 Sulail Fatima
Sulail Fatima Haiyan Zhou
Haiyan Zhou Yi Chen
Yi Chen Qinghang Liu
Qinghang Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 10 September 2024
Sec. Cell Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1450656
This article is part of the Research Topic Cell Death Programs in the Pathogenesis of Heart Disease View all 5 articles
Ferroptosis is a new form of regulated necrosis characterized by iron-dependent lipid peroxidation, leading to irreparable lipid damage, membrane permeabilization, and necrotic cell death. Ferroptosis has recently been implicated in the pathogenesis of multiple forms of heart disease such as myocardial infarction, cardiac hypertrophy, heart failure, and various cardiomyopathies. Important progress has also been made regarding how ferroptosis is regulated in vitro and in vivo as well as its role in cardiac homeostasis and disease pathogenesis. In this review, we discuss molecular mechanisms that regulates ferroptosis in the heart, including pathways leading to iron overload and lipid peroxidation as well as the roles of key organelles in this process. We also discuss recent findings pertaining to the new pathogenic role of ferroptosis in various forms of heart disease as well as genetic and pharmacologic strategies targeting ferroptosis in the heart.
Apoptotic and/or necrotic cell death has been implicated in multiple forms of heart disease, including ischemic myocardial injury, pathological remodeling, myocarditis, various forms of cardiomyopathy, and drug-induced cardiotoxicity (Del Re et al., 2019). Apoptosis is the most renowned form of regulated cell death mediated by death receptor or mitochondria dependent signaling pathways, which is characterized by cytosolic shrinkage, membrane blebbing, chromatin condensation, and DNA fragmentation, without loss of plasma membrane integrity (Danial and Korsmeyer, 2004). In contrast, necrosis had long been regarded as an unregulated process triggered by excessive pathological stress, characterized by cell swelling, plasma membrane rupture, cell lysis, and inflammatory response (Edinger and Thompson, 2004). However, this notion has been overturned by emerging evidence revealing that necrosis can also occur in a highly regulated and genetically controlled manner, termed “regulated necrosis”. Indeed, a number of regulated necrotic cell death modalities have recently been identified, including ferroptosis, necroptosis, pyroptosis, parthanatos, mitochondria-mediated necrosis, and other regulated necrotic processes (Del Re et al., 2019).
Ferroptosis is a newly identified form of regulated necrosis characterized by iron-dependent lipid peroxidation, leading to irreparable lipid damage, membrane permeabilization, and necrotic cell death (Dixon et al., 2012; Stockwell et al., 2017). Iron overload is a hallmark of ferroptosis, which promotes lipid peroxidation by producing hydroxyl and alkoxyl radicals through the Fenton reaction (Papanikolaou and Pantopoulos, 2005). Moreover, iron can also participate in enzymatic lipid peroxidation by promoting the activation of arachidonate lipoxygenase (ALOX) (Pu et al., 2022). Ferroptosis, regardless of the mechanisms of induction, is effectively inhibited by iron chelators, such as deferoxamine, indicating that iron is critically involved in the execution of ferroptosis. Moreover, cellular susceptibility to ferroptosis is closely regulated by iron metabolism, including its import, export, utilization, and storage (Tang D. et al., 2021). Accumulation of lipid peroxidation products is another hallmark of ferroptosis. Glutathione peroxidase 4 (GPX4), a glutathione (GSH)-dependent selenoenzyme, plays a crucial role in preventing ferroptosis by converting toxic lipid hydroperoxides to nontoxic lipid alcohols (Friedmann Angeli et al., 2014; Yang et al., 2014). Failure of GPX4 to clear lipid reactive oxygen species (ROS) leads to overwhelming lipid peroxidation and ferroptotic cell death (Friedmann Angeli et al., 2014; Yang et al., 2014). Apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as FSP1) has been identified as another key antioxidant protein that acts parallel to GPX4 in suppressing phospholipid peroxidation and ferroptosis (Bersuker et al., 2019; Doll et al., 2019). Moreover, the enzymes involved in the peroxidation of polyunsaturated fatty acids (PUFAs), such as acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and ALOXs, also play important roles in the induction of ferroptosis (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017).
Ferroptosis has recently been implicated in the pathogenesis of multiple forms of heart disease such as myocardial infarction, cardiac hypertrophy, heart failure, and various cardiomyopathies. New mechanistic insights have also been obtained regarding how ferroptosis is regulated in vitro and in vivo as well as its role in cardiac homeostasis and disease pathogenesis (Figure 1). Here, we review recent findings pertaining to the new pathogenic role of ferroptosis in various forms of heart disease as well as genetic and pharmacologic strategies that target ferroptosis in the heart. Molecular and cellular mechanisms of ferroptosis, especially pathways leading to iron overload and lipid peroxidation as well as new roles of key organelles, have recently been elucidated. Emerging evidence also reveals that ferroptosis contributes to the pathogenesis of acute cardiac injuries as well as chronic diseases by inducing cell death, inflammation, and tissue remodeling.
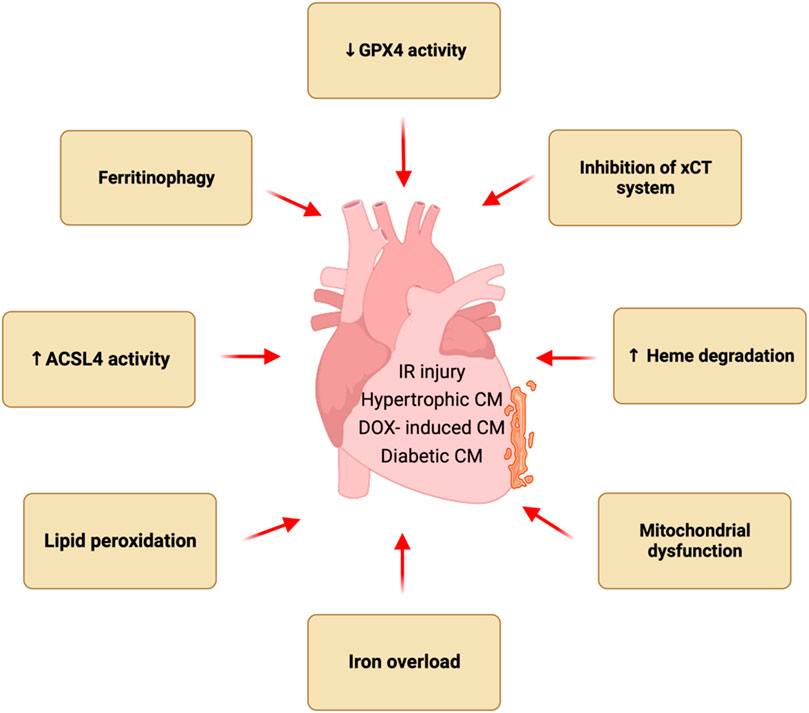
Figure 1. Ferroptosis in heart diseases. Ferroptosis has been implicated in the pathogenesis of multiple forms of heart disease such as myocardial infarction, cardiac hypertrophy, and various cardiomyopathies. Pathological stress induces ferroptotic cell death in the heart via multiple mechanisms such as iron overload, and lipid peroxidation, inhibition of xCT system, reduced GPX4 activity, increased ACSL4 activity, ferritinophagy, heme degradation, and mitochondrial dysfunction.
The absorption of diet iron involves several steps, including the uptake of iron from the intestinal lumen across the apical border of the villus and its transfer across the basolateral border to the plasma (Dev and Babitt, 2017). Extracellular iron in blood reversibly binds to transferrin (TF), a glycoprotein that is essential for the transport and cellular uptake of iron. Each transferrin molecule contains two binding sites for ferric ion. Iron-loaded transferrin is transported to the tissues, mainly erythroid marrow where it binds with transferrin receptor protein 1 (TFR1) and is internalized through clathrin-dependent endocytosis. The low pH environment in the endosome causes the release of ferric iron from the TF-TFR1 complex and, a transmembrane ferrireductase STEAP3 (six-transmembrane epithelial antigen of prostate) reduces ferric iron to ferrous iron. Next, DMT1 (divalent metal transporter 1) transports ferrous iron from the endosome into the cytosol (Bersuker et al., 2019). The carrier protein transferrin and TFR1 receptor are recycled back to the ECF and cell surface, respectively. Iron can be stored in the cytosol by the iron storage protein ferritin, which can chelate about 4,500 iron atoms (Chen et al., 2020). Iron enters the mitochondria via mitoferrin 1 and 2 where it participates in heme biosynthesis and hemoglobin production in developing erythroblasts (Shaw et al., 2006). Ferritin is also present in the mitochondria, termed mitochondrial ferritin (FTMT) (Santambrogio et al., 2007; Levi et al., 2021). Iron can be exported out of the cell by ferroportin-1 (FPN1, also known as SLC40A1) (Donovan et al., 2000; Azucenas et al., 2023). FPN1 is highly expressed in duodenal enterocytes, hepatocytes and macrophages (D’Anna et al., 2009).
In mammals, the iron regulatory proteins (IRPs; IRP1 and IRP2) are the central regulators of iron uptake, storage and export (Wang L. et al., 2019). In iron deficient states, IRPs bind to the iron response element (IRE) in the 3′UTR of target transcripts like TFR and DMT to stabilize the mRNA and increase translation of mRNA. At the same time, IRPs bind to the IRE in the 5′UTR of target transcripts such as ferritin and ferroportin to suppress translation of these proteins to combat iron deficiency. When iron levels are sufficient, the IRP system is under suppression. IRP1 contains Fe-S cluster which does not allow it to bind to IRE, and IRP2 is degraded by ubiquitin ligase which is sensitive to iron levels (Chen et al., 2020). Additionally, ferroportin levels are also regulated by hepcidin (Berezovsky et al., 2022). Hepcidin prevents iron efflux from the cells by binding to ferroportin and inducing endocytosis followed by the degradation (Charlebois et al., 2022).
Excess iron within the cell is stored in ferritin to prevent iron-mediated lipid peroxidation and ferroptosis. Under conditions of iron deficiency or high iron demand, ferritin undergoes autophagic lysosomal degradation to increase the labile iron content within the cells. However, elevated autophagy of ferritin, termed ferritinophagy, can induce iron overload and ferroptosis. The nuclear receptor coactivator 4 (NCOA4) serves as a specific cargo receptor for transporting ferritin to lysosomes for autophagic degradation (Mancias et al., 2014). Autophagy-related 5 and 7 (Atg5 and Atg7, respectively) genes are also critical for the formation of autophagosome during the process of ferritinophagy (Hou et al., 2016; Wen et al., 2019). The intracellular NCOA4 levels are regulated by cellular iron load. In conditions of high cellular iron levels, HERC2 (ECT and RLD domain-containing E3 ubiquitin protein ligase 2) facilitates the ubiquitination of NCOA4, marking it for degradation via the proteasome. This degradation limits NCOA4 availability, thereby reducing its ability to transport ferritin to lysosomes. In contrast, during iron deprivation, HERC2’s hold on NCOA4 weakens, allowing a pool of NCOA4 to remain unubiquitinated. This liberated NCOA4 can then bind to ferritin, facilitating its transport to lysosomes for degradation, consequently releasing iron for cellular utilization (Liu et al., 2020). It has been shown that NCOA4-mediated ferritinophagy was activated following pressure overload, leading to ferrous iron overload, increased lipid peroxidation, cardiomyocyte death, and ultimately heart failure in mice (Ito et al., 2021). Suppression of ferritinophagy by NCOA4 silencing protected the cells from iron overload and ferroptosis (Fang et al., 2021; Santana-Codina et al., 2021).
Heme is a crucial component of various biological processes like oxygen transport, electron transport, metabolism of drugs and toxins and signal transduction (Seiwert et al., 2020). Heme oxygenase (HO-1), a 32-kDa protein encoded by the Hmox1 gene, mediates the catabolism of heme into biliverdin, carbon monoxide (CO), and iron (Fe2+) (Cruse and Maines, 1988). Although HO-1 can elicit cytoprotective effects (Costa et al., 2020; Seiwert et al., 2020), excessive HO-1 activation can lead to iron overload, causing tissue damage and organ dysfunction (Miyamoto et al., 2022). In sickle cell disease, excess systemic heme has been shown to upregulate HO-1 expression and exacerbate iron overload, leading to cardiac ferroptosis and cardiomyopathy in mice (Menon et al., 2022). HO-1 upregulation has also been shown to promote iron overload in beta-thalassemia and anthracycline cardiotoxicity (Garcia-Santos et al., 2018; Fang et al., 2019). Importantly, a recent study showed that HO-1 silencing prevented simulated I/R-induced ferroptosis in cardiomyocytes (Miyamoto et al., 2022). Intriguingly, both pro- and anti-ferroptotic roles of HO-1 have been reported depending on cell types and pathological conditions (Chiang et al., 2018). To explain this discrepancy, accumulating evidence suggests that moderate activation of HO-1 elicits a cytoprotective effect whereas excessive and/or prolonged activation of HO-1 induces iron overload, leading to ferroptotic cell death (Chiang et al., 2018).
Free PUFAs, crucial substrates in lipid peroxidation, are incorporated into phospholipids by two pivotal enzymes: ACSL4 and LPCAT3. Inhibition of ACSL4 and LPCAT3 diminishes the availability of substrates necessary for lipid peroxidation, thus enhancing resistance to ferroptosis (Li and Li, 2020; Xu et al., 2020; Cui et al., 2021). During ferroptosis, PUFA derivatives within cellular membranes, such as the endoplasmic reticulum, mitochondria, lysosomes, and plasma membrane, undergo lipid peroxidation either via non-enzymatic Fenton reactions or enzymatic processes (Chen et al., 2021; Von Krusenstiern et al., 2023). Several enzyme systems are involved in lipid peroxidation, such as xanthine oxidase, cytochrome P450, NADPH oxidase, cyclooxygenases (COX), and lipoxygenase (LOX), many of which are iron dependent. LOX are iron containing nonheme dioxygenases, encoded by six ALOX genes ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3, which play an important role in ferroptosis (Mortensen et al., 2023).
The antiporter system Xc− is composed of two subunits, SLC7A11 and SLC3A2, and functions to import cystine into cells in exchange for glutamate. The cystine is degraded to cysteine, which is used to synthesize GSH (Liu et al., 2021). GPX4 is a selenoprotein that utilizes GSH to reduce lipid hydroperoxides, preventing lipid peroxidation and decreasing oxidative damage to the cells. There exist three isoforms of GPX4 localized to cytosol (cGPX4), nucleus (nGPX4) and mitochondria (mGPX4), respectively. GPX4 is unique among 8 known glutathione peroxides as it is the only enzyme capable of reducing oxidized fatty acids and cholesterol hydroperoxides. Mutations in GPX4 gene in humans led to cardiovascular, cerebrovascular, neuromuscular, or renal complications (Cheff et al., 2021). Deletion of GPX4, but not other GPX isoforms, caused embryonic lethality in mice (Yant et al., 2003; Yoo et al., 2012). Inducible ablation of GPX4 led to acute renal failure and early lethality in mice (Friedmann Angeli et al., 2014). Conditional deletion of GPX4 in neurons resulted in rapid onset of paralysis in the adult mice (Chen et al., 2015). GPX4 overexpression ameliorated, whereas GPX4 heterodeletion exaggerated myocardial ischemia/reperfusion (I/R) injury and doxorubicin-induced cardiomyopathy in mice (Miyamoto et al., 2022; Tadokoro et al., 2023).
Ferroptosis suppressor protein 1 (FSP1, also known as AIFM2) has been identified as another key suppressor of ferroptosis. It converts ubiquinone (Coenzyme Q10) to ubiquinol (Coenzyme QH2), which effectively sequesters lipid peroxyl radicals (Doll et al., 2019). FSP1 is primarily a cytosolic protein and gets translocated to the plasma membrane following myristoylation of its N-terminal. Apart from its role in modifying ubiquinone, FSP1 also reduces vitamin K to its hydroquinone form, which acts as a potent antioxidant against lipid peroxidation (Mishima et al., 2022). Additionally, FSP1 contributes to the reduction of α-tocopheryl radicals to α-tocopherol, which serves as an effective scavenger of the lipid radicals. Interestingly, it has been shown that FSP1 mediates resistance against ferroptosis by recruiting endosomal sorting complexes required for transport (ESCRT)-III for repairing the cell membrane (Zeng et al., 2022). The cells are also equipped with other antioxidants such as vitamin E, thioredoxin and peroxiredoxins (Llabani et al., 2019; Kuang et al., 2020; Hu et al., 2021). Interestingly, nitric oxide (NO·) generated by inducible nitric oxide synthase (iNOS) has been shown to substitute GPX4 inactivity and suppress ferroptosis in macrophages (Kapralov et al., 2020). The major pathways that regulate ferroptosis are illustrated in Figure 2.
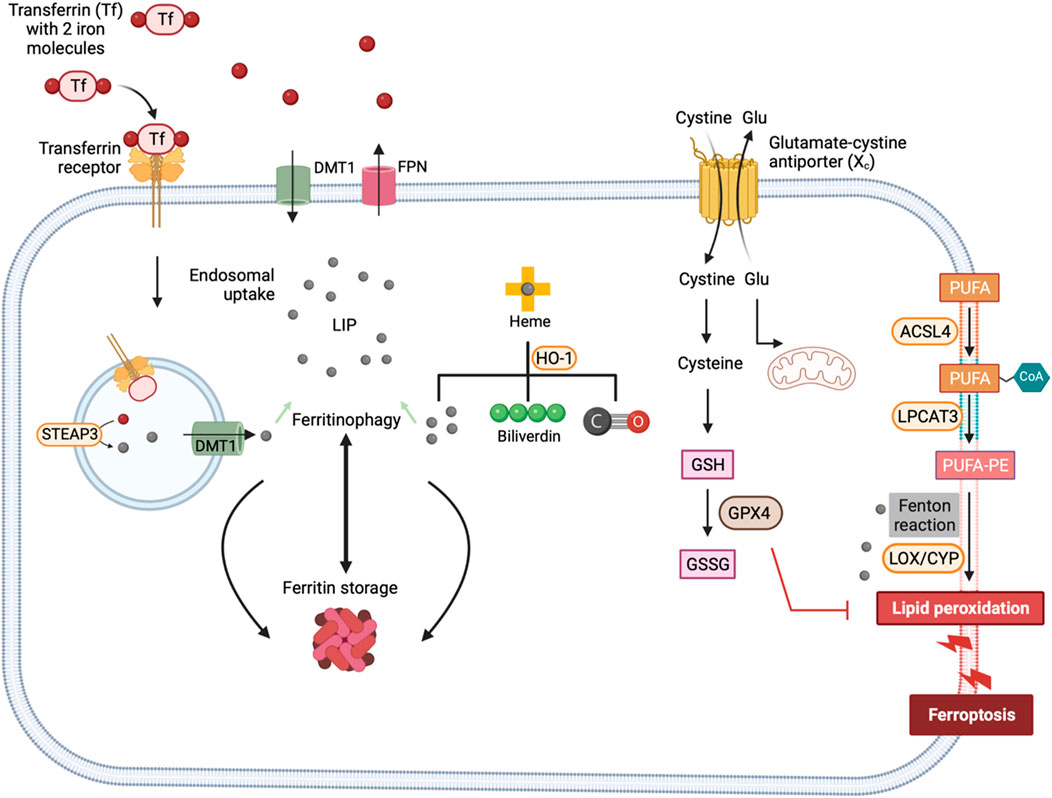
Figure 2. Mechanisms of ferroptosis. Ferroptotic cell death is triggered by iron-fueled excessive lipid peroxidation. Transferrin-TFR1 complex undergoes endocytosis and ferric iron is reduced to ferrous iron by the ferrireductase STEAP3. Iron is sequestered by ferritin or contribute to the LIP. HO-1 dependent heme degradation and NCOA4-mediated ferritinophagy also add iron to the LIP. Lipid peroxidation of PUFAs is mediated by the action of iron-dependent enzymes such as LOXs and CYP450 or iron catalyzed Fenton reactions. Glutamate cysteine exchanger mediates the exchange of extracellular cystine for intracellular glutamate. Once inside the cell, cystine is reduced to cysteine – a precursor for the synthesis of GSH. The activity of GPX4 depends on GSH to reduce lipid hydroperoxides and protect cell membranes from oxidative damage. ACSL4, Acyl-CoA synthetase long-chain family member 4; LPCAT3, Lysophosphatidylcholine acyltransferase 3; STEAP3, Six-Transmembrane Epithelial Antigen of the Prostate 3; DMT1, divalent metal transporter 1; LIP, labile iron pool; HO-1, hemeoxygenase-1; LOX, Lipoxygenase; CYP450, Cytochrome P450; GSH, Glutathione (reduced form); GSSG, Glutathione disulfide (oxidized form); GPX4, Glutathione peroxidase 4.
Ferroptotic cells exhibit various aberrant morphological and functional changes in mitochondria including, decrease in cristae, reduced membrane potential, increased permeability, increased iron, ROS and lipid peroxidation, and elevated DNA stress (Dixon et al., 2012; Gan, 2021). Mitochondria depletion prevented ferroptosis induced by cysteine-deprivation or erastin (Gao et al., 2019). Moreover, mitochondrial DNA depletion or mitochondrial ROS quenching inhibited ferroptosis induced by RSL3 (Jelinek et al., 2018; Oh et al., 2022). Multiple mechanisms may contribute to mitochondria iron overload in ferroptosis. For example, increased mitochondrial iron uptake through iron transporters, such as mitoferrin-1 (SLC25A37) and mitoferrin-2 (SLC25A28), can mediate mitochondrial iron overload (Paradkar et al., 2009; Hung et al., 2013). Moreover, cytosolic iron is translocated into mitochondria via the mitochondrial Ca2+ and Fe2+ uniporter (MCU) in photodynamic therapy-induced ferroptosis, leading to mitochondrial iron overload (Shui et al., 2021). Defective heme biosynthesis in mitochondria can lead to iron accumulation. Heme synthesis is a multistep process that involves a sequential action of at least eight enzymes in mammals. It begins in mitochondrial matrix where 5- aminolevulinic acid (ALA) is produced by the action of aminolevulinic acid synthase (ALAS). Disruption of ALAS-dependent heme synthesis can impair iron utilization and trigger ferroptosis (Paradkar et al., 2009). Moreover, under the influence of different stressors, HO-1 can be upregulated and even translocated to mitochondria (Bindu et al., 2011; Bansal et al., 2014). Indeed, we recently found that oxidative stress promoted HO-1 translocation and mitochondrial iron overload (Chen et al., 2023b). Within mitochondria, Fe2+ is utilized for heme and Fe-S cluster synthesis or stored in mitochondrial ferritin (MTFT). Several proteins involved in mitochondrial iron metabolism have been implicated in defense against ferroptosis. Iron-sulfur cluster assembly scaffold protein (ISCU), for instance, plays a critical role in Fe-S cluster synthesis and overexpression of ISCU suppresses ferroptosis (Du et al., 2019). A cysteine desulfurase NSF1, which catalyzes the abstraction of sulfur from amino acid l-cysteine also protects against ferroptosis by preventing in mitochondrial iron overload (Alvarez et al., 2017). Another important protein, frataxin (FXN), is responsible for transferring iron to ISCU for the assembly of Fe-S clusters. Decreased FXN levels, as seen in Friedreich’s ataxia, result in mitochondrial dysfunction, iron accumulation, and ferroptosis (Cotticelli et al., 2019). Additionally, ABCB7 and ABCB8, members of ATP binding cassette (ABC) transporter family, are involved in exporting Fe-S clusters from the mitochondria to the cytosol, although their role in ferroptosis has not been directly examined (Guo et al., 2022). MitoNEET (also known as CISD1), a redox sensitive Fe-S cluster protein, regulates mitochondrial iron metabolism and ROS balance by interacting with transferrin receptor and voltage-dependent anion channel (VDAC) (Furihata et al., 2018; Lipper et al., 2019). Loss of CISD1 facilitates erastin-induced ferroptosis by increasing iron accumulation and oxidative stress in cancer cells (Yuan et al., 2016).
Mitochondria generate a significant amount of ROS at multiple sites such as, electron transport chain and tricarboxylic acid cycle, which interact with Fe-S clusters to release free iron and promote ROS generation via the Fenton reaction. Therefore, the combination of high iron levels and potential for ROS generation make mitochondria an optimal site for ferroptosis. Accumulation of mitochondrial lipid ROS has been detected in cells undergoing ferroptosis, while mitochondria-targeted ROS scavengers can inhibit ferroptosis in various cell types (Yamada et al., 2020; Jiang et al., 2022). On the other hand, mitochondria are also equipped with numerous antioxidant systems to combat ferroptosis (Ali et al., 2022). Several mitochondria-associated antioxidant proteins, including GPX4, SOD2, and MGST1, play a crucial role in protecting mitochondria from oxidative damage during ferroptosis (Chen J. et al., 2022). The inner membrane of mitochondria also functions as a site for synthesizing Coenzyme Q (CoQ) - a redox-active cofactor essential for FSP1 activity that provides protection against ferroptosis. Mitochondrial dysfunction has been associated with reduced levels of CoQ, which increases ferroptosis susceptibility (Mourier et al., 2015; Kühl et al., 2017). Mitochondrial dihydroorotate dehydrogenase (DHODH) has been shown to suppress ferroptosis by oxidizing DHO to orotate by using CoQ as electron acceptor (Mao et al., 2021). The role of mitochondria in ferroptosis is illustrated in Figure 3.
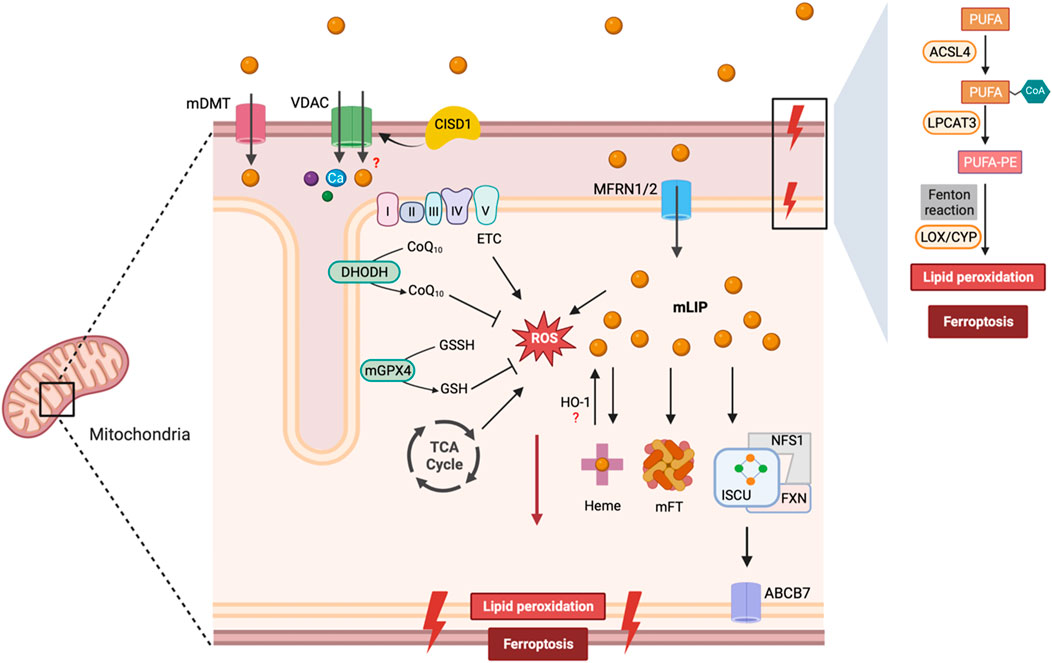
Figure 3. Mitochondrial regulation of ferroptosis. Iron is transported via mDT, MFRN1/2, and possibly VDAC into the mitochondria, where it is utilized for heme and Fe-S cluster synthesis. FXN transfers iron to ISCU and NFS1 serves as the sulfur donor for Fe-S cluster synthesis. Fe-S clusters are exported from the mitochondria to the cytosol via ABCB7 to allow the assembly of the cytosolic Fe-S containing proteins. Iron is sequestered by mitoferritin (mFT) or contributes to the labile iron pool within mitochondria (mLIP). Excess iron can lead to lipid peroxidation and ROS formation. ETC and TCA cycle also contribute to the mitochondrial ROS pool. GPX4 and DHODH represent two major antioxidant enzymes in mitochondria to prevent ferroptosis. Abbreviations: mDT, mitochondrial divalent transporter; VDAC, Voltage-Dependent Anion Channel; CISD1, CDGSH Iron Sulfur Domain 1; CoQ10, Coenzyme Q10; CoQ10H2, Coenzyme Q10 (reduced form); DHODH, Dihydroorotate Dehydrogenase; mGPX4, Mitochondrial Glutathione Peroxidase 4; GSSH, Oxidized Glutathione; GSH, Reduced Glutathione; MFRN1/2, Mitoferrin 1/2; mFT, Mitochondrial Ferritin; ISCU, Iron-Sulfur Cluster Scaffold Protein; FXN, Frataxin; ABCB7, ATP Binding Cassette Subfamily B Member 7; PUFA, Polyunsaturated Fatty Acids; ACSL4, Acyl-CoA Synthetase Long Chain Family Member 4; LPCAT3, Lysophosphatidylcholine Acyltransferase 3; LOX, Lipoxygenase; CYP, Cytochrome P450.
Accumulating evidence suggests that mitochondria-mediated ferroptosis contributes to the pathogenesis of heart disease. For example, mitochondria-mediated ferroptosis plays a key role in DOX-induced cardiomyopathy (Tadokoro et al., 2023). DOX downregulated GPX4 and induced excessive lipid peroxidation through DOX-Fe2+ complex in mitochondria, leading to mitochondria-dependent ferroptosis. Inhibiting ferroptosis by targeting mitochondrial-mediated pathways markedly attenuated DOX-induced cardiac toxicity (Fang et al., 2019). Mitochondria-mediated ferroptosis also mediates diabetic cardiomyopathy as well as catecholamine overload induced cardiomyopathy (Chen et al., 2023c; Chen et al., 2023a). Moreover, targeting mitochondrial ROS production effectively inhibits ferroptosis in cardiomyocytes (Sumneang et al., 2020; Chen et al., 2023a), offering a promising therapeutic option for the treatment of heart diseases by inhibiting ferroptosis.
Lysosomes play a crucial role in ferroptotic cell death through various mechanisms such as the activation of autophagy, release of lysosomal cathepsins, and accumulation of lysosomal iron or nitric oxide (Chen et al., 2021). Ablation of several autophagy related (ATG) genes, such as ATG3, ATG5, ATG6, ATG7, ATG8 and ATG13, has been shown to suppress ferroptosis, while activation of selective autophagy pathways prompts ferroptosis by targeting different cargoes. These include nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy, sequestosome 1-mediated SLC40A1 degradation, chaperone-mediated autophagy (CMA) of GPX4, lipophagy-dependent breakdown of lipid droplets and mitophagy-mediated mitochondrial degradation (Wu et al., 2019; Liu et al., 2020; Li et al., 2021; Rizzollo et al., 2021; Bengson et al., 2023). Moreover, signal transducer and activator of transcription 3 (STAT3) has been shown to mediate erastin-induced ferroptosis through activation of cathepsin B. In contrast, pharmacological inhibition of lysosomal enzymes such as cathepsins and vacuolar H+ ATPase suppresses erastin-induced ferroptosis (Gao et al., 2018). These findings suggest that lysosomal pathways are important mediators and potential molecular targets of ferroptosis.
Lysosome-mediated ferroptosis has been shown to mediate the pathogenesis of heart failure. Lysosomal function is essential for intracellular iron metabolism. Lysosomal damage promotes the accumulation of iron and lipid peroxides, leading to the activation of ferroptosis. Improving lysosomal ferroptosis protected against heart failure in a mouse model with cardiomyocyte-specific knockout of the mitochondrial translation factor p32 (Yagi et al., 2023).
The ferroptosis-inducing agents such as Erastin and RSL3 have been shown to trigger endoplasmic reticulum (ER) stress (Lee et al., 2018; Shin et al., 2018). ER stress plays a critical, yet complex role in regulating ferroptotic cell death via the eukaryotic translation initiation factor 2A (EIF2A)/activating transcription factor 4 (ATF4) pathway (Dixon et al., 2014). ATF4 inhibits ferroptosis by increasing the stability of GPX4 via HSPA5 upregulation or by promoting the expression of SLC7A11. Moreover, ER stress promotes membrane repair during ferroptosis via Ca2+-mediated ESCRT III activation. In contrast, ATF4 can upregulate ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) expression which in turn degrades GSH, thereby contributing to ferroptosis (Chen et al., 2017; Wang N. et al., 2019). ER can suppress PUFA-mediated ferroptosis by promoting the biosynthesis of MUFA primarily catalyzed by the ER enzyme - stearoyl-CoA desaturase (SCD) (Sen et al., 2023). ER also regulates ferroptosis sensitivity, potentially via STING1-dependent autophagy or mitochondrial fusion (Smith, 2021; Zhang Z. et al., 2022).
It has been shown that Inhibition of endoplasmic reticulum stress could alleviate ferroptosis and cell injury (Li W. et al., 2020). CHOP-mediated ER stress has also been shown to play an important role in I/R injury (Dixon et al., 2014). Moreover, iron overload in the ER triggers ferroptosis during cardiac I/R injury (Miyamoto et al., 2022). Therefore, these studies suggest that ERS induced by ferroptosis contributes to the pathogenesis of cardiac I/R injury.
Emerging evidence reveals that ferroptosis may crosstalk with other cell death pathways. Ferroptosis is a type of autophagy-dependent cell death (Zhou et al., 2020). Ferroptosis inducers promote the activation of autophagy, leading to the accumulation of autophagic vesicles. NCOA4-mediated autophagy, termed ferritinophagy, induces ferritin degradation and iron overload, promoting oxidative stress and ferroptosis. Elevated lipid peroxidation in ferroptosis also promotes GSDMD-mediated pyroptosis. Indeed, it has been shown that deletion of GPX4 led to lipid peroxidation-dependent caspase 11 and GSDMD cleavage (Kang et al., 2018). Elevated mitochondrial ROS during ferroptosis may also promote necroptosis, possibly by increasing the autophosphorylation of RIPK1 (Zhang et al., 2017). Moreover, the release of damage-associated molecular pattern molecules (DAMPs) from the plasma membrane pore is a common feature of necrotic cell death such as ferroptosis, pyroptosis, and necroptosis. The release of the DAMPs triggered by ferroptosis may further promote pyroptosis and necroptosis. The significance of ferroptosis-pyroptosis crosstalk in heart disease needs to be further investigated.
Myocardial ischemia/reperfusion (I/R) injury can occur during the restoration of blood supply to the acutely ischemic heart and contributes to the final infarct size (Zhang et al., 2023). Ferroptosis has recently been implicated in the pathogenesis of myocardial I/R injury (Han et al., 2023). I/R injury causes iron overload characterized by increased cardiac nonheme iron levels and ferritin expression (Fang et al., 2019). During I/R injury, there is also a time-dependent increase in ACSL4 levels with a concomitant decrease in GPX4 activity (Tang L.-J. et al., 2021). Pharmacological inhibition of ferroptosis with ferrostatin-1 or dexrazoxane has been shown to reduce cardiac infarct size following I/R (Fang et al., 2019). Moreover, inhibiting ferroptosis can also provide long-term benefits against I/R-induced cardiac remodeling and fibrosis. In patients undergoing coronary artery bypass grafting (CABG) surgery, infusion of an iron chelator deferoxamine also suppressed reperfusion-induced oxidative damage (Paraskevaidis et al., 2005).
Iron overload has been linked to endothelial dysfunction, impaired excitation-contraction coupling of cardiomyocytes, myocardial inflammation and tissue fibrosis, which all contribute to the development of HFpEF (Li et al., 2022). A recent study reveals that obesity-induced HFpEF leads to an upregulation in iNOS activity while reducing GPX4 activity. Further, treatment with an anti-diabetic agent, Imeglimin has been shown to prevent HFpEF by inhibiting myocardial production of iNOS and restoring myocardial expression of GPX4 (Kitakata et al., 2021). Tandem Mass Tag (TMT)-based proteomics studies reveal that ferroptotic metabolic pathways contribute to the development of HFpEF (Ma et al., 2022). Additionally, rats with HFpEF exhibited an increase in Fe2+ concentration and lipid peroxidation products, accompanied by increased expression of TFR1 and ACSL4 proteins. Moreover, there was a significant decrease in GSH concentrations and downregulation of xCT and FTH1 expression in HFpEF (Ma et al., 2022). These findings suggest that ferroptosis may contribute to the pathogenesis of HFpEF.
Dysregulation of iron metabolism and increased lipid peroxidation have been implicated in cardiac hypertrophic remodeling (Tang et al., 2019; Fan et al., 2022). Recent studies further revealed that ferroptosis plays a role in hypertrophic cardiomyopathy. It has been shown that xCT, a key regulator of ferroptosis, prevents cardiac hypertrophy by inhibiting ferroptosis (Zhang X. et al., 2022). xCT knockout aggravated angiotensin II (Ang II)-induced cardiac hypertrophy, fibrosis, and dysfunction (Zhang X. et al., 2022). Similarly, loss of ferritin H, a key iron storage protein, led to hypertrophic cardiomyopathy by inducing cardiac ferroptosis, which was rescued by overexpression of xCT (Fang et al., 2020). Moreover, overexpression of TRIM44, a deubiquitinase, promoted pressure overload-induced cardiac hypertrophy via activation of TLR4/NOX4-mediated ferroptosis (Wu et al., 2023).
Doxorubicin (DOX) induces cardiotoxicity, referred to as DOX-induced cardiomyopathy, which limits its clinical use as a chemotherapeutic agent (Rawat et al., 2021). The mechanism of DOX-induced cardiomyopathy remains incompletely understood, but recent studies have highlighted a prominent role of ferroptosis in pathogenesis. Fang et al. identified ferroptosis as a key mechanism for DOX-induced cardiomyopathy in mice (Fang et al., 2019). Importantly, inhibition of ferroptosis with ferrostatin-1 and dexrazoxane effectively attenuated DOX-induced cardiomyopathy. Mechanistically, they revealed that mitochondrial iron overload and lipid peroxidation play a key role in DOX-induced myocardial ferroptosis. Tadokoro et al. also showed that mitochondria-dependent ferroptosis plays a key role in DOX cardiomyopathy (Tadokoro et al., 2023). GPX4 expression was markedly downregulated during DOX cardiomyopathy, accompanied by increased lipid peroxidation in mitochondria. Importantly, transgenic overexpression of GPX4 ameliorated, whereas heterodeletion of GPX4 exacerbated DOX cardiomyopathy. Abe et al. further showed that DOX induces mitochondria-dependent ferroptosis by intercalating into mitochondrial DNA (mtDNA) (Abe et al., 2022). Moreover, DOX also disrupts heme synthesis and impairs iron utilization by downregulating 5′-aminolevulinate synthase 1 (Alas1), leading to mitochondrial iron overload and ferroptosis.
Ferritinophagy-mediated ferroptosis has been shown to contribute to the pathogenesis of septic cardiomyopathy (Li N. et al., 2020). Recent findings reveal a role of islet cell autoantigen 69 (ICA69)-STING signaling and transmembrane protein 43 (TMEM43) in lipopolysaccharide (LPS)-induced cardiomyocyte ferroptosis and cardiomyopathy. Ablation of ICA69 decreased STING trafficking and improved overall cardiac function by targeting LPS-induced ferroptosis. ICA69 levels are also positively correlated with the severity of sepsis in humans (Kong et al., 2022). Overexpression of TMEM43 inhibited LPS-induced ferroptosis with increased levels of SLC7A11 and GPX4, revealing a protective role of TMEM43 against sepsis-induced cardiomyopathy (Chen Z. et al., 2022).
A growing body of evidence highlights the role of ferroptosis in diabetic cardiomyopathy (DCM). The advanced glycation end-products (AGEs) that accumulate in cardiac tissue with the onset of diabetes, can induce ferroptosis as evident by increased MDA levels, upregulation of COX2, and downregulation of ferritin and SLC7A11. Moreover, activation of AMPK/NRF2 pathways with sulforaphane protects heart against AGE-induced ferroptosis (Wang X. et al., 2022). In contrast, Nrf2 signaling can also exert detrimental effect to the heart, particularly when autophagy is impaired such as in chronic in type 1 diabetes (Zang et al., 2020).
Ferroptosis has recently emerged as a potential contributor to radiation-induced cardiomyopathy (RICM) (Wang B. et al., 2020). Radiation exposure induces ROS production, which triggers lipid peroxidation and subsequent ferroptosis (Lei et al., 2020). Endothelial cell injury caused by radiation is an early event in RICM, leading to the release of cytokines and chemokines such as IL-6, IL-8, TGF-β, TNF-α, and IL-1β (Li X. et al., 2019; Li W et al., 2019; Wang C. et al., 2020). Increased ROS production and lipid peroxidation further contribute to endothelial cell damage, myocardial fibrosis, and cardiomyopathy (D’Oria et al., 2020; Jiang et al., 2021). Activation of the STING pathway and subsequent induction of interferon gamma and COX2 expression have been observed following radiation exposure (Lemos et al., 2020; Storozynsky and Hitt, 2020). Moreover, damaged endothelial cells release danger-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), which promote ferroptosis and inflammation (Dyer et al., 2018; Zhou et al., 2018; Green, 2019).
Ferroptosis as a potential target for the treatment of heart disease has been explored in various experimental models. Genetic or pharmacologic inhibition of ferroptosis has been shown to illicit cardioprotective effects in these studies (Table 1). Therefore, anti-ferroptosis therapies may hold a tremendous promise for the treatment of heart disease in humans. Several drugs currently in clinical use have been shown to target ferroptosis. For example, dexrazoxane (DXZ) has been used to treat doxorubicin-induced cardiotoxicity, which reverses DOX-induced ferroptosis mainly by chelating mitochondrial iron (Ichikawa et al., 2014). Several other iron chelators, including deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX) are clinically approved for managing iron overload-related diseases. In addition, N-acetylcysteine (NAC) has been shown to inhibit ferroptosis by targeting cysteine metabolism. NAC has been clinically shown to improve neurodegeneration-related symptoms by increasing cysteine levels and facilitating the synthesis of GSH (Monti et al., 2016). Notably, edaravone, a radical-trapping antioxidant clinically approved for treating acute ischemic stroke and amyotrophic lateral sclerosis, has been shown to inhibit ferroptosis under various pathological conditions (Homma et al., 2019). Thiazolidinediones (TZDs), such as rosiglitazone, pioglitazone, and troglitazone, are approved to treat adult type 2 diabetes, which have suppressing ferroptosis activity by selectively inhibiting ACSL4 (Doll et al., 2017). Of note, a screening of a library consisting of FDA-approved drugs has led to the successful identification of multiple ferroptosis inhibitors (Tan et al., 2024), which offers new therapeutic possibilities for the treatments of ferroptosis-related diseases.

Table 1. Targeting ferroptosis in experimental models of heart disease.
The clinical application of ferroptosis-related targets is still in its infancy. Iron metabolism-related indicators, such as serum iron, serum ferritin, transferrin and soluble transferrin receptors, have been used to monitor the progression of heart disease. For example, patients with elevated serum ferritin showed a higher incidence of acute myocardial infarction than those with reduced serum ferritin (Moradi et al., 2015). Moreover, elevated levels of soluble ferritin receptors corelates with a higher risk of coronary atherosclerotic heart disease (Braun et al., 2004). Hepcidin concentration has also been used to predict the risk of myocardial infarction or cardiovascular death (Zeller et al., 2018). Notably, elevated levels of ferritin and hepcidin were associated with a higher risk of heart failure in women (Klip et al., 2017). Other biomarkers of ferroptosis, such as lipid peroxidation products, might also be useful in monitoring the progression of heart disease.
Recent studies clearly demonstrate that ferroptosis contributes significantly to the pathogenesis of multiple forms of heart disease including acute cardiac injury and chronic disorders. Genetic or pharmacologic inhibition of ferroptosis showed beneficial effects under pathological conditions such as myocardial infarction and heart failure. Recent studies also provide new mechanistic insights into the regulatory mechanisms of ferroptosis, including new pathways that positively or negatively regulates ferroptosis signaling and the crosstalk between different subcellular compartments in orchestrating ferroptotic cell death. Numerous studies have demonstrated that cardiomyocytes undergo ferroptosis in response to pathological stress in vivo and in vivo. Ferroptosis of other cell types in the heart, including endothelial cells, smooth muscle cells and macrophages, has also been shown to play a role in the pathogenesis of certain forms of heart disease (Leng et al., 2021). The relative contribution of ferroptosis in different cell types to disease pathogenesis warrant further investigation under various disease conditions. Notably, interaction between ferroptosis and other modes of cell death, such as pyroptosis, necroptosis, and autophagy, increases the complexity of these pathways. Various cell death processes contribute to the loss of cardiac cells in heart diseases, and their specific roles in disease pathogensis need further investigation. It will be important to develop new diagnostic tools for assessing ferroptosis in vivo, given the lack of reliable and specific biomarkers for ferroptosis. Targeting ferroptosis represents an important therapeutic opportunity for the treatment of heart disease. Pharmacological inhibitors of the ferroptosis pathway have been developed for use in experimental settings, such as iron chelators and lipophilic antioxidants. However, given that adverse effects have been observed with these compounds (Miller et al., 2005; Tebbi et al., 2007), the identification of new molecular targets of the ferroptosis pathway and the development of novel ferroptosis inhibitors will be important for anti-ferroptosis therapies. Targeting the ferroptosis pathway represents a promising therapeutic strategy for various forms of heart disease, although these approaches warrant further investigation in clinical studies.
SF: Writing–original draft, Writing–review and editing. HZ: Writing–review and editing. YC: Investigation, Resources, Writing–review and editing. QL: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Institutes of Health (R01HL160767 and R01HL155035), American Heart Association (19TPA34850148), and the University of Washington Royalty Research Fund.
Figures were created with BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe K., Ikeda M., Ide T., Tadokoro T., Miyamoto H. D., Furusawa S., et al. (2022). Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal. 15, eabn8017. doi:10.1126/scisignal.abn8017
Ali M. Y., Oliva C. R., Flor S., Griguer C. E. (2022). Mitoferrin, cellular and mitochondrial iron homeostasis. Cells 11, 3464. doi:10.3390/cells11213464
Alvarez S. W., Sviderskiy V. O., Terzi E. M., Papagiannakopoulos T., Moreira A. L., Adams S., et al. (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. doi:10.1038/nature24637
Azucenas C. R., Ruwe T. A., Bonamer J. P., Qiao B., Ganz T., Jormakka M., et al. (2023). Comparative analysis of the functional properties of human and mouse ferroportin. Am. J. Physiol.-Cell Physiol. 324, C1110–C1118. doi:10.1152/ajpcell.00063.2023
Bansal S., Biswas G., Avadhani N. G. (2014). Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2, 273–283. doi:10.1016/j.redox.2013.07.004
Bengson E. F., Guggisberg C. A., Bastian T. W., Georgieff M. K., Ryu M.-S. (2023). Quantitative omics analyses of NCOA4 deficiency reveal an integral role of ferritinophagy in iron homeostasis of hippocampal neuronal HT22 cells. Front. Nutr. 10, 1054852. doi:10.3389/fnut.2023.1054852
Bersuker K., Hendricks J. M., Li Z., Magtanong L., Ford B., Tang P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi:10.1038/s41586-019-1705-2
Berezovsky B., Frýdlová J., Gurieva I., Rogalsky D. W., Vokurka M., Krijt J. (2022). Heart ferroportin protein content is regulated by heart iron concentration and systemic hepcidin expression. Int. J. Mol. Sci. 23, 5899. doi:10.3390/ijms23115899
Bindu S., Pal C., Dey S., Goyal M., Alam A., Iqbal M. S., et al. (2011). Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 286, 39387–39402. doi:10.1074/jbc.M111.279893
Braun S., Ndrepepa G., von Beckerath N., Vogt W., Schömig A., Kastrati A. (2004). Value of serum ferritin and soluble transferrin receptor for prediction of coronary artery disease and its clinical presentations. Atherosclerosis 174, 105–110. doi:10.1016/j.atherosclerosis.2004.01.011
Chang H., Wu R., Shang M., Sato T., Chen C., Shapiro J. S., et al. (2016). Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 8, 247–267. doi:10.15252/emmm.201505748
Charlebois E., Fillebeen C., Katsarou A., Rabinovich A., Wisniewski K., Venkataramani V., et al. (2022). A crosstalk between hepcidin and IRE/IRP pathways controls ferroportin expression and determines serum iron levels in mice. eLife 11, e81332. doi:10.7554/eLife.81332
Cheff D. M., Muotri A. R., Stockwell B. R., Schmidt E. E., Ran Q., Kartha R. V., et al. (2021). Development of therapies for rare genetic disorders of GPX4: roadmap and opportunities. Orphanet J. Rare Dis. 16, 446. doi:10.1186/s13023-021-02048-0
Chen J., Li X., Ge C., Min J., Wang F. (2022a). The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29, 467–480. doi:10.1038/s41418-022-00941-0
Chen L., Hambright W. S., Na R., Ran Q. (2015). Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 290, 28097–28106. doi:10.1074/jbc.M115.680090
Chen M.-S., Wang S.-F., Hsu C.-Y., Yin P.-H., Yeh T.-S., Lee H.-C., et al. (2017). CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 8, 114588–114602. doi:10.18632/oncotarget.23055
Chen X., Kang R., Kroemer G., Tang D. (2021). Organelle-specific regulation of ferroptosis. Cell Death Differ. 28, 2843–2856. doi:10.1038/s41418-021-00859-z
Chen X., Yu C., Kang R., Tang D. (2020). Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 8, 590226. doi:10.3389/fcell.2020.590226
Chen Y., Guo X., Zeng Y., Mo X., Hong S., He H., et al. (2023a). Ferroptosis contributes to catecholamine-induced cardiotoxicity and pathological remodeling. Free Radic. Biol. Med. 207, 227–238. doi:10.1016/j.freeradbiomed.2023.07.025
Chen Y., Guo X., Zeng Y., Mo X., Hong S., He H., et al. (2023b). Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 13, 15515. doi:10.1038/s41598-023-42760-4
Chen Y., Li S., Yin M., Li Y., Chen C., Zhang J., et al. (2023c). Isorhapontigenin attenuates cardiac microvascular injury in diabetes via the inhibition of mitochondria-associated ferroptosis through PRDX2-MFN2-ACSL4 pathways. Diabetes 72, 389–404. doi:10.2337/db22-0553
Chen Z., Cao Z., Gui F., Zhang M., Wu X., Peng H., et al. (2022b). TMEM43 protects against sepsis-induced cardiac injury via inhibiting ferroptosis in mice. Cells 11, 2992. doi:10.3390/cells11192992
Chiang S.-K., Chen S.-E., Chang L.-C. (2018). A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 20, 39. doi:10.3390/ijms20010039
Clarke G. D., Solis-Herrera C., Molina-Wilkins M., Martinez S., Merovci A., Cersosimo E., et al. (2017). Pioglitazone improves left ventricular diastolic function in subjects with diabetes. Diabetes Care 40, 1530–1536. doi:10.2337/dc17-0078
Costa D. L., Amaral E. P., Andrade B. B., Sher A. (2020). Modulation of inflammation and immune responses by heme oxygenase-1: implications for infection with intracellular pathogens. Antioxidants 9, 1205. doi:10.3390/antiox9121205
Cotticelli M. G., Xia S., Lin D., Lee T., Terrab L., Wipf P., et al. (2019). Ferroptosis as a novel therapeutic target for friedreich’s ataxia. J. Pharmacol. Exp. Ther. 369, 47–54. doi:10.1124/jpet.118.252759
Cruse I., Maines M. D. (1988). Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 263, 3348–3353. doi:10.1016/s0021-9258(18)69078-7
Cui Y., Zhang Y., Zhao X., Shao L., Liu G., Sun C., et al. (2021). ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain. Behav. Immun. 93, 312–321. doi:10.1016/j.bbi.2021.01.003
D’Anna M. C., Veuthey T. V., Roque M. E. (2009). Immunolocalization of ferroportin in healthy and anemic mice. J. Histochem. Cytochem. 57, 9–16. doi:10.1369/jhc.2008.951616
Dabkowski E. R., Williamson C. L., Hollander J. M. (2008). Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic. Biol. Med. 45, 855–865. doi:10.1016/j.freeradbiomed.2008.06.021
Danial N. N., Korsmeyer S. J. (2004). Cell death: critical control points. Cell 116, 205–219. doi:10.1016/S0092-8674(04)00046-7
Del Re D. P., Amgalan D., Linkermann A., Liu Q., Kitsis R. N. (2019). Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 99, 1765–1817. doi:10.1152/physrev.00022.2018
Dev S., Babitt J. L. (2017). Overview of iron metabolism in health and disease. Hemodial. Int. Int. Symp. Home Hemodial. 21 (Suppl. 1), S6–S20. doi:10.1111/hdi.12542
Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon S. J., Patel D. N., Welsch M., Skouta R., Lee E. D., Hayano M., et al. (2014). Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523. doi:10.7554/eLife.02523
Dixon S. J., Winter G. E., Musavi L. S., Lee E. D., Snijder B., Rebsamen M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi:10.1021/acschembio.5b00245
Doll S., Freitas F. P., Shah R., Aldrovandi M., Da Silva M. C., Ingold I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi:10.1038/s41586-019-1707-0
Doll S., Proneth B., Tyurina Y. Y., Panzilius E., Kobayashi S., Ingold I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi:10.1038/nchembio.2239
Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., et al. (2000). Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781. doi:10.1038/35001596
D’Oria R., Schipani R., Leonardini A., Natalicchio A., Perrini S., Cignarelli A., et al. (2020). The role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid. Med. Cell. Longev. 2020, 5732956. doi:10.1155/2020/5732956
Du J., Wang T., Li Y., Zhou Y., Wang X., Yu X., et al. (2019). DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 131, 356–369. doi:10.1016/j.freeradbiomed.2018.12.011
Dyer M. R., Chen Q., Haldeman S., Yazdani H., Hoffman R., Loughran P., et al. (2018). Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 8, 2068. doi:10.1038/s41598-018-20479-x
Edinger A. L., Thompson C. B. (2004). Death by design: apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 16, 663–669. doi:10.1016/j.ceb.2004.09.011
Fan X., Li A., Yan Z., Geng X., Lian L., Lv H., et al. (2022). From iron metabolism to ferroptosis: pathologic changes in coronary heart disease. Oxid. Med. Cell. Longev. 2022, 6291889. doi:10.1155/2022/6291889
Fang X., Cai Z., Wang H., Han D., Cheng Q., Zhang P., et al. (2020). Loss of cardiac ferritin H facilitates cardiomyopathy via slc7a11-mediated ferroptosis. Circ. Res. 127, 486–501. doi:10.1161/CIRCRESAHA.120.316509
Fang X., Wang H., Han D., Xie E., Yang X., Wei J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. 116, 2672–2680. doi:10.1073/pnas.1821022116
Fang Y., Chen X., Tan Q., Zhou H., Xu J., Gu Q. (2021). Inhibiting ferroptosis through disrupting the NCOA4–FTH1 interaction: a new mechanism of action. ACS Cent. Sci. 7, 980–989. doi:10.1021/acscentsci.0c01592
Feng Y., Madungwe N. B., Imam Aliagan A. D., Tombo N., Bopassa J. C. (2019). Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 520, 606–611. doi:10.1016/j.bbrc.2019.10.006
Friedmann Angeli J. P., Schneider M., Proneth B., Tyurina Y. Y., Tyurin V. A., Hammond V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi:10.1038/ncb3064
Furihata T., Takada S., Maekawa S., Mizushima W., Watanabe M., Takahashi H., et al. (2018). mitoNEET regulates mitochondrial iron homeostasis interacting with transferrin receptor. Cell Biol. doi:10.1101/330084
Gan B. (2021). Mitochondrial regulation of ferroptosis. J. Cell Biol. 220, e202105043. doi:10.1083/jcb.202105043
Gao H., Bai Y., Jia Y., Zhao Y., Kang R., Tang D., et al. (2018). Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556. doi:10.1016/j.bbrc.2018.07.078
Gao M., Yi J., Zhu J., Minikes A. M., Monian P., Thompson C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363. doi:10.1016/j.molcel.2018.10.042
Garcia-Santos D., Hamdi A., Saxova Z., Fillebeen C., Pantopoulos K., Horvathova M., et al. (2018). Inhibition of heme oxygenase ameliorates anemia and reduces iron overload in a β-thalassemia mouse model. Blood 131, 236–246. doi:10.1182/blood-2017-07-798728
Gonca E. (2017). Cardioprotective effect of zileuton: a 5-lipoxygenase inhibitor against myocardial ischemia/reperfusion injury. Turk. J. Thorac. Cardiovasc. Surg. 25, 273–281. doi:10.5606/tgkdc.dergisi.2017.13748
Green D. R. (2019). The coming decade of cell death research: five riddles. Cell 177, 1094–1107. doi:10.1016/j.cell.2019.04.024
Guo J., Zhou Y., Liu D., Wang M., Wu Y., Tang D., et al. (2022). Mitochondria as multifaceted regulators of ferroptosis. Life Metab. 1, 134–148. doi:10.1093/lifemeta/loac035
Han X., Zhang J., Liu J., Wang H., Du F., Zeng X., et al. (2023). Targeting ferroptosis: a novel insight against myocardial infarction and ischemia–reperfusion injuries. Apoptosis 28, 108–123. doi:10.1007/s10495-022-01785-2
Homma T., Kobayashi S., Sato H., Fujii J. (2019). Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp. Cell Res. 384, 111592. doi:10.1016/j.yexcr.2019.111592
Hou W., Xie Y., Song X., Sun X., Lotze M. T., Zeh H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Hu Q., Zhang Y., Lou H., Ou Z., Liu J., Duan W., et al. (2021). GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 12, 706. doi:10.1038/s41419-021-04008-9
Hung H.-I., Schwartz J. M., Maldonado E. N., Lemasters J. J., Nieminen A.-L. (2013). Mitoferrin-2-dependent mitochondrial iron uptake sensitizes human head and neck squamous carcinoma cells to photodynamic therapy. J. Biol. Chem. 288, 677–686. doi:10.1074/jbc.M112.422667
Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S. V., et al. (2014). Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630. doi:10.1172/JCI72931
Ito J., Omiya S., Rusu M.-C., Ueda H., Murakawa T., Tanada Y., et al. (2021). Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. eLife 10, e62174. doi:10.7554/eLife.62174
Jelinek A., Heyder L., Daude M., Plessner M., Krippner S., Grosse R., et al. (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 117, 45–57. doi:10.1016/j.freeradbiomed.2018.01.019
Jiang J.-J., Zhang G.-F., Zheng J.-Y., Sun J.-H., Ding S.-B. (2022). Targeting mitochondrial ROS-mediated ferroptosis by quercetin alleviates high-fat diet-induced hepatic lipotoxicity. Front. Pharmacol. 13, 876550. doi:10.3389/fphar.2022.876550
Jiang W., Xiong Y., Li X., Yang Y. (2021). Cardiac fibrosis: cellular effectors, molecular pathways, and exosomal roles. Front. Cardiovasc. Med. 8, 715258. doi:10.3389/fcvm.2021.715258
Kagan V. E., Mao G., Qu F., Angeli J. P. F., Doll S., Croix C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi:10.1038/nchembio.2238
Kang R., Zeng L., Zhu S., Xie Y., Liu J., Wen Q., et al. (2018). Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 24, 97–108. doi:10.1016/j.chom.2018.05.009
Kapralov A. A., Yang Q., Dar H. H., Tyurina Y. Y., Anthonymuthu T. S., Kim R., et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290. doi:10.1038/s41589-019-0462-8
Kitakata H., Endo J., Hashimoto S., Mizuno E., Moriyama H., Shirakawa K., et al. (2021). Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem. Biophys. Res. Commun. 572, 185–190. doi:10.1016/j.bbrc.2021.07.090
Klip I. T., Voors A. A., Swinkels D. W., Bakker S. J. L., Kootstra-Ros J. E., Lam C. S., et al. (2017). Serum ferritin and risk for new-onset heart failure and cardiovascular events in the community. Eur. J. Heart Fail. 19, 348–356. doi:10.1002/ejhf.622
Kong C., Ni X., Wang Y., Zhang A., Zhang Y., Lin F., et al. (2022). ICA69 aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Discov. 8, 187. doi:10.1038/s41420-022-00957-y
Kuang F., Liu J., Tang D., Kang R. (2020). Oxidative damage and antioxidant defense in ferroptosis. Front. Cell Dev. Biol. 8, 586578. doi:10.3389/fcell.2020.586578
Kühl I., Miranda M., Atanassov I., Kuznetsova I., Hinze Y., Mourier A., et al. (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6, e30952. doi:10.7554/eLife.30952
Lee Y.-S., Lee D.-H., Choudry H. A., Bartlett D. L., Lee Y. J. (2018). Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 16, 1073–1076. doi:10.1158/1541-7786.MCR-18-0055
Lei G., Zhang Y., Koppula P., Liu X., Zhang J., Lin S. H., et al. (2020). The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 30, 146–162. doi:10.1038/s41422-019-0263-3
Lemos H., Ou R., McCardle C., Lin Y., Calver J., Minett J., et al. (2020). Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J. Immunother. Cancer 8, e001182. doi:10.1136/jitc-2020-001182
Leng Y., Luo X., Yu J., Jia H., Yu B. (2021). Ferroptosis: a potential target in cardiovascular disease. Front. Cell Dev. Biol. 9, 813668. doi:10.3389/fcell.2021.813668
Levi S., Ripamonti M., Dardi M., Cozzi A., Santambrogio P. (2021). Mitochondrial ferritin: its role in physiological and pathological conditions. Cells 10, 1969. doi:10.3390/cells10081969
Li D., Li Y. (2020). The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 5, 108. doi:10.1038/s41392-020-00216-5
Li J., Liu J., Xu Y., Wu R., Chen X., Song X., et al. (2021). Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 17, 3361–3374. doi:10.1080/15548627.2021.1872241
Li N., Wang W., Zhou H., Wu Q., Duan M., Liu C., et al. (2020a). Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 160, 303–318. doi:10.1016/j.freeradbiomed.2020.08.009
Li Q., Zhao Z., Zhou X., Yan Y., Shi L., Chen J., et al. (2022). Ferroptosis: the potential target in heart failure with preserved ejection fraction. Cells 11, 2842. doi:10.3390/cells11182842
Li W., Feng G., Gauthier J. M., Lokshina I., Higashikubo R., Evans S., et al. (2019a). Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 129, 2293–2304. doi:10.1172/JCI126428
Li W., Li W., Leng Y., Xiong Y., Xia Z. (2020b). Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 39, 210–225. doi:10.1089/dna.2019.5097
Li X., Duan L., Yuan S., Zhuang X., Qiao T., He J. (2019b). Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J. Inflamm. 16, 11. doi:10.1186/s12950-019-0216-0
Lipper C. H., Stofleth J. T., Bai F., Sohn Y.-S., Roy S., Mittler R., et al. (2019). Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. 116, 19924–19929. doi:10.1073/pnas.1908271116
Liu J., Kuang F., Kroemer G., Klionsky D. J., Kang R., Tang D. (2020). Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem. Biol. 27, 420–435. doi:10.1016/j.chembiol.2020.02.005
Liu X., Zhang Y., Zhuang L., Olszewski K., Gan B. (2021). NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis. 8, 731–745. doi:10.1016/j.gendis.2020.11.010
Llabani E., Hicklin R. W., Lee H. Y., Motika S. E., Crawford L. A., Weerapana E., et al. (2019). Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 11, 521–532. doi:10.1038/s41557-019-0261-6
Ma S., He L.-L., Zhang G.-R., Zuo Q.-J., Wang Z.-L., Zhai J.-L., et al. (2022). Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn. Schmiedeb. Arch. Pharmacol. 395, 945–962. doi:10.1007/s00210-022-02243-1
Mancias J. D., Wang X., Gygi S. P., Harper J. W., Kimmelman A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. doi:10.1038/nature13148
Mao C., Liu X., Zhang Y., Lei G., Yan Y., Lee H., et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. doi:10.1038/s41586-021-03539-7
Menon A. V., Liu J., Tsai H. P., Zeng L., Yang S., Asnani A., et al. (2022). Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 139, 936–941. doi:10.1182/blood.2020008455
Miller E. R., Pastor-Barriuso R., Dalal D., Riemersma R. A., Appel L. J., Guallar E. (2005). Meta-Analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 142, 37–46. doi:10.7326/0003-4819-142-1-200501040-00110
Mishima E., Ito J., Wu Z., Nakamura T., Wahida A., Doll S., et al. (2022). A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783. doi:10.1038/s41586-022-05022-3
Miyamoto H. D., Ikeda M., Ide T., Tadokoro T., Furusawa S., Abe K., et al. (2022). Iron overload via heme degradation in the endoplasmic reticulum triggers ferroptosis in myocardial ischemia-reperfusion injury. JACC Basic Transl. Sci. 7, 800–819. doi:10.1016/j.jacbts.2022.03.012
Monti D. A., Zabrecky G., Kremens D., Liang T.-W., Wintering N. A., Cai J., et al. (2016). N-acetyl cysteine may support dopamine neurons in Parkinson’s disease: preliminary clinical and cell line data. PloS One 11, e0157602. doi:10.1371/journal.pone.0157602
Moradi M., Fariba F., Mohasseli A. S. (2015). Relation between the serum ferritin level and the risk for acute myocardial infarction. J. Res. Health Sci. 15, 147–151.
Mortensen M. S., Ruiz J., Watts J. L. (2023). Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cells 12, 804. doi:10.3390/cells12050804
Mourier A., Motori E., Brandt T., Lagouge M., Atanassov I., Galinier A., et al. (2015). Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 208, 429–442. doi:10.1083/jcb.201411100
Ni R., Cao T., Xiong S., Ma J., Fan G.-C., Lacefield J. C., et al. (2016). Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 90, 12–23. doi:10.1016/j.freeradbiomed.2015.11.013
Oh S.-J., Ikeda M., Ide T., Hur K. Y., Lee M.-S. (2022). Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 8, 414. doi:10.1038/s41420-022-01199-8
Papanikolaou G., Pantopoulos K. (2005). Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 202, 199–211. doi:10.1016/j.taap.2004.06.021
Paradkar P. N., Zumbrennen K. B., Paw B. H., Ward D. M., Kaplan J. (2009). Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 29, 1007–1016. doi:10.1128/MCB.01685-08
Paraskevaidis I. A., Iliodromitis E. K., Vlahakos D., Tsiapras D. P., Nikolaidis A., Marathias A., et al. (2005). Deferoxamine infusion during coronary artery bypass grafting ameliorates lipid peroxidation and protects the myocardium against reperfusion injury: immediate and long-term significance. Eur. Heart J. 26, 263–270. doi:10.1093/eurheartj/ehi028
Pu F., Chen F., Zhang Z., Shi D., Zhong B., Lv X., et al. (2022). Ferroptosis as a novel form of regulated cell death: implications in the pathogenesis, oncometabolism and treatment of human cancer. Genes Dis. 9, 347–357. doi:10.1016/j.gendis.2020.11.019
Rawat P. S., Jaiswal A., Khurana A., Bhatti J. S., Navik U. (2021). Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 139, 111708. doi:10.1016/j.biopha.2021.111708
Rizzollo F., More S., Vangheluwe P., Agostinis P. (2021). The lysosome as a master regulator of iron metabolism. Trends biochem. Sci. 46, 960–975. doi:10.1016/j.tibs.2021.07.003
Rocha V. C. J., França L. S. D. A., De Araújo C. F., Ng A. M., De Andrade C. M., Andrade A. C., et al. (2016). Protective effects of mito-TEMPO against doxorubicin cardiotoxicity in mice. Cancer Chemother. Pharmacol. 77, 659–662. doi:10.1007/s00280-015-2949-7
Santambrogio P., Biasiotto G., Sanvito F., Olivieri S., Arosio P., Levi S. (2007). Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 55, 1129–1137. doi:10.1369/jhc.7A7273.2007
Santana-Codina N., Gikandi A., Mancias J. D. (2021). “The role of NCOA4-mediated ferritinophagy in ferroptosis,” in Ferroptosis: mechanism and diseases. Editors A. F. Florez,, and H. Alborzinia (Cham: Springer International Publishing), 41–57. doi:10.1007/978-3-030-62026-4_4
Seiwert N., Wecklein S., Demuth P., Hasselwander S., Kemper T. A., Schwerdtle T., et al. (2020). Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 11, 787. doi:10.1038/s41419-020-02950-8
Sen U., Coleman C., Sen T. (2023). Stearoyl coenzyme A desaturase-1: multitasker in cancer, metabolism, and ferroptosis. Trends Cancer 9, 480–489. doi:10.1016/j.trecan.2023.03.003
Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., et al. (2006). Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100. doi:10.1038/nature04512
Shin D., Kim E. H., Lee J., Roh J.-L. (2018). Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454–462. doi:10.1016/j.freeradbiomed.2018.10.426
Shui S., Zhao Z., Wang H., Conrad M., Liu G. (2021). Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 45, 102056. doi:10.1016/j.redox.2021.102056
Smith J. A. (2021). STING, the endoplasmic reticulum, and mitochondria: is three a crowd or a conversation? Front. Immunol. 11, 611347. doi:10.3389/fimmu.2020.611347
Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Storozynsky Q., Hitt M. M. (2020). The impact of radiation-induced DNA damage on cGAS-STING-mediated immune responses to cancer. Int. J. Mol. Sci. 21, 8877. doi:10.3390/ijms21228877
Sumneang N., Siri-Angkul N., Kumfu S., Chattipakorn S. C., Chattipakorn N. (2020). The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 680, 108241. doi:10.1016/j.abb.2019.108241
Ta N., Qu C., Wu H., Zhang D., Sun T., Li Y., et al. (2022). Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 119, e2117396119. doi:10.1073/pnas.2117396119
Tadokoro T., Ikeda M., Ide T., Deguchi H., Ikeda S., Okabe K., et al. (2020). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747. doi:10.1172/jci.insight.132747
Tadokoro T., Ikeda M., Ide T., Deguchi H., Ikeda S., Okabe K., et al. (2023). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 8, e169756. doi:10.1172/jci.insight.169756
Tan Q., Wu D., Lin Y., Ai H., Xu J., Zhou H., et al. (2024). Identifying eleven new ferroptosis inhibitors as neuroprotective agents from FDA-approved drugs. Bioorg. Chem. 146, 107261. doi:10.1016/j.bioorg.2024.107261
Tang D., Chen X., Kang R., Kroemer G. (2021a). Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125. doi:10.1038/s41422-020-00441-1
Tang L.-J., Luo X.-J., Tu H., Chen H., Xiong X.-M., Li N.-S., et al. (2021b). Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn. Schmiedeb. Arch. Pharmacol. 394, 401–410. doi:10.1007/s00210-020-01932-z
Tang M., Huang Z., Luo X., Liu M., Wang L., Qi Z., et al. (2019). Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radic. Biol. Med. 134, 445–457. doi:10.1016/j.freeradbiomed.2019.01.052
Tebbi C. K., London W. B., Friedman D., Villaluna D., De Alarcon P. A., Constine L. S., et al. (2007). Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric hodgkin’s disease. J. Clin. Oncol. 25, 493–500. doi:10.1200/JCO.2005.02.3879
Von Krusenstiern A. N., Robson R. N., Qian N., Qiu B., Hu F., Reznik E., et al. (2023). Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol. 19, 719–730. doi:10.1038/s41589-022-01249-3
Wang B., Wang H., Zhang M., Ji R., Wei J., Xin Y., et al. (2020a). Radiation-induced myocardial fibrosis: mechanisms underlying its pathogenesis and therapeutic strategies. J. Cell. Mol. Med. 24, 7717–7729. doi:10.1111/jcmm.15479
Wang C., Yuan W., Hu A., Lin J., Xia Z., Yang C., et al. (2020b). Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 22, 175–184. doi:10.3892/mmr.2020.11114
Wang L., Liu X., You L., Ci Y., Chang S., Yu P., et al. (2019a). Hepcidin and iron regulatory proteins coordinately regulate ferroportin 1 expression in the brain of mice. J. Cell. Physiol. 234, 7600–7607. doi:10.1002/jcp.27522
Wang N., Zeng G.-Z., Yin J.-L., Bian Z.-X. (2019b). Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 519, 533–539. doi:10.1016/j.bbrc.2019.09.023
Wang X., Chen X., Zhou W., Men H., Bao T., Sun Y., et al. (2022a). Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 12, 708–722. doi:10.1016/j.apsb.2021.10.005
Wang Z., Yao M., Jiang L., Wang L., Yang Y., Wang Q., et al. (2022b). Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed. Pharmacother. 154, 113572. doi:10.1016/j.biopha.2022.113572
Wen Q., Liu J., Kang R., Zhou B., Tang D. (2019). The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283. doi:10.1016/j.bbrc.2019.01.090
Wu L., Jia M., Xiao L., Wang Z., Yao R., Zhang Y., et al. (2023). TRIM-containing 44 aggravates cardiac hypertrophy via TLR4/NOX4-induced ferroptosis. J. Mol. Med. 101, 685–697. doi:10.1007/s00109-023-02318-3
Wu Z., Geng Y., Lu X., Shi Y., Wu G., Zhang M., et al. (2019). Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. 116, 2996–3005. doi:10.1073/pnas.1819728116
Xu Y., Li X., Cheng Y., Yang M., Wang R. (2020). Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 34, 16262–16275. doi:10.1096/fj.202001758R
Yagi M., Do Y., Hirai H., Miki K., Toshima T., Fukahori Y., et al. (2023). Improving lysosomal ferroptosis with NMN administration protects against heart failure. Life Sci. Alliance 6, e202302116. doi:10.26508/lsa.202302116
Yamada Y., Takano Y., Satrialdi , Abe J., Hibino M., Harashima H. (2020). Therapeutic strategies for regulating mitochondrial oxidative stress. Biomolecules 10, 83. doi:10.3390/biom10010083
Yang W. S., SriRamaratnam R., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi:10.1016/j.cell.2013.12.010
Yant L. J., Ran Q., Rao L., Van Remmen H., Shibatani T., Belter J. G., et al. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502. doi:10.1016/S0891-5849(02)01360-6
Yoo S.-E., Chen L., Na R., Liu Y., Rios C., Van Remmen H., et al. (2012). Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827. doi:10.1016/j.freeradbiomed.2012.02.043
Yuan H., Li X., Zhang X., Kang R., Tang D. (2016). CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 478, 838–844. doi:10.1016/j.bbrc.2016.08.034
Zang H., Wu W., Qi L., Tan W., Nagarkatti P., Nagarkatti M., et al. (2020). Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes 69, 2720–2734. doi:10.2337/db19-1176
Zeller T., Altay A., Waldeyer C., Appelbaum S., Ojeda F., Ruhe J., et al. (2018). Prognostic value of iron-homeostasis regulating peptide hepcidin in coronary heart disease-evidence from the large AtheroGene study. Biomolecules 8, 43. doi:10.3390/biom8030043
Zeng F., Chen X., Deng G. (2022). The anti-ferroptotic role of FSP1: current molecular mechanism and therapeutic approach. Mol. Biomed. 3, 37. doi:10.1186/s43556-022-00105-z
Zhang J., Ding W., Liu J., Wan J., Wang M. (2023). Scavenger receptors in myocardial infarction and ischemia/reperfusion injury: the potential for disease evaluation and therapy. J. Am. Heart Assoc. 12, e027862. doi:10.1161/JAHA.122.027862
Zhang X., Zheng C., Gao Z., Chen H., Li K., Wang L., et al. (2022a). SLC7A11/xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc. Drugs Ther. 36, 437–447. doi:10.1007/s10557-021-07220-z
Zhang Y., Su S. S., Zhao S., Yang Z., Zhong C.-Q., Chen X., et al. (2017). RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329. doi:10.1038/ncomms14329
Zhang Z., Zhou H., Ouyang X., Dong Y., Sarapultsev A., Luo S., et al. (2022b). Multifaceted functions of STING in human health and disease: from molecular mechanism to targeted strategy. Signal Transduct. Target. Ther. 7, 394. doi:10.1038/s41392-022-01252-z
Zhou B., Liu J., Kang R., Klionsky D. J., Kroemer G., Tang D. (2020). Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 66, 89–100. doi:10.1016/j.semcancer.2019.03.002
Keywords: ferroptosis, iron overload, lipid peroxidation, signaling, heart disease
Citation: Fatima S, Zhou H, Chen Y and Liu Q (2024) Role of ferroptosis in the pathogenesis of heart disease. Front. Physiol. 15:1450656. doi: 10.3389/fphys.2024.1450656
Received: 17 June 2024; Accepted: 30 August 2024;
Published: 10 September 2024.
Edited by:
Sean Michael Wilson, Loma Linda University, United StatesReviewed by:
Yongnan Li, Lanzhou University, ChinaCopyright © 2024 Fatima, Zhou, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinghang Liu, cWNsaXVAdS53YXNoaW5ndG9uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.