 Nanette Christie
Nanette Christie Peri A. Tobias
Peri A. Tobias Sanushka Naidoo1
Sanushka Naidoo1 Carsten Külheim
Carsten Külheim- 1Department of Genetics, Forestry and Agricultural Biotechnology Institute, Genomics Research Institute, University of Pretoria, Pretoria, South Africa
- 2Department of Plant and Food Sciences, Faculty of Agriculture and Environment, University of Sydney, NSW, Australia
- 3Research School of Biology, College of Medicine, Biology and Environment, Australian National University, Canberra, ACT, Australia
Eucalyptus grandis is a commercially important hardwood species and is known to be susceptible to a number of pests and pathogens. Determining mechanisms of defense is therefore a research priority. The published genome for E. grandis has aided the identification of one important class of resistance (R) genes that incorporate nucleotide binding sites and leucine-rich repeat domains (NBS-LRR). Using an iterative search process we identified NBS-LRR gene models within the E. grandis genome. We characterized the gene models and identified their genomic arrangement. The gene expression patterns were examined in E. grandis clones, challenged with a fungal pathogen (Chrysoporthe austroafricana) and insect pest (Leptocybe invasa). One thousand two hundred and fifteen putative NBS-LRR coding sequences were located which aligned into two large classes, Toll or interleukin-1 receptor (TIR) and coiled-coil (CC) based on NB-ARC domains. NBS-LRR gene-rich regions were identified with 76% organized in clusters of three or more genes. A further 272 putative incomplete resistance genes were also identified. We determined that E. grandis has a higher ratio of TIR to CC classed genes compared to other woody plant species as well as a smaller percentage of single NBS-LRR genes. Transcriptome profiles indicated expression hotspots, within physical clusters, including expression of many incomplete genes. The clustering of putative NBS-LRR genes correlates with differential expression responses in resistant and susceptible plants indicating functional relevance for the physical arrangement of this gene family. This analysis of the repertoire and expression of E. grandis putative NBS-LRR genes provides an important resource for the identification of novel and functional R-genes; a key objective for strategies to enhance resilience.
Introduction
Eucalyptus grandis W. Hill ex Maiden (Flooded Gum) is an Australian myrtaceous forest species that is grown for timber-related products in many parts of the world (Myburg et al., 2014). Eucalypts are susceptible to a range of co-evolved and exotic pests and pathogens (Whyte et al., 2011), which can inhibit growth, reduce production and have significant effects on local economies (Coutinho et al., 1998; FAO, 2010). Therefore, determining the genetic basis for resistance to biotic stress is an urgent research priority with the goal of developing resilient forestry. As an important commercial species, E. grandis was selected as one of the first woody plants for genome sequencing (Myburg et al., 2011). The annotated draft genome for E. grandis, from a 17 year old inbred clone, BRASUZ1 (genome size of 640 Mbp, 11 haploid chromosomes), was released in 2011 with more comprehensive annotation data published in 2014 (Myburg et al., 2014). The availability of genomic sequence data for E. grandis makes it a useful model plant for the study of defense responses to current and emerging pathogens across a range of species within the Myrtaceae family.
Current understanding of plant defense responses is largely derived from research on crop plants and model species. After preformed defenses, such as cutin, waxes (Kolattukudy, 1980), and cell walls, a first line of defense is recognition and response to common pathogen-associated molecular patterns (Monaghan and Zipfel, 2012). Plants also exhibit pest (Aggarwal et al., 2014) and pathogen-specific responses mediated by resistance (R) genes (Ellis et al., 2000). The recognition proteins, encoded by R-genes, are predominantly intracellular receptors with high target specificity. Recognition and subsequent nucleotide binding leads to a cascade of responses that disrupt pathogenicity (Bernoux et al., 2011). Host-specific pathogens are known to deliver effector molecules into plant cells to disrupt host defense and increase pathogen virulence (Voegele and Mendgen, 2003). Resistant host plants harbor specific recognition receptors to counter pathogen effectors and initiate downstream immune response in a gene-for-gene manner (Flor, 1971). Models for pathogen perception describe direct and indirect recognition, thereby directing contrasting R-gene evolution (Jones and Dangl, 2006). Direct recognition involves the binding of receptors to effectors with demonstrated evidence from Linum usitatissimum (flax) response to Melampsora lini (flax rust; Dodds et al., 2006). Indirect recognition, appears to be more common than direct effector recognition (Kaschani et al., 2010; Bhattacharjee et al., 2011), and involves recognition of effector modified host proteins in what is termed the “guard hypothesis” (Jones and Dangl, 2006). Recently the recognition of effector modified host decoy proteins was validated in Arabidopsis thaliana (Le Roux et al., 2015), confirming the extension of the guard hypothesis to include decoy strategies and suggesting important evolutionary responses in plants to evading pathogens (van der Hoorn and Kamoun, 2008). While direct recognition requires adaptive responses to fast evolving pathogens, thereby driving rapid R-gene evolution, indirect recognition involves the ongoing surveillance of host molecular integrity and has been shown to permit long-term maintenance of R-genes (Stahl et al., 1999). Several classes of R-genes have been identified, however the most abundant are characterized by a centrally located nucleotide-binding site (NBS) domain and a carboxy-terminus leucine rich repeat domain (LRR; DeYoung and Innes, 2006), the NBS-LRR gene family.
The NBS domain incorporates sub-domains that are highly conserved in NBS-LRR and are therefore likely to be good predictors of gene function (Tameling et al., 2006). Within this domain is the NBS P-loop structure, involved in adenosine triphosphate (ATP) hydrolysis (Walker et al., 1982), near the N-terminus, as well as two ARC domains [APAF-1 (apoptotic protease activating factor-1), R-proteins and CED-4 (Caenorhabditis elegans death-4 protein) van Ooijen et al., 2008], at the C-terminus (NB-ARC). The NB-ARC domain is known to be involved in activating the hypersensitive response (HR), an important plant defense strategy characterized by rapid programmed cell death of affected and surrounding cells, thereby blocking disease progression of biotrophic and hemi-biotrophic pathogens (Mur et al., 2008). The LRR protein motif occurs across organisms and the alpha/beta horseshoe fold is believed to be involved in protein-protein interactions (Matsushima and Miyashita, 2012). This predicted feature, as well as the observed diversifying selection (Michelmore and Meyers, 1998) in this region of plant NBS-LRRs, suggested a role in pathogen recognition. Evidence supporting this is scant however and therefore their function in many plants is still unclear (DeYoung and Innes, 2006). Other structures often present are the coiled-coil (CC) motif and the Toll or interleukin-1 receptor homology domain (TIR) at the amino-terminus (McHale et al., 2006). Recent evidence points to these regions having strategic roles in pathogen recognition and signaling (Chang et al., 2013; Williams et al., 2014). These fused domains, and chimeric variants of them, commonly occur across plant genomes, although the TIR domain is notably absent from most monocotyledons (Tarr and Alexander, 2009).
The published genome of E. grandis has permitted investigations into important gene families such as terpene synthases (Külheim et al., 2015), lignin biosynthesis genes (Carocha et al., 2015), and pathogenesis-related genes (Naidoo et al., 2014). These analyses indicate that E. grandis has expanded gene families in relation to other plants. Further insights have been the high representation of defense-related genes amongst recent domain expansions and tandem duplications (Kersting et al., 2015). Themes that emerge for NBS-LRR genes across other woody plant genomes include the clear evolutionary divergence between two the major classes (TIR and CC), high proportions of genomic clustering and the large numbers within this gene family, >1% of protein coding genes (Tobias and Guest, 2014).
Here we determine the potential gene complement, physical location and genomic arrangement of NBS-LRR genes within the E. grandis genome. We classify these genes based on amino acid sequences of conserved domains. Further to this, we take two serious biotic stressors of E. grandis, stem canker (C. austroafricana), and the Eucalyptus gall wasp (Leptocybe invasa) and review NBS-LRR gene expression from resistant and susceptible clones. Apart from the characterization of all potential complete and partial NBS-LRR genes, key questions addressed in this research are; (i) what does the physical arrangement for this gene family indicate, (ii) can the putative genes be validated by expression data, and (iii) are there patterns of NBS-LRR gene expression under biotic stress?
Materials and Methods
Identification of Putative NBS-LRR Genes
The E. grandis annotation information file that was released as part of Phytozome v7.0 (Egrandis_162_annotation_info.txt, http://phytozome.jgi.doe.gov) was used to identify an initial list of 902 NB-ARC Pfam domain (PF00931) containing proteins (Goodstein et al., 2012). The presence of the NBS domain in each of the identified protein sequences was confirmed with PfamScan (www.ebi.ac.uk/Tools/pfa/pfamscan/; e < 1e-04; Figure 1a). An E. grandis specific Hidden Markov Model (HMM) for the conserved NB-ARC protein domain was constructed with 150 sequences (PfamScan e < 1e-60) using HMMER (Eddy, 2010). The E. grandis specific HMM was used to search for additional NB-ARC domains within the E. grandis protein sequence data using hmmsearch (e < 1e-04; Figure 1b). As a complement to this list, a nucleotide search was conducted using Basic Local Alignment Search Tool (BLAST) in Phytozome with three NBS-LRR genes from distantly related species (A. thaliana gi|3309618|, Medicago truncatula gi|357495078|and Solanum tuberosum gi|323370546|). The BLAST search identified genes with expected match results (e-value) of < 1e-10 and alignments >500 bp. E. grandis peptide homologs, from the results of this search, were then extracted and included in the potential NBS-LRR gene list (Figure 1c). The final list contained gene models that incorporated both complete and partial NBS-LRR genes as well as genes that shared homology with disease resistance genes from other plant species but without a full NB-ARC domain. The total list (1487 sequences) of candidate NBS-LRR genes was used in physical cluster and expression analysis while a reduced list (1215 sequences), including only genes with full NB-ARC domains, was used in all other downstream analysis.

Figure 1. Flow diagram of the strategy (a–c) that was followed to identify putative NBS-LRR genes within the Eucalyptus grandis genome.
Identification of Conserved Motifs
PfamScan was used to determine, from the library of Pfam HMMs, whether the identified candidate sequences encoded NB-ARC, TIR and LRR domains. Additionally, sequences were identified as having CC motifs according to COILS software (Lupas et al., 1991; http://www.ch.embnet.org/software/COILS_form.html), with a threshold of 0.9. Further validation was conducted with Paircoil2 (McDonnell et al., 2006; http://paircoil2.csail.mit.edu/; p < 0.025). MEME version 4.10.1 (Multiple Expectation Maximization for Motif Elicitation; http://meme-suite.org/tools/meme; Bailey and Elkan, 1994) analysis was used to identify conserved motifs within CC-NBS-LRR (CNL) and TIR-NBS-LRR (TNL) classes. The NB-ARC and TIR domain amino acid sequences, extracted from full protein sequences of 132 CNL and 174 TNL, were independently examined. A maximum of 15 motifs was allowed with zero or one motif per sequence.
Alignment and Phylogenetic Analysis
Alignment and phylogenetic relationships for NB-ARC domains (extracted from full protein sequences) were conducted using Mega6 (Tamura et al., 2013). The analysis was based on 480 E. grandis NBS-LRRs [154 TNLs, 123 CNLs and 203 NBS-LRRs (NLs)] and 15 NB-ARC sequences from outlier species including A. thaliana (At5g66900, At4g26090, At3g46530, At5g43470, At5g45260, At5g45250, At1g27170, At1g72840, At4g16950, At5g46520, At3g44630, At1g56510, At5G40060; Meyers et al., 2003), Malus domestica (AFP82245.1) and Ktedonobacter racemifer (WP_007911379.1; Figure 3A; Figure S1). Phylogenies for all NB-ARC domains from TIR [240 TIR-NBS (TN), 2 TIR-CC-NBS (TCN), 154 TNL] and non-TIR [86 CC-NBS (CN), 123 CNL, 202 NBS (N), 204 NL, 1 BED zinc finger-CNL (BCNL)] derived gene models were also constructed (Figures S2, S3). Due to pairwise distance calculation problems, 55 TIR and 153 non-TIR sequences were removed from the analysis. The sequences were aligned using ClustalW (Thompson et al., 1994) with default parameters.
Physical Cluster Analysis
On reviewing the literature we determined an appropriate definition for gene clusters and superclusters that permitted the mapping and analysis of E. grandis putative NBS-LRR genes (Meyers et al., 2003; Kohler et al., 2008; Jupe et al., 2012). We defined a gene cluster as a genomic region containing at least three NBS-LRR genes, (i) with < 9 other genes between neighboring NBS-LRR genes and (ii) in which two neighboring NBS-LRR genes are < 250 kb apart (example Figure S4A). We defined a gene supercluster as a genomic region containing at least one NBS-LRR gene cluster and at least two additional NBS-LRR genes, (i) with < 99 other genes between neighboring NBS-LRR genes and (ii) in which two neighboring NBS-LRR genes are < 2500 kb (2.5 Mb) apart (example Figure S4B).
To test the validity of the supercluster definition, we determined the probability of randomly observing at least one cluster and two additional genes (three NBS-LRR clusters/genes) within 5 Mb of the E. grandis genome (640 Mb). However, the calculation, using a permutation approach (R script) as described below, assumed the clusters and genes (contributing to supercluster) were randomly distributed across the E. grandis genome (640 Mb), whereas this gene family are found in clusters. The approach did not take into account the span of clusters, on average 400 kb. The permutation test: (1) Distribute 535 clusters/genes (136 clusters + 399 singletons) randomly across 640 Mb (640,000 kb), (2) Calculate the number of times that a group of three clusters/genes span < 5000 kb and divide the result by the number of groups (535), (3) Repeat the previous two steps 1000 times and save the resulting probability every time, (4) Calculate the 95th percentile of the resulting distribution of probabilities. The probability of observing three NBS-LRR clusters/genes within 5 Mb for a random distribution of the 535 clusters/genes over the 640Mb is 41%.
A cluster was defined as CNL-type if no genes with TIR domains were present, but at least one gene had a CC motif. Clusters were defined as TNL-type if no genes with CC motifs, but at least one gene had a TIR domain. Mixed-type clusters have at least one gene with a CC motif and one gene with a TIR domain present, while other-type clusters have no genes with CC motifs or TIR domains [therefore only genes in classes leucine rich repeat (L), N and NL].
Visualization of NBS-LRR Genes on Chromosomes
The positions of the TN(L), CN(L), and N(L) classes of the NBS-LRR genes were visualized using Circos (Krzywinski et al., 2009). Gene models, physical clusters and superclusters were mapped to the 11 E. grandis chromosomes using base pair start positions in Mapchart 2.2 (Voorrips, 1994).
NBS-LRR Predicted Protein Structure
Peptide sequences of a representative predicted gene from TNL (Eucgr.C00020) and non-TNL (Eucgr.L01363) were submitted to the I-Tasser server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) to determine predicted protein structures.
NBS-LRR Gene Numbers in Other Plants
A comparative review of NBS-LRR gene predictions from sequenced plants was undertaken using Phytozome genome data and published NBS-LRR papers for; A. thaliana (Meyers et al., 2003), Oryza sativa (Zhou et al., 2004), Populus trichocarpa (Kohler et al., 2008), Hevea brasiliensis (Rahman et al., 2013), Malus × domestica (Arya et al., 2014), Solanum lycopersicum (Sato et al., 2012), Threobroma cacao (Argout et al., 2011), Manihot esculenta (Lozano et al., 2015), Vitis vinifera (Argout et al., 2011 suppl.) and S. tuberosum (Jupe et al., 2012). Numbers of complete (CNL, TNL, and NL classes) and incomplete NBS-LRR gene models (CN, TN, N classes) were graphed (Figure 5).
Biotic Stress Trials
Clones of E. grandis were challenged with the fungal stem canker pathogen (C. austroafricana) and leaf gall wasp (L. invasa) as described by Mangwanda et al. (2015) and Oates et al. (2015), respectively. Infected C. austroafricana stem samples were collected 3 days post inoculation and infested L. invasa leaf samples were collected 7 days post infestation. Total RNA was extracted and sent to the Beijing Genome Institute (BGI) for RNA-Sequencing using the Illumina Genome Analyzer with a 50 bp paired end module (Illumina, San Diego, CA).
RNA-Seq Data Analysis
RNA-Seq data was analyzed using the Galaxy workspace (Giardine et al., 2005; Blankenberg et al., 2010; Goecks et al., 2010). FASTQC v0.52 was used to verify RNA-Seq data quality. Reads were mapped to the E. grandis v1.1 genome assembly using Bowtie (Langmead et al., 2009) and Tophat2 v2.0.9 (Trapnell et al., 2009). Mapping was allowed over multiple domains, however the default Tophat setting discards reads mapped to more than 20 locations. Therefore, if more than 20 family members existed with identical domains, mapping to those domains would have been unlikely. We used a mix of reads (unique and multiple with < 20 family members) but applied fragment bias correction and multi read mapping correction in Cuffdiff (Trapnell et al., 2010; Roberts et al., 2011). The expression quantification step would have corrected for this multiple mapping to highly conserved domains based on unique reads mapped to other regions of the expressed transcripts. It is therefore expected that highly expressed genes would be accurately mapped. Although genes with low numbers of mapped reads may represent low expression of those genes or may indicate false mapping, the depth of sequencing in our experiments (36–40 million reads per sample) would have improved detection of lowly expressed genes.
Mapped reads were assembled into transcripts and fragments per kilobase of exon per million fragments mapped (FPKM) values were calculated with Cufflinks v2.1.1 (Trapnell et al., 2010). Quartile normalization was conducted in Cufflinks. Detailed analysis was as described by Mangwanda et al. (2015) and Oates et al. (2015). The Eucalyptus data sets supporting these results are available in the NCBI Gene Expression Omnibus repository for C. austroafricana challenge (GSE67554) and NCBI BioProject ID PRJNA305347 for L. invasa challenge.
NBS-LRR Transcript Expression Analysis
Expression profiles for the putative NBS-LRR genes were extracted from a transcriptome-wide expression matrix using a custom Python script. Expression data were only available for genes in E. grandis v1.1. Analysis of variance (ANOVA) for putative NBS-LRR genes from treatment, control, resistant and susceptible groups was performed in GenStat (v. 16.2.0.11713, VSN International, Hemel Hempstead, UK). The E. grandis datasets from both C. austroafricana and L. invasa were analyzed separately. Expression analysis was based on the log2 fold change of inoculated vs. control samples. Genes in resistant and susceptible plants were considered up or down-regulated if their log2 gene expression ratios were >1 or smaller than −1. Differential expression was determined by taking significant p-values (< 0.01) from the ANOVA analysis by treatment and comparing this data with fold change values. Heatmaps that depict gene expression [as log2 of the normalized read count (FPKM)] and accompanying hierarchical clustering of both gene expression and treatments, were drawn in R studio (v. 0.98.981, RStudio Team, 2015) using the gplots and RColorBrewer packages. Color breaks, using red, yellow and green, were non-linear and adjusted individually to each heatmap (e.g., a subsample of NBS-LRR genes that had a maximum log2 FPKM value of 2 had color breaks at 0.1, 0.5, and 2).
A locus for resistance to Puccinia psidii (myrtle rust) has been mapped on the Eucalyptus reference map (Mamani et al., 2010). EMBRA125, one of the flanking markers of the P. psidii rust resistance (Ppr1) locus is closely linked to marker ePT_640786 (Kullan et al., 2012). This marker sequence has an estimated position of 52,900,000 bp on chromosome 3 of the E. grandis reference genome sequence and overlaps with the position of supercluster C-3. We determine the expression within this cluster in response to C. austroafricana (see heatmap Figure 6D).
Results
Identification of Putative NBS-LRR Genes
Our search strategy identified 1487 putative NBS-LRR genes within the E. grandis genome (Table 1). Of these, 557 gene models appeared to be complete, incorporating both the NB-ARC and leucine-rich repeat (LRR) domains, and 660 additional gene models incorporated the NB-ARC domain but lacked LRRs (partial NBS-LRR). Figure 2 gives the positional and class distribution per chromosome of all the identified NBS-LRR genes with a full NB-ARC domain. Gene models with domain chimeras were identified (Table S1) including one BED zinc finger-CNL, one CTN and two TCN as well as many with duplicate or variant fused domains (112 sequences) such as CNNL, CNN, NNL, NLNL, NNLN, NN, TNNL, TNN, TNTN, TTN. Incomplete gene models (270) were also identified that were homologs of disease resistance genes from other plant species (Table 1).
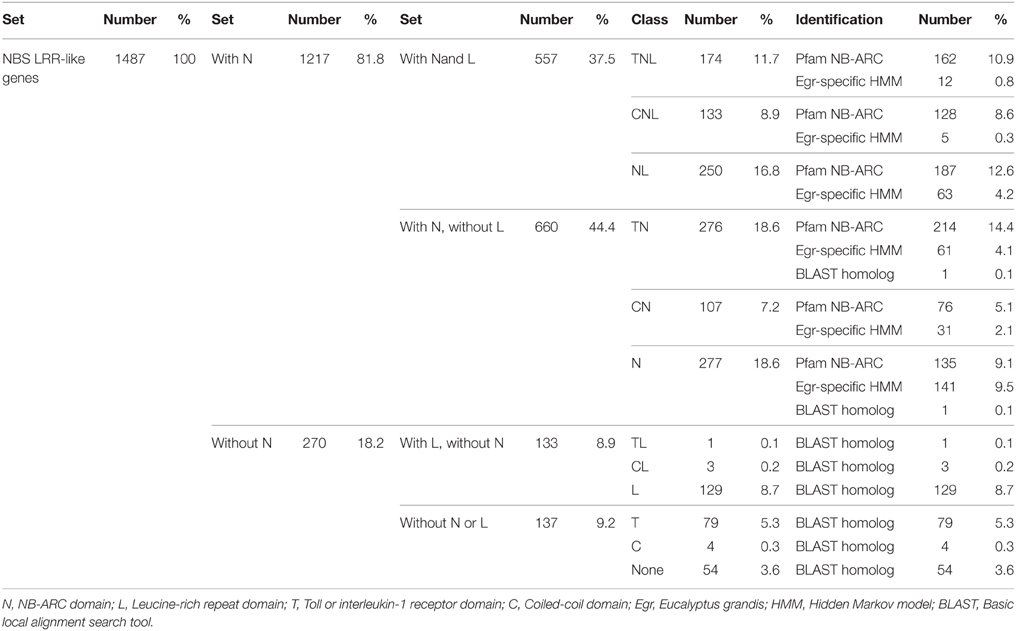
Table 1. The full set of nucleotide-binding site leucine-rich repeat (NBS-LRR) gene models, including complete, partial and incomplete genes, identified in the Eucalyptus grandis genome.
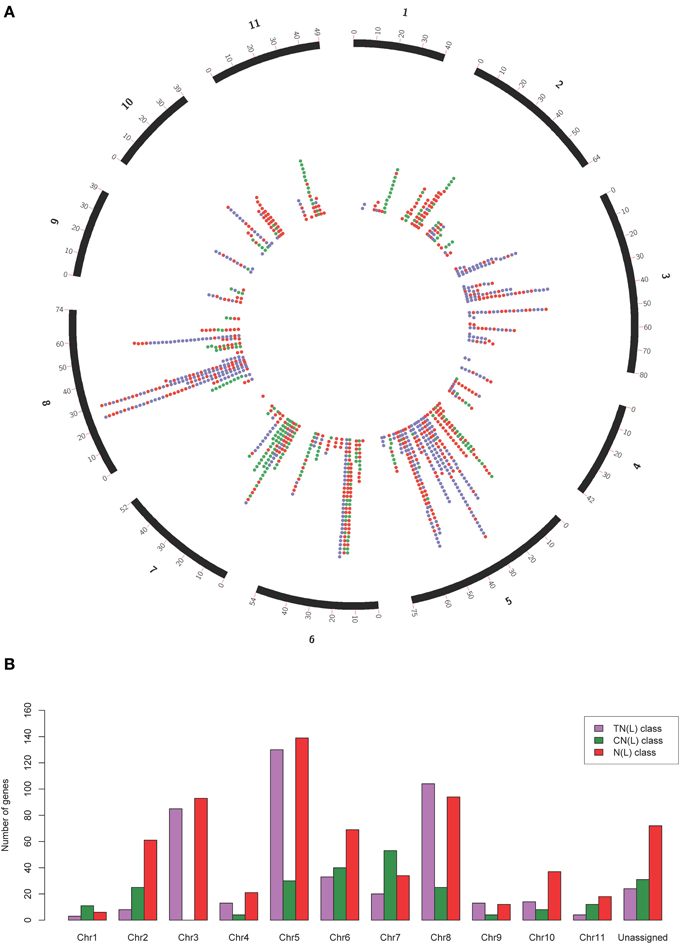
Figure 2. Circos plot (A) and class distribution (B) of the positions and numbers of complete and partial NBS-LRR genes (total = 1215) per chromosome identified within the Eucalyptus grandis genome. In the circos plot, purple dots represent genes in the TN(L) class, green dots represent genes in the CN(L) class and red dots represent genes in the N(L) class.
Domain Validation
The two major categories of putative NBS-LRR genes include sequences from the CNL class (which incorporate the amino acid tryptophan within the kinase 2 sub-domain) and sequences from the TNL class. The CNL class, identified as sequences containing the CC motif according to COILS, consisted of 132 complete NBS-LRR gene models. However, only 81 of these could be confirmed with Paircoil2 (p < 0.025). The TNL class consisted of 174 complete NBS-LRR gene models containing an N-terminal TIR domain. Out of the sequences in the NL class, 88 were physically located within CNL-type clusters and 107 within TNL-type clusters (Table S1). MEME analysis of NB-ARC domains from the mentioned classes [CNL, TNL, NL (within CNL-type clusters) and NL (within TNL-type clusters)] identified conserved motifs with homology to A. thaliana motifs (as identified in Meyers et al., 2003; Table S2). MEME analysis also identified motifs for TIR domains within all TN(L) containing sequences (Table S2).
Phylogenetic Analysis
The dataset of 557 complete NBS-LRR gene models was reduced to include all sequences for which pairwise distances between NB-ARC domain sequences could be calculated and for which only single NB-ARC domains were present. The evolutionary relatedness of 480 NB-ARC domains (123 CNL, 154 TNL, 203 NL) from complete NBS-LRR gene models separated into the two major clades: CNL and TNL (Figure 3A). Of the 203 NL sequences, 70 aligned with TNL and 133 with CNL NB-ARCs. The comparative numbers of gene models that aligned with TNL and CNL structures were 47% (224) and 53% (256), respectively. As expected, most of the genes within the CNL phylogenetic clade (61%) were physically located in CNL-type clusters, whereas only three genes (1%) were in TNL-type clusters (Figure 3B). Similarly, most genes within the TNL clade (74%) were physically located in TNL-type clusters, whereas one gene was in a CNL-type cluster (0.045%; Figure 3B).
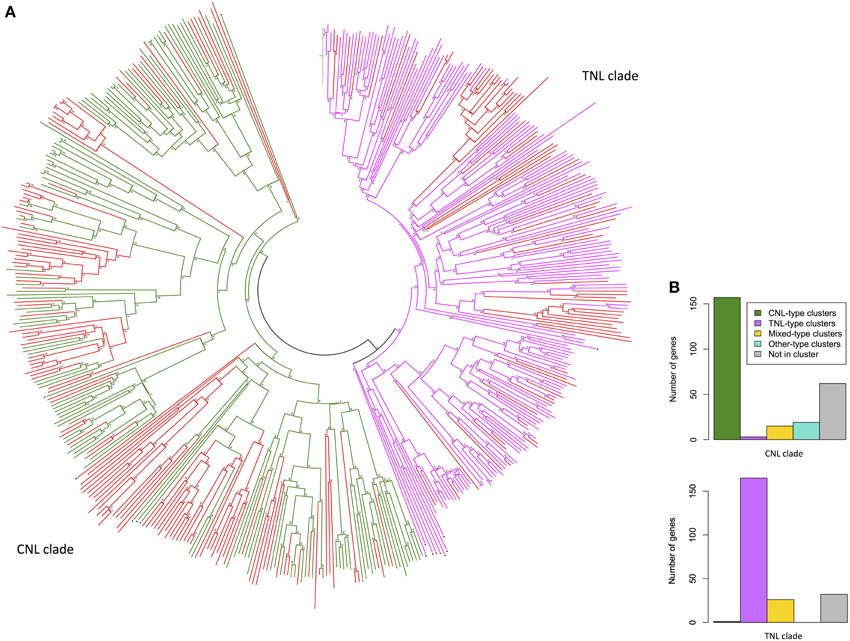
Figure 3. (A) Evolutionary relationships of Eucalyptus grandis NB-ARC domains from putative NBS-LRR genes. The evolutionary history was inferred using the Neighbor-Joining method. The optimal tree with the sum of branch length = 69.73913604 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The analysis involved 495 amino acid sequences (480 E. grandis). All ambiguous positions were removed for each sequence pair. There were a total of 1089 positions in the final dataset. Scale: 0.1 substitutions per site. Evolutionary analyses were conducted in MEGA6 (Tamura et al., 2013). (NL = red, CNL = green, TNL = pink, black triangle denotes outlier NB-ARC domains from other species). (B) Cluster type distribution of E. grandis NBS-LRR genes for the two major phylogenetic clades.
While generally the phylogenetic clustering of putative NBS-LRR genes supported recently evolved and syntenic duplications, on chromosomes, we also found occurrences of highly similar sequences from different chromosomes. Examples we identified included five similar CNL-like genes (Eucgr.F03324/Eucgr.E02964, Eucgr.J02649/ Eucgr.G00249, Eucgr.H05030/Eucgr.G00087, Eucgr.G00250/Eucgr.J02651, Eucgr. J02654/Eucgr.G00259) and four similar TNL-like genes (Eucgr.J02654/Eucgr. G00259, Eucgr.F04314/Eucgr.C02650, Eucgr.E01971/Eucgr.D01584, Eucgr.E01990/ Eucgr.D01592; Figure S1). Recent and close phylogenetic clustering from different chromosomes was also evident for partial (TN and CN) sequences (Figures S2, 3).
Cluster and Supercluster Analysis
We determined that 76% of putative NBS-LRR genes occurred within clusters and 71% within superclusters (Table S1). We identified 136 NBS-LRR gene clusters (on average 12 per chromosome) with an average of 8 genes per cluster. Clusters span regions from 13 to 2900 kb. We identified 32 NBS-LRR gene superclusters with an average of 33 genes. Superclusters span regions from 0.5 up to 18 Mb. TNL-type clusters occurred predominantly on chromosomes 3, 5, and 8 (21, 12, and 10 clusters, respectively; Figure 4A). CNL-type clusters were more evenly distributed with the largest superclusters on chromosomes 5, 6, 7, and 8 (10, 10, 8, and 7 clusters, respectively; Figure 4B). Out of the 20% of clusters that span the smallest genomic region, 63% were CNL-type while the 20% of clusters spanning the largest genomic region, 63% were TNL-type (Figure 4C). The largest TNL-type supercluster (SC-C-2), also the cluster spanning the largest genomic region (Figure 4D), is on chromosome 3 and consists of 93 genes.
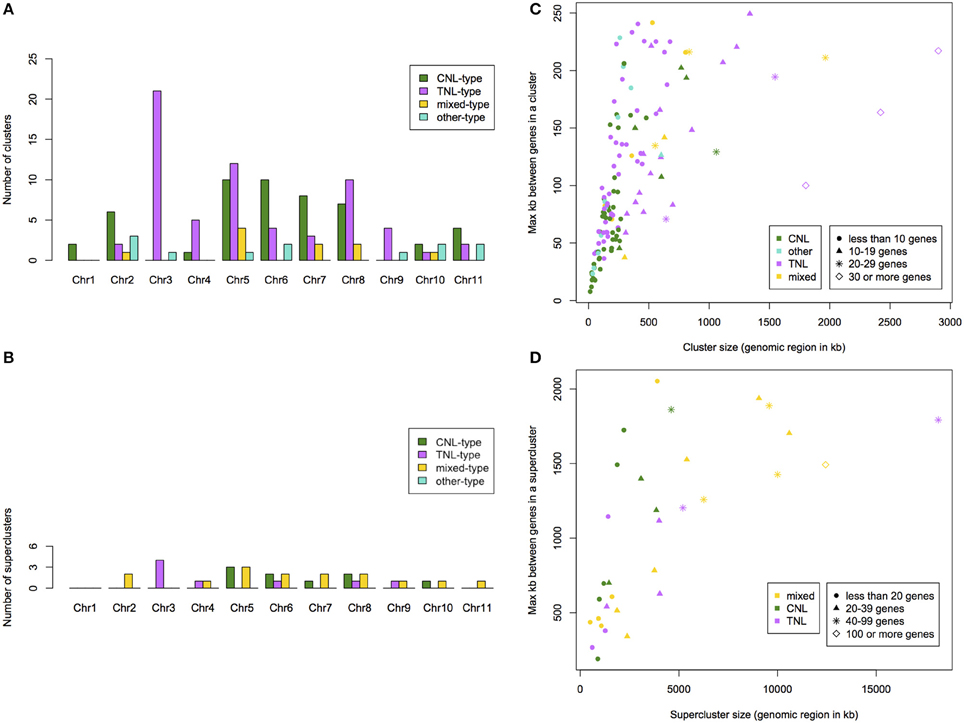
Figure 4. Summary per chromosome of the number of identified NBS-LRR gene clusters (A) and superclusters (B) within Eucalyptus grandis. Cluster (C) and supercluster (D) size (physical size and number of genes) and type of cluster relative to the maximum distance between genes in a cluster within the E. grandis genome.
Physical Map of the 11 E. grandis Chromosomes
Mapped chromosomal locations for all putative NBS-LRR genes, clusters and superclusters support the dense clustering for this gene family (Figure S5). Interestingly, though more TNL-like genes are present on chromosome 5 (Figure 2), a larger number of TNL-type clusters and superclusters are present on chromosome 3 (Figures 4A,B).
NBS-LRR Predicted Protein Structure
TNL (Eucgr.C00020) and CNL (Eucgr.L01363) protein structures were predicted using I-Tasser three dimensional modeling (Figure S6). The horseshoe fold of the LRR domain is indicated in the models. The GKT (Kinase 1) conserved subdomain is important in ATP hydrolysis while the hDD subdomain (Kinase 2) has a role in coordinating Mg2+ as a co-factor (Tameling et al., 2006). These two important sub-domains of NB-ARC, sometimes termed the Walker A and Walker B motifs (Walker et al., 1982), are identified within the models and designated A and B, respectively (Figure S6).
Comparison of NBS-LRR Gene Copy Numbers across Other Plants
When comparing the putative NBS-LRR gene results across other plant species (Figure 5) we found that the E. grandis genome harbored proportionately more putative TNL genes than all other species excepting A. thaliana. Interestingly, gene models that did not conform to the recognized structure of fused NB-ARC and LRR domains made up a large proportion of the total numbers of putative NBS-LRR genes within E. grandis and H. brasiliensis. Also, within the complete gene models, all plants excepting A. thaliana, appear to have large numbers of putative NL genes (lacking the amino terminal motif). E. grandis, M. domestica, Theobroma cacao, S. tuberosum, and O. sativa all had greater proportions of NL compared to putative CNL or TNL genes. The comparisons of gene numbers should, however, be treated with caution as they relate specifically to the version of genome available during the time of study and the search strategy employed.
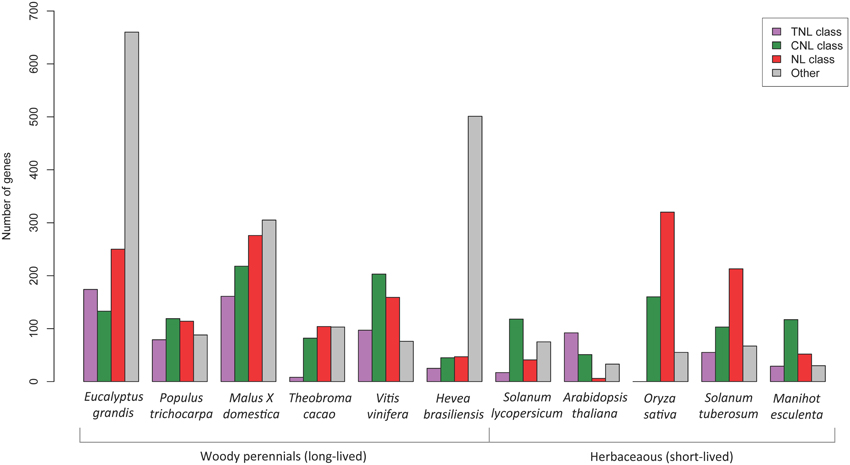
Figure 5. Comparison of putative NBS-LRR gene numbers determined in the genomes of different plant species. The “other” category (gray bars) include gene models in the TN, CN, and N classes.
Gene Expression
Gene expression was detected for 1037 and 1047 complete, partial and incomplete putative NBS-LRR genes in the C. austroafricana and L. invasa studies, respectively (Table S1). Of these, 1027 genes were shared between the two transcriptomes while 10 and 20 were unique to C. austroafricana and L. invasa studies, respectively. Of the predicted 557 complete NBS-LRR genes, expression was measured for 423 within both transcript sets. Predicted full sequences that were not expressed included 21 CNL, 59 NL, and 54 TNL genes (Table S1).
Differential expression (DE) of putative NBS-LRR genes was evident under the two treatments (Table 2). Of the differentially expressed transcripts, 6 and 1 were significantly up-regulated only in resistant plants in the C. austroafricana and L. invasa studies, respectively (ANOVA treatment; p < 0.01; Table S1). The single significantly up-regulated gene after L. invasa challenge was an incomplete gene with only a putative TIR domain (Eucgr.H02120). Up-regulated genes from C. austroafricana challenge included two incomplete genes with only a putative TIR domain (Eucgr.L01982, Eucgr.F03897), an incomplete gene model homologous to TIR-NBS-LRR (Eucgr.H03831), an incomplete gene with only a putative NB-ARC domain (Eucgr.H03112; notably both Eucgr.H03831 and Eucgr.H03112 were significantly down-regulated in susceptible plants), an incomplete gene with only a putative TIR and NB-ARC domain (Eucgr.E02315) and a putative complete NBS-LRR that aligned within the CNL clade (Eucgr.B00924; Table S1). Both biotic challenges involved significant down-regulation of genes with 53 and 15 gene models significantly DE only in resistant and 39 and 13 DE only in susceptible C. austroafricana and L. invasa studies, respectively.
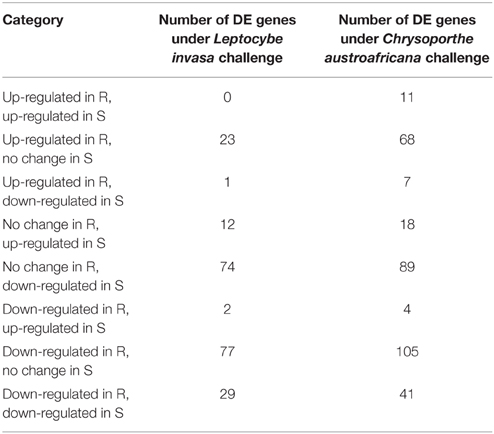
Table 2. Summary of significantly differentially expressed (DE) nucleotide-binding site leucine-rich repeat (NBS-LRR) genes under biotic stress challenge in resistant (R) and susceptible (S) Eucalyptus grandis genotypes.
We also identified the differential lack of transcripts for putative genes from resistant and susceptible plants. For the C. austroafricana challenge we found 26 gene transcripts lacking in susceptible plants and 11 in resistant plants. In the L. invasa challenge we found 23 gene transcripts lacking in susceptible plants and 49 in resistant plants (Table S1).
We identified physical clusters of significant DE for both treatments suggesting that the expression change of co-located genes may have functional relevance (Figure S5). We term these regions expression hotspots. Expression hotspots containing more than one significantly DE gene in C. austroafricana treated plants (28 in all) were located on chromosomes one to nine, with a large number of expressed clusters (11) on chromosome five (Table S1). A cluster of five adjacent CNL and NL genes on chromosome one (cluster A2, see Figure 6A) are significantly differentially expressed under C. austroafricana challenge, however a single putative CN gene (Eucgr.A02524), within the cluster, is not significantly up or down-regulated, though expression change is evident within heatmaps. Only six clusters of significantly DE genes were found in L. invasa treated plants. Interestingly, significant DE was determined for unassigned putative NBS-LRR genes suggesting important functional heterozygote allelic variants, for example Eucgr.L01982 (an incomplete putative TIR with log2 fold change up in resistant of 2.6) under C. austroafricana challenge. Much of the significant expression response by treatment was down-regulation of clusters of putative NBS-LRR genes, though notably two single incomplete NBS-LRR genes showed opposite regulation in resistant (up) and susceptible (down) genotypes under C. austroafricana challenge (Eucgr.H03112 and Eucgr.H03831; Table S1).
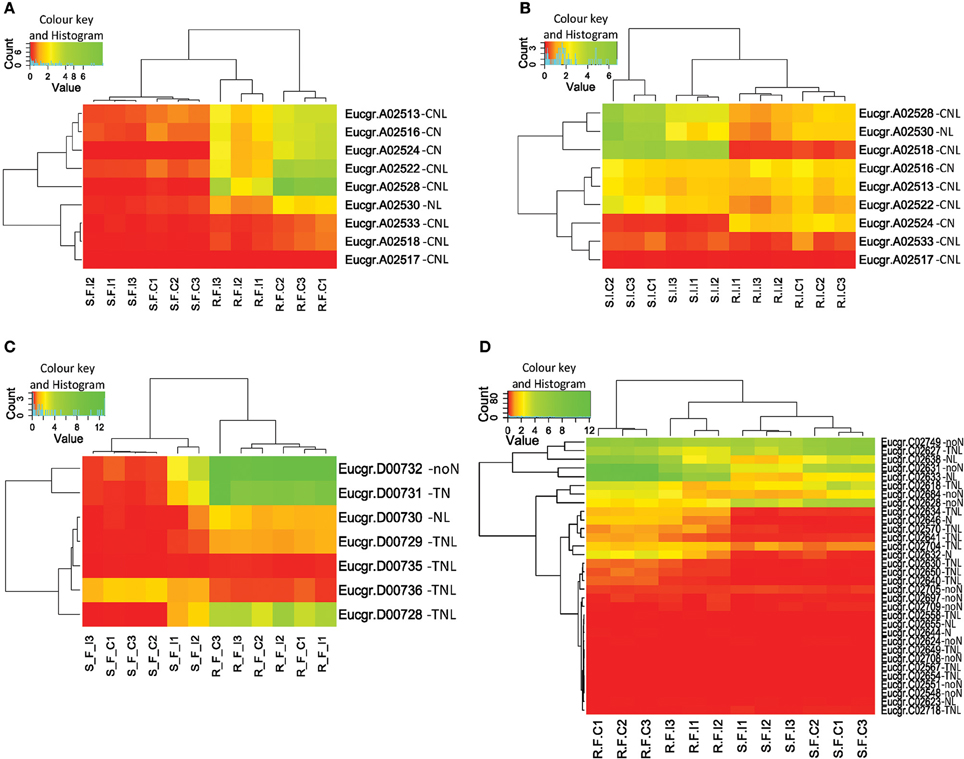
Figure 6. Heatmaps and dendrograms of putative NBS-LRR gene expression for Eucalyptus grandis clones. (A) Cluster A-2 under Chrysoporthe austroafricana challenge; (B) Cluster A-2 under Leptocybe invasa challenge; (C) Cluster D-1 under C. austroafricana challenge; (D) Supercluster C-3, genes within the Ppr1 locus (Puccinia psidii resistance gene 1), under C. austroafricana challenge. Red: 0–1 (very low–low expression), yellow: 1–10 (low to medium expression), green: 10–220 (medium to very high expression). S_F_C, susceptible, fungal treatment, control; S_F_I, susceptible, fungal treatment, inoculated; R_F_C, resistant, fungal treatment, control; R_F_I, resistant, fungal treatment, inoculated; S_I_C, susceptible, insect treatment, control; S_I_I, susceptible, insect treatment, infested; R_I_C, resistant, insect treatment, control; R_I_I, resistant, insect treatment, infested; N, NB-ARC domain; L, Leucine-rich repeat domain; T, Toll or interleukin-1 receptor domain; C, Coiled-coil domain; noN, no NB-ARC domain.
Differential expression between resistant and susceptible genotypes was evident for numerous gene models and clusters (Table S1), though change following treatment was not significant. Cluster A2 (Figure 6B) is an example of localized expression variation within a cluster following L. invasa challenge and provides an interesting contrast with expression following C. austroafricana challenge (Figure 6A). Within cluster D1 (Figure 6C) following C. austroafricana challenge and cluster E22 following L. invasa challenge, expression levels are higher in resistant plants but not statistically significant by treatment. On closer examination, the p-values based on genotype are highly significant for genes within the clusters (D1 and E22, see Table S1). We also identified fold change variation between susceptible and resistant plants where both are up or down-regulated despite no statistical significance by treatment. Here the difference in transcript abundance may be responsible for the observed resistant phenotype for example, an up-regulated TN gene model (Eucgr.E01683), which has a log2 fold change in resistant plants of 5.4 compared to 1.4 in susceptible plants.
Expression following C. austroafricana challenge within supercluster C3 indicates DE between susceptible and resistant plants (Figure 6D), though only two down-regulated genes (Eucgr.C02640 and C02631) in resistant plants are significant by treatment. The SC-C3 cluster correlates with the Ppr1 locus for resistance to P. psidii.
Discussion
A Recently Expanded TNL Class within Eucayptus grandis
We identified, within the E. grandis genome, 557 putative full-length NBS-LRR genes corresponding to the major class of plant resistance genes (Fluhr, 2001) and a further 660 gene models which incorporated the NB-ARC but lacked the LRR domain. Incomplete gene models (270) were also identified, that incorporated one or more domain(s) common to NBS-LRR genes, bringing the full gene list to 1487 (Table 1).
Of the identified complete NBS-LRR genes, we determined 174 TNL and 133 CNL (a ratio of ~ 6: 4) corresponding to the two major classes. A further 250 NL sequences that lacked CC or TIR domains were also identified, however the NB-ARC domains for these genes fell within the two clades, TNL (50) and CNL (123) indicating evolutionary relatedness. The numbers and proportions of sequences that lack an amino terminal CC or TIR motif nevertheless indicate a likely functional role for these genes, with differential gene expression in both biotic conditions supporting this (Figure 6). Notably genes lacking these motifs are also abundant across other species, except A. thaliana (Figure 5). Gene numbers for other woody species show a reversed ratio of TNL to CNL genes; P. trichocarpa 4: 6 (Kohler et al., 2008), Malus × domestica 4: 6 (Arya et al., 2014) and V. vinifera 3: 7 (Argout et al., 2011, Supplementary data). The comparative gene model numbers and proportions in E. grandis indicate a more recent TNL expansion, predominantly through tandem duplications (Figure 5). Although A. thaliana has similar TNL: CNL ratios (~6:4; Meyers et al., 2003), this appears to be uncommon amongst both woody and herbaceous species. The TIR domain is an important component of innate immunity across species through self-association and ligand specific protein-protein interactions (Ve et al., 2014). Evidence for the requirement of TIR heterodimer associations to form functional pathogen recognition complexes have been characterized in L. usitatissimum (flax; Ravensdale et al., 2012) and A. thaliana (Williams et al., 2014) TNL proteins. Perhaps heterodimerization of E. grandis TNL gene products occurs in a similar manner helping to explain the remarkable adaptability of this species.
Our phylogenetic analysis indicated an expected clear evolutionary divergence of the TNL and CNL groups (Figure 3A) as determined for other species (Meyers et al., 2003; Kohler et al., 2008). Clades of highly related sequences, based on phylogenies, show numerous examples of complete and partial NBS-LRR genes, which are closely associated physically (on chromosomes). These indicate recent duplications of existing genes, potentially adding variants to the plant resistance gene repertoire. The comparative numbers of full gene models, including genes lacking CC or TIR domains, that aligned with TNL and CNL NB-ARC structures were 47% (224) and 53% (256), respectively, based on pairwise distances of the reduced set.
Defense-Related Domains Associated with NBS-LRR Genes in E. grandis
Other notable domains often associated with NBS-LRR gene models are the BED zinc finger (PF02892; after BEAF and DREF from Drosophila melanogaster peptides) and other DNA binding domains such as MYB and WRKY transcription factors. These domains are largely involved in regulating transcription. We only determined one NBS-LRR gene model with the BED fused domain in E. grandis (Eucgr.F00886), compared to 34 in P. trichocarpa (Kohler et al., 2008; Germain and Séguin, 2011). We also identified a fused MYB-like DNA binding domain (PF00249) within an incomplete NBS-LRR gene model (Eucgr.H02157). MYB transcription factors have been found to interact with WRKY transcription factors bound to activated CNL recognition proteins in barley (Chang et al., 2013), thereby initiating defense gene expression. A. thaliana has a notable example of a TNL fused to a WRKY domain within Resistance to Ralstonia solanacearum gene 1 (RRS1-R; Le Roux et al., 2015). This fused DNA binding domain is believed to act as a decoy target for pathogen secreted molecules attempting to evade basal immune responses (Sarris et al., 2015). Our search, similarly to the NBS-LRR scrutiny of the P. trichocarpa genome (Kohler et al., 2008), did not identify any WRKY fused domains however the presence of BED and MYB fused domains may indicate similar decoys to RRS1-R. There are 174 MYB and 87 MYB-related transcription factors computationally annotated within the E. grandis genome (Jin et al., 2014). We also identified Jacalin-like lectin domains (PF01419), within four incomplete NBS-LRR genes (Eucgr.F01690, Eucgr.I02520, Eucgr.I02547, and Eucgr.I02558). These domains, which bind carbohydrates, are involved in innate immunity across a broad range of organisms (Rüdiger and Gabius, 2002). The sharing of this domain with NB-ARC and TIR domains, as determined in this study, suggests a role in pathogen recognition.
Physical Clusters of NBS-LRR Genes in E. grandis
Apart from an ancient lineage-specific whole-genome duplication event around 110 Ma, E. grandis has a very high rate of retained tandem duplications (34%; Myburg et al., 2014). These more recent gene expansions are likely to be due to imperfect pairing during meiosis, segmental duplication and gene conversions (Ober, 2010) and are notably evident for large gene families within E. grandis (Külheim et al., 2015; Li et al., 2015). Tandem duplications are three to five times more highly represented in E. grandis than other species of the rosids (Kersting et al., 2015). Domain expansions, whereby modular protein coding regions are extended, are also more prevalent in tandemly duplicated genes, generally, and stress-related genes, in particular, within E. grandis (Kersting et al., 2015). NBS-LRR gene regions have also been identified as being over-represented in presence/absence variation of exons in A. thaliana likely due to relaxed selection in duplicated genic regions (Bush et al., 2014). To retain duplicated genes over time, they must have functional relevance. However, multiple gene copies also enable mutations to be incorporated leading to functional divergence. The clustering and “birth and death” of NBS-LRR genes through evolutionary time has been suggested as a mechanism for highly diversified responses to pathogen evolution (Michelmore and Meyers, 1998).
Most putative NBS-LRR genes within E. grandis are located within clusters (76%) and in superclusters (71%; Table S1). Only 24% of E. grandis putative NBS-LRR genes are singletons in comparison to P. trichocarpa (37%) and A. thaliana (26.8%; (Kohler et al., 2008)). Though clusters of NBS-LRR genes have previously been defined in other species (at regions ~ 250 kb; Meyers et al., 2003; Ameline-Torregrosa et al., 2008; Tan and Wu, 2012), superclusters are less well defined. Nevertheless, so called “megaclusters” have been discussed for A. thaliana, in which NBS-LRR genes are within (20 centiMorgan) regions known as major resistance complexes (Holub, 2001). Within both Brachypodium distachyon and M. truncatula two large, extended clusters were determined by relaxing the definition of cluster (to a region spanning 430 kb for B. distachyon and undefined for M. truncatula; Ameline-Torregrosa et al., 2008; Tan and Wu, 2012). The biological significance of these superclusters is yet to be determined but such expansive regions would certainly aid in mispairing during recombination, an important mechanism for increasing genetic diversity (Friedman and Baker, 2007).
Clusters were predominantly classed as either TNL-type or CNL-type with fewer mixed-type clusters. Homogeneous clusters are expected due to the mode of gene expansion through tandem duplications. Heterogeneous clusters however, though rarer than homogeneous ones (Figure 4), are also a feature within A. thaliana and are suggested to be the result of segmental duplication (Leister, 2004), though stress-related transposition may also be involved (Lisch, 2013). Clustering may facilitate co-expression of important functionally related genes (Michalak, 2008). Pairs of NBS-LRR genes are often required for effective resistance and these genes often occur within clusters (Sinapidou et al., 2004). In rice (Ashikawa et al., 2008) and A. thaliana (Williams et al., 2014) tandemly residing NBS-LRR genes are co-expressed and form functional heterodimers which can interact with pathogen secreted molecules leading to plant resistance. It is therefore likely that clustering in E. grandis facilitates beneficial co-transcription in a similar manner with evidence from differential expression cluster hotspots supporting this assertion, for example Eucgr.D00731 and Eucgr.D00732 (Figure 6C).
While the phylogenetic clustering of E. grandis putative NBS-LRR genes generally supported recent evolution of tandem duplications, we also found occurrences of highly similar sequences from different chromosomes. Potential mechanisms for such translocations include segmental duplication (Leister, 2004) and the activity of transposable elements (Lisch, 2013). The proposed benefit of maintaining duplicated sequences on different chromosomes is that allele homogenization through recombination and gene conversion, likely for tandemly duplicated sequences, is reduced. Termed “recombinational isolation” (Leister, 2004) these “accidental” translocations, evident within E. grandis putative NBS-LRR genes, are likely to play an important role in gene evolution.
Partial NBS-LRR Genes Were Differentially Expressed Under Biotic Challenge
Co-ordination of resistance gene expression, translation and activation is highly regulated in plants in order to avoid unnecessary cellular damage when no threat is present (Boller and Felix, 2009; Källman et al., 2013). Regulation is therefore likely to be complex and only one aspect of the response can be ascertained from messenger RNA expression at single time-points post inoculation. Nevertheless, expression was identified for 1037 and 1047 complete, partial and incomplete putative NBS-LRR genes in the C. austroafricana and L. invasa studies, respectively. Of these, 423 putative complete NBS-LRR genes were present within both experiments. Of interest, we noted differential regulation of many chimeric variant putative genes including, but not limited to; Eucgr.B02976 (CNNL), Eucgr.B01237 (CNNL), Eucgr.A01678 (NNL), Eucgr.C02641(TNNNL; Table S1).
The differential lack of putative gene transcripts from resistant and susceptible plants is interesting and may suggest that these alleles are absent in these individuals. Significant differential expression under challenge was evident for complete, partial and incomplete genes indicating that they are important biologically or that they are the result of residual expression due to physical proximity to important functional genes. Expression of incomplete putative NBS-LRR genes has been found in M. esculenta (cassava; Lozano et al., 2015) and these are suggested to be relics from incomplete tandem gene expansions. The cassava study, in contrast to our study, found only a single partial putative NBS-LRR gene differentially expressed in response to Cassava Bacterial Blight.
Though genes within clusters are physically close, on chromosomes, not all genes within expressed clusters show significant DE (Figure 6) suggesting that certain genes do indeed have functional relevance and that these are not merely transcriptionally active zones. Interestingly much of the significant expression response identified was down-regulation of clusters of putative NBS-LRR genes, though notably two single incomplete NBS-LRR genes showed opposite regulation in resistant (up) and susceptible (down) genotypes under C. austroafricana challenge (Eucgr.H03112 and Eucgr.H03831). Effector suppression of resistance gene expression may be occurring as identified in other plant pathogen interactions (Houterman et al., 2008). A further interesting possibility is that the up-regulation and translation of partial genes may be involved in regulatory interactions by blocking functional heterodimers. The interaction of single domain microproteins dimerizing with a protein target can have a dominant negative regulatory effect as determined in transcription factor regulation in plants (Eguen et al., 2015).
Differential Expression of Physically Clustered Genes Suggest Functional Relevance
Comparison of resistant and susceptible genotypes (Figure 6; Table S1), suggests that genotype variation is worth investigating further, in particular for certain loci which harbor numerous DE putative NBS-LRR genes, such as the A2 and D1 clusters. Fold change variation in putative NBS-LRR genes between susceptible and resistant plants was also observed, where both are up or down-regulated, despite lack of statistical significance by treatment. These variations may account for the observed resistant phenotype due to differential log2 fold change. While no NBS-LRR genes have yet been cloned for E. grandis, the clustered expression patterns indicate that co-located genes, for example cluster D1, may have functional significance, as similarly determined for rice (Ashikawa et al., 2008), flax (Ravensdale et al., 2012) and A. thaliana (Saucet et al., 2015) where important receptor pairs form functional dimers.
Conclusion
Our scrutiny of the E. grandis genome for putative NBS-LRR genes has identified an extensive gene family with some differences to other woody plant species. Notably the TNL genes have recently expanded and E. grandis has a higher ratio of TNL to CNL compared to other woody plants. Retention of NBS-LRR genes in clusters and superclusters is an important feature for all plants, however E. grandis maintains a smaller percentage of single genes compared to P. trichocarpa and A. thaliana. Both these features are likely to be the result of recent tandem duplications, however selective pressure to retain higher numbers of TNL-like genes, occurring in their genomic arrangements, must play an important role. We found that a large proportion of putative NBS-LRR genes are expressed, including 423 complete gene models, and are therefore transcriptionally active. We further identified clusters where several putative complete and partial NBS-LRR genes were differentially expressed under challenge and this was specific to the type of challenge (fungal or insect). The clustering of putative NBS-LRR genes correlates with differential expression responses for many clusters suggesting functional relevance for the physical arrangement of this gene family. The findings of this study therefore provide researchers with a resource to investigate specificity of host response to biotic stress in an important forest species.
Author Contributions
NC and PT shared the lead author duties for this manuscript. NC carried out much of the data analysis and aspects of the manuscript drafting. PT conducted early data and gene expression analysis and much of the manuscript writing. SN and CK were involved in the design and co-ordination of the study and conducted some of the expression analysis and writing. All authors read and approved the final manuscript.
Funding
Top up scholarships were generously provided for PT from the University of Sydney and Rural Industries Research and Development Corporation, Australia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge Dr Sham V. Nair for his support, guidance and encouragement in the early stages of the study. Thank you to Professor Robert Park and Professor David Guest who made helpful comments on the draft article. We thank the reviewers for insightful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.01238
Table S1. Full list of Eucalyptus grandis putative NBS-LRR genes sorted by position on the genome. Information per gene includes the chromosomal position, class, physical cluster and phylogeny clade membership, identification method, raw expression data, log2 fold change values and ANOVA results (p-values). S_F_C, susceptible, fungal treatment, control; S_F_I, susceptible, fungal treatment, inoculated; R_F_C, resistant, fungal treatment, control; R_F_I, resistant, fungal treatment, inoculated; S_I_C, susceptible, insect treatment, control; S_I_I, susceptible, insect treatment, infested; R_I_C, resistant, insect treatment, control; R_I_I, resistant, insect treatment, infested.
Table S2. Conserved amino acid sequences for NB-ARC and TIR motifs from MEME analysis with CNL-like and TNL-like gene models in Eucalyptus grandis (Eg) and Arabidopsis thaliana (At; Meyers et al., 2003). The expected amino acid tryptophan (W) is identified in the Kinase 2 subdomain for CNL sequences–underlined.
Figure S1. Neighbor joining tree of 480 Eucalyptus grandis NB-ARC domains from complete NBS-LRR genes. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The analysis involved 495 amino acid sequences (480 E. grandis). All ambiguous positions were removed for each sequence pair.
Figure S2. Neighbor joining tree of 616 Eucalyptus grandis NB-ARC domains from all non-TIR NBS-LRR-like genes. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The analysis involved 631 amino acid sequences (616 E. grandis). All ambiguous positions were removed for each sequence pair.
Figure S3. Neighbor joining tree of 396 Eucalyptus grandis NB-ARC domains from all TIR NBS-LRR-like genes. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The analysis involved 411 amino acid sequences (396 E. grandis). All ambiguous positions were removed for each sequence pair.
Figure S4. The definition of a (A) cluster and a (B) supercluster is illustrated using a region (starting at 13 Mb and ending at 18 Mb) on chromosome 4.
Figure S5. Physical locations for all complete, partial, and incomplete NBS-LRR gene models that were expressed under challenge of Chrysoporthe austroafricana and Leptocybe invasa on Eucalyptus grandis chromosomes (Mapchart). Variation in means from treatment (ANOVA) were identified based on significance *p < 0.01, **p < 0.001, ***p < 0.0001 (*** are also underlined) and log2 gene expression ratios greater than 1 or smaller than −1 for resistant and susceptible plants. Color distinguishes between different classes (TNL = pink, CNL = green, NL = red, incomplete NL = black, BLAST homolog non-NL = black). Scale bar = Mb. Cluster and supercluster regions are indicated and E. grandis gene IDs are provided.
Figure S6. NB-ARC-LRR fused domains (A) and TIR-NB-ARC-LRR fused domains (B). Conserved amino acid sequences are indicated with lines (top). The GKT (Kinase 1) conserved motif is recognized as a P-loop structure important in ATP hydrolysis while the hDD is also well conserved in NB-ARC domains (Kinase 2) as important in co-ordinating Mg2+ as a co-factor (Tameling et al., 2006). These two important sub-domains of NB-ARC are sometimes termed the Walker A and Walker B motifs (Walker et al., 1982) and are identified as A and B, respectively, within the I-Tasser protein structures (bottom) for a representative CNL (Eucgr.L01363) and TNL (Eucgr.C00020) sequence from the Eucalyptus grandis genome.
References
Aggarwal, R., Subramanyam, S., Zhao, C., Chen, M.-S., Harris, M. O., and Stuart, J. J. (2014). Avirulence effector discovery in a plant galling and plant parasitic arthropod, the hessian fly (Mayetiola destructor). PLoS ONE 9:e100958. doi: 10.1371/journal.pone.0100958
Ameline-Torregrosa, C., Wang, B.-B., O'Bleness, M. S., Deshpande, S., Zhu, H., Roe, B., et al. (2008). Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21. doi: 10.1104/pp.107.104588
Argout, X., Salse, J., Aury, J.-M. M., Guiltinan, M. J., Droc, G., Gouzy, J., et al. (2011). The genome of Theobroma cacao. Nat. Genet. 43, 101–108. doi: 10.1038/ng.736
Arya, P., Kumar, G., Acharya, V., and Singh, A. K. (2014). Genome-wide identification and expression analysis of NBS-encoding genes in Malus x domestica and expansion of NBS genes family in Rosaceae. PLoS ONE 9:e107987. doi: 10.1371/journal.pone.0107987
Ashikawa, I., Hayashi, N., Yamane, H., Kanamori, H., Wu, J., Matsumoto, T., et al. (2008). Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180, 2267–2276. doi: 10.1534/genetics.108.095034
Bailey, T. L., and Elkan, C. (1994). “Fitting a mixture model by expectation maximization to discover motifs in biopolymers,” in Proceedings International Conference on Intelligent Systems for Molecular Biology; ISMB. International Conference on Intelligent Systems for Molecular Biology (Menlo Park, CA: AAAI Press), 28–36.
Bernoux, M., Ellis, J. G., and Dodds, P. N. (2011). New insights in plant immunity signaling activation. Curr. Opin. Plant Biol. 14, 512–518. doi: 10.1016/j.pbi.2011.05.005
Bhattacharjee, S., Halane, M. K., Kim, S. H., and Gassmann, W. (2011). Pathogen effectors target Arabidopsis EDS1 and alter its interactions with immune regulators. Science 334, 1405–1408. doi: 10.1126/science.1211592
Blankenberg, D., Kuster, G., Von Coraor, N., Ananda, G., Lazarus, R., Mangan, M., et al. (2010). Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 26, 1–21. doi: 10.1002/0471142727.mb1910s89
Boller, T., and Felix, G. (2009). A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 60, 379–406. doi: 10.1146/annurev.arplant.57.032905.105346
Bush, S. J., Castillo-Morales, A., Tovar-corona, J. M., Chen, L., Kover, P. X., and Urrutia, A. O. (2014). Presence-absence variation in A. thaliana is primarily associated with genomic signatures consistent with relaxed selective constraints. Mol. Biol. Evol. 31, 59–69. doi: 10.1093/molbev/mst166
Carocha, V., Soler, M., Hefer, C., Cassan-wang, H., Fevereiro, P., Myburg, A. A., et al. (2015). Genome-wide analysis of the lignin toolbox of Eucalyptus grandis. New Phytol. 4, 1297–1313. doi: 10.1111/nph.13313
Chang, C., Yu, D., Jiao, J., Jing, S., Schulze-Lefert, P., and Shen, Q.-H. (2013). Barley MLA immune receptors directly interfere with antagonistically acting transcription factors to initiate disease resistance signaling. Plant Cell 25, 1158–1173. doi: 10.1105/tpc.113.109942
Coutinho, T. A., Wingfield, M. J., Alfenas, A. C., and Crous, P. W. (1998). Special report eucalyptus rust: a disease with the potential for serious international implications. Plant Dis. 82, 819–825. doi: 10.1094/PDIS.1998.82.7.819
DeYoung, B. J., and Innes, R. W. (2006). Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 7, 1243–1249. doi: 10.1038/ni1410
Dodds, P. N., Lawrence, G. J., Catanzariti, A.-M., Teh, T., Wang, C.-I., Ayliffe, M. A., et al. (2006). Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. U.S.A. 103, 8888–8893. doi: 10.1073/pnas.0602577103
Eddy, S. R. (2010). HMMER User's Guide. Available online at: HMMER website: ftp://selab.janelia.org/pub/software/hmmer/CURRENT/Userguide.pdf (Accessed December 10, 2014).
Eguen, T., Straub, D., Graeff, M., and Wenkel, S. (2015). MicroProteins: small size – big impact. Trends Plant Sci. 20, 477–482. doi: 10.1016/j.tplants.2015.05.011
Ellis, J., Dodds, P., and Pryor, T. (2000). Structure, function and evolution of plant disease resistance genes. Curr. Opin. Plant Biol. 3, 278–284. doi: 10.1016/S1369-5266(00)00080-7
Flor, H. H. (1971). Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. doi: 10.1146/annurev.py.09.090171.001423
Fluhr, R. (2001). Sentinels of disease. Plant resistance genes. Plant Physiol. 127, 1367–1374. doi: 10.1104/pp.010763
Friedman, A. R., and Baker, B. J. (2007). The evolution of resistance genes in multi-protein plant resistance systems. Curr. Opin. Genet. Dev. 17, 493–499. doi: 10.1016/j.gde.2007.08.014
Germain, H., and Séguin, A. (2011). Innate immunity: has poplar made its BED? New Phytol. 189, 678–687. doi: 10.1111/j.1469-8137.2010.03544.x
Giardine, B., Riemer, C., and Hardison, R. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455. doi: 10.1101/gr.4086505
Goecks, J., Nekrutenko, A., and Taylor, J. (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86. doi: 10.1186/gb-2010-11-8-r86
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Holub, E. B. (2001). The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2, 516–527. doi: 10.1038/35080508
Houterman, P. M., Cornelissen, B. J. C., and Rep, M. (2008). Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 4:e1000061. doi: 10.1371/journal.ppat.1000061
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, 1182–1187. doi: 10.1093/nar/gkt1016
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Jupe, F., Pritchard, L., Etherington, G. J., MacKenzie, K., Cock, P. J., Wright, F., et al. (2012). Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genomics 13:75. doi: 10.1186/1471-2164-13-75
Källman, T., Chen, J., Gyllenstrand, N., and Lagercrantz, U. (2013). A significant fraction of 21-nucleotide small RNA originates from phased degradation of resistance genes in several perennial species. Plant Physiol. 162, 741–754. doi: 10.1104/pp.113.214643
Kaschani, F., Shabab, M., Bozkurt, T., Shindo, T., Schornack, S., Gu, C., et al. (2010). An effector-targeted protease contributes to defense against Phytophthora infestans and is under diversifying selection in natural hosts. Plant Physiol. 154, 1794–1804. doi: 10.1104/pp.110.158030
Kersting, A. R., Mizrachi, E., Bornberg-Bauer, E., and Myburg, A. A. (2015). Protein domain evolution is associated with reproductive diversification and adaptive radiation in the genus Eucalyptus. New Phytol. 206, 1328–1336. doi: 10.1111/nph.13211
Kohler, A., Rinaldi, C., Duplessis, S., Baucher, M., Geelen, D., Duchaussoy, F., et al. (2008). Genome-wide identification of NBS resistance genes in Populus trichocarpa. Plant Mol. Biol. 66, 619–636. doi: 10.1007/s11103-008-9293-9
Kolattukudy, P. E. (1980). Biopolyester membranes of plants: cutin and suberin reviewed work. Science 208, 990–1000. doi: 10.1126/science.208.4447.990
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Külheim, C., Padovan, A., Hefer, C., Krause, S. T., Köllner, T. G., Myburg, A. A., et al. (2015). The Eucalyptus terpene synthase gene family. BMC Genomics 16:450. doi: 10.1186/s12864-015-1598-x
Kullan, A. R., van Dyk, M. M., Hefer, C. A., Jones, N., Kanzler, A., and Myburg, A., a (2012). Genetic dissection of growth, wood basic density and gene expression in interspecific backcrosses of Eucalyptus grandis and E. urophylla. BMC Genet. 13:60. doi: 10.1186/1471-2156-13-60
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
Le Roux, C., Huet, G., Jauneau, A., Camborde, L., Trémousaygue, D., Kraut, A., et al. (2015). A receptor pair with an integrated decoy converts pathogen disabling of transcription factors to immunity. Cell 161, 1074–1088. doi: 10.1016/j.cell.2015.04.025
Leister, D. (2004). Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance genes. Trends Genet. 20, 116–122. doi: 10.1016/j.tig.2004.01.007
Li, Q., Yu, H., Cao, P. B., Fawal, N., Mathe, C., Azar, S., et al. (2015). Explosive tandem and segmental duplications of multigenic families in Eucalyptus grandis. Genome Biol. Evol. 7, 1068–1081. doi: 10.1093/gbe/evv048
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Lozano, R., Hamblin, M. T., Prochnik, S., and Jannink, J.-L. (2015). Identification and distribution of the NBS-LRR gene family in the Cassava genome. BMC Genomics 16:360. doi: 10.1186/s12864-015-1554-9
Lupas, A., Van Dyke, M., and Stock, J. (1991). Predicting coiled coils from protein sequences. Science 252, 1162–1164. doi: 10.1126/science.252.5009.1162
Mamani, E. M. C., Bueno, N. W., Faria, D. A., Guimarães, L. M. S., Lau, D., Alfenas, A. C., et al. (2010). Positioning of the major locus for Puccinia psidii rust resistance (Ppr1) on the Eucalyptus reference map and its validation across unrelated pedigrees. Tree Genet. Genomes 6, 953–962. doi: 10.1007/s11295-010-0304-z
Mangwanda, R., Myburg, A. A., and Naidoo, S. (2015). Transcriptome and hormone profiling reveals Eucalyptus grandis defence responses against Chrysoporthe austroafricana. BMC Genomics 16:319. doi: 10.1186/s12864-015-1529-x
Matsushima, N., and Miyashita, H. (2012). Leucine-Rich Repeat (LRR) domains containing intervening motifs in plants. Biomolecules 2, 288–311. doi: 10.3390/biom2020288
McDonnell, A. V., Jiang, T., Keating, A. E., and Berger, B. (2006). Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22, 356–358. doi: 10.1093/bioinformatics/bti797
McHale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212. doi: 10.1186/gb-2006-7-4-212
Meyers, B. C., Kozik, A., Griego, A., Kuang, H., and Michelmore, R. W. (2003). Genome-wide analysis of NBS-LRR – encoding genes in arabidopsis. Plant Cell 15, 809–834. doi: 10.1105/tpc.009308
Michalak, P. (2008). Co-expression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics 91, 243–248. doi: 10.1016/j.ygeno.2007.11.002
Michelmore, R. W., and Meyers, B. C. (1998). Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 8, 1113–1130.
Monaghan, J., and Zipfel, C. (2012). Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 15, 349–357. doi: 10.1016/j.pbi.2012.05.006
Mur, L. A. J., Kenton, P., Lloyd, A. J., Ougham, H., and Prats, E. (2008). The hypersensitive response; the centenary is upon us but how much do we know? J. Exp. Bot. 59, 501–520. doi: 10.1093/jxb/erm239
Myburg, A., Grattapaglia, D., Tuskan, G., Jenkins, J., Schmutz, J., Mizrachi, E., et al. (2011). The Eucalyptus grandis Genome Project: genome and transcriptome resources for comparative analysis of woody plant biology. BMC Proc. 5:I20. doi: 10.1186/1753-6561-5-S7-I20
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature 510, 356–362. doi: 10.1038/nature13308
Naidoo, S., Külheim, C., Zwart, L., Mangwanda, R., Oates, C. N., Visser, E. A., et al. (2014). Uncovering the defence responses of Eucalyptus to pests and pathogens in the genomics age. Tree Physiol. 34, 931–943. doi: 10.1093/treephys/tpu075
Oates, C. N., Külheim, C., Myburg, A. A., Slippers, B., and Naidoo, S. (2015). The transcriptome and terpene profile of Eucalyptus grandis reveals mechanisms of defense against the insect pest, Leptocybe invasa. Plant Cell Physiol. 56, 1418–1428. doi: 10.1093/pcp/pcv064
Ober, D. (2010). Gene duplications and the time thereafter - examples from plant secondary metabolism. Plant Biol. (Stuttg). 12, 570–577. doi: 10.1111/j.1438-8677.2009.00317.x
Rahman, A. Y. A., Usharraj, A. O., Misra, B. B., Thottathil, G. P., Jayasekaran, K., Feng, Y., et al. (2013). Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics 14:75. doi: 10.1186/1471-2164-14-75
Ravensdale, M., Bernoux, M., Ve, T., Kobe, B., Thrall, P. H., Ellis, J. G., et al. (2012). Intramolecular interaction influences binding of the Flax L5 and L6 resistance proteins to their AvrL567 ligands. PLoS Pathog. 8:e1003004. doi: 10.1371/journal.ppat.1003004
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., and Pachter, L. (2011). Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22. doi: 10.1186/gb-2011-12-3-r22
RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc. Available online at: http://www.rstudio.com
Rüdiger, H., and Gabius, H. J. (2002). Plant lectins: occurrence, biochemistry, functions and applications. Glycoconj. J. 18, 589–613. doi: 10.1023/A:1020687518999
Sarris, P. F., Duxbury, Z., Huh, S. U., Ma, Y., Segonzac, C., Sklenar, J., et al. (2015). A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell 161, 1089–1100. doi: 10.1016/j.cell.2015.04.024
Sato, S., Tabata, S., Hirakawa, H., Asamizu, E., Shirasawa, K., Isobe, S., et al. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Saucet, S. B., Ma, Y., Sarris, P., Furzer, O. J., Sohn, K. H., and Jones, J. D. G. (2015). Two linked pairs of Arabidopsis TNL resistance genes independently confer recognition of bacterial effector AvrRps4. Nat. Commun. 6. doi: 10.1038/ncomms7338
Sinapidou, E., Williams, K., Nott, L., Bahkt, S., Tör, M., Crute, I., et al. (2004). Two TIR:NB:LRR genes are required to specify resistance to Peronospora parasitica isolate Cala2 in Arabidopsis. Plant J. 38, 898–909. doi: 10.1111/j.1365-313X.2004.02099.x
Stahl, E. A., Dwyer, G., Mauricio, R., Kreitman, M., and Bergelson, J. (1999). Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400, 667–671. doi: 10.1038/23260
Tameling, W. I. L., Vossen, J. H., Albrecht, M., Lengauer, T., Berden, J. A., Haring, M. A., et al. (2006). Mutations in the NB-ARC domain of I-2 that impair ATP hydrolysis cause autoactivation. Plant Physiol. 140, 1233–1245. doi: 10.1104/pp.105.073510
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tan, S., and Wu, S. (2012). Genome wide analysis of nucleotide-binding site disease resistance genes in Brachypodium distachyon. Comp. Funct. Genomics 2012:418208. doi: 10.1155/2012/418208
Tarr, D. E. K., and Alexander, H. M. (2009). TIR-NBS-LRR genes are rare in monocots: evidence from diverse monocot orders. BMC Res. Notes 2:197. doi: 10.1186/1756-0500-2-197
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tobias, P. A., and Guest, D. I. (2014). Tree immunity: growing old without antibodies. Trends Plant Sci. 19, 367–370. doi: 10.1016/j.tplants.2014.01.011
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
van der Hoorn, R. A. L., and Kamoun, S. (2008). From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell 20, 2009–2017. doi: 10.1105/tpc.108.060194
van Ooijen, G., Mayr, G., Kasiem, M. M. A., Albrecht, M., Cornelissen, B. J. C., and Takken, F. L. W. (2008). Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383–1397. doi: 10.1093/jxb/ern045
Ve, T., Williams, S. J., and Kobe, B. (2014). Structure and function of Toll/interleukin-1 receptor/resistance protein (TIR) domains. Apoptosis 20, 250–261. doi: 10.1007/s10495-014-1064-2
Voegele, R. T., and Mendgen, K. (2003). Rust haustoria: nutrient uptake and beyond. New Phytol. 159, 93–100. doi: 10.1046/j.1469-8137.2003.00761.x
Voorrips, R. E. (1994). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Walker, J. E., Saraste, M., Runswick, M. J., and Gay, N. J. (1982). Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951.
Whyte, G., Howard, K., Hardy, G., and Burgess, T. (2011). Foliar pests and pathogens of Eucalyptus dunnii plantations in southern Queensland. Aust. For. 74, 161–169. doi: 10.1080/00049158.2011.10676359
Williams, S. J., Sohn, K. H., Wan, L., Bernoux, M., Sarris, P. F., Segonzac, C., et al. (2014). Structural basis for assembly and function of a heterodimeric plant immune receptor. Science 344, 299–303. doi: 10.1126/science.1247357
Keywords: Eucalyptus grandis, NB-ARC, NBS-LRR, resistance genes, gene clusters
Citation: Christie N, Tobias PA, Naidoo S and Külheim C (2016) The Eucalyptus grandis NBS-LRR Gene Family: Physical Clustering and Expression Hotspots. Front. Plant Sci. 6:1238. doi: 10.3389/fpls.2015.01238
Received: 04 November 2015; Accepted: 20 December 2015;
Published: 12 January 2016.
Edited by:
Stéphane Hacquard, Max Planck Institute for Plant Breeding Research, GermanyReviewed by:
Hugo Germain, Université du Québec à Trois-Rivières, CanadaAnnegret Kohler, Institut National de la Recherche Agronomique, Centre de Nancy, France
Copyright © 2016 Christie, Tobias, Naidoo and Külheim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peri A. Tobias, cGVyaS50b2JpYXNAc3lkbmV5LmVkdS5hdQ==