 Dawson Stroik
Dawson Stroik Zachery R. Gregorich
Zachery R. Gregorich Farhan Raza3
Farhan Raza3 Wei Guo
Wei Guo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 10 April 2024
Sec. Striated Muscle Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1385821
This article is part of the Research TopicState-of-the-Art Muscle Physiology Research: From Single Molecules and Cells to Living OrganismsView all 4 articles
The giant protein titin is an essential component of muscle sarcomeres. A single titin molecule spans half a sarcomere and mediates diverse functions along its length by virtue of its unique domains. The A-band of titin functions as a molecular blueprint that defines the length of the thick filaments, the I-band constitutes a molecular spring that determines cell-based passive stiffness, and various domains, including the Z-disk, I-band, and M-line, serve as scaffolds for stretch-sensing signaling pathways that mediate mechanotransduction. This review aims to discuss recent insights into titin’s functional roles and their relationship to cardiac function. The role of titin in heart diseases, such as dilated cardiomyopathy and heart failure with preserved ejection fraction, as well as its potential as a therapeutic target, is also discussed.
Titin (also known as connectin) constitutes the so-called third filament component of muscle sarcomeres (Maruyama, 1976; Wang et al., 1979). With a molecular weight of ∼3–4 MDa (in humans), titin is the largest protein expressed in mammals, extending from the Z-disk to the M-band where the N- and C-termini are anchored, respectively. Titin is divided into four portions based on the orientation of the molecule within the half sarcomere. These portions, which include the Z-disk, A-band, I-band, and M-line, determine titin’s functional roles. Titin’s A-band serves as a molecular blueprint that determines the length of the thick filament and helps to properly orient it within the sarcomere (Whiting et al., 1989; Tonino et al., 2017; Bennett et al., 2020). Titin functions as a molecular spring that gives rise to passive tension mainly through three extensible segments located in the I-band: the tandem immunoglobulin (Ig)-like domain, the repeating PEVK [proline (P), glutamate (E), valine (V) and lysine (K)] domain, and N2B domain (Labeit and Kolmerer, 1995). In addition to these segments, the binding of chaperones to the N2A domain also regulates its spring-like properties directly through binding to this domain and indirectly via modulation of N2A phosphorylation (Lun et al., 2014; Lanzicher et al., 2020). These elastic segments function as a spring that supports early diastolic recoil and late diastolic resistance to stretch (Granzier and Labeit, 2002; Granzier and Labeit, 2004; Hamdani and Paulus, 2013; Guo and Sun, 2018). In effect, titin generates a stretch-resisting force that functions to restore sarcomere resting or “slack” length (Linke et al., 1996). Several different mechanisms regulate titin-based passive stiffness, chief among which is alterative splicing in the I-band portion of titin (Forbes et al., 2005). In the heart, splicing in this portion of the molecule produces two cardiac-specific N2BA and N2B (arise from alternative splicing) titin isoforms that have been studied in great depth and will be the focus of this review (Zou et al., 2015; Chen et al., 2018). Splicing has also been shown to produce smaller Novex-1-3 isoforms while the Cronos isoform is produced from a unique promoter (Bang et al., 2001; Zou et al., 2015). However, the function(s) of these isoforms in cardiac structure and function, particularly in cardiomyopathy, is presently unclear. Titin also serves as a molecular signaling mediator that activates stretch-sensitive signaling pathways to reshape the myocardium in response to sarcomere stress through protein complexes tethered in its Z-disk, I-band, and M-line portions (Granzier and Labeit, 2002; Granzier and Labeit, 2004; Granzier and Labeit, 2006; Linke, 2008; Linke and Krüger, 2010; Hamdani and Paulus, 2013; Linke and Hamdani, 2014; Guo and Sun, 2018). The aim of this review is to discuss the structure and functions of titin’s various domains, as well as how titin splicing and post-translational modifications (PTMs) contribute to these functions. We also discuss heart diseases in which titin plays a role, such as dilated cardiomyopathy (DCM), heart failure with preserved ejection fraction (HFpEF), and diabetic cardiomyopathy (DbCM) (Herman et al., 2012; Zile et al., 2015; Hopf et al., 2018). Lastly, we discuss the potential therapeutic approaches for the treatment of heart diseases by targeting titin.
The titin gene (TTN) is located on chromosome 2q31 in humans and encompasses 364 exons (a single non-coding exon followed by 363 coding exons) (Bang et al., 2001). This paper will only refer to the 363 coding exons. One titin protein molecule spans one-half of a sarcomere, transversing the Z-disk, I-band, A-band, and M-line with its N- and C-terminal portions anchored in the Z-disk and M-line, respectively (Figure 1) (Wang et al., 1979).
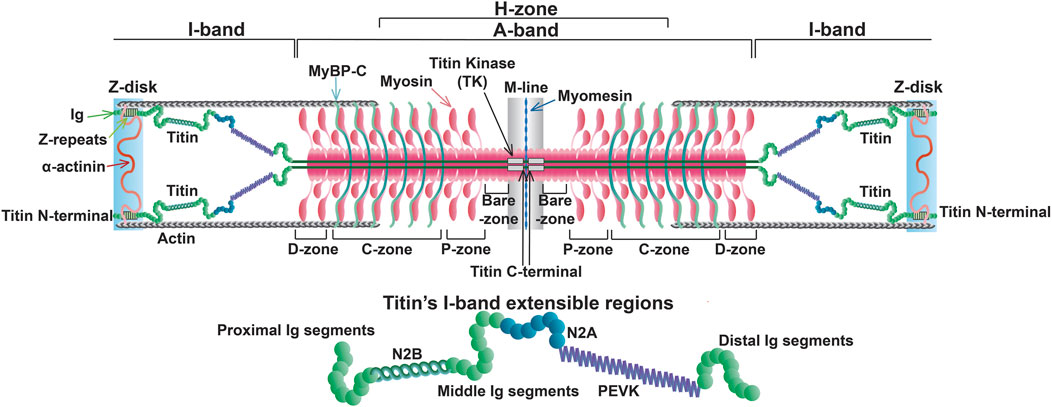
Figure 1. Sarcomere structure at slack length with full length titin.
Z-disk and Z-disk/I-band junction of titin is encoded by coding exons 1–28 and anchors the N-terminus of the protein in the Z-disk. This portion of the molecule consists primarily of consecutive immunoglobulin-like (Ig) domains and 45-residue repeat motifs termed Z-repeats (Figures 1, 2) (Gautel et al., 1996). The Ig-like domains are made of eight β-strands arranged in two sheets packed face-to-face making their structure similar to traditional Ig domains (Pfuhl and Pastore, 1995; Improta et al., 1996). The first and second Z-disk Ig-like domains (Z1 and Z2) in titin interact with telethonin (also known as T-cap) (Gregorio et al., 1998), which crosslinks titin molecules from adjacent sarcomeres in an anti-parallel arrangement (Zou et al., 2006). In addition, the Z-repeats have been shown to mediate interactions with the C-terminal domain of the actin-crosslinking protein α-actinin (Ohtsuka et al., 1997; Sorimachi et al., 1997; Joseph et al., 2001) (Figure 1). The interaction between titin and α-actinin provides strong anchoring of titin within the Z-disk through multiple Z-repeat/α-actinin bonds (Grison et al., 2017). Recent work has added to this list of Z-disk titin-interacting proteins, although many of these interactions remain to be validated and their functional significance is unclear (Rudolph et al., 2020; Filomena et al., 2021).
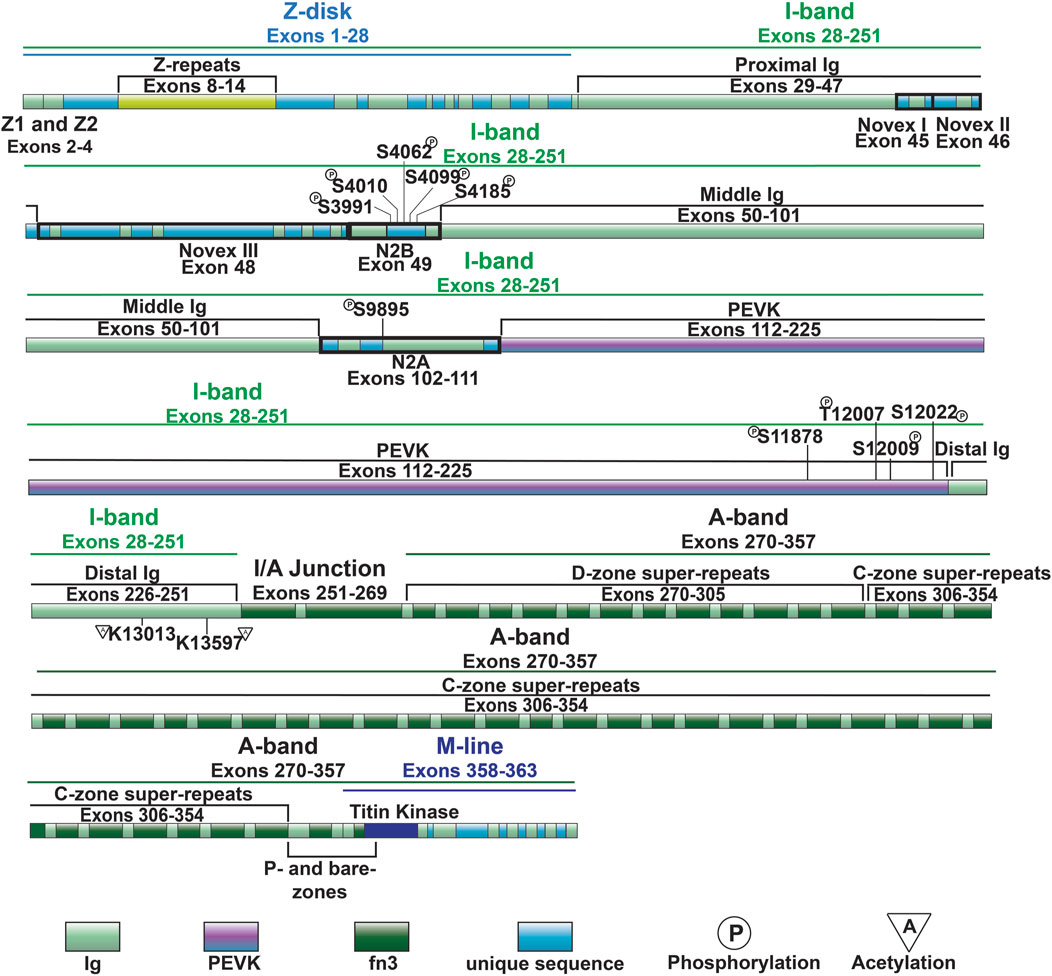
Figure 2. Full-length titin domain map with exons, sites of common I-band PTMs and A-band super-repeats noted.
I-band titin represents the extensible portion of the molecule and is encoded by exons 28–251, with the I/A-band junction encoded by exons 251–269 (Labeit and Kolmerer, 1995). The I-band portion of titin can be separated into six distinct regions: the proximal Ig-like domain (exons 29–47), cardiac-specific N2B segment (exon 49), middle Ig segment (exons 50–101), N2A segment (exons 102–111), PEVK region (exons 112–225), and distal Ig-like domain (exons 226–251) (Figures 1, 2) (Labeit and Kolmerer, 1995). As the names indicate, the proximal, middle, and distal Ig-like domains encode series of tandemly arranged Ig-like domains (Labeit and Kolmerer, 1995). On the other hand, the N2B and N2A segments are comprised of three and four Ig-like domains, respectively, each with additional unique sequences (572 amino acids in human N2B and 104 amino acids in human N2A) (Labeit and Kolmerer, 1995).
Titin’s A-band (encoded by exons 270–357) uniquely contains fibronectin-type III-like (fn3) domains that account for approximately 70% of the A-band portion (Figure 2) (Labeit et al., 1990; Labeit and Kolmerer, 1995). Together with the Ig-like domains, the fn3 domains form two distinct types of super-repeats that define two disparate zones in the A-band: the distal (D)- and central (C)-zones (Labeit and Kolmerer, 1995). The I/A-band junction and P-zone (exons 355–358) also contain fn3 domains, however, this zone does not follow the super-repeat structure found in the D- and C-zones (Labeit et al., 1997). The D-zone co-localizes with the tips of the myosin filaments at the border of the A-band and is composed of 6 repeats of Ig-fn3-fn3-Ig-fn3-fn3-fn3 (Figures 2, 3) (Labeit and Kolmerer, 1995). On the other hand, the C-zone has the pattern Ig-fn3-fn3-Ig-fn3-fn3-fn3-Ig-fn3-fn3-fn3 repeated 11 times and extends from the C-zone to the bare zone (Figures 2, 3) (Labeit and Kolmerer, 1995). The fn3 domains in titin consist of seven β-strands comprising two sheets forming a β-sandwich structure (Goll et al., 1998). It has been shown that the fn3 domain sequences in the A-band can be separated into three distinct fn3 sequence types, the arrangement of which defines distinct repeat units in the D- and C-zones, respectively (Fleming et al., 2023). Critically, these super-repeats serve as a template that regulates the length of the thick filament (Tonino et al., 2017), which will be discussed in greater detail in a later section.
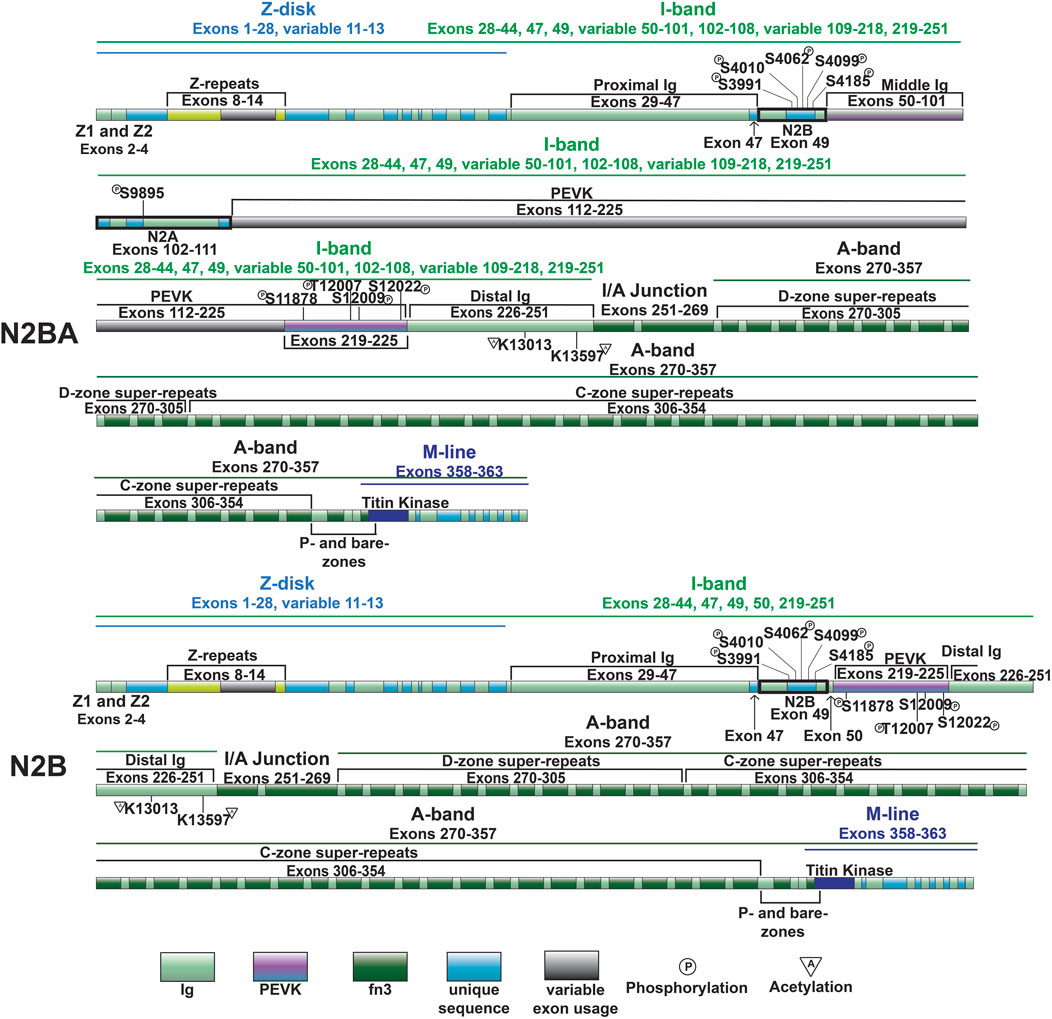
Figure 3. Domain map of cardiac titin isoforms—N2BA and N2B.
M-line titin is encoded by exons 358–363 (known as Mex1-Mex6) and tethers the C-terminus of the protein to the M-line (Kolmerer et al., 1996). In the M-line, titin molecules from adjacent half sarcomeres overlap, forming a continuous filament that stabilizes the sarcomere (Figure 1). A large proportion of cardiac titin expresses all six exons, Mex1-Mex6, unlike skeletal muscle, which often lacks Mex5 (Kolmerer et al., 1996). If Mex5 is not expressed in cardiac tissue, pathogenic phenotypes often develop (Marcello et al., 2022). Mex 1 encodes the titin kinase (TK), which is responsible for binding multiple different proteins (Kolmerer et al., 1996; Puchner et al., 2008). The role of these M-line regions change throughout development, and the absence of these exons results in embryonic lethality (Gotthardt et al., 2003). Under mechanical load, sarcomeres initially assemble but fail to grow laterally in the absence of a continuous titin filament and eventually disassemble, underscoring the important structural function of this portion in the beating heart (Mayans et al., 1998).
Titin is a functionally pleiotropic protein with specific functions mediated by its unique regions. Proper sarcomere formation requires titin, as myosin uses it as a blueprint that determines the length of the thick filament (Tonino et al., 2017). Titin also contains spring-like domains that grant it elasticity. These elastic properties allow titin to act as a molecular spring that contributes significantly to the passive stiffness of the myocardium (Gautel and Goulding, 1996; Linke et al., 1996; Tskhovrebova and Trinick, 1997; Linke et al., 1998; Trombitás et al., 1998). Titin also plays a role in mechanosignaling by serving as a scaffold for stretch-sensing protein complexes located in the Z-disk, I-band, and M-line (Linke and Krüger, 2010; Kötter et al., 2014a; van der Pijl et al., 2021). These functions will be discussed in greater detail below.
Early study of purified myosin assembled into “synthetic thick filaments” revealed that these filaments lacked the homogeneous length exhibited by thick filaments purified from muscle (HUXLEY, 1963). Observations such as this raised the question of how thick filament length is regulated in vivo. A critical finding came in 1989 when it was shown that the length of the large super-repeats in the C-zone of A-band titin matched the ∼43 nm spacing of myosin heads in the corresponding portion of the thick filament (Fürst et al., 1989). This finding, together with data providing evidence of interactions between titin and myosin (Murayama et al., 1989), lead to the hypothesis that titin determines the length of thick filaments (Whiting et al., 1989). Although this hypothesis remained contentious for many years, more recent evidence has confirmed that this is, in fact, the case (Tonino et al., 2017). Tonino et al. (2017) generated a novel mouse model in which the first two super-repeats of the C-zone were deleted. Structural studies confirmed that the length of the sarcomeric A-band was reduced in both cardiac and skeletal muscles from these mice (Tonino et al., 2017). Further support has been provided by analysis of the axial disposition of titin on the thick filament using super-resolution microscopy (Bennett et al., 2020). Thus, it can be concluded that A-band titin serves as a molecular blueprint for the sarcomeric A-band, thereby controlling the length of the thick filaments.
Passive stiffness/tension is the resistance to stretching that muscles exhibit when not actively contracting. In the heart, passive stiffness is determined by a variety of factors, including the cytoskeletal filaments, extracellular matrix (e.g., collagen), and titin, with the latter two contributing the greatest in the working range of cardiac sarcomeres (Linke et al., 1994; Granzier and Irving, 1995; Loescher et al., 2023). In particular, it has been shown that in the working range of cardiac sarcomeres (∼1.9–2.2 µm) titin-based stiffness predominates in the lower end of the working range and collagen-based stiffness at the higher end and beyond (Granzier and Irving, 1995). Titin-based stiffness arises from the reversible straightening of extensible domains in titin’s I-band (Gautel and Goulding, 1996; Linke et al., 1996; Tskhovrebova and Trinick, 1997; Linke et al., 1998; Trombitás et al., 1998). These extensible domains include the Ig and PEVK domains, as well as the N2B and N2A domains (Figure 1).
When the sarcomeres are at slack length, titin’s I-band exists in a folded conformation with the tandem Ig-like domains in a contracted configuration (Trombitás et al., 1995; Granzier et al., 1996). When stretched, the connecting segments between Ig-like domains straighten first, followed by extension of the PEVK domain (Gautel and Goulding, 1996; Linke et al., 1996; Tskhovrebova and Trinick, 1997; Linke et al., 1998; Trombitás et al., 1998) and N2B unique sequence (N2Bus) (Trombitás et al., 1999). Extension of the tandem Ig-like domains contributes to passive stiffness at the lower end of the working range of cardiac sarcomeres (<2.0 µm) (Trombitás et al., 1999). An early mechanism proposed to explain titin elasticity posited that the Ig-like domains themselves unfold, as was observed in isolated titin molecules (Erickson, 1994; Lu et al., 1998). A series of papers published in the late 1990s suggested that such unfolding in vivo would require non-physiological levels of force (Kellermayer et al., 1997; Rief et al., 1997; Tskhovrebova et al., 1997) and, thus, for many years it was thought that unfolding of the Ig-like domains did not occur naturally. However, the findings of a recent study suggest that Ig-like domain unfolding may occur at physiological sarcomere lengths and forces of 6–8 pN (Rivas-Pardo et al., 2016). These findings not only again raise the possibility that Ig-like domain unfolding occurs, but also put forward that refolding of these domains actively contributes to contraction (Rivas-Pardo et al., 2016). Additional studies will be necessary to confirm these findings.
The major mechanism regulating titin-based stiffness is alternative splicing of exons encoding Ig-like domains in the middle Ig segment (discussed in greater detail below) (Freiburg et al., 2000; Guo et al., 2010), however it has been found that the extensibility of domains such as the Ig-like domains can also be regulated by PTMs. For example, it has been shown that cryptic (i.e., buried) cysteine residues in the Ig-like domains can undergo reversible S-glutathionylation, which decreases titin-based passive stiffness by hindering refolding of these domains following stretch (Alegre-Cebollada et al., 2014; Loescher et al., 2020). Several CaMKII phosphorylation sites have been identified within Ig-like domains and the linker sequences between such domains in the I-band (Hamdani et al., 2013a). It has been proposed that phosphorylation of these sites may affect passive stiffness through a similar mechanism to oxidation (Hamdani et al., 2017), although whether this is the case remains to be determined. Besides PTMs, protein-protein interactions involving titin’s Ig-like domains have also been found to impact titin-based stiffness. Under stretching conditions, heat shock protein (HSP) 27 and αB-crystallin can bind to Ig-like domains and decrease passive stiffness (Kötter et al., 2014b). This interaction is believed to occur only upon unfolding of the Ig-like domains as HSP27 and αB-crystallin rarely localized to Ig-like domains in the longer and more compliant N2BA titin isoform, which consists of Ig-like domains that are less likely to unfold (Kötter et al., 2014b).
The PEVK region of titin is encoded by exons 112–225 in the I-band and is highly extensible (Gautel and Goulding, 1996; Linke et al., 1996). Extension of the PEVK is a major determinant of passive stiffness at cardiac sarcomere lengths greater than 2.0 µm (Trombitás et al., 1999). The elasticity of the PEVK domain is due, at least in part, to this domain acting as an entropic string (Linke et al., 2002). However, there are also charge contributions that impact stiffness, meaning that the spring-like feature of the region is not entirely entropically driven (Forbes et al., 2005). If regions of the PEVK are treated with an ionic solution, persistence length increases while Debye–Hückel length decreases, providing evidence that electrostatic interactions in this region also play an important part in determining the stiffness of the PEVK domain (Forbes et al., 2005).
Similar to the Ig-like domains of the middle Ig segment, control of PEVK stiffness can be regulated by alternative splicing of the exons encoding this domain (discussed below) (Freiburg et al., 2000; Guo et al., 2010), as well as through PTMs. Phosphorylation sites in the PEVK region of human titin include S11878 and S12022, which can both be phosphorylated by PKCα (Hidalgo et al., 2009) (Figure 4). PKCα-mediated phosphorylation of these sites increases passive tension (Hidalgo et al., 2009). Phosphorylation of additional residues in the PEVK domain, including T12869, S12871, and S12884 in mouse [corresponding to T12007, S12009, and S12022 in human titin, respectively (Figures 2, 3)], by CaMKII isoforms has also been described (Hamdani et al., 2013a). Intriguingly, treatment of single skinned mouse cardiomyocytes isolated from CaMKIIδ/γ double knockout mice with CaMKIIδ decreased titin-based passive stiffness (Hamdani et al., 2013a). Additional sites of CaMKII phosphorylation were also identified in other parts of the I-band, such as the N2Bus (Hamdani et al., 2013a). Given that the experimental design did not permit for the relative contributions of PEVK and N2B phosphorylation to passive stiffness to be assessed (Hamdani et al., 2013a), it is unclear whether the decrease was due to a dominant effect of N2B phosphorylation (discussed in the next section) or if the modulation of PEVK-associated titin stiffness is site-dependent.
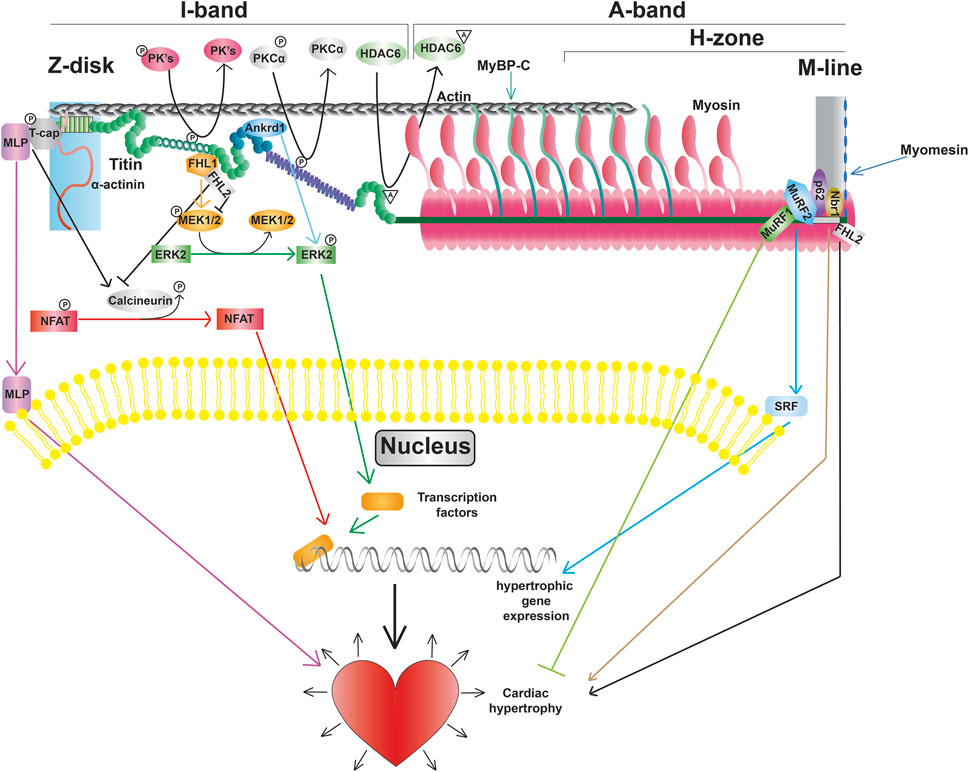
Figure 4. Titin modifications and signaling pathways, including promotion of transcription factors, altered gene expression, and cardiac hypertrophy.
Recent evidence indicates that acetylation of residues within the PEVK segment also modulates passive stiffness (Lin et al., 2022). Initially it was found that myofibril resting tension was elevated in mice deficient in histone deacetylase 6 (HDAC6) (Lin et al., 2022). Importantly, decreased titin stiffness following incubation with recombinant HDAC6 did not occur in isolated myofibril preparations from mice lacking 282 amino acids of the PEVK/Ig-like region of titin, indicating that acetylation of this region is responsible for acetylation-associated modulation of titin stiffness (Lin et al., 2022). Subsequently, mass spectrometry identified K13013 and K13597 as sites of acetylation downstream of the PEVK domain (Lin et al., 2022). Hdac6 deficient mice develop greater diastolic dysfunction in response to hypertension and aging, suggesting that HDAC6-mediated control of titin phosphorylation could be important for normal cardiac function and in response to stress and normal aging (Lin et al., 2022).
The cardiac specific N2B domain is encoded by exon 49 and contains three Ig-like domains and a 572 amino acid sequence unique to the domain (Figure 3) (Labeit and Kolmerer, 1995). Studies have shown the N2Bus contributes to passive stiffness via unfolding near the upper end of the physiological cardiac sarcomere length range (∼2.2 µm) (Helmes et al., 1999; Trombitás et al., 1999). The importance of N2B domain extensibility is underscored by the finding that deletion of the domain in mice leads to diastolic dysfunction secondary to increased extension of the remaining extensible elements (tandem Ig and PEVK domains) and increased titin stiffness (Radke et al., 2007). The N2A domain of the N2BA cardiac titin isoform (also expressed in the skeletal muscle N2A titin isoform) is another region of titin with elastic properties, characterized by four Ig-like domains and a 104 amino acid unique sequence (Labeit and Kolmerer, 1995). This domains unique α-helices surrounded by disordered region allow it to act as an entropic spring like the N2B domain (Lanzicher et al., 2020; van der Pijl et al., 2021).
The N2Bus is a well-known target for a variety of kinases. Common phosphorylation sites in the N2Bus include S3991, S4010, S4062, S4099, and S4185, among others (Figure 3). Each site can be phosphorylated by multiple different protein kinases such as PKA (Yamasaki et al., 2002), PKG (Krüger et al., 2009), PKD (Herwig et al., 2020), ERK2 (Perkin et al., 2015), and/or CaMKII (Hamdani et al., 2013a; Perkin et al., 2015) and dephosphorylated by the serine/threonine phosphotase PP5 (Krysiak et al., 2018). Phosphorylation of N2Bus by these different kinases decreases titin-based stiffness while dephosphorylation increases it (Yamasaki et al., 2002; Krüger et al., 2009; Perkin et al., 2015; Krysiak et al., 2018; Herwig et al., 2020). Although the mechanism whereby the phosphorylation of sites in the N2Bus produces a decrease in passive stiffness remains to be determined, it has been postulated that the introduction of a negatively charged group to the already negatively charged N2Bus improves extensibility by reducing the force required to extend this segment through increased intramolecular electrostatic repulsion (Koser et al., 2019). The N2A domain is also able to be phosphorylated by PKA at S9895, however it does not appear to contribute much, if at all, to titin’s passive stiffness. Instead, phosphorylation of the region helps mediate its protein interactions, which will be discussed in further detail in Section 3.4.2 (Lanzicher et al., 2020).
Stiffness of the N2Bus is also regulated by oxidative modifications. For example, it has been shown that disulfide bridges may form between cysteine residues in the N2Bus (Grützner et al., 2009). Although direct evidence for the formation of these disulfide bonds in the heart is lacking, treatment of isolated human heart myofibrils with the reducing agent thioredoxin reduced titin-based stiffness to a degree consistent with increased extensibility of the N2Bus (Grützner et al., 2009), providing circumstantial evidence for their existence.
As mentioned above, the most well-studied mechanism regulating titin extensibility involves alternative splicing in titin’s I-band region (Freiburg et al., 2000; Guo et al., 2010). The inclusion or exclusion of many exons in the middle Ig and PEVK domains dramatically affects the length of titin and, thus, its extensibility. The heart expresses two major classes of titin isoforms: a smaller and stiffer N2B isoform and larger more compliant N2BA isoforms (Figure 3) (Cazorla et al., 2000; Freiburg et al., 2000). The N2B isoform is stiffer due to exclusion of a greater number of exons in the middle Ig and PEVK domains while N2BA isoforms have greater extensibility because of inclusion of these exons and correspondingly longer Ig and PEVK domains. In a healthy adult human heart, the N2BA isoform comprises 30%–50% of titin proteins, while the N2B isoform makes up 50%–70% of titin proteins (Cazorla et al., 2000; Neagoe et al., 2002). The expression of cardiac titin isoforms has been shown to change during development and in pathological conditions (Bell et al., 2000; Neagoe et al., 2002; Wu et al., 2002; Warren et al., 2003; Makarenko et al., 2004; Nagueh et al., 2004; Opitz et al., 2004; Warren et al., 2004; Shapiro et al., 2007; Borbély et al., 2009; Hudson et al., 2011; Hamdani et al., 2013b; Zhu et al., 2017).
In 2012, we identified deletion of the gene encoding RNA binding motif protein 20 (RBM20) as the cause of altered titin splicing in rats deficient in titin splicing (Guo et al., 2012). This discovery made RBM20 the first factor known to regulate titin size through alternative splicing. In a follow up study, we determined that RBM20-mediated control of titin size occurs predominantly via exon exclusion although this was exon-dependent (Li et al., 2013). When present, RBM20 promotes exclusion of exons between 51 and 218 encoding the middle Ig and PEVK segments, thereby favoring the production of N2B titin (Figure 3) (Li et al., 2013). On the other hand, at reduced levels, or when RBM20 is absent, inclusion of these exons is favored, leading to the production of more compliant N2BA titin isoforms with longer middle Ig and PEVK segments (Figure 3) (Guo et al., 2012; Li et al., 2013). In the absence of RBM20, a giant N2BA isoform of titin is expressed (Borbély et al., 2009). Mechanistically, RBM20 promotes exon skipping by binding to sites downstream of target exons (Dauksaite and Gotthardt, 2018), although the precise mechanism has not been determined.
A growing list of additional splicing factors have been identified that can also regulate titin splicing in the heart. These factors include RBM24 (Liu et al., 2019), PTB4 (Dauksaite and Gotthardt, 2018), SLM2 (Boeckel et al., 2022), and RBPMS (Gan et al., 2023). Importantly, studies have only provided evidence for the regulation of select exons in titin for each of these factors and, apart from RBPMS (Gan et al., 2023), none of these factors have been shown to significantly switch titin size (Dauksaite and Gotthardt, 2018; Liu et al., 2019; Boeckel et al., 2022). RBPMS is involved in the inclusion of the N2B exon and loss of this protein in mice produced an apparent decrease in the size of both N2B and N2BA titin isoforms (Gan et al., 2023). This size change produces titin isoforms similar in size to those produced via N2B deletion (Gan et al., 2023). Other confirmed exon skipping events associated with loss of RBPMS were restricted to exons outside the I-band (Gan et al., 2023), which are generally constitutively expressed in cardiac titin. Thus, although exclusion of these exons produced a DCM-like phenotype in Rbpms deficient mice, the physiological significance of RBPMS-regulated titin splicing in the healthy heart is unclear at the present time. Therefore, RBM20 remains the major factor regulating alternative splicing of the TTN gene in health and disease.
Beyond titin’s role as a blueprint for the sarcomeric A-band and as a molecular spring responsible for myocardial passive stiffness, it also has an important role as an integrator and transducer of mechanical signals (Linke and Krüger, 2010; Kötter et al., 2014a; van der Pijl et al., 2021). Titin serves as a scaffold for a variety of mechano-sensitive signaling complexes localized in the Z-disk, I-band, and M-line. In addition, titin is also a substrate for signaling pathways enabling the refinement of passive stiffness through PTMs in regions such as the N2Bus and PEVK segment as discussed above (Figure 4).
The Z-disk of titin serves as a hub for pro-hypertrophic signaling pathways. Hypertrophic signaling at the Z-disk is regulated by the stress sensor MLP, which has been shown to bind to the titin-T-cap complex (Knöll et al., 2002). Study of mice lacking a single copy of the Mlp gene revealed that MLP plays an important role in localization of the phosphatase calcineurin to the Z-disk (Heineke et al., 2005). Moreover, transcriptional activation of NFAT was blunted in these mice following myocardial infarction demonstrating that the Z-disk-localized MLP-calcineurin complex is required for the stress-induced activation of the pro-hypertrophic calcineurin-NFAT signaling pathway (Heineke et al., 2005). In addition to activation of calcineurin-NFAT signaling, MLP itself can also translocate to the nucleus (Boateng et al., 2007). The finding that treatment with cell-permeable synthetic peptides containing the putative nuclear localization signal of MLP blocked not only nuclear translocation of the protein, but also increased protein accumulation in response to phenylephrine, suggests that nuclear shuttling of MLP is also important for adaptation to hypertrophic stimuli (Boateng et al., 2009). Interestingly, MLP and Ankrd1 together can transport PKCα to intercalated discs, which is often seen in DCM, and deletion of both proteins restores proper PKCα localization and function (Lange et al., 2016).
Beyond determining extensibility, titin’s I-band segments also serve as docking sites for protein complexes that mediate critical signaling events. The spring-like N2B domain interacts with FHL1, FHL2, and FHL3 (four and a half LIM protein 1, 2, and 3, respectively) (Lange et al., 2002; Sheikh et al., 2008). Interestingly, FHL1-mediated signaling has been shown to promote the activation of the pro-hypertrophic MAPK signaling cascade (Sheikh et al., 2008), while FHL2 opposes the hypertrophic response (Figure 4) (Kong et al., 2001). The functional role of FHL3 in cardiac muscle is not well understood, however, it has been shown to localize to titin’s N2B region and expression is severely deregulated in cardiomyopathies (Puchner et al., 2008). In addition to FHL proteins, I-band titin has also been shown to interact with proteins belonging to the muscle ankyrin repeat protein (MARP) family (Miller et al., 2003). One member of the MARP family, ANKRD1 (also known as CARP), has been implicated in cardiac hypertrophy due to upregulation during cardiogenesis and in response to hypertrophy stimuli (Kuo et al., 1999; Aihara et al., 2000). Similar to MLP, mechanical stimulation promotes translocation of ANKRD1 from the sarcomeres (where it co-localizes with the N2A domain of titin) to the nucleus (Miller et al., 2003). While a number of I-band protein interactors and signaling pathways are consistent between cardiac and skeletal muscle titin, there are some proteins, such as calpain-3, that only interact with the skeletal muscle titin N2A domain (Ojima et al., 2014).
A portion of M-line titin encodes a kinase, TK, that is located near the A-band/M-line junction (Labeit et al., 1992). Evidence from early studies suggested that TK plays an important role during muscle development (Mayans et al., 1998). Additional data suggested that this kinase could also be activated in response to mechanical strain making it a putative stress sensor (Puchner et al., 2008). However, more recent studies indicated that TK is a pseudokinase (Puchner et al., 2008), making it unlikely that the kinase activity of this domain is required for signal transduction in response to strain. Nevertheless, there is evidence that a portion of this domain can undergo reversible unfolding in response to stretch (Puchner et al., 2008), and TK does serve as an important scaffold for a variety of signaling proteins. In particular, the autophagy receptor Nbr1 has been shown to recruit p62 and MuRF2 to the TK domain (Lange et al., 2005). The E3 ubiquitin ligase MuRF1 can also interact with a sequence near the TK domain (Centner et al., 2001). Bogomolovas et al. showed that MuRF1 can ubiquitinate the TK domain in a stretch-dependent manner and this ubiquitination is required for recruitment of Nbr1 and p62 (Bogomolovas et al., 2021). Another location for E3 ubiquitin ligase binding on titin is at the C-terminal portion of the A-band at Ig141/Ig142/Fn3-132 (A168-170), which can bind MURF1, 2, and 3 (Müller et al., 2021). These pathways are currently thought to be important for breakdown of the sarcomeres (Bogomolovas et al., 2021), although further investigation will be required to determine whether this is the case.
Due to titin’s myriad functional roles in the heart, it is not surprising that disruption of these functions by things such as mutations is associated with the development of heart diseases. Indeed, titin has been implicated in a variety of heart muscle diseases among which dilated cardiomyopathy (DCM) and heart failure with preserved ejection fraction (HFpEF)/diabetic cardiomyopathy (DbCM) are the most prominent (Table 1) (Herman et al., 2012; Zile et al., 2015; Hopf et al., 2018). In the following section we will briefly discuss the contribution of titin to these diseases and provide updates regarding mechanism based on recently published studies.
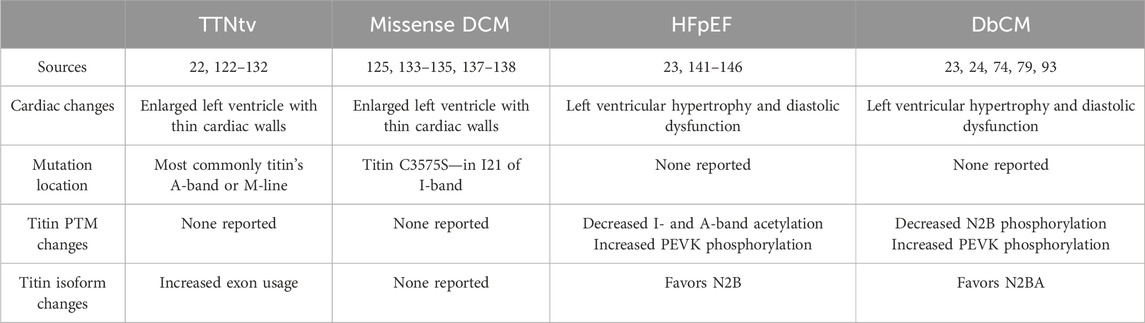
Table 1. Cardiac diseases associated with titin.
Titin truncating variants (TTNtvs) represent the most common cause of familial and sporadic DCM, accounting for upwards of 25% of familial cases and ∼15% of sporadic cases (Herman et al., 2012; Pugh et al., 2014; Roberts et al., 2015; Haggerty et al., 2019; Mazzarotto et al., 2020). Surprisingly, TTNtvs are also found in ∼1–2% of the general population (Pugh et al., 2014; Roberts et al., 2015; Schafer et al., 2017). Pathogenic TTNtvs are typically carried in the heterozygous state and are overrepresented in the A-band (Herman et al., 2012; Pugh et al., 2014; Roberts et al., 2015). This phenomenon is thought to be because exons encoding the A-band are constitutively expressed (Roberts et al., 2015). Over the years, a variety of hypotheses have been proposed to explain the pathogenicity of TTNtvs; however, the two most popular have been haploinsufficiency and the poison peptide hypothesis (Yotti et al., 2019). In the case of the former, titin production from the single unaffected allele is insufficient to maintain appropriate cardiac function, while the latter posits that the truncated protein acts in a manner that disrupts normal heart function. The findings of recent studies indicate that both mechanisms contribute to the development of DCM in TTNtv carriers.
One of the primary issues that has hindered acceptance of the poison peptide hypothesis has been the inability to detect truncated titin proteins in the hearts of DCM patients with TTNtvs (Roberts et al., 2015; Vikhorev et al., 2017). However, using a combination of gel electrophoresis and antibodies against the N- and C-termini of titin, McAfee et al. (2021) were able to detect appropriately sized TTNtvs in heart samples from patients with DCM caused by TTNtvs. An independent study from the Linke lab published at the same time made similar findings using similar methods (Fomin et al., 2021). Additionally, TTNtv cannot be properly ubiquitinated for degradation due to the M-line portion of titin not being expressed and have been detected in insoluble granules containing TTNtv in human iPSC-CMs (Huang et al., 2023). In addition to the poison peptide theory, Fomin et al. (2021) detected a decrease in total titin expression that correlated with decreased left ventricular ejection fraction, indicating that haploinsufficiency also plays a role in disease severity. Interestingly, they also found no difference in the ratio of M-band versus Z-disk titin based on immunofluorescence staining (Fomin et al., 2021), suggesting that the truncated proteins, which would be labeled by the Z-disk but not M-line antibodies, are not incorporated into the sarcomeres. Conversely, truncated titin proteins precipitated with skinned myofibril preparations from human DCM hearts as determined by gel electrophoresis (McAfee et al., 2021; Kellermayer et al., 2024). Moreover, using super-resolution microscopy, Kellermayer et al. (2024) detected reduced titin signal based on immunofluorescence staining of titin with N- and C-terminal antibodies however when TTNtv does assemble within the sarcomere, there is a 49-fold increase in disassembly (Bogomolovas et al., 2021). Considering this conflicting data, the question of whether truncated titin proteins are incorporated into the sarcomeres or if they exist in intracellular aggregates (Fomin et al., 2021) requires further investigation.
In addition to TTNtvs, a long list of missense variants in titin have also been identified in association with DCM, as well as peripartum cardiomyopathy (PPCM) (Merlo et al., 2013; Pugh et al., 2014; van Spaendonck-Zwarts et al., 2014; Begay et al., 2015; Akinrinade et al., 2019); however, evidence indicating a causal association between these variants and disease has been largely lacking. Recently, Domínguez et al. (2023) identified a case of familial DCM in a Spanish family carrying substitution of a highly conserved cysteine at position 3892 to serine. Linkage analysis produced a family-specific 2-point logarithm of the odds score of 3.96, which strongly supports that the variant is related to the phenotype (Domínguez et al., 2023). While prior studies of families carrying TTNtvs have implicated titin missense variants in disease (Gerull et al., 2002), this study provides the strongest evidence to date that missense variants in titin can cause DCM. Although the mechanism will require further investigation, limited evidence from circular dichroism spectroscopy suggests that this variant may destabilize the protein at physiological temperatures potentially causing disease through haploinsufficiency (Domínguez et al., 2023). Other missense mutations in titin have been associated with PPCM in mothers during late pregnancy and early post-partum (van Spaendonck-Zwarts et al., 2014). While there are many phenotypic similarities between DCM and PPCM, they differ in disease onset and the fact that a proportion of PPCM patients are able to fully recover, while DCM can only be managed (van Spaendonck-Zwarts et al., 2014). The mechanisms leading to the development of PPCM are not fully understood, but there are many similarities with DCM. Prior studies have shown that N2BA titin is increased in PPCM, similar to what has been shown in DCM, however the impact on passive force/tension is contradictory in the literature (van Spaendonck-Zwarts et al., 2014; Bollen et al., 2017). Also impacting passive tension, PKA- and PKC-mediated phosphorylation of titin at S4010 and S12022 are both significantly reduced in PPCM patients, while S12022 is unaffected in DCM patients (Bollen et al., 2017). Another titin contribution to PPCM is that the increased oxidative stress caused by pregnancy also contributes to the pathogenic phenotype (Bollen et al., 2014).
HFpEF and DbCM produce very similar cardiac phenotypes with cardiac hypertrophy, increased cardiac stiffness, and impaired diastolic function. Understanding the physiological mechanisms of HFpEF is one of the greatest unmet needs in cardiovascular disease due to a lack of effective treatments. Within 4 years of HFpEF diagnosis, the mortality rate is over 30% (Burkhoff, 2012). One of the biggest risk factors for HFpEF development is hypertension (Tam et al., 2017; Tadic et al., 2018). Granzier et al. showed that hypertensive mouse models favored the N2B titin isoform over the N2BA compared to control mice, which increases titin-based myocardial stiffness and decreases diastolic function (Hutchinson et al., 2015). As HFpEF develops, PTMs in titin’s elastic domains change to increase the overall titin-based myocardial stiffness. Patients with HFpEF often have increased PEVK phosphorylation at S11878 and decreased I- and A-band acetylation (Zile et al., 2015; Koser et al., 2022). There is also reduced N2B phosphorylation at S3991, S4080, and S4185 causing increased stiffness (see previous sections) (Zile et al., 2015; Koser et al., 2022). Further supporting altered titin PTMs, treatment of skinned cardiomyocytes isolated from a patient with HFpEF with PKA was able to restore passive force to near the healthy range (Borbély et al., 2005). Both titin isoform switching and altered PTMs play major roles in the increased titin-based myocardial stiffness found in HFpEF patients.
Like HFpEF, people suffering from DbCM often have impaired diastolic function because of altered PTMs in titin’s elastic I-band domains. Contrary to HFpEF, DbCM patients have the opposite effect in terms of titin isoform switching and promote N2BA expression (Hopf et al., 2018). The major alternative splicing factor RBM20 has increased expression in the presence of insulin, meaning that people who are insulin resistant have decreased RBM20 levels (Zhu et al., 2017). In turn, this promotes the expression of the N2BA titin isoform over N2B, decreasing titin-based myocardial stiffness. On the other hand, biopsies reveal those suffering from DbCM have altered N2B and I-band PTMs (Krüger et al., 2009; Zile et al., 2015; Lin et al., 2022). Patients with type 2 diabetes had decreased phosphorylation of S4099 (phosphorylated by PKG) in the N2B region and increased phosphorylation of S11878 (phosphorylated by PKCα) in the PEVK region (Krüger et al., 2009; Zile et al., 2015; Lin et al., 2022). The result of these altered PTMs is increased titin-based stiffness. Improving insulin sensitivity to modulate titin stiffness through targeting NRG-1 or via treatment with metformin reverses the altered PTMs of the N2B and PEVK domains found in diabetes patients, showing its therapeutic potential for diastolic dysfunction (Hopf et al., 2018).
Titin’s status as a causative agent in diseases such as DCM, as well as its role as an important modulator of myocardial passive stiffness (and by extension diastolic function), make it an appealing therapeutic target. In the case of DCM caused by variants in titin, two therapeutic strategies are currently being explored to target titin. The first is the use of antisense oligonucleotides (ASOs) to promote skipping of variant-containing exons has been explored as a potential strategy to treat pathogenic variants in titin that cause DCM (Gramlich et al., 2015). ASO-mediated strategies have shown promise in both mice and cultured cells (Gramlich et al., 2015; Hahn et al., 2019). Given that ASOs have already been approved for the treatment of other diseases (Crooke et al., 2021), such as Duchenne’s muscular dystrophy, ASO-based exon skipping strategies represent an accessible strategy to treat DCM caused by certain disruptive variants in titin. It has been predicted that upwards of 94 exons in titin may be suitable for therapeutic deletion to treat DCM-associated titin variants (Rodriguez-Polo and Behr, 2022). More recently, CRISPR-Cas9 genome editing was used to correct TTNtvs in human iPSC-CMs (Romano et al., 2022). Expansion of these results to model organisms such as mice will be an important step towards demonstrating the therapeutic potential of this strategy. In the case of idiopathic DCM, potential therapeutic approaches could involve targeting RBM20. Most people suffering from DCM have a shift in titin isoforms to favor the longer N2BA isoform paired with decreased ventricular passive stiffness and systolic dysfunction (LeWinter and Granzier, 2014). By increasing RBM20 expression through regulation of transcriptional factors, it would shift titin to favor the N2B isoform, restoring titin-based myocardial passive stiffness (Guo et al., 2012).
Diastolic dysfunction is a key element of heart diseases such as HFpEF and DbCM (Borlaug, 2014; Jia et al., 2016). Titin size changes resulting from alternative splicing plays a major role in myocardial stiffness, while PTMs in titin’s spring-like domain fine-tune myocardial stiffness, as discussed above (Forbes et al., 2005; Krüger et al., 2009; Raskin et al., 2012; Hamdani et al., 2013a; Hamdani et al., 2013b; Hidalgo et al., 2013; Kötter et al., 2013; Perkin et al., 2015; Krysiak et al., 2018; Herwig et al., 2020; Michel et al., 2020; Loescher et al., 2022). Thus, either manipulating titin size switching or altering PTMs in titin could reduce diastolic stiffness and, therefore, dysfunction. Given that RBM20 is the primary regulator of titin size through alternative splicing (Guo et al., 2012), this protein is an attractive target to modulate titin size and improve diastolic function. Reducing RBM20 expression through genetic editing or ASO-mediated RNA degradation has provided proof-of-concept evidence that increasing the expression of larger titin isoforms improves diastolic function in rodent models of HFpEF (Hutchinson et al., 2015; Hinze et al., 2016; Methawasin et al., 2016; Radke et al., 2021). It is important to point out that RBM20 also controls the splicing genes related to Ca2+-handling (Guo et al., 2012; Guo et al., 2018) and loss of RBM20-dependent splicing of these genes predisposes the heart to arrhythmia (Guo et al., 2012). Thus, targeting RBM20 expression to modulate titin size may have detrimental effects. The consensus sequence containing the core element UCUU has been shown to be the binding motif for RBM20 in titin mRNA (Maatz et al., 2014). Therefore, an alternative therapeutic option would be to design ASOs targeting the RBM20 binding sites on the titin pre-mRNA. Theoretically, such a strategy could reduce diastolic stiffness in HFpEF/DbCM by promoting exon inclusion and the expression of larger titin isoforms without affecting the splicing of other RBM20 target genes. Another option would be to combine both approaches. RBM20 binds many locations on titin and so designing ASO’s to inhibit RBM20 binding to titin mRNA poses difficulties. By reducing RBM20 expression, the titin ASO’s may lead to a greater increase in N2BA titin isoform expression. Nevertheless, the reduction in RBM20 expression must be highly regulated to prevent severe alterations in RBM20-mediated alternative splicing of other mRNA targets such as Ca2+-handling genes (Guo et al., 2012).
In addition to titin isoform switching, the modulation of titin-based stiffness through PTMs represents another potential treatment strategy to reduce diastolic stiffness in HFpEF and DbCM or increased diastolic stiffness in DCM. Studies show that patients with DbCM often display cardiac stiffening despite increased expression of more compliant N2BA titin (van Heerebeek et al., 2008). The potential mechanisms could be the increased phosphorylation in N2Bus and decreased phosphorylation in PEVK region (Yamasaki et al., 2002; Hidalgo et al., 2009; Krüger et al., 2009; Perkin et al., 2015; Krysiak et al., 2018; Herwig et al., 2020). Examination of titin phosphorylation in the hearts of diabetic patients revealed reduced phosphorylation of S4099 (in N2Bus) and increased phosphorylation of S11878 (in PEVK) that can account for increased passive stiffness (Hopf et al., 2018). Treating diabetic rats with NRG-1 (drug that improves glucose tolerance) restored diastolic function and titin phosphorylation to similar levels to wildtype through PKG- and PKCα-mediated phosphorylation (Hopf et al., 2018). NRG-1 treated rats also had an improvement in PKA-mediated phosphorylation, however not to the same extent (Hopf et al., 2018). Interestingly, diabetic rats treated with metformin (increases insulin sensitivity) have increased phosphorylation of titin S11878 in the PEVK (Hopf et al., 2018). It is noteworthy that there is conflicting data in the literature regarding the effect of metformin treatment on titin PTMs as others have shown little to no effect on PEVK phosphorylation but increased phosphorylation of PKA sites in the N2Bus (Slater et al., 2019). Studies also reveal that histone deacetylase 6 (HDAC6) can deacetylate K13013 and K13597 near the PEVK region (Figure 4) (Lin et al., 2022). Lack of HDAC6 or treated with HDAC6 inhibitors in mice increases myocardial stiffness (Lin et al., 2022). In addition to HDAC6, study found that deacetylase SIRT1 can reduce stiffness in rat cardiomyocytes (Abdellatif et al., 2021). Overall, these studies show that targeting PTMs in titin represents another therapeutic option to reduce diastolic stiffness in HFpEF/DbCM. Modulating titin stiffness through PTM’s may also be valuable as a supplement for other treatments. For example, regulation of titin PTMs and favored production of more compliant N2BA titin isoforms through ASOs could be used to synergistically decrease titin-based stiffeness in diseases with diastolic dysfunction, such as in DbCM. Therefore, it would be logical to conclude that regulating titin PTM’s could be especially beneficial in combination with therapies that also increase titin N2BA expression.
DS: Writing–review and editing, Writing–original draft. ZG: Writing–review and editing, Writing–original draft. FR: Writing–review and editing. YG: Writing–review and editing. WG: Writing–review and editing, Writing–original draft.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by the NIH NHLBI HL148733; the American Heart Association Foundation 19TPA3480072 and 23TPA1069731; the Wisconsin Alumni Research Foundation AAH4884; the University of Wisconsin Foundation AAH5964; and the USDA-NIFA Hatch grant (AAI4642); Department of Medicine (DOM) pilot grant (AAK1256).
YG would like to acknowledge NIH R01 HL109810. FR would like to acknowledge AHA Career Grant 23CDA1057697 and NIH/NCATS ICTR KL2TR002374-07 (FR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdellatif M., Trummer-Herbst V., Koser F., Durand S., Adão R., Vasques-Nóvoa F., et al. (2021). Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 13 (580), eabd7064. doi:10.1126/scitranslmed.abd7064
Aihara Y., Kurabayashi M., Saito Y., Ohyama Y., Tanaka T., Takeda S., et al. (2000). Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension 36 (1), 48–53. doi:10.1161/01.hyp.36.1.48
Akinrinade O., Heliö T., Lekanne Deprez R. H., Jongbloed J. D. H., Boven L. G., van den Berg M. P., et al. (2019). Relevance of titin missense and non-frameshifting insertions/deletions variants in dilated cardiomyopathy. Sci. Rep. 9 (1), 4093. doi:10.1038/s41598-019-39911-x
Alegre-Cebollada J., Kosuri P., Giganti D., Eckels E., Rivas-Pardo J. A., Hamdani N., et al. (2014). S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 156 (6), 1235–1246. doi:10.1016/j.cell.2014.01.056
Bang M. L., Centner T., Fornoff F., Geach A. J., Gotthardt M., McNabb M., et al. (2001). The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89 (11), 1065–1072. doi:10.1161/hh2301.100981
Begay R. L., Graw S., Sinagra G., Merlo M., Slavov D., Gowan K., et al. (2015). Role of titin missense variants in dilated cardiomyopathy. J. Am. Heart Assoc. 4 (11), e002645. doi:10.1161/JAHA.115.002645
Bell S. P., Nyland L., Tischler M. D., McNabb M., Granzier H., LeWinter M. M. (2000). Alterations in the determinants of diastolic suction during pacing tachycardia. Circ. Res. 87 (3), 235–240. doi:10.1161/01.res.87.3.235
Bennett P., Rees M., Gautel M. (2020). The axial alignment of titin on the muscle thick filament supports its role as a molecular ruler. J. Mol. Biol. 432 (17), 4815–4829. doi:10.1016/j.jmb.2020.06.025
Boateng S. Y., Belin R. J., Geenen D. L., Margulies K. B., Martin J. L., Hoshijima M., et al. (2007). Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am. J. Physiol. Heart Circ. Physiol. 292 (1), H259–H269. doi:10.1152/ajpheart.00766.2006
Boateng S. Y., Senyo S. E., Qi L., Goldspink P. H., Russell B. (2009). Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J. Mol. Cell Cardiol. 47 (4), 426–435. doi:10.1016/j.yjmcc.2009.04.006
Boeckel J. N., Möbius-Winkler M., Müller M., Rebs S., Eger N., Schoppe L., et al. (2022). SLM2 is A novel cardiac splicing factor involved in heart failure due to dilated cardiomyopathy. Genomics Proteomics Bioinforma. 20 (1), 129–146. doi:10.1016/j.gpb.2021.01.006
Bogomolovas J., Fleming J. R., Franke B., Manso B., Simon B., Gasch A., et al. (2021). Titin kinase ubiquitination aligns autophagy receptors with mechanical signals in the sarcomere. EMBO Rep. 22 (10), e48018. doi:10.15252/embr.201948018
Bollen I. A., Van Deel E. D., Kuster D. W., Van Der Velden J. (2014). Peripartum cardiomyopathy and dilated cardiomyopathy: different at heart. Front. Physiol. 5, 531. doi:10.3389/fphys.2014.00531
Bollen I. A. E., Ehler E., Fleischanderl K., Bouwman F., Kempers L., Ricke-Hoch M., et al. (2017). Myofilament remodeling and function is more impaired in peripartum cardiomyopathy compared with dilated cardiomyopathy and ischemic heart disease. Am. J. Pathol. 187 (12), 2645–2658. doi:10.1016/j.ajpath.2017.08.022
Borbély A., Falcao-Pires I., van Heerebeek L., Hamdani N., Edes I., Gavina C., et al. (2009). Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ. Res. 104 (6), 780–786. doi:10.1161/CIRCRESAHA.108.193326
Borbély A., van der Velden J., Papp Z., Bronzwaer J. G., Edes I., Stienen G. J., et al. (2005). Cardiomyocyte stiffness in diastolic heart failure. Circulation 111 (6), 774–781. doi:10.1161/01.CIR.0000155257.33485.6D
Borlaug B. A. (2014). The pathophysiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 11 (9), 507–515. doi:10.1038/nrcardio.2014.83
Burkhoff D. (2012). Mortality in heart failure with preserved ejection fraction: an unacceptably high rate. Eur. Heart J. 33 (14), 1718–1720. doi:10.1093/eurheartj/ehr339
Cazorla O., Freiburg A., Helmes M., Centner T., McNabb M., Wu Y., et al. (2000). Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ. Res. 86 (1), 59–67. doi:10.1161/01.res.86.1.59
Centner T., Yano J., Kimura E., McElhinny A. S., Pelin K., Witt C. C., et al. (2001). Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 306 (4), 717–726. doi:10.1006/jmbi.2001.4448
Chen Z., Song J., Chen L., Zhu C., Cai H., Sun M., et al. (2018). Characterization of TTN Novex splicing variants across species and the role of RBM20 in novex-specific exon splicing. Genes (Basel) 9 (2), 86. doi:10.3390/genes9020086
Crooke S. T., Baker B. F., Crooke R. M., Liang X. H. (2021). Antisense technology: an overview and prospectus. Nat. Rev. Drug Discov. 20 (6), 427–453. doi:10.1038/s41573-021-00162-z
Dauksaite V., Gotthardt M. (2018). Molecular basis of titin exon exclusion by RBM20 and the novel titin splice regulator PTB4. Nucleic Acids Res. 46 (10), 5227–5238. doi:10.1093/nar/gky165
Domínguez F., Lalaguna L., Martínez-Martín I., Piqueras-Flores J., Rasmussen T. B., Zorio E., et al. (2023). Titin missense variants as a cause of familial dilated cardiomyopathy. Circulation 147 (22), 1711–1713. doi:10.1161/CIRCULATIONAHA.122.062833
Erickson H. P. (1994). Reversible unfolding of fibronectin type III and immunoglobulin domains provides the structural basis for stretch and elasticity of titin and fibronectin. Proc. Natl. Acad. Sci. U. S. A. 91 (21), 10114–10118. doi:10.1073/pnas.91.21.10114
Filomena M. C., Yamamoto D. L., Carullo P., Medvedev R., Ghisleni A., Piroddi N., et al. (2021). Myopalladin knockout mice develop cardiac dilation and show a maladaptive response to mechanical pressure overload. Elife 10, e58313. doi:10.7554/eLife.58313
Fleming J. R., Müller I., Zacharchenko T., Diederichs K., Mayans O. (2023). Molecular insights into titin's A-band. J. Muscle Res. Cell Motil. 44 (4), 255–270. doi:10.1007/s10974-023-09649-1
Fomin A., Gärtner A., Cyganek L., Tiburcy M., Tuleta I., Wellers L., et al. (2021). Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 13 (618), eabd3079. doi:10.1126/scitranslmed.abd3079
Forbes J. G., Jin A. J., Ma K., Gutierrez-Cruz G., Tsai W. L., Wang K. (2005). Titin PEVK segment: charge-driven elasticity of the open and flexible polyampholyte. J. Muscle Res. Cell Motil. 26 (6-8), 291–301. doi:10.1007/s10974-005-9035-4
Freiburg A., Trombitas K., Hell W., Cazorla O., Fougerousse F., Centner T., et al. (2000). Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ. Res. 86 (11), 1114–1121. doi:10.1161/01.res.86.11.1114
Fürst D. O., Nave R., Osborn M., Weber K. (1989). Repetitive titin epitopes with a 42 nm spacing coincide in relative position with known A band striations also identified by major myosin-associated proteins. An immunoelectron-microscopical study on myofibrils. J. Cell Sci. 94 (Pt 1), 119–125. doi:10.1242/jcs.94.1.119
Gan P., Wang Z., Bezprozvannaya S., McAnally J. R., Tan W., Li H., et al. (2023). RBPMS regulates cardiomyocyte contraction and cardiac function through RNA alternative splicing. Cardiovasc Res. 120, 56–68. doi:10.1093/cvr/cvad166
Gautel M., Goulding D. (1996). A molecular map of titin/connectin elasticity reveals two different mechanisms acting in series. FEBS Lett. 385 (1-2), 11–14. doi:10.1016/0014-5793(96)00338-9
Gautel M., Goulding D., Bullard B., Weber K., Fürst D. O. (1996). The central Z-disk region of titin is assembled from a novel repeat in variable copy numbers. J. Cell Sci. 109 (Pt 11), 2747–2754. doi:10.1242/jcs.109.11.2747
Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., et al. (2002). Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 30 (2), 201–204. doi:10.1038/ng815
Goll C. M., Pastore A., Nilges M. (1998). The three-dimensional structure of a type I module from titin: a prototype of intracellular fibronectin type III domains. Structure 6 (10), 1291–1302. doi:10.1016/s0969-2126(98)00129-4
Gotthardt M., Hammer R. E., Hübner N., Monti J., Witt C. C., McNabb M., et al. (2003). Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J. Biol. Chem. 278 (8), 6059–6065. doi:10.1074/jbc.M211723200
Gramlich M., Pane L. S., Zhou Q., Chen Z., Murgia M., Schötterl S., et al. (2015). Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol. Med. 7 (5), 562–576. doi:10.15252/emmm.201505047
Granzier H., Helmes M., Trombitás K. (1996). Nonuniform elasticity of titin in cardiac myocytes: a study using immunoelectron microscopy and cellular mechanics. Biophys. J. 70 (1), 430–442. doi:10.1016/S0006-3495(96)79586-3
Granzier H., Labeit S. (2002). Cardiac titin: an adjustable multi-functional spring. J. Physiol. 541 (Pt 2), 335–342. doi:10.1113/jphysiol.2001.014381
Granzier H. L., Irving T. C. (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 68 (3), 1027–1044. doi:10.1016/S0006-3495(95)80278-X
Granzier H. L., Labeit S. (2004). The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ. Res. 94 (3), 284–295. doi:10.1161/01.RES.0000117769.88862.F8
Granzier H. L., Labeit S. (2006). The giant muscle protein titin is an adjustable molecular spring. Exerc Sport Sci. Rev. 34 (2), 50–53. doi:10.1249/00003677-200604000-00002
Gregorio C. C., Trombitás K., Centner T., Kolmerer B., Stier G., Kunke K., et al. (1998). The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 143 (4), 1013–1027. doi:10.1083/jcb.143.4.1013
Grison M., Merkel U., Kostan J., Djinović-Carugo K., Rief M. (2017). α-Actinin/titin interaction: a dynamic and mechanically stable cluster of bonds in the muscle Z-disk. Proc. Natl. Acad. Sci. U. S. A. 114 (5), 1015–1020. doi:10.1073/pnas.1612681114
Grützner A., Garcia-Manyes S., Kötter S., Badilla C. L., Fernandez J. M., Linke W. A. (2009). Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys. J. 97 (3), 825–834. doi:10.1016/j.bpj.2009.05.037
Guo W., Bharmal S. J., Esbona K., Greaser M. L. (2010). Titin diversity--alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 753675. doi:10.1155/2010/753675
Guo W., Schafer S., Greaser M. L., Radke M. H., Liss M., Govindarajan T., et al. (2012). RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18 (5), 766–773. doi:10.1038/nm.2693
Guo W., Sun M. (2018). RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys. Rev. 10 (1), 15–25. doi:10.1007/s12551-017-0267-5
Guo W., Zhu C., Yin Z., Wang Q., Sun M., Cao H., et al. (2018). Splicing factor RBM20 regulates transcriptional network of titin associated and calcium handling genes in the heart. Int. J. Biol. Sci. 14 (4), 369–380. doi:10.7150/ijbs.24117
Haggerty C. M., Damrauer S. M., Levin M. G., Birtwell D., Carey D. J., Golden A. M., et al. (2019). Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation 140 (1), 42–54. doi:10.1161/CIRCULATIONAHA.119.039573
Hahn J. K., Neupane B., Pradhan K., Zhou Q., Testa L., Pelzl L., et al. (2019). The assembly and evaluation of antisense oligonucleotides applied in exon skipping for titin-based mutations in dilated cardiomyopathy. J. Mol. Cell Cardiol. 131, 12–19. doi:10.1016/j.yjmcc.2019.04.014
Hamdani N., Bishu K. G., von Frieling-Salewsky M., Redfield M. M., Linke W. A. (2013b). Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 97 (3), 464–471. doi:10.1093/cvr/cvs353
Hamdani N., Herwig M., Linke W. A. (2017). Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys. Rev. 9 (3), 225–237. doi:10.1007/s12551-017-0263-9
Hamdani N., Krysiak J., Kreusser M. M., Neef S., Dos Remedios C. G., Maier L. S., et al. (2013a). Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ. Res. 112 (4), 664–674. doi:10.1161/CIRCRESAHA.111.300105
Hamdani N., Paulus W. J. (2013). Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation 128 (1), 5–8. doi:10.1161/CIRCULATIONAHA.113.003437
Heineke J., Ruetten H., Willenbockel C., Gross S. C., Naguib M., Schaefer A., et al. (2005). Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc. Natl. Acad. Sci. U. S. A. 102 (5), 1655–1660. doi:10.1073/pnas.0405488102
Helmes M., Trombitás K., Centner T., Kellermayer M., Labeit S., Linke W. A., et al. (1999). Mechanically driven contour-length adjustment in rat cardiac titin's unique N2B sequence: titin is an adjustable spring. Circ. Res. 84 (11), 1339–1352. doi:10.1161/01.res.84.11.1339
Herman D. S., Lam L., Taylor M. R., Wang L., Teekakirikul P., Christodoulou D., et al. (2012). Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366 (7), 619–628. doi:10.1056/NEJMoa1110186
Herwig M., Kolijn D., Lódi M., Hölper S., Kovács Á., Papp Z., et al. (2020). Modulation of titin-based stiffness in hypertrophic cardiomyopathy via protein kinase D. Front. Physiol. 11, 240. doi:10.3389/fphys.2020.00240
Hidalgo C., Hudson B., Bogomolovas J., Zhu Y., Anderson B., Greaser M., et al. (2009). PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ. Res. 105 (7), 631–638. 17 p following 8. doi:10.1161/CIRCRESAHA.109.198465
Hidalgo C. G., Chung C. S., Saripalli C., Methawasin M., Hutchinson K. R., Tsaprailis G., et al. (2013). The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIδ) phosphorylates cardiac titin's spring elements. J. Mol. Cell Cardiol. 54, 90–97. doi:10.1016/j.yjmcc.2012.11.012
Hinze F., Dieterich C., Radke M. H., Granzier H., Gotthardt M. (2016). Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J. Mol. Med. Berl. 94 (12), 1349–1358. doi:10.1007/s00109-016-1483-3
Hopf A. E., Andresen C., Kötter S., Isić M., Ulrich K., Sahin S., et al. (2018). Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can Be modulated by neuregulin-1. Circ. Res. 123 (3), 342–355. doi:10.1161/CIRCRESAHA.117.312166
Huang G., Bisaria A., Wakefield D. L., Yamawaki T. M., Luo X., Zhang J. A., et al. (2023). Titin-truncating variants in hiPSC cardiomyocytes induce pathogenic proteinopathy and sarcomere defects with preserved core contractile machinery. Stem Cell Rep. 18 (1), 220–236. doi:10.1016/j.stemcr.2022.11.008
Hudson B., Hidalgo C., Saripalli C., Granzier H. (2011). Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circ. Res. 109 (8), 858–866. doi:10.1161/CIRCRESAHA.111.246819
Hutchinson K. R., Saripalli C., Chung C. S., Granzier H. (2015). Increased myocardial stiffness due to cardiac titin isoform switching in a mouse model of volume overload limits eccentric remodeling. J. Mol. Cell Cardiol. 79, 104–114. doi:10.1016/j.yjmcc.2014.10.020
Huxley H. E. (1963). ELECTRON MICROSCOPE STUDIES ON THE STRUCTURE OF NATURAL AND SYNTHETIC PROTEIN FILAMENTS FROM STRIATED MUSCLE. J. Mol. Biol. 7, 281–308. doi:10.1016/s0022-2836(63)80008-x
Improta S., Politou A. S., Pastore A. (1996). Immunoglobulin-like modules from titin I-band: extensible components of muscle elasticity. Structure 4 (3), 323–337. doi:10.1016/s0969-2126(96)00036-6
Jia G., DeMarco V. G., Sowers J. R. (2016). Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12 (3), 144–153. doi:10.1038/nrendo.2015.216
Joseph C., Stier G., O'Brien R., Politou A. S., Atkinson R. A., Bianco A., et al. (2001). A structural characterization of the interactions between titin Z-repeats and the alpha-actinin C-terminal domain. Biochemistry 40 (16), 4957–4965. doi:10.1021/bi002739r
Kellermayer D., Tordai H., Kiss B., Török G., Péter D. M., Sayour A. A., et al. (2024). Truncated titin is structurally integrated into the human dilated cardiomyopathic sarcomere. J. Clin. Invest. 134 (2), e169753. doi:10.1172/JCI169753
Kellermayer M. S., Smith S. B., Granzier H. L., Bustamante C. (1997). Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 276 (5315), 1112–1116. doi:10.1126/science.276.5315.1112
Knöll R., Hoshijima M., Hoffman H. M., Person V., Lorenzen-Schmidt I., Bang M. L., et al. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111 (7), 943–955. doi:10.1016/s0092-8674(02)01226-6
Kolmerer B., Olivieri N., Witt C. C., Herrmann B. G., Labeit S. (1996). Genomic organization of M line titin and its tissue-specific expression in two distinct isoforms. J. Mol. Biol. 256 (3), 556–563. doi:10.1006/jmbi.1996.0108
Kong Y., Shelton J. M., Rothermel B., Li X., Richardson J. A., Bassel-Duby R., et al. (2001). Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation 103 (22), 2731–2738. doi:10.1161/01.cir.103.22.2731
Koser F., Hobbach A. J., Abdellatif M., Herbst V., Türk C., Reinecke H., et al. (2022). Acetylation and phosphorylation changes to cardiac proteins in experimental HFpEF due to metabolic risk reveal targets for treatment. Life Sci. 309, 120998. doi:10.1016/j.lfs.2022.120998
Koser F., Loescher C., Linke W. A. (2019). Posttranslational modifications of titin from cardiac muscle: how, where, and what for? FEBS J. 286 (12), 2240–2260. doi:10.1111/febs.14854
Kötter S., Andresen C., Krüger M. (2014a). Titin: central player of hypertrophic signaling and sarcomeric protein quality control. Biol. Chem. 395 (11), 1341–1352. doi:10.1515/hsz-2014-0178
Kötter S., Gout L., Von Frieling-Salewsky M., Müller A. E., Helling S., Marcus K., et al. (2013). Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res. 99 (4), 648–656. doi:10.1093/cvr/cvt144
Kötter S., Unger A., Hamdani N., Lang P., Vorgerd M., Nagel-Steger L., et al. (2014b). Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J. Cell Biol. 204 (2), 187–202. doi:10.1083/jcb.201306077
Krüger M., Kötter S., Grützner A., Lang P., Andresen C., Redfield M. M., et al. (2009). Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 104 (1), 87–94. doi:10.1161/CIRCRESAHA.108.184408
Krysiak J., Unger A., Beckendorf L., Hamdani N., von Frieling-Salewsky M., Redfield M. M., et al. (2018). Protein phosphatase 5 regulates titin phosphorylation and function at a sarcomere-associated mechanosensor complex in cardiomyocytes. Nat. Commun. 9 (1), 262. doi:10.1038/s41467-017-02483-3
Kuo H., Chen J., Ruiz-Lozano P., Zou Y., Nemer M., Chien K. R. (1999). Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 126 (19), 4223–4234. doi:10.1242/dev.126.19.4223
Labeit S., Barlow D. P., Gautel M., Gibson T., Holt J., Hsieh C. L., et al. (1990). A regular pattern of two types of 100-residue motif in the sequence of titin. Nature 345 (6272), 273–276. doi:10.1038/345273a0
Labeit S., Gautel M., Lakey A., Trinick J. (1992). Towards a molecular understanding of titin. EMBO J. 11 (5), 1711–1716. doi:10.1002/j.1460-2075.1992.tb05222.x
Labeit S., Kolmerer B. (1995). Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270 (5234), 293–296. doi:10.1126/science.270.5234.293
Labeit S., Kolmerer B., Linke W. A. (1997). The giant protein titin. Emerging roles in physiology and pathophysiology. Circ. Res. 80 (2), 290–294. doi:10.1161/01.res.80.2.290
Lange S., Auerbach D., McLoughlin P., Perriard E., Schäfer B. W., Perriard J. C., et al. (2002). Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J. Cell Sci. 115 (Pt 24), 4925–4936. doi:10.1242/jcs.00181
Lange S., Gehmlich K., Lun A. S., Blondelle J., Hooper C., Dalton N. D., et al. (2016). MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nat. Commun. 7, 12120. doi:10.1038/ncomms12120
Lange S., Xiang F., Yakovenko A., Vihola A., Hackman P., Rostkova E., et al. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science 308 (5728), 1599–1603. doi:10.1126/science.1110463
Lanzicher T., Zhou T., Saripalli C., Keschrumrus V., Smith Iii J. E., Mayans O., et al. (2020). Single-molecule force spectroscopy on the N2A element of titin: effects of phosphorylation and CARP. Front. Physiol. 11, 173. doi:10.3389/fphys.2020.00173
LeWinter M. M., Granzier H. L. (2014). Cardiac titin and heart disease. J. Cardiovasc Pharmacol. 63 (3), 207–212. doi:10.1097/FJC.0000000000000007
Li S., Guo W., Dewey C. N., Greaser M. L. (2013). Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 41 (4), 2659–2672. doi:10.1093/nar/gks1362
Lin Y. H., Major J. L., Liebner T., Hourani Z., Travers J. G., Wennersten S. A., et al. (2022). HDAC6 modulates myofibril stiffness and diastolic function of the heart. J. Clin. Invest. 132 (10), e148333. doi:10.1172/JCI148333
Linke W. A. (2008). Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 77 (4), 637–648. doi:10.1016/j.cardiores.2007.03.029
Linke W. A., Hamdani N. (2014). Gigantic business: titin properties and function through thick and thin. Circ. Res. 114 (6), 1052–1068. doi:10.1161/CIRCRESAHA.114.301286
Linke W. A., Ivemeyer M., Mundel P., Stockmeier M. R., Kolmerer B. (1998). Nature of PEVK-titin elasticity in skeletal muscle. Proc. Natl. Acad. Sci. U. S. A. 95 (14), 8052–8057. doi:10.1073/pnas.95.14.8052
Linke W. A., Ivemeyer M., Olivieri N., Kolmerer B., Rüegg J. C., Labeit S. (1996). Towards a molecular understanding of the elasticity of titin. J. Mol. Biol. 261 (1), 62–71. doi:10.1006/jmbi.1996.0441
Linke W. A., Krüger M. (2010). The giant protein titin as an integrator of myocyte signaling pathways. Physiol. (Bethesda) 25 (3), 186–198. doi:10.1152/physiol.00005.2010
Linke W. A., Kulke M., Li H., Fujita-Becker S., Neagoe C., Manstein D. J., et al. (2002). PEVK domain of titin: an entropic spring with actin-binding properties. J. Struct. Biol. 137 (1-2), 194–205. doi:10.1006/jsbi.2002.4468
Linke W. A., Popov V. I., Pollack G. H. (1994). Passive and active tension in single cardiac myofibrils. Biophys. J. 67 (2), 782–792. doi:10.1016/S0006-3495(94)80538-7
Liu J., Kong X., Zhang M., Yang X., Xu X. (2019). RNA binding protein 24 deletion disrupts global alternative splicing and causes dilated cardiomyopathy. Protein Cell 10 (6), 405–416. doi:10.1007/s13238-018-0578-8
Loescher C. M., Breitkreuz M., Li Y., Nickel A., Unger A., Dietl A., et al. (2020). Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc. Natl. Acad. Sci. U. S. A. 117 (39), 24545–24556. doi:10.1073/pnas.2004900117
Loescher C. M., Freundt J. K., Unger A., Hessel A. L., Kuhn M., Koser F., et al. (2023). Titin governs myocardial passive stiffness with major support from microtubules and actin and the extracellular matrix. Nat. Cardiovasc. Res. 2, 991–1002. doi:10.1038/s44161-023-00348-1
Loescher C. M., Hobbach A. J., Linke W. A. (2022). Titin (TTN): from molecule to modifications, mechanics, and medical significance. Cardiovasc Res. 118 (14), 2903–2918. doi:10.1093/cvr/cvab328
Lu H., Isralewitz B., Krammer A., Vogel V., Schulten K. (1998). Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 75 (2), 662–671. doi:10.1016/S0006-3495(98)77556-3
Lun A. S., Chen J., Lange S. (2014). Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat. Rec. Hob. 297 (9), 1615–1629. doi:10.1002/ar.22968
Maatz H., Jens M., Liss M., Schafer S., Heinig M., Kirchner M., et al. (2014). RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Invest. 124 (8), 3419–3430. doi:10.1172/JCI74523
Makarenko I., Opitz C. A., Leake M. C., Neagoe C., Kulke M., Gwathmey J. K., et al. (2004). Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ. Res. 95 (7), 708–716. doi:10.1161/01.RES.0000143901.37063.2f
Marcello M., Cetrangolo V., Savarese M., Udd B. (2022). Use of animal models to understand titin physiology and pathology. J. Cell Mol. Med. 26 (20), 5103–5112. doi:10.1111/jcmm.17533
Maruyama K. (1976). Connectin, an elastic protein from myofibrils. J. Biochem. 80 (2), 405–407. doi:10.1093/oxfordjournals.jbchem.a131291
Mayans O., van der Ven P. F., Wilm M., Mues A., Young P., Fürst D. O., et al. (1998). Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 395 (6705), 863–869. doi:10.1038/27603
Mazzarotto F., Tayal U., Buchan R. J., Midwinter W., Wilk A., Whiffin N., et al. (2020). Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 141 (5), 387–398. doi:10.1161/CIRCULATIONAHA.119.037661
McAfee Q., Chen C. Y., Yang Y., Caporizzo M. A., Morley M., Babu A., et al. (2021). Truncated titin proteins in dilated cardiomyopathy. Sci. Transl. Med. 13 (618), eabd7287. doi:10.1126/scitranslmed.abd7287
Merlo M., Sinagra G., Carniel E., Slavov D., Zhu X., Barbati G., et al. (2013). Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin. Transl. Sci. 6 (6), 424–428. doi:10.1111/cts.12116
Methawasin M., Strom J. G., Slater R. E., Fernandez V., Saripalli C., Granzier H. (2016). Experimentally increasing the compliance of titin through RNA binding motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 134 (15), 1085–1099. doi:10.1161/CIRCULATIONAHA.116.023003
Michel K., Herwig M., Werner F., Špiranec Spes K., Abeßer M., Schuh K., et al. (2020). C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 5 (22), e139910. doi:10.1172/jci.insight.139910
Miller M. K., Bang M. L., Witt C. C., Labeit D., Trombitas C., Watanabe K., et al. (2003). The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 333 (5), 951–964. doi:10.1016/j.jmb.2003.09.012
Müller E., Salcan S., Bongardt S., Barbosa D. M., Krüger M., Kötter S. (2021). E3-ligase knock down revealed differential titin degradation by autopagy and the ubiquitin proteasome system. Sci. Rep. 11 (1), 21134. doi:10.1038/s41598-021-00618-7
Murayama T., Nakauchi Y., Kimura S., Maruyama K. (1989). Binding of connectin to myosin filaments. J. Biochem. 105 (2), 323–326. doi:10.1093/oxfordjournals.jbchem.a122661
Nagueh S. F., Shah G., Wu Y., Torre-Amione G., King N. M., Lahmers S., et al. (2004). Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110 (2), 155–162. doi:10.1161/01.CIR.0000135591.37759.AF
Neagoe C., Kulke M., del Monte F., Gwathmey J. K., de Tombe P. P., Hajjar R. J., et al. (2002). Titin isoform switch in ischemic human heart disease. Circulation 106 (11), 1333–1341. doi:10.1161/01.cir.0000029803.93022.93
Ohtsuka H., Yajima H., Maruyama K., Kimura S. (1997). The N-terminal Z repeat 5 of connectin/titin binds to the C-terminal region of alpha-actinin. Biochem. Biophys. Res. Commun. 235 (1), 1–3. doi:10.1006/bbrc.1997.6534
Ojima K., Ono Y., Hata S., Noguchi S., Nishino I., Sorimachi H. (2014). Muscle-specific calpain-3 is phosphorylated in its unique insertion region for enrichment in a myofibril fraction. Genes cells. 19 (11), 830–841. doi:10.1111/gtc.12181
Opitz C. A., Leake M. C., Makarenko I., Benes V., Linke W. A. (2004). Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ. Res. 94 (7), 967–975. doi:10.1161/01.RES.0000124301.48193.E1
Perkin J., Slater R., Del Favero G., Lanzicher T., Hidalgo C., Anderson B., et al. (2015). Phosphorylating titin's cardiac N2B element by ERK2 or CaMKIIδ lowers the single molecule and cardiac muscle force. Biophys. J. 109 (12), 2592–2601. doi:10.1016/j.bpj.2015.11.002
Pfuhl M., Pastore A. (1995). Tertiary structure of an immunoglobulin-like domain from the giant muscle protein titin: a new member of the I set. Structure 3 (4), 391–401. doi:10.1016/s0969-2126(01)00170-8
Puchner E. M., Alexandrovich A., Kho A. L., Hensen U., Schäfer L. V., Brandmeier B., et al. (2008). Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. U. S. A. 105 (36), 13385–13390. doi:10.1073/pnas.0805034105
Pugh T. J., Kelly M. A., Gowrisankar S., Hynes E., Seidman M. A., Baxter S. M., et al. (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16 (8), 601–608. doi:10.1038/gim.2013.204
Radke M. H., Badillo-Lisakowski V., Britto-Borges T., Kubli D. A., Jüttner R., Parakkat P., et al. (2021). Therapeutic inhibition of RBM20 improves diastolic function in a murine heart failure model and human engineered heart tissue. Sci. Transl. Med. 13 (622), eabe8952. doi:10.1126/scitranslmed.abe8952
Radke M. H., Peng J., Wu Y., McNabb M., Nelson O. L., Granzier H., et al. (2007). Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc. Natl. Acad. Sci. U. S. A. 104 (9), 3444–3449. doi:10.1073/pnas.0608543104
Raskin A., Lange S., Banares K., Lyon R. C., Zieseniss A., Lee L. K., et al. (2012). A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J. Biol. Chem. 287 (35), 29273–29284. doi:10.1074/jbc.M112.372839
Rief M., Gautel M., Oesterhelt F., Fernandez J. M., Gaub H. E. (1997). Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276 (5315), 1109–1112. doi:10.1126/science.276.5315.1109
Rivas-Pardo J. A., Eckels E. C., Popa I., Kosuri P., Linke W. A., Fernández J. M. (2016). Work done by titin protein folding assists muscle contraction. Cell Rep. 14 (6), 1339–1347. doi:10.1016/j.celrep.2016.01.025
Roberts A. M., Ware J. S., Herman D. S., Schafer S., Baksi J., Bick A. G., et al. (2015). Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 7 (270), 270ra6. doi:10.1126/scitranslmed.3010134
Rodriguez-Polo I., Behr R. (2022). Exploring the potential of symmetric exon deletion to treat non-ischemic dilated cardiomyopathy by removing frameshift mutations in TTN. Genes (Basel) 13 (6), 1093. doi:10.3390/genes13061093
Romano R., Ghahremani S., Zimmerman T., Legere N., Thakar K., Ladha F. A., et al. (2022). Reading frame repair of TTN truncation variants restores titin quantity and functions. Circulation 145 (3), 194–205. doi:10.1161/CIRCULATIONAHA.120.049997
Rudolph F., Fink C., Hüttemeister J., Kirchner M., Radke M. H., Lopez Carballo J., et al. (2020). Deconstructing sarcomeric structure-function relations in titin-BioID knock-in mice. Nat. Commun. 11 (1), 3133. doi:10.1038/s41467-020-16929-8
Schafer S., de Marvao A., Adami E., Fiedler L. R., Ng B., Khin E., et al. (2017). Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 49 (1), 46–53. doi:10.1038/ng.3719
Shapiro B. P., Lam C. S., Patel J. B., Mohammed S. F., Kruger M., Meyer D. M., et al. (2007). Acute and chronic ventricular-arterial coupling in systole and diastole: insights from an elderly hypertensive model. Hypertension 50 (3), 503–511. doi:10.1161/HYPERTENSIONAHA.107.090092
Sheikh F., Raskin A., Chu P. H., Lange S., Domenighetti A. A., Zheng M., et al. (2008). An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 118 (12), 3870–3880. doi:10.1172/JCI34472
Slater R. E., Strom J. G., Methawasin M., Liss M., Gotthardt M., Sweitzer N., et al. (2019). Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J. Gen. Physiol. 151 (1), 42–52. doi:10.1085/jgp.201812259
Sorimachi H., Freiburg A., Kolmerer B., Ishiura S., Stier G., Gregorio C. C., et al. (1997). Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: implications for the nature of vertebrate Z-discs. J. Mol. Biol. 270 (5), 688–695. doi:10.1006/jmbi.1997.1145
Tadic M., Cuspidi C., Frydas A., Grassi G. (2018). The role of arterial hypertension in development heart failure with preserved ejection fraction: just a risk factor or something more? Heart Fail Rev. 23 (5), 631–639. doi:10.1007/s10741-018-9698-8
Tam M. C., Lee R., Cascino T. M., Konerman M. C., Hummel S. L. (2017). Current perspectives on systemic hypertension in heart failure with preserved ejection fraction. Curr. Hypertens. Rep. 19 (2), 12. doi:10.1007/s11906-017-0709-2
Tonino P., Kiss B., Strom J., Methawasin M., Smith J. E., Kolb J., et al. (2017). The giant protein titin regulates the length of the striated muscle thick filament. Nat. Commun. 8 (1), 1041. doi:10.1038/s41467-017-01144-9
Trombitás K., Freiburg A., Centner T., Labeit S., Granzier H. (1999). Molecular dissection of N2B cardiac titin's extensibility. Biophys. J. 77 (6), 3189–3196. doi:10.1016/S0006-3495(99)77149-3
Trombitás K., Greaser M., Labeit S., Jin J. P., Kellermayer M., Helmes M., et al. (1998). Titin extensibility in situ: entropic elasticity of permanently folded and permanently unfolded molecular segments. J. Cell Biol. 140 (4), 853–859. doi:10.1083/jcb.140.4.853
Trombitás K., Jin J. P., Granzier H. (1995). The mechanically active domain of titin in cardiac muscle. Circ. Res. 77 (4), 856–861. doi:10.1161/01.res.77.4.856
Tskhovrebova L., Trinick J. (1997). Direct visualization of extensibility in isolated titin molecules. J. Mol. Biol. 265 (2), 100–106. doi:10.1006/jmbi.1996.0717
Tskhovrebova L., Trinick J., Sleep J. A., Simmons R. M. (1997). Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature 387 (6630), 308–312. doi:10.1038/387308a0
van der Pijl R. J., Domenighetti A. A., Sheikh F., Ehler E., Ottenheijm C. A. C., Lange S. (2021). The titin N2B and N2A regions: biomechanical and metabolic signaling hubs in cross-striated muscles. Biophys. Rev. 13 (5), 653–677. doi:10.1007/s12551-021-00836-3
van Heerebeek L., Hamdani N., Handoko M. L., Falcao-Pires I., Musters R. J., Kupreishvili K., et al. (2008). Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117 (1), 43–51. doi:10.1161/CIRCULATIONAHA.107.728550
van Spaendonck-Zwarts K. Y., Posafalvi A., van den Berg M. P., Hilfiker-Kleiner D., Bollen I. A., Sliwa K., et al. (2014). Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. 35 (32), 2165–2173. doi:10.1093/eurheartj/ehu050
Vikhorev P. G., Smoktunowicz N., Munster A. B., Copeland O., Kostin S., Montgiraud C., et al. (2017). Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 7 (1), 14829. doi:10.1038/s41598-017-13675-8
Wang K., McClure J., Tu A. (1979). Titin: major myofibrillar components of striated muscle. Proc. Natl. Acad. Sci. U. S. A. 76 (8), 3698–3702. doi:10.1073/pnas.76.8.3698
Warren C. M., Jordan M. C., Roos K. P., Krzesinski P. R., Greaser M. L. (2003). Titin isoform expression in normal and hypertensive myocardium. Cardiovasc Res. 59 (1), 86–94. doi:10.1016/s0008-6363(03)00328-6
Warren C. M., Krzesinski P. R., Campbell K. S., Moss R. L., Greaser M. L. (2004). Titin isoform changes in rat myocardium during development. Mech. Dev. 121 (11), 1301–1312. doi:10.1016/j.mod.2004.07.003
Whiting A., Wardale J., Trinick J. (1989). Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 205 (1), 263–268. doi:10.1016/0022-2836(89)90381-1
Wu Y., Bell S. P., Trombitas K., Witt C. C., Labeit S., LeWinter M. M., et al. (2002). Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation 106 (11), 1384–1389. doi:10.1161/01.cir.0000029804.61510.02
Yamasaki R., Wu Y., McNabb M., Greaser M., Labeit S., Granzier H. (2002). Protein kinase A phosphorylates titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 90 (11), 1181–1188. doi:10.1161/01.res.0000021115.24712.99
Yotti R., Seidman C. E., Seidman J. G. (2019). Advances in the genetic basis and pathogenesis of sarcomere cardiomyopathies. Annu. Rev. Genomics Hum. Genet. 20, 129–153. doi:10.1146/annurev-genom-083118-015306
Zhu C., Yin Z., Tan B., Guo W. (2017). Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (9), 2363–2371. doi:10.1016/j.bbadis.2017.06.023
Zile M. R., Baicu C. F., Ikonomidis J. S., Stroud R. E., Nietert P. J., Bradshaw A. D., et al. (2015). Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 131 (14), 1247–1259. doi:10.1161/CIRCULATIONAHA.114.013215
Zou J., Tran D., Baalbaki M., Tang L. F., Poon A., Pelonero A., et al. (2015). An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of Titin in zebrafish. Elife 4, e09406. doi:10.7554/eLife.09406
Keywords: titin, passive stiffness, dilated cardiomyopathy (DCM), heart failure with preserved ejection fraction (HFpEF), diabetic cardiomyopathy (DbCM)
Citation: Stroik D, Gregorich ZR, Raza F, Ge Y and Guo W (2024) Titin: roles in cardiac function and diseases. Front. Physiol. 15:1385821. doi: 10.3389/fphys.2024.1385821
Received: 13 February 2024; Accepted: 25 March 2024;
Published: 10 April 2024.
Edited by:
Matthew J. Gage, University of Massachusetts Lowell, United StatesReviewed by:
Robbert Van Der Pijl, University of Arizona, United StatesCopyright © 2024 Stroik, Gregorich, Raza, Ge and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Guo, d2d1bzJAd2lzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.