly, network based strategies are largely determined by the biological question addressed and the prior knowledge available.
We anticipate that the increase in available experimental data, driven by new molecular techniques, will enrich existing databases. In addition, the shift from microarrays to next generation high-throughput sequencing technologies will provide further insights into genome scale functional networks of many species. Together with the increased sensitivity of high-resolution technologies enabling the acquisition of cell-specific transcriptome profiles, novel biological insights can be gained. The extensive accumulation of data will require further efforts for their storage, accessibility and processing. One of the common strategies for all co-expression network studies is the integration of disparate data sources for the biological interpretation of networks. As a result, the development of integrative web interfaces such as CressInt (Chen et al., 2015) are needed to facilitate the integration of available genomics data. Furthermore, the development of computational tools, such as machine learning based algorithms, although computationally intense, will support the optimal integration and exploitation of prioritization strategies (Radivojac et al., 2013). In such a scenario, the collaboration of bioinformaticians and biologists is highly desirable and will become increasingly important.
To fully describe the link between genotype and phenotype and to understand the underlying gene regulation, coordination of networks at different molecular levels (gene, protein, metabolite) is needed (Gaudinier et al., 2015). Additionally, genetically anchored gene expression profiles (eQTLs) have proven to be powerful tools to reveal causal regulatory variants. The genetical genomics approach provides a multifactorial design to study the simultaneous effect of gene perturbations. Kliebenstein (2012) demonstrated that shallow sequencing depth in transcriptomics experiments enables capturing most of their genomic information. The result of their study suggested that 10% of the transcripts would detain more than 80% of the information present in a variety of transcriptomics experiments. In another study, Li Y. et al. (2008) introduced the generalized genetical genomics design to optimally study genetic by environment interactions. These findings suggest that there is room for improvement in the design of transcript sequencing for large-scale factorial analysis in which the size of the population studied or the number of conditions to be tested can be increased in a cost-effective manner.
Co-expression networks are an attractive framework for gene interaction analysis and offer a diverse range of applications, from the gene functional annotation to the comparison of co-expression networks across species. Improved and enriched co-expression network analyses will further empower the predictive power of networks and their translational application by circumventing the need of additional extensive functional genomic and phenomic resources. This approach will further contribute to the elucidation of important biological processes and provide a valuable predictive tool for contemporary molecular breeding and crop engineering strategies.
Author Contributions
ES wrote the manuscript. HN, HH, and WL participated in the design and critical reviewing of the manuscript.
Funding
This publication was supported by the Dutch Technology Foundation STW, which is part of the Netherlands Organization for Scientific Research (NWO), and which is partly funded by the Ministry of Economic Affairs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This publication was supported by the Dutch Technology Foundation STW, which is part of the Netherlands Organization for Scientific Research (NWO), and which is partly funded by the Ministry of Economic Affairs.
References
Albert, R. (2005). Scale-free networks in cell biology. J. Cell Sci. 118, 4947–4957. doi: 10.1242/jcs.02714
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
Aoki, K., Ogata, Y., and Shibata, D. (2007). Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol. 48, 381–390. doi: 10.1093/pcp/pcm013
Ashburner, M., Ball, C. A., Blake, J. A., Bolstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Atias, O., Chor, B., and Chamovitz, D. A. (2009). Large-scale analysis of Arabidopsis transcription reveals a basal co-regulation network. BMC Syst. Biol. 3:86. doi: 10.1186/1752-0509-3-86
Aya, K., Suzuki, G., Suwabe, K., Hobo, T., Takahashi, H., Shiono, K., et al. (2011). Comprehensive network analysis of anther-expressed genes in rice by the combination of 33 laser microdissection and 143 spatiotemporal microarrays. PLoS ONE 6:e26162. doi: 10.1371/journal.pone.0026162
Ballouz, S., Verleyen, W., and Gillis, J. (2015). Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics 31, 2123–2130. doi: 10.1093/bioinformatics/btv118
Barabási, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Bar-Joseph, Z., Gitter, A., and Simon, I. (2012). Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 13, 552–564. doi: 10.1038/nrg3244
Bassel, G. W., Lan, H., Glaab, E., Gibbs, D. J., Gerjets, T., Krasnogor, N., et al. (2011). Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc. Natl. Acad. Sci. U.S.A. 108, 9709–9714. doi: 10.1073/pnas.1100958108
Becker, J. D., Takeda, S., Borges, F., Dolan, L., and Feijó, J. A. (2014). Transcriptional profiling of Arabidopsis root hairs and pollen defines an apical cell growth signature. BMC Plant Biol. 14:197. doi: 10.1186/s12870-014-0197-3
Belmonte, M. F., Kirkbride, R. C., Stone, S. L., Pelletier, J. M., Bui, A. Q., Yeung, E. C., et al. (2013). Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc. Natl. Acad. Sci. U.S.A. 110, E435–E444. doi: 10.1073/pnas.1222061110
Brady, S. M., Orlando, D. A., Lee, J.-Y., Koch, J., Dinneny, J. R., Mace, D., et al. (2007). A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801–806 doi: 10.1126/science.1146265
Brady, S. M., and Provart, N. J. (2009). Web-queryable large-scale data sets for hypothesis generation in plant biology. Plant Cell 21, 1034–1051. doi: 10.1105/tpc.109.066050
Brazma, A. (2003). ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 31, 68–71. doi: 10.1093/nar/gkg091
Breitling, R., Armengaud, P., Amtmann, A., and Herzyk, P. (2004). Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 573, 83–92. doi: 10.1016/j.febslet.2004.07.055
Brückner, A., Polge, C., Lentze, N., Auerbach, D., and Schlattner, U. (2009). Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 10, 2763–2788. doi: 10.3390/ijms10062763
Buettner, F., Natarajan, K. N., Casale, F. P., Proserpio, V., Scialdone, A., Theis, F. J., et al. (2015). Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 33, 155–160. doi: 10.1038/nbt.3102
Butte, A. J., and Kohane, I. S. (2000). Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 5, 415–426.
Carlson, M. R., Zhang, B., Fang, Z., Mischel, P. S., Horvath, S., and Nelson, S. F. (2006). Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7:40. doi: 10.1186/1471-2164-7-40
Caspi, R., Altman, T., Billington, R., Dreher, K., Foerster, H., Fulcher, C. A., et al. (2014). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 42, D459–D471. doi: 10.1093/nar/gkt1103
Chan, E. K., Rowe, H. C., Corwin, J. A., Joseph, B., and Kliebenstein, D. J. (2011). Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 9:e1001125. doi: 10.1371/journal.pbio.1001125
Chen, X., Ernst, K., Soman, F., Borowczak, M., and Weirauch, M. T. (2015). CressInt: a user-friendly web resource for genome-scale exploration of gene regulation in Arabidopsis thaliana. Curr. Plant Biol. 3–4, 48–55. doi: 10.1016/j.cpb.2015.09.001
Childs, K. L., Davidson, R. M., and Buell, C. R. (2011). Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS ONE 6:e22196. doi: 10.1371/journal.pone.0022196
Conant, G. C., and Wolfe, K. H. (2006). Functional partitioning of yeast co-expression networks after genome duplication. PLoS Biol. 4:e109. doi: 10.1371/journal.pbio.0040109
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Costa, M. C., Righetti, K., Nijveen, H., Yazdanpanah, F., Ligterink, W., Buitink, J., et al. (2015). A gene co-expression network predicts functional genes controlling the re-establishment of desiccation tolerance in germinated Arabidopsis thaliana seeds. Planta 242, 435–449. doi: 10.1007/s00425-015-2283-7
Cubillos, F. A., Coustham, V., and Loudet, O. (2012). Lessons from eQTL mapping studies: non-coding regions and their role behind natural phenotypic variation in plants. Curr. Opin. Plant Biol. 15, 192–198. doi: 10.1016/j.pbi.2012.01.005
Cubillos, F. A., Stegle, O., Grondin, C., Canut, M., Tisné, S., Gy, I., et al. (2014). Extensive cis-regulatory variation robust to environmental perturbation in Arabidopsis. Plant Cell 26, 4298–4310. doi: 10.1105/tpc.114.130310
De Bodt, S., Carvajal, D., Hollunder, J., Van Den Cruyce, J., Movahedi, S., and Inzé, D. (2010). CORNET: a user-friendly tool for data mining and integration. Plant Physiol. 152, 1167–1179. doi: 10.1104/pp.109.147215
De Smet, R., and Marchal, K. (2010). Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8, 717–729. doi: 10.1038/nrmicro2419
De Smet, R., and Van De Peer, Y. (2012). Redundancy and rewiring of genetic networks following genome-wide duplication events. Curr. Opin. Plant Biol. 15, 168–176. doi: 10.1016/j.pbi.2012.01.003
Dobzhansky, T. (1973). Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 35, 125–129. doi: 10.2307/4444260
Drost, D. R., Benedict, C. I., Berg, A., Novaes, E., Novaes, C. R., Yu, Q., et al. (2010). Diversification in the genetic architecture of gene expression and transcriptional networks in organ differentiation of Populus. Proc. Natl. Acad. Sci. U.S.A. 107, 8492–8497. doi: 10.1073/pnas.0914709107
Edgar, R., Domrachev, M., and Lash, A. E. (2001). Gene expression omnibus: NCBI gene hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Efroni, I., Ip, P. L., Nawy, T., Mello, A., and Birnbaum, K. D. (2015). Quantification of cell identity from single-cell gene expression profiles. Genome Biol. 16, 9. doi: 10.1186/s13059-015-0580-x
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Feltus, F. A., Ficklin, S. P., Gibson, S. M., and Smith, M. C. (2013). Maximising capture of gene co-expression network relationships through pre-clustering of input expression samples: an Arabidopsis case study. BMC Syst. Biol. 7:44. doi: 10.1186/1752-0509-7-44
Ficklin, S. P., and Feltus, F. A. (2011). Gene coexpression network alignment and conservation of gene modules between two grass species: maize and rice. Plant Physiol. 156, 1244–1256. doi: 10.1104/pp.111.173047
Ficklin, S. P., Luo, F., and Feltus, F. A. (2010). The association of multiple interacting genes with specific phenotypes in rice using gene coexpression networks. Plant Physiol. 154, 13–24. doi: 10.1104/pp.110.159459
Flassig, R. J., Heise, S., Sundmacher, K., and Klamt, S. (2013). An effective framework for reconstructing gene regulatory networks from genetical genomics data. Bioinformatics 29, 246–254. doi: 10.1093/bioinformatics/bts679
Fu, J., Keurentjes, J. J., Bouwmeester, H., America, T., Verstappen, F. W., Ward, J. L., et al. (2009). System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat. Genet. 41, 166–167. doi: 10.1038/ng.308
Fukushima, A., Nishizawa, T., Hayakumo, M., Hikosaka, S., Saito, K., Goto, E., et al. (2012). Exploring tomato gene functions based on coexpression modules using graph clustering and differential coexpression approaches. Plant Physiol. 158, 1487–1502. doi: 10.1104/pp.111.188367
Gansner, E. R., and North, S. C. (2000). An open graph visualization system and its applications to software engineering. Softw. Pract. Exp. 30, 1203–1233. doi: 10.1002/1097-024X(200009)
Gaudinier, A., Tang, M., and Kliebenstein, D. J. (2015). Transcriptional networks governing plant metabolism. Curr. Plant Biol. 3–4, 56–64. doi: 10.1016/j.cpb.2015.07.002
Gifford, M. L., Dean, A., Gutierrez, R. A., Coruzzi, G. M., and Birnbaum, K. D. (2008). Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad. Sci. U.S.A. 105, 803–808. doi: 10.1073/pnas.0709559105
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Hakala, K., Van Landeghem, S., Salakoski, T., Van De Peer, Y., and Ginter, F. (2015). Application of the EVEX resource to event extraction and network construction: Shared Task entry and result analysis. BMC Bioinformatics 16(suppl. 16):S3. doi: 10.1186/1471-2105-16-S16-S3
Hamada, K., Hongo, K., Suwabe, K., Shimizu, A., Nagayama, T., Abe, R., et al. (2011). OryzaExpress: an integrated database of gene expression networks and omics annotations in rice. Plant Cell Physiol. 52, 220–229. doi: 10.1093/pcp/pcq195
Hansen, B. G., Halkier, B. A., and Kliebenstein, D. J. (2008). Identifying the molecular basis of QTLs: eQTLs add a new dimension. Trends Plant Sci. 13, 72–77. doi: 10.1016/j.tplants.2007.11.008
Hansen, B. O., Vaid, N., Musialak-Lange, M., Janowski, M., and Mutwil, M. (2014). Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 5:394. doi: 10.3389/fpls.2014.00394
Higo, K., Ugawa, Y., Iwamoto, M., and Koregana, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Hruz, T., Laule, O., Szabo, G., Wessendorp, F., Bleuler, S., Oertle, L., et al. (2008). Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinformatics 2008, 1–5. doi: 10.1155/2008/420747
Iancu, O. D., Kawane, S., Bottomly, D., Searles, R., Hitzemann, R., and McWeeney, S. (2012). Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics 28, 1592–1597. doi: 10.1093/bioinformatics/bts245
Imoto, S., Higuchi, T., Goto, T., Tashiro, K., Kuhara, S., and Miyano, S. (2004). Combining microarrays and biological knowledge for estimating gene networks via bayesian networks. J. Bioinform. Comput. Biol. 2, 77–98. doi: 10.1142/S021972000400048X
Itkin, M., Heinig, U., Tzfadia, O., Bhide, A. J., Shinde, B., Carnedas, P. D., et al. (2013). Biosynthesis of Antinutritional alkaloids in Solanaceaous crops is mediated by clustered genes. Science 341, 175–179. doi: 10.1126/science.1240230
Jansen, R. C., and Nap, J.-P. (2001). Genetical genomics: the added value from segregation. Trends Genet. 11, 388–391. doi: 10.1016/S0168-9525(01)02310-1
Jeong, H., Mason, S. P., Barabasi, A. L., and Oltvai, Z. N. (2001). Lethality and centrality in protein networks. Nature 441, 41–42. doi: 10.1038/35075138
Jiménez-Gómez, J. M. (2014). Network types and their application in natural variation studies in plants. Curr. Opin. Plant Biol. 18, 80–86. doi: 10.1016/j.pbi.2014.02.010
Jimenez-Gomez, J. M., Wallace, A. D., and Maloof, J. N. (2010). Network analysis identifies ELF3 as a QTL for the shade avoidance response in Arabidopsis. PLoS Genet. 6:e1001100. doi: 10.1371/journal.pgen.1001100
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Joosen, R. V., Ligterink, W., Hilhorst, H. W., and Keurentjes, J. J. (2009). Advances in genetical genomics of plants. Curr. Genomics 10, 540–549. doi: 10.2174/138920209789503914
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kerwin, R. E., Jimenez-Gomez, J. M., Fulop, D., Harmer, S. L., Maloof, J. N., and Kliebenstein, D. J. (2011). Network quantitative trait loci mapping of circadian clock outputs identifies metabolic pathway-to-clock linkages in Arabidopsis. Plant Cell 23, 471–485. doi: 10.1105/tpc.110.082065
Keurentjes, J. J., Fu, J., Terpstra, I. R., Garcia, J. M., Van Den Ackerveken, G., Snoek, L. B., et al. (2007). Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. U.S.A. 104, 1708–1713. doi: 10.1073/pnas.0610429104
Khanin, R., and Wit, E. (2006). How scale-free networks are biological networks. J. Comput. Biol. 13, 810–818. doi: 10.1089/cmb.2006.13.810
Kliebenstein, D. (2009). Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol. 60, 93–114. doi: 10.1146/annurev.arplant.043008.092114
Kliebenstein, D. J. (2012). Exploring the shallow end; estimating information content in transcriptomics studies. Front. Plant Sci. 3:213. doi: 10.3389/fpls.2012.00213
Kliebenstein, D. J., West, M. A., Van Leeuwen, H., Loudet, O., Doerge, R. W., and St Clair, D. A. (2006). Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinformatics 7:308. doi: 10.1186/1471-2105-7-308
Kloosterman, B., Anithakumari, A. M., Chibon, P. Y., Oortwijn, M., Van Der Linden, G. C., Visser, R. G., et al. (2012). Organ specificity and transcriptional control of metabolic routes revealed by expression QTL profiling of source–sink tissues in a segregating potato population. BMC Plant Biol. 12:17. doi: 10.1186/1471-2229-12-17
Kourmpetis, Y. A., Van Dijk, A. D., Van Ham, R. C., and Ter Braak, C. J. (2011). Genome-wide computational function prediction of Arabidopsis proteins by integration of multiple data sources. Plant Physiol. 155, 271–281. doi: 10.1104/pp.110.162164
Krouk, G., Mirowski, P., Lecun, Y., Shasha, D. E., and Coruzzi, G. M. (2010). Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 11:R123. doi: 10.1186/gb-2010-11-12-r123
Kuzniar, A., Van Ham, R. C., Pongor, S., and Leunissen, J. A. (2008). The quest for orthologs: finding the corresponding gene across genomes. Trends Genet. 24, 539–551. doi: 10.1016/j.tig.2008.08.009
Lachowiec, J., Queitsch, C., and Kliebenstein, D. J. (2015). Molecular mechanisms governing differential robustness of development and environmental responses in plants. Ann. Bot. doi: 10.1093/aob/mcv151. [Epub ahead of print].
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., et al. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210. doi: 10.1093/nar/gkr1090
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lavenus, J., Goh, T., Guyomarc'h, S., Hill, K., Lucas, M., Voss, U., et al. (2015). Inference of the Arabidopsis lateral root gene regulatory network suggests a bifurcation mechanism that defines primordia flanking and central zones. Plant Cell 27, 1368–1388. doi: 10.1105/tpc.114.132993
Le, B. H., Cheng, C., Bui, A. Q., Wagmaister, J. A., Henry, K. F., Pelletier, J., et al. (2010). Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 8063–8070. doi: 10.1073/pnas.1003530107
Leal, L. G., López, C., and López-Kleine, L. (2014). Construction and comparison of gene co-expression networks shows complex plant immune responses. PeerJ 2:e610. doi: 10.7717/peerj.610
Lee, H. K., Hsu, A. K., Sajdak, J., Qin, J., and Pavlidis, P. (2004). Coexpression analysis of human genes across many microarray data sets. Genome Res. 14, 1085–1094. doi: 10.1101/gr.1910904
Lee, I., Ambaru, B., Thakkar, P., Marcotte, E. M., and Rhee, S. Y. (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat. Biotechnol. 28, 149–156. doi: 10.1038/nbt.1603
Lee, I., Seo, Y.-S., Coltrane, D., Hwang, S., Oh, T., Marcotte, E. M., et al. (2011). Genetic dissection of the biotic stress response using a genome-scale gene network for rice. Proc. Natl. Acad. Sci. U.S.A. 108, 18548–18553. doi: 10.1073/pnas.1110384108
Lee, T., Oh, T., Yang, S., Shin, J., Hwang, S., Kim, C. Y., et al. (2015a). RiceNet v2: an improved network prioritization server for rice genes. Nucleic Acids Res. 43, W122–W127. doi: 10.1093/nar/gkv253
Lee, T., Yang, S., Kim, E., Ko, Y., Hwang, S., Shin, J., et al. (2015b). AraNet v2: an improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucleic Acids Res. 43, D996–D1002. doi: 10.1093/nar/gku1053
Levy, S. F., and Siegal, M. L. (2008). Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol. 6:e264. doi: 10.1371/journal.pbio.0060264
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, G., Ma, Q., Tang, H., Paterson, A. H., and Xu, Y. (2009). QUBIC: a qualitative biclustering algorithm for analyses of gene expression data. Nucleic Acids Res. 37:e101–e101. doi: 10.1093/nar/gkp491
Li, M., Chen, J. E., Wang, J. X., Hu, B., and Chen, G. (2008). Modifying the DPClus algorithm for identifying protein complexes based on new topological structures. BMC Bioinformatics 9:398. doi: 10.1186/1471-2105-9-398
Li, Y., Breitling, R., and Jansen, R. C. (2008). Generalizing genetical genomics: getting added value from environmental perturbation. Trends Genet. 24, 518–524. doi: 10.1016/j.tig.2008.08.001
Li, Y., Pearl, S. A., and Jackson, S. A. (2015). Gene networks in plant biology: approaches in reconstruction and analysis. Trends Plant Sci. 20, 664–675. doi: 10.1016/j.tplants.2015.06.013
Ligterink, W., Joosen, R. V. L., and Hilhorst, H. W. M. (2012). Unravelling the complex trait of seed quality: using natural variation through a combination of physiology, genetics and -omics technologies. Seed Sci. Res. 22, S45–S52. doi: 10.1017/S0960258511000328
Lima-Mendez, G., and Van Helden, J. (2009). The powerful law of the power law and other myths in network biology. Mol. Biosyst. 5, 1482–1493. doi: 10.1039/b908681a
Liseron-Monfils, C., and Ware, D. (2015). Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 3–4, 30–39. doi: 10.1016/j.cpb.2015.11.001
Lysenko, A., Defoin-Platel, M., Hassani-Pak, K., Taubert, J., Hodgman, C., Rawlings, C. J., et al. (2011). Assessing the functional coherence of modules found in multiple-evidence networks from Arabidopsis. BMC Bioinformatics 12:203. doi: 10.1186/1471-2105-12-203
Ma, S., Shah, S., Bohnert, H. J., Snyder, M., and Dinesh-Kumar, S. P. (2013). Incorporating motif analysis into gene co-expression network reveals novel modular expression pattern and new signaling pathways. PLoS Genet 9:e1003840. doi: 10.1371/journal.pgen.1003840
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Mao, L., Van Hemert, J. L., Dash, S., and Dickerson, J. A. (2009). Arabidopsis gene co-expression network and its functional modules. BMC Bioinformatics 10:346. doi: 10.1186/1471-2105-10-346
Marbach, D., Costello, J. C., Küffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., et al. (2012). Wisdom of crowds for robust gene network inference. Nat. Meth. 9, 796–804. doi: 10.1038/nmeth.2016
Margolin, A. A., Nemenman, I., Basso, K., Wiggins, C., Stolovitzky, G., Favera, R. D., et al. (2006). ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7:S7. doi: 10.1186/1471-2105-7-S1-S7
Meyer, P. E., Lafitte, F., and Bontempi, G. (2008). minet: AR/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics 9:461. doi: 10.1186/1471-2105-9-461
Mochida, K., and Shinozaki, K. (2011). Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol. 52, 2017–2038. doi: 10.1093/pcp/pcr153
Morris, J. H., Knudsen, G. M., Verschueren, E., Johnson, J. R., Cimermancic, P., Greninger, A. L., et al. (2014). Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 9, 2539–2554. doi: 10.1038/nprot.2014.164
Movahedi, S., Van Bel, M., Heyndrickx, K. S., and Vandepoele, K. (2012). Comparative co-expression analysis in plant biology. Plant Cell Environ. 35, 1787–1798. doi: 10.1111/j.1365-3040.2012.02517.x
Movahedi, S., Van De Peer, Y., and Vandepoele, K. (2011). Comparative network analysis reveals that tissue specificity and gene function are important factors influencing the mode of expression evolution in Arabidopsis and rice. Plant Physiol. 156, 1316–1330. doi: 10.1104/pp.111.177865
Mutwil, M., Klie, S., Tohge, T., Giorgi, F. M., Wilkins, O., Campbell, M. M., et al. (2011). PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23, 895–910. doi: 10.1105/tpc.111.083667
Mutwil, M., Usadel, B., Schütte, M., Loraine, A., Ebenhöh, O., and Persson, S. (2010). Assembly of an interactive correlation network for the Arabidopsis genome using a novel heuristic clustering algorithm. Plant Physiol. 152, 29–43. doi: 10.1104/pp.109.145318
Nelson, T., Gandotra, N., and Tausta, S. L. (2008). Plant cell types: reporting and sampling with new technologies. Curr. Opin. Plant Biol. 11, 567–573. doi: 10.1016/j.pbi.2008.06.006
Netotea, S., Sundell, D., Street, N. R., and Hvidsten, T. R. (2014). ComPlEx: conservation and divergence of co-expression networks in A. thaliana, Populus and O. sativa. BMC Genomics 15:106. doi: 10.1186/1471-2164-15-106
Obayashi, T., Kinoshita, K., Nakai, K., Shibaoka, M., Hayashi, S., Saeki, M., et al. (2007). ATTED-II: a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 35, D863–D869. doi: 10.1093/nar/gkl783
Obayashi, T., Okamura, Y., Ito, S., Tadaka, S., Aoki, Y., Shirota, M., et al. (2014). ATTED-II in 2014: evaluation of gene coexpression in agriculturally important plants. Plant Cell Physiol. 55, e6. doi: 10.1093/pcp/pct178
Obertello, M., Shrivastava, S., Katari, M. S., and Coruzzi, G. M. (2015). Cross-species network analysis uncovers conserved nitrogen-regulated network modules in rice. Plant Physiol. 168, 1830–1843. doi: 10.1104/pp.114.255877
Opgen-Rhein, R., and Strimmer, K. (2007). From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst. Biol. 1:37. doi: 10.1186/1752-0509-1-37
Palaniswamy, S. K., James, S., Sun, H., Lamb, R. S., Davuluri, R. V., and Grotewold, E. (2006). AGRIS and AtRegNet. A platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol 140, 818–829. doi: 10.1104/pp.105.072280
Parkinson, H. (2004). ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 33, D553–D555. doi: 10.1093/nar/gki056
Pavlopoulos, G. A., Secrier, M., Moschopoulos, C. N., Soldatos, T. G., Kossida, S., Aerts, J., et al. (2011). Using graph theory to analyze biological networks. BioData Min. 4:10. doi: 10.1186/1756-0381-4-10
Petretto, E., Mangion, J., Dickens, N. J., Cook, S. A., Kumaran, M. K., Lu, H., et al. (2006). Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2:e172. doi: 10.1371/journal.pgen.0020172
Polanski, K., Rhodes, J., Hill, C., Zhang, P., Jenkins, D. J., Kiddle, S. J., et al. (2014). Wigwams: identifying gene modules co-regulated across multiple biological conditions. Bioinformatics 30, 962–970. doi: 10.1093/bioinformatics/btt728
Proost, S., Van Bel, M., Vaneechoutte, D., Van De Peer, Y., Inzé, D., Mueller-Roeber, B., et al. (2015). PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 43, D974–D981. doi: 10.1093/nar/gku986
Provero, P. (2002). Gene networks from DNA microarray data: centrality and lethality. arXiv preprint cond-mat/0207345.
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Radivojac, P., Clark, W. T., Oron, T. R., Schnoes, A. M., Wittkop, T., Sokolov, A., et al. (2013). A large-scale evaluation of computational protein function prediction. Nat. Methods 10, 221–227. doi: 10.1038/nmeth.2340
Ransbotyn, V., Yeger-Lotem, E., Basha, O., Acuna, T., Verduyn, C., Gordon, M., et al. (2014). A combination of gene expression ranking and co-expression network analysis increases discovery rate in large-scale mutant screens for novel Arabidopsis thaliana abiotic stress genes. Plant Biotechnol. J. 13, 501–513. doi: 10.1111/pbi.12274
Rhee, S. Y., and Mutwil, M. (2014). Towards revealing the functions of all genes in plants. Trends Plant Sci. 19, 212–221. doi: 10.1016/j.tplants.2013.10.006
Righetti, K., Vu, J. L., Pelletier, S., Vu, B. L., Glaab, E., Lalanne, D., et al. (2015). Inference of longevity-related genes from a robust coexpression network of seed maturation identifies regulators linking seed storability to biotic defense-related pathways. Plant Cell 27, 2692–2708. doi: 10.1105/tpc.15.00632
Rivera, C. G., Vakil, R., and Bader, J. S. (2010). NeMo: network Module identification in Cytoscape. BMC Bioinformatics 11(suppl. 1):S61. doi: 10.1186/1471-2105-11-S1-S61
Rockman, M. V., and Kruglyak, L. (2006). Genetics of global gene expression. Nat. Rev. Genet. 7, 862–872. doi: 10.1038/nrg1964
Rung, J., and Brazma, A. (2013). Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 14, 89–99. doi: 10.1038/nrg3394
Saito, K., Hirai, M. Y., and Yonekura-Sakakibara, K. (2008). Decoding genes with coexpression networks and metabolomics - ‘majority report by precogs’. Trends Plant Sci. 13, 36–43. doi: 10.1016/j.tplants.2007.10.006
Saito, R., Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., Lotia, S., et al. (2012). A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076. doi: 10.1038/nmeth.2212
Sato, Y., Namiki, N., Takehisa, H., Kamatsuki, K., Minami, H., Ikawa, H., et al. (2013). RiceFREND: a platform for retrieving coexpressed gene networks in rice. Nucleic Acids Res. 41, D1214–D1221. doi: 10.1093/nar/gks1122
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Silva, A. T., Ribone, P. A., Chan, R. L., Ligterink, W., and Hilhorst, H. W. (2016). A predictive co-expression network identifies novel genes controlling the seed-to-seedling phase transition in Arabidopsis thaliana. Plant Physiol. 170, 2218–2231. doi: 10.1104/pp.15.01704
Slane, D., Kong, J., Berendzen, K. W., Kilian, J., Henschen, A., Kolb, M., et al. (2014). Cell type-specific transcriptome analysis in the early Arabidopsis thaliana embryo. Development 141, 4831–4840. doi: 10.1242/dev.116459
Song, L., Langfelder, P., and Horvath, S. (2012). Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinformatics 13:328. doi: 10.1186/1471-2105-13-328
Srinivasasainagendra, V., Page, G. P., Mehta, T., Coulibaly, I., and Loraine, A. E. (2008). CressExpress: a tool for large-scale mining of expression data from Arabidopsis. Plant Physiol. 147, 1004–1016. doi: 10.1104/pp.107.115535
Stuart, J. M., Segal, E., Koller, D., and Kim, S. K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. doi: 10.1126/science.1087447
Szakonyi, D., Van Landeghem, S., Baerenfaller, K., Baeyens, L., Blomme, J., Casanova-Sáez, R., et al. (2015). The KnownLeaf literature curation system captures knowledge about Arabidopsis leaf growth and development and facilitates integrated data mining. Curr. Plant Biol. 2, 1–11. doi: 10.1016/j.cpb.2014.12.002
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2014). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Tamada, Y., Kim, S., Bannai, H., Imoto, S., Tashiro, K., Kuhara, S., et al. (2003). Estimating gene networks from gene expression data by combining Bayesian network model with promoter element detection. Bioinformatics 19, ii227–ii236. doi: 10.1093/bioinformatics/btg1082
Tang, F., Lao, K., and Surani, M. A. (2011). Development and applications of single-cell transcriptome analysis. Nat. Methods 8, S6–S11. doi: 10.1038/nmeth.1557
Terpstra, I. R., Snoek, L. B., Keurentjes, J. J., Peeters, A. J., and Van Den Ackerveken, G. (2010). Regulatory network identification by genetical genomics: signaling downstream of the Arabidopsis receptor-like kinase ERECTA. Plant Physiol. 154, 1067–1078. doi: 10.1104/pp.110.159996
Thimm, O., Bläsing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313X.2004.02016.x
Tzfadia, O., Amar, D., Bradbury, L. M., Wurtzel, E. T., and Shamir, R. (2012). The MORPH algorithm: ranking candidate genes for membership in Arabidopsis and tomato pathways. Plant Cell 24, 4389–4406. doi: 10.1105/tpc.112.104513
Tzfadia, O., Diels, T., De Meyer, S., Vandepoele, K., Aharoni, A., and Van De Peer, Y. (2015). CoExpNetViz: comparative co-expression networks construction and visualization tool. Front. Plant Sci. 6:1194. doi: 10.3389/fpls.2015.01194
Ulitsky, I., and Shamir, R. (2009). Identifying functional modules using expression profiles and confidence-scored protein interactions. Bioinformatics 25, 1158–1164. doi: 10.1093/bioinformatics/btp118
Usadel, B., Obayashi, T., Mutwil, M., Giorgi, F. M., Bassel, G. W., Tanimoto, M., et al. (2009). Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ. 32, 1633–1651. doi: 10.1111/j.1365-3040.2009.02040.x
Vandepoele, K., Quimbaya, M., Casneuf, T., De Veylder, L., and Van De Peer, Y. (2009). Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol. 150, 535–546. doi: 10.1104/pp.109.136028
Vashishtha, S., Broderick, G., Craddock, T. J., Fletcher, M. A., and Klimas, N. G. (2015). Inferring broad regulatory biology from time course data: have we reached an upper bound under constraints typical of in vivo studies? PLoS ONE 10:e0127364. doi: 10.1371/journal.pone.0127364
Vignes, M., Vandel, J., Allouche, D., Ramadan-Alban, N., Cierco-Ayrolles, C., Schiex, T., et al. (2011). Gene regulatory network reconstruction using bayesian networks, the dantzig selector, the lasso and their meta-analysis. PLoS ONE 6:e29165. doi: 10.1371/journal.pone.0029165
Wang, J., Yu, H., Weng, X., Xie, W., Xu, C., Li, X., et al. (2014). An expression quantitative trait loci-guided co-expression analysis for constructing regulatory network using a rice recombinant inbred line population. J. Exp. Bot. 65, 1069–1079. doi: 10.1093/jxb/ert464
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P., et al. (2010). The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220. doi: 10.1093/nar/gkq537
Wei, C. H., Harris, B. R., Li, D., Berardini, T. Z., Huala, E., Kao, H. Y., et al. (2012). Accelerating literature curation with text-mining tools: a case study of using PubTator to curate genes in PubMed abstracts. Database 2012:bas041. doi: 10.1093/database/bas041
Wei, H., Yordanov, Y. S., Georgieva, T., Li, X., and Busov, V. (2013). Nitrogen deprivation promotes Populus root growth through global transcriptome reprogramming and activation of hierarchical genetic networks. New Phytol. 200, 483–497. doi: 10.1111/nph.12375
Wentzell, A. M., Rowe, H. C., Hansen, B. G., Ticconi, C., Halkier, B. A., and Kliebenstein, D. J. (2007). Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet. 3, 1687–1701. doi: 10.1371/journal.pgen.0030162
Werhli, A. V., and Husmeier, D. (2008). Gene regulatory network reconstruction by bayesian integration of prior knowledge and/or different experimental conditions. J. Bioinform. Comput. Biol. 6, 543–572. doi: 10.1142/S0219720008003539
West, M. A., Kim, K., Kliebenstein, D. J., Van Leeuwen, H., Michelmore, R. W., Doerge, R. W., et al. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175, 1441–1450. doi: 10.1534/genetics.106.064972
Windram, O., Madhou, P., McHattie, S., Hill, C., Hickman, R., Cooke, E., et al. (2012). Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell 24, 3530–3557. doi: 10.1105/tpc.112.102046
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2:e718. doi: 10.1371/journal.pone.0000718
Wolfe, C. J., Kohane, I. S., and Butte, A. J. (2005). Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics 6:227. doi: 10.1186/1471-2105-6-227
Yang, X., Ye, C. Y., Bisaria, A., Tuskan, G. A., and Kalluri, U. C. (2011). Identification of candidate genes in Arabidopsis and Populus cell wall biosynthesis using text-mining, co-expression network analysis and comparative genomics. Plant Sci. 181, 675–687. doi: 10.1016/j.plantsci.2011.01.020
Yim, W. C., Yu, Y., Song, K., Jang, C. S., and Lee, B.-M. (2012). PLANEX: the plant co-expression database. BMC Plant Biol. 13:83. doi: 10.1186/1471-2229-13-83
Zarrineh, P., Fierro, A. C., Sánchez-Rodríguez, A., De Moor, B., Engelen, K., and Marchal, K. (2011). COMODO: an adaptive coclustering strategy to identify conserved coexpression modules between organisms. Nucleic Acids Res. 39, e41. doi: 10.1093/nar/gkq1275
Zhan, J., Thakare, D., Ma, C., Lloyd, A., Nixon, N. M., Arakaki, A. M., et al. (2015). RNA sequencing of laser-capture microdissected compartments of the maize kernel identifies regulatory modules associated with endosperm cell differentiation. Plant Cell 27, 513–531. doi: 10.1105/tpc.114.135657
Zhang, B., and Horvath, S. (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17. doi: 10.2202/1544-6115.1128
Keywords: co-expression, gene expression, gene networks, gene prioritization, transcriptomics
Citation: Serin EAR, Nijveen H, Hilhorst HWM and Ligterink W (2016) Learning from Co-expression Networks: Possibilities and Challenges. Front. Plant Sci. 7:444. doi: 10.3389/fpls.2016.00444
Received: 14 January 2016; Accepted: 21 March 2016;
Published: 08 April 2016.
Edited by:
David Toubiana, Ben Gurion University, IsraelReviewed by:
Staffan Persson, University of Melbourne, AustraliaOren Tzfadia, Weizmann Institute of Science, Israel
Copyright © 2016 Serin, Nijveen, Hilhorst and Ligterink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilco Ligterink, d2lsY28ubGlndGVyaW5rQHd1ci5ubA==