 Stephen A. Goff1*
Stephen A. Goff1* Matthew Vaughn2
Matthew Vaughn2 Sheldon McKay1 Eric Lyons1
Sheldon McKay1 Eric Lyons1 Ann E. Stapleton3,4 Damian Gessler1 Naim Matasci1 Liya Wang5
Ann E. Stapleton3,4 Damian Gessler1 Naim Matasci1 Liya Wang5 Matthew Hanlon2 Andrew Lenards1 Andy Muir1 Nirav Merchant1 Sonya Lowry1
Matthew Hanlon2 Andrew Lenards1 Andy Muir1 Nirav Merchant1 Sonya Lowry1 Stephen Mock2
Stephen Mock2 Matthew Helmke1
Matthew Helmke1 Adam Kubach2 Martha Narro1 Nicole Hopkins1 David Micklos6 Uwe Hilgert6 Michael Gonzales2 Chris Jordan2 Edwin Skidmore1 Rion Dooley2 John Cazes2 Robert McLay2 Zhenyuan Lu5 Shiran Pasternak5 Lars Koesterke2 William H. Piel7 Ruth Grene8
Adam Kubach2 Martha Narro1 Nicole Hopkins1 David Micklos6 Uwe Hilgert6 Michael Gonzales2 Chris Jordan2 Edwin Skidmore1 Rion Dooley2 John Cazes2 Robert McLay2 Zhenyuan Lu5 Shiran Pasternak5 Lars Koesterke2 William H. Piel7 Ruth Grene8 Christos Noutsos5
Christos Noutsos5 Karla Gendler2
Karla Gendler2 Xin Feng5,9 Chunlao Tang5 Monica Lent1 Seung-Jin Kim1 Kristian Kvilekval10 B. S. Manjunath10,27 Val Tannen11 Alexandros Stamatakis12 Michael Sanderson13
Xin Feng5,9 Chunlao Tang5 Monica Lent1 Seung-Jin Kim1 Kristian Kvilekval10 B. S. Manjunath10,27 Val Tannen11 Alexandros Stamatakis12 Michael Sanderson13 Stephen M. Welch14 Karen A. Cranston15 Pamela Soltis16 Doug Soltis17
Stephen M. Welch14 Karen A. Cranston15 Pamela Soltis16 Doug Soltis17 Brian O’Meara18 Cecile Ane19,20
Brian O’Meara18 Cecile Ane19,20 Tom Brutnell21 Daniel J. Kleibenstein22 Jeffery W. White23
Tom Brutnell21 Daniel J. Kleibenstein22 Jeffery W. White23 James Leebens-Mack24 Michael J. Donoghue25 Edgar P. Spalding26 Todd J. Vision28 Christopher R. Myers32 David Lowenthal29 Brian J. Enquist13 Brad Boyle13
James Leebens-Mack24 Michael J. Donoghue25 Edgar P. Spalding26 Todd J. Vision28 Christopher R. Myers32 David Lowenthal29 Brian J. Enquist13 Brad Boyle13 Ali Akoglu30 Greg Andrews29 Sudha Ram31 Doreen Ware5 Lincoln Stein5,9 and Dan Stanzione2
Ali Akoglu30 Greg Andrews29 Sudha Ram31 Doreen Ware5 Lincoln Stein5,9 and Dan Stanzione2
- 1 BIO5 Institute, University of Arizona, Tucson, AZ, USA
- 2 Texas Advanced Computer Center, University of Texas, Austin, TX, USA
- 3 Department of Biology, University of North Carolina, Wilmington, NC, USA
- 4 Department of Marine Sciences, University of North Carolina, Wilmington, NC, USA
- 5 Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
- 6 DNA Learning Center, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
- 7 Yale Peabody Museum, Yale University, New Haven, CT, USA
- 8 Department of Plant Pathology, Physiology and Weed Science, Virginia Tech University, Blacksburg, VA, USA
- 9 Ontario Center for Cancer Research, Toronto, ON, Canada
- 10 Center for Bio-image Informatics, University of California, Santa Barbara, CA, USA
- 11 Department of Computer and Information Science, University of Pennsylvania, Philadelphia, PA, USA
- 12 Scientific Computing Group, Heidelberg Institute for Theoretical Studies, Heidelberg, Germany
- 13 Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ, USA
- 14 Department of Agronomy, Kansas State University, Manhattan, KS, USA
- 15 National Evolutionary Synthesis Center (NESCent), Durham, NC, USA
- 16 Florida Museum of Natural History, University of Florida, Gainesville, FL, USA
- 17 Department of Biology, University of Florida, Gainesville, FL, USA
- 18 Department of Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN, USA
- 19 Department of Statistics, University of Wisconsin, Madison, WI, USA
- 20 Department of Botany, University of Wisconsin, Madison, WI, USA
- 21 Boyce Thompson Institute for Plant Research, Cornell University, Ithaca, NY, USA
- 22 Department of Plant Sciences, University of California, Davis, CA, USA
- 23 Arid-Land Agricultural Research Center, United States Department of Agriculture-Agricultural Research Service, Maricopa, AZ, USA
- 24 Department of Plant Biology, University of Georgia, Athens, GA, USA
- 25 Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT, USA
- 26 Department of Botany, University of Wisconsin, Madison, WI, USA
- 27 Electrical and Computer Engineering, University of California, Santa Barbara, CA, USA
- 28 Department of Biology, University of North Carolina, Chapel Hill, NC, USA
- 29 Department of Computer Science, University of Arizona, Tucson, AZ, USA
- 30 Department of Electrical and Computer Engineering, University of Arizona, Tucson, AZ, USA
- 31 Eller School of Business, University of Arizona, Tucson, AZ, USA
- 32 Department of Physics, Cornell University, Ithaca, NY, USA
The iPlant Collaborative (iPlant) is a United States National Science Foundation (NSF) funded project that aims to create an innovative, comprehensive, and foundational cyberinfrastructure in support of plant biology research (PSCIC, 2006). iPlant is developing cyberinfrastructure that uniquely enables scientists throughout the diverse fields that comprise plant biology to address Grand Challenges in new ways, to stimulate and facilitate cross-disciplinary research, to promote biology and computer science research interactions, and to train the next generation of scientists on the use of cyberinfrastructure in research and education. Meeting humanity’s projected demands for agricultural and forest products and the expectation that natural ecosystems be managed sustainably will require synergies from the application of information technologies. The iPlant cyberinfrastructure design is based on an unprecedented period of research community input, and leverages developments in high-performance computing, data storage, and cyberinfrastructure for the physical sciences. iPlant is an open-source project with application programming interfaces that allow the community to extend the infrastructure to meet its needs. iPlant is sponsoring community-driven workshops addressing specific scientific questions via analysis tool integration and hypothesis testing. These workshops teach researchers how to add bioinformatics tools and/or datasets into the iPlant cyberinfrastructure enabling plant scientists to perform complex analyses on large datasets without the need to master the command-line or high-performance computational services.
What is iPlant?
iPlant is a cyberinfrastructure development project designed to create the foundation to support the computational needs of the research community and facilitate progress toward solutions of major problems in plant biology. This cyberinfrastructure foundation must support a diverse group of plant science researchers and bring together experts in various fields of biology and computer science. The platform created by iPlant helps researchers use tools and data more easily and efficiently, gain access to high-performance computing (HPC) when it is needed, and provide interoperable software analysis and large data access in a sustainable fashion. The cyberinfrastructure platform is useful to scientists at all levels of expertise ranging from students to traditional biology researchers and computational biology experts. An initial release of the iPlant cyberinfrastructure is available and enhancements are being released on a regular basis.
Why the Research Community Needs Cyberinfrastructure Now
Consider the following example. Tara1, a plant biology researcher, wants to know how varieties of a major crop species can be developed to better suit a changing environment. She is coordinating a collaborative project to address this question by identifying and analyzing small molecules, drought responsive regulatory/signaling pathways, and key epigenetic events in plants with a long history of adaption to limited water. To do this, her global team generates, and uses, molecular genetics, transcriptomic (including small RNA expression) and metabolite profiling data from related genotypes/varieties within the species under study, and data from closely related species that differ in their tolerance to drought.
Tara takes advantage of iPlant’s cyberinfrastructure, which helps her generate predicted functions for her team’s candidate pathways and modes of their regulation. Data are integrated and candidate genes are selected based on their association with specific regulatory/signaling and metabolic pathways and physiological tolerance traits from small-scale field trials. Tara infers putative roles for these genes in cellular function and environmental adaption, and uses her working hypotheses to set priorities for her team’s large-scale experimental tests. These large-scale studies further validate correlations between drought-tolerance and specific genomic responses, allowing Tara and her team to prioritize the genes and their variants for use in new crop variety development.
Like Tara, many plant scientists today are uniquely positioned to address some of the world’s most pressing societal, economic, and environmental challenges. From feeding an expanding human population to creating new forms of renewable energy, advances in plant science promise to deliver new solutions to urgent problems. These challenges will be addressed through breeding efforts based on modern molecular analysis techniques, through a better understanding of the evolution of important plant traits, and through better predictions of the environment’s impact on plant physiology. Biology is a data-driven and data-intensive science (Smith et al., 2011a). Biologists are inundated with new data, from ever-cheaper DNA sequence data to complex traits, species relationships, environmental impacts and responses, and molecular phenotypes. Plant science data range in scope from complete genome sequences of individual plant varieties to geospatial maps of plant species distribution across the entire biosphere (Hughes, 2006; Armstead et al., 2009). These data vary in scale from the results published in a single journal article to data entries in enormous databases. Analytical methods are being developed at an accelerating pace – but data sets are not necessarily easy to integrate and tools to analyze these data are often inaccessible or poorly scalable. The data integration problem is larger than a single lab can handle, and the solution requires cross-disciplinary approaches with expertise from computer science, information science, and the life sciences. Investment in the creation of the existing analysis tools and datasets has been significant and must be leveraged by iPlant (Benfey et al., 2010; Buell and Last, 2010; Cook and Varshney, 2010; Edwards and Batley, 2010; Hirayama and Shinozaki, 2010; Paterson et al., 2010; Pichersky and Gerats, 2011; Proost et al., 2011). Use of analysis tools in isolation contributes to the lack of experimental verifiability/reproducibility for computational analyses. This article describes how iPlant’s cyberinfrastructure addresses these profound needs, and how researchers like Tara will benefit from the cyberinfrastructure.
Cyberinfrastructure (CI), as defined by the NSF in their CI Vision report (Atkins et al., 2003) includes the use of HPC, use of large shared data storage, and the establishment of collaborations and virtual organizations around shared analysis tools and analyzed data. Traditional bioinformatics focuses on solutions to individual problems. The CI approach is to provide a foundation from which bioinformatics work can proceed efficiently in a collaborative environment. The iPlant CI for plant biology (or life sciences in general) is leveraging the computational and storage infrastructure created by hundreds of millions of dollars in NSF investments such as the TeraGrid (now XSEDE). The iPlant CI is focused on developing the comprehensive platform to support data analysis tools and data storage useful for plant biology research and subsequent applications. iPlant’s CI platform provides methods for leveraging physical resources, integrating tools, and integrating data. This platform will be sustainable and species-independent. Other efforts in CI development such as the Department of Energy’s Systems Biology Knowledgebase (Gregurick, 2010) and the European Life Sciences Infrastructure for Biological Information (ELIXIR, 2010) have overlapping synergistic goals. These efforts are being coordinated with iPlant’s CI development where appropriate and mutually beneficial. Plant biologists like Tara are being empowered to use HPC and integrated tools and data in collaborative research projects without becoming computational experts.
Collaborations and Grand Challenges
The iPlant CI is a platform designed to enable researchers to make progress toward solutions of Grand Challenge problems; these are questions fundamental to plant biology and are too large for any single lab to tackle in isolation. The Grand Challenge focus is a mandate for plant science CI development, and enables plant scientists to coordinate cross-disciplinary research efforts. A yearlong series of iPlant-sponsored workshops, meetings, and white papers culminated in the iPlant Board of Directors, an independently chosen group of community members, prioritizing two Grand Challenge focus efforts:
The iPlant Tree of Life (iPToL) Project: To build scalable tools to permit the generation of phylogenetic trees containing all green plant species (∼500,000 taxa), decorated with additional data (e.g., phenotypic traits), and analyzed efficiently to facilitate discovery, and
The iPlant Genotype-to-Phenotype (iPG2P) Project: To provide scalable analytical tools, data integration, and data storage systems to facilitate the prediction of a plant’s phenotype given the plant’s genetic makeup and sufficient environmental information about where it is grown and the conditions under which it is grown.
Phylogenetic trees help biologists understand the tempo and mode of the evolution of individual plant species and related groups of species, the evolution of plant genomes, the progression of plant development, and the distribution and interactions of organisms in communities and ecosystems. Phylogenetic methods are being used to identify and predict responses to a changing global climate (Yesson and Culham, 2006a,b; Faith, 2008; Willis et al., 2008; Donoghue et al., 2009; Hendry et al., 2010; Thuiller et al., 2011).
Understanding the association between a specific genotype, either a single genetic trait or a set of genes or pathways, and a measured phenotypic trait, is a shared goal across all the life sciences: plant, animal, fungal, and microbial. For plant biologists it is particularly important to incorporate environmental interactions into the association of traits and phenotypes because the environment has a tremendous impact on observed plant phenotypes. The aforementioned example in drought response research would benefit from both Grand Challenge projects described above because Tara’s team would use an iPToL generated species phylogeny and gene family evolutionary relationships overlaid on a common phylogenetic tree to identify, across species, homologs of genes or small RNAs whose responses correlated with superior drought-tolerance in their field trials. She could then use the environmental interaction analyses from the iPG2P project’s tools to investigate potential cellular and whole-plant contributions to drought-tolerance physiology.
To facilitate the two major Grand Challenge Projects outlined above, iPlant is supporting and collaborating with a variety of complementary smaller projects. These projects include a high-throughput image analysis platform for automated phenotyping, cloud computing development to provide use of virtual machine images, and semantic web technology development to facilitate web-based data and tool discovery. Several smaller CI development projects, collectively called “Seed Projects,” are supported by iPlant to provide the initial development of CI for plant nutrition, plant adaptation, forest tree biology, and botanical geospatial diversity. These projects complement the Grand Challenge focus and provide CI support across the diverse disciplines of plant biology. Specific tools are being developed that support reproducibility of bioinformatics analysis, scientific networking for phylogenetics researchers based on specific clades of interest, standardized storage of genome sequence information, provenance tracking of analysis, the use of graphics processing units for life science analysis, and facilitation of modern plant breeding. These efforts are described in detail below. The general philosophy of iPlant’s development effort is to take advantage of the numerous existing analysis tools and data sets by adapting them to iPlant’s foundational CI platform rather than re-developing tools and support systems (Galperin and Cochrane, 2011; Gaudet et al., 2011). (see Box 1 for a summary of the integrated tools.) The iPlant CI project serves as both a process for gathering user requirements and as a platform for providing access to resources in a uniform fashion, improving usability through consistent access models, and tracking provenance – all of which are essential for making computational experiments transparent, verifiable, and sustainable. Above all, the iPlant CI is extensible and is designed to grow with the needs of the plant science research community. Researchers like those collaborating with Tara will help define the user requirements that drive iPlant development.
Box 1. Current CI services available (and more coming online regularly).
• Bioinformatics software available through the iPlant Discovery Environment
– Data Importers
– Sequence alignments and phylogenetic tree building
– Phylogenetic and evolutionary analyses
– QTL mapping and genome-wide association studies
– Ultrahigh-throughput sequence processing
– Functional analyses
– Clustering and network analyses
– Variant detection and annotation
– RNAseq analyses
– ChIP-seq studies
– Utility tools and scripts
– Full list at https://pods.iplantcollaborative.org/wiki/display/DEman0p4/Tools + list
• Access to collaboration tools
– Public and private wiki spaces, Mailing lists
– Video conferencing setup and support
• Data hosting – Access to mirroring, backup, and recovery services at petascale
• Web and application hosting
• Access to persistent virtual machines
– Algorithm development
– Software prototyping
• Command-line access to production and experimental supercomputers, archive systems
• Access to an online bug tracking and issue system
• Git/svn code hosting within iPlant and through SourceForge and GitHub
The iPlant Cyberinfrastructure Organization and Architecture
The IPlant Discovery Environment
The primary graphical user interface to iPlant is the discovery environment (DE). The DE provides a web interface and a platform to access the computing, data storage, and analysis application resources provided by iPlant. The DE is designed to facilitate data exploration and scientific discovery by integrating analytical tools as modular components that may be used individually or in workflows, accessing iPlant’s data store, and seamlessly running tools on local or HPC nodes depending on the throughput and resource needs of the analysis. In addition, the DE will employ provenance tracking of both primary and derived files to track and reproduce experiments, and collaboration tools enabling users to share data, workflows, analysis results, and data visualizations.
What is behind the Screen – iPlant’s CI Features
The overarching goal of iPlant’s CI is to help biologists like Tara and her team to effectively allocate their time toward answering biological research questions, rather than dealing with computing resource details, learning new analysis software with each new question, or converting data between file types. iPlant’s CI makes several aspects of computation easier including: (i) data management, (ii) analysis management and execution, (iii) computational scalability, (iv) large dataset sharing, and (v) large dataset processing. Depending on the type of user (biologist, bioinformaticist, programmer), these features are already available via a web interface, RESTful services (see Box 2 for definitions and links), and underlying application programming interfaces (APIs). However, each future release of iPlant’s software will integrate additional features and make them more visible, accessible, and easy to use; furthermore, iPlant utilizes community feedback to make iterative improvements. To enhance collaboration and allow researchers to build on previous discoveries rather than duplicate efforts, the iPlant CI supports sharing of analyses and workflows when users desire to do so. Data, analysis tools, analysis workflows and results visualization are all supported by HPC and cloud computing resources. These pillars of functionality provide quality analyses, security, life cycle management, governance, provenance for data, and sustainability. The CI components range from the person analyzing data to the computer chip executing the analysis (see Figure 1).
Box 2. Definitions, abbreviations, and acronyms.
iPlant – The iPlant Collaborative
CI – Cyberinfrastructure
DE – iPlant’s Discovery Environment
Tools – Data analysis methods that accept specific data types as inputs, do an operation and return the results of the operation as outputs.
iRODS – Integrated Rule-Oriented Data Management System – www.irods.org
PSCIC – NSF Plant Science Cyberinfrastructure Collaborative
HPC – high-performance computing
HTC – high-throughput computing
TeraGrid – NSF’s open scientific grid computing project that includes 11 partners:
Indiana, LONI, NCAR, NCSA, NICS, ORNL, PSC, Purdue, SDSC, TACC and UC/ANL. See https://www.teragrid.org/
XSEDE – eXtreme Science and Engineering Discovery Environment, the TeraGrid sites after July 1, 2011
Elixir – European life science infrastructure for biological information. See http://www.elixir-europe.org/page.php
iPG2P – iPlant Genotype-to-Phenotype Grand Challenge project
iPToL – iPlant Tree of Life grand challenge project
RESTful Services – Representational State Transfer – a key design idiom that embraces a stateless client-server architecture in which the web services are viewed as resources and can be identified by their URLs. See http://www.oracle.com/technetwork/articles/javase/index-137171.html
API – application programming interface, allows computational access to the software or services
Metadata – Data that describes or provides information on other data or data sets
SSWAP – simple semantic web architecture and protocol – http://sswap.info/
Taverna – Workflow engine for biological analysis – http://www.taverna.org.uk/
Kepler – Open-source scientific workflow engine – https://kepler-project.org/
Pegasus – Workflow management system – http://pegasus.isi.edu/wms/
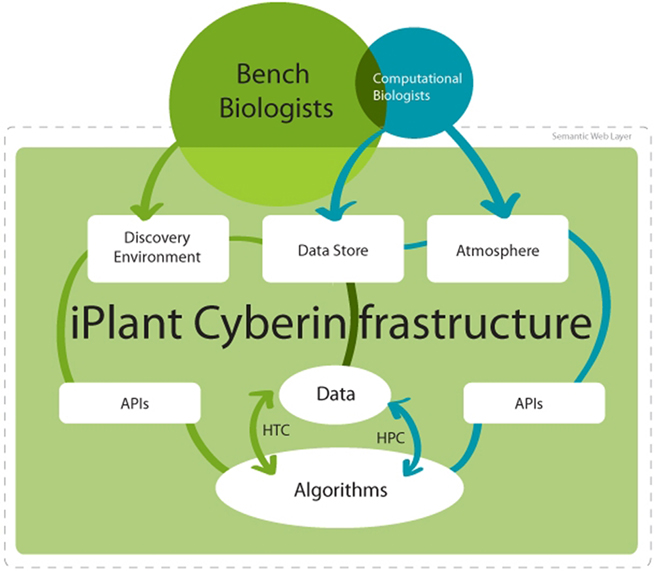
Figure 1. Overview of access options for iPlant’s major cyberinfrastructure components. iPlant’s CI consists of several major systems, each of which provides a set of computational resources for different types of users. The basic level of services provides access to data and data analysis algorithms. For large datasets, there are two general paradigms for computing: high-throughput computing (HTC) and high-performance computing (HPC). The major difference is that HTC provides rapid access to many different data types requiring discreet computation while HPC provides access to many interlinked compute nodes for tightly coupled parallel computation. Data resides in iPlant’s Data Store, a cloud-based distributed system for storing and sharing large quantities of data that are automatically replicated between iPlant’s major sites. iPlant resources can be accessed directly or through APIs. iPlant’s Discovery Environment is a web-based system and provides functionality to manage data, add new algorithms and tools, and run analyses on appropriate computational resources. Atmosphere is iPlant’s on-demand cloud computing resource that allows users to launch virtual machines, install complex software of their choice, connect to iPlant’s Data Store and other compute resources, and share cloud resources with collaborators. Together, iPlant’s CI proves a wealth of interconnected computational and data management resources to users with different needs and diverse levels of computational expertise.
The DE provides access to a range of bioinformatics tools and workflows using a high-level portal for users who do not want to interact directly with the lower-level infrastructure such as the command-line on a UNIX or Linux system. Scientific analysis tools and supported workflows in the current release of the DE include trait evolution analysis on phylogenetic trees [continuous and discrete ancestral character estimation, phylogenetic independent contrasts (PIC)], ultrahigh-throughput DNA and RNA sequence analysis (pre-processing, variant detection, transcript abundance), analysis of gene duplication patterns as compared to species trees (tree reconciliation), and taxonomic name resolution, which assists in finding alternative spellings or variant names for species lists. (see Box 1).
The iPlant Cyberinfrastructure Architecture
As in all life science research, plant biology data and analytical methods evolve rapidly. iPlant’s CI uses a modular design to be:
1. Flexible for changing data and new analytical tools.
2. Extensible for accommodating varying analytical workflows and visualization methods.
3. Scalable for increasing data volume and compute cycle needs.
4. Upgradable to provide more robust analysis solutions.
iPlant’s CI makes use of both iPlant-owned hardware resources and NSF TeraGrid (now XSEDE) hardware. This approach leverages the massive computational and data storage systems created with NSF funding at the Texas Advanced Computing Center (TACC) and other XSEDE service providers. Data are replicated between Texas, San Diego and the University of Arizona via the iRODS (integrated Rule-Oriented Data Management System; Rajasekar et al., 2006) to provide reliable, replicable, and scalable storage for very large data sets. The iRODS software has been adopted as the data management middleware for the iPlant Collaborative and provides the facilities for data federation, data replication, quota management and access control. Data federation is a method of linking data from two or more physically different locations and making the access appear as if the data was co-located. This is an example of synergy between NSF-funded projects, since iRODS and iPlant are both NSF-funded projects. Tara and her collaborators will be able to store data in a simple file format and will be unaware that this integrated data is in different physical locations.
Access to the iPlant CI hardware is provided by a software layer of core services (see Figure 2). An API (Public API) layer creates a unified, consistent way to access the diverse resources contained in the layers beneath it. These APIs allow bioinformatics experts and software developers to embed iPlant resources in their own scripts and tools. Running on top of this layer is an application layer that provides various interfaces for users. The primary graphical user interface is the web-based iPlant DE. The iPlant CI allows users to access resources at any layer. For expert users, direct command-line access to the compute and storage resources is available. For bioinformaticians, direct access to the API allows the embedding of iPlant compute, data, and analysis resources directly into their own scripts and workflows, or the creation of their own interfaces at the application layer for their own users and communities. Finally, any user can access resources through the DE or other interfaces available at the application layer.
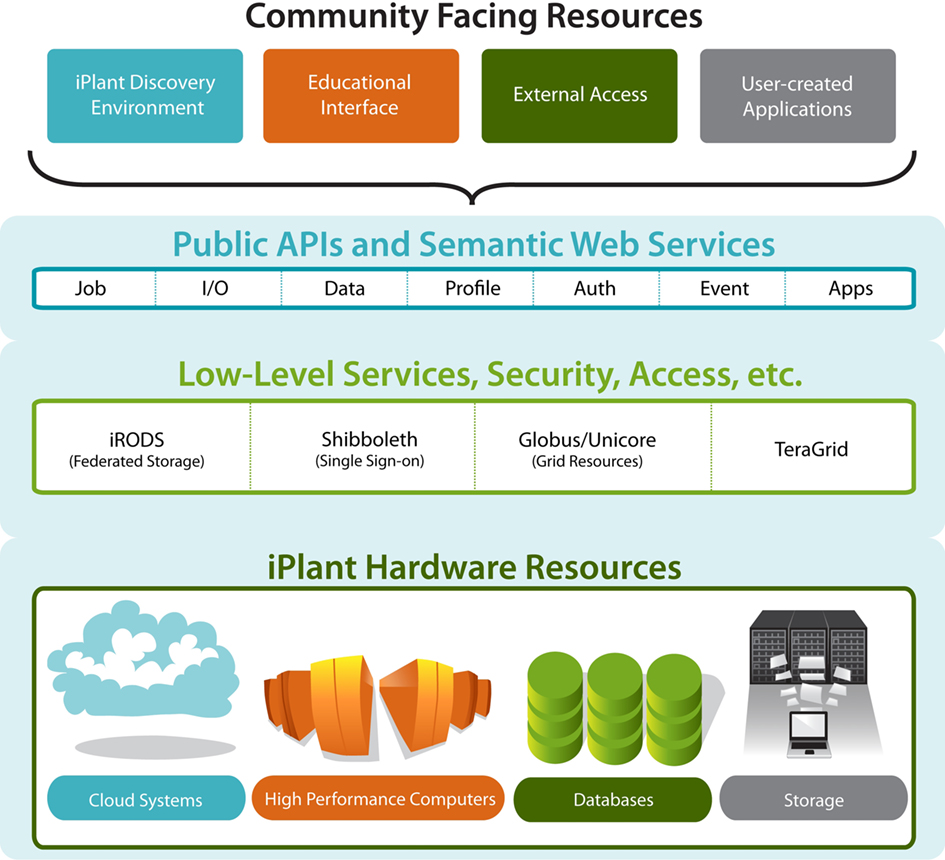
Figure 2. An illustration of the multilayered iPlant cyberinfrastructure platform. The three primary building blocks of the iPlant CI are depicted with overlying community access options. Individual blocks are comprised of distinct software and hardware components that collectively provide the integration and communication capabilities with the adjacent blocks. This architecture facilitates the rapid development of applications and user interfaces on a robust underlying foundation, and provides unified secure access to scalable storage and the computational grid infrastructure.
The iPlant APIs
iPlant’s layered approach for the APIs present a generic, RESTful set of interfaces for basic actions like file and data operations, user authentication, tool integration, compute job invocation, and event monitoring. On top of these Foundational APIs is the semantic awareness interface, based on simple semantic web architecture and protocol (SSWAP2; Gessler et al., 2009; Nelson et al., 2010). It is made up of interfaces for metadata-driven workflow construction and orchestration, extraction and management of metadata for data and tools, and interactions with federated data sources for integration of third-part hosted data. Metadata is simply data that describes and gives information about other data. From these small discrete operations, complex analytical workflows can be constructed. Each of these Foundational APIs is exposed via HTTP, and can be used by web applications like the DE, workflow management applications such as Taverna (Oinn et al., 2004; Hull et al., 2006; Kawas et al., 2006; Krabbenhoft et al., 2008; Lanzen and Oinn, 2008; Li et al., 2008; Wassink et al., 2009; Kano et al., 2010), Pegasus (Deelman et al., 2004), and Kepler (Altintas et al., 2004; Ludascher et al., 2006), various third party Web applications, and other RESTful web services, or by user-written scripts.
The iPlant input/output (IO) API provides a simple interface to bring data in and out of the storage repository. It allows users to directly import, export, and organize files, and mark those data with additional metadata descriptions. The iPlant IO API may be used to retrieve data from remote data resources for subsequent processing, transfer users’ files to remote Internet accessible locations, and manage permissions on files and directories. The iPlant DATA API facilitates the translation and transformation of data between different file formats. File formats are described by simple metadata information and by computer code that executes pairwise translation between formats and versions using information provided by file format developers. Data operations can be performed in-place on user data, reducing the need to move files.
Bioinformatics application developers and research software users can take advantage of iPlant Foundational APIs to manage tools and applications. These APIs provide an interface with which to describe the properties and parameters of an analytical application, to identify applications with specific properties or capabilities, and to run instances of those applications on HPC resources. The parameters for invoking a particular software analysis program are described in a simple metadata language. This metadata description is stored, allowing the analysis software to be identified and used by other researchers. Potential users can search for analysis programs to run and receive detailed, programmatically interpretable information about how to invoke them via the JOB API. In addition, the JOB API permits retrieving the state of a running job as well as its outputs and associated submission metadata. Output files can be automatically sent to the user’s home directory, where they are available via the IO API. The APPS and JOB APIs interact with the Semantic API, described below, to automatically create Resource Description Graphs and Resource Invocation Graphs, so all applications developed under this system become semantically discoverable and usable. The innovative feature of this system is that numerous bioinformatics applications may be discovered and used via a single, easy-to-learn interface that is compatible with today’s advanced web-based application technologies.
Other iPlant Foundational APIs include:
1. An event management system that permits both users and computational applications to publish and subscribe to notifications about the status of various activities in the iPlant CI.
2. An authentication service that provides federated access to iPlant services without explicitly transmitting user credentials.
3. An auditing service that allows the tracking of resource use and access patterns by iPlant.
4. A profile service that creates computer-readable summaries of user profile data.
Bioinformatics experts on Tara’s collaborating team will be able to use the iPlant APIs to connect their analysis software to the iPlant CI, and non-experts will take advantage of the APIs through a user-friendly interface.
Atmosphere, iPlant’s Cloud Computing Service
While the large-scale cluster and storage resources provided by the iPlant CI are suitable for many applications, some existing applications need a dedicated server to provide their own interface, a local database, or persistent services. To provide a reliable home for all of these applications, and integration with the iPlant CI, the iPlant team created cloud-style services called Atmosphere. Atmosphere provides users with an image of a virtual machine, which is a completely isolated operating system (Smith and Nair, 2005). iPlant provides many different types of virtual machine images, from basic Linux to Linux with complex analytical software stacks pre-installed and configured. Running instances may be modified and used to create new images for sharing additional software stacks. In addition iPlant’s virtual machines are configured to retrieve and store data from iPlant’s iRODS data repository to provide long-term storage of cloud-accessible data. By enabling researchers to access cloud resources where testing can be done more easily and safely using machines that can be built, rebuilt, or removed in minutes, Atmosphere will accelerate the pace of scientific discovery by plant scientists who are developing new tools and algorithms. Users of Atmosphere can create cloud instances for development, host tools within iPlant with custom interfaces to any user community, or provide custom tools integrated through the iPlant API. Collaborators on Tara’s team could use iPlant’s Atmosphere to share software analysis routines and process datasets reproducibly even when the data are stored only at specific distant locations, such as at field sites.
Private cloud computing solutions typically provide access via only Infrastructure as a Service and/or Platform as a Service (see Box 3). iPlant’s Atmosphere lowers the entry barrier by also providing a third level of service, Software as a Service. Atmosphere’s full cloud services include Infrastructure as a Service with advanced APIs, Platform as a Service with capabilities for developing and deploying software applications to public users, and Software as a Service with preconfigured, frequently used analysis routines, relevant algorithms, and data sets in an available on-demand environment (Box 3).
Box 3. Services provided by iPlant.
Infrastructure as a Service (IaaS). Atmosphere provides cloud infra-structure managers with the ability to dynamically manage computing resources, network resources and user resources, such as allocation of virtual cores on a per-user basis, allocation of memory on a per-user basis, quota management on the total number of CPU hours, amount of memory, amount of storage, and the number of instances created by a specific user.
Platform as a Service (PaaS). Atmosphere provides tool developers with the ability to create resources on-demand using an intuitive rich web graphical user interface. Each virtual machine is an isolated, fully independent computing environment with the ability to utilize persistent storage. Atmosphere’s PaaS facilitates the deployment of applications without the cost and complexity of buying and managing the underlying hardware and software. Atmosphere’s PaaS provides all the facilities required to support a complete life cycle of building and delivering web applications and services entirely available from the Internet.
Software as a Service (SaaS). Atmosphere’s software services allow tool users to access the applications/tools provided by specific tool developers as well as those provided within iPlant’s cyberinfrastruc-ture. To provide an intuitive research environment for the biologist, Atmosphere uses an application catalog accessible by an intuitive user interface. The key benefit of Atmosphere’s SaaS is the speed and ease with which it provides a fully configured environment for tool users. Atmosphere is modeled after the familiar application-style interface, much like the Apple iPad/iPhone or the Android Operating System, users select analysis tool icons to launch from an application catalog. Atmosphere provides additional convenience while working with the analysis tools, such as sending notifications, providing usage statistics, detailed information, and advanced management features for users of any level of technical expertise.
API Service. In addition to the three major service models described above, Atmosphere also provides open-source APIs for deeper inte-gration with other software and services. The Atmosphere APIs are HTTP-based Remote Procedure Calls (see http://en.wikipedia.org/wiki/Hypertext_Transfer_Protocol for additional details). Atmosphere’s APIs include functionality providing notifications of changes in a user’s resources, management of VMIs, and external web service connec-tions when a virtual instance changes state. iPlant is currently working on additional functionality to improve Atmosphere including integration of Atmosphere with iPlant DE, the ability to access large public data sets, automation for bundling of multiple VMIs, improved application catalog features (searching, collaboration, and personalization), and save specific VMIs for later use. For more details on Atmosphere, please see the tutorial: https://pods.iplantcollaborative.org/wiki/display/atmosphere/Demo+with+picture+walkthrough.
The iPlant Semantic Web
The World Wide Web is a system of linked hypertext documents that cannot easily be computationally processed to discover and extract information useful to plant research biologists. The vision of iPlant’s semantic web effort is to enable computer programs to create more biologically relevant connections between web documents and facilitate the use of web-based information in plant science research. The iPlant semantic web effort is designed to link genomic data with phylogenetic, evolutionary, proteomic, and metabolomic data, and so forth. This semantic web approach is appropriate where data connectivity is unclear, where connections may be unknown at design time, where data is contributed by multiple, independent sources (such as the members of Tara’s collaborating team), and where value and context are also subject to change.
Many biological research applications are available as web services. Software designed to use a series of protocols over the internet greatly expands the potential user base. The semantic web and web services both deliver important functionality, but they currently exist as separate technologies. The semantic web lacks formal web service protocols, while web services lack the explicit semantics and formal logic of the semantic web. iPlant is leveraging a novel hybrid approach that integrates aspects of the semantic web and web services into a single semantic web services protocol and architecture called SSWAP (Gessler et al., 2009; Nelson et al., 2010). The iPlant semantic web effort has developed an API and a software development kit that allows web service providers to describe how their services work in a language amenable to machine reasoning. This is done using the industry standard Web Ontology Language (OWL; Mcguinness and Van Harmelen, 2011). The semantic web approach allows service requestors to discover data and analysis services with a high degree of connectivity, and is achieved by re-purposing existing peer-reviewed community ontologies for semantic web services use. The iPlant Semantic Web Architecture3 differs from other semantic web platforms by using automated machine reasoners at the time a service is requested. This feature means that there is no need for parties to pre-agree on domain vocabularies or be limited by existing static data models. The platform handles the necessary conversions in a fashion that is transparent to both the developer and end user. The only specialized operation required by individual web sites is their own mapping of their idiosyncratic schemas into and out of a shared, public semantic.
The iPlant Grand Challenge Projects – The Scientific Focus of iPlant Development
The IPlant Tree of Life Grand Challenge Project
iPlant tree of life was created to facilitate understanding of the evolutionary relationships of green plant species, and to use this knowledge to gain insight into the evolution of specific plant traits. A major iPToL objective is to create a CI that facilitates the generation of very large phylogenetic trees (500,000 taxa) on a regular basis, and visualize and interact with those trees. However, particular sub-challenges needed to be addressed and resolved:
1. Most of the relevant biological data to generate and decorate phylogenetic trees are not yet collected, and a large fraction of what has been collected is not sufficiently organized.
2. Hundreds of phylogenetic analysis tools exist, probably enough to address most biological questions. However, those analysis tools are not necessarily interoperable and frequently use inconsistent data formats.
3. Some phylogenetic tree construction tools take too much time to run with even a relatively small number of taxa.
4. Phylogenetic tree visualization tools do not scale to large tree sizes (from tens to hundreds of thousands of taxa).
Discussion of the CI needed to address these challenges resulted in several defined deliverables:
1. A scientific networking site for gathering existing phylogenetic data.
2. A system to host relevant data without an NCBI home.
3. A system with a common user interface and API to access data and analytical tools.
4. Enhanced phylogenetic tree construction tools that run in HPC environments.
5. A phylogenetic tree visualization tool with dynamic scaling running on a system where RAM resources are guaranteed.
iPtol Large Phylogenetic Trees
It is estimated that there are ∼500,000 species of green plants (Viridophytes). Yet, until recently, only about 1% of these species were assigned positions in a phylogenetic tree. Much larger trees are now being constructed, one containing approximately 10% of green plant species (Smith et al., 2011b). iPToL working group members are making construction of such large trees more tractable by optimizing existing tree construction methods such as Maximum Likelihood with RAxML (Stamatakis et al., 2005, 2008; Stamatakis, 2006; Stamatakis and Ott, 2008) and Neighbor Joining with NINJA/WindJammer (Saitou and Nei, 1987; Wheeler, 2009; Mclay et al., 2011). A new version of RAxML (RAxML-Light version v105) makes it possible to compute trees that require 1TB of RAM with 20,000,000 sites and about 1,500 taxa on approximately 700 CPUs in less than 48 h (computing the likelihood on a single alignment). iPToL is also working to improve the infrastructure for creating very large phylogenetic trees through the use of HPC so large analyses can be performed in a timely fashion. iPToL working group efforts are focused on improving the existing phylogenetic analysis software through the addition of checkpointing, by code parallelization, and by code refactoring in HPC-friendly languages. The original tree creation software could require months of runtime and large amounts of computer memory to analyze the relationships of several thousand species. The enhanced analysis software speeds the analysis by two orders of magnitude while decreasing the memory requirement. iPlant is providing support to create large phylogenetic trees and regularly update these large trees as new data become available. Tara’s collaborating group will benefit from comparative analysis of related drought-sensitive and drought-tolerant plant groups.
Another way to make inference of large phylogenetic trees more tractable is to simplify access to these improved codes so they can run efficiently by any user on scalable high-end computational resources. To accomplish this, iPlant is supporting the ongoing development of the CIPRES Science Gateway project4 (Miller et al., 2010), which has developed software and tools for deploying analyses on XSEDE HPC resources through a browser interface. The CIPRES Science Gateway group is integrating community tools for sequence alignment and tree inference.
Visualizing and manipulating large phylogenetic trees is also computationally challenging and is facilitated by the iPlant CI. A large tree viewer has been developed that is capable of displaying evolutionary relationships for up to 500,000 species with branch lengths included. Biologists can browse the large tree, zoom in to specific groups of related species, select species of interest, and annotate specific species with additional data. Resolution-dependent renderings, or semantic zooming techniques, are used to display an appropriate level of detail.
iPtol Tree Reconciliation
The evolutionary history of genes and gene families is often more complex than species phylogenetic relationships due to processes such as gene duplication and gene loss, interspecies hybridization and horizontal gene transfer events. Reconciling gene trees with species trees provides insight into how gene families have evolved and helps to identify orthologous (diverged by speciation) versus paralogous (diverged by duplication) genes when comparing different species. To reconcile species trees and the gene trees, iPlant developed an analytical pipeline that uses a known species phylogeny to guide the generation of an optimized gene tree. This gene tree is then used to infer duplication events and identify paralogous genes and orthologous genes. The current implementation of the analysis pipeline is based on MUSCLE (Edgar, 2004) to align sequences, and TreeBeST (Vilella et al., 2009), a Maximum Likelihood method. The tree reconciliation service uses a version of the Ensembl Compara database (Vilella et al., 2009) that was extended to model reconciled gene/species trees. iPlant’s Tree Reconciliation Service offers searches of the reconciled tree data from a variety of entry points, including gene names, gene ontology (GO) terms, and BLAST searches. The search results are highly visual in nature, and an interactive tree viewer forms a central part of the interface. iPlant is also developing an alternative gene/species tree reconciliation method based on the Bayesian approach employed in PrIME-GSR (Akerborg et al., 2009). These two methods are being used to analyze the growing amount of data coming from the 1,000 plant transcriptome project, which has the goal of sequencing expressed genes from 1,000 phylogenetically diverse species5. The analytical pipeline from this project will be made available to the plant science community and will benefit Tara’s collaborating team by providing them with comparative tools to study abiotic stress across a wide range of species.
iPtol Trait Evolution
Trait Evolution is a post-tree analysis approach that provides the scientific community with the ability to make inferences about evolutionary processes. Trait Evolution uses phylogenetic relationships to more accurately interpret trait data gathered from multiple species. Many methods and software are used to associate trait variation with phylogenetic relationships. However, the available software analysis programs do not scale to the magnitude of DNA sequencing and phylogenetic data now available. In some cases, the analysis programs were written for phylogenetic trees with fewer than a thousand species, and do not handle computer memory management, are not optimized for speed, or are simply not designed to handle the data volume that underlie the large phylogenetic trees currently available. Even well-designed analysis software programs can be too slow for real-time application. The iPToL Trait Evolution group is developing an infrastructure to support trait analysis of very large trees.
The Ancestral Character Estimation software allows researchers to estimate the state of an ancestral character and its associated uncertainty given a set of observations and the species’ phylogeny. Both continuous and discrete characters are supported and estimated using a Maximum Likelihood implementation written in R (Paradis et al., 2004). For continuous characters, like height or yield, the ancestral values and their 95% confidence intervals are obtained, whereas for discrete characters like flower color or leaf type, the proportional likelihoods of the possible states are reported. In both cases, the estimates can be plotted on a phylogenetic tree to visualize the character’s evolution.
Phylogenetic independent contrasts (Felsenstein, 1985, 2008; Ackerly and Reich, 1999; Stone et al., 2011) uses information about the phylogeny of organisms to test for correlated evolutionary changes in two or more traits. PIC is a statistically based approach that uses the phylogenetic tree and evolutionary branch lengths as a guide to determine whether two or more quantitative characters are evolutionarily correlated. By using a phylogeny, it avoids being misled by correlations that are due to the inheritance of similar characters, rather than adaptive changes. For example, PIC was used to evaluate leaf characters such as life span and specific area, among others, in light of alternative plant phylogenies and found a strong correlation between these characters, indicating convergence rather than commonality by descent (Ackerly and Reich, 1999). However, this same study showed that other traits such as leaf life span and lamina area are not correlated when phylogeny is taken into account. Tara’s team could use PIC to determine if two or more metabolite changes observed during drought responses are evolving independently.
The iPlant Taxonomic Name Resolution Service
The integration of disparate plant data sources is done through the matching of taxon names. However, this methodology assumes that names have been standardized. Unfortunately, this assumption is rarely attained in even the most highly curated datasets. The digitalization of biodiversity data is leading to the proliferation of erroneous taxon names. This “names problem” is increasingly becoming the fundamental challenge in integrating disparate massive data sources and impeding the progress of biodiversity science. Incorrect names and bad taxonomy presents a fundamental problem to comparative biology. For example, ecological studies encompass large numbers of species, conservation decisions are based on data from multiple sources, molecular analyses increasingly link sequence data from multiple organisms and taxa – all require accurate species names, and the correct matching of names among data sets. If uncorrected, lack of standardization of species names can lead to gross overestimations of species richness and mismatched observations.
The Taxonomic Name Resolution Service (TNRS) is a tool and service under development designed to reconcile misspelled taxonomic names with standardized versions and to convert synonyms to accepted names. The TNRS accepts a list of plant species names as input and compares names to a standardized list. The tool finds and returns exact matches and close matches, and provides the submitter with an opportunity to choose which match is most appropriate. The TNRS uses exact and fuzzy algorithms to return suggestions for the canonical spelling of names submitted based on the Tropicos database6 at the Missouri Botanical Gardens7. Use of the Global Names Index Name Parser8,9 (Patterson et al., 2010) for name decomposition and analysis combined with Taxamatch10 for fuzzy matching enables the TNRS to return a more complete resolution solution. Based on the edit distance between the submitted name and the matched name, an algorithm calculates an overall match score. This score enables ranking the results and presenting an ordered list of possible matches rated by probability. By default, the highest ranked item in a fuzzy match is returned, but users may also select a lower ranked item as the proper match. The TNRS addresses hyphenated infraspecific names by searching the database for a properly hyphenated string that matches, then proceeding with the unhyphenated version of the original string if none is found. The matching algorithm also handles accented characters (Ú, Ó, Ü, etc.) by searching for both accented and plain ascii representations. iPlant developers are fine-tuning TNRS with more optimization techniques to speed up SQL queries, database indexing, and the database server configuration.
The TNRS is a collaborative effort between iPlant and the Botanical Information and Ecology Network (BIEN), a working group supported by the National Center for Ecological Analysis and Synthesis (Reichman, 2004), and the Missouri Botanical Garden. BIEN is working closely with the Missouri Botanical Garden to create a global plant information database using data found in several well-known plant biodiversity databases as well as hundreds of smaller but important sources. A central challenge of this initiative is the standardization of taxonomic information from numerous data sets collected by different researchers at different times and places. By eliminating spelling and digitization errors and merging synonymous names, the TNRS reduces duplications in data, allows for more efficient storage and searching, and ensures biologically meaningful cross-linkages among data sets. The TNRS will ultimately enable more accurate and comprehensive analyses since all name data for each species will be found in single location. Future releases of the TNRS will incorporate additional name resources, such as the International Plant Names Index11, broadening the base against which submitted names can be compared.
My-plant Scientific Networking Site
My-Plant.org is a scientific networking and collaboration portal for the plant phylogenetics research community (Hanlon et al., 2010). My-Plant.org provides researchers with a site to connect with other researchers and to facilitate new collaborations and wider communication. My-Plant is also designed to facilitate data assembly for phylogenetic trees. My-Plant.org is a hierarchical network, connecting members to each other, to groups with common interests, and to the content these groups co-create. The network is based on clades.
Volunteers from the community manage clades, organizing the clade and providing direction for development of the clade. Therefore, the phylogenetic tree on which the network is structured reflects only those clades with user community support. As the user interest grows and a base of users for a particular clade becomes apparent, new clades can be added to the network at any time.
My-Plant.org is not only for scientists. Anyone interested in plants and in connecting with others who have similar interests will benefit from My-Plant.org. Educators and students will find the phylogenetic structure of the network useful for teaching and learning about various aspects of plant science. Enthusiasts will have a forum for sharing their knowledge and insights while also being able to connect with other plant enthusiasts and research scientists. My-Plant.org presents members with a unique mechanism for connecting with those who share their interests and to work with citizen scientists.
The iPlant Genotype-to-Phenotype Project
The overarching goal of the iPlant Genotype-to-Phenotype (iPG2P) project is to create CI that facilitates the efficient identification of genetic control mechanisms and environmental impacts on specific plant traits of interest. Elucidating the relationship between plant genotypes and the resultant phenotypes in complex (e.g., non-constant) environments is one of the foremost challenges in plant biology. The basic genotype-to-phenotype challenge is simply stated as “Given the genomic and environmental information about a given plant growing in a specific environment, predict its characteristics using computational approaches.” This is essentially what Tara’s collaborating team is attempting to do with abiotic stress responses. Plant phenotypes are often determined by very intricate interactions between genetic control mechanisms and environmental variables. In a world where the environment is undergoing rapid, anthropogenic change, predicting altered plant responses is central to studies of plant adaptation, ecological genomics, crop improvement activities (ranging from international agriculture to renewable biofuels), plant physiology (photosynthesis, stress responses, etc.), plant development, and many more related plant attributes. A concerted attack on the genotype-to-phenotype problem requires the combined and integrated efforts of specialists in functional-, quantitative-, and computational genetics/genomics, bioinformatics, modeling, plant physiology, computer science (for topics ranging from HPC to data visualization), etc. CI innovations are needed to facilitate collaborations across this diversity of disciplines. The iPG2P project identified a number of high priority focus areas where progress is needed to facilitate discovery. Advances in DNA/RNA sequencing will have the greatest impact on plant science in the next few years. Working groups are focused on ultrahigh-throughput DNA and RNA sequence data, statistical and predictive modeling, data integration, visual analytics, and virtual genotype and molecular phenotype data.
Support for Ultrahigh-throughput DNA/RNA Sequencing
iPlant’s sequence analysis effort enables users to upload DNA or RNA sequencing data from their desktop, a remote server, or from the NCBI Sequence Read Archive, then view, manage, and perform basic analysis on the data in a user-centric workspace. Data management capabilities include annotation with metadata and pre-processing sequence data to remove non-biological sequence production artifacts (e.g., linkers, primers, etc.). Scientists are able to perform basic analytical workflows using their post-processed sequence data in a relatively short period of time and without complex command-line utilities.
The first workflow supports DNA sequence data and allows users to detect single nucleotide polymorphisms (SNPs) in a test sequence compared to a reference sequence. This workflow is called variant detection. The input of the workflow is a library of short read data and a reference sequence and the output is a list of SNP differences. The second workflow supports RNA sequence data and provides transcript quantification relative to a reference genome. Initially, users will be able to choose various reference genomes (thale cress – Arabidopsis thaliana and Arabidopsis lyrata, maize – Zea mays, bunch grass – Brachypodium distachyon, rice – Oryza sativa cv. Nipponbare, and O. sativa indica, poplar – Populus trichocarpa, Sorghum bicolor, and grape – Vitis vinifera) as the basis for their analyses. Reference genome data and related annotations are provided via integration with model organism database providers. Users are able to download data outputs derived at specific stages of the workflow. These output files include processed FASTA/FASTQ sequences, genome alignments in SAM/BAM format (Sequence Alignment/Map, Binary Alignment/Map; Li et al., 2009) as well as tabular representations of the outputs of the two workflows. A third workflow supports ChIP-seq experiments to identify regions of histone modification and the locations of transcription factors and other chromatin binding factors. This workflow is based around the peak-calling software PeakRanger (Feng et al., 2011), which combines high accuracy with excellent performance. This workflow can be executed on iPlant cloud computing resources. Additional workflows are under development to allow discovery of novel RNA transcripts, comparative gene expression (RNAseq) analyses, and automated functional annotation of discovered polymorphisms.
One clearly pressing need within the plant science research community is a strategy and mechanism to store and analyze resequenced genomes from numerous plant varieties. Ultimately resequenced genomes should be stored permanently in GenBank/EMBL/DDBJ, but for efficient analysis, a mechanism for temporary storage and analysis near HPC facilities would be beneficial. Storage of resequenced genomes within the TeraGrid system would be an ideal solution to allow analysis and storage to be done under an NSF-supported umbrella and is supported by iPlant. Smaller analysis results files could then be moved efficiently. It would also be beneficial to the research community to adopt a standard format for storage and analysis of resequenced genomes from a variety of species. Such a standard would simplify cross-species analysis and comparative genomics. Standards for such storage are under active discussion.
Genome and Transcriptome Assembly
As described above, genomics studies are being revolutionized by advances in next generation sequencing technologies. Whole-genome and transcriptome sequencing are now much more accessible to the average researcher, but they are developing arts that, despite the large volumes of data that can be produced, may still fail to provide a clear, scientifically interpretable result. Assembly requires access to substantial computing resources, complex primary and evaluative workflows, and effective means of parameterization. iPlant is developing a set of component-based workflows for de novo and reference-guided genomic and RNA assemblies that will run on the TACC’s high-performance cluster systems with access to iPlant’s storage and analytical software infrastructure.
In the genome assembly efforts, best practices derived from the Plantagora project12 are being distilled into the workflows available in the iPlant CI. The Plantagora project was designed to study how these new DNA sequencing technologies could be analyzed to achieve the highest quality assemblies. Simulated reads from several different plant genomes of different sizes were created that mimicked either 454 or Illumina reads, with varying paired end spacing distances. Thousands of datasets of reads were created by the Plantagora project and these test data were assembled with different software assemblers, including Newbler, Abyss, and SOAP de novo, and the resulting assemblies were evaluated by an extensive battery of metrics chosen for these studies.
Also in development are workflows to facilitate evaluation of assembly quality and to perform first-pass automated functional annotation of newly assembled genomes and transcriptomes. These will include generation of N50 or NG50 charts and tables, discovery and annotation of repeat content, comparative BLAT to reference species (Kent, 2002), and BLASTX-based gene prediction. These workflows will provide higher quality initial genome assemblies and allow researchers like Tara’s collaborators to devote more time to basic research.
Genome Services
To manage and serve published plant genome data, iPlant has partnered with CoGe (Freeling et al., 2008; Lyons et al., 2008, 2011; Tang et al., 2011) to modularize CoGe’s genome data model. CoGe’s genome data model supports storage of multiple genomes in any state of assembly and annotation, and currently houses 12,000 genomes from 10,000 organisms, including all publically available plant cellular and organelle genomes. This partnership creates an iPlant genome services module that provides access to all plant genomes through a combination of file-based repositories located in the iPlant data cloud and a RESTful API. Genome services facilitates ultrahigh-throughput sequencing and other sequence-oriented analyses within the iPlant CI.
Statistical Modeling and Inference
The efficiency of both forward and reverse genetics studies is not as high as initially predicted or desired. Only a small percentage of plant genes have laboratory or field-based evidence for their functions, and multigene families, non-orthologous gene displacement (Koonin et al., 1996), lateral gene transfer, and several other natural biological processes make functional assignments more difficult. The vast majority of genes are classified computationally. Sequencing of whole genomes and marker analysis of specific varieties provides an opportunity to associate genetic variation with trait variation using statistical approaches. Likewise, statistically based tools can be used to infer links between genetic variation and biochemical or regulatory networks. Many such statistical analysis tools already exist, but in some cases do not efficiently scale to the number of genetic variables (e.g., tens of millions of SNPs) or to the number of genes controlling complex traits. To address this scalability challenge, the iPG2P Modeling Working Group is focused on creating modeling tools capable of taking advantage of CI and HPC. A modeling framework to support the construction, parameter estimation, sensitivity analysis, and utilization of models is under active development. Over the near-to-intermediate term, components of ecophysiological models will increasingly employ the results of gene-based network studies, thus enhancing their application in breeding and research projects like the abiotic stress project Tara’s team is focused on.
Graphical Processing Units and General Linear Models
The Statistical Inference Working Group identified and prioritized general classes of statistical genetics methods that will be supported by the iPlant CI. These include general linear models (GLMs), Mixed Models, Machine Learning, and Bayesian approaches. GLMs, are most pertinent to the widest cross-section of plant biologists, and are being addressed first. A test implementation of GLMs developed by iPlant serves as a reference for optimization and parallelization of the GLM algorithm on alternative architectures. The iPlant team has developed a multiple-SNP forward-regression version of general linear modeling and improved the performance of single-SNP forward-regression on graphics processing units. The current software implementations achieved a significant speedup over a previous version of the code written in C. This speedup includes all information transfer steps to and from the GPU in the host computer. In the multiple-GPU version of the code, the source code will be specifically optimized to take advantage of the GPU features on the TACC computing cluster. Future work from the Statistical Inference group will include solutions on how to view and explore the large (2.5E + 6 points) multidimensional data sets expected to emerge from genetic association studies as well as how to make the results of such analyses more accessible to the general research community.
High-Throughput Image Analysis Platform – Phytobisque
Although the identification of genetic variation is advancing rapidly due to enhancements in and decreasing costs of DNA sequencing technology, phenotyping is still very difficult and even becoming more expensive. In an effort to provide more balanced support for both genotyping and phenotyping, iPlant is leveraging the BISQUE software system to build an efficient, scalable platform to analyze plant-related images in the context of phenotype analysis. BISQUE is the Bio-Image Semantic Query User Environment (Kvilekval et al., 2010) and was developed at the Center for Bio-Image Informatics at University of California Santa Barbara. Created for the exchange, exploration, and analysis of biological images, BISQUE supports the needs of imaging researchers worldwide, providing everything from basic image capture to advanced querying and algorithm-based analysis. The plant-oriented adaptation of BISQUE, called PhytoBISQUE, extends the platform by offering integration with iPlant’s authentication, cloud storage, and high-performance grid computing infrastructure. It includes a software development kit and API for creation and deployment of new algorithms and workflows to facilitate collaborations between biological science researchers and experts in machine vision and image processing. To illustrate the capabilities of the system to researchers like Tara’s team, it is configured with sample data and algorithms designed to assay phenotypes such as directional root-tip growth or comparisons of seed size differences (Miller et al., 2007; Spalding, 2009, 2010; Wang et al., 2009).
The iPlant Collaborative Seed Projects: from DNA to the Globe
Seed Projects are an additional way for iPlant to receive collaboration requests from the community, with the goal of developing Grand Challenge projects in new areas by mid 2012. The Seed Project strategy was developed in response to community feedback requesting a streamlined process for engaging with iPlant, though holding Grand Challenge Workshops remains an option. Seed Projects are intended to broaden the community iPlant serves and the CI it is building by describing additional plant biology challenges that require computational solutions. The working groups at iPlant’s 2010 Conference were invited to submit small Seed Projects that included a CI-related deliverable. As a result, iPlant is now collaborating on four Seed Projects: Botanical Geospatial Diversity, Plant Adaptation to Environment, Plant Nutrition, and Forest Tree Biology. In addition, CI support for geospatially referenced data was a major, common need for advancing plant science research in these areas; therefore, iPlant formed a community-led geographic information system (GIS) working group to collaborate with iPlant to scope and develop its GIS infrastructure. The CI envisioned and being built by iPlant will help researchers utilize data and models that span scales ranging from molecular and cellular to whole organism to ecosystems, thus enabling understanding of plant biology from DNA to the globe.
The Generation Challenge Program’s Integrated Breeding Platform and iPlant
iPlant and the Integrated Breeding Platform project funded by the Bill & Melinda Gates Foundation have created a mutually beneficial collaboration with coordinated development efforts. iPlant benefits by having a close interaction with the highly experienced breeders from several of the centers of the Consultative Group on International Agricultural Research (CGIAR) that are geographically dispersed around the world and concentrate on agricultural research for food security and development. The IBP benefits by being able to focus immediately on the creation and development of breeding tools specific for their needs, building on the iPlant CI platform that many plant biology researchers will use for discovery research. iPlant collaborators can benefit by gaining access to the users in the CGIAR and academic research organizations interested in supporting the humanitarian applications of the IBP, and to rich resources of biological data that will be accessible through the iPlant CI for collaborative biological research. Taken together, this coordinated effort should be universally advantageous to the plant science community.
The CGIAR and other partners have been working on developing the International Crop Information System and a computerized field book system for maize breeding for over a decade. IBP managers have expressed their desire and willingness to update and merge these applications to be compatible with iPlant’s design and to be scalable to the CGIAR’s new needs. The first step in the collaboration will be developing a basic field notebook system and a statistical analysis pipeline based on existing R scripts. These will be refined and improved modules added in a staged collaboration. Representatives of USDA and AAFC have agreed that the Workbench would provide a valuable tool for breeding and agricultural research in the public sector in developed and developing countries.
Reproducible Bioinformatics Analysis
Computational analysis experiments in biology are often difficult to reproduce because versions of data sets may have changed, software used in the original experiment cannot be reconstructed, or the input parameters for an experiment may not be captured or sufficiently documented by the original analysis team. A set of tools called Rex (for Reproducible experiments; Perianayagam et al., 2010) has been developed that enables a researcher to record an analysis experiment and archive it in detail, replay the recorded experiments, run new experiments on a recorded apparatus, and compare recorded experiments. Recording an experiment is as simple as prefixing the experiment with the record tool. To replay an experiment, one just needs to run the replay tool on the archive containing the experiment. The Rex tools can be used to capture an experiment for posterity (including all code used, the versions of data and software used, the input and output data, and the execution environment and parameters), move a recorded experiment to a different host system and replay it there, run a new experiment with different input parameters or data sets, and compare two experiments to see where and why they differ.
iPlant’s Education Program
The Next Generation of Scientists – Education and Training in iPlant
Creating interest in research science and building research capacity in the next generation of citizens are key aspects of iPlant’s strategy. Biology is undergoing a paradigm shift, from a data-limited to a data-rich state, from hypotheses limited by data to data-limited by hypotheses, and from reductionism to systems biology. A number of lessons from past education efforts can be applied to advance biology education: (1) student–scientists partnerships are essential; scientists have to care about the data students generate; (2) with students as active co-investigators, the collaborative projects should have the potential to lead to publication.; (3) individual classroom experiments should scale up to distributed projects; (4) the analysis needs of distributed projects should create a seamless transition from educational interfaces and tools to research. The iPlant education team has focused on a few projects with potential for broad national impact. The key goal is to create education projects unified with the DE and built to support the iPToL and iPG2P Grand Challenge Teams. To do this, the iPlant education team developed computational tools that can be integrated with classroom research projects. The first education application is named the DNA Subway (see Figure 3). It was developed with 25 collaborators at 11 institutions and launched in March 2010; by December 2010 there were 603 registered users and 11,000 visits.
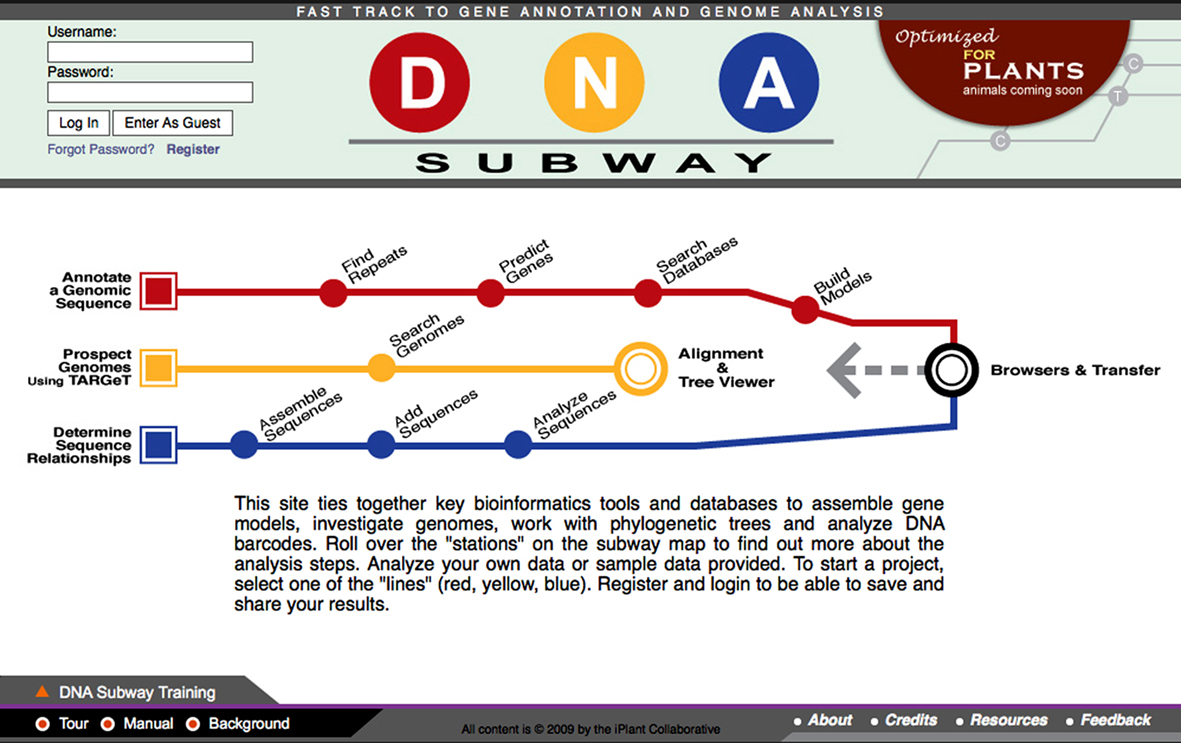
Figure 3. The DNA Subway. The DNA Subway is an Educational DE, a simplified workflow for gene annotation and comparison. The subway lines are the Red Line (predicts and annotates genes in contigs less than 150kb), the Yellow Line (identifies homologs in other sequenced genomes), and the Blue Line (supports phylogenetics analysis and DNA barcode analysis). Additional “lines” are under development as new tools are integrated into the iPlant CI that can be consumed by a web-based DE.
The second major education project is DNA Barcoding (Hebert et al., 2003; Hollingsworth, 2009), which fits with objectives of the iPToL Grand Challenge. This project combines lab experimentation with bioinformatics. The Barcoding project uses simplified kits for plant DNA extraction and target gene amplification followed by inexpensive commercial sequencing ($3.00–3.50 per sample). The DNA Subway Blue Line handles the data analysis. DNA Barcoding using plants provides opportunities for both student research and publication of novel sequences, completing the loop from understanding gene structure to understanding data gathering.
The next steps for iPlant education include incorporation of modules for computation of PICs to study trait evolution and the analysis of gene expression (RNAseq) datasets. These tools will integrate iPToL and iPG2P tools directly into education projects, ultimately developing educational interfaces directly atop the DE API.
Integration of Analysis Tools into the iPlant CI
iPlant is supporting the integration of numerous existing analysis tools and datasets into the iPlant CI by developing tools and training to facilitate this integration. The majority of analysis tools iPlant will provide within the DE will be existing tools rather than newly generated tools. There are thousands of existing tools and data sources as described in the annual Nucleic Acids Research issue on databases and software (Galperin and Cochrane, 2011) and in journals such as BioInformatics. If necessary, iPlant will add checkpointing or parallelization to existing tools or refactor them to provide scalability. The process of integrating an existing analysis tool into the iPlant DE will require creating a structured text description that will enable the tool or data source to be recognized and used within the iPlant DE. Up-to-date tutorials for tool integration are available at the iPlant wiki. iPlant supports a semi-automated, biologist-friendly integration process driven by the completion of a form description of the software tool or data source rather than a manually created description. This form-driven approach will significantly lower the barrier to integration by members of the broad research community.
Ongoing Challenges and How to Get Involved
As a cyberinfrastructure project, iPlant is providing an advanced computational, networking, and collaboration framework, but this is only the beginning. As the CI matures, iPlant will evolve into a hub for biologists, bioinformaticians, and computer scientists. This mature CI will provide numerous opportunities to the computing community to initiate collaborations and projects beneficial to biologists. The iPlant CI will provide a location to examine which tools and workflows plant researchers are using most frequently. Computing researchers will see which components are in highest demand and which may need better algorithms. The CI itself will provide the support necessary for data format conversions and output handling that would normally need to be built into a stand-alone software tool. The iPlant CI will be a marketplace to distribute ideas on better tools, workflows, algorithms, and ontologies to the plant biology research community. The iPlant project is eager for analysis tool developers to integrate their products with the DE through the iPlant APIs. Tool integration and development workshops are being planned and integration training will be available both online and in person to facilitate community contributions.
Who makes up the iPlant Collaborative?
The iPlant Collaborative is you, the members of the science community, plus the participants of the iPlant teams developing the foundational cyberinfrastructure. iPlant Working Groups, which arose from the Grand Challenge and Seed Projects, had only a handful of members in April 2009, but now include more than 70 faculty, postdocs, and graduate students at over 40 institutions. The user base of the iPlant CI grew from zero in March 2010 to more than 800 unique users in October 2010, and bridging activities with other infrastructure and service projects introduced iPlant to hundreds of other users.
Each and every community participant is essential in building the CI to support plant science research. Why? Without community support and input, iPlant will not have the expertise and resources to write, modify, and integrate all the tools/datasets/databases/file formats that are required for a comprehensive plant sciences cyberinfrastructure. How will the CI work? Through powerful APIs, user-friendly integration and authoring tools, and a capable computing infrastructure, the community is empowered to bring their own innovative algorithms, analyses, and best practices into an environment where everyone can easily make use of them. As talented developers build tools in this foundational CI, a tipping point will be reached where the iPlant CI becomes the place to share and make use of computational biology tools and data in the plant sciences.
iPlant welcomes and encourages your participation – to ensure the CI meets the needs of working biologists, to ensure that the solutions are scientifically valid, and to grow the CI faster through participation of experienced developers and researchers. You and your colleagues can participate in building the CI by integrating useful analysis tools, by creating new types of analyses, and by integrating different data sources. Contact iPlant with your needs and ideas – the only requirement is to have test data and specific analyses in mind. Even if you are not a developer, you can write tutorials or combine existing tools into new and innovative workflows. You can attend workshops that combine science synthesis with tool integration and workflow creation, then take these back to your institutions and teach your friends and colleagues how to use the CI capabilities. Finally, you can simply provide feedback: tell iPlant what works, what does not work, and how to improve the existing CI and applications. The iPlant Collaborative is building a community-powered and community-empowering cyberinfrastructure. Anyone can contribute – everyone can benefit. The iPlant team encourages you to get involved now.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The iPlant Collaborative is funded by a grant from the National Science Foundation Plant Cyberinfrastructure Program (#DBI-0735191).
Footnotes
- ^Tara is a fictional persona in this use case example.
- ^http://sswap.info/
- ^http://www.iplantcollaborative.org/communities/developers/SemanticWeb
- ^http://www.phylo.org/portal2
- ^http://www.onekp.com
- ^http://www.tropicos.org
- ^http://www.mobot.org
- ^https://github.com/GlobalNamesArchitecture/biodiversity
- ^http://gni.globalnames.org/
- ^http://code.google.com/p/taxamatch-webservice/
- ^http://www.ipni.org
- ^http://www.plantagora.org/about_plantagora/
References
Ackerly, D. D., and Reich, P. B. (1999). Convergence and correlations among leaf size and function in seed plants: a comparative test using independent contrasts. Am. J. Bot. 86, 1272.
Akerborg, O., Sennblad, B., Arvestad, L., and Lagergren, J. (2009). Simultaneous Bayesian gene tree reconstruction and reconciliation analysis. Proc. Natl. Acad. Sci. U.S.A. 106, 5714–5719.
Altintas, I., Berkley, C., Jaeger, E., Jones, M., Ludascher, B., and Mock, S. (2004). “Kepler: an extensible system for design and execution of scientific workflows,” in 16th International Conference on Scientific and Statistical Database Management, Santorini Island.
Armstead, I., Huang, L., Ravagnani, A., Robson, P., and Ougham, H. (2009). Bioinformatics in the orphan crops. Brief. Bioinformatics 10, 645–653.
Atkins, D. E., Droegemeier, K. K., Feldman, H., Garcia-Molina, H., Klein, M. L., Messerschmitt, D. G., Messina, P., Ostriker, J. P., and Wright, M. H. (2003). Revolutionizing Science and Engineering Through Cyberinfrastructure. Report of the national science foundation blue-ribbon advisory panel on cyberinfrastructure. Arlington, VA: Office of Cyberinfrastructure, The National Science Foundation.
Benfey, P. N., Bennett, M., and Schiefelbein, J. (2010). Getting to the root of plant biology: impact of the Arabidopsis genome sequence on root research. Plant J. 61, 992–1000.
Buell, C. R., and Last, R. L. (2010). Twenty-first century plant biology: impacts of the Arabidopsis genome on plant biology and agriculture. Plant Physiol. 154, 497–500.
Cook, D. R., and Varshney, R. K. (2010). From genome studies to agricultural biotechnology: closing the gap between basic plant science and applied agriculture. Curr. Opin. Plant Biol. 13, 115–118.
Deelman, E., Blythe, J., Gil, Y., Kesselman, C., Mehta, G., Patil, S., Su, M., Vahi, K., and Livny, M. (2004). “Pegasus: mapping scientific workflows onto the grid,” in 2nd European Across Grids Conference, Nicosia.
Donoghue, M. J., Yahara, T., Conti, E., Cracraft, J., Crandall, K. A., Faith, D. P., Häuser, C., Hendry, A. P., Joly, C., Kogure, K., Lohmann, L. G., MagallÓn, S. A., Moritz, C., Tillier, S., Zardoya, R., Prieur-Richard, A.-H., Larigauderie, A., and Walther, B. A. (2009). bioGENESIS: Providing an Evolutionary Framework for Biodiversity Science. DIVERSITAS Report No. 6, 52. Sydney.
Edgar, R. C. (2004). Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113.
Edwards, D., and Batley, J. (2010). Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 8, 2–9.
ELIXIR. (2010). The European Life Sciences Infrastructure for Biological Information. Available at: http://www.elixir-europe.org/page.php?page=home
Faith, D. P. (2008). Threatened species and the potential loss of phylogenetic diversity: conservation scenarios based on estimated extinction probabilities and phylogenetic risk analysis. Conserv. Biol. 22, 1461–1470.
Felsenstein, J. (2008). Comparative methods with sampling error and within-species variation: contrasts revisited and revised. Am. Nat. 171, 713–725.
Feng, X., Grossman, R., and Stein, L. (2011). Peakranger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics 12, 139.
Freeling, M., Lyons, E., Pedersen, B., Alam, M., Ming, R., and Lisch, D. (2008). Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 18, 1924–1937.
Galperin, M. Y., and Cochrane, G. R. (2011). The 2011 nucleic acids research database issue and the online molecular biology database collection. Nucleic Acids Res. 39, D1–D6.
Gaudet, P., Bairoch, A., Field, D., Sansone, S. A., Taylor, C., Attwood, T. K., Bateman, A., Blake, J. A., Bult, C. J., Cherry, J. M., Chisholm, R. L., Cochrane, G., Cook, C. E., Eppig, J. T., Galperin, M. Y., Gentleman, R., Goble, C. A., Gojobori, T., Hancock, J. M., Howe, D. G., Imanishi, T., Kelso, J., Landsman, D., Lewis, S. E., Mizrachi, I. K., Orchard, S., Ouellette, B. F., Ranganathan, S., Richardson, L., Rocca-Serra, P., Schofield, P. N., Smedley, D., Southan, C., Tan, T. W., Tatusova, T., Whetzel, P. L., White, O., and Yamasaki, C. (2011). Towards biodbcore: a community-defined information specification for biological databases. Nucleic Acids Res. 39, D7–D10.
Gessler, D. D., Schiltz, G. S., May, G. D., Avraham, S., Town, C. D., Grant, D., and Nelson, R. T. (2009). SSWAP: a simple semantic web architecture and protocol for semantic web services. BMC Bioinformatics 10, 309.
Gregurick, S. (2010). DOE Systems Biology Knowledgebase Implementation Plan. Washington, DC: The US Department of Energy, Office of Biological and Environmental Research.
Hanlon, M. R., Mock, S., Nuthulapati, P., Gonzales, M. B., Soltis, P., Soltis, D., Majure, L. C., Payton, A., Mishler, B., Tremblay, S., Madsen, T., Olmstead, R., Mccourt, R., Wojciechowski, M., and Merchant, N. (2010). “My-plant.org: a phylogenetically structured social network for the plant sciences,” in Gateway Computing Environments (GCE), New Orleans, LA, 1–8.
Hebert, P. D., Cywinska, A., Ball, S. L., and Dewaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321.
Hendry, A. P., Lohmann, L. G., Conti, E., Cracraft, J., Crandall, K. A., Faith, D. P., Hauser, C., Joly, C. A., Kogure, K., Larigauderie, A., Magallon, S., Moritz, C., Tillier, S., Zardoya, R., Prieur-Richard, A. H., Walther, B. A., Yahara, T., and Donoghue, M. J. (2010). Evolutionary biology in biodiversity science, conservation, and policy: a call to action. Evolution 64, 1517–1528.
Hirayama, T., and Shinozaki, K. (2010). Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J. 61, 1041–1052.
Hollingsworth, M. A. (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 106, 12794–12797.
Hughes, S. (2006). Opinion piece: genomics and crop plant science in Europe. Plant Biotechnol. J. 4, 3–5.
Hull, D., Wolstencroft, K., Stevens, R., Goble, C., Pocock, M. R., Li, P., and Oinn, T. (2006). Taverna: a tool for building and running workflows of services. Nucleic Acids Res. 34, W729–W732.
Kano, Y., Dobson, P., Nakanishi, M., Tsujii, J., and Ananiadou, S. (2010). Text mining meets workflow: linking U-compare with Taverna. Bioinformatics 26, 2486–2487.
Kawas, E., Senger, M., and Wilkinson, M. D. (2006). Biomoby extensions to the Taverna workflow management and enactment software. BMC Bioinformatics 7, 523.
Koonin, E. V., Mushegian, A. R., and Bork, P. (1996). Non-orthologous gene displacement. Trends Genet. 12, 334–336.
Krabbenhoft, H. N., Moller, S., and Bayer, D. (2008). Integrating ARC grid middleware with Taverna workflows. Bioinformatics 24, 1221–1222.
Kvilekval, K., Fedorov, D., Obara, B., Singh, A., and Manjunath, B. S. (2010). Bisque: a platform for bioimage analysis and management. Bioinformatics 26, 544–552.
Lanzen, A., and Oinn, T. (2008). The Taverna interaction service: enabling manual interaction in workflows. Bioinformatics 24, 1118–1120.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., and Durbin, R. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079.
Li, P., Castrillo, J. I., Velarde, G., Wassink, I., Soiland-Reyes, S., Owen, S., Withers, D., Oinn, T., Pocock, M. R., Goble, C. A., Oliver, S. G., and Kell, D. B. (2008). Performing statistical analyses on quantitative data in Taverna workflows: an example using R and maxdBrowse to identify differentially-expressed genes from microarray data. BMC Bioinformatics 9, 2334.
Ludascher, B., Altintas, I., Berkley, C., Higgins, D., Jaeger, E., Jones, M., Lee, E. A., Tao, J., and Zhao, Y. (2006). Scientific workflow management and the Kepler system. Concurr. Comput. 18, 1039–1065.
Lyons, E., Freeling, M., Kustu, S., and Inwood, W. (2011). Using genomic sequencing for classical genetics in E. coli K12. PLoS ONE 6, e16717.
Lyons, E., Pedersen, B., Kane, J., Alam, M., Ming, R., Tang, H., Wang, X., Bowers, J., Paterson, A., Lisch, D., and Freeling, M. (2008). Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781.
Mcguinness, D. L., and Van Harmelen, F. (2011). OWL. Available at: http://www.w3.org/TR/owl2-overview
Mclay, R., Stanzione, D., Mckay, S. J., and Wheeler, T. (2011). “A scalable parallel implementation of the neighbor joining algorithm for phylogenetic trees,” in ICCABS Conference, Orlando, FL.
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 1–8.
Miller, N. D., Parks, B. M., and Spalding, E. P. (2007). Computer-vision analysis of seedling responses to light and gravity. Plant J. 52, 374–381.
Nelson, R. T., Avraham, S., Shoemaker, R. C., May, G. D., Ware, D., and Gessler, D. D. (2010). Applications and methods utilizing the simple semantic web architecture and protocol (SSWAP) for bioinformatics resource discovery and disparate data and service integration. BioData Min. 3, 3.
Oinn, T., Addis, M., Ferris, J., Marvin, D., Senger, M., Greenwood, M., Carver, T., Glover, K., Pocock, M. R., Wipat, A., and Li, P. (2004). Taverna: a tool for the composition and enactment of bioinformatics workflows. Bioinformatics 20, 3045–3054.
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290.
Paterson, A. H., Freeling, M., Tang, H., and Wang, X. (2010). Insights from the comparison of plant genome sequences. Annu. Rev. Plant Biol. 61, 349–372.
Patterson, D. J., Cooper, J., Kirk, P. M., Pyle, R. L., and Remsen, D. P. (2010). Names are key to the big new biology. Trends Ecol. Evol. (Amst.) 25, 686–691.
Perianayagam, S., Andrews, G. R., and Hartman, J. H. (2010). “Rex: a toolset for reproducing software experiments,” in Proceedings 2010 IEEE International Conference on Bioinformatics and Biomedicine, Hong Kong, 613–667.
Pichersky, E., and Gerats, T. (2011). The plant genome: an evolutionary perspective on structure and function. Plant J. 66, 1–3.
Proost, S., Pattyn, P., Gerats, T., and Van De Peer, Y. (2011). Journey through the past: 150 million years of plant genome evolution. Plant J. 66, 58–65.
PSCIC. (2006). Plant Science Cyberinfrastructure Collaborative. Available at: http://www.nsf.gov/publications/pub_summ.jsp?ods_key=nsf06594
Rajasekar, A., Wan, M., Moore, R., and Schroeder, W. (2006). “A prototype rule-based distributed data management system,” in Proceedings of High Performance Distributed Computing workshop on Next Generation Distributed Data Management, Paris.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Smith, A., Balazinska, M., Baru, C., Gomelsky, M., Mclennan, M., Rose, L., Smith, B., Stewart, E., and Kolker, E. (2011a). Biology and data-intensive scientific discovery in the beginning of the 21st century. OMICS 15, 209–212.
Smith, S. A., Beaulieu, J. M., Stamaktakis, A., and Donoghue, M. J. (2011b). Understanding angiosperm diversification using small and large phylogenetic trees. Am. J. Bot. 98, 404–4414.
Spalding, E. P. (2009). Computer vision as a tool to study plant development. Methods Mol. Biol. 553, 317–326.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690.
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771.
Stamatakis, A., Ludwig, T., and Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463.
Stamatakis, A., and Ott, M. (2008). Efficient computation of the phylogenetic likelihood function on multi-gene alignments and multi-core architectures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3977–3984.
Stone, G. N., Nee, S., and Felsenstein, J. (2011). Controlling for non-independence in comparative analysis of patterns across populations within species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 1410–1424.
Tang, H., Lyons, E., Pedersen, B., Schnable, J. C., Paterson, A. H., and Freeling, M. (2011). Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12, 102.
Thuiller, W., Lavergne, S., Roquet, C., Boulangeat, I., Lafourcade, B., and Araujo, M. B. (2011). Consequences of climate change on the tree of life in Europe. Nature 470, 531–534.
Vilella, A. J., Severin, J., Ureta-Vidal, A., Heng, L., Durbin, R., and Birney, E. (2009). Ensemblcompara genetrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335.
Wang, L., Uilecan, I. V., Assadi, A. H., Kozmik, C. A., and Spalding, E. P. (2009). Hypotrace: image analysis software for measuring hypocotyl growth and shape demonstrated on Arabidopsis seedlings undergoing photomorphogenesis. Plant Physiol. 149, 1632–1637.
Wassink, I., Rauwerda, H., Neerincx, P. B., Van Der Vet, P. E., Breit, T. M., Leunissen, J. A., and Nijholt, A. (2009). Using R in Taverna: RShell v1.2. BMC Res. Notes 2, 138.
Wheeler, T. J. (2009). Large-scale neighbor-joining with NINJA. Lect. Notes Comput. Sci. 5724, 375–389.
Willis, C. G., Ruhfel, B., Primack, R. B., Miller-Rushing, A. J., and Davis, C. C. (2008). Phylogenetic patterns of species loss in Thoreau’s woods are driven by climate change. Proc. Natl. Acad. Sci. U.S.A. 105, 17029–17033.
Yesson, C., and Culham, A. (2006a). Phyloclimatic modeling: combining phylogenetics and bioclimatic modeling. Syst. Biol. 55, 785–802.
Keywords: cyberinfrastructure, bioinformatics, plant biology, computational biology
Citation: Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, Matasci N, Wang L, Hanlon M, Lenards A, Muir A, Merchant N, Lowry S, Mock S, Helmke M, Kubach A, Narro M, Hopkins N, Micklos D, Hilgert U, Gonzales M, Jordan C, Skidmore E, Dooley R, Cazes J, McLay R, Lu Z, Pasternak S, Koesterke L, Piel WH, Grene R, Noutsos C, Gendler K, Feng X, Tang C, Lent M, Kim S-J, Kvilekval K, Manjunath BS, Tannen V, Stamatakis A, Sanderson M, Welch SM, Cranston KA, Soltis P, Soltis D, O’Meara B, Ane C, Brutnell T, Kleibenstein DJ, White JW, Leebens-Mack J, Donoghue MJ, Spalding EP, Vision TJ, Myers CR, Lowenthal D, Enquist BJ, Boyle B, Akoglu A, Andrews G, Ram S, Ware D, Stein L and Stanzione D (2011) The iPlant collaborative: cyberinfrastructure for plant biology. Front. Plant Sci. 2:34. doi: 10.3389/fpls.2011.00034
Received: 21 May 2011; Paper pending published: 01 June 2011;
Accepted: 11 July 2011; Published online: 25 July 2011.
Edited by:
Gane Ka-Shu Wong, University of Alberta, CanadaReviewed by:
Michael Deyholos, University of Alberta, CanadaSean W. Graham, University of British Columbia, Canada
Copyright: © 2011 Goff, Vaughn, McKay, Lyons, Stapleton, Gessler, Matasci, Wang, Hanlon, Lenards, Muir, Merchant, Lowry, Mock, Helmke, Kubach, Narro, Hopkins, Micklos, Hilgert, Gonzales, Jordan, Skidmore, Dooley, Cazes, McLay, Lu, Pasternak, Koesterke, Piel, Grene, Noutsos, Gendler, Feng, Tang, Lent, Kim, Kvilekval, Manjunath, Tannen, Stamatakis, Sanderson, Welch, Cranston, Soltis, Soltis, O’Meara, Ane, Brutnell, Kleibenstein, White, Leebens-Mack, Donoghue, Spalding, Vision, Myers, Lowenthal, Enquist, Boyle, Akoglu, Andrews, Ram, Ware, Stein and Stanzione. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Stephen A. Goff, iPlant Collaborative, BIO5 Institute, University of Arizona, Tucson, AZ 85721, USA. e-mail:c2dvZmZAaXBsYW50Y29sbGFib3JhdGl2ZS5vcmc=