 Xianbin Yu
Xianbin Yu Guangyong Zheng1
Guangyong Zheng1 Xin-Guang Zhu
Xin-Guang Zhu- 1Group of Plant System Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 2Department of Computational Regulatory Genomics, CAS-MPG Partner Institutes for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 3Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
- 4Group of Plant System Biology, Institute of Plant Physiology and Ecology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
Photosynthesis is one of the most important biological processes on the earth. So far, though the molecular mechanisms underlying photosynthesis is well understood, however, the regulatory networks of photosynthesis are poorly studied. Given the current interest in improving photosynthetic efficiency for greater crop yield, elucidating the detailed regulatory networks controlling the construction and maintenance of photosynthetic machinery is not only scientifically significant but also holding great potential in agricultural application. In this study, we first identified transcription factors (TFs) related to photosynthesis through the TRAP approach using position weight matrix information. Then, for TFs related to photosynthesis, interactions between them and their targets were also determined by the ARACNE approach. Finally, a gene regulatory network was established by combining TF-targets information generated by these two approaches. Topological analysis of the regulatory network suggested that (a) the regulatory network of photosynthesis has a property of “small world”; (b) there is substantial coordination mediated by transcription factors between different components in photosynthesis.
Introduction
In recent years, more and more research shows that improving photosynthetic efficiency is a major viable approach to further increase crop productivity for enhanced food and fuel production (Zhu et al., 2010). In this aspect, much research has been devoted to study the molecular mechanism of photosynthesis and identify potential options to further optimize the photosynthetic machinery (Zhu et al., 2010). A number of engineering targets have indeed been identified, such as increasing expression of SBPase (Lefebvre et al., 2005) and manipulation of recovery speed from photo-protected state (Zhu et al., 2004) etc. Great efforts are now undertaken to engineer these targets in different crops to improve photosynthetic efficiency.
In most of the current research on improving photosynthesis, individual components that can potentially increase photosynthesis were identified and used as targets for engineering. This approach has generated certain success, as in the case of SBPase where its over-expression increased photosynthesis and biomass production (Lefebvre et al., 2005; Zhu et al., 2007). However, this approach usually does not consider the inherent interaction between different components of photosynthetic machinery. Much evidence however suggested that there is substantial interaction among different components of photosynthesis. As a result, altering the expression level of one gene might generate changes in the expression level of many other photosynthetic genes simultaneously. For example, decrease of the expression level of Rubisco small subunit led to changes in phosphoribulokinase activity in Calvin-Benson cycle in tobacco (Hudson et al., 1992). Knock down of Rieske FeS led to decrease of the concentrations of the cytochrome b6f complex and Rubisco in tobacco (Price et al., 1998). In C4 plants, mutation of Zmhcf136 caused loss of PSII complexes and grana thylakoid in mesophyll cells and simultaneous changes in expression patterns in the bundle sheath cells and mesophyll cells, including the differential levels of several C4 genes (e.g., PEPC, CA etc) (Covshoff et al., 2008). Over expression of C4 PEPC in rice also changed the expression level of Rubisco and FBPase (Agarie et al., 2002).
The close interaction among photosynthetic components is also reflected in the coordinated expressions of different components of photosynthetic machinery. In most C3 plants, surveys of photosynthetic physiological parameters showed that the maximal rate of Rubisco-limited photosynthesis (Vcmax) and the maximal rate of RuBP-limited photosynthesis (Jmax) strongly correlated with each other (Wullschleger, 1993) suggesting that expression of the genes underlying these parameters should be highly coordinated. In maize, a typical C4 plant, the bundle sheath and mesophyll cells have distinct patterns of protein accumulation, e.g., compared to mesophyll thylakoids, the thylakoid in the bundle sheath cells have a 55% reduced PSII content, unchanged ATP Synthase content, and a 65% increased PSI and 33% increased cytb6f contents (Majeran et al., 2008). These stable coordinated expression of different components of photosynthesis in C4 plants also suggested that they were regulated by delicate regulatory networks.
The close coordination of expression of photosynthetic components has great physiological significance, in particular, it is critical for maintaining a high resource use efficiency of photosynthesis under different conditions. This has been demonstrated in a number of cases. For example, under elevated CO2 conditions, the expression level of soluble rbcs protein is gradually decreased in rice (Chen et al., 2005), which is consistent with the required adjustment for enhanced photosynthetic light and nitrogen use efficiency (Long et al., 2004). Therefore, understanding the molecular mechanisms underlying these coordinated changes in expression of different components in photosynthesis under different conditions will not only be of scientific significance but also have great agricultural application potentials.
Unfortunately, so far, only a limited number of transcription factors related to regulation of photosynthetic genes have been discovered, mainly through traditional forward genetic studies (Saibo et al., 2009). The research on construction of genetic regulatory network of photosynthesis using high throughput transcriptomic and genomic data are mostly lacking. This is in sharp contrast to the rapid progresses in construction of regulatory networks related to other plant processes, e.g., the circadian clock and flowering control (Keurentjes et al., 2007; Ma et al., 2007; Thai et al., 2007; Long et al., 2008; Lee et al., 2010).
In this study, we aim to construct genetic regulatory network using transcriptomic and genomic data. Specifically, we combined two existing bioinformatics algorithms, the TRAP (Roider et al., 2007) and the ARACNE (Basso et al., 2005; Margolin et al., 2006) approaches to construct gene regulatory network. We used Arabidopsis thaliana as the model species because its genomic sequences are available and there are large amount of transcriptomic data. The TRAP algorithm was developed to predict downstream target genes of TFs through calculating binding affinity between transcription factors and DNA fragments, which has been shown to have high accuracy in previous studies (Roider et al., 2007). ARACNE is a software developed to find assocition relationship between genes based on mutual information using expression data (Basso et al., 2005; Margolin et al., 2006). This study identified a number of new candidate transcription factors as regulators of photosynthesis, together with some TFs reported earlier. Though the resulting genetic regulatory network is still small-scale, the network already showed the “small world” property (Braha and Bar-Yam, 2004). We also found evidences suggesting that TFs played a crucial role in coordinating expression of different components in photosynthesis.
Materials and Methods
Workflow of the Whole Project
The workflow of our study was showed in Figure 1A. We first applied the TRAP algorithm to calculate the binding affinities between TF and DNA fragments in promoter regions of Arabidopsis genes. Then TFs involved in photosynthesis were identified through a modified Fisher's test. After that, we further identified the interaction between these TFs and their target gene using the ARACNE algorithm based on transcriptomics data. Finally, TF-target pairs identified by both algorithms were selected as edges of the gene regulatory network of photosynthesis in Arabidopsis. Here we describe in detail the major algorithms involved in this workflow.
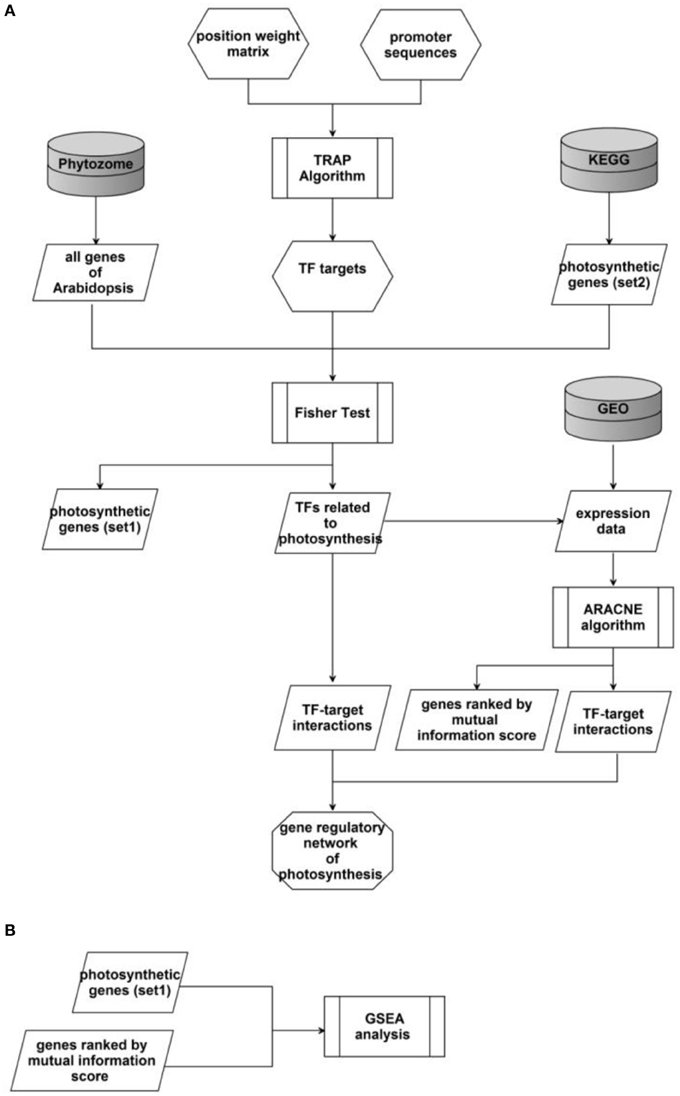
Figure 1. Schema of the whole study. (A) The workflow of the whole project. (B) The schema of GSEA analysis for a particular TF.
Collecting and grouping photosynthetic genes
We first collected pathways (map00195 and map00710) associated with photosynthesis for Arabidopsis from the KEGG database (http://www.genome.jp/kegg/). The genes contained in these pathways were used as photosynthetic genes. These genes include all enzymes involved in the Calvin-Benson cycle, ATP synthesis, components involved in electron transfer, the light reactions and genes related to C4 photosynthesis. Though Arabidopsis did not operate C4 photosynthesis, all C4 photosynthesis related genes exist in Arabidopsis. Altogether, 124 photosynthesis-related genes or isoforms were used in our study. These genes were categorized into following groups according to their biological functions, i.e., the Calvin Cycle (CC), Photosystem II (PSII), Photosystem I (PSI), Light Harvest Complex (LHC), Photosynthesis Electronic Transport (PET), Cytb6/f, F-type ATPase (FTA), and C4 related genes (C4).
Predicting interactions between TFS and candidate genes using the TRAP algorithm
In this study, we defined the genomic region from upstream 1000 bp to downstream 500 bp from the transcription start site (TSS) of a gene as the promoter region. The promoter region sequences of all Arabidopsis genes were downloaded from the Phytozome database (http://www.phytozome.net). Then we collected all plant transcription factors and their corresponding position weight matrices (PWMs) from the TRANSFAC database (Matys et al., 2003). As a result, 124 TFs and their corresponding PWMs were obtained for further study. The promoter region sequences and PWMs were used as input of the Transcription Factor Affinity Prediction (TRAP) algorithm to predict interactions between TFs and candidate genes.
Given a PWM (with length W) of certain TF and a promoter sequence (with length L) of a potential targeting gene, the binding affinity score (N) is computed as the sum of contributions from all possible sites (l) in the promoter sequence through followed equation:
Where, pl is the binding probability of site l, and R0 is defined by followed equation:
Here, K(S0) is an equilibrium constant. In Equation (2), the energy E is set to be zero, and [TF] denotes the concentration of the transcription factor (Roider et al., 2007). Given a transcription factor, β El(λ) describes its binding energies where the parameter λ is used to scale the mismatch energies in units of thermal energy (Roider et al., 2007).
Based on Equation (2), R0 is a constant for a TF at a given concentration. Therefore, we only need to consider the influence of parameter λ on binding affinity score. In practice, we used a series values of λ (i.e., 0.5, 0.6, 0.65, 0.7, 0.75, 0.8, 0.9, with 0.7 being the default value) to calculate the score. Given a certain value of λ, its impacts on the score was evaluated through counting the number of overlapped genes in top 1000 candidates compared with prediction result with a λ of 0.7.
Identifying transcription factors involved in photosynthesis
We first tested the impacts of modifying λ on prediction of TF and their targets. We found that varying λ did not influence the top 1000 genes much. Therefore, in this study, we used a λ value of 0.7. With this, we calculated binding affinities between these 124 TFs and their target genes with TRAP. Then, we used a modified Fisher's test to identify TFs whose target genes were significantly enriched in photosynthesis: (1) during calculation of the binding affinity scores, we set a series of values spanning from 0.5 to 5.0 with a step size of 0.1 as score cutoff. When a gene's binding affinity score was higher than the cutoff, it was regarded as a potential target of a certain TF. (2) A modified Fisher's test was conducted to calculate the p-value and judge whether targets were enriched in photosynthesis gene set among the background gene set. For each TF, a series of p-values were calculated based on different cutoffs values, and then the minimal p-value was selected. When the p-value was less than 0.001, the TF was regarded as a TF targeting photosynthesis genes.
Identifying TF-target pairs from microarray data
Microarray data of mature leaves in Arabidopsis (generated by the Affymetrix GPL198 platform) were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Altogether 391 experiments with 5626 chips were obtained (Data Sheet 3). The ARACNE algorithm, which uses mutual information between TF and their target genes, requires differential expression for involved genes (Basso et al., 2005; Margolin et al., 2006). We therefore selected experiments in which more than half of the interested TFs have a relative high expression diversity, i.e., for which the coefficient of variation in the expression values is higher than 0.1. After this filtering step, we obtained 23 experiments with a total of 454 chips. These chips were normalized across experiments using “normalizeBetweenArrays” implemented in R package “limma” (Smyth, 2005). In this study, some TFs' PWMs were not derived from Arabidopsis. For these TFs, we identified their best orthologs in Arabidopsis through querying their sequences against the Arabidopsis protein database with the BLASTP program. After that, we used the ARACNE algorithm, which was developed to identify correlated gene pairs based on mutual information (Basso et al., 2005; Margolin et al., 2006), to identify TF-target pairs with a threshold p-value of 10−7. After that, for each TF, its target genes were put into a list, where genes were ranked in a descending order based on the value of mutual information.
Coherence analysis of two methods for photosynthetic genes
For a given TF, a ranked targeted gene list based on mutual information was constructed by the ARACNE algorithm. Then the software GSEA (Mootha et al., 2003; Subramanian et al., 2005) was utilized to detect whether the TF's target genes identified by the TRAP algorithm are enriched in this ranked gene list (Figure 1B).
Network properties of the regulatory network
The final genetic regulatory network was constructed by integrating the TF-target relationship predicted with both methods. We calculated a number of network properties for this network: (1) the degree of a node, which is the number of edges connected to node; (2) the diameter of a network, which is the longest value of all the shortest paths in the network; (3) clustering coefficient of a node, which is the ratio of existing edges linking a node's neighbors to each other to the maximum possible number of such edges among the neighbors of a node.
Results and Discussion
A Feasible Strategy for Identification of TFS Related to Photosynthesis
In this study, we first tested whether prediction results of the TRAP algorithm is sensitive to parameter λ. As shown in data sheet 1 (see Supplemental Data), for most of the transcription factors, more than 90% of the top 1000 genes generated by different λ value were identical. Results of ten selected TFs are showed in Figure 2. These results suggest that predictions from the TRAP algorithm is not sensitive to the parameter λ. Hence, in this study, we use the default value (0.7) of λ to calculate the binding affinity between TFs and candidate genes in Arabidopsis. Subsequently, a modified Fisher's test is utilized to identify TFs related to photosynthesis (p-value < 0.001). In total, we identified 13 TFs that were related to photosynthesis (Table 1), among which 8 TFs have been reported earlier to have close relation to the regulation of photosynthesis (Table 1). Additionally, RITA-1, although it was not reported as a regulator of photosynthesis, its ortholog in Arabidopsis, bZIP9, can form a heterodimer with bZIP25 (Opaque-2) and bZIP63 (CPRF-2), which play an important role in regulation of light-induced genes (annotated in UniproKB) (http://www.uniprot.org). So it is possible that RITA-1 is also an important regulator of photosynthesis. Therefore, the strategy using in our study to recognize TFs involved in photosynthesis is feasible, since 9 out of 13 TFs (around 70%) are verified as important regulators of photosynthesis.
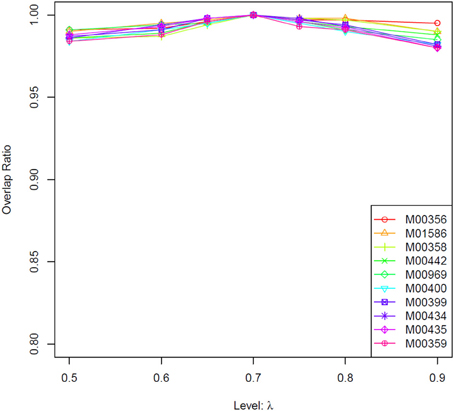
Figure 2. Comparison results of different values of parameter λ for the TRAP algorithm. We used a number of values of λ, i.e., 0.5, 0.6, 0.65, 0.75, 0.8, 0.9, to calculate the binding affinity between TFs and all the genes in Arabidopsis genome using TRAP. Then for each TF, we chose the top 1000 target genes with high binding affinity. We further calculated the overlap between this gene set and the top_1000_genes identified under λ = 0.7 (the default value). The horizontal axis denotes the used values of λ, while vertical axis denotes the ratio of overlap between these two gene sets. The result shows that TRAP is not sensitive to the parameter value of λ. Here the overlaps for only two TFs' results are showed (see the Supplemental Table 1 for all results). M00503, ATHB-5, from Arabidopsis thaliana; M00356, bZIP910, bZIP transcription factor from Antirrhinum majus.
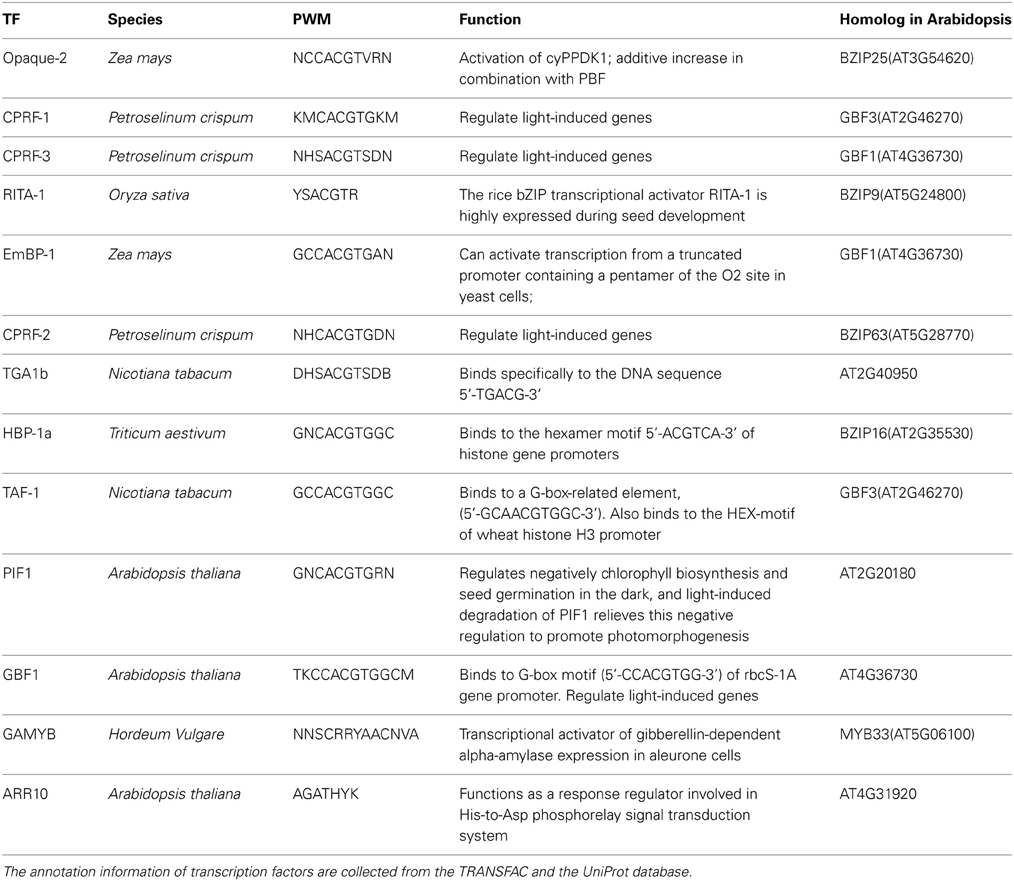
Table 1. Summary of TFs related to photosynthesis.
Coherence Analysis of Photosynthetic Gene Sets
After identifying TFs related to photosynthesis by the TRAP algorithm, homologies of these TFs in Arabidopsis were collected through the BLASTP program in NCBI (www.ncbi.nlm.nih.gov). As a result, 10 TFs in Arabidopsis and their corresponding targets (set1) were obtained. For each of these 10 TFs, we used the ARACNE software (Basso et al., 2005; Margolin et al., 2006) to calculate mutual information between the TF and all genes in Arabidopsis. Then genes associated with the TF were ranked based on the mutual information score. After that, for each TF, a coherence analysis is conducted between targets identified by the TRAP and the ranking generated by the ARACNE software (Table 2). As showed in the Table 2, genes of set1 are significantly enriched on top of the rankings for almost TFs except HBP-1a. These results suggested that the TFs regulating photosynthetic gene sets identified by TRAP and ARACNE were consistent with each other.
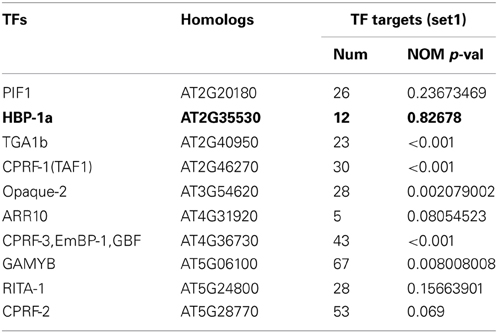
Table 2. Results of coherence analysis for TF targets identified by the TRAP and ARACNE algorithm.
Identified TFS Related to Photosynthesis Regulation
Table 1 lists the TFs identified by the TRAP algorithm. Among these TFs, many belong to the family of G-box binding protein [22]. G-box is a cis-acting element, 5… CACGTG… 3, identified in promoters of many plant genes (Williams et al., 1992). G-box binding proteins is a big gene family, while CPRF-1, CPRF-2, CPRF-3, EmBP1, TAF1, Opaque-2, GBF1 are members of the family (Siberil et al., 2001) (Table 1). CPRF-3 and EmBP1 are homologs of Arabidopsis GBF1 while CPRF-1 and TAF-1 are homologs of Arabidopsis GBF3 (Table 1). GBF1 exists in the nuclei of tomato and Arabidopsis and can interact with the G-box motif in promoters of many rbcs isoforms; while GBF3 shares the same binding motif with GBF1 (Giuliano et al., 1988). Earlier reports have shown that GBF1 and HY5 form DNA-binding heterodimer at rbcs-1a promoter; but different from HY5, GBF1 is a negative regulator of rbcs-1a [24]. GBF1-HY5 heterodimer is also a positive regulator of CAB1, which promotes accumulation of chlorophyll in cells and plays an important role in blue-light-induced photo-morphogenesis (Singh et al., 2012). Previous study also suggested that in Arabidopsis the Pro-rich activation domain of GBF1 can interact with GLK2 and GLK1, which regulates chloroplast development in diverse plant species (Tamai et al., 2002). These evidences suggested that GBF1 is an important TF regulating photosynthesis. Another identified G-box binding protein CPRF-2 can form heterodimer with CPRF1 or CPRF3 (Armstrong et al., 1992). Therefore, these 6 G-box TFs are potentially important regulators of photosynthesis. Opaque-2 (O2), another G-box binding factor, is also identified as an important regulator controlling expression of photosynthetic genes (Table 1). Opaque-2 has been reported to control the expression of a cytosolic form of pyruvate orthophosphate dikinase-1 (cyPPDK1) (Maddaloni et al., 1996), a key enzyme in the C4 photosynthesis. In the o2 mutant maize, expression of multiple photosynthetic genes were are down-regulated, included PEPC and ME (Hartings et al., 2011). By the way, EmBP-1 can bind to the same site as Opaque-2 in the same promoters, but inhibit the regulated transcription of these promoters of Opaque-2. (Carlini et al., 1999). We also identified PIF1, which is a negative regulator of chlorophyll biosynthesis and seed germination in darkness, light-induced degradation of PIF1 can relieve this negative regulation and promote photomorphogenesis (Moon et al., 2008).
Properties of the Gene Regulatory Network of Photosynthesis
A gene regulatory network of photosynthesis was established by combining TF-target pairs identified by both the TRAP and the ARACNE algorithm (Figure 3A). Then topology analysis was conducted by the “NetworkAnalyzer” module (Assenov et al., 2008) of the cytoscape software (http://www.cytoscape.org/). Topology analysis of the network showed that it has a diameter of 6; the average number of neighboring nodes and average length of shortest path are 4 and 3 respectively (Figures 4A,B). In addition, distribution of node degree in the network follows a power-law distribution (Figure 4C). These results indicate that the photosynthetic network is a scale free network, i.e., most of the components in the photosynthesis are regulated by relatively small number of regulators, i.e., TF in this case. These small number of regulators are regarded as hubs of the photosynthetic regulatory network and may play crucial role in coordination of expression of genes involved in photosynthesis. In the regulatory network that we obtained, the components of PSI, PET and PSII parts are co-regulated by the TFs of TGA1b, PIF1, Opaque-2, and CPRF-2 (Figure 3B). Furthermore, genes from Calvin cycle and F-Type ATP synthase are also co-regulated TF GAMYB and CPRF-2 (Figure 3B). Detailed reverse genetics studies should be conducted to study these coordination and their physiological significance.
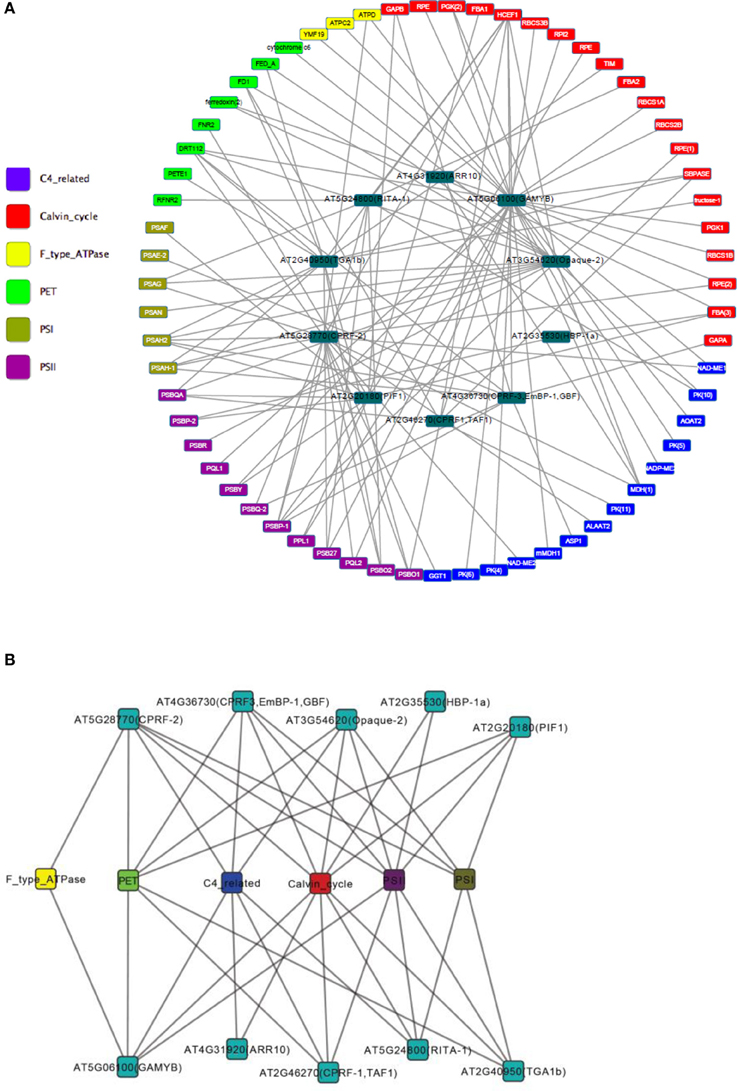
Figure 3. Gene regulatory network of photosynthesis in Arabidopsis. (A) Nodes in different colors represent genes involved in different sections of photosynthesis. We have divided the photosynthesis system into the following sections: ATPase, enzymes related to C4 photosynthesis, Calvin cycle, light harvesting complex, photosynthetic electron transfer, photosynthesis I, and photosystem II. (B) Transcription factor-components network. Photosynthesis genes are assigned to components. And a transcription factor and a component are linked if the transcription factor linked to one of the genes in the component in network (A).
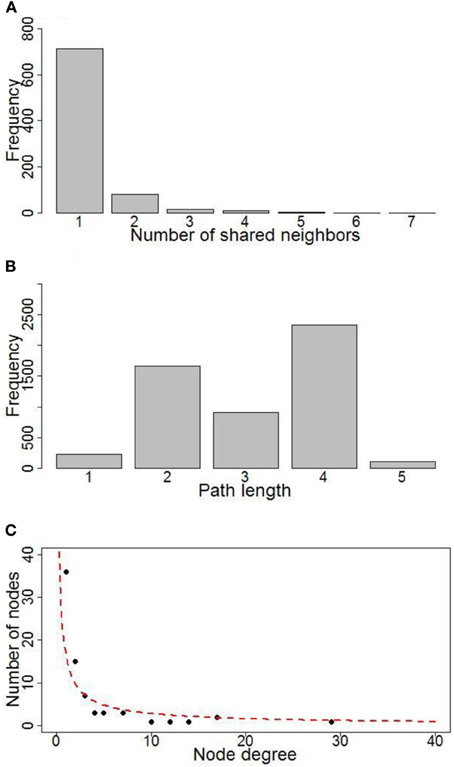
Figure 4. Topology property of the photosynthetic network. (A) Distribution of neighbors for nodes; (B) Distribution of shortest path length for the network; (C) Plot of number of nodes VS. nodes' degree.
Conclusions
In this study, a genetic regulatory network of photosynthesis in Arabidopsis was constructed through combining the genomic sequence, TF binding information and gene expression data. We identified a number of novel transcription factors related to photosynthesis. The identified network follows a scale-free property. The potential hubs in the network coordinating various components of photosynthesis were also identified. Transgenic experiments are undergoing now to test the consequence of manipulating expression of these TFs on photosynthetic performances.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank other laboratory members of Plant System Biology for stimulating discussions. Funding for authors' research is from the Bill and Melinda Gates Foundation (Grant No. OPP1014417), Ministry of Science and Technology of China (Grant No. 2011DFA31070), and the Young Talent Frontier Program of Shanghai Institutes for Biology Sciences/Chinese Academy of Sciences (Grant No. 09Y1C11501).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00273/abstract
References
Agarie, S., Miura, A., Sumikura, R., Tsukamoto, S., Nose, A., Arima, S., et al. (2002). Overexpression of C4 PEPC caused O2-insensitive photosynthesis in transgenic rice plants. Plant Sci. 162, 257–265. doi: 10.1016/S0168-9452(01)00572-6
Armstrong, G. A., Weisshaar, B., and Hahlbrock, K. (1992). Homodimeric and heterodimeric leucine zipper proteins and nuclear factors from parsley recognize diverse promoter elements with ACGT cores. Plant Cell 4, 525–537. doi: 10.1105/tpc.4.5.525
Assenov, Y., Ramirez, F., Schelhorn, S. E., Lengauer, T., and Albrecht, M. (2008). Computing topological parameters of biological networks. Bioinformatics 24, 282–284. doi: 10.1093/bioinformatics/btm554
Basso, K., Margolin, A. A., Stolovitzky, G., Klein, U., Dalla-Favera, R., and Califano, A. (2005). Reverse engineering of regulatory networks in human B cells. Nat. Genet. 37, 382–390. doi: 10.1038/ng1532
Braha, D., and Bar-Yam, Y. (2004). Topology of large-scale engineering problem-solving networks. Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 69:016113. doi: 10.1103/Physreve.69.016113
Carlini, L. E., Ketudat, M., Parsons, R. L., Prabhakar, S., Schmidt, R. J., and Guiltinan, M. J. (1999). The maize EmBP-1 orthologue differentially regulates opaque2-dependent gene expression in yeast and cultured maize endosperm cells. Plant Mol. Biol. 41, 339–349. doi: 10.1023/A:1006338727053
Chen, G. Y., Yong, Z. H., Liao, Y., Zhang, D. Y., Chen, Y., Zhang, H. B., et al. (2005). Photosynthetic acclimation in rice leaves to free-air CO2 enrichment related to both ribulose-1,5-bisphosphate carboxylation limitation and ribulose-1,5-bisphosphate regeneration limitation. Plant Cell Physiol. 46, 1036–1045. doi: 10.1093/pcp/pci113
Covshoff, S., Majeran, W., Liu, P., Kolkman, J. M., van Wijk, K. J., and Brutnell, T. P. (2008). Deregulation of maize C4 photosynthetic development in a mesophyll cell-defective mutant. Plant Physiol. 146, 1469–1481. doi: 10.1104/pp.107.113423
Giuliano, G., Pichersky, E., Malik, V. S., Timko, M. P., Scolnik, P. A., and Cashmore, A. R. (1988). An evolutionarily conserved protein binding sequence upstream of a plant light-regulated gene. Proc. Natl. Acad. Sci. U.S.A. 85, 7089–7093. doi: 10.1073/pnas.85.19.7089
Hartings, H., Lauria, M., Lazzaroni, N., Pirona, R., and Motto, M. (2011). The Zea mays mutants opaque-2 and opaque-7 disclose extensive changes in endosperm metabolism as revealed by protein, amino acid, and transcriptome-wide analyses. BMC Genomics 12:41. doi: 10.1186/1471-2164-12-41
Hudson, G. S., Evans, J. R., von Caemmerer, S., Arvidsson, Y. B., and Andrews, T. J. (1992). Reduction of ribulose-1,5-bisphosphate carboxylase/oxygenase content by antisense RNA reduces photosynthesis in transgenic tobacco plants. Plant Physiol. 98, 294–302. doi: 10.1104/pp.98.1.294
Keurentjes, J. J., Fu, J., Terpstra, I. R., Garcia, J. M., van den Ackerveken, G., Snoek, L. B., et al. (2007). Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. U.S.A. 104, 1708–1713. doi: 10.1073/pnas.0610429104
Lee, I., Ambaru, B., Thakkar, P., Marcotte, E. M., and Rhee, S. Y. (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat. Biotechnol. 28, 149–156. doi: 10.1038/nbt.1603
Lefebvre, S., Lawson, T., Zakhleniuk, O. V., Lloyd, J. C., Raines, C. A., and Fryer, M. (2005). Increased sedoheptulose-1,7-bisphosphatase activity in transgenic tobacco plants stimulates photosynthesis and growth from an early stage in development. Plant Physiol. 138, 451–460. doi: 10.1104/pp.104.055046
Long, S. P., Ainsworth, E. A., Rogers, A., and Ort, D. R. (2004). Rising atmospheric carbon dioxide: plants face the future. Annu. Rev. Plant Biol. 55, 591–628. doi: 10.1146/annurev.arplant.55.031903.141610
Long, T. A., Rady, S. M., and Benfey, P. N. (2008). Systems approaches to identifying gene regulatory networks in plants. Ann. Rev. Cell Dev. Biol. 24, 81–103. doi: 10.1146/annurev.cellbio.24.110707.175408
Ma, S., Gong, Q., and Bohnert, H. J. (2007). An Arabidopsis gene network based on the graphical Gaussian model. Genome Res. 17, 1614–1625. doi: 10.1101/gr.6911207
Maddaloni, M., Donini, G., Balconi, C., Rizzi, E., Gallusci, P., Forlani, F., et al. (1996). The transcriptional activator Opaque-2 controls the expression of a cytosolic form of pyruvate orthophosphate dikinase-1 in maize endosperms. Mol. Gen. Genet. 250, 647–654.
Majeran, W., Zybailov, B., Ytterberg, A. J., Dunsmore, J., Sun, Q., and van Wijk, K. J. (2008). Consequences of C4 differentiation for chloroplast membrane proteomes in maize mesophyll and bundle sheath cells. Mol. Cell. Proteomics 7, 1609–1638. doi: 10.1074/mcp.M800016-MCP200
Margolin, A. A., Wang, K., Lim, W. K., Kustagi, M., Nemenman, I., and Califano, A. (2006). Reverse engineering cellular networks. Nat. Protoc. 1, 662–671. doi: 10.1038/nprot.2006.106
Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl, R., et al. (2003). TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31, 374–378. doi: 10.1093/nar/gkg108
Moon, J., Zhu, L., Shen, H., and Huq, E. (2008). PIF1 directly and indirectly regulates chlorophyll biosynthesis to optimize the greening process in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 105, 9433–9438. doi: 10.1073/pnas.0803611105
Mootha, V. K., Lindgren, C. M., Eriksson, K. F., Subramanian, A., Sihag, S., Lehar, J., et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. doi: 10.1038/ng1180
Price, G. D., Caemmerer, S. V., Evans, J. R., Siebke, K., Anderson, J. M., and Badger, M. R. (1998). Photosynthesis is strongly reduced by antisense suppression of chloroplastic cytochrome bf complex in transgenic tobacco. Funct. Plant Biol. 25, 445–452.
Roider, H. G., Kanhere, A., Manke, T., and Vingron, M. (2007). Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics 23, 134–141. doi: 10.1093/bioinformatics/btl565
Saibo, N. J. M., Lourenco, T., and Oliveira, M. M. (2009). Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann. Bot. 103, 609–623. doi: 10.1093/aob/mcn227
Siberil, Y., Doireau, P., and Gantet, P. (2001). Plant bZIP G-box binding factors. Modular structure and activation mechanisms. Eur. J. Biochem. 268, 5655–5666. doi: 10.1046/j.0014-2956.2001.02552.x
Singh, A., Ram, H., Abbas, N., and Chattopadhyay, S. (2012). Molecular Interactions of GBF1 with HY5 and HYH Proteins during Light-mediated Seedling Development in Arabidopsis thaliana. J. Biol. Chem. 287, 25995–26009. doi: 10.1074/jbc.M111.333906
Smyth, G. K. (2005). “Limma: linear models for microarray data,” in Bioinformatics and Computational Biology Solutions Using R and Bioconductor, eds R. Gentleman, V. Carey, W. Huber, R. Irizarry, and S. Dudoit (New York, NY: Springer), 397–420. doi: 10.1007/0-387-29362-0_23
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Tamai, H., Iwabuchi, M., and Meshi, T. (2002). Arabidopsis GARP transcriptional activators interact with the Pro-rich activation domain shared by G-box-binding bZIP factors. Plant Cell Physiol. 43, 99–107. doi: 10.1093/pcp/pcf011
Thai, M. T., Cai, Z., and Du, D. Z. (2007). Genetic networks: processing data, regulatory network modelling and their analysis. Optim. Methods Softw. 22, 169–185. doi: 10.1080/10556780600881860
Williams, M. E., Foster, R., and Chua, N. H. (1992). Sequences flanking the hexameric G-box core CACGTG affect the specificity of protein binding. Plant Cell 4, 485–496. doi: 10.1105/tpc.4.4.485
Wullschleger, S. D. (1993). Biochemical limitations to carbon assimilation in C(3) plants - a retrospective analysis of the a/Ci curves from 109 species. J. Exp. Bot. 44, 907–920. doi: 10.1093/Jxb/44.5.907
Zhu, X. G., de Sturler, E., and Long, S. P. (2007). Optimizing the distribution of resources between enzymes of carbon metabolism can dramatically increase photosynthetic rate: a numerical simulation using an evolutionary algorithm. Plant Physiol. 145, 513–526. doi: 10.1104/pp.107.103713
Zhu, X. G., Long, S. P., and Ort, D. R. (2010). Improving photosynthetic efficiency for greater yield. Annu Rev. Plant Biol. 61, 235–261. doi: 10.1146/annurev-arplant-042809-112206
Keywords: photosynthesis, transcription factors, regulatory network, small world, coordination
Citation: Yu X, Zheng G, Shan L, Meng G, Vingron M, Liu Q and Zhu X-G (2014) Reconstruction of gene regulatory network related to photosynthesis in Arabidopsis thaliana. Front. Plant Sci. 5:273. doi: 10.3389/fpls.2014.00273
Received: 05 December 2013; Accepted: 25 May 2014;
Published online: 13 June 2014.
Edited by:
Bernd Mueller-Roeber, University of Potsdam, GermanyReviewed by:
Gerrit T. S. Beemster, University of Antwerp, BelgiumMing Chen, Zhejiang University, China
Copyright © 2014 Yu, Zheng, Shan, Meng, Vingron, Liu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-Guang Zhu, Group of Plant System Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, 320 Yueyang Road, Shanghai 200031, China e-mail:emh1eGluZ3VhbmdAcGljYi5hYy5jbg==