 Jiao Lu
Jiao Lu Sichong Qian
Sichong Qian Zheng Sun
Zheng Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 24 June 2024
Sec. Integrative Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1405569
Histone deacetylases (HDAC) catalyze the removal of acetylation modifications on histones and non-histone proteins, which regulates gene expression and other cellular processes. HDAC inhibitors (HDACi), approved anti-cancer agents, emerge as a potential new therapy for heart diseases. Cardioprotective effects of HDACi are observed in many preclinical animal models of heart diseases. Genetic mouse models have been developed to understand the role of each HDAC in cardiac functions. Some of the findings are controversial. Here, we provide an overview of how HDACi and HDAC impact cardiac functions under physiological or pathological conditions. We focus on in vivo studies of zinc-dependent classical HDACs, emphasizing disease conditions involving cardiac hypertrophy, myocardial infarction (MI), ischemic reperfusion (I/R) injury, and heart failure. In particular, we review how non-biased omics studies can help our understanding of the mechanisms underlying the cardiac effects of HDACi and HDAC.
The classic zinc-dependent histone deacetylase (HDACs) are categorized into class I, class II (IIa & IIb), and class IV based on the sequence homology (Yang and Seto, 2008). HDAC1, 2, 3, 8 are class I HDACs. HDAC4, 5, 7, 9, are class IIa HDACs. HDAC6 and HDAC10 are class IIb HDACs. HDAC11 is class IV HDAC. The NAD-dependent class III HDACs are sirtuins with distinct catalytic mechanisms, which will not be discussed in this review. Based on the chemical structure, HDAC inhibitors (HDACi) of zinc-dependent HDACs can be categorized into several families (Yang and Seto, 2008). Trichostatin A (TSA), givinostat (ITF2357), CG200745, MPT0E014, and vorinostat (SAHA) belong to the hydroxamic acid chemical class and are pan-HDAC inhibitors that inhibit all classic HDACs. MS275/Entinostat, mocetinostat, and N-(2-aminophenyl)-4-{[benzyl(2-hydroxyethyl)amino]methyl} benzamide (K-183) belong to the benzamide class and inhibit class I HDACs. Valproic acid (VPA) and butyrate belong to the short-chain fatty acid class, which inhibit class I and II HDACs (Kitagawa et al., 2007; Lai et al., 2012; Maes et al., 2013; Jung et al., 2017) (Table 1). HDACi have shown the potential to attenuate pathological cardiac remodeling as a promising therapeutic strategy.
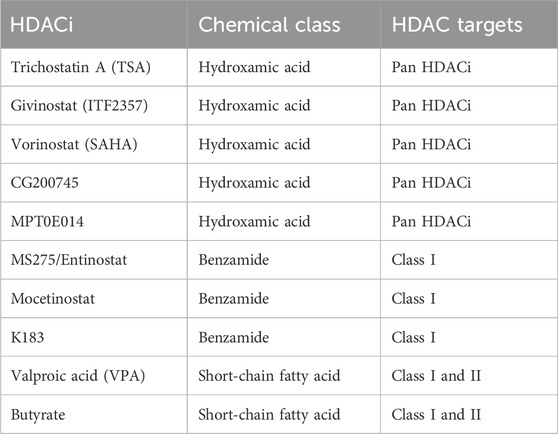
Table 1. Chemical class and HDAC targets of cited HDACi.
Genetic mouse models have been constructed to study the roles of each HDAC, including cardiomyocyte-specific HDAC depletion, whole-body or non-cardiac HDAC loss-of-function, HDAC transgenic overexpression, and the virus-based knockdown (KD) or overexpression (OE) of HDACs. This review focuses on how HDACi administration or HDAC genetic manipulation affects cardiac and vascular function in healthy and diseased conditions in vivo. We focus on disease conditions involving cardiac hypertrophy, myocardial infarction (MI), ischemic reperfusion (I/R) injury, and heart failure. In particular, we review how non-biased omics studies can help our understanding of the mechanisms underlying the cardiac effects of HDACi and HDAC.
TSA shows cardioprotective effects in several studies. TSA is protective against ischemia injury of the heart. Chronic TSA treatment reduced myocardial infarct size and improved contractile function in mice with MI (Zhang et al., 2012a; Zhang et al., 2012b) (Table 2). Acute TSA administration 24 h before cardiac ischemia alleviated HDAC4 OE-induced infarct size, cell death, and contractile dysfunctions of the mice (Zhang L. et al., 2018). TSA protects the mouse heart from the doxorubicin-induced increase in left ventricle internal diameter (LVID) and impairment in ejection fraction (EF) (Song et al., 2018).
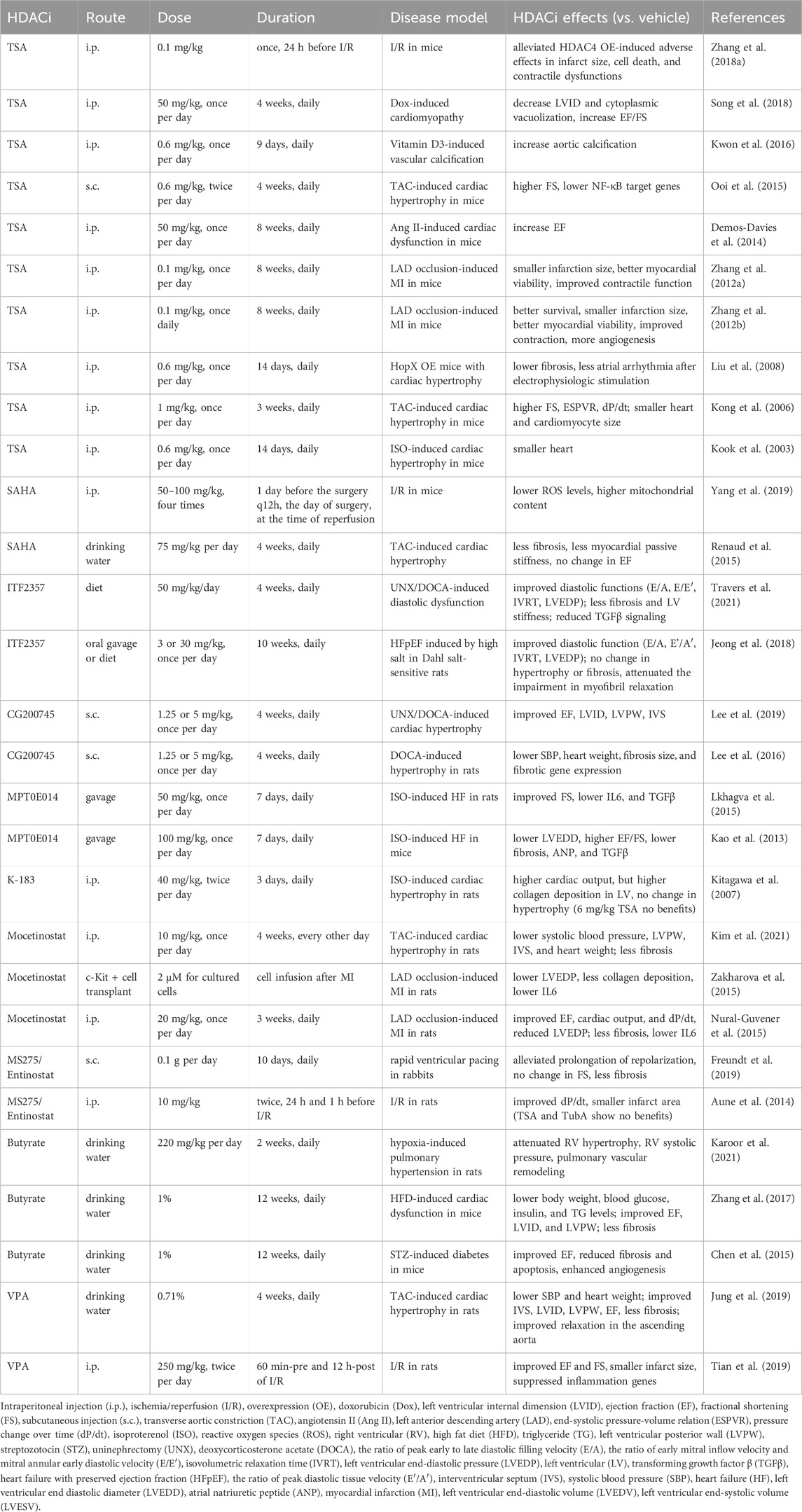
Table 2. The effects of HDACi on cardiac functions.
The cardioprotective effects of TSA have also been investigated in cardiac hypertrophy models. Angiotensin II (Ang II)-induced EF decline in mice was abolished by treatment of TSA (Demos-Davies et al., 2014) (Table 2). TSA also reduced atrial arrhythmia duration after electrophysiologic stimulation and reduced atrial fibrosis in mice overexpressing homeodomain-only protein, a transgenic model of cardiac hypertrophy (Liu et al., 2008). In transverse aortic constriction (TAC)-induced cardiac hypertrophy in mice, TSA preserved fractional shortening (FS), the end-systolic pressure-volume relation, and the pressure change over time (dP/dt), as well as inhibited cardiac hypertrophy (Kong et al., 2006). TSA attenuated the increase in wall thickness and prevented FS decline induced by TAC, as well as suppressed NFκB target genes (Ooi et al., 2015). This is in line with the attenuated cardiac hypertrophy induced by isoproterenol infusion (Kook et al., 2003). While most studies have shown that TSA is protective for the heart, its effect appears opposite in the vascular system. A study reported that TSA significantly enhanced Pi-induced calcium deposition in the aorta and enhanced vitamin D3-induced vascular calcification (Kwon et al., 2016).
SAHA was studied in I/R or hypertrophic mouse models. In I/R injury, SAHA decreased the loss of mitochondrial membrane potential and decreased cytosolic reactive oxygen species (ROS) levels (Yang et al., 2019) (Table 2). In another study, SAHA reduced fibrosis and passive stiffness of the left ventricle myocardium after TAC, but had no effect on cardiac hypertrophy and systolic function (Renaud et al., 2015). ITF2357 (givinostat) can improve diastolic function in models of diastolic dysfunction, including the uninephrectomy (UNX)/deoxycorticosterone acetate (DOCA)-induced diastolic dysfunction model, and the heart failure with preserved ejection fraction (HFpEF) model induced by high salt in Dahl salt-sensitive rats (Jeong et al., 2018; Travers et al., 2021).
Cardiac effects have been characterized for several new HDACis, including CG200745, MPT0E014, and K-183. The pan-HDACi CG200745 attenuated cardiac hypertrophy and fibrosis in DOCA-induced hypertensive rats (Lee et al., 2016; Lee et al., 2019). MPT0E014 improved systolic function and lowered TGFβ levels in isoproterenol-induced heart failure in both mice and rats (Kao et al., 2013; Lkhagva et al., 2015). In isoproterenol-induced cardiac hypertrophy in rats, K-183 attenuated the decrease of cardiac output, even though it did not ameliorate cardiac hypertrophy or collagen production. In comparison, TSA did not show beneficial effects in this study (Kitagawa et al., 2007).
Mocetinostat, a class I HDACi, attenuated cardiac hypertrophy in a TAC-induced hypertrophic model in rats (Kim et al., 2021) (Table 2). It also improved cardiac function in rats with MI, including improved EF and decreased scar size and fibrosis (Nural-Guvener et al., 2015). Furthermore, infusion of Mocetinostat treated c-Kit + cells resulted in a better outcome in myocardial infarcted rats, with reduced left ventricular end-diastolic pressure (LVEDP) and less collagen deposition (Zakharova et al., 2015). Another class I HDACi MS275/Entinostat alleviated the prolonged repolarization in rabbits subjected to rapid ventricular pacing (Freundt et al., 2019). In addition, Entinostat improved dP/dt and reduced infarct size in I/R hearts from rats, whereas TSA and TubA did not show protection (Aune et al., 2014).
Butyrate and VPA, as common class I & II HDACi, were investigated in various disease conditions. In hypoxia-induced pulmonary hypertension in rats, treatment with butyrate attenuated right ventricular (RV) hypertrophy, systolic pressure increase in RV, and pulmonary vascular remodeling (Karoor et al., 2021). In high-fat diet (HFD)-induced cardiac dysfunction in mice, treatment with butyrate lowered body weight, blood glucose, insulin, and TG levels, attenuated cardiac hypertrophy, and improved contractile function (Zhang et al., 2017). Butyrate improved EF, reduced fibrosis and apoptosis, and enhanced angiogenesis in streptozotocin (STZ)-induced diabetic mice (Chen et al., 2015). VPA considerably attenuated systolic blood pressure and hypertrophy, improved cardiac function, and improved relaxation in the ascending aorta in TAC-induced cardiac hypertrophy in rats (Jung et al., 2019) (Table 2). VPA also improved cardiac function and reduced infarct size in rats with I/R injury (Tian et al., 2019). In summary, most studies on HDACis reported protective effects of HDACis on cardiac function. The cardioprotective effects of HDACis are widely observed in cardiac hypertrophy, MI, I/R, and heart failure.
HDACi can have many systemic effects or HDAC-independent effects. Genetic mouse models with cardiomyocyte-specific HDAC loss-of-function have served as a valuable tool for studying the functions of HDACs in the heart. Cardiac-specific double knockout (KO) of HDAC1 and HDAC2 with α-myosin heavy chain (αMHC)-Cre in mice caused postnatal lethality, cardiac arrhythmias, severe ventricular dilation, and heart failure (Montgomery et al., 2007) (Table 3). In comparison, a single gene deletion of either HDAC1 or HDAC2 in the heart with αMHC-Cre displayed no gross cardiac abnormalities (Montgomery et al., 2007). HDAC1 KD by intramyocardial small interfering RNA (siRNA) injection showed recovered cardiac function, improved myocardial injury and inflammation, and less cardiomyocyte apoptosis in a septic mouse model (Nong et al., 2022) (Table 3). Similarly, KD of HDAC1 via intravenous injection of adeno-associated virus (AAV)-siRNA carrying cardiomyocyte-specific cTNT (cardiac troponin T) promotor improved cardiac function, restored lipid contents, and reduced myocardial injury as well as inflammation in a coronary heart disease model of rats (Zhou et al., 2022). Cardiomyocyte-specific KO of HDAC2 with αMHC-Cre alleviated ultrastructural disorganization of atrial myocytes in a disease model of atrial fibrillation (Scholz et al., 2019). Cardiomyocyte-specific KO of HDAC3 with αMHC-Cre in mice caused cardiac hypertrophy, elevated myocardial triglyceride content, and lethal heart failure (Montgomery et al., 2008) (Table 3). In contrast, postnatal myocyte-specific KO of HDAC3 with myosin creatine kinase (MCK)-Cre only displayed lethal heart failure on HFD (Sun et al., 2011). Both studies suggest that HDAC3 is critical in regulating cardiac hypertrophy and lipid metabolism. The role of HDAC3 during heart development has also been investigated. Mouse embryos depleted for HDAC3 in the second heart field with Isl1-Cre [mainly in RV and outflow tract (Cai et al., 2003)] or Mef2C-Cre [in anterior heart field (Verzi et al., 2005)] is lethal, along with ascending aortic dilatation, double outlet right ventricle, and aberrant valve development (Lewandowski et al., 2015). HDAC3 KO in the developing epicardium with Wt1-Cre led to hypoplasia of the ventricular myocardial wall and reduced epicardium-derived cells in mice, suggesting that HDAC3 promotes myocardial growth during development (Jang et al., 2022).
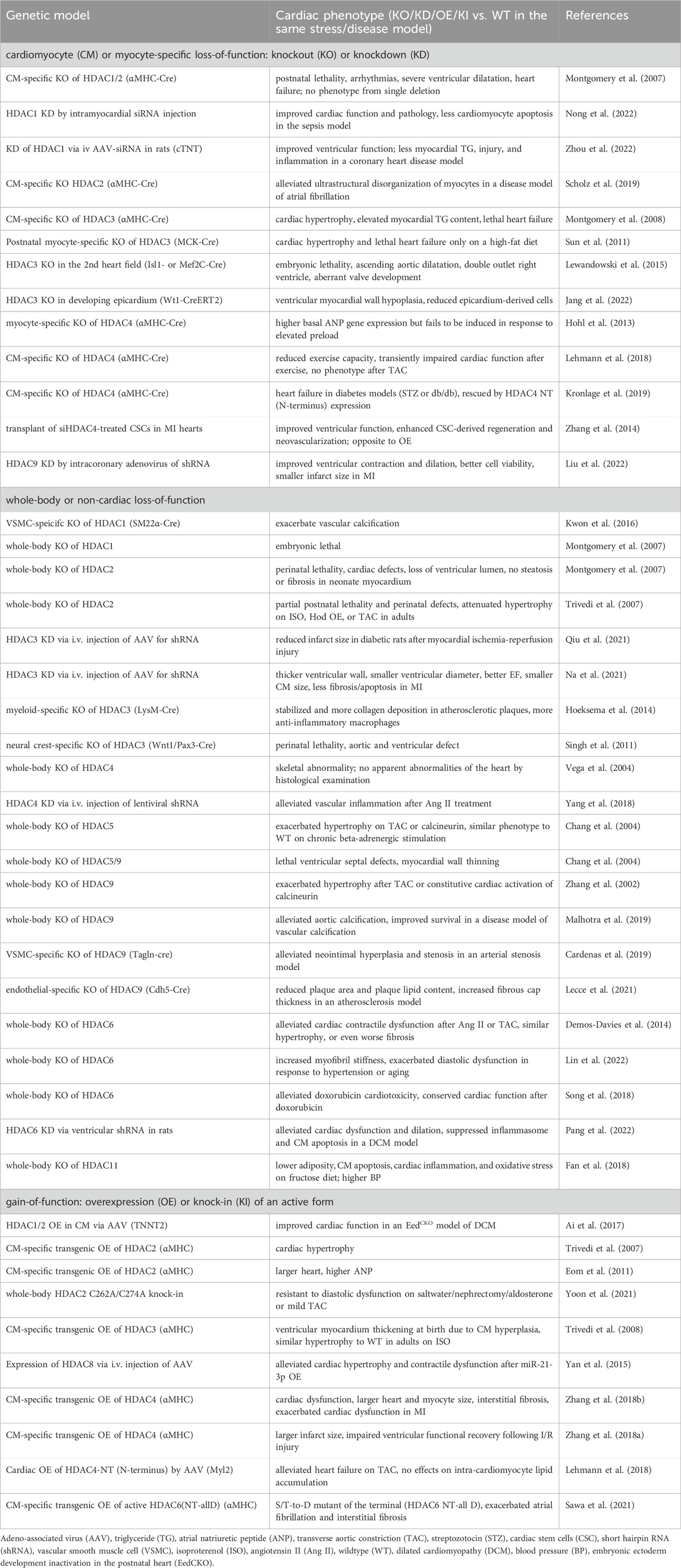
Table 3. The effects of HDAC genetic manipulations on cardiac functions.
Mice with cardiac-specific HDAC4 depletion using αMHC-Cre show elevated basal cardiac atrial natriuretic peptide (ANP) gene expression but a failure of the ANP gene expression activation by cardiac pressure overload (Hohl et al., 2013) (Table 3). The cardiac HDAC4 KO mice also show reduced exercise capacity and transiently impaired cardiac function after exercise but have normal phenotypic changes in response to pressure overload (Lehmann et al., 2018). In another study, mice with cardiac KO of HDAC4 developed heart failure in diabetes models (STZ or db/db), which was rescued by the expression of the N-terminal proteolytic fragment of HDAC4 (Kronlage et al., 2019), suggesting that the N-terminal fragment of HDAC4 plays a role in protecting the diabetic heart from failure. In contrast, transplanting siHDAC4-treated cardiac stem cells (CSC) in MI hearts improved cardiac function and enhanced CSC-derived myocardial regeneration and neovascularization, which is opposite to the effects caused by HDAC4 overexpression (Zhang et al., 2014). HDAC9 KD by intracoronary delivery of adenoviruses carrying HDAC9-short hairpin RNA (shRNA) restored cardiac function in MI and reduced infarct size (Liu et al., 2022). In summary, most studies using HDAC cardiac-specific KO mouse models reported detrimental effects on the heart, whereas virus-mediated KD can improve cardiac function only under specific conditions.
While whole-body KO of HDAC1 is embryonic lethal, whole-body KO of HDAC2 (exons 2-4 deletion) is perinatal lethal, with abnormal morphology of right ventricle, interventricular septum thickening, and bradycardia (Montgomery et al., 2007) (Table 3). In another mouse model of HDAC2 whole-body KO (exons 9–14 deletion), partial lethality was observed along with thickened ventricular wall perinatally. However, when HDAC2 KO mice were subject to hypertrophy stimuli, there was attenuated hypertrophy compared to wild type (WT) in response to isoproterenol, homeobox gene Hod overexpression, or pressure overload. In contrast, transgenic overexpressing HDAC2 in cardiomyocytes led to cardiac hypertrophy, indicating that HDAC2 is both necessary and sufficient for hypertrophic responses (Trivedi et al., 2007). In the aforementioned HDAC2 whole-body KO models, both exons 2-4 deletion of HDAC2 and exons 9–14 deletion target the catalytic domain that is required for the enzymatic activity of HDAC2. The lethality and hypertrophy observed in both models suggest that the deacetylase activity of HDAC2 is critical for the normal function of the heart. In addition to the whole-body KO of HDAC1, the vascular-specific role of HDAC1 was also investigated. Vascular smooth muscle cell (VSMC)-specific KO of HDAC1 by SM22α-Cre exacerbated vascular calcification (Kwon et al., 2016). This study suggests that targeting HDAC1 in vasculature may be a therapeutic strategy for calcification-induced vascular diseases. HDAC3 KD via intravenous injection of AAV-shRNA in STZ-induced diabetic rats reduced infarct size after myocardial I/R injury (Qiu et al., 2021). Similarly, another study found that HDAC3 KD, through intravenous injection of AAV-shRNA, attenuated heart failure induced by MI in mice (Na et al., 2021). In addition to the systemic KD of HDAC3, the tissue-specific KO of HDAC3 also provides insights into the function of HDAC3 in the heart and vascular system. Myeloid-specific KO of HDAC3 with LysM-Cre resulted in stabilized atherosclerotic plaques and more anti-inflammatory macrophages (Hoeksema et al., 2014). Neural crest-specific KO of HDAC3 with Wnt1/Pax3-Cre caused perinatal lethality associated with aortic and ventricular defects (Singh et al., 2011).
Whole-body KO of HDAC4 resulted in skeletal abnormality but no obvious morphological abnormalities of the heart (Vega et al., 2004) (Table 3). HDAC4 KD in mice via intravenous injection of lentiviral shRNA alleviated vascular inflammation after Ang II treatment, indicating that HDAC4 may be a therapeutic target for vascular inflammatory diseases (Yang et al., 2018). In contrast to the not obvious cardiac phenotype by whole-body HDAC4 KO, the changes by HDAC5 or HDAC9 whole-body KO were more evident. Whole-body KO of HDAC5 alone exacerbated hypertrophy upon TAC or calcineurin activation in mice (Chang et al., 2004). Similarly, mice with whole-body KO of HDAC9 alone developed cardiac hypertrophy by 8 months old and are more susceptible to pressure overload-induced hypertrophy (Zhang et al., 2002). These two studies suggest the important role of HDAC5 and HDAC9 in suppressing hypertrophic signaling in the heart. In addition, whole-body KO of both HDAC5 and HDAC9 worsened the outcome, resulting in lethal ventricular septal defects and myocardial wall thinning (Chang et al., 2004). In summary, these studies suggest the redundant functions of HDAC5 and HDAC9 in controlling cardiac development and function. In terms of the role of HDAC9 in the vasculature, one study reported that whole-body HDAC9 KO alleviated aortic calcification and improved survival in a disease model of vascular calcification (Malhotra et al., 2019). VSMC-specific HDAC9 KO with Tagln-Cre alleviated neointimal hyperplasia and stenosis in an arterial stenosis model by carotid artery ligation (Cardenas et al., 2019). Endothelial-specific KO of HDAC9 with Cdh5-Cre showed reduced plaque area and plaque lipid content, and increased fibrous cap thickness in an atherosclerosis model, suggesting that targeting HDAC9 may aid in the treatment of atherosclerosis (Lecce et al., 2021) (Table 3). These three studies provide evidence of a beneficial outcome by depleting HDAC9 in the vasculature, which is opposite to that in the heart.
Whole-body KO of HDAC6 preserved cardiac systolic function in response to Ang II or TAC, despite cardiac hypertrophy (Demos-Davies et al., 2014) (Table 3). HDAC6 KD in rats using lentiviral shRNA injected into the left ventricular cavity also demonstrated alleviated systolic dysfunction in a dilated cardiomyopathy (DCM) model (Pang et al., 2022). However, whole-body HDAC6 KO exacerbated diastolic dysfunction in response to hypertension or aging, along with increased cardiac myofibril stiffness (Lin et al., 2022). These studies suggest that it is possible to have opposite effects on diastolic versus systolic functions from the same genetic manipulation. Additionally, whole-body KO of HDAC6 also alleviated doxorubicin-induced cardiotoxicity, shown by conserved cardiac function after doxorubicin treatment (Song et al., 2018). Finally, whole-body HDAC11 deficiency led to lower adiposity, cardiac inflammation and oxidative stress, but higher blood pressure on the fructose diet (Fan et al., 2018).
In summary, the widely observed detrimental effects on the heart resulting from whole-body HDAC KO align with the observations from HDAC cardiac-specific KO. Specifically, the detrimental effects on the heart are observed in whole-body KO of HDAC1, HDAC2, HDAC4, HDAC5, HDAC9, or HDAC6, as well as in cardiac-specific KO of HDAC1, HDAC2, HDAC3, or HDAC4 (Table 3). Some beneficial effects of whole-body HDAC KD are also observed in some cases. For instance, whole-body KD of HDAC3, HDAC4, or HDAC6 was shown to be protective against ischemic injury, vascular inflammation, or DCM, respectively (Yang et al., 2018; Na et al., 2021; Qiu et al., 2021; Pang et al., 2022). Cardiac-specific KD of HDAC1 or HDAC9 was found cardioprotective in a sepsis model and a coronary heart disease model, or MI model, respectively (Liu et al., 2022; Nong et al., 2022; Zhou et al., 2022). In contrast to cardiac phenotypes, a few lines of evidence suggest a protective role of HDAC depletion in the vascular system. For example, VSMC-specific KO of HDAC9 is protective in an arterial stenosis model, and endothelial cells-specific KO of HDAC9 is protective in an atherosclerosis model (Cardenas et al., 2019; Lecce et al., 2021). These results suggest the distinct roles of HDAC in the heart versus the vasculature.
Overexpression of HDAC1 and 2 in cardiomyocytes via intraperitoneal injection of AAV9 with TNNT2 promoter improved cardiac function in a DCM model created by depletion of EED (embryonic ectoderm development) (Ai et al., 2017) (Table 3). Cardiomyocyte-specific transgenic OE of HDAC2 with αMHC promoter resulted in cardiac hypertrophy (Trivedi et al., 2007; Eom et al., 2011). HDAC2 can be post-translationally modified by S-nitrosylation on C262A and C274A sites by neuronal nitric oxide synthase pathway, which promotes HDAC2 activity in the nucleus. Abolishing the S-nitrosylation in mice with HDAC2 C262A/C274A knock-in resulted in resistance to diastolic dysfunction induced by salt water/nephrectomy/aldosterone or mild TAC (Yoon et al., 2021). Together, these studies suggest that overexpressing HDAC2 activates hypertrophic signaling. Cardiomyocyte-specific transgenic OE of HDAC3 with αMHC displayed ventricular myocardium thickening at birth due to cardiomyocyte hyperplasia, but there was no further increase in cardiac hypertrophy in adults under isoproterenol stimulation compared with WT (Trivedi et al., 2008). This result suggests that HDAC3 regulates cardiomyocyte proliferation during heart development, and it plays distinct roles in development versus adulthood. While miR-21-3p suppressed cardiac hypertrophy, overexpression of HDAC8 via AAV9 attenuated its suppressing effect on hypertrophy (Yan et al., 2015). Transgenic OE of HDAC4 in cardiomyocytes with αMHC promoter impaired cardiac contraction and exacerbated contractile dysfunction on MI, as well as impaired ventricular functional recovery following I/R injury (Zhang L. et al., 2018; Zhang LX. et al., 2018), suggesting that activation of HDAC4 is a crucial regulator in ischemic injury. Cardiac OE of HDAC4-NT (N-terminus) by intravenous injection of AAV9 with the cytomegalovirus (CMV)-enhanced myosin light chain promoter (Myl2) protected against the heart failure on TAC (Lehmann et al., 2018), suggesting that HDAC4-NT can promote cardiac function. However, cardiomyocyte-specific transgenic OE of active HDAC6 with αMHC promoter exacerbated atrial fibrillation and interstitial fibrosis (Sawa et al., 2021), suggesting a role of HDAC6 catalytic activity in atrial fibrillation. In summary, HDAC OE in the heart can lead to different results depending on the specific HDAC. HDAC2, 3, and 4 are involved in hypertrophic signaling in the heart, as shown by the common phenotype of cardiac hypertrophy in response to cardiomyocyte-specific OE of HDAC2, 3 or 4 (Trivedi et al., 2007; Trivedi et al., 2008; Eom et al., 2011; Zhang L. et al., 2018).
Multi-omics approaches provide non-biased insights into the effects of HDACi and HDACs functions. Microarray analysis on ventricular myocardium in dogs identified transcriptomic changes in response to NVS050, mocetinostat, and SAHA. The NVS050 and mocetinostat-induced differentially expressed genes (DEGs) are enriched in protein transport/trafficking and localization, which might contribute to the delayed cardiac repolarization in response to these HDACi (Spence et al., 2016) (Table 4). Microarray on H9c2 rat cardiomyocytes treated by CBHA or TSA found canonical TGFβ, TNF-α, IFNγ, and IL-6 networks were downregulated by both HDACi (Majumdar et al., 2012). ChIP-sequencing identified increased histone acetylation of NFκB target genes in TAC-induced hypertrophic hearts, which were attenuated by TSA (Ooi et al., 2015). Mass spectrometry analysis on a mouse model of diastolic dysfunction suggested that ITF2357/Givinostat treatment suppressed cardiac fibrosis by blocking extracellular matrix deposition. RNA-sequencing further revealed suppression of cardiac fibroblast activation by ITF2357/Givinostat treatment (Travers et al., 2021). Mass spectrometry unraveled cardiac 20S acetylome in murine hearts, and HDAC inhibition via SAHA and valproate induced the acetylation of five identified lysine residues, correlating with the enhanced 20S proteasomal activity, which suggests a new strategy of improving the proteolytic function in treating heart failure (Wang et al., 2013). A novel HDACi compound 106 in a mouse model of Friedreich ataxia, a neurodegenerative and cardiac disease, reversed most DEGs in these mice to WT levels (Rai et al., 2008). Proteomics and microarray on Rhein-treated primary human ventricular cardiac fibroblasts found that Rhein inhibited collagen contraction and increased the abundance of SMAD7, indicating Rhein may serve as a potential anti-fibrotic agent to treat cardiac fibrosis (Barbosa et al., 2020). In summary, the omics studies with HDACis indicate the anti-inflammatory and anti-fibrotic role of HDACi in cardiac diseases.
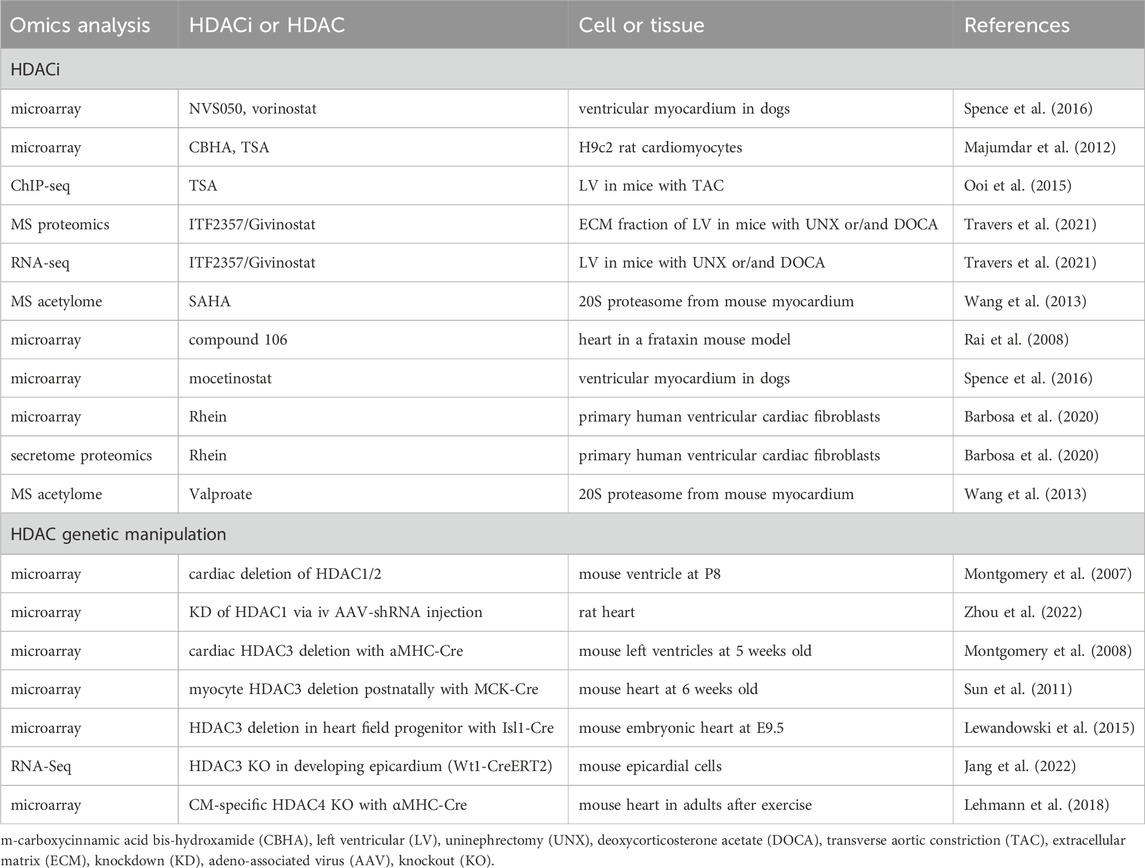
Table 4. Omics analysis on HDACi or HDAC genetic manipulations in the heart.
Microarray analysis on HDAC1 and HDAC2 depleted hearts identified that upregulated genes in KO compared with WT are those involved in cell structure and motility, particularly those encoding cytoskeletal proteins or ion channels (Montgomery et al., 2007) (Table 4). Another miRNA microarray analysis revealed that HDAC1 inhibition via shRNA led to miR-182 overexpression, which suppresses TGF-β/Smad pathway to alleviate coronary heart disease (Zhou et al., 2022). Microarray on HDAC3-depleted hearts with αMHC-Cre at 5 weeks old found DEGs in cardiac metabolism, including downregulated genes in glucose metabolism and upregulated genes in fatty acid uptake, fatty acid oxidation, and mitochondrial uncoupling (Montgomery et al., 2008). However, postnatal HDAC3 depletion in the heart with MCK-Cre showed downregulated expression of mitochondrial genes and genes in lipid metabolism (Sun et al., 2011). These two studies suggest HDAC3 is a crucial regulator of cardiac energy metabolism. During cardiac development, HDAC3 deletion in the second heart field progenitor cells activates TGF-β signaling pathway, which mediates enhanced endothelial-to-mesenchymal transition and extracellular matrix remodeling, as identified by microarray (Lewandowski et al., 2015). This result suggests that HDAC3 suppresses TGF-β in the developing heart. Additionally, RNA-seq analysis in HDAC3-deficient epicardial cells found decreased expression of FGF9 (fibroblast growth factor 9) and IGF2 (insulin-like growth factor 2), while supplementation of FGF9 or IGF2 rescued cardiomyocyte proliferation defects, suggesting that HDAC3 promotes myocardial growth by activating FGF9 and IGF2 (Jang et al., 2022). Increased Nr4a1 that activates hexosamine biosynthetic pathway was identified by microarray in HDAC4-depleted hearts after exercise, whereas the impaired exercise and cardiac function can be rescued by re-expression of HDAC4 N-terminal proteolytic fragment, suggesting a cardioprotective role of HDAC4 that may involve the regulation of the hexosamine biosynthetic pathway (Lehmann et al., 2018). In summary, the omics analysis in HDAC KO or KD hearts identified pathways involved in cardiac development or metabolism. Future work to further investigate the underlying molecular mechanisms by which HDACs regulate cardiac function will be necessary to facilitate the development of improved therapies to treat cardiac diseases.
Accumulating evidence suggests cardioprotective effects of different HDACi. However, a few studies reported no change or adverse effects. For example, TSA showed no benefits in an isoproterenol-induced cardiac hypertrophy model in rats (Kitagawa et al., 2007) or in a rat model of I/R (Aune et al., 2014), while a number of other studies reported cardioprotection of TSA against isoproterenol or TAC-induced cardiac hypertrophy in mice, I/R injury in mice, or MI injury in mice (Kook et al., 2003; Kong et al., 2006; Zhang et al., 2012a; Zhang et al., 2012b; Ooi et al., 2015; Zhang L. et al., 2018). In a different study, administration of TSA exacerbated aortic calcification in a model of vitamin D3-induced vascular calcification (Kwon et al., 2016). The differences in administration conditions or the disease models may underlie the discrepancies. Therefore, it is critical to dissect how each HDACi impacts cardiac function under specific conditions, which might be dependent on the dose, route, duration, or disease models. Other adverse side effects of HDACi in clinical trials include disruption of the gastrointestinal and hematologic systems (Subramanian et al., 2010). Novel delivery methods to enrich HDACi in the heart might help reduce some side effects on other organs. However, arrhythmias were observed in some trials (Shah et al., 2007). A more comprehensive characterization of HDACi toxicity for chronic treatment will be necessary to evaluate their potential as a therapy for heart diseases.
Genetic manipulations of HDACs can have inconsistent effects as HDACi. Overall, there is more evidence supporting the detrimental effects of complete HDAC depletion on the heart, which is opposite to the overall cardioprotective effects of HDACi. First, this discrepancy could partially be attributed to differential developmental effects. Genetic KO of HDAC in the heart using constitutional Cre recombinase might disrupt developmental processes, whereas the administration of HDACi in adult animals would not. Inducible HDACs KO in adult animals in future studies can efficiently address this issue due to confounding developmental defects. Second, differential gene dosages might also contribute to the opposing effects of genetic HDAC depletion versus HDAC inhibition by HDACi. While a complete loss-of-function of HDAC might be detrimental to the heart, partial inhibition of HDACs by HDACi can be cardioprotective. It is possible that the abundance or function of HDACs needs to be tightly regulated according to the physiological context. For example, both cardiomyocyte-specific KO of HDAC3 and cardiomyocyte-specific transgenic OE of HDAC3 can lead to cardiac hypertrophy (Montgomery et al., 2008; Trivedi et al., 2008; Sun et al., 2011). Future studies comparing cardiac phenotypes by different abundance of HDACs in the heart can address such dose-dependent effects of HDACs. Third, previous studies have revealed the deacetylase-independent functions of HDACs (Sun et al., 2013). Thus, dissecting the deacetylase-independent versus deacetylase-dependent functions of HDAC will help provide additional insights into the different effects of genetic HDAC KO versus HDACi. Last, HDACi acts on the whole body, which makes it possible that the cardioprotection can be indirectly conferred through other organs in the body. In this case, future studies to further explore more tissue-specific roles of HDAC outside of the heart will help to understand how HDACs in other organs indirectly regulate cardiac function.
Omics analysis is a powerful tool for non-biased profiling of molecular changes caused by HDAC KO or HDACi treatment. However, the data does not distinguish direct versus indirect effects and does not suggest a causal role of gene expression changes in phenotypical changes. Further functional studies are warranted to dissect to what degree certain molecular changes are responsible for the observed cardiac phenotypic changes. Finally, it is also important to take into account how different HDACs can regulate or compensate one another in the heart. Future studies addressing the functional interaction of HDACs or simultaneous targeting of a combination of HDACs can be helpful in understanding their roles in heart diseases.
JL: Writing–original draft, Writing–review and editing. SQ: Writing–original draft. ZS: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The investigators are supported by the American Heart Association (AHA30970064) and the National Institutes of Health (R01HL153320).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ai S., Peng Y., Li C., Gu F., Yu X., Yue Y., et al. (2017). EED orchestration of heart maturation through interaction with HDACs is H3K27me3-independent. Elife 6, e24570. doi:10.7554/eLife.24570
Aune S. E., Herr D. J., Mani S. K., Menick D. R. (2014). Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J. Mol. Cell. Cardiol. 72, 138–145. doi:10.1016/j.yjmcc.2014.03.005
Barbosa D. M., Fahlbusch P., Herzfeld de Wiza D., Jacob S., Kettel U., Al-Hasani H., et al. (2020). Rhein, a novel Histone Deacetylase (HDAC) inhibitor with antifibrotic potency in human myocardial fibrosis. Sci. Rep. 10 (1), 4888. doi:10.1038/s41598-020-61886-3
Cai C.-L., Liang X., Shi Y., Chu P.-H., Pfaff S. L., Chen J., et al. (2003). Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 5 (6), 877–889. doi:10.1016/s1534-5807(03)00363-0
Cardenas C. L. L., Kessinger C. W., Chou E., Ghoshhajra B., Ashish S. Y., Das S., et al. (2019). HDAC9 complex inhibition improves smooth muscle–dependent stenotic vascular disease. JCI insight 4 (2), e124706. doi:10.1172/jci.insight.124706
Chang S., McKinsey T. A., Zhang C. L., Richardson J. A., Hill J. A., Olson E. N. (2004). Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24 (19), 8467–8476. doi:10.1128/MCB.24.19.8467-8476.2004
Chen Y., Du J., Zhao Y. T., Zhang L., Lv G., Zhuang S., et al. (2015). Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 14, 99–13. doi:10.1186/s12933-015-0262-8
Demos-Davies K. M., Ferguson B. S., Cavasin M. A., Mahaffey J. H., Williams S. M., Spiltoir J. I., et al. (2014). HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am. J. Physiology-Heart Circulatory Physiology 307 (2), H252–H258. doi:10.1152/ajpheart.00149.2014
Eom G. H., Cho Y. K., Ko J.-H., Shin S., Choe N., Kim Y., et al. (2011). Casein kinase-2α1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 123 (21), 2392–2403. doi:10.1161/CIRCULATIONAHA.110.003665
Fan X.-D., Wan L.-L., Duan M., Lu S. (2018). HDAC11 deletion reduces fructose-induced cardiac dyslipidemia, apoptosis and inflammation by attenuating oxidative stress injury. Biochem. biophysical Res. Commun. 503 (2), 444–451. doi:10.1016/j.bbrc.2018.04.090
Freundt J. K., Frommeyer G., Spieker T., Wötzel F., Grotthoff J. S., Stypmann J., et al. (2019). Histone deacetylase inhibition by Entinostat for the prevention of electrical and structural remodeling in heart failure. BMC Pharmacol. Toxicol. 20, 16–19. doi:10.1186/s40360-019-0294-x
Hoeksema M. A., Gijbels M. J., Van den Bossche J., van der Velden S., Sijm A., Neele A. E., et al. (2014). Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol. Med. 6 (9), 1124–1132. doi:10.15252/emmm.201404170
Hohl M., Wagner M., Reil J.-C., Müller S.-A., Tauchnitz M., Zimmer A. M., et al. (2013). HDAC4 controls histone methylation in response to elevated cardiac load. J. Clin. investigation 123 (3), 1359–1370. doi:10.1172/JCI61084
Jang J., Song G., Pettit S. M., Li Q., Song X., Cai C.-L., et al. (2022). Epicardial HDAC3 promotes myocardial growth through a novel microRNA pathway. Circulation Res. 131 (2), 151–164. doi:10.1161/CIRCRESAHA.122.320785
Jeong M. Y., Lin Y. H., Wennersten S. A., Demos-Davies K. M., Cavasin M. A., Mahaffey J. H., et al. (2018). Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 10 (427), eaao0144. doi:10.1126/scitranslmed.aao0144
Jung D. E., Park S. B., Kim K., Kim C., Song S. Y. (2017). CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci. Rep. 7 (1), 10921. doi:10.1038/s41598-017-11094-3
Jung H., Lee E., Kim I., Song J., Kim G. (2019). Histone deacetylase inhibition has cardiac and vascular protective effects in rats with pressure overload cardiac hypertrophy. Physiological Res. 68 (5), 727–737. doi:10.33549/physiolres.934110
Kao Y.-H., Liou J.-P., Chung C.-C., Lien G.-S., Kuo C.-C., Chen S.-A., et al. (2013). Histone deacetylase inhibition improved cardiac functions with direct antifibrotic activity in heart failure. Int. J. Cardiol. 168 (4), 4178–4183. doi:10.1016/j.ijcard.2013.07.111
Karoor V., Strassheim D., Sullivan T., Verin A., Umapathy N. S., Dempsey E. C., et al. (2021). The short-chain fatty acid butyrate attenuates pulmonary vascular remodeling and inflammation in hypoxia-induced pulmonary hypertension. Int. J. Mol. Sci. 22 (18), 9916. doi:10.3390/ijms22189916
Kim G. J., Jung H., Lee E., Chung S. W. (2021). Histone deacetylase inhibitor, mocetinostat, regulates cardiac remodelling and renin-angiotensin system activity in rats with transverse aortic constriction-induced pressure overload cardiac hypertrophy. Rev. Cardiovasc. Med. 22 (3), 1037–1045. doi:10.31083/j.rcm2203113
Kitagawa Y., Tamura Y., Shimizu J., Nakajima-Takenaka C., Taniguchi S., Uesato S., et al. (2007). Effects of a novel histone deacetylase inhibitor, N-(2-aminophenyl) benzamide, on a reversible hypertrophy induced by isoproterenol in in situ rat hearts. J. Pharmacol. Sci. 104 (2), 167–175. doi:10.1254/jphs.fp0070091
Kong Y., Tannous P., Lu G., Berenji K., Rothermel B. A., Olson E. N., et al. (2006). Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113 (22), 2579–2588. doi:10.1161/CIRCULATIONAHA.106.625467
Kook H., Lepore J. J., Gitler A. D., Lu M. M., Yung W. W.-M., Mackay J., et al. (2003). Cardiac hypertrophy and histone deacetylase–dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. investigation 112 (6), 863–871. doi:10.1172/JCI19137
Kronlage M., Dewenter M., Grosso J., Fleming T., Oehl U., Lehmann L. H., et al. (2019). O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation 140 (7), 580–594. doi:10.1161/CIRCULATIONAHA.117.031942
Kwon D.-H., Eom G. H., Ko J. H., Shin S., Joung H., Choe N., et al. (2016). MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 7 (1), 10492. doi:10.1038/ncomms10492
Lai M.-J., Huang H.-L., Pan S.-L., Liu Y.-M., Peng C.-Y., Lee H.-Y., et al. (2012). Synthesis and biological evaluation of 1-arylsulfonyl-5-(N-hydroxyacrylamide) indoles as potent histone deacetylase inhibitors with antitumor activity in vivo. J. Med. Chem. 55 (8), 3777–3791. doi:10.1021/jm300197a
Lecce L., Xu Y., V’Gangula B., Chandel N., Pothula V., Caudrillier A., et al. (2021). Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J. Clin. Investigation 131 (15), e131178. doi:10.1172/JCI131178
Lee E., Lee H. A., Kim M., Do G. Y., Cho H. M., Kim G. J., et al. (2019). Upregulation of C/EBPβ and TSC2 by an HDAC inhibitor CG200745 protects heart from DOCA-induced hypertrophy. Clin. Exp. Pharmacol. Physiology 46 (3), 226–236. doi:10.1111/1440-1681.13022
Lee E., Song M.-J., Lee H.-A., Kang S.-H., Kim M., Yang E. K., et al. (2016). Histone deacetylase inhibitor, CG200745, attenuates cardiac hypertrophy and fibrosis in DOCA-induced hypertensive rats. Korean J. physiology Pharmacol. 20 (5), 477–485. doi:10.4196/kjpp.2016.20.5.477
Lehmann L. H., Jebessa Z. H., Kreusser M. M., Horsch A., He T., Kronlage M., et al. (2018). A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 24 (1), 62–72. doi:10.1038/nm.4452
Lewandowski S. L., Janardhan H. P., Trivedi C. M. (2015). Histone deacetylase 3 coordinates deacetylase-independent epigenetic silencing of transforming growth factor-β1 (TGF-β1) to orchestrate second heart field development. J. Biol. Chem. 290 (45), 27067–27089. doi:10.1074/jbc.M115.684753
Lin Y.-H., Major J. L., Liebner T., Hourani Z., Travers J. G., Wennersten S. A., et al. (2022). HDAC6 modulates myofibril stiffness and diastolic function of the heart. J. Clin. Investigation 132 (10), e148333. doi:10.1172/JCI148333
Liu F., Di Y., Ma W., Kang X., Li X., Ji Z. (2022). HDAC9 exacerbates myocardial infarction via inactivating Nrf2 pathways. J. Pharm. Pharmacol. 74 (4), 565–572. doi:10.1093/jpp/rgab065
Liu F., Levin M. D., Petrenko N. B., Lu M. M., Wang T., Yuan L. J., et al. (2008). Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 45 (6), 715–723. doi:10.1016/j.yjmcc.2008.08.015
Lkhagva B., Lin Y.-K., Kao Y.-H., Chazo T.-F., Chung C.-C., Chen S.-A., et al. (2015). Novel histone deacetylase inhibitor modulates cardiac peroxisome proliferator-activated receptors and inflammatory cytokines in heart failure. Pharmacology 96 (3-4), 184–191. doi:10.1159/000438864
Maes K., Menu E., Van Valckenborgh E., Van Riet I., Vanderkerken K., De Bruyne E. (2013). Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 5 (2), 430–461. doi:10.3390/cancers5020430
Majumdar G., Adris P., Bhargava N., Chen H., Raghow R. (2012). Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells. BMC genomics 13, 709–720. doi:10.1186/1471-2164-13-709
Malhotra R., Mauer A. C., Lino Cardenas C. L., Guo X., Yao J., Zhang X., et al. (2019). HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 51 (11), 1580–1587. doi:10.1038/s41588-019-0514-8
Montgomery R. L., Davis C. A., Potthoff M. J., Haberland M., Fielitz J., Qi X., et al. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes & Dev. 21 (14), 1790–1802. doi:10.1101/gad.1563807
Montgomery R. L., Potthoff M. J., Haberland M., Qi X., Matsuzaki S., Humphries K. M., et al. (2008). Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. investigation 118 (11), 3588–3597. doi:10.1172/JCI35847
Na J., Jin H., Wang X., Huang K., Sun S., Li Q., et al. (2021). The crosstalk of HDAC3, microRNA-18a and ADRB3 in the progression of heart failure. Cell & Biosci. 11 (1), 31–15. doi:10.1186/s13578-020-00523-y
Nong R., Qin C., Lin Q., Lu Y., Li J. (2022). Down-regulated HDAC1 and up-regulated microRNA-124-5p recover myocardial damage of septic mice. Bioengineered 13 (3), 7168–7180. doi:10.1080/21655979.2022.2034583
Nural-Guvener H., Zakharova L., Feehery L., Sljukic S., Gaballa M. (2015). Anti-fibrotic effects of class I HDAC inhibitor, mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int. J. Mol. Sci. 16 (5), 11482–11499. doi:10.3390/ijms160511482
Ooi J. Y., Tuano N. K., Rafehi H., Gao X.-M., Ziemann M., Du X.-J., et al. (2015). HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 10 (5), 418–430. doi:10.1080/15592294.2015.1024406
Pang X., Guan Q., Lin X., Chang N. (2022). Knockdown of HDAC6 alleviates ventricular remodeling in experimental dilated cardiomyopathy via inhibition of NLRP3 inflammasome activation and promotion of cardiomyocyte autophagy. Cell Biol. Toxicol. 39, 2365–2379. doi:10.1007/s10565-022-09727-z
Qiu Z., Ming H., Lei S., Zhou B., Zhao B., Yu Y., et al. (2021). Roles of HDAC3-orchestrated circadian clock gene oscillations in diabetic rats following myocardial ischaemia/reperfusion injury. Cell death Dis. 12 (1), 43. doi:10.1038/s41419-020-03295-y
Rai M., Soragni E., Jenssen K., Burnett R., Herman D., Coppola G., et al. (2008). HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PloS one 3 (4), e1958. doi:10.1371/journal.pone.0001958
Renaud L., Harris L. G., Mani S. K., Kasiganesan H., Chou J. C., Baicu C. F., et al. (2015). HDACs regulate miR-133a expression in pressure overload–induced cardiac fibrosis. Circ. Heart Fail. 8 (6), 1094–1104. doi:10.1161/CIRCHEARTFAILURE.114.001781
Sawa Y., Matsushita N., Sato S., Ishida N., Saito M., Sanbe A., et al. (2021). Chronic HDAC6 activation induces atrial fibrillation through atrial electrical and structural remodeling in transgenic mice. Int. Heart J. 62 (3), 616–626. doi:10.1536/ihj.20-703
Scholz B., Schulte J. S., Hamer S., Himmler K., Pluteanu F., Seidl M. D., et al. (2019). HDAC (histone deacetylase) inhibitor valproic acid attenuates atrial remodeling and delays the onset of atrial fibrillation in mice. Circulation Arrhythmia Electrophysiol. 12 (3), e007071. doi:10.1161/CIRCEP.118.007071
Shah M. H., Villalona-Calero M. A., Marcucci G., Byrd J. C., Grever M. R., de Bono J. (2007). HDAC inhibitors and cardiac safety. Clin. Cancer Res. 13 (3), 1068–1069. doi:10.1158/1078-0432.CCR-06-1715
Singh N., Trivedi C. M., Lu M., Mullican S. E., Lazar M. A., Epstein J. A. (2011). Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circulation Res. 109 (11), 1240–1249. doi:10.1161/CIRCRESAHA.111.255067
Song R., Yang Y., Lei H., Wang G., Huang Y., Xue W., et al. (2018). HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving α-tubulin acetylation. J. Mol. Cell. Cardiol. 124, 58–69. doi:10.1016/j.yjmcc.2018.10.007
Spence S., Deurinck M., Ju H., Traebert M., McLean L., Marlowe J., et al. (2016). Histone deacetylase inhibitors prolong cardiac repolarization through transcriptional mechanisms. Toxicol. Sci. 153 (1), 39–54. doi:10.1093/toxsci/kfw104
Subramanian S., Bates S. E., Wright J. J., Espinoza-Delgado I., Piekarz R. L. (2010). Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3 (9), 2751–2767. doi:10.3390/ph3092751
Sun Z., Feng D., Fang B., Mullican S. E., You S.-H., Lim H.-W., et al. (2013). Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 52 (6), 769–782. doi:10.1016/j.molcel.2013.10.022
Sun Z., Singh N., Mullican S. E., Everett L. J., Li L., Yuan L., et al. (2011). Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J. Biol. Chem. 286 (38), 33301–33309. doi:10.1074/jbc.M111.277707
Tian S., Lei I., Gao W., Liu L., Guo Y., Creech J., et al. (2019). HDAC inhibitor valproic acid protects heart function through Foxm1 pathway after acute myocardial infarction. EBioMedicine 39, 83–94. doi:10.1016/j.ebiom.2018.12.003
Travers J. G., Wennersten S. A., Peña B., Bagchi R. A., Smith H. E., Hirsch R. A., et al. (2021). HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation 143 (19), 1874–1890. doi:10.1161/CIRCULATIONAHA.120.046462
Trivedi C. M., Lu M. M., Wang Q., Epstein J. A. (2008). Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. J. Biol. Chem. 283 (39), 26484–26489. doi:10.1074/jbc.M803686200
Trivedi C. M., Luo Y., Yin Z., Zhang M., Zhu W., Wang T., et al. (2007). Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 13 (3), 324–331. doi:10.1038/nm1552
Vega R. B., Matsuda K., Oh J., Barbosa A. C., Yang X., Meadows E., et al. (2004). Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119 (4), 555–566. doi:10.1016/j.cell.2004.10.024
Verzi M. P., McCulley D. J., De Val S., Dodou E., Black B. L. (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 287 (1), 134–145. doi:10.1016/j.ydbio.2005.08.041
Wang D., Fang C., Zong N. C., Liem D. A., Cadeiras M., Scruggs S. B., et al. (2013). Regulation of acetylation restores proteolytic function of diseased myocardium in mouse and human. Mol. Cell. Proteomics 12 (12), 3793–3802. doi:10.1074/mcp.M113.028332
Yan M., Chen C., Gong W., Yin Z., Zhou L., Chaugai S., et al. (2015). miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc. Res. 105 (3), 340–352. doi:10.1093/cvr/cvu254
Yang D., Xiao C., Long F., Su Z., Jia W., Qin M., et al. (2018). HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc. Res. 114 (7), 1016–1028. doi:10.1093/cvr/cvy051
Yang J., He J., Ismail M., Tweeten S., Zeng F., Gao L., et al. (2019). HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 130, 36–48. doi:10.1016/j.yjmcc.2019.03.008
Yang X.-J., Seto E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9 (3), 206–218. doi:10.1038/nrm2346
Yoon S., Kim M., Lee H., Kang G., Bedi K., Margulies K. B., et al. (2021). S-nitrosylation of histone deacetylase 2 by neuronal nitric oxide synthase as a mechanism of diastolic dysfunction. Circulation 143 (19), 1912–1925. doi:10.1161/CIRCULATIONAHA.119.043578
Zakharova L., Nural-Guvener H., Feehery L., Popovic-Sljukic S., Gaballa M. A. (2015). Transplantation of epigenetically modified adult cardiac c-Kit+ cells retards remodeling and improves cardiac function in ischemic heart failure model. Stem Cells Transl. Med. 4 (9), 1086–1096. doi:10.5966/sctm.2014-0290
Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002). Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110 (4), 479–488. doi:10.1016/s0092-8674(02)00861-9
Zhang L., Chen B., Zhao Y., Dubielecka P. M., Wei L., Qin G. J., et al. (2012a). Inhibition of histone deacetylase-induced myocardial repair is mediated by c-kit in infarcted hearts. J. Biol. Chem. 287 (47), 39338–39348. doi:10.1074/jbc.M112.379115
Zhang L., Du J., Yano N., Wang H., Zhao Y. T., Dubielecka P. M., et al. (2017). Sodium butyrate protects against high fat diet-induced cardiac dysfunction and metabolic disorders in type II diabetic mice. J. Cell. Biochem. 118 (8), 2395–2408. doi:10.1002/jcb.25902
Zhang L., Qin X., Zhao Y., Fast L., Zhuang S., Liu P., et al. (2012b). Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J. Pharmacol. Exp. Ther. 341 (1), 285–293. doi:10.1124/jpet.111.189910
Zhang L., Wang H., Zhao Y., Wang J., Dubielecka P. M., Zhuang S., et al. (2018a). Myocyte-specific overexpressing HDAC4 promotes myocardial ischemia/reperfusion injury. Mol. Med. 24, 37–10. doi:10.1186/s10020-018-0037-2
Zhang L. X., DeNicola M., Qin X., Du J., Ma J., Tina Zhao Y., et al. (2014). Specific inhibition of HDAC4 in cardiac progenitor cells enhances myocardial repairs. Am. J. Physiology-Cell Physiology 307 (4), C358–C372. doi:10.1152/ajpcell.00187.2013
Zhang L. X., Du J., Zhao Y. T., Wang J., Zhang S., Dubielecka P. M., et al. (2018b). Transgenic overexpression of active HDAC4 in the heart attenuates cardiac function and exacerbates remodeling in infarcted myocardium. J. Appl. Physiology 125 (12), 1968–1978. doi:10.1152/japplphysiol.00006.2018
Keywords: histone deacetylase, histone deacetylase inhibitor, cardiac hypertrophy, myocardial infarction, ischemic reperfusion, heart failure, genetic mouse models, multi-omics
Citation: Lu J, Qian S and Sun Z (2024) Targeting histone deacetylase in cardiac diseases. Front. Physiol. 15:1405569. doi: 10.3389/fphys.2024.1405569
Received: 23 March 2024; Accepted: 31 May 2024;
Published: 24 June 2024.
Edited by:
Carlos Fernandez-Patron, University of Alberta, CanadaReviewed by:
Shafeeq Ahmed Mohammed, University Hospital Zürich, SwitzerlandCopyright © 2024 Lu, Qian and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng Sun, emhlbmcuc3VuQGJjbS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.