 Qi Wang1†Jie Zhang1†Chenhao Liu1Chuanjian Ru1Qian Qian1Minghuan Yang1
Qi Wang1†Jie Zhang1†Chenhao Liu1Chuanjian Ru1Qian Qian1Minghuan Yang1 Shanchun Yan1*
Shanchun Yan1* Wei Liu2*
Wei Liu2* Guirong Wang2
Guirong Wang2- 1Key Laboratory of Sustainable Forest Ecosystem Management-Ministry of Education, Northeast Forestry University, Harbin, China
- 2Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
Alternative splicing is an essential post-transcriptional regulatory mechanism that diversifies gene function by generating multiple protein isoforms from a single gene and act as a crucial role in insect environmental adaptation. Olfaction, a key sense for insect adaptation, relies heavily on the antennae, which are the primary olfactory organs expressing most of the olfactory genes. Despite the extensive annotation of olfactory genes within insect antennal tissues facilitated by high-throughput sequencing technology advancements, systematic analyses of alternative splicing are still relatively less. In this study, we focused on the oriental fruit fly (Bactrocera dorsalis), a significant pest of fruit crops. We performed a detailed analysis of alternative splicing in its antennae by utilizing the full-length transcriptome of its antennal tissue and the insect’s genome. The results revealed 8600 non-redundant full-length transcripts identified in the oriental fruit fly antennal full-length transcriptome, spanning 4,145 gene loci. Over 40% of these loci exhibited multiple isoforms. Among these, 161 genes showed sex-biased isoform switching, involving seven different types of alternative splicing. Notably, events involving alternative transcription start sites (ATSS) and alternative transcription termination sites (ATTS) were the most common. Of all the genes undergoing ATSS and ATTS alternative splicing between male and female, 32 genes were alternatively spliced in protein coding regions, potentially affecting protein function. These genes were categorized based on the length of the sex-biased isoforms, with the highest difference in isoform fraction (dIF) associated with the ATSS type, including genes such as BdorABCA13, BdorCAT2, and BdorTSN3. Additionally, transcription factor binding sites for doublesex were identified upstream of both BdorABCA13 and BdorCAT2. Besides being expressed in the antennal tissues, BdorABCA13 and BdorCAT2 are also expressed in the mouthparts, legs, and genitalia of both female and male adults, suggesting their functional diversity. This study reveals alternative splicing events in the antennae of Bactrophora dorsalis from two aspects: odorant receptor genes and other types of genes expressed in the antennae. This study not only provides a research foundation for understanding the regulation of gene function by alternative splicing in the oriental fruit fly but also offers new insights for utilizing olfaction-based behavioral manipulation techniques to manage this pest.
1 Introduction
Alternative splicing (AS) is an essential post-transcriptional regulatory mechanism and enables the generation of structurally variable transcripts from a single gene (Verta and Jacobs, 2022). This mechanism is widespread in insects, greatly enriching the diversity of insect mRNAs and proteins. For instance, in the Drosophila melanogaster genome, the proportion of alternative splicing events is as high as 40% (Stolc et al., 2004), in which Dscam alone can generate up to 38,016 different isoforms through alternative splicing (Schmucker et al., 2000; Wojtowicz et al., 2004). Insects exhibit remarkable adaptability to their environment, and alternative splicing plays a crucial role in regulating various aspects of their adaptability. Regarding temperature adaptation, D. melanogaster timeless produces two isoforms under low temperature conditions, which regulate the circadian rhythm of D. melanogaster in that environment (Martin Anduaga et al., 2019). In the context of chemical sensing, there are at least eight different isoforms of acj6 in Drosophila, which play distinct roles in modulating odorant receptor genes (Bai and Carlson, 2010). In terms of immune response, the Dscam in Anopheles gambiae generates different isoforms through alternative splicing that can specifically bind to different pathogens, aiding in infection defense (Smith et al., 2011). Additionally, in the realm of pesticide resistance, the sodium ion channel gene in Nilaparvata lugens exhibits multiple isoforms, with significant variations in their sensitivity to neonicotinoid insecticides (Sun et al., 2020).
Sexual dimorphism arises from the long-term adaptation of insects to their environment, with alternative splicing playing a pivotal role in this adaptation. Within a single species, males and females share the majority of their genome, yet they often encounter divergent selective pressures (Andersson, 1994). These pressures can lead to significant sexual conflicts when they affect traits that have a shared genetic basis (Parker and Partridge, 1998; Bonduriansky and Chenoweth, 2009). Sex-biased alternative splicing serves as a mechanism to alleviate these conflicts, allowing for the retention of beneficial exons while splicing out harmful ones (Rogers et al., 2021). In the genomes of insects, there is a notable presence of sex-biased alternative splicing (Telonis-Scott et al., 2009; Hartmann et al., 2011; Brown et al., 2014). Isoform switching, generated by sex-biased alternative splicing events (Xing et al., 2020), plays an essential role in sex determination (Verhulst et al., 2010; Verhulst et al., 2013), development (Chang et al., 2011), and behavioral patterns (Sato et al., 2019). Therefore, the systematic understanding of sex-biased alternative splicing can hold promising prospects for pest control techniques. For instance, by manipulating genes involved in insect sex determination pathways through alternative splicing, researchers have developed female-specific release of insects carrying Dominant Lethals (fsRIDL) and precision guided Sterile Insect Technology (pgSIT) for pests control (Fu et al., 2007; Siddall et al., 2022). Additionally, targeting sex-specific alternative splicing of Actin-4 has been used to weaken the flight ability of female mosquitoes for pest management (Labbé et al., 2012).
The accuracy of alternative splicing analysis is contingent upon the capability to accurately identify transcript splice sites and their specific combinations. Eukaryotic transcriptome analysis primarily relies on next-generation RNA sequencing (RNA-Seq) (Mutz et al., 2013), which involves extensive computational assembly of short reads. While RNA-Seq is adept at identifying splice sites, deducing the precise combinations of splice-site usage presents a significant challenge (Steijger et al., 2013; Wang et al., 2016). Technologies for full-length transcriptome sequencing, such as isoform sequencing (Iso-Seq), produce longer reads that eliminate the need for assembly, thus providing direct evidence of transcript structures. This advancement substantially enhances the accuracy of predicting splice site combinations in transcripts (Au et al., 2013; Tilgner et al., 2014; Treutlein et al., 2014; Gordon et al., 2015), outperforming RNA-Seq in the analysis of alternative splicing (Wang et al., 2016). Iso-Seq technology can overcome the presence of repeated sequences in insect genomes (Sproul et al., 2023), particularly addressing the issue of low accuracy in short-read assembly caused by tandem repeats (Tørresen et al., 2019). Presently, Iso-Seq has been employed in the analysis of alternative splicing events across various insects (Jia et al., 2018; Hong et al., 2020; Ouyang et al., 2021; Xing et al., 2022; Zheng et al., 2023).
The Oriental fruit fly (Bactrocera dorsalis) represents a major pest affecting fruit trees globally, inflicting damage on over 150 different types of fruit crops (Christenson and Foote, 1960). Male lures and protein baits are considered environmentally friendly control methods for managing B. dorsalis, taking advantage of the sexual dimorphism in its chemical sensing behavior (Tan and Nishida, 2012; Pagadala Damodaram et al., 2014; Zhang et al., 2023). Currently, in B. dorsalis, odorant receptor genes are the main molecular targets of its behavioral regulators (Liu et al., 2018); in addition, studies have found that other types of genes expressed in insect chemosensory organs are also involved in the behavioral regulation of insects. For instance, takeout influences insect feeding behavior, biological rhythms, male courtship behavior and chemoreceptive behavior (Sarov-Blat et al., 2000; Dauwalder et al., 2002; Bohbot and Vogt, 2005; Meunier et al., 2007); P450 can serve as an odor-degrading enzyme and participate in the host plant recognition in Locusta migratoria (Wu et al., 2022); CncC can regulate the expression of odor-degrading enzymes in L. migratoria antennae (Wu et al., 2023). The antennae serve as the primary chemical sensory organs in insects, playing a key role in tactile sensing, olfaction, and taste (Lv et al., 2020). Thus, accurately and effectively analyzing alternative splicing events in the antennae is crucial for understanding the behavioral differences between males and females and for developing novel behavioral manipulation techniques for B. dorsalis. However, obtaining full-length transcriptome data from antennal tissues remains challenging due to the high RNA library requirements (>300 ng) for full-length transcriptome sequencing (Pacific Biosciences, 2023). Currently, most studies aiming to acquire full-length transcriptome data rely on pooling multiple samples or using tissues with larger volumes to gather sufficient RNA (Shields et al., 2021; Li et al., 2022). Despite these efforts, antennae, which have relatively lower RNA content, still lack comprehensive full-length transcriptome data specifically needed for research on behavioral molecular targets in B. dorsalis.
This study used the antennal full-length transcriptome and whole genome of B. dorsalis to systematically analyze sex-biased alternative splicing within its antennae. Initially, isoforms and gene loci in the B. dorsalis antennal full-length transcriptome were identified and quantified. Gene loci containing more than one isoform were then selected for further analysis. Subsequently, sex-biased alternative splicing events within these gene loci were annotated. Among the two most prevalent alternative splicing patterns, genes undergoing isoform switching, which exhibit differential protein-coding regions between female-biased and male-biased isoforms, were identified. These genes were categorized based on the length of the sex-biased isoforms. Additionally, within each category, isoform-switching genes that have the strongest sex preference and also have binding sites for transcription factors related to sexual differentiation in their upstream regulatory regions were further screened. Finally, the expression patterns of the screened genes and their conservation across closely related species were analyzed. These findings can provide a research foundation for understanding of functional differences between sexes in B. dorsalis genes regulated by alternative splicing and offer new insights for utilizing olfactory-guided behavioral control techniques in managing B. dorsalis.
2 Materials and methods
2.1 Insect rearing and sample collection
The population of B. dorsalis for this experiment was reared at the Shenzhen Institute of Genomics, Chinese Academy of Agricultural Sciences. The rearing conditions were 26 ± 1°C, 60%–70% relative humidity and a 14 L: 10 D (light: dark) cycle (darkness started at 20:00). The larvae were fed an artificial diet (Ren et al., 2021; Ren et al., 2023). Mature larvae were gathered and placed into small plastic jars filled with moist sand to allow for pupation. The pupae were then collected and transferred to Petri dishes until the adults emerged. Upon emergence, adults were separated by sex; the adult diet consisted of a 1:1 mixture of yeast and sugar in dry powder form, and a separate water container was provided. The female and male adults were reared until sexual maturity (12 days old) and then 500 individuals of each sex were collected for subsequent antennal tissue collection. The antennae were collected separately from both female and male B. dorsalis. In total, 500 pairs of female and male B. dorsalis antennae were collected, immediately frozen in liquid nitrogen, and stored at −80°C for ISO-Seq analysis (Supplementary Figure S1A).
2.2 Identification of full-length transcripts in B.dorsalis antennal full-length transcriptome and comparison with B.dorsalis antennal RNA-Seq transcripts
2.2.1 Identification of non-redundant full-length transcripts in B.dorsalis antennal full-length transcriptome
The Iso-Seq library was prepared according to the Isoform Sequencing protocol (Iso-Seq) using the Clontech SMARTer PCR cDNA Synthesis Kit and the BluePippin Size Selection System protocol as described by Pacific Biosciences (PN 100-092-800-03). Sequence data were processed using the SMRTlink 6.0 software (https://www.pacb.com/support/software-downloads/). Circular consensus sequence (CCS) was generated from subread BAM files by CCS software, and the minimum quality was set to 0.9 (-min-rq 0.9). CCS. bam files were output, which were then classified into full length and non-full length reads using Lima, removing polyA using Isoseq3 refine. The Isoseq3 cluster program was used to obtain high-quality full-length consensus sequences. The Gmap version 2021-08-25 (Wu and Watanabe, 2005) was used to align the high-quality full-length consensus sequences to previously available B. dorsalis reference genome (National Bioinformatics Center GSA database, PRJCA020830) (parameters: -f samse -n 0 -B 5). The ToFu software in cDNA-Cupcake v29.0.0 (https://github.com/Magdoll/cDNA_Cupcake) was used to collapse redundant transcripts and merge female and male antennal full-Length transcriptome, obtaining non-redundant full-length transcripts of B. dorsalis antennae (Supplementary Figure S1B).
2.2.2 Quality control and classification of non-redundant full-length transcripts in B.dorsalis antennal full-length transcriptome
Using Sqanti3 v3-5.1 (Tardaguila et al., 2018), combined with the previously available B. dorsalis reference genome and high-quality RNA-Seq data of B. dorsalis antennae, the B. dorsalis antennal non-redundant full-length transcripts were subjected to quality control, classification, and removal of potential artifacts (parameters: all_canonical: canonical, RTS_stage: FALSE, min_cov: 3), obtaining final full-length non-redundant transcript sequences and annotation files supported by B. dorsalis genome and B. dorsalis antennal RNA-Seq reads.
2.2.3 Assembly of RNA-Seq reads of B.dorsalis antennae
Using Hisat2 v2.2.1 (Kim et al., 2019) default parameters, the high-quality RNA-Seq reads of B. dorsalis antennae were aligned to B. dorsalis reference genome, and the transcripts were assembled using Stringtie v2.2.1 (Pertea et al., 2015) (parameters: -rf -e) and Taco v0.6.2 (Niknafs et al., 2017) default parameters.
2.2.4 Comparison of the length, GC content and splicing sites of non-redundant full-length transcripts and RNA-Seq transcripts in the antennae of B.dorsalis
According to the annotation files of full-length transcriptome and RNA-Seq transcriptome of B. dorsalis antennae, the gene loci detected by both transcriptome were screened using Gffread (Pertea and Pertea, 2020). The length and GC content of full-length transcripts and RNA-Seq transcripts in these gene loci were counted separately using Seqkit (Shen et al., 2016). The splicing sites of full-length transcripts and RNA-Seq transcripts in these gene loci were compared using Gffcompare (Pertea and Pertea, 2020).
2.3 Identification, functional enrichment analysis of multi-isoform genes in B.dorsalis antennal full-length transcriptome
2.3.1 Functional annotation of genes identified in B.dorsalis antennal full-length transcriptome
Blastp v2.12.0 was used to compare all genes identified in B. dorsalis antennal full-length transcriptome with the NR library and the best alignment results of each gene were extracted. Interproscan v1.8.0 (Quevillon et al., 2005) (parameters: -dp -goterms -pa) was used to perform GO annotation on these genes; the KEGG website (https://www.genome.jp/kaas-bin/kaas_main) was used to perform KEGG annotation on these genes.
2.3.2 Screening and functional enrichment analysis of mult-isoform genes in B.dorsalis antennal full-length transcriptome
Mult-isoform gene IDs were obtained based on the annotation file of the full-length transcriptome of the B. dorsalis antennae. Using the R package ClusterProfiler (Wu et al., 2021), all genes identified in the full-length transcriptome of B. dorsalis antennae were used as background to perform GO and KEGG functional enrichment analysis on multi-isoform genes. In the KEGG enrichment analysis, pathways with p. adjust <0.05 and qvalue <0.2 were defined as significantly enriched; in the GO enrichment analysis, GO terms with p. adjust <0.001 and qvalue <0.2 were defined as significantly enriched.
2.4 Identification of sex-biased alternative splicing events and isoform-switching genes in B.dorsalis antennal full-length transcriptome
2.4.1 Quantification of non-redundant full-length transcripts in B.dorsalis antennal full-length transcriptome
Using the annotation files of B. dorsalis antennal full-length transcriptome and the high-quality RNA-Seq data of B. dorsalis antennae already available in the laboratory, we used Salmon v1.10.0 (Patro et al., 2017) quasi-mapping-based mode to quantify transcripts in B. dorsalis antennal full-length transcriptome.
2.4.2 Identification of sex-biased alternative splicing and isoform-switching genes in B.dorsalis antennal full-length transcriptome
The R package IsoformSwitchAnalyzeR (Vitting-Seerup and Sandelin, 2019) was used to perform isoform switch analysis of multi-isoform genes identified in the full-length transcriptome of B. dorsalis antennae. The input to this package is a quantitative file from salmon, a full-length transcripts nucleotide sequence file, a full-length transcriptome annotation file, and a grouping file containing sample IDs and corresponding sexes. IsoformSwitchAnalyzeR evaluates isoform usage through isoform fraction (IF), which is the ratio of the expression level of a specific isoform to the expression level of its parent gene (isoform_expression/gene_expression). Differential isoform usage (DIU) is quantified as the difference in isoform fraction (dIF), calculated as IF_Female - IF_Male, to measure effect size. The IsoformSwitchTestDEXSeq () function was used to identify DIUs between sexes based on the dIF threshold. Isoforms that meet the criteria of |dIF| > 0.1, FDR <0.05, and TPM mean values greater than one in both females and males were defined as significantly sex-biased isoforms. If a gene contains at least two sex-biased isoforms with opposite effect sizes, it is considered to have undergone sex-biased isoform switching. The analyzeORF() function was used to obtain the longest open reading frame (ORF) of sex-biased isoforms, and the extractSequence () function was used to extract the nucleotide and amino acid sequences of the longest ORF of sex-biased isoforms. Based on the ORF nucleotide and amino acid sequences of sex-biased isoforms, CPC2 (http://cpc2.gao-lab.org/) was used to predict the coding potential of sex-biased isoforms, and the Pfam and SignalP databases in Interpro (https://www.ebi.ac.uk/interpro/search/sequence/) were used to predict the protein domains and signal peptides of sex-biased isoforms. IUPred3 (https://iupred.elte.hu/) was used to predict the intrinsically disordered regions (IDRs) of sex-biased splicing isoforms. The analyzeCPC2(), analyzeSignalP (), analyzePFAM(), and analyzeIUPred2A () functions were used to integrate the prediction results. If all sex-biased isoforms in an isoform switching gene have TPM mean values greater than 20 in at least one sex and there are differences in the protein coding regions between male-biased and female-biased isoforms, they were considered key isoform switching genes for subsequent analysis. The R package clusterProfiler was used to perform GO enrichment analysis on key isoform switching genes, and GO terms with p. adjust <0.05 and qvalue <0.2 were defined as significantly enriched.
2.5 The expression regulation prediction, expression pattern analysis and closely related species comparison of BdorABCA13 and BdorCAT2
2.5.1 Prediction of upstream transcription factor binding sites of BdorABCA13, BdorCAT2 and BdorTSN3
For the genes BdorABCA13, BdorCAT2, and BdorTSN3 identified within the Bactrocera dorsalis antennal full-length transcriptome, the transcription start site corresponding to the longest isoform was selected. The sequence from 5000bp upstream of the transcription start site to the start codon of the same isoform’s ORF was selected from the B. dorsalis reference genome sequence file as the candidate upstream regulatory region. The core transcription factor binding sites for Drosophila melanogaster, comprising 160 entries, were downloaded from the Jaspar database (https://jaspar2022.genereg.net/). PWMscan (Ambrosini et al., 2018) was used to align the D. melanogaster transcription factor binding sites to the candidate upstream regulatory region of each gene (p-value Cut-off = 1e-5). The amino acid sequence of the D. melanogaster transcription factors corresponding to the aligned transcription factor binding sites were found in the Uniprot database (https://www.uniprot.org/). Orthofinder v2.5.4 (Emms and Kelly, 2019) default parameters and Reciprocal Best Hit BLAST V.2 process (https://www.protocols.io/view/reciprocal-best-hit-blast-x54v9rezv3eq/) were used to find the homologous genes of the above-mentioned D. melanogaster transcription factors in the B. dorsalis reference genome. The genes with TPM >20 in female or male antennae were filtered as candidate transcription factor genes.
2.5.2 Differential expression gene analysis of upstream transcription factor genes of BdorABCA13, BdorCAT2 and BdorTSN3 between sexes
According to the alignment results of B. dorsalis antennal RNA-Seq reads to B. dorsalis reference genome, combined with the annotation file of the B. dorsalis reference genome, FeatureCounts v2.0.1 (Liao et al., 2014) (parameters: -p -t CDS -g gene_id) was used to quantify the annotated genes in B. dorsalis reference genome, and only genes with TPM >0 were retained. The R package DESeq2 (Love et al., 2014) was used to perform differential expression gene analysis between female and male antennae, and genes that met the condition |log2FoldChange| > 0.58, Benjamini–Hochberg (BH) adjusted p < 0.05 were considered significantly differentially expressed genes between female and male antennae. The expression changes of the upstream transcription factor genes of BdorABCA13, BdorCAT2 and BdorTSN3 between female and male antennae of B. dorsalis were extracted from the differential expression gene analysis results.
2.5.3 Analysis of the expression pattern of BdorABCA13 and BdorCAT2 in peripheral nervous tissues of B.dorsalis
The laboratory has previously obtained the RNA-Seq data of the peripheral nervous tissues of B. dorsalis, including the antennae, mouthparts, frontlegs, midlegs, hindlegs and genitalia of the female and male adult flies. Using Hisat2 v2.2.1 default parameters, the RNA-Seq reads of the peripheral nervous tissues of B. dorsalis were aligned to B. dorsalis reference genome, and the genes annotated in B. dorsalis reference genome were quantified using FeatureCounts v2.0.1 (parameters: -p -t CDS -g gene_id), and the raw counts and TPM values were calculated. The expression of BdorABCA13 and BdorCAT2 was extracted from the TPM expression matrix of the peripheral nervous tissues of B. dorsalis. The R package DESeq2 was used to perform differential expression gene analysis between sexes in the adult antenna, mouthparts, frontleg, midleg, hindleg and genitalia, and genes that met the condition |log2FoldChange| > 0.58, Benjamini–Hochberg (BH) adjusted p < 0.05 were considered significantly differentially expressed genes between female and male. The expression changes of BdorABCA13, BdorCAT2 and BdorTSN3 between female and male of B. dorsalis peripheral nervous tissues were extracted from the differential expression gene analysis results.
2.5.4 Phylogenetic tree construction of ABCA13 and CAT2
The protein sequences of ABCA13 and CAT2 from Anastrepha ludens, A. obliqua, B. cucurbitae, B. latifrons, B. neohumeralis, B. oleae, B. tryoni, Ceratitis capitata, Rhagoletis pomonella, R. zephyria and D. melanogaster were collected from the NCBI database (https://www.ncbi.nlm.nih.gov/). These sequences were aligned using MAFFT v7.490 (Katoh et al., 2002) with the default parameters for multiple sequence alignment. Following the alignment, the maximum likelihood phylogenetic trees for ABCA13 and CAT2 were constructed separately using IQ-TREE v2.2.0.3 (Nguyen et al., 2015) with the parameters: -bb 1000 -nt 20 -m MFP --bnni.
2.5.5 Reverse Transcription PCR (RT-PCR) validation of BdorABCA13 and BdorCAT2 isoforms
60 male adults and 60 female adults of B. dorsalis were selected, their antennae were collected between 9 a.m. and 11 a.m. (mixed samples of males and females), and the collected antennae were quickly frozen in liquid nitrogen. Total RNA was extracted from collected antennae using TriZol (Invitrogen, Carlsbad, CA, United States). Using approximately 1 µg of total RNA as a template, template cDNA of RT-PCR was synthesized using HiScript® III first Strand cDNA Synthesis (+gDNAwiper) Kit (Vazyme, Nanjing, China). Adjust the concentration of template cDNA to about 500ng/ul and store at −20°C. According to transcriptome analysis results, both BdorABCA13 and BdorCAT2 have two isoforms, and there are distinct differences in the 5′regions between different isoforms of the same gene. However, the Open Reading Frame (ORF) and 3′ Untranslated Region (3′ UTR) sequences and structures exhibit high similarity (Figures 4B, C). To design specific primers, in each isoform, we set the forward primer in the 5′UTR and the reverse primer in the ORF, and verified the existence of alternative splicing by detecting the 5′region of the isoform with obvious differences (Figure 5A; Supplementary Table S4). The specific primers for 5’ region of each isoform were used for amplification under the following conditions: 98°C for 3 min; followed by 35 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 15 s, and extension at 72°C for 1 min; with a final extension step at 72°C for 10 min; the samples were then held at 12°C. The PCR products of each isoform were then cloned into blunt-end vectors, and 24 single-clonal colonies were screened from each product to identify whether the PCR cloned sequence is consistent with the sequence obtained from transcriptome analysis.
2.5.6 Quantitative Real-Time PCR (qRT-PCR) validation of BdorABCA13 and BdorCAT2 isoform switch in B.dorsalis antennae
60 male adults and 60 female adults of B. dorsalis were selected, their antennae were collected between 9 a.m. and 11 a.m. (separated samples of males and females). and the collected antennae were quickly frozen in liquid nitrogen. Total RNA was extracted from collected antennae using TriZol (Invitrogen, Carlsbad, CA, United States). cDNA was synthesized using about 1 µg of total RNA as a template and HiScript® III first Strand cDNA Synthesis (+gDNAwiper) Kit (Vazyme, Nanjing, China) as a qRT-PCR template, and stored at −20°C (Supplementary Figure S5A). Based on the transcript sequences obtained from transcriptome analysis, NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) was used to design specific primers, and BdorActin2 were used as endogenous control (Shen et al., 2010) (Supplementary Table S5). The PCR volume was 10 μL, including 5 μL 12× Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China), 0.5 μL of each primer (10 μM), 1 μL cDNA, and 4 μL RNase-free water. PCR was performed on a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA). Before formal experiments, dissociation curve analysis was performed to ensure the specificity of amplification (Supplementary Figure S5B; Supplementary Table S6). Relative gene expression levels for each sample were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001). A total of three biological replicates were performed. Statistical analysis of qPCR results was performed using Graph Pad Prism (Version 8.0.1). All replicate data results are expressed as mean ± standard error. The original data were first tested for normality using the Shapiro-Wilk test, and non-normally distributed data were transformed using log (x+1). Unpaired t-test was used to compare the expression levels of BdorABCA13 and BdorCAT2 isoforms in the male and female antennae of B. dorsalis. The difference is significant when <0.05.
3 Results
3.1 Identification of full-length transcripts in B.dorsalis antennal full-length transcriptomes and comparison with RNA-Seq transcripts
Isoform sequencing was performed using RNA obtained from the male and female antennae of B. dorsalis, and the original subreads obtained were 18,374,257 and 12,437,865, respectively, each with a length exceeding 2500 bp. These reads were then processed using Isoseq3 workflow, involving steps such as adapter removal, classification, and clustering, resulting in 10,218 and 6,893 full-length (FL) reads, respectively (Supplementary Table S1). The FL reads were then aligned to the B. dorsalis reference genome using Gmap. Redundancy was further removed using Cupcake ToFU, and artifacts were eliminated using SQANTI3. In total, 8,600 non-redundant full-length transcripts were obtained, covering 4,145 gene loci (Supplementary Table S7). These transcripts can be categorized into eight types, ranked from highest to lowest proportion: Novel isoform with at least one new splicing site (NNC), Matches all splicing sites perfectly (FSM), Novel isoform with a new combination of known splice sites (NIC), Matches the reference splicing sites partially (ISM), Novel isoform in the intergenic region (Intergenic), Fusion, Overlaps introns and exons (Genic), and Novel isoform antisense to an annotated gene (Antisense) (Figures 1A, B). Additionally, a comparison between the ISO-Seq transcriptome and the RNA-Seq transcriptome for shared gene loci revealed that the ISO-Seq transcriptome had a peak transcript length around 2–3 kb, which is longer than the 1–2 kb observed in the RNA-Seq transcriptome (Figure 1C). Regarding GC content, ISO-Seq transcriptome had a peak value of 0.4, slightly lower than the 0.43-0.47 observed in RNA-Seq transcriptome (Figure 1C). In terms of splice junction structures, ISO-Seq transcriptome provided more accurate combinations of splice sites, with only 24.8% of ISO-Seq transcriptome junctions fully reconstructed by RNA-Seq reads, an additional 29.3% were partially reconstructed, and over 40% of the combinations remained unreconstructed (Figure 1D). In summary, utilizing antennal ISO-Seq transcriptome in conjunction with the genome enables effective discovery of novel alternative splicing events and significantly enhances the quality of identified transcripts.
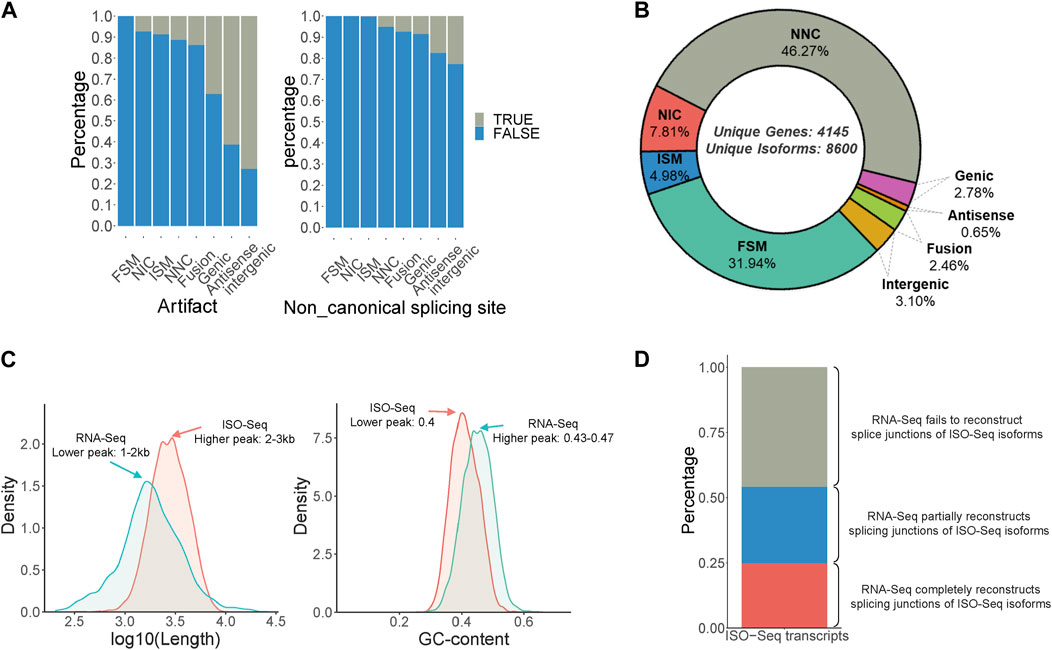
Figure 1. Identification of full-Length transcripts in B. dorsalis antennal full-length transcriptome. (A) Quality control of non-redundant full-length transcripts in B. dorsalis antennal full-length transcriptome. Quality control metrics include artifact proportion and non-canonical splicing junction proportion. (B) Classification of non-redundant full-length transcripts and the proportion of each category. FSM: Matches all SJ perfectly. ISM: Matches the reference SJs partially. NIC: Novel isoform with a new combination of known splice sites. NNC: Novel isoform with at least a new splicing site. Antisense: Novel isoform is anti-sense to an annotated gene. Genic: Overlaps introns and exons. Intergenic: Novel isoform is in the intergenic region. (C) The distribution of length and GC content of ISO-Seq transcripts and RNA-Seq transcripts among the gene loci detected by both ISO-Seq transcriptome and RNA-Seq transcriptome of B. dorsalis antennae. (D) The proportion of ISO-Seq transcripts that can be reconstructed by RNA-Seq transcripts in gene loci detected by both RNA-Seq transcripts and ISO-Seq transcripts. RNA-Seq completely reconstructs splicing junctions of ISO-Seq isoforms means that the number of exons and splicing junctions of transcripts reconstructed by RNA-Seq reads are completely consistent with ISO-Seq transcripts; RNA-Seq partially reconstructs splicing junctions of ISO-Seq isoforms means that the number of exons of transcripts reconstructed by RNA-Seq reads is less than that of ISO-Seq transcripts, but the splicing junctions are consistent with ISO-Seq transcripts; RNA-Seq fails to reconstruct splice junctions of ISO-Seq isoforms means that the splicing junctions of transcripts reconstructed by RNA-Seq reads are inconsistent with ISO-Seq transcripts.
3.2 Identification and functional annotation of multi-isoform genes, and analysis of alternative splicing in odorant receptor genes within the B.dorsalis antennal full-length transcriptome
Within the context of the full-length transcriptome of B. dorsalis antennae, we screened for genes with multiple isoforms and identified a total of 1,704 genes, which account for 41.1% of all gene loci. Remarkably, some single genes were found to contain up to 45 isoforms. Among these genes, the highest proportion consists of single genes containing two to three isoforms, representing 29.3% of all gene loci (Figure 2A). To further understand the potential role of these multi-isoform genes in the physiological processes of B. dorsalis, we performed functional annotation on the genes identified from the antennal full-length transcriptome through GO enrichment analysis (with a significance level of p < 0.001) and KEGG enrichment analysis (with a significance level of p < 0.05) specifically for the multi-isoform genes (Supplementary Figure S2A). The GO enrichment results revealed that multi-isoform genes significantly enriched in 37 GO terms. Among these, 29 were related to biological processes, with protein phosphorylation being the most prominent. Additionally, six terms were associated with cellular components, with the plasma membrane region being the most enriched. Two terms were related to molecular functions, with the most significant enrichment in cytoskeletal protein binding (see Figure 2B). Furthermore, the KEGG enrichment analysis demonstrated that multi-isoform genes significantly enriched in 14 KEGG pathways, including pathways related to neuronal functions, such as dopaminergic synapse, cholinergic synapse, and calcium signaling pathway (Figure 2C). Multi-isoform genes obtained from B. dorsalis antennal full-length transcriptome were quantified using B. dorsalis antennal RNA-Seq reads, and alternative splicing analysis were performed on these genes using exon-based method. In total, we annotated 1,602 genes that exhibit alternative splicing events, covering seven types of such events: alternative 3′ end (A3), alternative 5’ end (A5), alternative transcription start sites (ATSS), alternative transcription termination site (ATTS), exon skipping (ES), intron retention (IR), and mutually exclusive exons (MES) (Supplementary Table S8). Regarding odorant receptor (OR) genes, we identified two OR genes (BdorOR63a.1 and BdorOR10a and the odorant co-receptor (ORCO) gene BdorORCO have multiple isoforms. BdorOR63a.1 was identified with three isoforms, one of which underwent RI and A5 alternative splicing events without altering the protein coding regions (Supplementary Figure S3A). BdorOR10a was identified with three isoforms, and two of these isoforms exhibited RI and A5 alternative splicing events compared to the constitutive splicing transcript (Supplementary Figure S3B). BdorORCO was identified with nine isoforms, revealing A3, A5, RI, and SE alternative splicing events, resulting in three distinct protein coding regions (Supplementary Figure S3C).
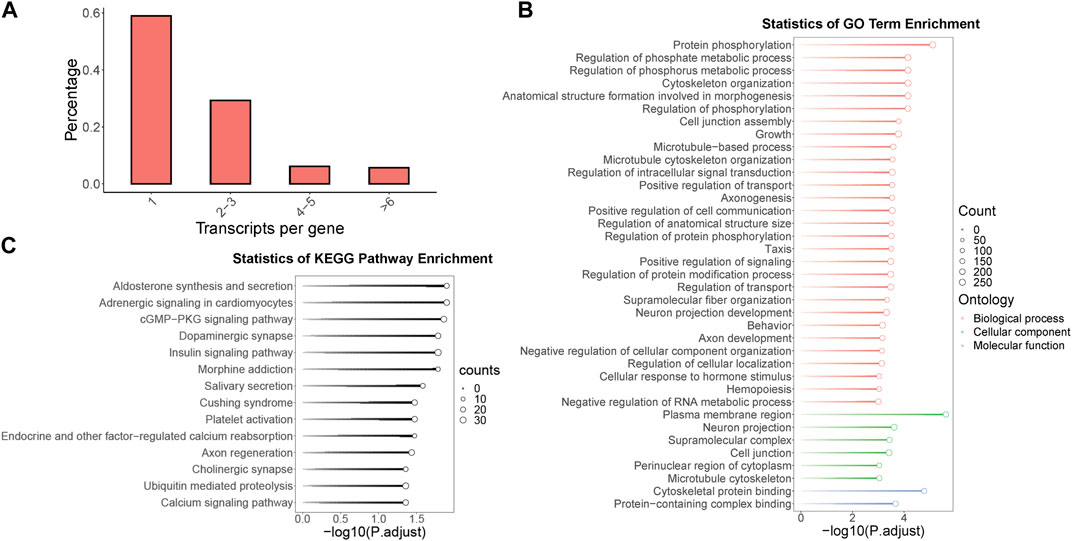
Figure 2. The identification, functional annotation of multi-isoform genes in B. dorsalis antennal full-length transcriptome. (A) The distribution of isoform numbers in gene loci identified by the full-length transcriptome of B. dorsalis antennae. (B) KEGG enrichment analysis of multi-isoform genes. (C) GO enrichment analysis of multi-isoform genes.
3.3 Identification of sex-biased alternative splicing and isoform-switching genes in B.dorsalis antennal full-length transcriptome
Sex-biased alternative splicing was screened based on the differences in isoform splicing sites and expression patterns between males and females. A total of 775 sex-biased alternative splicing events were annotated, of which 506 produced isoform switches, covering 161 gene loci. These isoform switches were attributed to seven types of alternative splicing: A3, A5, ATSS, ATTS, ES, IR, and MES (Supplementary Table S9). Notably, ATSS and ATTS were the most prevalent, accounting for 31% and 33% of all sex-biased alternative splicing events, respectively, with ATSS being more common in females and ATTS more common in males (Figure 3A). Among the 161 genes mentioned above, we further screened genes with high expression of sex-biased alternative splicing transcripts (TPM>20) and sex-biased ATSS or ATTS alternative splicing occurring in the protein coding region as key isoform-switching genes for subsequent analysis, and finally obtained 32 key genes (Supplementary Table S9). To determine the potential functions of key isoform-switching genes, we performed GO enrichment analysis on these genes. The results showed that the five significantly enriched GO terms were all related to transporter activity (Figure 3B). According to the length of sex-biased isoforms, we divided the above 32 genes into four groups: male prefer shorter isoform, male prefer longer isoform, female prefer shorter isoform, and female prefer longer isoform. We ranked the male-biased and female-biased isoforms among the 32 genes by descending dIF value and identified the genes with the strongest sex-biased isoform in each of the four categories. We found that the genes with the strongest sex bias in these categories all involved ATSS alternative splicing. The gene with the strongest male-biased isoform in the case of male prefer shorter isoform was ATP-binding cassette sub-family A member 13, BdorABCA13, and the gene with the strongest male-biased isoform in the case of male prefer longer isoform was Cationic amino acid transporter 2, BdorCAT2 (Figure 3C); the gene with the strongest female-biased isoform in the case of female prefer shorter isoform was Cationic amino acid transporter 2, BdorCAT2, and the gene with the strongest female-biased isoform in the case of female prefer longer isoform was Tension-3, BdorTSN3 (Figure 3D).
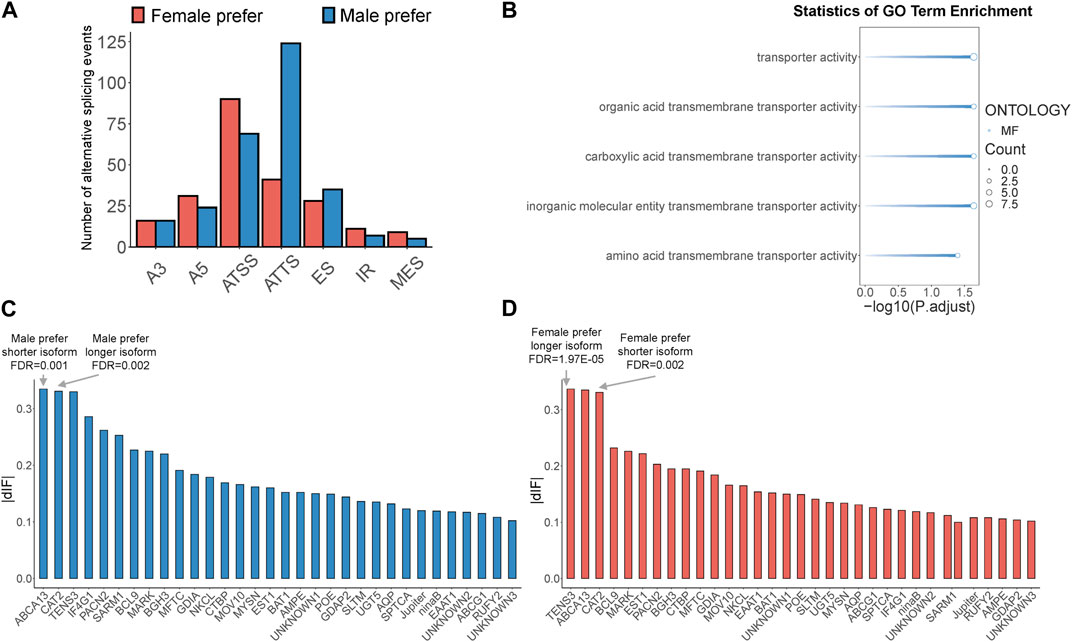
Figure 3. Identification of sex-biased alternative splicing and isoform-switching genes in B. dorsalis antennal full-length transcriptome. (A) The distribution of sex-biased splicing events in isoform-switching genes. A3: Alternative 3′ acceptor site. A5: Alternative 5′ donor site. ATSS: Alternative Transcription Start Site. ATTS: Alternative Transcription Termination Site. ES: Exon Skipping. IR: Intron Retention. MES: Multiple Exon Skipping. (B) GO enrichment analysis of key isoform-switching genes. (C) The distribution of dIF values of male-biased isoforms in key isoform-switching genes. (D) The distribution of dIF values of female-biased isoforms in key isoform-switching genes.
3.4 Prediction of expression regulation, analysis of expression patterns, and comparison with closely related species for BdorABCA13 and BdorCAT2
To further elucidate the relationship between the identified key isoform-switching genes and regulation related to sexual differentiation, we predicted transcription factors in the upstream regulatory regions of BdorABCA13, BdorCAT2, and BdorTSN3. The transcription start site corresponding to the longest isoform, as derived from the B. dorsalis antennal full-length transcriptome for each gene, was selected. The sequence from 5000 bp upstream of the transcription start site to the start codon of the same isoform’s ORF was extracted from the B. dorsalis reference genome sequence file as the candidate upstream regulatory region. Using the transcription factor binding sites information of D. melanogaster in the public database, the analysis was performed according to the workflow shown in the figure (Supplementary Figure S1C). A total of 17 transcription factor binding sites were identified (Supplementary Table S3), among which only doublesex is associated with sex-specific differentiation, exhibiting the highest expression level among the three sex-biased transcription factors (padj = 3.71E-15, logFC = 0.595) (Figure 4A; Supplementary Table S10). The doublesex binding site was found exclusively in the upstream regions of BdorABCA13 and BdorCAT2, suggesting that the sex-biased alternative splicing of these two genes might be regulated by doublesex. The upstream doublesex binding site of BdorABCA13 is located at 336bp-347bp upstream of the start codon of the longest isoform, and is located in the intron of the 5′UTR (Figure 4B). The upstream doublesex binding site of BdorCAT2 is located at 248bp-259bp upstream of the transcription start site of the longest isoform (Figure 4C). To validate the alternative splicing and isoform switches of BdorABCA13 and BdorCAT2, we confirmed the presence of their isoform structures using RT-PCR. Additionally, we quantified the expression of isoforms of these two genes in the female and male antennae of B. dorsalis using qRT-PCR. Given the high similarity between ORF regions and 3′ UTR regions of the isoforms of these two genes, we selectively amplified the 5′ end fragment with noticeable differences. The results revealed that all isoforms of BdorABCA13 and BdorCAT2 exist and exhibit significant differences between males and females, consistent with the findings from the transcriptome analysis (Figures 5A, B). BdorABCA13 was expressed in all peripheral nervous tissues, and had relatively high expression level in the antennae, forelegs and hindlegs; in the genitalia, BdorABCA13 had significantly higher expression in male than in female (log2FoldChange = 1.77, P. adj = 5.55E-34), and no significant sex difference in other peripheral nervous tissues (Figure 4D; Supplementary Table S10). BdorCAT2 was expressed in all peripheral nervous tissues, and had the highest expression level in the antennae; in the mouthparts, BdorCAT2 had significantly higher expression in female than in male (log2FoldChange = −1.79, P. adj = 5.79E-27), and no significant sex difference in other tissues (Figure 4E; Supplementary Table S10). Therefore, we speculate that BdorABCA13 and BdorCAT2 can play roles in multiple peripheral nervous tissues. To verify whether the alternative splicing of these two genes is conserved among the Tephritidae species, we collected the protein sequence data of the above two genes from 11 Diptera species (10 Tephritidae species and D. melanogaster) from the NCBI database (Supplementary Table S2), and constructed phylogenetic trees respectively. The results showed that except for R. zephyria, CAT2 of other Tephritidae species and D. melanogaster had two isoforms, and the splicing type was ATSS (Figure 4F; Supplementary Figure S4A); for ABCA13, D. melanogaster had four isoforms; in Tephritidae species, only B. dorsalis and B. correcta had two isoforms of ABCA13, and the alternative splicing type was ATSS, while other Tephritidae species had only one isoform (Figure 4G; Supplementary Figure S4B).
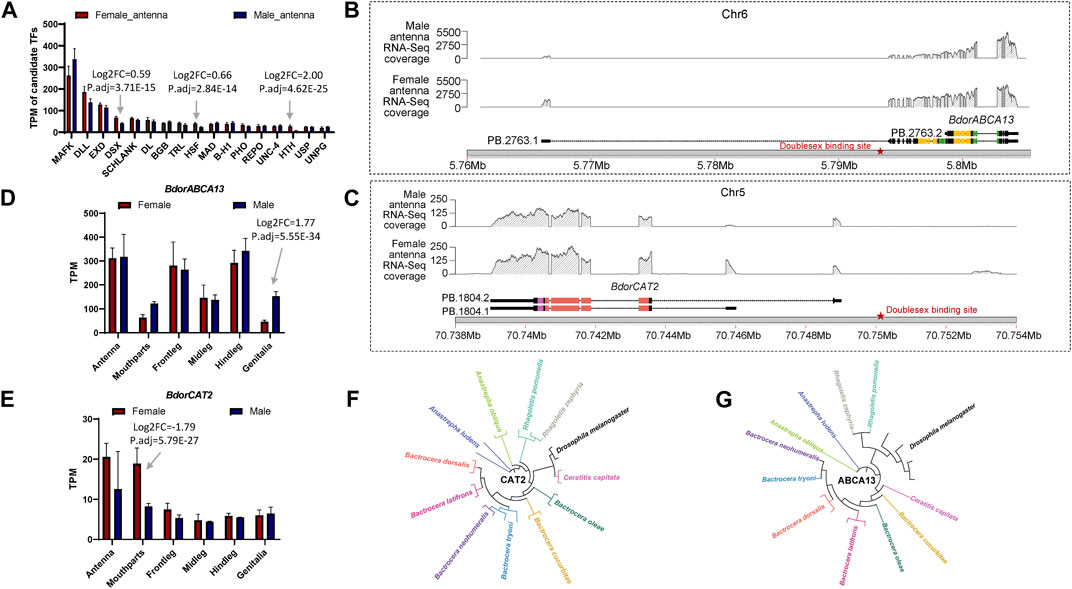
Figure 4. The expression regulation prediction, expression pattern analysis and closely related species comparison of BdorABCA13 and BdorCAT2. (A) Statistics of the TPM values of the transcription factors that bind to the upstream regulatory regions of BdorABCA13 and BdorCAT2. DSX, HSF and HTH have significant differences in expression between female and male. (B) Isoform structure, RNA-Seq coverage and location on B. dorsalis reference genome of the two isoforms of BdorABCA13. Orange represents the ABC2-membrane-3 domain. Green represents the ABC-tran-domain. Red stars indicate the predicted doublesex transcription factor binding sites. (C) Isoform structure, RNA-Seq coverage and location on B. dorsalis reference genome of the two isoforms of BdorCAT2. Purple represents the AA-permease-C domain. Red represents the AA-permease-2 domain. Red stars indicate the predicted doublesex transcription factor binding sites. (D) The expression pattern of BdorABCA13 in B. dorsalis peripheral nervous tissues. (E) The expression pattern of BdorCAT2 in B. dorsalis peripheral nervous tissues. (F) The phylogenetic tree of D. melanogaster CAT2 and CAT2 in 11 Tephritidae species. (G) The phylogenetic tree of D. melanogaster ABCA13 and ABCA13 in 11 Tephritidae species.
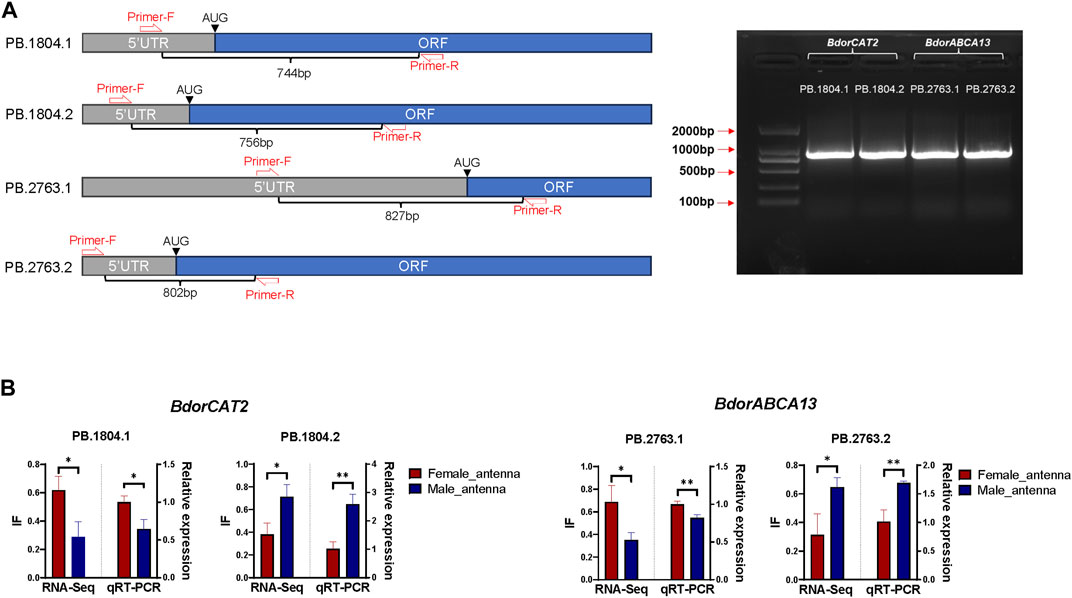
Figure 5. Validation of isoform switch of BdorCAT2 and BdorABCA13. (A) Reverse Transcription PCR (RT-PCR) primer setting positions and gel map of BdorCAT2 and BdorABCA13 isoforms. (B) RNA-Seq quantification and Quantitative Real-Time PCR (qRT-PCR) verification of BdorCAT2 and BdorABCA13 isoforms in male and female antennae of B. dorsalis.
4 Discussion
Alternative splicing contributes to the diversification of gene functions and is prevalent in insects displaying sexual dimorphism. Several studies have comprehensively annotated the sex-biased alternative splicing events in Drosophila species using microarrays and next-generation sequencing technologies (McIntyre et al., 2006; Chang et al., 2011; Singh and Agrawal, 2023), revealing that this phenomenon is evolutionarily conserved (Gibilisco et al., 2016). Chemoreception plays a crucial role in the environmental adaptation of B. dorsalis, with reported sexual dimorphism in chemosensory behavior. For example, methyl eugenol can only attract B. dorsalis males, and benzothiazole and 1-octen-3-ol can induce oviposition behavior of female B. dorsalis after mating, with potential molecular targets for behavioral regulation identified through these phenomena (Miyazaki et al., 2018; Xu et al., 2023). However, current research primarily focuses on differences in gene expression between sexes and has not extensively explored post-transcriptional regulatory processes such as alternative splicing. The antennae serve as the primary chemosensory organs in B. dorsalis. Investigating sex-biased alternative splicing in the antennae of B. dorsalis will enhance our understanding of the molecular mechanisms underlying chemosensory behavioral dimorphism and identify new molecular targets for behavioral regulation in B. dorsalis. This study successfully acquired the full-length antennal transcriptome of B. dorsalis through isoform sequencing technology and created a comprehensive, non-redundant full-length transcripts of B. dorsalis antennae by integrating next-generation RNA sequencing, isoform sequencing, and bioinformatics methods. The full-length transcriptome was utilized to thoroughly annotate sex-biased alternative splicing events and isoform-switching genes in B. dorsalis antennae, and to predict the upstream transcription factors of BdorABCA13 and BdorCAT2, which experienced ATSS type sex-biased alternative splicing. This research provides high-quality ISO-Seq data of B. dorsalis antennae and establishes a foundation for the development of new behavioral manipulation technologies for B. dorsalis.
Previous studies have shown that tissue-specific full-length transcriptome can improve the quality of genome annotation. For example, in the full-length transcriptome of silkworm silk gland and human bone marrow, 77% and 60%–70% of transcript structures not identified in the original genome annotation, respectively (Deslattes Mays et al., 2019; Chen et al., 2020). In this study, 31.94% of the full-length transcripts from the B. dorsalis antennal full-length transcriptome perfectly matched all splicing sites with the reference genes in the B. dorsalis reference genome annotation. The remaining approximately 68% were not identified in the current genome annotation file, suggesting that these transcripts represent either new transcripts of known genes or entirely new genes. This indicates that compared to the existing genome annotation, utilizing the antennal full-length transcriptome allows for a more accurate acquisition of the transcript structure of genes expressed in the antennal tissue, which aids in inferring their potential functions. Among all the new transcripts identified by the B. dorsalis antennal full-length transcriptome, those classified as NNC type transcripts constituted the highest proportion. This suggests that, compared to the B. dorsalis reference genome annotation, the antennal full-length transcriptome revealed more new types of splice sites. This may be caused by two reasons: firstly, the B. dorsalis reference genome annotation used in this study did not provide the structure information of the gene UTR region, while the transcripts obtained by the antennal full-length transcriptome retained the UTR region, and the gene structure obtained was more complete; secondly, for a single gene locus, the antennal full-length transcriptome can obtain more comprehensive alternative splicing transcript structures. Having complete information on gene and transcript structures provides a solid foundation for researching gene functions.
Previous studies have indicated that transcripts derived from full-length transcriptome technologies like ISO-Seq offer superior advantages in terms of length and structural integrity compared to those obtained from RNA-Seq technology (Sharon et al., 2013). This study screened the gene loci detected by both antennal RNA-Seq transcriptome and antennal ISO-Seq transcriptome of B. dorsalis, and compared the RNA-Seq transcripts and ISO-Seq transcripts in these gene loci from three aspects: transcript length, GC content and splice junction structure. In terms of transcript length, the length of transcripts in ISO-Seq transcriptome was longer than that of RNA-Seq transcripts, which was consistent with previous studies in insects, such as ISO-Seq transcripts of Agasicles hygrophila, about 39% of which were longer than 2kb, while only 2.31% of RNA-Seq transcripts were longer than 2 kb (Jia et al., 2018); in terms of GC content, the GC content of ISO-Seq transcripts was slightly lower than that of RNA-Seq transcripts, and previous studies have shown that the GC content of gene UTR regions was lower than that of CDS regions (Jaksik and Rzeszowska-Wolny, 2012), and ISO-Seq transcripts could obtain more complete UTR region structures, which might lead to lower average GC content of ISO-Seq transcripts; in terms of splice junction structure, only about 24.8% of ISO-Seq transcripts had splice junction structures that were completely consistent with RNA-Seq transcripts, which might be caused by misassembled transcripts generated during the RNA-Seq transcript assembly process (Steijger et al., 2013). Correct splice structure is essential for gene function research, and ISO-Seq can provide more accurate structural information for gene function research than RNA-Seq.
Several previous studies have reported alternative splicing events of insect odorant receptor genes, which may imply that isoforms have different roles in olfactory-guided behavioral responses (Robertson et al., 2003; Cattaneo, 2018; Garczynski et al., 2019). This study found that BdorOR63a.1 and BdorOR10a, two olfactory receptor genes, undergo alternative splicing. The relationship between alternative splicing of odorant receptor genes and their functional changes is still poorly understood, but some studies have found that changes in the C-terminal of amino acid sequence of odorant receptors affect ligand specificity (Hill et al., 2015); in addition, this study also found that the odorant co-receptor gene BdorORCO undergoes alternative splicing, and previous studies have found that ORCO in D. melanogaster also has two isoforms, but the protein coding regions of the two isoforms are the same (Matthews et al., 2015). The relationship between ORCO structure and function is still unknown, but its C-terminal structure is thought to be involved in receptor-receptor protein interactions (Miller and Tu, 2008). The impact of alternative splicing on the function of odorant receptor genes and odorant co-receptor genes warrants further experimental investigation.
Alternative transcription start sites (ATSS) are important forms of alternative splicing, which can further assist post-transcriptional regulation by affecting mRNA stability and protein translation efficiency, and are widely present in the genomes of many species (Rach et al., 2009; Yoon et al., 2019). ATSS can affect important life activities of insects. ATSS may be related to insect growth and development, such as the alcohol dehydrogenase (Adh) gene of D. melanogaster uses different promoters to produce two isoforms with different transcription start sites, one mainly expressed in the embryo and larval stages, and the other mainly expressed in the adult stage (Corbin and Maniatis, 1989); the Hunchback (Hb) of D. melanogaster also produces two isoforms with different transcription start sites, and they play roles in different stages of embryonic development (Margolis et al., 1995). This study identified 506 sex-biased alternative splicing events in the antennal tissue of Bactrocera dorsalis, with ATSS accounting for 31% of these events. This phenomenon of ATSS having a relatively high proportion in alternative splicing events has also been found in other species (Reyes and Huber, 2018; Chen et al., 2022). Currently, there are a lack of studies on the relationship between ATSS events and inter-sexual differentiation of gene functions in insects. However, we speculate that ATSS may play a role in the sex-specific development process of B. dorsalis antennal tissue, though its precise function requires further investigation.
In insects, doublesex coordinate regulatory network of genes (Arbeitman et al., 2016), and this network is thought to be related to sexual dimorphism in insects (Lee et al., 2002; Iijima et al., 2019; Han et al., 2022). In this study, we found binding sites of doublesex transcription factors in the upstream regulatory regions of both BdorABCA13 and BdorCAT2 genes. Therefore, we speculate that the alternative splicing of these two genes may be regulated by doublesex. ATP-binding cassette transporters are divided into full-transporters and half-transporters. Full-transporters have two nucleotide-binding domains and two transmembrane domains, and can function directly; half-transporters only contain one nucleotide-binding domain and one transmembrane domain, and can only function when forming homologous or heterologous dimers with another half-transporter (Hou et al., 2000; Jones and George, 2004; Hollenstein et al., 2007). This study found that the female-biased isoform of BdorABCA13 is a full-transporter, while the male-biased isoform is a half-transporter (Figure 4C). In fruit flies, this gene is thought to be involved in lipid transport (Dermauw and Van Leeuwen, 2014). Studies have shown that there are differences in lipid metabolism between female and male insects. During adult development, females need to consume a large amount of lipids to generate oocytes, while males need to store a lot of lipids for flight energy (Gilbert and Chino, 1974). Therefore, we speculate that the sex-biased isoform switch of BdorABCA13 is related to the differences in lipid metabolism between females and males. The full-transporter form in females can significantly improve the lipid transport efficiency, to meet the high lipid metabolism demand of female B. dorsalis. BdorCAT2 gene contains two isoforms, both isoforms have no difference in domain structure, but the female-biased isoform has a shorter first intron than the male-biased isoform (Figure 4B). Long introns increase the transcription cost of genes (Castillo-Davis et al., 2002; Swinburne et al., 2008). Studies have found that CAT2 is related to the nutritional signal transduction in the reproductive process of female insects (Attardo et al., 2006). We speculate that the shorter first intron of the female-biased isoform of BdorCAT2 is to enhance the transcription efficiency of the gene, so that it can play a faster role in the nutritional signal transduction.
This study used the full-length transcriptome and whole genome of B. dorsalis to systematically analyze sex-biased alternative splicing within its antennae. First, isoforms and gene loci were identified and quantified in the full-length transcriptome of the B. dorsalis antennae. Gene loci containing more than one isoform were then selected for further analysis. Next, sex-biased alternative splicing events within these gene loci were annotated. Among the two most prevalent alternative splicing patterns, isoform-switching genes that have differential protein-coding regions between female-biased and male-biased isoforms were identified, and they were categorized based on the length of the sex-biased isoforms. Additionally, BdorABCA3 and BdorCAT2 were screened from the above isoform-switching genes, both of which have doublesex transcription factor binding sites upstream of the genes. Finally, the expression patterns and conservation among closely related species of these two genes were analyzed. This study provided high-quality full-length transcriptome data of B. dorsalis antennae, which laid the foundation for the development of novel behavioral regulators of B. dorsalis.
Data availability statement
The data presented in this study can be found in the Genome Sequence Archive repository (https://ngdc.cncb.ac.cn/gsa/), accession number CRA015403.
Ethics statement
This article presents research on animals that do not require ethical approval for their study.
Author contributions
QW: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Resources, Software, Validation, Visualization. JZ: Writing–review and editing, Investigation, Resources. QW and JZ contributed equally and are co-first authors of the study. CL: Investigation, Resources, Writing–review and editing, Validation. CR: Writing–review and editing, Investigation, Resources, Validation. QQ: Writing–review and editing, Investigation, Resources, Validation. MY: Resources, Validation, Visualization, Writing–review and editing. SY: Funding acquisition, Methodology, Writing–review and editing. WL: Funding acquisition, Methodology, Writing–review and editing. GW: Funding acquisition, Methodology, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Shenzhen Science and Technology Program (Grant No. KQTD20180411143628272), special funds for science technology innovation and industrial development of Shenzhen Dapeng New District (Grant No. PT202101-02), and the National Key Research and Development Program of China (Grant No. 2022YFD1700201).
Acknowledgments
The authors thank the support from the Shenzhen Science and Technology Program (Grant No. KQTD20180411143628272), special funds for science technology innovation and industrial development of Shenzhen Dapeng New District (Grant No. PT202101-02), and the National Key Research and Development Program of China (Grant No. 2022YFD1700201).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2024.1384426/full#supplementary-material
References
Ambrosini G., Groux R., Bucher P. (2018). PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinforma. Oxf. Engl. 34 (14), 2483–2484. doi:10.1093/bioinformatics/bty127
Arbeitman M. N., New F. N., Fear J. M., Howard T. S., Dalton J. E., Graze R. M. (2016). Sex differences in Drosophila somatic gene expression: variation and regulation by doublesex. G3 (Bethesda, Md.) 6 (7), 1799–1808. doi:10.1534/g3.116.027961
Attardo G. M., Hansen I. A., Shiao S. H., Raikhel A. S. (2006). Identification of two cationic amino acid transporters required for nutritional signaling during mosquito reproduction. J. Exp. Biol. 209 (Pt 16), 3071–3078. doi:10.1242/jeb.02349
Au K. F., Sebastiano V., Afshar P. T., Durruthy J. D., Lee L., Williams B. A., et al. (2013). Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. U. S. A. 110 (50), E4821–E4830. doi:10.1073/pnas.1320101110
Bai L., Carlson J. R. (2010). Distinct functions of acj6 splice forms in odor receptor gene choice. J. Neurosci. 30 (14), 5028–5036. doi:10.1523/JNEUROSCI.6292-09.2010
Bohbot J., Vogt R. G. (2005). Antennal expressed genes of the yellow fever mosquito (Aedes aegypti L.); characterization of odorant-binding protein 10 and takeout. Insect Biochem. Mol. Biol. 35 (9), 961–979. doi:10.1016/j.ibmb.2005.03.010
Bonduriansky R., Chenoweth S. F. (2009). Intralocus sexual conflict. Trends Ecol. Evol. 24 (5), 280–288. doi:10.1016/j.tree.2008.12.005
Brown J. B., Boley N., Eisman R., May G. E., Stoiber M. H., Duff M. O., et al. (2014). Diversity and dynamics of the Drosophila transcriptome. Nature 512 (7515), 393–399. doi:10.1038/nature12962
Castillo-Davis C. I., Mekhedov S. L., Hartl D. L., Koonin E. V., Kondrashov F. A. (2002). Selection for short introns in highly expressed genes. Nat. Genet. 31 (4), 415–418. doi:10.1038/ng940
Cattaneo A. M. (2018). Current status on the functional characterization of chemosensory receptors of Cydia pomonella (Lepidoptera: tortricidae). Front. Behav. Neurosci. 12, 189. doi:10.3389/fnbeh.2018.00189
Chang P. L., Dunham J. P., Nuzhdin S. V., Arbeitman M. N. (2011). Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC genomics 12, 364. doi:10.1186/1471-2164-12-364
Chen Q., Lin X., Tang W., Deng Q., Wang Y., Lin Y., et al. (2022). Transcriptomic complexity in strawberry fruit development and maturation revealed by nanopore sequencing. Front. plant Sci. 13, 872054. doi:10.3389/fpls.2022.872054
Chen T., Sun Q., Ma Y., Zeng W., Liu R., Qu D., et al. (2020). A transcriptome atlas of silkworm silk glands revealed by PacBio single-molecule long-read sequencing. Mol. Genet. genomics MGG 295 (5), 1227–1237. doi:10.1007/s00438-020-01691-9
Christenson L. D., Foote R. H. (1960). Biology of fruit flies. Annu. Rev. Entomology 5, 171–192. doi:10.1146/annurev.en.05.010160.001131
Corbin V., Maniatis T. (1989). The role of specific enhancer-promoter interactions in the Drosophila Adh promoter switch. Genes and Dev. 3 (12B), 2191–2120. doi:10.1101/gad.3.12b.2191
Dauwalder B., Tsujimoto S., Moss J., Mattox W. (2002). The Drosophila takeout gene is regulated by the somatic sex-determination pathway and affects male courtship behavior. Genes and Dev. 16 (22), 2879–2892. doi:10.1101/gad.1010302
Dermauw W., Van Leeuwen T. (2014). The ABC gene family in arthropods: comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 45, 89–110. doi:10.1016/j.ibmb.2013.11.001
Deslattes Mays A., Schmidt M., Graham G., Tseng E., Baybayan P., Sebra R., et al. (2019). Single-Molecule real-time (SMRT) full-length RNA-sequencing reveals novel and distinct mRNA isoforms in human bone marrow cell subpopulations. Genes 10 (4), 253. doi:10.3390/genes10040253
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi:10.1186/s13059-019-1832-y
Fu G., Condon K. C., Epton M. J., Gong P., Jin L., Condon G. C., et al. (2007). Female-specific insect lethality engineered using alternative splicing. Nat. Biotechnol. 25 (3), 353–357. doi:10.1038/nbt1283
Garczynski S. F., Cattaneo A. M., Walker W. B. (2019). Alternative splicing of the CpomOR53 gene produces three transcripts expressed in codling moth (Lepidoptera: tortricidae) antennae. J. Econ. entomology 112 (2), 991–996. doi:10.1093/jee/toy370
Gibilisco L., Zhou Q., Mahajan S., Bachtrog D. (2016). Alternative splicing within and between Drosophila species, sexes, tissues, and developmental stages. PLoS Genet. 12 (12), e1006464. doi:10.1371/journal.pgen.1006464
Gilbert L. I., Chino H. (1974). Transport of lipids in insects. J. lipid Res. 15 (5), 439–456. doi:10.1016/s0022-2275(20)36764-x
Gordon S. P., Tseng E., Salamov A., Zhang J., Meng X., Zhao Z., et al. (2015). Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PloS one 10 (7), e0132628. doi:10.1371/journal.pone.0132628
Han C., Peng Q., Sun M., Jiang X., Su X., Chen J., et al. (2022). The doublesex gene regulates dimorphic sexual and aggressive behaviors in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 119 (37), e2201513119. doi:10.1073/pnas.2201513119
Hartmann B., Castelo R., Miñana B., Peden E., Blanchette M., Rio D. C., et al. (2011). Distinct regulatory programs establish widespread sex-specific alternative splicing in Drosophila melanogaster. RNA (New York, N.Y.) 17 (3), 453–468. doi:10.1261/rna.2460411
Hill S. R., Majeed S., Ignell R. (2015). Molecular basis for odorant receptor tuning: a short C-terminal sequence is necessary and sufficient for selectivity of mosquito Or8. Insect Mol. Biol. 24 (4), 491–501. doi:10.1111/imb.12176
Hollenstein K., Frei D. C., Locher K. P. (2007). Structure of an ABC transporter in complex with its binding protein. Nature 446 (7132), 213–216. doi:10.1038/nature05626
Hong F., Mo S. H., Lin X. Y., Niu J., Yin J., Wei D. (2020). The PacBio full-length transcriptome of the tea aphid as a reference resource. Front. Genet. 11, 558394. doi:10.3389/fgene.2020.558394
Hou Y., Cui L., Riordan J. R., Chang X. (2000). Allosteric interactions between the two non-equivalent nucleotide binding domains of multidrug resistance protein MRP1. J. Biol. Chem. 275 (27), 20280–20287. doi:10.1074/jbc.M001109200
Iijima T., Yoda S., Fujiwara H. (2019). The mimetic wing pattern of Papilio polytes butterflies is regulated by a doublesex-orchestrated gene network. Commun. Biol. 2, 257. doi:10.1038/s42003-019-0510-7
Jaksik R., Rzeszowska-Wolny J. (2012). The distribution of GC nucleotides and regulatory sequence motifs in genes and their adjacent sequences. Gene 492 (2), 375–381. doi:10.1016/j.gene.2011.10.050
Jia D., Wang Y., Liu Y., Hu J., Guo Y., Gao L., et al. (2018). SMRT sequencing of full-length transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt). Sci. Rep. 8 (1), 2197. doi:10.1038/s41598-018-20181-y
Jones P. M., George A. M. (2004). The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. life Sci. CMLS 61 (6), 682–699. doi:10.1007/s00018-003-3336-9
Katoh K., Misawa K., Kuma K., Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids Res. 30 (14), 3059–3066. doi:10.1093/nar/gkf436
Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi:10.1038/s41587-019-0201-4
Labbé G. M., Scaife S., Morgan S. A., Curtis Z. H., Alphey L. (2012). Female-specific flightless (fsRIDL) phenotype for control of Aedes albopictus. PLoS neglected Trop. Dis. 6 (7), e1724. doi:10.1371/journal.pntd.0001724
Lee G., Hall J. C., Park J. H. (2002). Doublesex gene expression in the central nervous system of Drosophila melanogaster. J. neurogenetics 16 (4), 229–248. doi:10.1080/01677060216292
Li W., Wang X., Jiang P., Yang M., Li Z., Huang C., et al. (2022). A full-length transcriptome and gene expression analysis of three detoxification gene families in a predatory stink bug, Picromerus lewisi. Front. physiology 13, 1016582. doi:10.3389/fphys.2022.1016582
Liao Y., Smyth G. K., Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl. 30 (7), 923–930. doi:10.1093/bioinformatics/btt656
Liu H., Chen Z. S., Zhang D. J., Lu Y. Y. (2018). BdorOR88a modulates the responsiveness to methyl eugenol in mature males of Bactrocera dorsalis (hendel). Front. physiology 9, 987. doi:10.3389/fphys.2018.00987
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25 (4), 402–408. doi:10.1006/meth.2001.1262
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Lv D., Zhao H., Zhao X. P., Zhao M., Zhang H. B., Zhao G. S., et al. (2020). Ultrastructural observation of female adults antennae, maxipalp and ovipositor sensilla of Megastigmus sabinae. J. Environ. Entomology 42 (4), 1028–1036. doi:10.3969/j.issn.1674-0858.2020.04.30
Margolis J. S., Borowsky M. L., Steingrímsson E., Shim C. W., Lengyel J. A., Posakony J. W. (1995). Posterior stripe expression of hunchback is driven from two promoters by a common enhancer element. Dev. Camb. Engl. 121 (9), 3067–3077. doi:10.1242/dev.121.9.3067
Martin Anduaga A., Evantal N., Patop I. L., Bartok O., Weiss R., Kadener S. (2019). Thermosensitive alternative splicing senses and mediates temperature adaptation in Drosophila. eLife 8, e44642. doi:10.7554/eLife.44642
Matthews B. B., Dos Santos G., Crosby M. A., Emmert D. B., St Pierre S. E., Gramates L. S., et al. (2015). Gene model annotations for Drosophila melanogaster: impact of high-throughput data. G3 (Bethesda, Md.) 5 (8), 1721–1736. doi:10.1534/g3.115.018929
McIntyre L. M., Bono L. M., Genissel A., Westerman R., Junk D., Telonis-Scott M., et al. (2006). Sex-specific expression of alternative transcripts in Drosophila. Genome Biol. 7 (8), R79. doi:10.1186/gb-2006-7-8-R79
Meunier N., Belgacem Y. H., Martin J. R. (2007). Regulation of feeding behaviour and locomotor activity by takeout in Drosophila. J. Exp. Biol. 210 (Pt 8), 1424–1434. doi:10.1242/jeb.02755
Miller R., Tu Z. (2008). Odorant receptor c-terminal motifs in divergent insect species. J. Insect Sci. 8 (1), 1–10. doi:10.1673/031.008.5301
Miyazaki H., Otake J., Mitsuno H., Ozaki K., Kanzaki R., Chui-Ting Chieng A., et al. (2018). Functional characterization of olfactory receptors in the Oriental fruit fly Bactrocera dorsalis that respond to plant volatiles. Insect Biochem. Mol. Biol. 101, 32–46. doi:10.1016/j.ibmb.2018.07.002
Mutz K. O., Heilkenbrinker A., Lönne M., Walter J. G., Stahl F. (2013). Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 24 (1), 22–30. doi:10.1016/j.copbio.2012.09.004
Nguyen L. T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi:10.1093/molbev/msu300
Niknafs Y. S., Pandian B., Iyer H. K., Chinnaiyan A. M., Iyer M. K. (2017). TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. methods 14 (1), 68–70. doi:10.1038/nmeth.4078
Ouyang H., Wang X., Zheng X., Lu W., Qin F., Chen C. (2021). Full-length SMRT transcriptome sequencing and SSR analysis of Bactrocera dorsalis (hendel). Insects 12 (10), 938. doi:10.3390/insects12100938
Pacific Biosciences (2023). Procedure and checklist - preparing whole genome and metagenome sequencing libraries using SMRTbell prep Kit 3.0. Available at: https://www.pacb.com/wp-content/uploads/Procedure-Checklist-Preparing-Whole-Genome-and-Metagenome-Sequencing-Libraries-Using-SMRTbell-Prep-Kit-3.0.pdf.
Pagadala Damodaram K. J., Kempraj V., Aurade R. M., Venkataramanappa R. K., Nandagopal B., Verghese A., et al. (2014). Oviposition site-selection by Bactrocera dorsalis is mediated through an innate recognition template tuned to γ-octalactone. PloS one 9 (1), e85764. doi:10.1371/journal.pone.0085764
Parker G. A., Partridge L. (1998). Sexual conflict and speciation. Philosophical Trans. R. Soc. Lond. Ser. B, Biol. Sci. 353 (1366), 261–274. doi:10.1098/rstb.1998.0208
Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. methods 14 (4), 417–419. doi:10.1038/nmeth.4197
Pertea G., Pertea M. (2020). GFF utilities: GffRead and GffCompare. F1000Research 9, ISCB Comm J-304. doi:10.12688/f1000research.23297.2
Pertea M., Pertea G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., et al. (2005). InterProScan: protein domains identifier. Nucleic acids Res. 33 (Web Server issue), W116–W120. doi:10.1093/nar/gki442
Rach E. A., Yuan H. Y., Majoros W. H., Tomancak P., Ohler U. (2009). Motif composition, conservation and condition-specificity of single and alternative transcription start sites in the Drosophila genome. Genome Biol. 10 (7), R73. doi:10.1186/gb-2009-10-7-r73
Ren C., Zhang J., Yuan J. X., Wuyun Q., Yan S. C., Liu W., et al. (2023). Light intensity regulates the sexual behaviors of oriental fruit fly Bactrocera dorsalis under laboratory conditions. J. Integr. Agric. 22 (9), 2772–2782. doi:10.1016/j.jia.2023.04.025
Ren L., Ma Y., Xie M., Lu Y., Cheng D. (2021). Rectal bacteria produce sex pheromones in the male oriental fruit fly. Curr. Biol. CB 31 (10), 2220–2226.e4. doi:10.1016/j.cub.2021.02.046
Reyes A., Huber W. (2018). Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic acids Res. 46 (2), 582–592. doi:10.1093/nar/gkx1165
Robertson H. M., Warr C. G., Carlson J. R. (2003). Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 100 (Suppl. 2), 14537–14542. doi:10.1073/pnas.2335847100
Rogers T. F., Palmer D. H., Wright A. E. (2021). Sex-specific selection drives the evolution of alternative splicing in birds. Mol. Biol. Evol. 38 (2), 519–530. doi:10.1093/molbev/msaa242
Sarov-Blat L., So W. V., Liu L., Rosbash M. (2000). The Drosophila takeout gene is a novel molecular link between circadian rhythms and feeding behavior. Cell 101 (6), 647–656. doi:10.1016/s0092-8674(00)80876-4
Sato K., Ito H., Yokoyama A., Toba G., Yamamoto D. (2019). Partial proteasomal degradation of Lola triggers the male-to-female switch of a dimorphic courtship circuit. Nat. Commun. 10 (1), 166. doi:10.1038/s41467-018-08146-1
Schmucker D., Clemens J. C., Shu H., Worby C. A., Xiao J., Muda M., et al. (2000). Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101 (6), 671–684. doi:10.1016/s0092-8674(00)80878-8
Sharon D., Tilgner H., Grubert F., Snyder M. (2013). A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31 (11), 1009–1014. doi:10.1038/nbt.2705
Shen G. M., Jiang H. B., Wang X. N., Wang J. J. (2010). Evaluation of endogenous references for gene expression profiling in different tissues of the oriental fruit fly Bactrocera dorsalis (Diptera: Tephritidae). BMC Mol. Biol. 11, 76. doi:10.1186/1471-2199-11-76
Shen W., Le S., Li Y., Hu F. (2016). SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS one 11 (10), e0163962. doi:10.1371/journal.pone.0163962
Shields E. J., Sorida M., Sheng L., Sieriebriennikov B., Ding L., Bonasio R. (2021). Genome annotation with long RNA reads reveals new patterns of gene expression and improves single-cell analyses in an ant brain. BMC Biol. 19 (1), 254. doi:10.1186/s12915-021-01188-w
Siddall A., Harvey-Samuel T., Chapman T., Leftwich P. T. (2022). Manipulating insect sex determination pathways for genetic pest management: opportunities and challenges. Front. Bioeng. Biotechnol. 10, 867851. doi:10.3389/fbioe.2022.867851
Singh A., Agrawal A. F. (2023). Two forms of sexual dimorphism in gene expression in Drosophila melanogaster: their coincidence and evolutionary genetics. Mol. Biol. Evol. 40 (5), msad091. doi:10.1093/molbev/msad091
Smith P. H., Mwangi J. M., Afrane Y. A., Yan G., Obbard D. J., Ranford-Cartwright L. C., et al. (2011). Alternative splicing of the Anopheles gambiae Dscam gene in diverse Plasmodium falciparum infections. Malar. J. 10, 156. doi:10.1186/1475-2875-10-156
Sproul J. S., Hotaling S., Heckenhauer J., Powell A., Marshall D., Larracuente A. M., et al. (2023). Analyses of 600+ insect genomes reveal repetitive element dynamics and highlight biodiversity-scale repeat annotation challenges. Genome Res. 33 (10), 1708–1717. doi:10.1101/gr.277387.122
Steijger T., Abril J. F., Engström P. G., Kokocinski F., Hubbard T. J., Guigó R., et al. (2013). Assessment of transcript reconstruction methods for RNA-seq. Nat. methods 10 (12), 1177–1184. doi:10.1038/nmeth.2714
Stolc V., Gauhar Z., Mason C., Halasz G., van Batenburg M. F., Rifkin S. A., et al. (2004). A gene expression map for the euchromatic genome of Drosophila melanogaster. Sci. (New York, N.Y.) 306 (5696), 655–660. doi:10.1126/science.1101312
Sun H., Du Y., Liu Z., Dong K. (2020). Distinct functional properties of sodium channel variants are associated with usage of alternative exons in Nilaparvata lugens. Insect Biochem. Mol. Biol. 118, 103292. doi:10.1016/j.ibmb.2019.103292
Swinburne I. A., Miguez D. G., Landgraf D., Silver P. A. (2008). Intron length increases oscillatory periods of gene expression in animal cells. Genes Dev. 22 (17), 2342–2346. doi:10.1101/gad.1696108
Tan K. H., Nishida R. (2012). Methyl eugenol: its occurrence, distribution, and role in nature, especially in relation to insect behavior and pollination. J. insect Sci. (Online) 12, 56. doi:10.1673/031.012.5601
Tardaguila M., de la Fuente L., Marti C., Pereira C., Pardo-Palacios F. J., Del Risco H., et al. (2018). SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 28 (3), 396–411. Advance online publication. doi:10.1101/gr.222976.117
Telonis-Scott M., Kopp A., Wayne M. L., Nuzhdin S. V., McIntyre L. M. (2009). Sex-specific splicing in Drosophila: widespread occurrence, tissue specificity and evolutionary conservation. Genetics 181 (2), 421–434. doi:10.1534/genetics.108.096743
Tilgner H., Grubert F., Sharon D., Snyder M. P. (2014). Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. U. S. A. 111 (27), 9869–9874. doi:10.1073/pnas.1400447111
Tørresen O. K., Star B., Mier P., Andrade-Navarro M. A., Bateman A., Jarnot P., et al. (2019). Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic acids Res. 47 (21), 10994–11006. doi:10.1093/nar/gkz841
Treutlein B., Gokce O., Quake S. R., Südhof T. C. (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 111 (13), E1291–E1299. doi:10.1073/pnas.1403244111
Verhulst E. C., Beukeboom L. W., van de Zande L. (2010). Maternal control of haplodiploid sex determination in the wasp Nasonia. Sci. (New York, N.Y.) 328 (5978), 620–623. doi:10.1126/science.1185805
Verhulst E. C., Lynch J. A., Bopp D., Beukeboom L. W., van de Zande L. (2013). A new component of the Nasonia sex determining cascade is maternally silenced and regulates transformer expression. PloS one 8 (5), e63618. doi:10.1371/journal.pone.0063618
Verta J. P., Jacobs A. (2022). The role of alternative splicing in adaptation and evolution. Trends Ecol. Evol. 37 (4), 299–308. doi:10.1016/j.tree.2021.11.010
Vitting-Seerup K., Sandelin A. (2019). IsoformSwitchAnalyzeR: analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinforma. Oxf. Engl. 35 (21), 4469–4471. doi:10.1093/bioinformatics/btz247
Wang B., Tseng E., Regulski M., Clark T. A., Hon T., Jiao Y., et al. (2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7, 11708. doi:10.1038/ncomms11708
Wojtowicz W. M., Flanagan J. J., Millard S. S., Zipursky S. L., Clemens J. C. (2004). Alternative splicing of Drosophila Dscam generates axon guidance receptors that exhibit isoform-specific homophilic binding. Cell 118 (5), 619–633. doi:10.1016/j.cell.2004.08.021
Wu H., Liu J., Liu Y., Abbas M., Zhang Y., Kong W., et al. (2022). CYP6FD5, an antenna-specific P450 gene, is potentially involved in the host plant recognition in Locusta migratoria. Pesticide Biochem. physiology 188, 105255. doi:10.1016/j.pestbp.2022.105255
Wu H., Wang H., Li R., Liu Y., Zhang Y., Chen N., et al. (2023). Transcription factor CncC regulates the expression of antennal CYP6MU1 gene responsible for trans-2-hexen-1-al and nonanal recognition in Locusta migratoria. Pesticide Biochem. physiology 196, 105620. doi:10.1016/j.pestbp.2023.105620
Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. Camb. (Mass.) 2 (3), 100141. doi:10.1016/j.xinn.2021.100141
Wu T. D., Watanabe C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21 (9), 1859–1875. doi:10.1093/bioinformatics/bti310
Xing L., Wu Q., Xi Y., Huang C., Liu W., Wan F., et al. (2022). Full-length codling moth transcriptome atlas revealed by single-molecule real-time sequencing. Genomics 114 (2), 110299. doi:10.1016/j.ygeno.2022.110299
Xing Y., Yang W., Liu G., Cui X., Meng H., Zhao H., et al. (2020). Dynamic alternative splicing during mouse preimplantation embryo development. Front. Bioeng. Biotechnol. 8, 35. doi:10.3389/fbioe.2020.00035
Xu L., Jiang H. B., Yu J. L., Pan D., Tao Y., Lei Q., et al. (2023). Two odorant receptors regulate 1-octen-3-ol induced oviposition behavior in the oriental fruit fly. Commun. Biol. 6 (1), 176. doi:10.1038/s42003-023-04551-5
Yoon Y., Klomp J., Martin-Martin I., Criscione F., Calvo E., Ribeiro J., et al. (2019). Embryo polarity in moth flies and mosquitoes relies on distinct old genes with localized transcript isoforms. eLife 8, e46711. doi:10.7554/eLife.46711
Zhang J., Zhang Y., Liu W., Yan S. C., Wang G. R. (2023). Chemical communication mechanism and strategy for attractant development in Bactrocera dorsalis (Diptera: tetriphitidae). Acta Entomol. Sin. 66 (1), 108–120. doi:10.16380/j.kcxb.2023.01.014
Keywords: Bactrocera dorsalis, antennae, full-length transcripts, sex-biased differential alternative splicing, isoform switch
Citation: Wang Q, Zhang J, Liu C, Ru C, Qian Q, Yang M, Yan S, Liu W and Wang G (2024) Identification of antennal alternative splicing by combining genome and full-length transcriptome analysis in Bactrocera dorsalis. Front. Physiol. 15:1384426. doi: 10.3389/fphys.2024.1384426
Received: 09 February 2024; Accepted: 29 May 2024;
Published: 17 June 2024.
Edited by:
Frederique Hilliou, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceCopyright © 2024 Wang, Zhang, Liu, Ru, Qian, Yang, Yan, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanchun Yan, eWFuc2hhbmNodW5AMTI2LmNvbQ==; Wei Liu, bGl1d2VpMTFAY2Fhcy5jbg==
†These authors have contributed equally to this work and share first authorship