 Mary B. Rowland
Mary B. Rowland Patrick E. Moore
Patrick E. Moore Robert N. Correll
Robert N. Correll
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 12 January 2024
Sec. Cell Physiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1304669
This article is part of the Research Topic Cell Death Programs in the Pathogenesis of Heart Disease View all 5 articles
The endoplasmic reticulum (ER) is a tightly regulated organelle that requires specific environmental properties to efficiently carry out its function as a major site of protein synthesis and folding. Embedded in the ER membrane, ER stress sensors inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) serve as a sensitive quality control system collectively known as the unfolded protein response (UPR). In response to an accumulation of misfolded proteins, the UPR signals for protective mechanisms to cope with the cellular stress. Under prolonged unstable conditions and an inability to regain homeostasis, the UPR can shift from its original adaptive response to mechanisms leading to UPR-induced apoptosis. These UPR signaling pathways have been implicated as an important feature in the development of cardiac fibrosis, but identifying effective treatments has been difficult. Therefore, the apoptotic mechanisms of UPR signaling in cardiac fibroblasts (CFs) are important to our understanding of chronic fibrosis in the heart. Here, we summarize the maladaptive side of the UPR, activated downstream pathways associated with cell death, and agents that have been used to modify UPR-induced apoptosis in CFs.
Understanding the signaling cascades responsible for CF apoptosis could uncover ways to ameliorate chronic cardiac fibrosis (Matsumoto et al., 1996; Han et al., 2009; Groenendyk et al., 2016; Ren et al., 2021). Fibroblasts secrete and maintain tissue extracellular matrix (ECM) and, when differentiated into myofibroblasts, they enhance the matrix to aid in cell migration, communication, and wound healing (Galbraith and Sheetz, 1998; Brown et al., 2007; Jellis et al., 2010; van Nieuwenhoven and Turner, 2013). ER stress results from many cardiac pathologies, and homeostasis is maintained by signaling through the three arms of the UPR: IRE1, PERK, and ATF6 (Figure 1) (Minamino and Kitakaze, 2010; Arrieta et al., 2018). While the UPR is typically protective, under prolonged stressful conditions, such as oxidative stress, proteotoxicity, or impaired calcium signaling, it may shift to maladaptive signaling resulting in programmed cell death (Liang et al., 2012; Muchowicz et al., 2015; Losada et al., 2020).
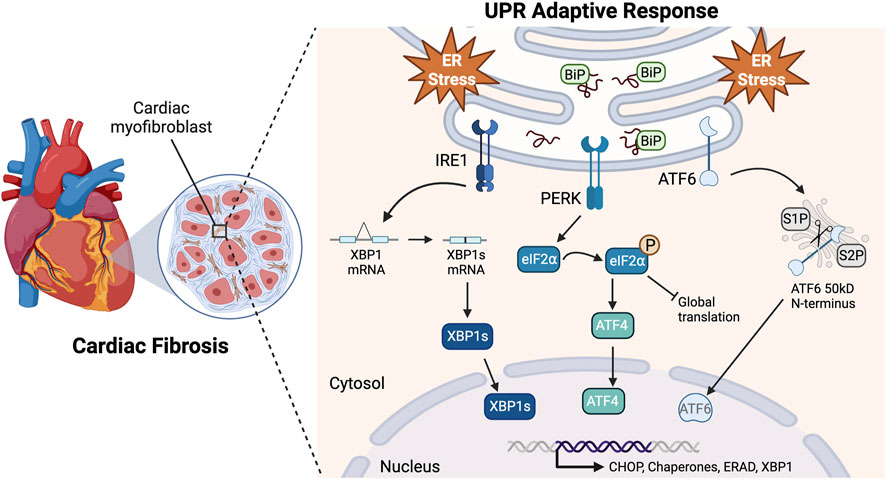
FIGURE 1. Adaptive UPR signaling pathways. Upon accumulation of misfolded proteins and ER stress, BiP (GRP78) disassociates from the UPR sensors to act as a chaperone. This allows IRE1 homodimerization causing the RNase domain to splice XBP1 mRNA resulting in the expression of XBP1s. PERK also homodimerizes, resulting in kinase activity that phosphorylates eIF2⍺. This leads to a global translation block and a reading frame shift in ATF4 that allows it to escape the block. ATF6 translocates to the Golgi apparatus where it is cleaved by proteases S1P and S2P, releasing its 50kD N-terminus. These pathways lead to the translocation of transcriptionally active XBP1s, ATF4, and N-ATF6 to the nucleus to upregulate UPR genes in response to ER stress Created with BioRender.com.
UPR-induced apoptosis may be divided into three phases that include initiation, commitment, and execution (Szegezdi et al., 2006a). Unresolvable stress initiates signaling, causing a commitment to apoptosis by upregulation of CCAAT enhancer-binding protein C/EBP homologous protein (CHOP) by all three arms, and an execution phase through downstream caspase activation (Matsumoto et al., 1996; Zinszner et al., 1998; Szegezdi et al., 2006b; Yang et al., 2020). The mechanisms that contribute to each phase have not been well characterized in CFs. Most studies have been performed in vitro, leaving many unanswered questions, such as how UPR signaling contributes to in vivo replacement, interstitial, and perivascular fibrosis (Factor et al., 1991; Aoki et al., 2011; Dai et al., 2012). Here, we review the known maladaptive pathways of the UPR, the current literature available about the role of the UPR in CF apoptosis, and areas of expansion needed in this field.
Maladaptive downstream effectors of the UPR, such as Jun-N-terminal kinase (JNK) and the B-cell lymphoma-2 (Bcl-2)/Bax ratio, have been assessed as a standard indicator for cell death in CFs (Tian et al., 2002; Zhao and Eghbali-Webb, 2002; Mayorga et al., 2004; Lai et al., 2009; Ghavami et al., 2012a; Ghavami et al., 2012b; Parra-Flores et al., 2021). Ghavami, et al. investigated maladaptive UPR signaling in the context of the mevalonate cascade, a pathway important in cholesterol synthesis (Ghavami et al., 2012a). Clinically, statins, which inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA), have been observed to decrease cardiac remodeling and activate apoptosis (Ghavami et al., 2012a). It was reported that inhibiting HMG-CoA with simvastatin simultaneously activated apoptosis and the UPR in human atrial fibroblasts and could be reversed by exposure to exogenous mevalonate (Ghavami et al., 2012a). This was supported with evidence that IRE1, cleaved ATF6, phosphorylated PERK, and CHOP expression increased upon simvastatin treatment. Spliced X-box binding protein 1 (XBP1) had the most significant increase in expression compared to all the UPR components examined (Ghavami et al., 2012a). This was further substantiated by characterization of maladaptive downstream molecules of IRE1, such as JNK, p53-upregulated modulator of apoptosis (PUMA), NOXA, and Bcl-2/Bax (Ghavami et al., 2012a). There was a significant increase in Bax, PUMA, NOXA, and caspase-3/7/9 expression with a simultaneous decrease in Bcl-2 and Mcl-1 (Ghavami et al., 2012a). The decrease in Bcl-2 was reversed by JNK inhibition, indicating that JNK signaling may be responsible for additional promotion of apoptotic effects which the authors suggest is limited by autophagic flux (Ghavami et al., 2012a). By performing a 3-(4,5dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay for cell viability, the data suggested that an increase in IRE1 expression was accompanied with an increase in human atrial fibroblast death (Ghavami et al., 2012a). The authors did not investigate the direct relationship between ATF6 activation and CHOP-mediated apoptosis, leaving it undetermined if ATF6 signaling orchestrated the observed maladaptive cell death pathways. Further clarifying the association between CF apoptosis and the Bcl-2/Bax ratio, Ghavami et al. exposed rat ventricular myofibroblasts to the trans-fatty acids (TFAs), vaccenic acid (VA) and elaidic acid (EA), at concentrations of 200 and 400 µM (Ghavami et al., 2012b). These treatments resulted in a significant increase in the percentage of apoptosis and a significant decrease in cell viability (Ghavami et al., 2012b). This was corroborated with a significant decrease in Bcl-2/Bax ratio (Ghavami et al., 2012b). Although TFAs are known ER stressors and UPR inducers, they are reported to not all have the same effect on cellular homeostasis and additional work is required to establish that specific TFAs (such as VA and EA) induce CF apoptosis through maladaptive UPR mechanisms (Oteng and Kersten, 2020).
Most of the work investigating the UPR in CF cell death has been completed in vitro, so more in vivo experiments are necessary to understand the broader physiological impacts of these pathways. This is particularly relevant when investigating CFs because they are mechanosensitive while functioning in an environment with repeated movement and a precise matrix composition and stiffness. In a recent study by Parra-Flores and collaborators, an in vivo model of ischemia/reperfusion (I/R), a known activator of the UPR, was used (Zhang et al., 2017; Parra-Flores et al., 2021). They saw a decrease in neonatal CF viability and an increase in apoptotic signaling following I/R; these effects were recovered by antioxidant exposure (Parra-Flores et al., 2021). This was associated with toll-like receptor 4 activation, a known regulator of IRE1 and XBP1 (Martinon et al., 2010; Parra-Flores et al., 2021). Further analyses showed decreases in pro-caspase 3/9 expression, decreases in p38 MAPK and JNK phosphorylation, and an increase in the Bcl-2/Bax ratio (Parra-Flores et al., 2021). Because all these components are important downstream players in maladaptive IRE1 signaling, expanding this work could clarify how the UPR may orchestrate pathways leading to CF apoptosis (Wang and Ron, 1996; Griffiths et al., 2001; Zong et al., 2001; Hitomi et al., 2004; Kato et al., 2012; Parra-Flores et al., 2021).
Irreversible ER stress causes IRE1 dimerization, resulting in recruitment of tumor necrosis factor receptor associated factor 2 (TRAF2) and apoptotic-signaling-kinase 1 (ASK1) that leads to maladaptive signaling (Urano et al., 2000; Nishitoh et al., 2002; Luo et al., 2008). The most referenced maladaptive downstream player of IRE1, JNK, activates apoptotic pathways and phosphorylates the anti-apoptotic family of Bcl-2 proteins (Lei and Davis, 2003; Wei et al., 2008; Shimizu et al., 2010; Kato et al., 2012). The upregulation of CHOP by the IRE1-TRAF2-ASK1 complex increases apoptotic proteins, such as Bcl-2-like 11 (BIM) and death receptor 5 (DR5), while simultaneously suppressing anti-apoptotic gene expression such as Bcl-2 (Wang and Ron, 1996; McCullough et al., 2001; Ghosh et al., 2012; Jung et al., 2015). The IRE1-TRAF2-ASK1 complex also activates caspases, such as caspase-12 and caspase-3, required for apoptosis (Yoneda et al., 2001; Hitomi et al., 2004).
The significance of maladaptive downstream signalers of IRE1 in CF apoptosis can be supported through the work done by Feng and collaborators (Feng et al., 2018). These authors showed that the elevated levels of BiP, CHOP, PUMA, and caspase-3 protein resulting from transverse aortic constriction (TAC) could be significantly reduced when these mice were treated with hydrogen sulfide (H2S) (Feng et al., 2018). It was unclear if these expression levels were specifically due to CFs or other cardiac cell types, but these effects were supported in vitro through H2S attenuation of hydrogen peroxide (H2O2)-mediated apoptosis in isolated human CFs (Feng et al., 2018).
The Bcl-2/Bax ratio is regulated downstream of IRE1 and influences CF apoptosis (Mayorga et al., 2004; Shemorry et al., 2019). siRNA knockdown of Bcl-2 resulted in an increase in CF apoptosis (Mayorga et al., 2004). JNK phosphorylation can inhibit Bcl-2 function and reduce the cell’s ability to properly regulate Ca2+ homeostasis in the ER and increase mitochondrial Ca2+ uptake (Murphy et al., 1996; He et al., 1997; Lei and Davis, 2003; Scorrano et al., 2003; Wei et al., 2008). Indeed, IRE1 can also activate JNK through TRAF2 and ASK1 leading to disinhibition of Bax/Bak by Bcl-2, and enabling cytochrome c release from the mitochondria (Gorman et al., 2012). Mitochondrial-mediated apoptosis was associated with shifts in Bcl-2, Bax, or caspase expression (Tian et al., 2002). Tian et al. treated rat CFs with inflammatory cytokines, which induced nitric oxide-mediated apoptosis (Tian et al., 2002). This exposure was reported alongside a significant 3.5-fold increase in Bax, a 2.5-fold increase in caspase-3 expression, and a 7-fold increase in caspase-3 activity (Tian et al., 2002). Meanwhile, Lai and others showed that higher doses of norepinephrine up to 100 µM significantly increased apoptosis and decreased the number of viable rat CFs (Lai et al., 2009). This exposure was reported to be associated with an increase in Bax mRNA expression and caspase-3 activity, indicating cytotoxicity and the activation of apoptotic pathways (Lai et al., 2009). Although these authors did not identify the upstream mediators of these results, it has been demonstrated that norepinephrine can induce UPR signaling, specifically ATF6 and IRE1, in HepG2 cells, human fat explants, and 3T3-L1 mouse adipocytes (Lai et al., 2009; Abdullahi et al., 2020).
Interestingly, Zhao and collaborators looked at differences by sex of rat CFs to apoptotic stimuli (Zhao and Eghbali-Webb, 2002). They found that following 15 min of hypoxia, isolated CFs from males had a steeper increase in JNK expression relative to females in comparison to each of their basal levels, but females had an overall higher basal JNK expression compared to male CFs (Zhao and Eghbali-Webb, 2002). Evidence in rodents and humans supports that there is less cardiac remodeling and fibrosis in females (Kessler et al., 2019). Because sex differences are associated with human cardiovascular disease progression and outcomes, this perspective of CF cell death is worth exploring further. Of particular significance when considering CFs in vitro is the absence of intrinsic, sex-based hormonal signaling, such as estrogen, a known contributor to enhanced wound repair. With this, the lack of complex sex specific characteristics in cell culture should be taken into consideration. Other tissue types, such as the kidney, have also shown sex differences in UPR signaling through increased ER stress markers and apoptosis in tunicamycin-treated male mice, compared to females (Hodeify et al., 2013). Using the perspective taken by Zhao, et al., the potential role of UPR signaling through JNK and its apoptotic effects in CFs could further clarify factors contributing to the observed sex differences in cardiac outcomes.
The serine/threonine kinase activity of PERK phosphorylates eukaryotic translation initiation factor 2a (eIF2⍺) (Koumenis et al., 2002; Cui et al., 2011). This attenuates overall translation while increasing the translation of specific mRNAs such as activating transcription factor 4 (ATF4), critical for the transcription of CHOP (Fawcett et al., 1999; Blais et al., 2004). CHOP upregulates three important components of UPR-induced apoptosis which are tribbles-related protein 3 (TRB3), DR5, and growth arrest and DNA damage-inducible gene 34 (GADD34) (Marciniak et al., 2004; Yamaguchi and Wang, 2004; Ohoka et al., 2005). TRB3 prevents proliferation, transcription, and differentiation signaling by binding Akt and inhibiting phosphorylation (Du et al., 2003). Apoptotic signaling is enhanced through DR5 by the activation of caspases (Tanner and Grisanti, 2021). When ER stress cannot be reversed, GADD34 binds protein phosphatase-1⍺ to dephosphorylate eIF2⍺ and remove the translation block which is associated with apoptosis (Brush et al., 2003; Choy et al., 2015; Collier et al., 2015). Pro-apoptotic signaling through CHOP also influences Bax/Bak and outer mitochondrial membrane permeabilization by reducing Bcl-2 and increasing BIM (Puthalakath et al., 2007; Luna-Vargas and Chipuk, 2016; Zhou et al., 2019).
PERK may orchestrate maladaptive effects in CFs because multiple downstream PERK signaling molecules, such as CHOP, ATF4, DR5, and Bcl-2, have been shown to influence CF apoptosis (Mayorga et al., 2004; Humeres et al., 2014; Sokolova et al., 2017; Feng et al., 2018; Olivares-Silva et al., 2021; Tanner and Grisanti, 2021). Since all three arms of the UPR regulate CHOP signaling, defining which branch has the most impact on CF apoptosis is critical for fully explaining the role of CHOP in this process (Yang et al., 2020). Recently, Olivares-Silva and others reported that ER stress induced by tunicamycin, ischemia, and I/R increased CHOP protein expression in neonatal Sprague Dawley rat CFs (Olivares-Silva et al., 2021). This was associated with an increase in apoptosis and reduction of viability in a time and concentration-dependent manner (Olivares-Silva et al., 2021). Similarly, Humeres and collaborators isolated CFs from neonatal Sprague-Dawley rats and induced ER stress through thapsigargin treatments (Humeres et al., 2014). This increased GRP78, protein disulfide-isomerase (PDI), ATF4, and CHOP protein levels while simultaneously decreasing cell viability in a time and concentration-dependent manner (Humeres et al., 2014). Work by Feng et al. found that exposure to H2O2 resulted in a significant decrease in human CF viability and an increase in apoptosis in a dosage dependent manner, which occurred concurrently with an increase in CHOP expression and could be ameliorated by exposure to H2S (Feng et al., 2018). Together, these investigations provide evidence that downstream mediators of PERK signaling, such as CHOP and ATF4, modulate CF cell viability.
Sokolova and coauthors recently found that palmitate (PA), a saturated fatty acid found in plasma, induced ER stress in adult mouse CFs (Sokolova et al., 2017). PA increased the gene expression of CHOP and ATF4 while also increasing CF apoptosis and decreasing CF contractile function (Sokolova et al., 2017). Annexin V-fluorescein isothiocyanate (FITC) labeling detected a significant increase in early-stage apoptosis while propidium iodide binding to nuclear DNA did not show a significant late-stage apoptosis/necrosis when treated with PA (Sokolova et al., 2017). This is particularly interesting because it has been reported that PERK induction of CHOP is more significant in the later stages of apoptosis, as indicated by propidium iodide staining, in other cell types (Lu et al., 2017; Liu and Zhang, 2020). It may be useful to apply these apoptotic stage analyses to evaluate the timing of maladaptive signaling within each of the UPR branches. Further, determining the time point of each switch between adaptive and maladaptive UPR signaling could have clinical applications in treatments targeting these mechanisms.
PERK is a known upstream mediator of DR5 (Lu et al., 2014). Tanner and others showed that DR5 signaling was correlated with proliferation in inactivated ventricular fibroblasts but apoptosis in activated ventricular myofibroblasts (Tanner and Grisanti, 2021). This was evident through a significant increase in DR5 expression, caspase 3/7 activity, and apoptosis, measured by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, in myofibroblasts compared to fibroblasts (Tanner and Grisanti, 2021). This was further substantiated in vivo by Masson’s Trichome staining, which showed that after isoproterenol injection there was an increase in fibrosis in DR5 gene-deleted mice compared to control mice (Tanner and Grisanti, 2021). Studies such as this that compare UPR signaling in fibroblasts and myofibroblasts could help to clarify the roles of these signaling pathways in CF cell death.
Elevated ATF6 expression, due to either disease processes or viral transduction, is associated with increased cellular apoptosis (Morishima et al., 2011; Tan et al., 2020). In colorectal cancer cells, the ATF6 transcriptionally active N-terminus increased GRP78, DDIT3 (which encodes CHOP), and EIF2AK3 (which encodes PERK) gene expression and significantly increased apoptotic cells (Spaan et al., 2019). Similarly, overexpression of ATF6 increased CHOP and Bax mRNA and protein levels, decreased Bcl-2 expression, and significantly increased the rate of apoptosis (Huang et al., 2018). Depletion of ATF6 decreased CHOP expression and increased Bcl-2 expression, resulting in a decrease of apoptosis (Xiong et al., 2017). Upon ATF6 silencing, pro-apoptotic effects of hydroxycamptothecin on fibroblasts was significantly weakened (Wei et al., 2018; Yao et al., 2019; Tao et al., 2021). Further examination of the PERK/p-eLF2α/ATF4 pathway could elucidate ATF6’s role in apoptosis, as it has been reported that eIF2⍺ phosphorylation and ATF4 activation are necessary for ATF6 activation (Teske et al., 2011).
Little work has been done to examine the role of ATF6 signaling in CF apoptosis. ATF6 activity is generally protective in the heart, but it is unclear how its signaling affects CF activity and survival (Toko et al., 2010; Glembotski et al., 2019). Data presented by Toko and collaborators showed that inhibition of ATF6 with 4-(2-aminoethyl) benzenesulfonyl fluoride or knockdown of ATF6 with siRNA decreased cardiac function, increased myocardial infarction mortality rate, and increased cardiomyocyte apoptosis in mice (Toko et al., 2010). However, ATF6 is also known to transcriptionally upregulate the important UPR-mediated proapoptotic molecule, CHOP (Yoshida et al., 2000; Yang et al., 2020).
The role of ATF6 in CF function has recently been expanded on in Stauffer, et al. (Stauffer et al., 2020). It was reported that pharmacologic activation of ATF6 using compound 147 in murine ventricular fibroblasts resulted in a decrease in fibroblast activation and contraction while the opposite was seen in siRNA knockdown of ATF6 (Stauffer et al., 2020). However, ATF6 effects on apoptosis and cell viability were not measured (Stauffer et al., 2020). Because CHOP upregulation is not entirely dependent on ATF6 signaling, many experiments did not consider the potential contribution of the ATF6 pathway in their analyses. Because ATF6 has not been fully explored and its significance to maladaptive UPR signaling in CFs is unclear, it is a plausible target for further investigation.
Fibroblasts have unique characteristics and gene expression patterns that are organ-specific (Lindner et al., 2012). In most tissues, fibroblasts undergo apoptosis following scar formation (Desmouliere et al., 1995). However, in the heart, activated and ⍺SMA-expressing cardiac myofibroblasts have been found in the infarct scar up to 17 years following an initial cardiac event (Willems et al., 1994). Continuous presence of myofibroblasts results in excessive synthesis and secretion of ECM components causing ventricular stiffness and heart failure (van den Borne et al., 2010). Elucidating the specific regulatory mechanisms that allow CFs to elude apoptosis more frequently than other tissue fibroblasts could identify key features of how the UPR may contribute to chronic cardiac fibrosis.
A possible explanation for CF apoptosis evasion is through distinctive extrinsic pro-survival conditions, such as integrin-mediated transduction, paracrine factors, or a specific composition of ECM network resulting from the electrical and mechanical stimuli in the heart (Huebener et al., 2008; Cai et al., 2019; Titus et al., 2021). Another explanation for enhanced apoptotic resistance is that the augmented pro-survival pathways are more magnified than the maladaptive signals, causing an increase in fibroblast viability (Bea et al., 2022). CFs could also have heightened resistance to apoptosis through response to intrinsic signaling and continual autophagy (Zeglinski et al., 2016). It has been shown that activated cardiac myofibroblasts are more resistant to apoptosis than quiescent CFs, indicating enhanced pro-survival molecular mechanisms in those cells (Lagares et al., 2017; Hinz and Lagares, 2020). Some of the suggested cytoprotective molecular mechanisms aiding in apoptosis avoidance include canonical Transforming Growth Factor β (TGFβ) signaling, a decrease in Bax and caspase expression, and an increase in Bcl-2 expression (Mayorga et al., 2004; Anuka et al., 2013; Vivar et al., 2013; Olivares-Silva et al., 2021). The UPR is a potential candidate to investigate CF survival due to its crosstalk with TGFβ signaling and its response to Bcl-2 upregulation (Mayorga et al., 2004; Vivar et al., 2013; Olivares-Silva et al., 2021).
ER stress and the UPR are contributors to various cardiac pathologies such as hypertrophy, ventricular dysfunction, and heart failure (Park et al., 2012). There is a significant amount of literature describing the relevance of the UPR in apoptosis in other cell types or in fibroblasts of other tissues (Shi et al., 2013; Hong et al., 2015; Tang et al., 2016; Delbrel et al., 2018; Pibiri et al., 2020). UPR regulation of CF cell death is relatively understudied in the context of reducing pathological cardiac fibrosis. Downstream effectors of IRE1 such as Bax, Bcl-2, PUMA, JNK, and caspase-3 have been reported to be involved in CF apoptosis (Tian et al., 2002; Mayorga et al., 2004; Lai et al., 2009; Ghavami et al., 2012a; Ghavami et al., 2012b; Feng et al., 2018; Parra-Flores et al., 2021). Research has also affirmed that activation of ATF6 upregulates CHOP, but the extent to which this regulates CF apoptosis has not been explored (Yoshida et al., 2000; Ghavami et al., 2012a; Yang et al., 2020). All three UPR arms upregulate CHOP, but the PERK pathway is essential for CHOP expression in comparison to IRE1 and ATF6 (Humeres et al., 2014; Sokolova et al., 2017; Olivares-Silva et al., 2021). Therefore, this may suggest that PERK-ATF4-CHOP signaling is the most influential UPR arm in CF apoptosis.
Much of the work reviewed did not explicitly identify the specific UPR arm influencing CF cell death and instead looked at their downstream mediators. Expanding these investigations can determine if these downstream molecules are signaled to through a specific UPR pathway or an alternative upstream mechanism. Additionally, studies investigating UPR-mediated CF apoptosis use a variety of methods to induce cellular stress that may result in different ER stress mechanisms being activated and variations in severity that could obfuscate our understanding. Future work exploring these mechanisms will provide a better understanding of chronic fibrosis, CF apoptotic resistance, and potential pharmacological manipulations that might provide new therapies for various cardiovascular pathologies.
MR: Conceptualization, Writing–original draft, Writing–review and editing. PM: Writing–original draft, Writing–review and editing. RC: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Department of Biological Sciences and College of Arts and Sciences at the University of Alabama and an award from the Dale/Roy family to RC.
Figure 1 was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdullahi A., Wang V., Auger C., Patsouris D., Amini-Nik S., Jeschke M. G. (2020). Catecholamines induce endoplasmic reticulum stress via both alpha and beta receptors. Shock 53, 476–484. doi:10.1097/SHK.0000000000001394
Anuka E., Yivgi-Ohana N., Eimerl S., Garfinkel B., Melamed-Book N., Chepurkol E., et al. (2013). Infarct-induced steroidogenic acute regulatory protein: a survival role in cardiac fibroblasts. Mol. Endocrinol. 27, 1502–1517. doi:10.1210/me.2013-1006
Aoki T., Fukumoto Y., Sugimura K., Oikawa M., Satoh K., Nakano M., et al. (2011). Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. -Comparison between preserved and reduced ejection fraction heart failure. Circ. J. 75, 2605–2613. doi:10.1253/circj.cj-11-0568
Arrieta A., Blackwood E. A., Glembotski C. C. (2018). ER protein quality control and the unfolded protein response in the heart. Curr. Top. Microbiol. Immunol. 414, 193–213. doi:10.1007/82_2017_54
Bea A., Valero J. G., Irazoki A., Lana C., López-Lluch G., Portero-Otín M., et al. (2022). Cardiac fibroblasts display endurance to ischemia, high ROS control and elevated respiration regulated by the JAK2/STAT pathway. FEBS J. 289, 2540–2561. doi:10.1111/febs.16283
Blais J. D., Filipenko V., Bi M., Harding H. P., Ron D., Koumenis C., et al. (2004). Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell Biol. 24, 7469–7482. doi:10.1128/MCB.24.17.7469-7482.2004
Brown R. D., Jones G. M., Laird R. E., Hudson P., Long C. S. (2007). Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem. Biophys. Res. Commun. 362, 200–205. doi:10.1016/j.bbrc.2007.08.003
Brush M. H., Weiser D. C., Shenolikar S. (2003). Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell Biol. 23, 1292–1303. doi:10.1128/MCB.23.4.1292-1303.2003
Cai W., Zhong S., Zheng F., Zhang Y., Gao F., Xu H., et al. (2019). Angiotensin II confers resistance to apoptosis in cardiac myofibroblasts through the AT1/ERK1/2/RSK1 pathway. IUBMB Life 71, 261–276. doi:10.1002/iub.1967
Choy M. S., Yusoff P., Lee I. C., Newton J. C., Goh C. W., Page R., et al. (2015). Structural and functional analysis of the GADD34:PP1 eIF2α phosphatase. Cell Rep. 11, 1885–1891. doi:10.1016/j.celrep.2015.05.043
Collier A. E., Wek R. C., Spandau D. F. (2015). Translational repression protects human keratinocytes from UVB-induced apoptosis through a discordant eIF2 kinase stress response. J. Invest. Dermatol 135, 2502–2511. doi:10.1038/jid.2015.177
Cui W., Li J., Ron D., Sha B. (2011). The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D. Biol. Crystallogr. 67, 423–428. doi:10.1107/S0907444911006445
Dai Z., Aoki T., Fukumoto Y., Shimokawa H. (2012). Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 60, 416–421. doi:10.1016/j.jjcc.2012.06.009
Delbrel E., Soumare A., Naguez A., Label R., Bernard O., Bruhat A., et al. (2018). HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 8, 17939. doi:10.1038/s41598-018-36063-2
Desmouliere A., Redard M., Darby I., Gabbiani G. (1995). Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 146, 56–66.
Du K., Herzig S., Kulkarni R. N., Montminy M. (2003). TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300, 1574–1577. doi:10.1126/science.1079817
Factor S. M., Butany J., Sole M. J., Wigle E. D., Williams W. C., Rojkind M. (1991). Pathologic fibrosis and matrix connective tissue in the subaortic myocardium of patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 17, 1343–1351. doi:10.1016/s0735-1097(10)80145-7
Fawcett T. W., Martindale J. L., Guyton K. Z., Hai T., Holbrook N. J. (1999). Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 339 (Pt 1), 135–141. doi:10.1042/0264-6021:3390135
Feng A., Ling C., Xin-Duo L., Bing W., San-Wu W., Yu Z., et al. (2018). Hydrogen sulfide protects human cardiac fibroblasts against H(2)O(2)-induced injury through regulating autophagy-related proteins. Cell Transpl. 27, 1222–1234. doi:10.1177/0963689718779361
Galbraith C. G., Sheetz M. P. (1998). Forces on adhesive contacts affect cell function. Curr. Opin. Cell Biol. 10, 566–571. doi:10.1016/s0955-0674(98)80030-6
Ghavami S., Cunnington R. H., Yeganeh B., Davies J. J. L., Rattan S. G., Bathe K., et al. (2012b). Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim. Biophys. Acta 1823, 2274–2286. doi:10.1016/j.bbamcr.2012.09.008
Ghavami S., Yeganeh B., Stelmack G. L., Kashani H. H., Sharma P., Cunnington R., et al. (2012a). Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 3, e330. doi:10.1038/cddis.2012.61
Ghosh A. P., Klocke B. J., Ballestas M. E., Roth K. A. (2012). CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One 7, e39586. doi:10.1371/journal.pone.0039586
Glembotski C. C., Rosarda J. D., Wiseman R. L. (2019). Proteostasis and beyond: ATF6 in ischemic disease. Trends Mol. Med. 25, 538–550. doi:10.1016/j.molmed.2019.03.005
Gorman A. M., Healy S. J., Jager R., Samali A. (2012). Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol. Ther. 134, 306–316. doi:10.1016/j.pharmthera.2012.02.003
Griffiths G. J., Corfe B. M., Savory P., Leech S., Esposti M. D., Hickman J. A., et al. (2001). Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene 20, 7668–7676. doi:10.1038/sj.onc.1204995
Groenendyk J., Lee D., Jung J., Dyck J. R. B., Lopaschuk G. D., Agellon L. B., et al. (2016). Inhibition of the unfolded protein response mechanism prevents cardiac fibrosis. PLoS One 11, e0159682. doi:10.1371/journal.pone.0159682
Han D., Lerner A. G., Vande Walle L., Upton J. P., Xu W., Hagen A., et al. (2009). IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575. doi:10.1016/j.cell.2009.07.017
He H., Lam M., McCormick T. S., Distelhorst C. W. (1997). Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol. 138, 1219–1228. doi:10.1083/jcb.138.6.1219
Hinz B., Lagares D. (2020). Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 16, 11–31. doi:10.1038/s41584-019-0324-5
Hitomi J., Katayama T., Taniguchi M., Honda A., Imaizumi K., Tohyama M. (2004). Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 357, 127–130. doi:10.1016/j.neulet.2003.12.080
Hodeify R., Megyesi J., Tarcsafalvi A., Mustafa H. I., Hti Lar Seng N. S., Price P. M. (2013). Gender differences control the susceptibility to ER stress-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 304, F875–F882. doi:10.1152/ajprenal.00590.2012
Hong Y. H., Uddin M. H., Jo U., Kim B., Song J., Suh D. H., et al. (2015). ROS accumulation by PEITC selectively kills ovarian cancer cells via UPR-mediated apoptosis. Front. Oncol. 5, 167. doi:10.3389/fonc.2015.00167
Huang J., Wan L., Lu H., Li X. (2018). High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol. Med. Rep. 17, 6483–6489. doi:10.3892/mmr.2018.8658
Huebener P., Abou-Khamis T., Zymek P., Bujak M., Ying X., Chatila K., et al. (2008). CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 180, 2625–2633. doi:10.4049/jimmunol.180.4.2625
Humeres C., Montenegro J., Varela M., Ayala P., Vivar R., Letelier A., et al. (2014). 4-Phenylbutyric acid prevent cytotoxicity induced by thapsigargin in rat cardiac fibroblast. Toxicol Vitro 28, 1443–1448. doi:10.1016/j.tiv.2014.07.013
Jellis C., Martin J., Narula J., Marwick T. H. (2010). Assessment of nonischemic myocardial fibrosis. J. Am. Coll. Cardiol. 56, 89–97. doi:10.1016/j.jacc.2010.02.047
Jung K. J., Min K. J., Bae J. H., Kwon T. K. (2015). Carnosic acid sensitized TRAIL-mediated apoptosis through down-regulation of c-FLIP and Bcl-2 expression at the post translational levels and CHOP-dependent up-regulation of DR5, Bim, and PUMA expression in human carcinoma caki cells. Oncotarget 6, 1556–1568. doi:10.18632/oncotarget.2727
Kato H., Nakajima S., Saito Y., Takahashi S., Katoh R., Kitamura M. (2012). mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 19, 310–320. doi:10.1038/cdd.2011.98
Kessler E. L., Rivaud M. R., Vos M. A., van Veen T. A. B. (2019). Sex-specific influence on cardiac structural remodeling and therapy in cardiovascular disease. Biol. Sex. Differ. 10, 7. doi:10.1186/s13293-019-0223-0
Koumenis C., Naczki C., Koritzinsky M., Rastani S., Diehl A., Sonenberg N., et al. (2002). Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell Biol. 22, 7405–7416. doi:10.1128/MCB.22.21.7405-7416.2002
Lagares D., Santos A., Grasberger P. E., Liu F., Probst C. K., Rahimi R. A., et al. (2017). Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 9, eaal3765. doi:10.1126/scitranslmed.aal3765
Lai K. B., Sanderson J. E., Yu C. M. (2009). High dose norepinephrine-induced apoptosis in cultured rat cardiac fibroblast. Int. J. Cardiol. 136, 33–39. doi:10.1016/j.ijcard.2008.04.022
Lei K., Davis R. J. (2003). JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100, 2432–2437. doi:10.1073/pnas.0438011100
Liang C. P., Han S., Li G., Tabas I., Tall A. R. (2012). Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes 61, 2609–2620. doi:10.2337/db11-1415
Lindner D., Zietsch C., Becher P. M., Schulze K., Schultheiss H. P., Tschöpe C., et al. (2012). Differential expression of matrix metalloproteases in human fibroblasts with different origins. Biochem. Res. Int. 2012, 875742. doi:10.1155/2012/875742
Liu C., Zhang A. (2020). ROS-mediated PERK-eIF2α-ATF4 pathway plays an important role in arsenite-induced L-02 cells apoptosis via regulating CHOP-DR5 signaling. Environ. Toxicol. 35, 1100–1113. doi:10.1002/tox.22946
Losada A., Berlanga J. J., Molina-Guijarro J. M., Jiménez-Ruiz A., Gago F., Avilés P., et al. (2020). Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharmacol. 172, 113744. doi:10.1016/j.bcp.2019.113744
Lu M., Lawrence D. A., Marsters S., Acosta-Alvear D., Kimmig P., Mendez A. S., et al. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101. doi:10.1126/science.1254312
Lu X., Li C., Li C., Li P., Fu E., Xie Y., et al. (2017). Heat-labile enterotoxin-induced PERK-CHOP pathway activation causes intestinal epithelial cell apoptosis. Front. Cell Infect. Microbiol. 7, 244. doi:10.3389/fcimb.2017.00244
Luna-Vargas M. P., Chipuk J. E. (2016). The deadly landscape of pro-apoptotic BCL-2 proteins in the outer mitochondrial membrane. FEBS J. 283, 2676–2689. doi:10.1111/febs.13624
Luo D., He Y., Zhang H., Yu L., Chen H., Xu Z., et al. (2008). AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 283, 11905–11912. doi:10.1074/jbc.M710557200
Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi:10.1101/gad.1250704
Martinon F., Chen X., Lee A. H., Glimcher L. H. (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418. doi:10.1038/ni.1857
Matsumoto M., Minami M., Takeda K., Sakao Y., Akira S. (1996). Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 395, 143–147. doi:10.1016/0014-5793(96)01016-2
Mayorga M., Bahi N., Ballester M., Comella J. X., Sanchis D. (2004). Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J. Biol. Chem. 279, 34882–34889. doi:10.1074/jbc.M404616200
McCullough K. D., Martindale J. L., Klotz L. O., Aw T. Y., Holbrook N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 21, 1249–1259. doi:10.1128/MCB.21.4.1249-1259.2001
Minamino T., Kitakaze M. (2010). ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 48, 1105–1110. doi:10.1016/j.yjmcc.2009.10.026
Morishima N., Nakanishi K., Nakano A. (2011). Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via induction of WW domain binding protein 1. J. Biol. Chem. 286, 35227–35235. doi:10.1074/jbc.M111.233502
Muchowicz A., Firczuk M., Wachowska M., Kujawa M., Jankowska-Steifer E., Gabrysiak M., et al. (2015). SK053 triggers tumor cells apoptosis by oxidative stress-mediated endoplasmic reticulum stress. Biochem. Pharmacol. 93, 418–427. doi:10.1016/j.bcp.2014.12.019
Murphy A. N., Bredesen D. E., Cortopassi G., Wang E., Fiskum G. (1996). Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. U. S. A. 93, 9893–9898. doi:10.1073/pnas.93.18.9893
Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., et al. (2002). ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355. doi:10.1101/gad.992302
Ohoka N., Yoshii S., Hattori T., Onozaki K., Hayashi H. (2005). TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255. doi:10.1038/sj.emboj.7600596
Olivares-Silva F., Espitia-Corredor J., Letelier A., Vivar R., Parra-Flores P., Olmedo I., et al. (2021). TGF-β1 decreases CHOP expression and prevents cardiac fibroblast apoptosis induced by endoplasmic reticulum stress. Toxicol Vitro 70, 105041. doi:10.1016/j.tiv.2020.105041
Oteng A. B., Kersten S. (2020). Mechanisms of action of trans fatty acids. Adv. Nutr. 11, 697–708. doi:10.1093/advances/nmz125
Park C. S., Cha H., Kwon E. J., Sreenivasaiah P. K., Kim D. H. (2012). The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 421, 578–584. doi:10.1016/j.bbrc.2012.04.048
Parra-Flores P., Espitia-Corredor J., Espinoza-Pérez C., Queirolo C., Ayala P., Brüggendieck F., et al. (2021). Toll-like receptor 4 activation prevents rat cardiac fibroblast death induced by simulated ischemia/reperfusion. Front. Cardiovasc Med. 8, 660197. doi:10.3389/fcvm.2021.660197
Pibiri M., Sulas P., Camboni T., Leoni V. P., Simbula G. (2020). α-Lipoic acid induces Endoplasmic Reticulum stress-mediated apoptosis in hepatoma cells. Sci. Rep. 10, 7139. doi:10.1038/s41598-020-64004-5
Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., et al. (2007). ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349. doi:10.1016/j.cell.2007.04.027
Ren J., Bi Y., Sowers J. R., Hetz C., Zhang Y. (2021). Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 18, 499–521. doi:10.1038/s41569-021-00511-w
Scorrano L., Oakes S. A., Opferman J. T., Cheng E. H., Sorcinelli M. D., Pozzan T., et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139. doi:10.1126/science.1081208
Shemorry A., Harnoss J. M., Guttman O., Marsters S. A., Kőműves L. G., Lawrence D. A., et al. (2019). Caspase-mediated cleavage of IRE1 controls apoptotic cell commitment during endoplasmic reticulum stress. Elife 8, e47084. doi:10.7554/eLife.47084
Shi K., Wang D., Cao X., Ge Y. (2013). Endoplasmic reticulum stress signaling is involved in mitomycin C (MMC)-induced apoptosis in human fibroblasts via PERK pathway. PLoS One 8, e59330. doi:10.1371/journal.pone.0059330
Shimizu S., Konishi A., Nishida Y., Mizuta T., Nishina H., Yamamoto A., et al. (2010). Involvement of JNK in the regulation of autophagic cell death. Oncogene 29, 2070–2082. doi:10.1038/onc.2009.487
Sokolova M., Vinge L. E., Alfsnes K., Olsen M. B., Eide L., Kaasbøll O. J., et al. (2017). Palmitate promotes inflammatory responses and cellular senescence in cardiac fibroblasts. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 234–245. doi:10.1016/j.bbalip.2016.11.003
Spaan C. N., Smit W. L., van Lidth de Jeude J. F., Meijer B. J., Muncan V., van den Brink G. R., et al. (2019). Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis. 10, 490. doi:10.1038/s41419-019-1729-4
Stauffer W. T., Blackwood E. A., Azizi K., Kaufman R. J., Glembotski C. C. (2020). The ER unfolded protein response effector, ATF6, reduces cardiac fibrosis and decreases activation of cardiac fibroblasts. Int. J. Mol. Sci. 21, 1373. doi:10.3390/ijms21041373
Szegezdi E., Duffy A., O'Mahoney M. E., Logue S. E., Mylotte L. A., O'brien T., et al. (2006b). ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem. Biophys. Res. Commun. 349, 1406–1411. doi:10.1016/j.bbrc.2006.09.009
Szegezdi E., Logue S. E., Gorman A. M., Samali A. (2006a). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi:10.1038/sj.embor.7400779
Tan J. H., Cao R. C., Zhou L., Zhou Z. T., Chen H. J., Xu J., et al. (2020). ATF6 aggravates acinar cell apoptosis and injury by regulating p53/AIFM2 transcription in Severe Acute Pancreatitis. Theranostics 10, 8298–8314. doi:10.7150/thno.46934
Tang J., Ge Y., Yang L., Xu X., Sui T., Ge D., et al. (2016). ER stress via CHOP pathway is involved in FK506-induced apoptosis in rat fibroblasts. Cell Physiol. Biochem. 39, 1965–1976. doi:10.1159/000447894
Tanner M. A., Grisanti L. A. (2021). A dual role for death receptor 5 in regulating cardiac fibroblast function. Front. Cardiovasc Med. 8, 699102. doi:10.3389/fcvm.2021.699102
Tao J., Chen H., Li X., Wang J. (2021). The role of activating transcription factor 6 in hydroxycamptothecin-induced fibroblast autophagy and apoptosis. J. Orthop. Surg. Res. 16, 1. doi:10.1186/s13018-020-02056-z
Teske B. F., Wek S. A., Bunpo P., Cundiff J. K., McClintick J. N., Anthony T. G., et al. (2011). The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 22, 4390–4405. doi:10.1091/mbc.E11-06-0510
Tian B., Liu J., Bitterman P. B., Bache R. J. (2002). Mechanisms of cytokine induced NO-mediated cardiac fibroblast apoptosis. Am. J. Physiol. Heart Circ. Physiol. 283, H1958–H1967. doi:10.1152/ajpheart.01070.2001
Titus A. S., Venugopal H., Ushakumary M. G., Wang M., Cowling R. T., Lakatta E. G., et al. (2021). Discoidin domain receptor 2 regulates AT1R expression in angiotensin II-stimulated cardiac fibroblasts via fibronectin-dependent integrin-β1 signaling. Int. J. Mol. Sci. 22, 9343. doi:10.3390/ijms22179343
Toko H., Takahashi H., Kayama Y., Okada S., Minamino T., Terasaki F., et al. (2010). ATF6 is important under both pathological and physiological states in the heart. J. Mol. Cell Cardiol. 49, 113–120. doi:10.1016/j.yjmcc.2010.03.020
Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi:10.1126/science.287.5453.664
van den Borne S. W., Diez J., Blankesteijn W. M., Verjans J., Hofstra L., Narula J. (2010). Myocardial remodeling after infarction: the role of myofibroblasts. Nat. Rev. Cardiol. 7, 30–37. doi:10.1038/nrcardio.2009.199
van Nieuwenhoven F. A., Turner N. A. (2013). The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 58, 182–188. doi:10.1016/j.vph.2012.07.003
Vivar R., Humeres C., Ayala P., Olmedo I., Catalán M., García L., et al. (2013). TGF-β1 prevents simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis by activation of both canonical and non-canonical signaling pathways. Biochim. Biophys. Acta 1832, 754–762. doi:10.1016/j.bbadis.2013.02.004
Wang X. Z., Ron D. (1996). Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272, 1347–1349. doi:10.1126/science.272.5266.1347
Wei Y., Li C., Zhang Y., He H., Zhang G., Hao X., et al. (2018). Hydroxycamptothecin mediates antiproliferative effects through apoptosis and autophagy in A549 cells. Oncol. Lett. 15, 6322–6328. doi:10.3892/ol.2018.8107
Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. doi:10.1016/j.molcel.2008.06.001
Willems I. E., Havenith M. G., De Mey J. G., Daemen M. J. (1994). The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 145, 868–875.
Xiong Y., Chen H., Lin P., Wang A., Wang L., Jin Y. (2017). ATF6 knockdown decreases apoptosis, arrests the S phase of the cell cycle, and increases steroid hormone production in mouse granulosa cells. Am. J. Physiol. Cell Physiol. 312, C341–C353. doi:10.1152/ajpcell.00222.2016
Yamaguchi H., Wang H. G. (2004). CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502. doi:10.1074/jbc.M406933200
Yang H., Niemeijer M., van de Water B., Beltman J. B. (2020). ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience 23, 100860. doi:10.1016/j.isci.2020.100860
Yao Z., Zheng W., Zhang X., Xiong H., Qian Y., Fan C. (2019). Hydroxycamptothecin prevents fibrotic pathways in fibroblasts in vitro. IUBMB Life 71, 653–662. doi:10.1002/iub.2013
Yoneda T., Imaizumi K., Oono K., Yui D., Gomi F., Katayama T., et al. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940. doi:10.1074/jbc.M010677200
Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., et al. (2000). ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 20, 6755–6767. doi:10.1128/MCB.20.18.6755-6767.2000
Zeglinski M. R., Davies J. J. L., Ghavami S., Rattan S. G., Halayko A. J., Dixon I. M. C. (2016). Chronic expression of Ski induces apoptosis and represses autophagy in cardiac myofibroblasts. Biochim. Biophys. Acta 1863, 1261–1268. doi:10.1016/j.bbamcr.2016.03.027
Zhang C., Tang Y., Li Y., Xie L., Zhuang W., Liu J., et al. (2017). Unfolded protein response plays a critical role in heart damage after myocardial ischemia/reperfusion in rats. PLoS One 12, e0179042. doi:10.1371/journal.pone.0179042
Zhao X., Eghbali-Webb M. (2002). Gender-related differences in basal and hypoxia-induced activation of signal transduction pathways controlling cell cycle progression and apoptosis, in cardiac fibroblasts. Endocrine 18, 137–145. doi:10.1385/ENDO:18:2:137
Zhou W., Ilamycin E., Fang H., Wu Q., Wang X., Liu R., et al. (2019). Ilamycin E, a natural product of marine actinomycete, inhibits triple-negative breast cancer partially through ER stress-CHOP-Bcl-2. Int. J. Biol. Sci. 15, 1723–1732. doi:10.7150/ijbs.35284
Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., et al. (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995. doi:10.1101/gad.12.7.982
Keywords: unfolded protein response, er stress, cardiac fibroblast, fibrosis, cell death, apoptosis
Citation: Rowland MB, Moore PE and Correll RN (2024) Regulation of cardiac fibroblast cell death by unfolded protein response signaling. Front. Physiol. 14:1304669. doi: 10.3389/fphys.2023.1304669
Received: 29 September 2023; Accepted: 21 December 2023;
Published: 12 January 2024.
Edited by:
Fiona Murray, University of Aberdeen, United KingdomReviewed by:
Yongnan Li, Lanzhou University, ChinaCopyright © 2024 Rowland, Moore and Correll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert N. Correll, cm5jb3JyZWxsMUB1YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.