 Zain AlShanableh
Zain AlShanableh Evan C. Ray
Evan C. Ray- Renal-Electrolyte Division, UPMC and University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Hypertension is associated with increased risk of cardiovascular disease and death. Evidence suggests that Mg2+ depletion contributes to hypertension. It is estimated that 25% or more of the United States population experiences chronic, latent Mg2+ depletion. This review explores mechanisms by which Mg2+ influences blood pressure, modifying risk of hypertension and complicating its treatment. Mechanisms addressed include effects upon i) sympathetic tone, via the modulation of N-methyl-D-aspartate (NMDA) receptor and N-type Ca2+ channel activity, influencing catecholamine release from sympathetic nerve endings; ii) vascular tone, via alteration of L-type Ca2+ and endothelial nitric oxide synthase (eNOS) activity and prostacyclin release; iii) renal K+ handling, influencing systemic K+ balance and potentially indirectly influencing blood pressure; iv) aldosterone secretion from the adrenal cortex; and v) modulation of pro-hypertensive inflammatory processes in dendritic cells and macrophages, including activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and stimulation of isolevuglandin (IsoLG) production. Discovery of these mechanisms has furthered our understanding of the pathogenesis of hypertension, with implications for treatment and has highlighted the role of Mg2+ balance in hypertension and cardiovascular disease.
Introduction
Magnesium is an essential ion and is required for normal health, including the cardiovascular system (de Baaij et al., 2015). Numerous studies have explored the association between Mg2+ and blood pressure, and evidence suggests that Mg2+ depletion contributes to hypertension. This review will focus on the effect of Mg2+ on blood pressure and hypertension and will discuss different mechanisms by which Mg2+ influences blood pressure.
Hypertension is widely prevalent. In the United States, it affects 119.9 million adults - nearly half the population (United States Centers for Disease Control and Prevention, 2015). Hypertension increases the risk of cardiovascular disease and stroke (Fuchs and Whelton, 2020). In 2020, it was estimated that cardiovascular disease contributed to around 19,000,000 deaths globally (Tsao et al., 2022).
Systemic Mg2+ depletion is common. Reports estimate that 25% or more of the population of the United States experiences chronic, latent Mg2+ depletion (Lowenstein and Stanton, 1986; Rosanoff et al., 2022). Mg2+ depletion is under-appreciated clinically, partly because reference ranges for plasma Mg2+ in clinical laboratories are based upon population distribution rather than healthy levels. Study groups in the United States (Costello et al., 2016) and in Germany (Micke et al., 2021), have independently recommended an evidence-based lower limit of normal for serum Mg2+ of 2.07 mg/dL (0.85 mmol/L). However, a 2022 study found that in 41 out of 43 medical centers in 16 countries employ a lower limit beneath this recommended threshold (Rosanoff et al., 2022). Prevalence of Mg2+ depletion appears even higher in individuals with hypertension, as intracellular Mg2+ levels are lower in hypertensive individuals than control individuals (Resnick et al., 1984; Touyz et al., 1992). Plasma Mg2+, a less sensitive indicator of Mg2+ deficiency, was found to be lower in hypertensive individuals with elevated renin but not in other hypertensive individuals (Resnick et al., 1983).
Dietary Mg2+ insufficiency is a common contributor to systemic Mg2+ depletion. The United States estimated average requirement (EAR) for Mg2+ is 255 mg/day for women aged 19–30 years, increasing to 265 mg/day for women aged ≥31 years. For men aged 19–30 the EAR is 330 mg/day, increasing to 350 mg/day for men aged ≥31 years (Rosanoff et al., 2012). According to the United States National Health and Nutrition Examination Survey (NHANES) 2013–2016 report, this EAR was not met in nearly half (48%) of the U.S. population (USDA Agricultural Research Service, 2019).
Commonly prescribed medications also contribute to systemic Mg2+ depletion (Ray et al., 2023). Given the contribution of Mg2+ depletion to cardiovascular disease (Kolte et al., 2014); it is particularly concerning that treatment with a first-line therapy for hypertension, thiazide-type diuretics, promotes urinary Mg2+ wasting and systemic Mg2+ depletion (Hollifield, 1986).
Evidence for a relationship between Mg2+ and hypertension
The earliest findings of an effect of Mg2+ upon blood pressure were reported more than 100 years ago, when Kenneth Blackfan (subsequently famous for his description of Diamond Blackfan anemia) and Charles McKhann described “a rapid fall in blood pressure” in children with glomerulonephritis and severely elevated blood pressure (Blackfan and Mills, 1923; Blackfan and McKhann, 1931). Studies much later would seek to understand the circumstances under which Mg2+ can attenuate hypertension.
Several studies have explored the relationship between dietary Mg2+ and blood pressure in experimental animals, with mixed results. In rats, some studies show increased blood pressure with dietary Mg2+ restriction (Berthelot and Esposito, 1983; Altura et al., 1984; Laurant et al., 1999; Murasato et al., 1999; Carlin Schooley and Franz, 2002; Blache et al., 2006), others do not (Itokawa et al., 1974; Overlack et al., 1987; Lowney et al., 1988; Luthringer et al., 1988; Evans et al., 1989; Liu et al., 1994; Laurant et al., 1997; Tomiyasu et al., 1998). No doubt these discrepancies reflect differences in experimental details such as strains used, severity of dietary Mg2+ restriction, and duration. In mice, dietary Mg2+ deficiency was shown to stimulate salt-sensitive increase in blood pressure in DBA but not C57Bl/6J mice (Kumagai et al., 2021). However, in C56Bl/6J mice, a Mg2+-restricted diet did increase blood pressure raising-effects of sympathetic stimulation. In mice of the sv129 background, dietary Mg2+ restriction increased blood pressure by 21 days until sacrifice at 5 weeks (Pitzer Mutchler et al., 2023). Intravenous Mg2+ infusion in rats attenuates increases in blood pressure resulting from angiotensin II (Atarashi et al., 1990) or sympathetic nerve stimulation (Shimosawa et al., 2004).
Observational studies have explored the correlation between circulating Mg2+ in humans and blood pressure. A structured review and subgroup analysis of observational studies explored the association between dietary Mg2+ intake and blood pressure. Findings suggested an inverse relationship between dietary Mg2+ intake and blood pressure, though heterogeneity in study methods complicated interpretation (Mizushima et al., 1998). In an observational study of 1,000 ambulatory hypertensive patients, hypomagnesemia was associated with worsened hypertension, as indicated by a greater number of prescribed anti-hypertensive medications (Whang et al., 1982). Plasma Mg2+ levels are lower in individuals with untreated elevated systolic blood pressure and diastolic blood pressure than in normotensive controls (Rodríguez-Moran and Guerrero-Romero, 2014; Rodríguez-Ramírez et al., 2015).
Numerous human clinical trials have examined the effects of Mg2+ supplementation in management of hypertension. A meta-analysis by Zhang et al., pooled 24 randomized controlled trials (RCTs) with a total of 2,028 participants. They concluded that supplementation of Mg2+ at a mean dose of 368 mg/day for a median period of 3 months resulted in 2 mmHg reduction in SBP (95% CI, 0.43–3.58 mmHg; p = 0.01) and 1.78 mmHg reduction in DBP (CI, 0.73–2.82 mmHg; p = 0.001) (Zhang et al., 2016). Another meta-analysis by Dibaba et al., reviewed 11 RCTs, including 543 participants. Mg2+ supplementation of 365–450 mg/day for a mean of 3.6 months significantly reduced SBP by a mean of 4.18 mmHg (standard mean difference: −0.20; 95% CI: −0.37, −0.03) and DBP by a mean of 2.27 mmHg (standard mean difference: −0.27; 95% CI: −0.52, −0.03) (Dibaba et al., 2017). Rosanoff et al., conducted a meta-analysis of 49 clinical trials that stratified study participants into the following groups: 1) untreated hypertensives 2) uncontrolled hypertensives 3) controlled hypertensives 4) normotensive subjects. They found that a Mg2+ dose of ≥240 mg/day decreases BP in treated but uncontrolled hypertensive individuals, and a dose of >600 mg/day lowers BP in untreated hypertensives. There was no change in BP in individuals who were normotensive, had controlled HTN, or were Mg2+-replete (Rosanoff et al., 2021). A meta-analysis of seven RCTs examining hypertensive individuals with diabetes found that Mg2+ supplementation reduced systolic blood pressure by 5.78 and diastolic blood pressure by 2.5 mmHg (Asbaghi et al., 2021). This is of particular interest, as diabetic patients tend to be Mg2+ depleted (Ray et al., 2023).
Together, these findings suggest that systemic Mg2+ depletion promotes increased blood pressure in patients with hypertension.
Mechanisms influencing blood pressure
Because of its vast physiologic effects, Mg2+ depletion likely influences blood pressure via multiple mechanisms, as discussed below and summarized in Figure 1.
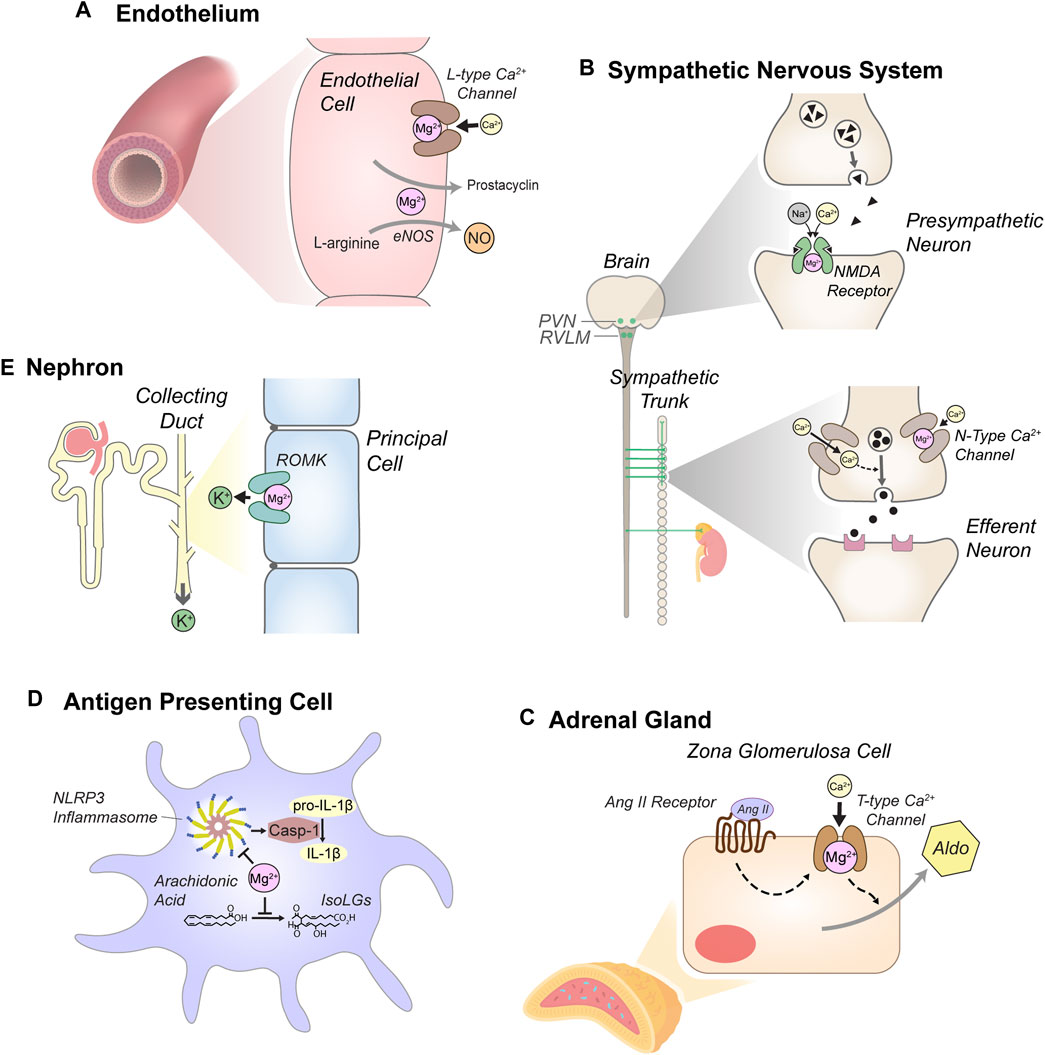
Figure 1. Anti-hypertensive effects of Mg2+. (A) Mg2+ reduces vascular tone through actions in endothelial cells, including blockade of L-type Ca2+ channels and by supporting secretion of prostacyclin and nitric oxide (NO). L-type Ca2+ channel blockade and other mechanisms also reduce intracellular Ca2+ in myocytes, attenuating cell contraction (not depicted). (B) Mg2+ attenuates sympathetic tone. Blockade of N-methyl-D-aspartate (NMDA) receptors in the paraventricular nucleus (PVN) of the hypothalamus and the rostral ventrolateral medulla (RVLM) attenuates pre-sympathetic neuron activity. In sympathetic ganglia, Mg2+ reduces N-type Ca2+ channel activity, modulating stimulation of efferent sympathetic neurons. (C) Mg2+ reduces aldosterone secretion in the adrenal cortex. Mg2+ blockade of T-type Ca2+ channels in zona glomerulosa cells modulates stimulation of aldosterone (Aldo) secretion by angiotensin II (Ang II). (D) Mg2+-depletion stimulates antigen presenting cells (dendritic cells and monocytes). Mg2+ depletion enhances expression of NLR family pyrin domain containing 3 (NLRP3), a key component of the inflammasome, which activates caspase-1 (Casp-1). Casp-1 stimulates production of pro-inflammatory cytokines, including IL-1β. Mg2+ depletion also enhances production of isolevuglandins (IsoLGs), reactive aldehydes that promote pro-hypertensive inflammation. (E) Mg2+ reduces urinary K+ excretion through blockade of the renal outer medullary K+ channel (ROMK) in the distal nephron, attenuating systemic K+-depletion. Although K+-depletion can stimulate the thiazide-sensitive NaCl cotransporter (NCC), this mechanism does not appear to contribute to increased blood pressure in the context of systemic Mg2+ depletion (see text).
Vascular tone
Effects of Mg2+ on vascular tone may contribute to its influence upon blood pressure. Empiric evidence for an effect of Mg2+ upon vascular tone is demonstrated by the observations that decreased extracellular Mg2+ or systemic Mg2+ depletion in animals produces vasospasm and reduces microvascular blood flow (Altura and Turlapaty, 1982; Altura et al., 1983; Altura et al., 1984). Humans given an acute MgSO4 infusion exhibit increased renal blood flow, despite reduced blood pressure (Nadler et al., 1987).
Vascular smooth muscle constriction is stimulated when Ca2+ enters smooth muscle cytosol via L-type voltage-gated Ca2+ channels. Intracellular Ca2+ stimulates phospholipase C and production of diacylglycerol (DG) and inositol 1,4,5-trisphosphate (IP3). IP3 activates the IP3 receptor, releasing Ca2+ from the sarcoplasmic reticulum. Cytosolic Ca2+ then binds to calmodulin, activating myosin light chain kinase (MLCK). Activated MLCK phosphorylates the myosin light chain, stimulating interaction of actin and myosin, and eliciting cell contraction (Webb, 2003).
Intracellular Mg2+ attenuates myocyte contraction via several mechanisms. Mg2+ diminishes cellular Ca2+ entry via L-type voltage gated Ca2+ channels (Zhang et al., 2007; Sharma et al., 2012). Intracellular Mg2+ inhibits Ca2+-stimulated Ca2+-release from the sarcoplasmic reticulum, at least in cardiac muscle (Dunnett and Nayler, 1978). The sarcoplasmic reticulum Ca2+-ATPase, which is required to return released Ca2+ to intracellular stores, requires Mg2+ for activity (Hasselbach et al., 1981). Consequently, low Mg2+ conditions prolong elevation of intracellular Ca2+ following release from intracellular stores (Gasallaherraiz et al., 1995). Thus, Mg2+ reduces smooth muscle contraction.
Prostacyclin release: Effects of systemic Mg2+ status upon vascular tone may be mediated, in part, by effects on prostacyclin release. Prostacyclin (PGI2) is recognized to have important systemic vasodilatory effects (Zhao and Richardson, 1990). In the kidney, prostacyclin is critical for maintaining vasodilation and blood flow in the context of extrarenal vasoconstriction. Mice lacking prostacyclin synthase exhibit hypertension, thickening of the aortic medial and adventitial layers, and nephrosclerosis (Yokoyama et al., 2002). In humans, a repeat polymorphism in the promoter region of the prostacyclin synthase gene was found to reduce prostacyclin synthase transcription and to be associated with increased odds of hypertension (Iwai et al., 1999).
Mg2+ modulates vascular prostacyclin release. In cultured vascular endothelial cells or smooth muscle cells, increased extracellular Mg2+ stimulated prostacyclin secretion (Briel et al., 1987; Satake et al., 2004). In the rat deoxycorticosterone acetate (DOCA)-salt model of hypertension, a Mg2+-enriched diet significantly increased PGI2 levels (Laurant et al., 1992). Infusion of MgSO4 into humans enhanced urinary excretion of immunoreactive 6-ketoprostaglandin F1α (6-keto-PGF1a), a stable break-down product of PGI2, while reducing blood pressure (Nadler et al., 1987). The importance of prostaglandin synthesis in this blood pressure effect was demonstrated by the observation that cyclooxygenase inhibition prevented the decrease in blood pressure and increase in renal blood flow. Moreover, the Mg2+-stimulated increase in PGI2 release was blocked by the calcium channel blocker, nifedipine, suggesting that the influence of Mg2+ on cyclooxygenase is dependent upon Ca2+ entry into cells.
Nitric oxide metabolism: Mg2+ also influences vascular tone through effects on nitric oxide (NO). Nitric oxide is an endogenous vasodilator produced in endothelial cells from L-arginine by endothelial NO synthase (eNOS) (Rees et al., 1989; Gamboa et al., 2007). In cultured endothelial cells, NO production was roughly 3-fold higher in cells grown in high (5 mM, or 12 mg/dL) than in control (1 mM, or 2.4 mg/dL) extracellular Mg2+ (Maier et al., 2004). This finding was attributed to an observed increase in eNOS protein abundance in cells grown in high Mg2+.
Altered NO release may contribute to effects of Mg2+ upon NO signaling. In mouse aorta and mesenteric vessels, endothelium-dependent, Mg2+-induced arterial relaxation is attenuated by blockade of eNOS activity with N (gamma)-nitro-L-arginine methyl ester (L-NAME) (Kudryavtseva et al., 2024). In canine coronary arteries, Mg2+-free conditions attenuated acetylcholine and ADP-stimulated, NO-dependent reduction in arterial tone (Pearson et al., 1998). The Ca2+ ionophore A23187, which induces endothelial NO release independently of receptor-mediated signaling mechanisms, reduced arterial tension in a Mg2+-independent fashion. Bradykinin-stimulated vascular relaxation, which occurs via endothelium-dependent, but NO-independent mechanisms, was unaffected (Pearson et al., 1998). These findings suggest that Mg2+ is required for stimulation of NO release but not for NO-stimulated relaxation. Evidence that Mg2+ influences NO signaling in humans comes from a study examining flow-mediated vasodilation of the brachial artery (FMD), a process that is at least partly NO mediated (Green et al., 2014). Oral Mg2+ supplementation in individuals with coronary artery disease significantly improved FMD and exercise tolerance (Shechter et al., 2000).
Mg2+ likely also promotes relaxation via additional, NO-independent pathways. Blockade of SK and IK Ca2+-activated K+ channels, which participate in endothelium-derived relaxation factor-stimulated arterial relaxation, blunted Mg2+-dependent arterial relaxation additively with eNOS inhibition (Kudryavtseva et al., 2024).
Together, these observations suggest that systemic Mg2+ status could influence blood pressure through multiple effects on vascular tone.
Sympathetic tone
Mg2+ exerts an inhibitory effect on the sympathetic nervous system, whereas Mg2+ deficiency stimulates sympathetic tone.
Effects of Mg2+ upon the sympathetic nervous system are mediated, in part, through modulation of N-methyl-D-aspartate (NMDA) receptor activity. The NMDA receptor is a Ca2+-selective ion channel that opens in response to NMDA and L-Glutamate (L-Glu) (Dingledine et al., 1999; Kagiyama et al., 2001). NMDA receptor activity in the rostral ventrolateral medulla (RVLM) and hypothalamic paraventricular nucleus (PVN) increase sympathetic outflow and blood pressure (Dampney et al., 2003; Li and Pan, 2017). NMDA receptor activity is negatively regulated by Mg2+ (Dingledine et al., 1999). Thus, attenuation of NMDA receptor activity by Mg2+ may be expected to reduce blood pressure.
In support of this hypothesis, Kagiyama et al., studied the effect of Mg2+ in the RVLM upon blood pressure. Injection of magnesium sulfate (MgSO4) into the RVLM exerted a dose-dependent attenuation of increased blood pressure occurring in response to NMDA injection (Kagiyama et al., 2001).
Mg2+ also negatively influences sympathetic tone through modulation of catecholamine release from peripheral nerve endings and by blocking N-type Ca2+ channels at nerve endings (Shimosawa et al., 2004). In neuronally differentiated PC12 cells, N-type Ca2+ channel activity was decreased by elevated extracellular Mg2+ and increased by reduced extracellular Mg2+. With cytosolic Ca2+ being a major stimulus for catecholamine release, low extracellular Mg2+ buffer enhanced norepinephrine release from the periarterial plexus of the mesenteric artery compared to control or high Mg2+ buffer. Urinary catecholamine excretion was found to be more than two-fold higher in Mg2+ deficient rats than control rats (Murasato et al., 1999). Mg2+ infusion also attenuated sympathetically mediated reflex tachycardia following hydralazine infusion (Shimosawa et al., 2004). In addition to reducing norepinephrine release from sympathetic neurons, Mg2+ also increased norepinephrine uptake in isolated adrenergic nerve granules, suggesting an influence upon norepinephrine reuptake in the synaptic cleft (von Euler and Lishajko, 1963; von Euler and Lishajko, 1973).
Effects of Mg2+ on the sympathetic nervous system have also been demonstrated in human subjects. James et al., studied the effect of MgSO4 infusion upon simulation of catecholamine release and blood pressure in response to endotracheal intubation. In controls, intubation rapidly increased circulating epinephrine levels, norepinephrine levels, and systolic blood pressure. MgSO4 infusion attenuated the increase in each of these (James et al., 1989).
Thus, Mg2+ modulates peripheral sympathetic nervous system activity, reducing blood pressure.
Effects on K+ and Na+ handling
Systemic Mg2+ status could influence blood pressure indirectly, through effects on handling of K+ and Na+.
Bodily K+ balance influences blood pressure. Several meta-analyses of clinical trials find that K+ supplementation reduces blood pressure in hypertensive individuals (van Bommel and Cleophas, 2012; Poorolajal et al., 2017; Filippini et al., 2020). The influence of K+ upon blood pressure is likely mediated by multiple mechanisms, including effects upon vascular tone and upon extracellular fluid volume. In Dahl salt-sensitive rats, a high K+-diet promotes vascular relaxation (Raij et al., 1988). Fluid volume effects may occur secondary to enhanced tubular Na+ reabsorption in the context of systemic K+ depletion. In the kidney’s proximal convoluted tubule (PCT), a low K+ diet enhanced protein abundance of the Na+/H+ exchanger, type 3 (NHE3), promoting Na+/H+ exchange (Shirley et al., 1990; Soleimani et al., 1990; Elkjær et al., 2002). In the distal convoluted tubule (DCT), K+ deficiency stimulates phosphorylation-mediated activation of sodium-chloride cotransporter (NCC) through modulation of the WNK/SPAK/OSR1 (with no lysine/SPS1-related proline-alanine-rich kinase/oxidative stress-responsive kinase 1) signal transduction pathway (Terker et al., 2015).
Mg2+ depletion promotes K+ depletion. Because serum K+ represents only 2% of total body K+, even when plasma or serum K+ is not appreciably reduced, intracellular and total body K+ stores can be depleted (Patrick, 1977; Brown, 1984). In rats subjected to dietary Mg2+ restriction, intramuscular K+ declined (Macintyre and Davidsson, 1958; Manitius and Epstein, 1963; Whang and Welt, 1963; Ginn et al., 1967; Dørup and Clausen, 1993). This was associated with decreased whole body K+ following prolonged (60-day) dietary Mg2+ restriction (Whang and Welt, 1963). In human subjects given a low Mg2+ diet, urinary K+ excretion increased overall and total exchangeable K+ decreased (Shils, 1969). Intracellular Mg2+ depletion is thought to promote urinary K+ excretion through loss of voltage-dependent blockade of the outer medullary K+ channel (ROMK) in the kidney tubule, enhancing tubular K+ secretion (Huang and Kuo, 2007). Additionally, systemic Mg2+ depletion increases circulating aldosterone levels (discussed below), enhancing urinary K+ excretion in exchange for Na+ reabsorption.
Taken together, these findings suggest the hypothesis that Mg2+ depletion could contribute to urinary Na+ retention and increased blood pressure by stimulating NCC activity in the DCT. Surprisingly, rats given a low Mg2+ diet exhibit reduced NCC expression (Fanestil et al., 1999). Ferdaus et al. confirmed these findings in mice and found that dietary Mg2+ depletion reduced both total and phosphorylated NCC protein abundance in the kidney. NCC mRNA levels were unchanged, suggesting post-transcriptional effects on NCC expression. The hypothesis that dietary Mg2+ restriction may stimulate NCC degradation was supported by the observation that kidney-specific deletion of the ubiquitin ligase NEDD4-2 blocked downregulation of NCC by dietary Mg2+ restriction. Dietary Mg2+ depletion even blocked the ability of a K+-restricted diet to increase total and phosphorylated-NCC protein abundance, providing further evidence that the influence of Mg2+ depletion on blood pressure is not NCC-mediated.
Systemic K+ depletion may influence Na+ handling in other portions of the nephron, such as the thick ascending loop of Henle. The Na-K-Cl co-transporter (NKCC2) in the thick ascending limb (TAL) is also modulated by intracellular WNK/SPAK/OSR1 pathway (Moriguchi et al., 2005; Rinehart et al., 2005; Liu et al., 2011; Richardson et al., 2011; Park et al., 2013; Terker et al., 2018; Marcoux et al., 2019). Given the ability of systemic K+ status to influence the WNK/SPAK/OSR1 pathway, it seems likely that differences in Mg2+ homeostasis may influence this pathway via changes in systemic K+, but we are unaware of data directly exploring this hypothesis.
Mg2+ could also influence Na+ reabsorption in the TAL through modulation of the calcium-sensing receptor (CaSR). Activation of the CaSR on the basolateral surface of cortical TAL cells reduces apical K+ channel activity (Wang et al., 1996). Impaired cellular K+ efflux impairs Na+ and Cl− reabsorption through NKCC2, producing a loop diuretic-like effect. Mg2+, like Ca2+, can activate the CaSR, which may explain the earlier observation that intravenous Mg2+ infusion can decrease TAL Na+ reabsorption (Ploth et al., 1976). Whether changes in plasma Mg2+ within the physiologic range modulate CaSR activity and TAL NaCl reabsorption remains unclear.
Renin-angiotensin-aldosterone system
Systemic Mg2+ status may also influence Na+ and K+ handling via modulation of the renin-angiotensin-aldosterone system. In laboratory animals, dietary Mg2+ restriction appears to stimulate aldosterone levels. Sapna et al. found that in rats, 6 days on low Mg2+ chow resulted in serum aldosterone of 205.0 ± 66.2 pg/mL, which was not significantly higher than 138.1 ± 80.8 pg/mL seen on control chow (Sapna et al., 2006). However. Laurant et al. observed an increase in plasma aldosterone in rats given Mg2+-deficient diet for two and 21 weeks (Laurant et al., 1999). Stimulation of aldosterone levels by dietary Mg2+ restriction seems to occur independently of extracellular fluid volume status, as dietary Mg2+ depletion continued to stimulate increased serum aldosterone even in animals given a high Na+ diet (Solounias and Schwartz, 1975).
Acute intravenous Mg2+ administration also reduces aldosterone levels. A study examining six “healthy,” normotensive volunteers infused MgSO4 at a rate of 0.6 mg/h (5 mEq/hr) and found that plasma aldosterone levels decreased to 4 ± 0.8 ng/dL (111 ± 22 pmol/L) compared with 6 ± 0.2 ng/dL (166 ± 5.5 pmol/L) in controls (p < 0.05) (Ichihara et al., 1993). This occurred despite an increase in plasma renin activity. Corica et al. infused 3 gm (24 mEq) of MgSO4 into “healthy” volunteers and observed reduced serum aldosterone from 18.97 ± 11 ng/dL (526 ± 305 pmol/L) to 6.34 ± 5 ng/dL (176 ± 139 pmol/L) (Corica et al., 1996). This effect did not appear to be fluid volume mediated, as atrial natriuretic peptide levels did not change, and a control infusion with isotonic saline had no significant impact on aldosterone levels. Thus, Mg2+ sulfate infusion reduces aldosterone levels in humans, at least acutely.
Despite these observations, oral supplementation studies in humans have largely failed to demonstrate reduction in circulating aldosterone. One study gave 365 mg (15 mmol) of Mg2+, as Mg2+ aspartate, to 17 subjects for 4 weeks (Cappuccio et al., 1985). Neither blood pressure nor aldosterone changed compared with participants receiving placebo. Participants had a mean baseline serum Mg2+ level of 2.16 mg/dL (0.89 mmol/L), as compared to a normal reference range for Mg2+ of 1.82–2.32 mg/dL (0.75–0.96 mmol/L) from the U.S. National Health and Nutrition Examination Survey I study (Lowenstein and Stanton, 1986). In another study of 15 untreated hypertensive individuals given 600 mg/day of Mg2+ in the form of Mg2+ oxide, blood pressure decreased, but no difference in aldosterone levels was observed (Sanjuliani et al., 1996). In a third study that provided 600 mg of Mg2+ daily as Mg2+ oxide to 17 subjects, Mg2+ decreased blood pressure but again failed to significantly reduce plasma aldosterone (Haga, 1992). In this study, baseline serum Mg2+ levels were 1.88–1.91 mg/dL (0.77–0.79 mmol/L). In these three studies, mean baseline aldosterone levels ranged from roughly 9–13 ng/dL (240–260 pmol/L). This is on the lower side of the reference range of 5–30 ng/dL (140–830 pmol/L) determined in healthy adults on an unrestricted Na+ diet (Al-Dujaili and Edwards, 1978). Thus, although Mg2+ supplementation did not reduce aldosterone levels, these findings may be influenced by the observation that study participants exhibited neither Mg2+-depletion nor elevated aldosterone (e.g., from extracellular fluid volume depletion) at baseline.
Few studies have examined the response of aldosterone to oral Mg2+ supplementation in humans in the context of an aldosterone secreting stimulus, such as extracellular fluid volume depletion or a dietary K+ challenge. An exception is a study that measured aldosterone changes in response to an hour of exercise in nine men (Golf et al., 1984). Exercise increased plasma aldosterone from 9.4 ± 5.0 ng/dL (260 ± 140 pmol/L) to 19.1 ± 13.7 ng/dL (530 ± 370 pmol/L), perhaps secondary to either extracellular fluid volume depletion or increased plasma K+. Two weeks of daily oral supplementation with 360 mg (15 mmol) Mg2+ as Mg2+ aspartate abrogated this increase in aldosterone, leading to aldosterone levels before and after exercise that were 13.7 ± 3.2 (380 ± 90 pmol/L) and 11.9 ± 7.9 ng/dL (330 ± 220 pmol/L), respectively. Whether oral Mg2+ supplementation influences aldosterone secretion in response to thiazide diuretics used for hypertension, which both deplete Mg2+ and stimulate aldosterone secretion through fluid volume contraction, seems likely, though unreported.
What are the mechanisms by which Mg2+ may modulate aldosterone secretion? This effect could be mediated by a direct influence upon aldosterone-secreting zona glomerulosa cells or via modulation of upstream components the renin-angiotensin-aldosterone system. A direct effect upon adrenal function is suggested by studies showing that Mg2+ exerted a voltage-dependent blockade of inwardly rectifying K+ and Ca2+ channels in adrenal glomerulosa cells (Vassilev et al., 1992; Lotshaw and Li, 1996). Activity of each of these channel types modulates aldosterone secretion. Furthermore, in cultured adrenal cells, higher extracellular Mg2+ reduced basal aldosterone secretion (Antonipillai et al., 1997). Extracellular Mg2+ also attenuates stimulation of aldosterone secretion by angiotensin II. In adrenal cells in culture, increased extracellular Mg2+ reduced angiotensin II-stimulated aldosterone release (Atarashi et al., 1989; Antonipillai et al., 1997). This effect was also observed in vivo, as rats given an infusion of angiotensin II in combination with Mg2+ sulfate exhibited diminished plasma aldosterone, as compared with rats given angiotensin II alone (Atarashi et al., 1990). Evidence for an influence of Mg2+ on angiotensin II-mediated aldosterone secretion in humans is provided by a study showing that 3 weeks on a very low (<1 mEq/day) Mg2+ diet augmented angiotensin II-stimulated aldosterone secretion (Rude et al., 1989). This increase in aldosterone secretion was partially rescued by acute intravenous Mg2+ repletion with Mg2+ sulfate. Together, these studies suggest that extracellular Mg2+ decreases sensitivity of adrenal glomerulosa cells to angiotensin II-stimulated aldosterone secretion.
Studies examining the influence of Mg2+ on upstream components of the renin-angiotensin-aldosterone system are more mixed. In laboratory rats, 14 weeks on a Mg2+-deficient diet resulted in no difference in angiotensin II levels (Jin et al., 2013). However, another study examining rats on a Mg2+-deficient diet for 6 days found increased angiotensin II, as well as increased plasma renin activity (Sapna et al., 2006). A third study examining dietary Mg2+-restriction in rats found increased plasma renin activity at 2 weeks but not at 21 weeks (Laurant et al., 1999). In dogs given a low Mg2+ diet, plasma renin activity did not increase at any of several time-points through 90 days, although plasma aldosterone excretion did increase (Helber et al., 1972). In humans, serum Mg2+ was found to correlate directly with renin activity (Lind et al., 1989). In contrast, acute Mg2+ sulfate infusion increased plasma renin activity (Ichihara et al., 1993).
Taken together, these studies suggest that systemic Mg2+ depletion promotes aldosterone secretion without necessarily stimulating increased renin or angiotensin II levels. Whether this increase in aldosterone is reversible or leads to persistent aldosterone secretion (e.g., by promoting adrenal hyperplasia) remains unexplored. Although mechanisms discussed above suggest that systemic Mg2+ depletion should promote tubular reabsorption of Na+, modulation of extracellular fluid volume by Mg2+ has not been described. In a study examining the impact of dietary Mg2+ depletion upon blood pressure and fluid volume in mice, although a Mg2+-deficient diet increased blood pressure, it did not increase body fluid content, as measured using quantitative magnetic resonance (Pitzer Mutchler et al., 2023). However, Na+ overload promotes hypertension via mechanisms that may be Mg2+-sensitive, discussed below.
Pro-hypertensive inflammatory processes
Na+ can increase blood pressure via at least two pro-inflammatory mechanisms (Kirabo, 2017). First, high Na+ diet increases oxidative stress in antigen-presenting cells (APCs). Peroxidation of arachidonic acid forms isolevuglandins (IsoLGs), γ ketoaldehydes capable of covalently modifying endogenous proteins. Modified proteins are presented at the APC surface, stimulating inflammation. Genetic prevention of IsoLG formation or pharmacologic scavenging of IsoLGs prevents salt-induced hypertension in mouse models (Kirabo et al., 2014; Barbaro et al., 2017). Second, high Na+ diet increases expression of NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3), a key component of the inflammasome, in the renal medulla and other tissues (Zhu et al., 2016). The NLRP3 inflammasome catalyzes the production and secretion of the proinflammatory cytokines, IL-1β and IL-18 (Elijovich et al., 2021). Genetic or pharmacologic impairment of the NLRP3 inflammasome prevented blood pressure increases in mouse models of hypertension (Wang et al., 2014; Pitzer et al., 2022).
A Mg2+-deficient diet activates these hypertension-promoting inflammatory processes. In laboratory animals, dietary Mg2+ deficiency stimulates leukocytosis and circulating inflammatory cytokine levels (Weglicki et al., 1992; Malpuech-Brugère et al., 2000; Van Orden et al., 2006). Oral Mg2+ supplementation in humans suppresses circulating C-reactive protein (Mazidi et al., 2018). In mice experiencing hypertension in response to dietary Mg2+ restriction, circulating IL-1β levels increase (Pitzer Mutchler et al., 2023). NLRP3 and IsoLG positivity in splenic and renal dendritic cells increase to levels comparable to a high Na+ diet. Whether hypertension induced by dietary Mg2+ depletion is dependent upon activation of the NLRP3 inflammasome or production of IsoLGs was not examined, but these findings are consistent with a contribution of Mg2+ depletion to hypertension-promoting inflammation. Interesting questions remain regarding the mechanisms by which dietary Mg2+ restriction induces inflammation and whether dietary Mg2+ supplementation protects against high salt diet-mediated inflammation and hypertension.
Clinical implications
The likely contributions of Mg2+-deficiency to increased blood pressure and to other aspects of cardiovascular disease suggest that optimal management of hypertension should include attention to Mg2+ balance. Clinicians should have a high index of suspicion for Mg2+-depletion in hypertensive patients, given that 1) dietary Mg2+ deficiency is common (USDA Agricultural Research Service, 2019), 2) hypertension is associated with Mg2+ depletion, even in untreated patients (Rodríguez-Moran and Guerrero-Romero, 2014; Rodríguez-Ramírez et al., 2015), 3) common comorbidities (such as diabetes mellitus) are also associated with Mg2+ depletion (Ray et al., 2023), and 4) commonly prescribed medications promote Mg2+ deficiency, including thiazide-type and loop diuretics, and proton pump inhibitors (Ray et al., 2023). Clinicians should not rely solely upon measurement of plasma Mg2+ levels for determination of Mg2+ depletion, since 1) less than 1% of bodily Mg2+ resides in the plasma, so that plasma Mg2+ levels do not faithfully reflect bodily Mg2+ stores, and 2) “normal” reference ranges typically used for plasma Mg2+ are likely inappropriately low (Costello et al., 2016; Micke et al., 2021). Clinicians should consider prescription of a well-absorbed oral Mg2+ supplement. Well-absorbed supplements include most organic salts and possibly the chloride salt, as discussed elsewhere (Ray et al., 2023). Clinicians should have a low threshold for prescribing agents that oppose urinary Mg2+-wasting, such as K+ and Mg2+-sparing diuretics (e.g., spironolactone or amiloride), and SGLT2 inhibitors (Ray et al., 2020). It is likely that improved Mg2+ balance associated with these agents contributes to improved cardiovascular benefits associated with their use (Ray, 2020).
Conclusion
Mg2+ depletion likely promotes increased blood pressure via numerous mechanisms, described above, including effects on the sympathetic nervous system, vascular tone, the RAAS system, systemic Na+ and K+ balance, and inflammatory processes. Given the widespread prevalence of Mg2+ depletion and the tendency of some approaches to treating hypertension to induce Mg2+-depletion, attention to systemic Mg2+ deficiency has the potential to improve clinical management of hypertension and cardiovascular outcomes.
Author contributions
ZA: Writing–original draft, writing–review and editing. ER: Conceptualization, funding acquisition, writing–original draft, writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Carl W. Gottschalk Research Scholar Grant from the American Society of Nephrology supported salary for ECR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Dujaili E., Edwards C. (1978). The development and application of a direct radioimmunoassay for plasma aldosterone using 125I-labeled ligand—comparison of three methods. J. Clin. Endocrinol. Metab. 46, 105–113. doi:10.1210/jcem-46-1-105
Altura B. M., Altura B. T., Carella A. (1983). Magnesium deficiency-induced spasms of umbilical vessels: relation to preeclampsia, hypertension, growth retardation. Science 221, 376–378. doi:10.1126/science.6867714
Altura B. M., Altura B. T., Gebrewold A., Ising H., Günther T. (1984). Magnesium deficiency and hypertension: correlation between magnesium-deficient diets and microcirculatory changes in situ. Science 223, 1315–1317. doi:10.1126/science.6701524
Altura B. M., Turlapaty P. D. (1982). Withdrawal of magnesium enhances coronary arterial spasms produced by vasoactive agents. Br. J. Pharmacol. 77, 649–659. doi:10.1111/j.1476-5381.1982.tb09343.x
Antonipillai I., Hong H., Horton R. (1997). Magnesium modulates ouabain action on angiotensin II-induced aldosterone synthesis in vitro. Magnes. Res. 10, 307–313.
Asbaghi O., Hosseini R., Boozari B., Ghaedi E., Kashkooli S., Moradi S. (2021). The effects of magnesium supplementation on blood pressure and Obesity measure Among type 2 diabetes patient: a systematic review and meta-analysis of randomized controlled trials. Biol. Trace Elem. Res. 199, 413–424. doi:10.1007/s12011-020-02157-0
Atarashi K., Matsuoka H., Takagi M., Sugimoto T. (1989). Magnesium ion: a possible physiological regulator of aldosterone production. Life Sci. 44, 1483–1489. doi:10.1016/0024-3205(89)90327-5
Atarashi K., Takagi M., Matsuoka H., Sugimoto T. (1990). Effects of magnesium on changes in blood pressure and plasma aldosterone induced by angiotensin II. Am. J. Hypertens. 3, 488–490. doi:10.1093/ajh/3.6.488
Barbaro N. R., Foss J. D., Kryshtal D. O., Tsyba N., Kumaresan S., Xiao L., et al. (2017). Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 21, 1009–1020. doi:10.1016/j.celrep.2017.10.002
Berthelot A., Esposito J. (1983). Effects of dietary magnesium on the development of hypertension in the spontaneously hypertensive rat. J. Am. Coll. Nutr. 2, 343–353. doi:10.1080/07315724.1983.10719931
Blache D., Devaux S., Joubert O., Loreau N., Schneider M., Durand P., et al. (2006). Long-term moderate magnesium-deficient diet shows relationships between blood pressure, inflammation and oxidant stress defense in aging rats. Free Radic. Biol. Med. 41, 277–284. doi:10.1016/j.freeradbiomed.2006.04.008
Blackfan K., Mills C. (1923). The effect of two per cent magnesium Sulphate Solution on cerebral Symptoms in acute nephritis. Tr. Am. Pediat. Soc. 35, 197.
Blackfan K. D., McKhann C. F. (1931). Acute glomerular nephritis in children: treatment of the cerebral manifestations. JAMA 97, 1052–1055. doi:10.1001/jama.1931.02730150008003
Briel R., Lippert T., Zahradnik H. (1987). Changes in blood coagulation, thrombocyte function and vascular prostacyclin synthesis caused by magnesium sulfate. Geburtshilfe Frauenheilkd. 47, 332–336. doi:10.1055/s-2008-1035831
Brown R. S. (1984). Potassium homeostasis and clinical implications. Am. J. Med. 77, 3–10. doi:10.1016/s0002-9343(84)80002-9
Cappuccio F., Markandu N., Beynon G., Shore A., Sampson B., MacGregor G. (1985). Lack of effect of oral magnesium on high blood pressure: a double blind study. Br. Med. J. Clin. Res. Ed. 291, 235–238. doi:10.1136/bmj.291.6490.235
Carlin Schooley M., Franz K. B. (2002). Magnesium deficiency during pregnancy in rats increases systolic blood pressure and plasma nitrite. Am. J. Hypertens. 15, 1081–1086. doi:10.1016/s0895-7061(02)03064-9
Corica F., Allegra A., Ientile R., Buemi M., Cucinotta G., Bonanzinga S., et al. (1996). Effects of the intravenous magnesium administration on aldosterone and atrial natriuretic factor plasma concentrations. Nephron 73, 739–741. doi:10.1159/000189187
Costello R. B., Elin R. J., Rosanoff A., Wallace T. C., Guerrero-Romero F., Hruby A., et al. (2016). Perspective: the Case for an evidence-based reference Interval for serum magnesium: the time has come. Adv. Nutr. 7, 977–993. doi:10.3945/an.116.012765
Dampney R. A. L., Horiuchi J., Tagawa T., Fontes M. A. P., Potts P. D., Polson J. W. (2003). Medullary and supramedullary mechanisms regulating sympathetic vasomotor tone. Acta Physiol. Scand. 177, 209–218. doi:10.1046/j.1365-201X.2003.01070.x
de Baaij J. H., Hoenderop J. G., Bindels R. J. (2015). Magnesium in man: implications for health and disease. Physiol. Rev. 95, 1–46. doi:10.1152/physrev.00012.2014
Dibaba D. T., Xun P., Song Y., Rosanoff A., Shechter M., He K. (2017). The effect of magnesium supplementation on blood pressure in individuals with insulin resistance, prediabetes, or noncommunicable chronic diseases: a meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 106, 921–929. doi:10.3945/ajcn.117.155291
Dingledine R., Borges K., Bowie D., Traynelis S. F. (1999). The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61.
Dørup I., Clausen T. (1993). Correlation between magnesium and potassium contents in muscle: role of Na(+)-K+ pump. Am. J. Physiol. 264, C457–C463. doi:10.1152/ajpcell.1993.264.2.C457
Dunnett J., Nayler W. G. (1978). Calcium efflux from cardiac sarcoplasmic reticulum: effects of calcium and magnesium. J. Mol. Cell Cardiol. 10, 487–498. doi:10.1016/0022-2828(78)90369-3
Elijovich F., Kleyman T. R., Laffer C. L., Kirabo A. (2021). Immune mechanisms of dietary salt-induced hypertension and kidney disease: Harry Goldblatt Award for early Career Investigators 2020. Hypertension 78, 252–260. doi:10.1161/HYPERTENSIONAHA.121.16495
Elkjær M.-L., Kwon T.-H., Wang W., Nielsen J., Knepper M. A., Frøkiær J., et al. (2002). Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am. J. Physiol. Ren. Physiol. 283, F1376–F1388. doi:10.1152/ajprenal.00186.2002
Evans G. H., Weaver C. M., Harrington D. D., Babbs C. F. (1989). Dietary magnesium does not affect blood pressure in spontaneously hypertensive rats. Clin. Exp. Hypertens. A 11, 619–632. doi:10.3109/10641968909035364
Fanestil D. D., Hyde R. H., Blakely P., Vaughn D. A. (1999). Dietary magnesium, not calcium, regulates renal thiazide receptor. J. Am. Soc. Nephrol. 10, 458–463. doi:10.1681/asn.v103458
Filippini T., Naska A., Kasdagli M. I., Torres D., Lopes C., Carvalho C., et al. (2020). Potassium intake and blood pressure: a dose-response meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 9, e015719. doi:10.1161/JAHA.119.015719
Fuchs F. D., Whelton P. K. (2020). High blood pressure and cardiovascular disease. Hypertension 75, 285–292. doi:10.1161/HYPERTENSIONAHA.119.14240
Gamboa A., Shibao C., Diedrich A., Choi L., Pohar B., Jordan J., et al. (2007). Contribution of endothelial nitric oxide to blood pressure in humans. Hypertension 49, 170–177. doi:10.1161/01.HYP.0000252425.06216.26
Gasallaherraiz J., Rhee S., Isales C. M. (1995). Calcium sensitive Probes for the measurement of intracellular calcium: effects of buffer system and magnesium concentration. Biochem. Biophys. Res. Commun. 214, 373–388. doi:10.1006/bbrc.1995.2298
Ginn H. E., Cade R., McCallum T., Fregley M. (1967). Aldosterone secretion in magnesium-deficient rats. Endocrinology 80, 969–971. doi:10.1210/endo-80-5-969
Golf S. W., Happel O., Graef V., Seim K. E. (1984). Plasma aldosterone, cortisol and electrolyte concentrations in physical exercise after magnesium supplementation. J. Clin. Chem. Clin. Biochem. 22, 717–721. doi:10.1515/cclm.1984.22.11.717
Green D. J., Dawson E. A., Groenewoud H. M. M., Jones H., Thijssen D. H. J. (2014). Is flow-mediated Dilation nitric oxide mediated? Hypertension 63, 376–382. doi:10.1161/HYPERTENSIONAHA.113.02044
Haga H. (1992). Effects of dietary magnesium supplementation on diurnal variations of blood pressure and plasma Na+, K(+)-ATPase activity in essential hypertension. Jpn. Heart J. 33, 785–800. doi:10.1536/ihj.33.785
Hasselbach W., Fassold E., Migala A., Rauch B. (1981). Magnesium dependence of sarcoplasmic reticulum calcium transport. Fed. Proc. 40, 2657–2661.
Helber A., Hayduk K., Nowaczynski W., Brecht H., Küchel O., Genest J. (1972). Effect of experimentally altered plasma-magnesium concentration on aldosterone secretion in dogs. Res. Exp. Med. 157, 336–346. doi:10.1007/BF01852077
Hollifield J. W. (1986). Thiazide treatment of hypertension: effects of thiazide diuretics on serum potassium, magnesium, and ventricular ectopy. Am. J. Med. 80, 8–12. doi:10.1016/0002-9343(86)90335-9
Huang C. L., Kuo E. (2007). Mechanism of hypokalemia in magnesium deficiency. J. Am. Soc. Nephrol. 18, 2649–2652. doi:10.1681/ASN.2007070792
Ichihara A., Suzuki H., Saruta T. (1993). Effects of magnesium on the renin-angiotensin-aldosterone system in human subjects. J. Lab. Clin. Med. 122, 432–440. doi:10.5555/uri:pii:002221439390132I
Itokawa Y., Tanaka C., Fujiwara M. (1974). Changes in body temperature and blood pressure in rats with calcium and magnesium deficiencies. J. Appl. Physiol. 37, 835–839. doi:10.1152/jappl.1974.37.6.835
Iwai N., Katsuya T., Ishikawa K., Mannami T., Ogata J., Higaki J., et al. (1999). Human prostacyclin synthase gene and hypertension: the Suita Study. Circulation 100, 2231–2236. doi:10.1161/01.cir.100.22.2231
James M. F., Beer R. E., Esser J. D. (1989). Intravenous magnesium sulfate inhibits catecholamine release associated with tracheal intubation. Anesth. Analg. 68, 772–776. doi:10.1213/00000539-198906000-00015
Jin K., Kim T. H., Kim Y. H., Kim Y. W. (2013). Additional antihypertensive effect of magnesium supplementation with an angiotensin II receptor blocker in hypomagnesemic rats. Korean J. Intern Med. 28, 197–205. doi:10.3904/kjim.2013.28.2.197
Kagiyama S., Tsuchihashi T., Phillips M. I., Abe I., Matsumura K., Fujishima M. (2001). Magnesium decreases arterial pressure and inhibits cardiovascular responses induced by N-methyl-D-aspartate and metabotropic glutamate receptors stimulation in rostral ventrolateral medulla. J. Hypertens. 19, 2213–2219. doi:10.1097/00004872-200112000-00015
Kirabo A. (2017). A new paradigm of sodium regulation in inflammation and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 313, R706-R710–R710. doi:10.1152/ajpregu.00250.2017
Kirabo A., Fontana V., de Faria A. P., Loperena R., Galindo C. L., Wu J., et al. (2014). DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Invest 124, 4642–4656. doi:10.1172/JCI74084
Kolte D., Vijayaraghavan K., Khera S., Sica D. A., Frishman W. H. (2014). Role of magnesium in cardiovascular diseases. Cardiol. Rev. 22, 182–192. doi:10.1097/CRD.0000000000000003
Kudryavtseva O., Lyngsø K. S., Jensen B. L., Dimke H. (2024). Nitric oxide, endothelium-derived hyperpolarizing factor, and smooth muscle-dependent mechanisms contribute to magnesium-dependent vascular relaxation in mouse arteries. Acta Physiol. 240, e14096. doi:10.1111/apha.14096
Kumagai A., Takeda S., Sohara E., Uchida S., Iijima H., Itakura A., et al. (2021). Dietary magnesium insufficiency induces salt-sensitive hypertension in mice associated with reduced kidney catechol-o-methyl transferase activity. Hypertension 78, 138–150. doi:10.1161/HYPERTENSIONAHA.120.16377
Laurant P., Dalle M., Berthelot A., Rayssiguier Y. (1999). Time-course of the change in blood pressure level in magnesium-deficient Wistar rats. Br. J. Nutr. 82, 243–251. doi:10.1017/s0007114599001427
Laurant P., Moussard C., Alber D., Henry J., Berthelot A. (1992). In vivo and in vitro magnesium effects on aortic prostacyclin generation in DOCA-salt hypertensive rats. Prostagl. Leukot. Essent. Fat. Acids 47, 183–186. doi:10.1016/0952-3278(92)90236-c
Laurant P., Robin S., Berthelot A. (1997). Magnesium deficiency increases vasoconstrictor activity without affecting blood pressure of aged spontaneously hypertensive rats. Magnes. Res. 10, 107–117.
Li D.-P., Pan H.-L. (2017). Glutamatergic regulation of hypothalamic presympathetic neurons in hypertension. Curr. Hypertens. Rep. 19, 78–87. doi:10.1007/s11906-017-0776-4
Lind L., Wide L., Sörensen O. H., Ljunghall S. (1989). An association between mineral metabolism and the renin-aldosterone system in human hypertension. J. Hum. Hypertens. 3, 137–140.
Liu D. T., Turner S. W., Wang M. X., Whitworth J. A. (1994). Effects of dietary magnesium on blood pressure and vascular lesions in hypertensive rats. Pathology 26, 365–369. doi:10.1080/00313029400169022
Liu Z., Xie J., Wu T., Truong T., Auchus R. J., Huang C. L. (2011). Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum. Mol. Genet. 20, 855–866. doi:10.1093/hmg/ddq525
Lotshaw D. P., Li F. (1996). Angiotensin II activation of Ca(2+)-permeant nonselective cation channels in rat adrenal glomerulosa cells. Am. J. Physiol. 271, C1705–C1715. doi:10.1152/ajpcell.1996.271.5.C1705
Lowenstein F. W., Stanton M. F. (1986). Serum magnesium levels in the United States, 1971-1974. J. Am. Coll. Nutr. 5, 399–414. doi:10.1080/07315724.1986.10720143
Lowney P., Gershwin M. E., Hurley L. S., Stern J. S., Keen C. L. (1988). The effect of variable magnesium intake on potential factors influencing endurance capacity. Biol. Trace Elem. Res. 16, 1–18. doi:10.1007/BF02795329
Luthringer C., Rayssiguier Y., Gueux E., Berthelot A. (1988). Effect of moderate magnesium deficiency on serum lipids, blood pressure and cardiovascular reactivity in normotensive rats. Br. J. Nutr. 59, 243–250. doi:10.1079/bjn19880031
Macintyre I., Davidsson D. (1958). The production of secondary potassium depletion, sodium retention, nephrocalcinosis and hypercalcaemia by magnesium deficiency. Biochem. J. 70, 456–462. doi:10.1042/bj0700456
Maier J. A., Bernardini D., Rayssiguier Y., Mazur A. (2004). High concentrations of magnesium modulate vascular endothelial cell behaviour in vitro. Biochim. Biophys. Acta 1689, 6–12. doi:10.1016/j.bbadis.2004.02.004
Malpuech-Brugère C., Nowacki W., Daveau M., Gueux E., Linard C., Rock E., et al. (2000). Inflammatory response following acute magnesium deficiency in the rat. Biochim. Biophys. Acta Mol. Basis Dis. 1501, 91–98. doi:10.1016/s0925-4439(00)00018-1
Manitius A., Epstein F. H. (1963). Some observations on the influence of a magnesium-deficient diet on rats, with special reference to renal concentrating ability. J. Clin. Invest 42, 208–215. doi:10.1172/JCI104707
Marcoux A. A., Slimani S., Tremblay L. E., Frenette-Cotton R., Garneau A. P., Isenring P. (2019). Regulation of Na(+)-K(+)-Cl(-) cotransporter type 2 by the with no lysine kinase-dependent signaling pathway. Am. J. Physiol. Cell Physiol. 317, C20-C30–c30. doi:10.1152/ajpcell.00041.2019
Mazidi M., Rezaie P., Banach M. (2018). Effect of magnesium supplements on serum C-reactive protein: a systematic review and meta-analysis. Arch. Med. Sci. 14, 707–716. doi:10.5114/aoms.2018.75719
Micke O., Vormann J., Kraus A., Kisters K. (2021). Serum magnesium: time for a standardized and evidence-based reference range. Magnes. Res. 34, 84–89. doi:10.1684/mrh.2021.0486
Mizushima S., Cappuccio F., Nichols R., Elliott P. (1998). Dietary magnesium intake and blood pressure: a qualitative overview of the observational studies. J. Hum. Hypertens. 12, 447–453. doi:10.1038/sj.jhh.1000641
Moriguchi T., Urushiyama S., Hisamoto N., Iemura S., Uchida S., Natsume T., et al. (2005). WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 280, 42685–42693. doi:10.1074/jbc.M510042200
Murasato Y., Harada Y., Ikeda M., Nakashima Y., Hayashida Y. (1999). Effect of magnesium deficiency on autonomic circulatory regulation in conscious rats. Hypertension 34, 247–252. doi:10.1161/01.hyp.34.2.247
Nadler J. L., Goodson S., Rude R. K. (1987). Evidence that prostacyclin mediates the vascular action of magnesium in humans. Hypertension 9, 379–383. doi:10.1161/01.hyp.9.4.379
Overlack A., Zenzen J. G., Ressel C., Müller H. M., Stumpe K. O. (1987). Influence of magnesium on blood pressure and the effect of nifedipine in rats. Hypertension 9, 139–143. doi:10.1161/01.hyp.9.2.139
Park H. J., Curry J. N., McCormick J. A. (2013). Regulation of NKCC2 activity by inhibitory SPAK isoforms: KS-SPAK is a more potent inhibitor than SPAK2. Am. J. Physiol. Ren. Physiol. 305, F1687–F1696. doi:10.1152/ajprenal.00211.2013
Patrick J. (1977). Assessment of body potassium stores. Kidney Int. 11, 476–490. doi:10.1038/ki.1977.65
Pearson P. J., Evora P. R., Seccombe J. F., Schaff H. V. (1998). Hypomagnesemia inhibits nitric oxide release from coronary endothelium: protective role of magnesium infusion after cardiac operations. Ann. Thorac. Surg. 65, 967–972. doi:10.1016/s0003-4975(98)00020-4
Pitzer A., Elijovich F., Laffer C. L., Ertuglu L. A., Sahinoz M., Saleem M., et al. (2022). DC ENaC-dependent inflammasome activation contributes to salt-sensitive hypertension. Circ. Res. 131, 328–344. doi:10.1161/CIRCRESAHA.122.320818
Pitzer Mutchler A., Huynh L., Patel R., Lam T., Bain D., Jamison S., et al. (2023). The role of dietary magnesium deficiency in inflammatory hypertension. Front Physiol 14, 1167904. doi:10.3389/fphys.2023.1167904
Ploth D. W., Sawin L. L., DiBona G. F. (1976). Effect of magnesium on rat nephron sodium reabsorption: a segmental analysis. Am. J. Physiol. 230, 398–402. doi:10.1152/ajplegacy.1976.230.2.398
Poorolajal J., Zeraati F., Soltanian A. R., Sheikh V., Hooshmand E., Maleki A. (2017). Oral potassium supplementation for management of essential hypertension: a meta-analysis of randomized controlled trials. PLoS One 12, e0174967. doi:10.1371/journal.pone.0174967
Raij L., Lüscher T. F., Vanhoutte P. M. (1988). High potassium diet augments endothelium-dependent relaxations in the Dahl rat. Hypertension 12, 562–567. doi:10.1161/01.hyp.12.6.562
Ray E., Mohan K., Ahmad S., Wolf M. T. F. (2023). Physiology of a Forgotten electrolyte-magnesium Disorders. Adv. Kidney Dis. Health 30, 148–163. doi:10.1053/j.akdh.2022.12.001
Ray E. C. (2020). Evolving understanding of cardiovascular protection by SGLT2 inhibitors: focus on renal protection, myocardial effects, uric acid, and magnesium balance. Curr. Op. Pharmacol. 54, 11–17. doi:10.1016/j.coph.2020.06.001
Ray E. C., Boyd-Shiwarski C. R., Liu P., Novacic D., Cassiman D. (2020). SGLT2 inhibitors for treatment of Refractory hypomagnesemia: a Case report of 3 patients. Kidney Med. 2, 359–364. doi:10.1016/j.xkme.2020.01.010
Rees D., Palmer R., Moncada S. (1989). Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. U. S. A. 86, 3375–3378. doi:10.1073/pnas.86.9.3375
Resnick L. M., Gupta R. K., Laragh J. H. (1984). Intracellular free magnesium in erythrocytes of essential hypertension: relation to blood pressure and serum divalent cations. Proc. Natl. Acad. Sci. U. S. A. 81, 6511–6515. doi:10.1073/pnas.81.20.6511
Resnick L. M., Laragh J. H., Sealey J. E., Alderman M. H. (1983). Divalent cations in essential hypertension: relations between serum ionized calcium, magnesium, and plasma renin activity. N. Engl. J. Med. 309, 888–891. doi:10.1056/NEJM198310133091504
Richardson C., Sakamoto K., de los Heros P., Deak M., Campbell D. G., Prescott A. R., et al. (2011). Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J. Cell Sci. 124, 789–800. doi:10.1242/jcs.077230
Rinehart J., Kahle K. T., de Los Heros P., Vazquez N., Meade P., Wilson F. H., et al. (2005). WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl-cotransporters required for normal blood pressure homeostasis. Proc. Natl. Acad. Sci. U. S. A. 102, 16777–16782. doi:10.1073/pnas.0508303102
Rodríguez-Moran M., Guerrero-Romero F. (2014). Hypomagnesemia and prehypertension in otherwise healthy individuals. Eur. J. Intern Med. 25, 128–131. doi:10.1016/j.ejim.2013.08.706
Rodríguez-Ramírez M., Simental-Mendía L. E., González-Ortiz M., Martínez-Abundis E., Madero A., Brito-Zurita O., et al. (2015). Prevalence of prehypertension in Mexico and its association with hypomagnesemia. Am. J. Hypertens. 28, 1024–1030. doi:10.1093/ajh/hpu293
Rosanoff A., Costello R. B., Johnson G. H. (2021). Effectively prescribing oral magnesium therapy for hypertension: a Categorized systematic review of 49 clinical trials. Nutrients 13, 195. doi:10.3390/nu13010195
Rosanoff A., Weaver C. M., Rude R. K. (2012). Suboptimal magnesium status in the United States: are the health consequences underestimated? Nutr. Rev. 70, 153–164. doi:10.1111/j.1753-4887.2011.00465.x
Rosanoff A., West C., Elin R. J., Micke O., Baniasadi S., Barbagallo M., et al. (2022). Recommendation on an updated standardization of serum magnesium reference ranges. Eur. J. Nutr. 61, 3697–3706. doi:10.1007/s00394-022-02916-w
Rude R., Manoogian C., Ehrlich L., DeRusso P., Ryzen E., Nadler J. (1989). Mechanisms of blood pressure regulation by magnesium in man. Magnesium 8, 266–273.
Sanjuliani A. F., de Abreu Fagundes V. G., Francischetti E. A. (1996). Effects of magnesium on blood pressure and intracellular ion levels of Brazilian hypertensive patients. Int. J. Cardiol. 56, 177–183. doi:10.1016/0167-5273(96)02716-7
Sapna S., Ranjith S. K., Shivakumar K. (2006). Cardiac fibrogenesis in magnesium deficiency: a role for circulating angiotensin II and aldosterone. Am. J. Physiol. Heart Circ. Physiol. 291, H436–H440. doi:10.1152/ajpheart.01185.2005
Satake K., Lee J. D., Shimizu H., Uzui H., Mitsuke Y., Yue H., et al. (2004). Effects of magnesium on prostacyclin synthesis and intracellular free calcium concentration in vascular cells. Magnes. Res. 17, 20–27.
Sharma N., Cho D. H., Kim S. Y., Bhattarai J. P., Hwang P. H., Han S. K. (2012). Magnesium sulfate suppresses L-type calcium currents on the basilar artery smooth muscle cells in rabbits. Neurol. Res. 34, 291–296. doi:10.1179/1743132812Y.0000000016
Shechter M., Sharir M., Labrador M. J. P., Forrester J., Silver B., Merz C. N. B. (2000). Oral magnesium therapy improves endothelial function in patients with coronary artery disease. Circulation 102, 2353–2358. doi:10.1161/01.cir.102.19.2353
Shils M. E. (1969). Experimenal human magnesium depletion. Medicine 48, 61. doi:10.1097/00005792-196901000-00003
Shimosawa T., Takano K., Ando K., Fujita T. (2004). Magnesium inhibits norepinephrine release by blocking N-type calcium channels at peripheral sympathetic nerve endings. Hypertension 44, 897–902. doi:10.1161/01.HYP.0000146536.68208.84
Shirley D., Zewde T., Walter S. (1990). Renal function in normal and potassium-depleted rats before and after preparation for micropuncture experimentation. Pflügers Arch. 416, 74–79. doi:10.1007/BF00370225
Soleimani M., Bergman J. A., Hosford M. A., McKinney T. D. (1990). Potassium depletion increases luminal Na+/H+ exchange and basolateral Na+:CO3=:HCO3- cotransport in rat renal cortex. J. Clin. Invest 86, 1076–1083. doi:10.1172/JCI114810
Solounias B. M., Schwartz R. (1975). The effect of magnesium deficiency on serum aldosterone in rats fed two levels of sodium. Life Sci. 17, 1211–1217. doi:10.1016/0024-3205(75)90129-0
Terker A. S., Castañeda-Bueno M., Ferdaus M. Z., Cornelius R. J., Erspamer K. J., Su X. T., et al. (2018). With no lysine kinase 4 modulates sodium potassium 2 chloride cotransporter activity in vivo. Am. J. Physiol. Ren. Physiol. 315, F781-F790–f790. doi:10.1152/ajprenal.00485.2017
Terker A. S., Zhang C., McCormick J. A., Lazelle R. A., Zhang C., Meermeier N. P., et al. (2015). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21, 39–50. doi:10.1016/j.cmet.2014.12.006
Tomiyasu T., Chishaki A., Nakamura M. (1998). Magnesium deficiency in adult rats promotes the induction of ventricular tachycardia by the administration of epinephrine. Heart Vessels 13, 122–131. doi:10.1007/BF01747829
Touyz R. M., Milne F. J., Reinach S. G. (1992). Intracellular Mg2+, Ca2+, Na2+ and K+ in platelets and erythrocytes of essential hypertension patients: relation to blood pressure. Clin. Exp. Hypertens. A 14, 1189–1209. doi:10.3109/10641969209038200
Tsao C. W., Aday A. W., Almarzooq Z. I., Alonso A., Beaton A. Z., Bittencourt M. S., et al. (2022). Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation 145, e153–e639. doi:10.1161/CIR.0000000000001052
United States Centers for Disease Control and Prevention (2015). Hypertension. National Center for Health Statistics. Available at http://www.cdc.gov/nchs/fastats/hypertension.htm.
USDA Agricultural Research Service (2019). Usual nutrient intake from food and beverages, by gender and age, what We Eat in America, NHANES 2013–2016. Food Surv. Res. Group Beltsv. (MD).
van Bommel E., Cleophas T. (2012). Potassium treatment for hypertension in patients with high salt intake: a meta-analysis. Int. J. Clin. Pharmacol. Ther. 50, 478–482. doi:10.5414/CP201724
Van Orden R., Eggett D. L., Franz K. B. (2006). Influence of graded magnesium deficiencies on white blood cell counts and lymphocyte subpopulations in rats. Magnes. Res. 19, 93–101.
Vassilev P. M., Kanazirska M. V., Quinn S. J., Tillotson D. L., Williams G. H. (1992). K+ channels in adrenal zona glomerulosa cells. I. Characterization of distinct channel types. Am. J. Physiol. 263, E752–E759. doi:10.1152/ajpendo.1992.263.4.E752
von Euler U., Lishajko F. (1963). Effect of adenine nucleotides on catecholamine release and uptake in isolated adrenergic nerve granules. Acta Physiol. Scand. 59, 454–461. doi:10.1111/j.1748-1716.1963.tb02761.x
von Euler U., Lishajko F. (1973). Effects of Mg2+ and Ca2+ on noradrenaline release and uptake in adrenergic nerve granules in differential media. Acta Physiol. Scand. 89, 415–422. doi:10.1111/j.1748-1716.1973.tb05536.x
Wang Q., So A., Nussberger J., Ives A., Bagnoud N., Shäefer S., et al. (2014). Renin-dependent hypertension in mice requires the NLRP3-inflammasome. J. Hypertens. 3, 2167–1095. 10001. doi:10.4172/2167-1095.1000187
Wang W., Lu M., Hebert S. C. (1996). Cytochrome P-450 metabolites mediate extracellular Ca (2+)-induced inhibition of apical K+ channels in the TAL. Am. J. Physiol. Cell Physiol. 271, C103–C111. doi:10.1152/ajpcell.1996.271.1.C103
Webb R. C. (2003). Smooth muscle contraction and relaxation. Adv. Physiol. Educ. 27, 201–206. doi:10.1152/advances.2003.27.4.201
Weglicki W. B., Phillips T. M., Freedman A. M., Cassidy M. M., Dickens B. F. (1992). Magnesium-deficiency elevates circulating levels of inflammatory cytokines and endothelin. Mol. Cell Biochem. 110, 169–173. doi:10.1007/BF02454195
Whang R., Chrysant S., Dillard B., Smith W., Fryer A. (1982). Hypomagnesemia and hypokalemia in 1,000 treated ambulatory hypertensive patients. J. Am. Coll. Nutr. 1, 317–322. doi:10.1080/07315724.1982.10719001
Whang R., Welt L. (1963). Observations in experimental magnesium depletion. J. Clin. Invest 42, 305–313. doi:10.1172/JCI104717
Yokoyama C., Yabuki T., Shimonishi M., Wada M., Hatae T., Ohkawara S., et al. (2002). Prostacyclin-deficient mice Develop Ischemic renal Disorders, including nephrosclerosis and renal Infarction. Circulation 106, 2397–2403. doi:10.1161/01.cir.0000034733.93020.bc
Zhang J., Berra-Romani R., Sinnegger-Brauns M. J., Striessnig J., Blaustein M. P., Matteson D. R. (2007). Role of Cav1.2 L-type Ca2+ channels in vascular tone: effects of nifedipine and Mg2+. Am. J. Physiol. Heart Circ. Physiol. 292, H415–H425. doi:10.1152/ajpheart.01214.2005
Zhang X., Li Y., Del Gobbo L. C., Rosanoff A., Wang J., Zhang W., et al. (2016). Effects of magnesium supplementation on blood pressure: a meta-analysis of randomized double-blind Placebo-controlled trials. Hypertension 68, 324–333. doi:10.1161/HYPERTENSIONAHA.116.07664
Zhao W. G., Richardson J. S. (1990). Prostacyclin, thromboxane A2, and hypertension. Clin. Invest Med. 13, 343–352.
Keywords: magnesium, hypertension, aldosterone, NLPR3 inflammasome, isolevuglandins (IsoLG)
Citation: AlShanableh Z and Ray EC (2024) Magnesium in hypertension: mechanisms and clinical implications. Front. Physiol. 15:1363975. doi: 10.3389/fphys.2024.1363975
Received: 31 December 2023; Accepted: 25 March 2024;
Published: 10 April 2024.
Edited by:
Ji-Bin Peng, University of Alabama at Birmingham, United StatesReviewed by:
Mohammed Zubaerul Ferdaus, Vanderbilt University Medical Center, United StatesKenichi Goto, Kyushu University, Japan
Yujiro Maeoka, Hiroshima University, Japan
Copyright © 2024 AlShanableh and Ray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evan C. Ray, cmF5ZWNAdXBtYy5lZHU=