
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 19 July 2024
Sec. Clinical and Translational Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1401822
 Tadashi Nakajima1*
Tadashi Nakajima1* Shuntaro Tamura1Reika Kawabata-Iwakawa2Hideki Itoh3Hiroshi Hasegawa1Takashi Kobari1Shun Harasawa4
Shuntaro Tamura1Reika Kawabata-Iwakawa2Hideki Itoh3Hiroshi Hasegawa1Takashi Kobari1Shun Harasawa4 Akiko Sekine4
Akiko Sekine4 Masahiko Nishiyama5
Masahiko Nishiyama5 Masahiko Kurabayashi1Keiji Imoto6
Masahiko Kurabayashi1Keiji Imoto6 Yoshiaki Kaneko1
Yoshiaki Kaneko1 Yosuke Nakatani1
Yosuke Nakatani1 Minoru Horie7Hideki Ishii1
Minoru Horie7Hideki Ishii1Introduction: KCNQ1 and KCNE1 form slowly activating delayed rectifier potassium currents (IKs). Loss-of-function of IKs by KCNQ1 variants causes type-1 long QT syndrome (LQTS). Also, some KCNQ1 variants are reported to cause epilepsy. Segment 4 (S4) of voltage-gated potassium channels has several positively-charged amino acids that are periodically aligned, and acts as a voltage-sensor. Intriguingly, KCNQ1 has a neutral-charge glutamine at the third position (Q3) in the S4 (Q234 position in KCNQ1), which suggests that the Q3 (Q234) may play an important role in the gating properties of IKs. We identified a novel KCNQ1 Q234K (substituted for a positively-charged lysine) variant in patients (a girl and her mother) with LQTS and epileptiform activity on electroencephalogram. The mother had been diagnosed with epilepsy. Therefore, we sought to elucidate the effects of the KCNQ1 Q234K on gating properties of IKs.
Methods: Wild-type (WT)-KCNQ1 and/or Q234K-KCNQ1 were transiently expressed in tsA201-cells with KCNE1 (E1) (WT + E1-channels, Q234K + E1-channels, and WT + Q234K + E1-channels), and membrane currents were recorded using whole-cell patch-clamp techniques.
Results: At 8-s depolarization, current density (CD) of the Q234K + E1-channels or WT + Q234K + E1-channels was significantly larger than the WT + E1-channels (WT + E1: 701 ± 59 pA/pF; Q234K + E1: 912 ± 50 pA/pF, p < 0.01; WT + Q234K + E1: 867 ± 48 pA/pF, p < 0.05). Voltage dependence of activation (VDA) of the Q234K + E1-channels or WT + Q234K + E1-channels was slightly but significantly shifted to depolarizing potentials in comparison to the WT + E1-channels ([V1/2] WT + E1: 25.6 ± 2.6 mV; Q234K + E1: 31.8 ± 1.7 mV, p < 0.05; WT + Q234K + E1: 32.3 ± 1.9 mV, p < 0.05). Activation rate of the Q234K + E1-channels or WT + Q234K + E1-channels was significantly delayed in comparison to the WT + E1-channels ([half activation time] WT + E1: 664 ± 37 ms; Q234K + E1: 1,417 ± 60 ms, p < 0.01; WT + Q234K + E1: 1,177 ± 71 ms, p < 0.01). At 400-ms depolarization, CD of the Q234K + E1-channels or WT + Q234K + E1-channels was significantly decreased in comparison to the WT + E1-channels (WT + E1: 392 ± 42 pA/pF; Q234K + E1: 143 ± 12 pA/pF, p < 0.01; WT + Q234K + E1: 209 ± 24 pA/pF, p < 0.01) due to delayed activation rate and depolarizing shift of VDA.
Conclusion: The KCNQ1 Q234K induced IKs gain-of-function during long (8-s)-depolarization, while loss of-function during short (400-ms)-depolarization, which indicates that the variant causes LQTS, and raises a possibility that the variant may also cause epilepsy. Our data provide novel insights into the functional consequences of charge addition on the Q3 in the S4 of KCNQ1.
Long QT syndrome (LQTS) is a genetic disorder characterized by QT prolongation on electrocardiogram (ECG) and polymorphic ventricular tachycardia, torsades de pointes, and ventricular fibrillation, leading to syncope and sudden cardiac death (Nakajima et al., 2021). At least 17 causative genes have been identified thus far, although not all appear to cause LQTS (Adler et al., 2020). Most causative genes of LQTS encode cardiac ion channels or their auxiliary subunits. The first three identified genes, which account for approximately 90% of genetically affected LQTS patients, encode α-subunits of cardiac voltage-gated ion channels: KCNQ1 for type-1 LQTS, KCNH2 for type-2 LQTS, and SCN5A for type-3 LQTS.
KCNQ1 belongs to the voltage-gated potassium channel superfamily and encodes the pore-forming α-subunit of slowly activating delayed rectifier potassium channels (IKs); KCNQ1 interacts with KCNE1, an auxiliary subunit, and forms IKs (Barhanin et al., 1996; Sanguinetti et al., 1996). KCNQ1 is expressed in both the heart and brain (Goldman et al., 2009; Rannals et al., 2016). In the heart, loss-of-function of IKs can prolong cardiac action potential duration (APD), thus prolonging the QT interval on ECG. Multiple loss-of-function mechanisms in IKs, including trafficking defects, producing non-functional channels, altered channel gating properties and a combination thereof, underlie LQTS (Yamashita et al., 2001; Peroz et al., 2008). Notably, the altered gating properties of IKs can cause both loss- and gain-of-function, thus they can be associated with not only LQTS but also other arrhythmic disorders, such as short QT syndrome and atrial fibrillation (Chen et al., 2003; Bellocq et al., 2004; Hong et al., 2005; Bartos et al., 2011; Hasegawa et al., 2014; Zhou et al., 2019). On the other hand, there have been several reports that KCNQ1 variants may also be associated with epilepsy as a brain/cerebral phenotype (Goldman et al., 2009; Tiron et al., 2015).
The α-subunits of voltage-gated ion channels have six transmembrane domains: segment (S) 1-S6. The first four transmembrane segments (S1-S4) form a voltage-sensing domain (VSD), the remaining two segments (S5-S6) form a pore domain (PD), and the central PD is surrounded by four VSDs that respond to membrane depolarization. S4 in the VSD typically consists of several positively-charged amino acids (AAs) that are periodically aligned in every three AAs, and is postulated to function as a voltage-sensor (Nakajima et al., 2019; Wu and Larsson, 2020). Therefore, variants in the S4 of genes encoding cardiac voltage-gated ion channels, such as KCNQ1, KCNH2 and SCN5A, can confer unique gating properties in each channel, and it is therefore possible that they may confer unique phenotypic manifestations (Nakajima et al., 1999; Hasegawa et al., 2014; Nakajima et al., 2015).
The AA alignment of the S4 of KCNQ1 is unique from that of shaker-like potassium channels (Wu et al., 2010; Sun and MacKinnon, 2017) (Figure 1A). It consists of R1 (arginine at the first position of positively-charged AAs in the S4) (or R228: arginine at position 228 in KCNQ1), R2 (or R231), Q3 (or Q234), R4 (or R237), H5 (or H240), and R6 (or R243). Notably, unlike shaker-like potassium channels, the third position in the S4 of KCNQ1 is not a positively-charged AA but a neutral-charge AA, glutamine (Q3 or Q234) (Figure 1A). This suggests that the neutral-charge at the third position in the S4 of KCNQ1 may play an important role in the gating properties of IKs.
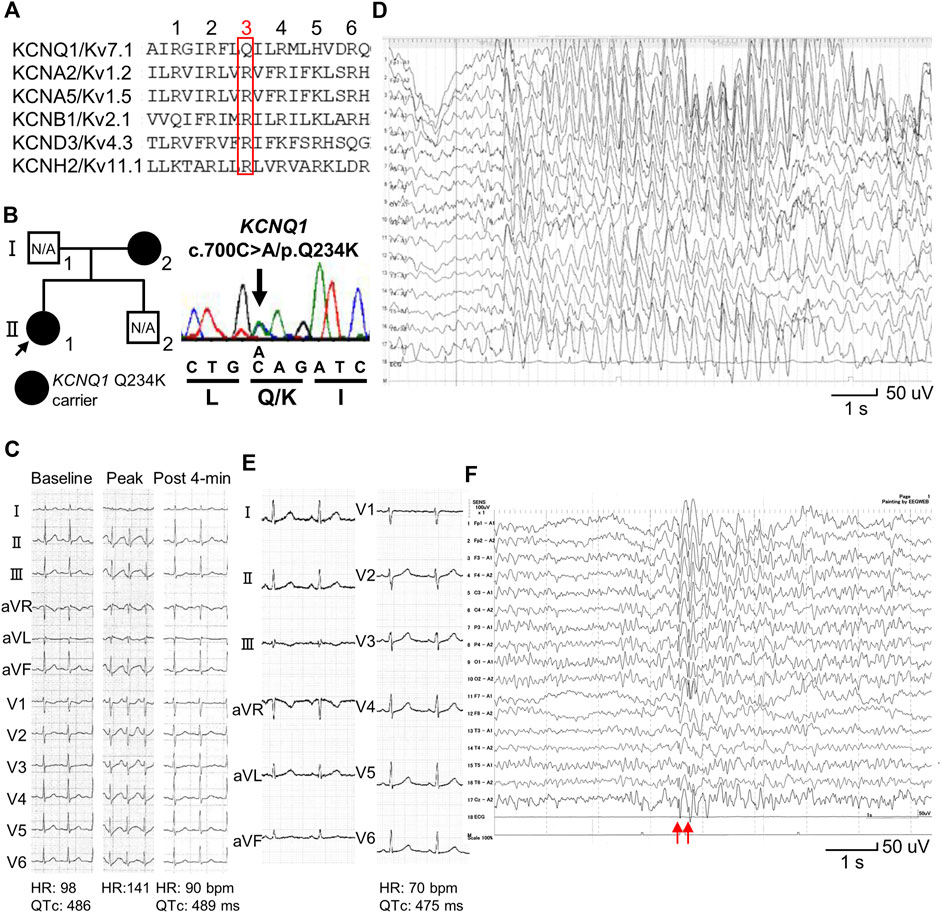
Figure 1. Identification of a KCNQ1 Q234K variant in patients with long QT syndrome and epileptiform activity on electroencephalogram. (A) Alignment of segment 4 (S4) of voltage-gated potassium channels. The third position of amino acids that are periodically aligned is surrounded by a red box. The third position (Q3) of S4 in KCNQ1 corresponds to the position Q234 in KCNQ1. (B) Pedigree harboring the KCNQ1 Q234K variant (left panel). Arrow indicates the proband. Black filled symbols indicate heterozygous carriers of the KCNQ1 Q234K variant. N/A indicates not assessed. Electropherogram of part of KCNQ1 exon 5 of the proband (II-1) (right panel). (C) 12-lead ECGs of an exercise stress test using a treadmill of the proband (II-1). Those before exercise (baseline) (left panel), at peak exercise (middle panel), and at recovery phase of 4-min (right panel) are shown. (D) Electroencephalogram (EEG) of the proband (Ⅱ-1). It showed frequent 3 Hz spike-and-wave complexes lasting several seconds on high amplitude background slow waves. (E) 12-lead ECG of the proband’s mother (Ⅰ-2). (F) EEG of the proband’s mother (Ⅰ-2). It showed generalized 5–6 Hz spike-and-wave complexes (red arrows), followed by slow waves.
We recently identified a novel KCNQ1 variant, Q234K (substituted for a positively-charged lysine) (or Q3K), in patients (a girl and her mother) with LQTS and epileptiform activity on electroencephalogram (EEG). The mother had been diagnosed with epilepsy. This attracted our interest in how the variant affects IKs and clinical phenotypes. Therefore, we sought to elucidate the effects of the KCNQ1 Q234K variant on the gating properties of KCNQ1 channels and IKs using patch-clamp techniques.
This study was approved by the Gunma University Ethical Review Board for Medical Research Involving Human Subjects (approval number: HS 2017–15). Written informed consents of the subjects (the proband and her mother) were obtained from the proband’s mother. The Genomic DNA was extracted from peripheral blood lymphocytes. Target panel sequencing of 72 genes, including inherited arrhythmia syndrome-related genes, was performed using the proband’s sample as previously described (Nakajima et al., 2020a). Confirmation of nucleotide substitutions in the proband and her mother was performed using Sanger sequencing. Exon 5 of KCNQ1 (NM_000218.3) was analyzed as previously described (Imai et al., 2014).
Full-length cDNA encoding human wild-type (WT)-KCNQ1 subcloned into a pIRES2-EGFP expression vector (WT-KCNQ1 cDNA) was kindly provided by Prof. Jacques Barhanin (Laboratories of Excellence, Ion Channel Science and Therapeutics, Nice, France). Q234K-KCNQ1 cDNA was constructed using a Quick Change II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, United States). Full-length cDNA encoding human KCNE1 subcloned into the pCDNA3.1 expression vector (KCNE1 cDNA) was obtained as described previously (Wu et al., 2014).
For electrophysiological characterization of WT-KCNQ1 (WT alone) channels and Q234K-KCNQ1 (Q234K alone) channels, 0.5 μg of WT-KCNQ1 cDNA alone or 0.5 μg of Q234K-KCNQ1 cDNA alone was transiently transfected into tsA201-cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States). For electrophysiological characterization of WT-KCNQ1+KCNE1 (WT + E1) channels and Q234K-KCNQ1+KCNE1 (Q234K + E1) channels, 0.5 μg of WT-KCNQ1 cDNA or 0.5 μg of Q234K-KCNQ1 cDNA in combination with 0.5 μg KCNE1 cDNA was transiently transfected. For electrophysiological characterization of WT-KCNQ1+Q234K-KCNQ1+KCNE1 (WT + Q234K + E1) channels, 0.25 μg of WT-KCNQ1 cDNA plus 0.25 μg of Q234K-KCNQ1 cDNA in combination with 0.5 μg KCNE1 cDNA was transiently transfected. Transfected cells were maintained in DMEM at 37°C for 42–54 h before current recordings as described previously (Nakajima et al., 2022). Cells that exhibited green fluorescence were chosen for the current recordings.
Membrane currents were recorded using whole-cell patch-clamp techniques at room temperature (23°C–25°C). The bath solution contained (in mmol/L) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 glucose and 10 HEPES (pH 7.4 with NaOH), and the pipette solution contained (in mmol/L) 140 KAsp, 1 MgCl2, 4 MgATP, 10 EGTA and 10 HEPES (pH 7.3 with KOH). The electrode resistance ranged from 2.0 to 3.0 MΩ. Data acquisition was carried out using an Axopatch 200B amplifier and pCLAMP10.3 software (Molecular Devices, Sunnyvale, CA, United States). Currents were acquired at 20–50 kHz, and low pass-filtered at 5 kHz using an analog-to-digital interface (Digidata 1440A acquisition system, Molecular Devices). Current densities at each test potential were obtained by dividing the expressed currents by cell capacitance.
All pulse protocols are shown in each of Figures or Figure legends. The voltage dependence of activation was fitted with a Boltzmann function of the following form: y = 1–1/{1+exp[(Vm-V1/2)/K]}, where y is the relative current, Vm is the membrane potential, V1/2 is the voltage at which half of the channels are available to open, and K is the slope factor. The time course of activation was fitted with a single exponential function of the following form: I(t)/Imax = A0+A1exp(-t/τ), and the time course of deactivation was fitted with a single exponential function of the following form: I(t)/Imax = A0+A1[1-exp(-t/τ)], where A and τ refer to the amplitude and time constant, respectively, I refers to the current, and t refers to the time.
To avoid potential endogenous current contamination, recordings from the cells exhibiting peak outward current amplitudes of <0.8 nA were excluded from the analyses.
In order to understand how the changes of the channel kinetics or current density in IKs might affect APD or QT interval, we mimicked the experimental data for the WT or the Q234K model and simulated action potentials across the transmural myocardium or ECG (Itoh et al., 2009; Kojima et al., 2020). O’Hara and Rudy model was available for this simulation (O'Hara et al., 2011), and we searched for appropriate parameters according to the experimental data under the Q234K heterozygous condition. For the analysis of IKs, we used the experimental data in which KCNQ1 would be co-expressed with KCNE1. We modified a parameter associated with Hodgkin-Huxley model in IKs and simulated the Q234K + WT + E1 model as shown below.
To simulate the transmural myocardium and pseudo-ECG, we linearly connected 165 myocardial cells through epicardial (1–60), mid-myocardial (61–105), and endocardial cells (106–165). The cells were stimulated in the endocardial cell for 3 min at the cycle length of 1,000 ms. The pseudo-ECG was simulated using Gima and Rudy model (Gima and Rudy, 2002).
All data are expressed as mean ± standard error, and statistical comparisons were tested using the unpaired Student’s t-test with p < 0.05 considered to be statistically significant. In some figures, the standard error bars are smaller than the data symbols.
A 13-year-old female (II-1-1) (Figure 1B) was referred to our institution because QT prolongation on her ECG had been pointed out at medical check-up in junior high school. Her 12-lead ECG showed QTc prolongation (QTc: 486 ms) (Figure 1C). She experienced an episode of syncope several months ago, but the cause was unknown. Transthoracic echocardiography revealed no structural heart diseases. An exercise stress test using a treadmill did not induce any ventricular arrhythmias, but induced QTc prolongation at peak exercise and at recovery phase of 4-min (QTc: 489 ms) (Figure 1C). Her EEG showed frequent spike-and-wave complexes (Figure 1D), although there was no epileptic seizure while recording EEG.
12-lead ECG of her mother (I-2) (Figure 1B) also showed QTc prolongation (QTc: 475 ms) (Figure 1E). She had experienced several episodes of syncope since she was 1 year of age. Prodromal symptoms, such as blinking her eyes, were present before syncope, and her EEGs recorded repeatedly showed spike-and-wave complexes, which led to a diagnosis of epilepsy at 3 years of age. Sodium valproate had been prescribed for many years, but she discontinued treatment on her own judgment several years ago. She had been asymptomatic since she was 4 years of age. Her EEG recorded at age 39 also showed spike-and-wave complexes (Figure 1F).
Target panel sequencing of 72 genes, including inherited arrhythmia syndrome-related genes, was performed in the proband (II-1) (Figure 1B). A novel KCNQ1 Q234K variant was identified and validated using Sanger sequencing (Figure 1B). Sanger sequencing of the proband’s mother (I-2) (Figure 1B) revealed that she harbored the same variant. This variant was not found in gnomAD (https://gnomad.broadinstitute.org/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and was deleterious in SIFT (https://sift.bii.a-star.edu.sg/) and probably damaging in PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
We first compared the electrophysiological properties of the WT alone channels and Q234K alone channels. The current density of the Q234K alone channels, measured at the end of a 2-s depolarizing potential of 100 mV, was not different from that of the WT alone channels (Table 1) (Figures 2A,B), indicating that trafficking of the Q234K alone channels might not be impaired. However, the voltage dependence of activation (VDA) of the Q234K alone channels was significantly shifted to positive potentials (≈5.8 mV) in comparison to the WT alone channels (Table 1) (Figure 2B).
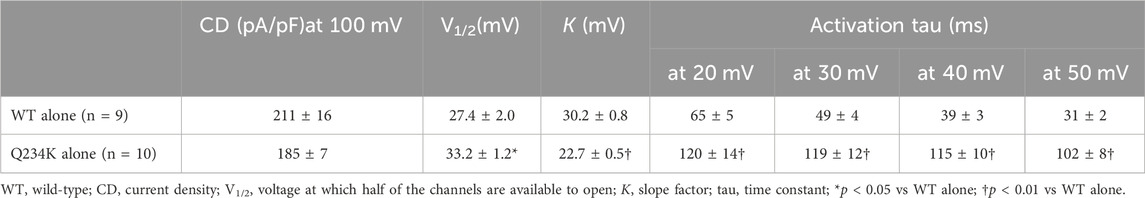
Table 1. Activation parameters of the WT alone and Q234K alone obtained by a 2-s depolarizing pulse protocol.
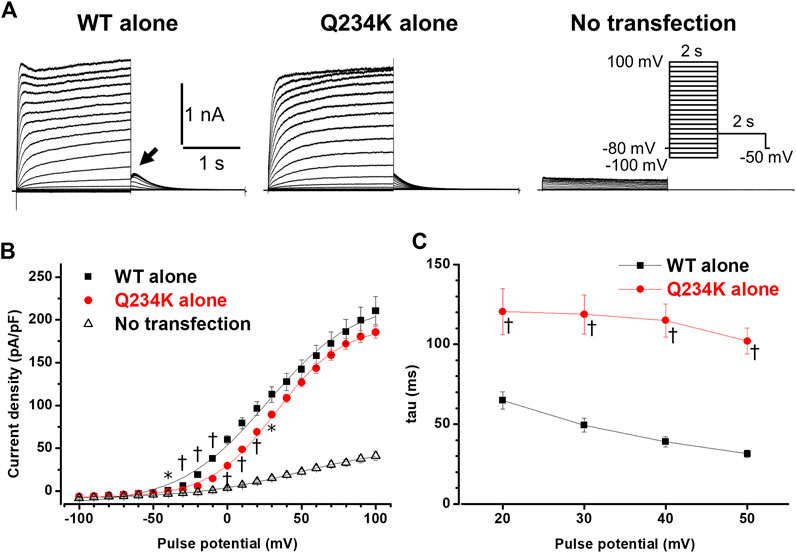
Figure 2. Gating abnormalities of the Q234K alone channels obtained by a 2-s depolarization pulse protocol. (A) Representative currents, obtained by the pulse protocol in the inset, from a cell transfected with wild-type (WT) KCNQ1 cDNA alone (WT alone) (left panel), a cell transfected with Q234K KCNQ1 cDNA alone (Q234K alone) (middle panel), and a non-transfected cell (No transfection) (right panel). The arrow in the left panel indicates the hook. (B) The current-voltage relationships, measured at the end of 2-s depolarizing pulses, of the WT alone (black filled squares, n = 9), Q234K alone (red filled circles, n = 10) and no transfection (black open triangles, n = 6). * indicates p < 0.05 vs WT + E1 and † indicates p < 0.01 vs WT + E1. (C) The time constants (taus) of initial 200-ms activating currents of the WT alone (black filled squares, n = 9) and Q234K alone (red filled circles, n = 10) fitted with a single exponential function between 20 mV and 50 mV in 10 mV increments.
The activating currents were fitted with a single exponential function. The time constants of the Q234K alone channels were significantly larger than those of the WT alone channels, indicating that the Q234K alone channels exhibited a delay of the activation rate in comparison to the WT alone channels (Table 1) (Figures 2A,C). Notably, the WT alone channels exhibited inactivation at higher depolarizing potentials while the Q234K alone channels did not exhibit apparent inactivation (Figure 2A). The absence of inactivation in the Q234K alone channels might, at least in part, contribute to the delayed activation rate in comparison to the WT alone channels.
Tail currents of the WT alone channels, recorded at −50 mV after depolarizing pulses, exhibited hooks (Figure 2A), the initial component of which indicated that the current increase may have resulted from recovery from inactivation, as described previously (Tristani-Firouzi and Sanguinetti, 1998; Franqueza et al., 1999), whereas those of the Q234K alone channels did not exhibit apparent hooks (Figure 2A). The hooks were also present at repolarizing potentials after depolarization in the WT alone channels, but not in the Q234K alone channels (Figure 3A). The presence of the hook in the tail currents of the WT alone channels suggested that these channels inactivated during depolarization and recovered from inactivation during repolarization, which is compatible with the notion that the WT alone channels inactivate during depolarization (Tristani-Firouzi and Sanguinetti, 1998). On the other hand, the lack of a hook in the tail currents of the Q234K alone channels suggested that these channels did not inactivate during depolarization or that they recovered from inactivation very quickly. The disappearance of the hook has also been observed in other variants of KCNQ1 (Franqueza et al., 1999; Moreno et al., 2017).
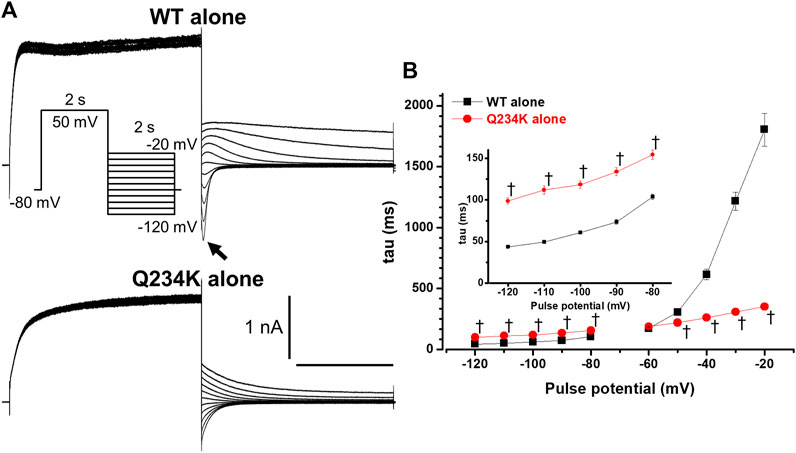
Figure 3. The KCNQ1 Q234K mutation affected deactivation rate in a voltage-dependent manner. (A) Representative currents of the WT alone (upper panel) and Q234K alone (lower panel) obtained by the pulse protocol in the inset. The arrow in the upper panel indicates the hook. (B) The time constants (taus) of deactivating currents of the WT alone (black filled squares, n = 11) and Q234K alone (red filled circles, n = 12) fitted with a single exponential function between −120 mV and −20 mV in 10 mV increments. The tau at −70 mV was deleted because −70 mV was close to the reversal potential. The inset shows an expanded scale of taus between −120 mV and −80 mV † indicates p < 0.01 vs WT + E1.
The deactivation rate was assessed by fitting deactivating currents with a single exponential function. In the WT alone channels, an initial increase in the hook was not included in the fitting procedure (Figure 3A). Intriguingly, the time constants of the Q234K alone channels were significantly larger at hyperpolarizing potentials (below the reversal potential) but were significantly smaller at depolarizing potentials (over the reversal potential) in comparison to the WT alone channels (Table 2) (Figure 3B), indicating that the deactivation rate of the Q234K alone channels was decelerated at hyperpolarizing potentials but accelerated at depolarizing potentials.
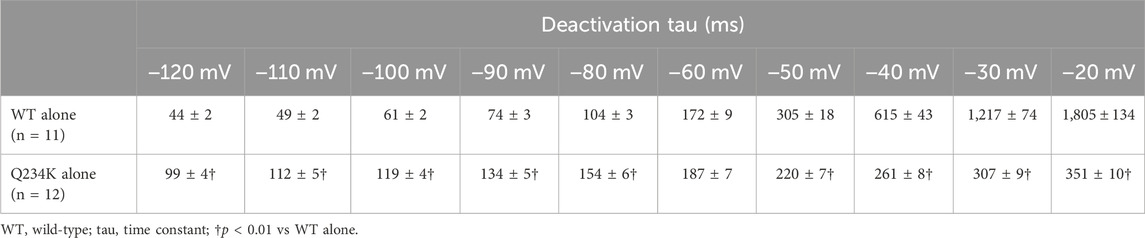
Table 2. Deactivation time constants of the WT alone and Q234K alone.
The interaction of KCNE1 with KCNQ1 forms IKs and dramatically affects WT alone channels. Therefore, we compared the electrophysiological properties of the WT-KCNQ1+KCNE1 (WT + E1) channels and Q234K-KCNQ1+KCNE1 (Q234K + E1) channels. The current density, measured at the end of the 2-s depolarizing potential of 100 mV, of the Q234K + E1 channels was not different from that of the WT + E1 channels (Table 3) (Figures 4A,B). The VDA of the Q234K + E1 channels was significantly shifted to positive potentials (≈8.5 mV) in comparison to the WT + E1 channels (Table 3) (Figure 4B).

Table 3. Activation parameters and current densities, measured at various depolarizing potentials and durations, of the WT + E1, Q234K + E1, and WT + Q234K + E1 obtained by a 2-s depolarizing pulse protocol.
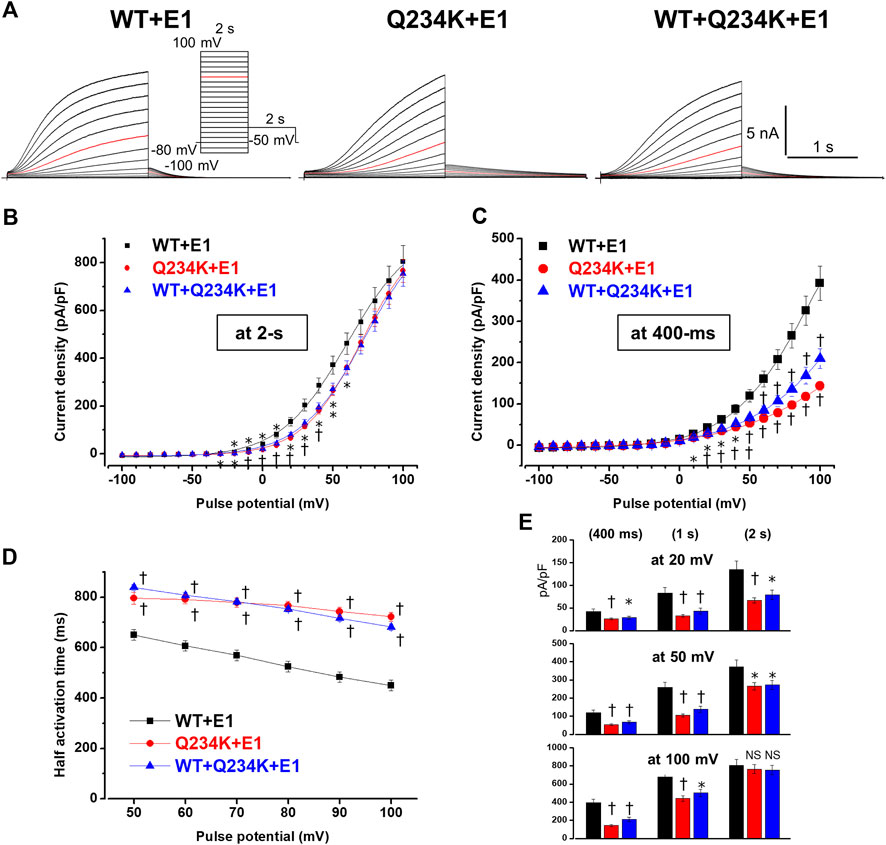
Figure 4. Gating abnormalities of the Q234K + E1 channels and WT + Q234K + E 1 channels revealed by the 2-s depolarization pulse protocol. (A) Representative currents of WT + E1 channels (left panel), Q234K + E1 channels (middle panel) and WT + Q234K + E1 channels (right panel) obtained by the pulse protocol in the inset. Red current tracings indicate the currents recorded at a depolarizing potential of 50 mV. (B) and (C) The current-voltage relationships of the WT + E1 channels (black filled squares, n = 14), Q234K + E1 channels (red filled circles, n = 14), and WT + Q234K + E1 channels (blue filled triangles, n = 15) at the end of 2-s (B) and 400-ms (C) depolarizing potentials. * indicates p < 0.05 vs WT + E1 and † indicates p < 0.01 vs WT + E1. (D) The half activation time during 2-s depolarizing pulses of the WT + E1 channels (black filled squares, n = 14), Q234K + E1 channels (red filled circles, n = 14), and WT + Q234K + E1 channels (blue filled triangles, n = 15). (E) The current densities of the WT + E1 channels (black bars, n = 14), Q234K + E1 channels (red bars, n = 14), and WT + Q234K + E1 channels (blue bars, n = 15) measured at various depolarizing potentials (20 mV, 50 mV, and 100 mV) and various pulse durations (400-ms, 1-s, and 2-s).
Because activating currents during depolarizing pulses for both channels could not be fitted with a single or double exponential function, we assessed the half activation time (HAT) during the 2-s depolarizing pulses. The HAT of the Q234K + E1 channels was significantly larger than that of the WT + E1 channels (Table 3) (Figures 4A,D), indicating that the activation rate of the Q234K + E1 channels was significantly decelerated in comparison to the WT + E1 channels.
We also assessed the current densities at 2-s, 1-s, and 400-ms depolarizing pulses of various depolarizing potentials (20 mV, 50 mV, and 100 mV). Although the current density of the Q234K + E1 channels, measured at the 2-s depolarizing potential of 100 mV, was comparable to that of the WT + E1 channels as mentioned above, those of the Q234K + E1 channels, measured at the 2-s depolarizing potentials of 20 mV and 50 mV, were smaller than those of the WT + E1 channels, mainly due to the depolarizing shift of the VDA. Notably, the current densities, measured at 400-ms and 1-s depolarizing potentials of 100 mV, of the Q234K + E1 channels were significantly smaller than those of the WT + E1 channels (Table 3) (Figures 4C,E), mainly due to the delayed activation rate in the Q234K + E1 channels. On the other hand, the current densities of the Q234K + E1 channels, measured at 400-ms and 1-s depolarizing potentials of 50 mV and 20 mV, were smaller than those of the WT + E1 channels (Table 3) (Figures 4C,E), due to both the depolarizing shift of the VDA and delayed activation rate in the Q234K + E1 channels.
Because 2-s depolarizing pulses appeared to be insufficient to fully activate both channels, 8-s depolarizing pulses were employed. The current density, measured at the end of the 8-s depolarizing potential of 100 mV, of the Q234K + E1 channels was significantly larger than that of the WT + E1 channels (Table 4) (Figures 5A,B). The VDA of the Q234K + E1 channels was still slightly but significantly shifted to positive potentials (≈6.2 mV) in comparison to the WT + E1 channels (Table 4) (Figure 5C).
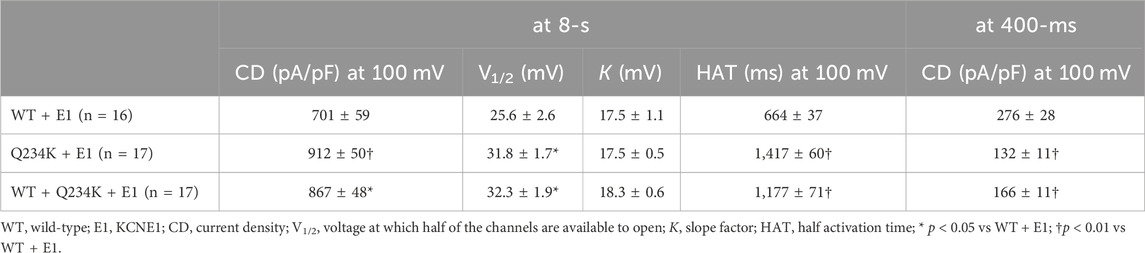
Table 4. Activation parameters of the WT + E1, Q234K + E1, and WT + Q234K + E1 obtained by an 8-s depolarizing pulse protocol.
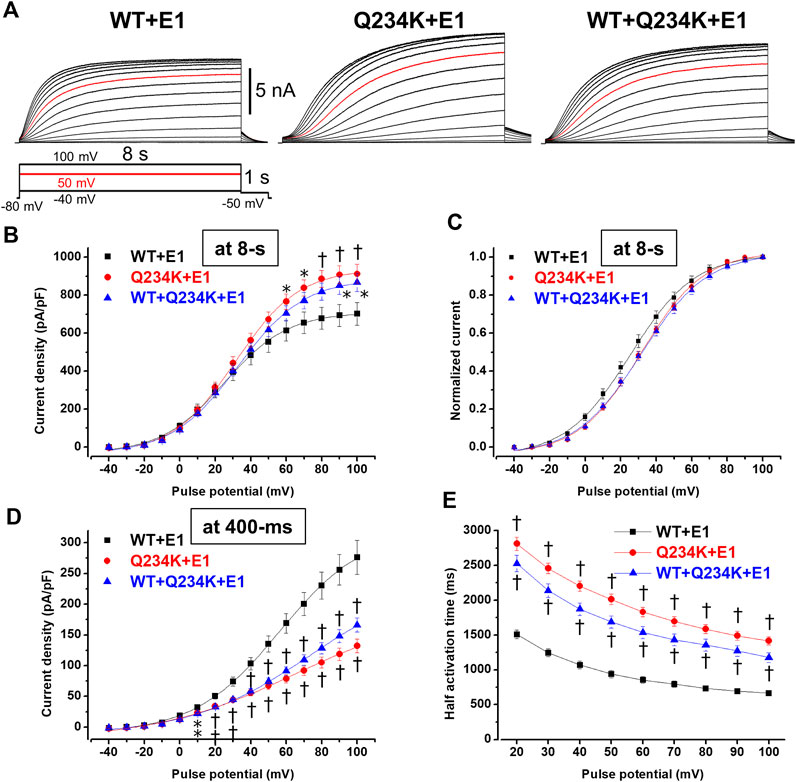
Figure 5. Gating abnormalities of the Q234K + E1 channels and WT + Q234K + E1 channels revealed by an 8-s depolarization pulse protocol. (A) Representative currents of the WT + E1 channels (left panel), Q234K + E1 channels (middle panel), and WT + Q234K + E1 channels (right panel) obtained by the pulse protocol in the inset. 8-s depolarizing pulses were applied from −40 mV to 100 mV in 10 mV increments. Red current tracings indicate currents obtained at a depolarizing potential of 50 mV. (B) The current-voltage relationships of the WT + E1 channels (black filled squares, n = 16), Q234K + E1 channels (red filled circles, n = 17), and WT + Q234K + E1 channels (blue filled triangles, n = 17) at the end of 8-s depolarizing potentials. * indicates p < 0.05 vs WT + E1 and † indicates p < 0.01 vs WT + E1. (C) Normalized current-voltage relationships of (B). (D) The current-voltage relationships of the WT + E1 channels (black filled squares, n = 16), Q234K + E1 channels (red filled circles, n = 17), and WT + Q234K + E1 channels (blue filled triangles, n = 17) at the end of 400-ms depolarizing potentials. (E) Half activation times of the WT + E1 channels (black filled squares, n = 16), Q234K + E1 channels (red filled circles, n = 17), and WT + Q234K + E1 channels (blue filled triangles, n = 17).
In contrast, the current density, measured at a 400-ms depolarizing potential of 100 mV, of the Q234K + E1 channels was significantly smaller than that of the WT + E1 channels (Table 4) (Figures 5A,D), as obtained by the 2-s depolarizing pulse protocol. The HAT of the Q234K + E1 channels was significantly larger than that of the WT + E1 channels (Table 4) (Figure 4E), indicating that the activation rate of the Q234K + E1 channels was significantly slower than that of the WT + E1 channels, as obtained by the 2-s depolarizing pulse protocol. Like these, using the 8-s depolarizing pulse protocol, we obtained almost the same results as the 2-s depolarizing pulse protocol, except for an increased current density in the Q234K + E1 channels.
The deactivation rates were assessed by fitting deactivating currents during repolarizing pulses with a single exponential function (Figure 6A). The time constants of the Q234K + E1 channels were significantly larger than those of the WT + E1 channels (Table 5) (Figure 6B), indicating that the Q234K + E1 channels caused a significant delay of the deactivation rate.
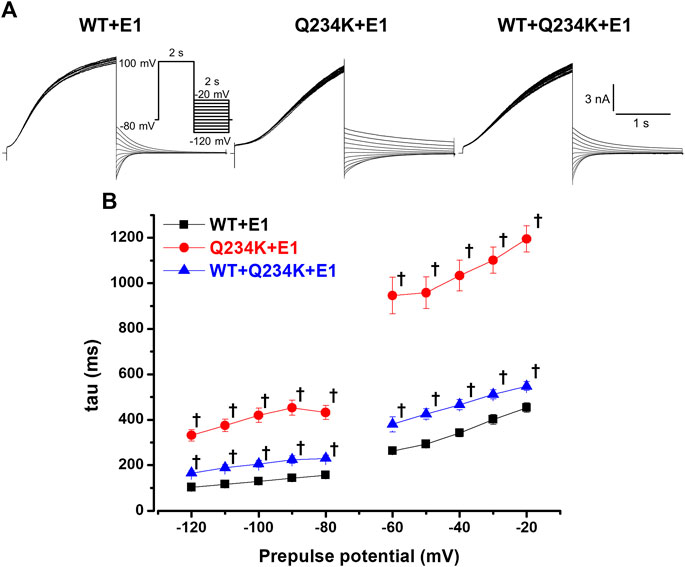
Figure 6. Delayed deactivation rates in the Q234K + E1 channels and WT + Q234K + E1 channels. (A) Representative currents of the WT + E1 (left panel), Q234K + E1 (middle panel), and WT + Q234K + E1 (right panel) obtained by the pulse protocol in the inset. (B) Time constants (taus) of deactivating currents of the WT + E1 channels (black filled squares, n = 13), Q234K + E1 channels (red filled circles, n = 12), and WT + Q234K + E1 channels (blue filled triangles, n = 14) fitted with a single exponential function between −120 mV and −20 mV in 10 mV increments. The tau at −70 mV was deleted because −70 mV was close to the reversal potential. † indicates p < 0.01 vs WT + E1.
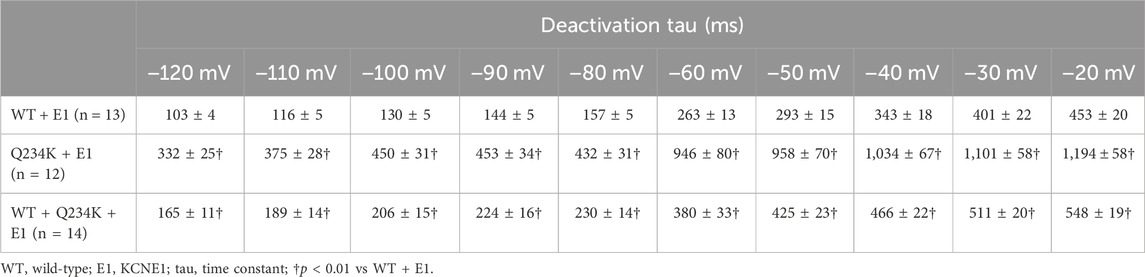
Table 5. Deactivation time constants of the WT + E1, Q234K + E1, and WT + Q234K + E1.
Since the patients in this study harbored the KCNQ1 Q234K variant in a heterozygous manner, we characterized the electrophysiological properties of the WT-KCNQ1+Q234K-KCNQ1+KCNE1 (WT + Q234K + E1) channels.
At the end of the 8-s depolarizing potential of 100 mV, the current density of the WT + Q234K + E1 channels was significantly larger than that of the WT + E1 channels (Table 4) (Figures 5A,B). The VDA of the WT + Q234K + E1 channels was slightly but significantly shifted to positive potentials (≈6.7 mV) in comparison to the WT + E1 channels (Table 4) (Figure 5C). The HATs, obtained by both the 2-s and 8-s depolarizing pulse protocols, of the WT + Q234K + E1 channels were significantly larger than those of the WT + E1 channels (Tables 3 and 4) (Figures 4A,D, 5A,E), indicating that the activation rate of the WT + Q234K + E1 channels was significantly slower than the WT + E1 channels. Using both the 2-s and 8-s depolarizing pulse protocols, the current densities, measured at 400-ms depolarizing potential of 100 mV, of the WT + Q234K + E1 channels were significantly smaller than those of the WT + E1 channels (Tables 3 and 4) (Figures 4A,C, 5A,D).
The time constants of deactivation of the WT + Q234K + E1 channels were significantly larger than those of the WT + E1 channels (Table 5) (Figure 6B), indicating that the deactivation rate of the WT + Q234K + E1 channels was slower than the WT + E1 channels.
The IKs (WT + E1 channels) have been reported to display frequency-dependent current accumulation (Romey et al., 1997). Therefore, we examined whether the Q234K + E1 channels and WT + Q234K + E1 channels display frequency-dependent current accumulation during the physiological range of ventricular APD. For this purpose, repeated 400-ms depolarizing pulses at 1 Hz were applied. The WT + E1 channels displayed frequency-dependent current accumulation, while the Q234K + E1 channels and the WT + Q234K + E1 channels displayed weak frequency-dependent current accumulation in comparison to the WT + E1 channels (Table 6) (Figures 7A,B).
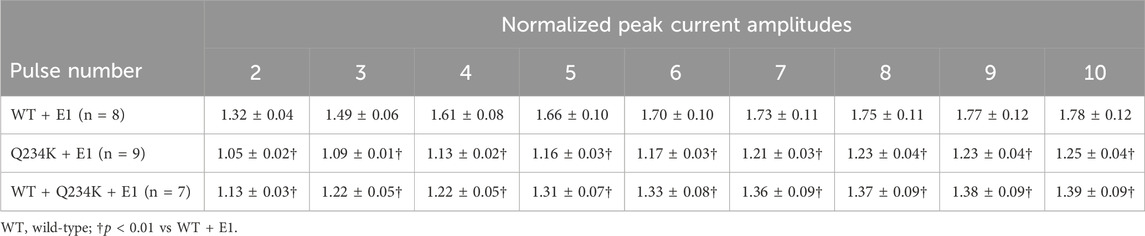
Table 6. Normalized peak current amplitudes of the WT + E1, Q234K + E1, and WT + Q234K + E1 obtained by 400-ms repetitive depolarizing pulses of 50 mV applied by 1 Hz.
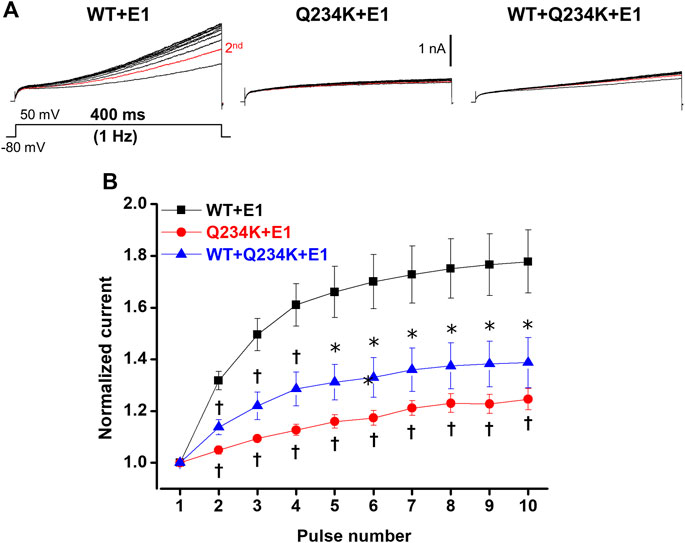
Figure 7. Impaired frequency-dependent accumulation of the Q234K + E1 channels and WT + Q234K + E1 channels. (A) Representative currents of the WT + E1 channels (left panel), Q234K + E1 channels (middle panel), and WT + Q234K + E1 channels (right panel) obtained by the pulse protocol in the inset. Red current tracings indicate currents obtained by the second pulse. (B) Normalized current amplitudes at 400-ms depolarizing pulse of 50 mV obtained by 10 times repeated pulses at 1 Hz * indicates p < 0.05 vs WT + E1 and † indicates p < 0.01 vs WT + E1.
In order to elucidate how IKs with the Q234K would affect the action potential or QT interval, we focused on three characteristics of the Q234K channel following the experimental data; the shift of the voltage dependence of activation toward the depolarizing side, the slower deactivation time curse, and the larger current density. The modification of the time constant,
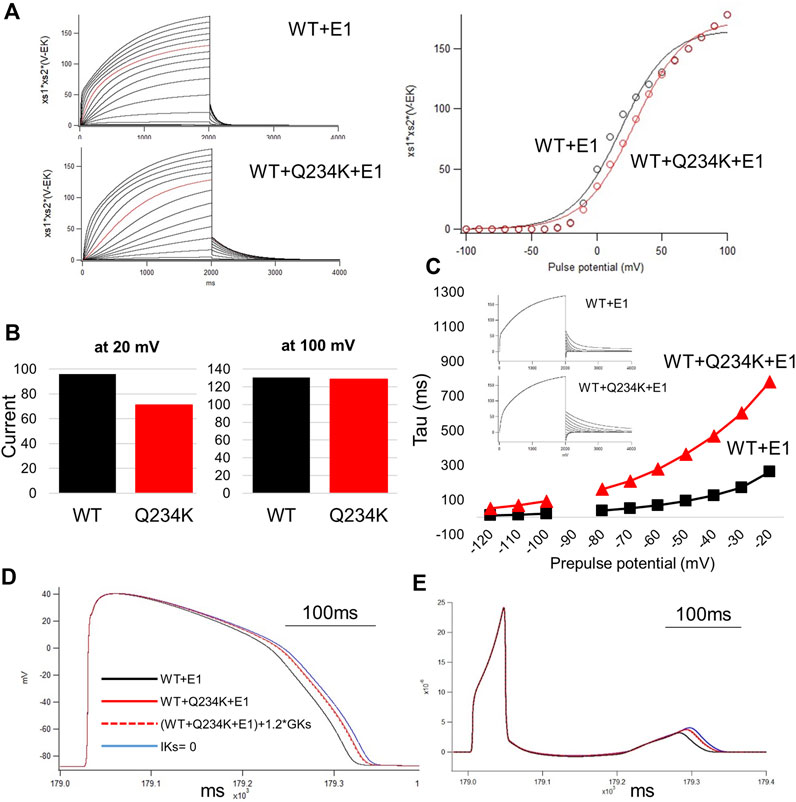
Figure 8. The simulation study of the WT and Q234K heterozygous model. The O’Hara and Rudy model was available in this study. (A) Current-voltage relationships for amplitudes of steady-state currents at the end of 2-s depolarizing pulses. Current traces were either WT + E1 or WT + Q234K + E1 and red lines were recorded at a depolarizing pulse of 50 mV (left panel). Black open circles = WT + E1, red open circles = WT + Q234K + E1. The voltage clamp protocol was same as shown in Figure 4A. (B) The currents at the depolarizing pulse of 20 mV or 100 mV. WT = WT + E1, Q234K = WT + Q234K + E1. The current of the Q234K model was smaller than the WT at the depolarizing pulse of 20 mV while currents were similar at the depolarizing pulse of 100 mV. (C) The time constant of deactivation. The voltage clamp protocol was same as shown in Figure 6A and the insets were current traces at those times. (D) The simulated action potentials in mid-myocardial cell which was 90th from the epicardium. Black line = WT + E1, red solid line = WT + Q234K + E1 without the modification of the current density, red dotted line = WT + Q234K + E1 with the increased current density, and blue line = a model with abolished IKs. (E) Pseudo-ECGs of WT or Q234K-heterozygous carrier. The QT interval of Q234K heterozygous model were between WT and the model without IKs. The modification of the current density in the Q234K model almost did not affect QT interval.
The S4 of α-subunits of voltage-gated potassium channels typically consists of several positively-charged AAs aligned periodically in every three AAs, and is postulated to function as a voltage-sensor. It is widely accepted that the S4 in the VSD senses depolarizing voltage changes across the membrane and moves outwards within the membrane. This outward movement leads to conformational changes in the VSD that trigger structural rearrangements in the PD (VSD-PD coupling or electromechanical coupling), which induces channel opening (Kuenze et al., 2019; Nakajo, 2019; Wu and Larsson, 2020). Unlike Shaker-like voltage-gated potassium channels (Kv1.2), the alignment of positively-charged AAs in the S4 of KCNQ1 is unique in terms of having a neutral-charge AA, glutamine (Q) at the third position (Q3) (Wu et al., 2010; Cui, 2016; Sun and MacKinnon, 2017).
We identified a novel KCNQ1 Q234K (or Q3R) variant in the S4, a substitution for positively-charged lysine, in patients with LQTS and epileptiform activity on EEG. It was expected that an addition of a positive charge on Q3, a critical position of S4, might provide unique gating abnormalities of IKs thus resulting in unique phenotypic manifestations.
The interaction of KCNE1 with KCNQ1 slows the channel activation rate, increases voltage-dependent current amplitude, shifts the VDA to depolarizing potentials, and eliminates channel inactivation (Barhanin et al., 1996; Sanguinetti et al., 1996; Wu and Larsson, 2020). However, how KCNE1 associates with and functionally interacts with KCNQ1 remains unknown.
KCNE1 is suggested to directly bind to the pore, particularly S6, of the KCNQ1 channels to control KCNQ1 gating, and to have contact with S4 and the extracellular end of S1 in the KCNQ1 channel (Wu and Larsson, 2020). An interaction between KCNE1 and KCNQ1 may separate the voltage-sensor movements of the IKs (WT + E1 channels) into two steps. The first step involves S4 gating charge movement, and the second step involves a slow voltage-sensor movement opening the channel. It is assumed that KCNQ1 alone channels can open after the first step, whereas WT + E1 channels only open after both steps (Barro-Soria et al., 2014; Wu and Larsson, 2020). On the other hand, Nakajo et al. assumed that there might be three states in IKs channel gating: closed state (S4 in the down state and PD closed), intermediate stage (S4 in the up state and PD closed), and open state (S4 in the full up state and PD open) (Nakajo, 2019).
When we focused on the activation rate of IKs, the KCNQ1 Q234K variant induced a marked delay in the activation rate of IKs. Although the mechanism underlying the marked delay in the activation rate is unclear, there may be several possible explanations: the Q234K might affect the first step or the second step of the S4 movements, or the conformational change of the gate via VSD-PD coupling, or a combination thereof. It is also undetermined which state the Q234K affects: transition from a closed state to an intermediate state, from an intermediate state to a closed state, or a combination of the two. It is noteworthy that the electrophysiological properties of the WT + Q234K + E1 channels are generally more similar to those of the Q234K + E1 channels than the WT + E1 channels. However, the deactivation kinetics of the WT + Q234K + E1 channels resemble those of the WT + E1 channels (Figure 6), which may weaken the degree of the gain-of-function of IKs. These findings suggest that the Q234K subunits in heterotetrameric channels may not equally affect the transition from the closed state to the intermediate state and the transition from the intermediate state to the open state, and vice versa, in the VSD-PD coupling. To clarify these matters, voltage-clamp fluorometry experiments in combination with ionic current recordings may be required.
Functional changes in the KCNQ1 channels induced by amino acid substitutions in the S4 of KCNQ1 have been reported (Table 7) (Nakajo and Kubo, 2007; Panaghie and Abbott, 2007; Shamgar et al., 2008; Wu et al., 2010). In particular, substitutions of positively-charged arginines for negatively-charged glutamic acids (R1E and R2E) in the N-terminal half (before Q3) of S4 are susceptible to becoming constitutively open in the absence and presence of KCNE1, indicating that basic residues in the N-terminal half of S4 stabilize the resting state (Wu et al., 2010). In contrast, substitutions of basic residues for glutamic acids (R4E and H5E) in the C-terminal half (after Q3) of S4 are susceptible to shifting the VDA to depolarizing potentials in the absence and presence of KCNE1, indicating that basic residues in the C-terminal half of S4 stabilize the activated state (Wu et al., 2010).
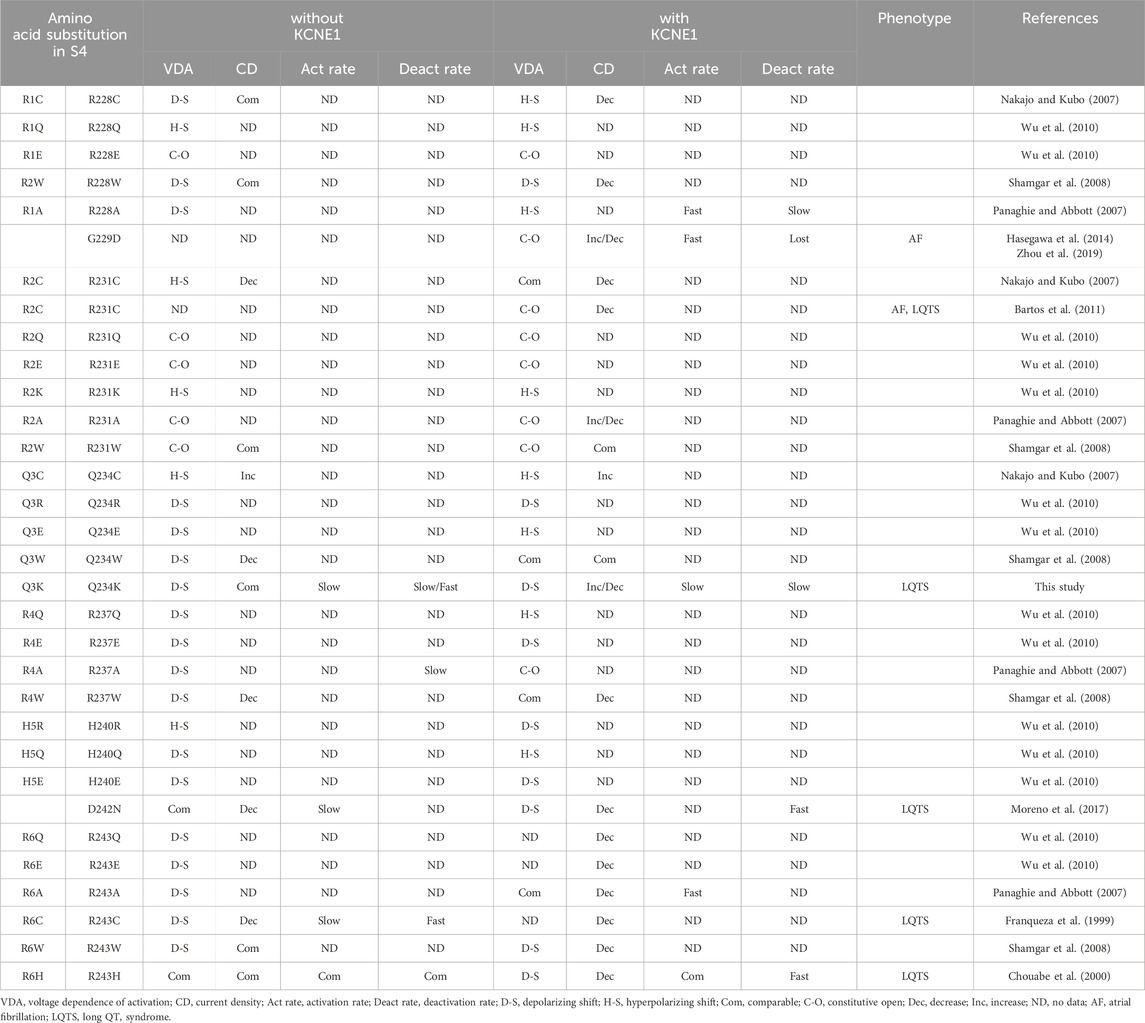
Table 7. Functional changes caused by substitutions of amino acids and disease-causing variants in S4 of KCNQ1.
Regarding neutral-charge glutamine (Q3), the effects of amino acid substitutions on the channel function appear to differ according to the substituted amino acids and the absence or presence of KCNE1 (Table 7). A substitution for cysteine (Q3C or Q234C) shifted the VDA to hyperpolarizing potentials and increased the current density in the absence and presence of KCNE1 (Nakajo and Kubo, 2007). A substitution for tryptophan (Q3W or Q234W) shifted the VDA to depolarizing potentials and decreased the current density in the absence of KCNE1, but did not affect the VDA and current density in the presence of KCNE1 (Shamgar et al., 2008). A substitution for positively-charged arginine (Q3R or Q234R) shifted the VDA to depolarizing potentials in the absence and presence of KCNE1 (Wu et al., 2010). In contrast, a substitution for negatively-charged glutamic acid (Q3E or Q234E) shifted the VDA to depolarizing potentials in the absence of KCNE1, while it shifted the VDA to hyperpolarizing potentials in the presence of KCNE1, suggesting that a positive charge at Q3 would favor the resting state (Wu et al., 2010).
Several pathogenic variants in the S4 of KCNQ1 have been reported (Table 7). KCNQ1 G229D and R231C variants, located in the N-terminal half of S4, caused constitutively open IKs and were associated with atrial fibrillation and/or LQTS (Bartos et al., 2011; Hasegawa et al., 2014; Zhou et al., 2019). In contrast, KCNQ1 D242N, R243C and R243H variants, located in the C-terminal half of S4, strongly shifted the VDA to depolarizing potentials or produced low IKs and are thus associated with LQTS (Franqueza et al., 1999; Chouabe et al., 2000; Park et al., 2005; Moreno et al., 2017). In the case of the R243H variant, reduced phosphaditylinositol-4,5-bisphosphate (PIP2) affinity of the mutant may also be involved in reduced IKs (Park et al., 2005). Like these, variants in the N-terminal half (before Q3) of S4 generally cause constitutively open IKs and are associated with atrial fibrillation and/or LQTS, whereas variants in the C-terminal half (after Q3) of S4 generally shift the VDA to depolarizing potentials thus producing low IKs and are associated with LQTS. Huang et al. reported several other variants, I227L, Q234P, L236P, and L236R in S4 associated with LQTS (Huang et al., 2018). Cell surface expression levels were not reduced, but current densities were remarkably reduced in the I227L and Q234P channels, while both the cell surface expression levels and current densities were reduced in the L236P and L236R channels.
We found that the Q3K (or Q234K) shifted the VDA to depolarizing potentials in the absence and presence of KCNE1, which resembles the effects of Q3R (or Q234R) (Wu et al., 2010), although the effects were weaker than those of the Q234R. Thus, our data support the notion that a positive charge at position 234 in KCNQ1 favors the resting state (Wu et al., 2010). Notably, we found, for the first time, that a single missense variant in the S4, Q3K (or Q234K), markedly delayed both activation and deactivation rates in the presence of KCNE1, although the activation and deactivation rates were rarely assessed in previous experiments of AA substitutions in the S4. Moreover, in the presence of KCNE1, the Q3K (or Q234K) paradoxically induced a gain-of-function during long-depolarization, even though the Q234K + E1 channels favor the resting state, whereas it induced a loss-of-function during short-depolarization (the physiological range of ventricular APD), which is due to the delayed activation rate and depolarizing shift of the VDA. Our data emphasize the importance of assessing not only the VDA but also the activation and deactivation rates of IKs to reveal the association between altered channel function and phenotypic manifestation.
In patients with the KCNQ1 Q234K variant in this study, one allele contains KCNQ1 Q234K and the other contains KCNQ1 WT, we therefore assessed the WT + Q234K + E1 channels. The WT + Q234K + E1 channels displayed an increased current density at the end of 8-s depolarizing potentials (gain-of-function of IKs), depolarizing shift of the VDA, and decelerated activation and deactivation rates. However, at 400-ms depolarizing potentials, the physiological range of the ventricular APD, the WT + Q234K + E1 channels displayed a decreased current density (loss-of-function of IKs) in comparison to the WT + E1 channels. Furthermore, contrary to our expectation, the WT + Q234K + E1 channels displayed a weak frequency-dependent current accumulation, despite their delayed deactivation rates. These findings indicate that the WT + Q234K + E1 channels induce a loss-of-function of IKs during the physiological range of ventricular APD, which underlies QT prolongation. The simulation study could prove that electrophysiological properties of the WT + Q234K + E1 obtained by patch-clamp experiments can prolong ventricular APD and QT interval.
Both KCNQ1 and KCNE1 are reported to be expressed in the brain, where KCNQ1 interacts with KCNE1 (Goldman et al., 2009; Rannals et al., 2016), and there have been several reports that KCNQ1 variants are associated with both LQTS and epilepsy (Goldman et al., 2009; Tiron et al., 2015). In addition to the LQTS phenotype, both the proband and her mother showed epileptiform activity on EEG, and her mother had been diagnosed with epilepsy, although the causes of their syncopal events are unknown. In any case, their EEGs showed apparent abnormalities compatible with epilepsy.
In the brain cells, potassium channels function as setting the resting membrane potential, reducing excitability as well as controlling the duration, shape and firing frequency of action potentials (Greene and Hoshi, 2017). Therefore, IKs loss-of-function by the KCNQ1 Q234K variant in the brain cells may induce hyperexcitability of the brain cells. However, in case of long-lasting firings (that mimic a long-depolarization), IKs gain-of-function may act in the direction of preventing firings. It may be conceivable that the instability of membrane potential of the brain cells may be associated with epileptiform activity (or epilepsy). Moreover, the fact that KCND3 variants which cause unique electrophysiological abnormalities (both gain- and loss-of-function) of Ito are associated with a novel cardiocerebral channelopathy may also suggest the association between unique electrophysiological properties of IKs and epileptiform activity (or epilepsy) (Takayama et al., 2019; Nakajima et al., 2020b).
We identified a novel KCNQ1 Q234K (or Q3K) variant in the S4 of KCNQ1 in patients with LQTS and epileptiform activity on EEG. Our functional analyses revealed that the variant displayed unique gating abnormalities of IKs: The variant induced an increased current density (gain-of-function) during a long-depolarization, while a decreased current density (loss-of-function) during a short-depolarization (the physiological range of ventricular APD) due to a delayed activation rate, depolarizing shift of the VDA, and weak frequency-dependent current accumulation, which can be associated with LQTS. Our data provide novel insights into the functional consequences of charge addition on Q3 in the S4 of KCNQ1.
The association of the KCNQ1 Q234K variant and epileptiform activity (or epilepsy) remains a matter of speculation. Further studies are needed to clarify this issue.
Regarding the simulation study, there are a few limitations. First, we failed to find the modification of any coefficients in O’Hara and Rudy model which match all experimental data in this study. In order to evaluate whether the change of parameters in the mutant channel could prolong or shorten QT interval, we focused on the important three characteristics underlying the mutant channel. Second, the V1/2 of activation of the Hodkin-Huxley model was 18.3 mV and was quite different as compared with 62.8 mV in the experimental data. The experimental data was conducted under the room temperature at 23°C–25°C while the human ventricle data of O’Hara and Rudy model was studied under the body temperature at 37°C. According to Q10 theory, this difference of baseline temperature could affect the kinetics of IKs current. We focused on relative differences of the channel kinetics as compared with WT because the purpose of the simulation study in this study was to elucidate whether the Q234K model corresponding to the experimental study could prolong or shorten the APD or QT interval as compared with WT.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author. The original contributions presented in the study are publicly available. These data can be found here: https://gnomad.broadinstitute.org/gene/ENSG00000053918?dataset=gnomad_r4, https://www.ncbi.nlm.nih.gov/clinvar/?term=KCNQ1%5Bgene%5D&redir=gene, https://sift.bii.a-star.edu.sg/, http://genetics.bwh.harvard.edu/ggi/pph2/a6b96a0ed66a5f1c4a4af7e58c3eebd9138f4989/9873417.html.
The studies involving humans were approved by Gunma University Ethical Review Board for Medical Research Involving Human Subjects (approval number: HS 2017–15). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
TN: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing–original draft. ST: Conceptualization, Investigation, Resources, Writing–original draft. RK-I: Formal Analysis, Investigation, Methodology, Software, Writing–original draft. HIt: Investigation, Formal Analysis, Methodology, Software, Writing–review and editing. HH: Investigation, Resources, Writing–original draft. TK: Investigation, Resources, Writing–original draft. SH: Investigation, Resources, Writing–original draft. AS: Writing–review and editing, Investigation, Resources, MN: Supervision, Writing–review and editing. MK: Supervision, Writing–review and editing. KI: Writing–review and editing, Supervision. YK: Supervision, Writing–review and editing, Investigation, Resources. YN: Writing–review and editing, Investigation, Resources. MH: Supervision, Writing–review and editing. HIs: Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (Grant Number: 23K06845 to TN) and by MEXT Program for supporting the introduction of the new sharing system (Grant Number: JPMXS0420600120).
We thank Prof. Jacques Barhanin (Laboratories of Excellence, Ion Channel Science and Therapeutics, Nice, France) for kindly providing us with the Full-length cDNA encoding human wild-type (WT) KCNQ1 subcloned into a pIRES2-EGFP expression vector. We also thank Miki Matsui, Saori Fujimoto, Yohei Morishita, Yuichi Uosaki, Yoko Yokoyama, Hiroko Matsuda, Pinjie Bao, and Saori Umezawa for their helpful technical assistances.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adler A., Novelli V., Amin A. S., Abiusi E., Care M., Nannenberg E. A., et al. (2020). An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141 (6), 418–428. doi:10.1161/CIRCULATIONAHA.119.043132
Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384 (6604), 78–80. doi:10.1038/384078a0
Barro-Soria R., Rebolledo S., Liin S. I., Perez M. E., Sampson K. J., Kass R. S., et al. (2014). KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat. Commun. 5, 3750. doi:10.1038/ncomms4750
Bartos D. C., Duchatelet S., Burgess D. E., Klug D., Denjoy I., Peat R., et al. (2011). R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm 8 (1), 48–55. doi:10.1016/j.hrthm.2010.09.010
Bellocq C., van Ginneken A. C., Bezzina C. R., Alders M., Escande D., Mannens M. M., et al. (2004). Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109 (20), 2394–2397. doi:10.1161/01.CIR.0000130409.72142.FE
Chen Y. H., Xu S. J., Bendahhou S., Wang X. L., Wang Y., Xu W. Y., et al. (2003). KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299 (5604), 251–254. doi:10.1126/science.1077771
Chouabe C., Neyroud N., Richard P., Denjoy I., Hainque B., Romey G., et al. (2000). Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res. 45 (4), 971–980. doi:10.1016/s0008-6363(99)00411-3
Cui J. (2016). Voltage-dependent gating: novel insights from KCNQ1 channels. Biophys. J. 110 (1), 14–25. doi:10.1016/j.bpj.2015.11.023
Franqueza L., Lin M., Shen J., Splawski I., Keating M. T., Sanguinetti M. C. (1999). Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274 (30), 21063–21070. doi:10.1074/jbc.274.30.21063
Gima K., Rudy Y. (2002). Ionic current basis of electrocardiographic waveforms: a model study. Circ. Res. 90 (8), 889–896. doi:10.1161/01.res.0000016960.61087.86
Goldman A. M., Glasscock E., Yoo J., Chen T. T., Klassen T. L., Noebels J. L. (2009). Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci. Transl. Med. 1 (2), 2ra6. doi:10.1126/scitranslmed.3000289
Greene D. L., Hoshi N. (2017). Modulation of Kv7 channels and excitability in the brain. Cell Mol. Life Sci. 74 (3), 495–508. doi:10.1007/s00018-016-2359-y
Hasegawa K., Ohno S., Ashihara T., Itoh H., Ding W. G., Toyoda F., et al. (2014). A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm 11 (1), 67–75. doi:10.1016/j.hrthm.2013.09.073
Hong K., Piper D. R., Diaz-Valdecantos A., Brugada J., Oliva A., Burashnikov E., et al. (2005). De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 68 (3), 433–440. doi:10.1016/j.cardiores.2005.06.023
Huang H., Kuenze G., Smith J. A., Taylor K. C., Duran A. M., Hadziselimovic A., et al. (2018). Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 4 (3), eaar2631. doi:10.1126/sciadv.aar2631
Imai M., Nakajima T., Kaneko Y., Niwamae N., Irie T., Ota M., et al. (2014). Novel KCNQ1 splicing mutation in patients with forme fruste LQT1 aggravated by hypokalemia. J. Cardiol. 64 (2), 121–126. doi:10.1016/j.jjcc.2013.11.014
Itoh H., Sakaguchi T., Ding W. G., Watanabe E., Watanabe I., Nishio Y., et al. (2009). Latent genetic backgrounds and molecular pathogenesis in drug-induced long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2 (5), 511–523. doi:10.1161/CIRCEP.109.862649
Kojima A., Fukushima Y., Itoh H., Imoto K., Matsuura H. (2020). A computational analysis of the effect of sevoflurane in a human ventricular cell model of long QT syndrome: importance of repolarization reserve in the QT-prolonging effect of sevoflurane. Eur. J. Pharmacol. 883, 173378. doi:10.1016/j.ejphar.2020.173378
Kuenze G., Duran A. M., Woods H., Brewer K. R., McDonald E. F., Vanoye C. G., et al. (2019). Upgraded molecular models of the human KCNQ1 potassium channel. PLoS One 14 (9), e0220415. doi:10.1371/journal.pone.0220415
Moreno C., Oliveras A., Bartolucci C., Munoz C., de la Cruz A., Peraza D. A., et al. (2017). D242N, a K(V)7.1 LQTS mutation uncovers a key residue for I(Ks) voltage dependence. J. Mol. Cell Cardiol. 110, 61–69. doi:10.1016/j.yjmcc.2017.07.009
Nakajima T., Dharmawan T., Kawabata-Iwakawa R., Tamura S., Hasegawa H., Kobari T., et al. (2020a). Biophysical defects of an SCN5A V1667I mutation associated with epinephrine-induced marked QT prolongation. J. Cardiovasc Electrophysiol. 31 (8), 2107–2115. doi:10.1111/jce.14575
Nakajima T., Furukawa T., Hirano Y., Tanaka T., Sakurada H., Takahashi T., et al. (1999). Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc Res. 44 (2), 283–293. doi:10.1016/s0008-6363(99)00195-9
Nakajima T., Kaneko Y., Dharmawan T., Kurabayashi M. (2019). Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: the implication of closed-state inactivation. Channels (Austin) 13 (1), 331–343. doi:10.1080/19336950.2019.1649521
Nakajima T., Kaneko Y., Saito A., Ota M., Iijima T., Kurabayashi M. (2015). Enhanced fast-inactivated state stability of cardiac sodium channels by a novel voltage sensor SCN5A mutation, R1632C, as a cause of atypical Brugada syndrome. Heart Rhythm 12 (11), 2296–2304. doi:10.1016/j.hrthm.2015.05.032
Nakajima T., Kawabata-Iwakawa R., Kaneko Y., Hamano S. I., Sano R., Tamura S., et al. (2020b). Novel cardiocerebral channelopathy associated with a KCND3 V392I mutation. Int. Heart J. 61 (5), 1049–1055. doi:10.1536/ihj.20-203
Nakajima T., Kawabata-Iwakawa R., Tamura S., Hasegawa H., Kobari T., Itoh H., et al. (2022). Novel CACNA1C R511Q mutation, located in domain Ⅰ-Ⅱ linker, causes non-syndromic type-8 long QT syndrome. Plos One 17 (7), e0271796. doi:10.1371/journal.pone.0271796
Nakajima T., Tamura S., Kurabayashi M., Kaneko Y. (2021). Towards mutation-specific precision medicine in atypical clinical phenotypes of inherited arrhythmia syndromes. Int. J. Mol. Sci. 22 (8), 3930. doi:10.3390/ijms22083930
Nakajo K. (2019). Gating modulation of the KCNQ1 channel by KCNE proteins studied by voltage-clamp fluorometry. Biophys. Physicobiol 16, 121–126. doi:10.2142/biophysico.16.0_121
Nakajo K., Kubo Y. (2007). KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 130 (3), 269–281. doi:10.1085/jgp.200709805
O’Hara T., Virag L., Varro A., Rudy Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7 (5), e1002061. doi:10.1371/journal.pcbi.1002061
Panaghie G., Abbott G. W. (2007). The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 129 (2), 121–133. doi:10.1085/jgp.200609612
Park K. H., Piron J., Dahimene S., Merot J., Baro I., Escande D., et al. (2005). Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96 (7), 730–739. doi:10.1161/01.RES.0000161451.04649.a8
Peroz D., Rodriguez N., Choveau F., Baro I., Merot J., Loussouarn G. (2008). Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 586 (7), 1785–1789. doi:10.1113/jphysiol.2007.148254
Rannals M. D., Hamersky G. R., Page S. C., Campbell M. N., Briley A., Gallo R. A., et al. (2016). Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron 90 (1), 43–55. doi:10.1016/j.neuron.2016.02.021
Romey G., Attali B., Chouabe C., Abitbol I., Guillemare E., Barhanin J., et al. (1997). Molecular mechanism and functional significance of the MinK control of the KvLQT1 channel activity. J. Biol. Chem. 272 (27), 16713–16716. doi:10.1074/jbc.272.27.16713
Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384 (6604), 80–83. doi:10.1038/384080a0
Shamgar L., Haitin Y., Yisharel I., Malka E., Schottelndreier H., Peretz A., et al. (2008). KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS One 3 (4), e1943. doi:10.1371/journal.pone.0001943
Sun J., MacKinnon R. (2017). Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169 (6), 1042–1050. doi:10.1016/j.cell.2017.05.019
Takayama K., Ohno S., Ding W. G., Ashihara T., Fukumoto D., Wada Y., et al. (2019). A de novo gain-of-function KCND3 mutation in early repolarization syndrome. Heart Rhythm 16 (11), 1698–1706. doi:10.1016/j.hrthm.2019.05.033
Tiron C., Campuzano O., Perez-Serra A., Mademont I., Coll M., Allegue C., et al. (2015). Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure 25, 65–67. doi:10.1016/j.seizure.2015.01.003
Tristani-Firouzi M., Sanguinetti M. C. (1998). Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510 (1), 37–45. doi:10.1111/j.1469-7793.1998.037bz.x
Wu D., Pan H., Delaloye K., Cui J. (2010). KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys. J. 99 (11), 3599–3608. doi:10.1016/j.bpj.2010.10.018
Wu J., Naiki N., Ding W. G., Ohno S., Kato K., Zang W. J., et al. (2014). A molecular mechanism for adrenergic-induced long QT syndrome. J. Am. Coll. Cardiol. 63 (8), 819–827. doi:10.1016/j.jacc.2013.08.1648
Wu X., Larsson H. P. (2020). Insights into cardiac IKs (KCNQ1/KCNE1) channels regulation. Int. J. Mol. Sci. 21 (24), 9440. doi:10.3390/ijms21249440
Yamashita F., Horie M., Kubota T., Yoshida H., Yumoto Y., Kobori A., et al. (2001). Characterization and subcellular localization of KCNQ1 with a heterozygous mutation in the C terminus. J. Mol. Cell Cardiol. 33 (2), 197–207. doi:10.1006/jmcc.2000.1300
Keywords: epilepsy, epileptiform activity, IKs, KCNQ1, long QT syndrome, segment 4, voltage-sensor
Citation: Nakajima T, Tamura S, Kawabata-Iwakawa R, Itoh H, Hasegawa H, Kobari T, Harasawa S, Sekine A, Nishiyama M, Kurabayashi M, Imoto K, Kaneko Y, Nakatani Y, Horie M and Ishii H (2024) Novel KCNQ1 Q234K variant, identified in patients with long QT syndrome and epileptiform activity, induces both gain- and loss-of-function of slowly activating delayed rectifier potassium currents. Front. Physiol. 15:1401822. doi: 10.3389/fphys.2024.1401822
Received: 16 March 2024; Accepted: 10 June 2024;
Published: 19 July 2024.
Edited by:
Christina Maria Pabelick, Mayo Clinic, United StatesReviewed by:
Kazuharu Furutani, Tokushima Bunri University, JapanCopyright © 2024 Nakajima, Tamura, Kawabata-Iwakawa, Itoh, Hasegawa, Kobari, Harasawa, Sekine, Nishiyama, Kurabayashi, Imoto, Kaneko, Nakatani, Horie and Ishii. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tadashi Nakajima, dG5ha2FqaW1AZ3VubWEtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.