 Zikang Hao
Zikang Hao Kerui Liu2
Kerui Liu2 Ping Chen
Ping Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 08 June 2023
Sec. Exercise Physiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1193031
This article is part of the Research TopicSpotlight on Aging: Anthropological Factors Impacting Physiology, Prevention and Management of Aging ConditionsView all 8 articles
Disproportionate to the severity of Alzheimer’s disease (AD) and the huge number of patients, the exact treatment and prevention of AD is still being explored. With increasing ageing, the search for means to prevent and treat AD has become a high priority. In the search for AD, it has been suggested that exercise may be one of the more effective and less costly means of preventing and treating AD, and therefore a large part of current research is aimed at exploring the effectiveness of exercise in the prevention and treatment of AD. However, due to the complexity of the specific pathogenesis of AD, there are multiple hypotheses and potential mechanisms for exercise interventions in AD that need to be explored. This review therefore specifically summarises the hypotheses of the interaction between exercise and AD from a molecular perspective, based on the available evidence from animal models or human experiments, and explores them categorised according to the pathologies associated with AD: exercise can activate a number of signalling pathways inhibited by AD (e.g., Wnt and PI3K/Akt signalling pathways) and reactivate the effects of downstream factors regulated by these signalling pathways, thus acting to alleviate autophagic dysfunction, relieve neuroinflammation and mitigate Aβ deposition. In addition, this paper introduces a new approach to regulate the blood-brain barrier, i.e., to restore the stability of the blood-brain barrier, reduce abnormal phosphorylation of tau proteins and reduce neuronal apoptosis. In addition, this paper introduces a new concept.” Motor factors” or “Exerkines”, which act on AD through autocrine, paracrine or endocrine stimulation in response to movement. In this process, we believe there may be great potential for research in three areas: (1) the alleviation of AD through movement in the brain-gut axis (2) the prevention and treatment of AD by movement combined with polyphenols (3) the continued exploration of movement-mediated activation of the Wnt signalling pathway and AD.
One of the direct effects of the improvement in living standards is the increase in the average age of human survival, which has increased by almost 5.6 years compared to 50 years ago. This has triggered another phenomenon, namely, the aging of the population, with an increase in the number of people older than 60 years old (Rudnicka et al., 2020). It is estimated that by 2030, 500 million people will be affected by neurodegenerative diseases, which are inextricably linked to aging, with dementia accounting for a significant proportion of the population, and Alzheimer’s disease (AD) can be observed to account for more than 65% of the population (Hou et al., 2019). AD not only affects patients’ memory and higher cognitive abilities, but is also potentially lethal, and according to a cross-sectional survey from the United States, AD is the seventh most lethal disease in the nation. In addition to the physical suffering of patients, AD plays an equally detrimental role in socioeconomic development, with global investment in AD care reaching $82.8 billion as of 2015, and estimates suggesting that this figure could rise to $2 trillion by 2030 (Gaugler et al., 2022).
AD is a neurodegenerative disease with insidious onset and progressive development, characterized by cognitive impairment, psycho-behavioral abnormalities and reduced social life functions (Dubois et al., 2021). Generally, a family tendency to develop AD is called familial AD, while a non-familial tendency is called sporadic Alzheimer’s disease (Andrews et al., 2020). The World Health Organization reports that approximately 50 million people worldwide are currently living with dementia, with Alzheimer’s disease being the most common type. Possible risk factors for Alzheimer’s disease include increased age, female gender, low education level, smoking, midlife hypertension and obesity, hearing impairment, traumatic brain injury, lack of exercise, social isolation, diabetes and depressive disorders.
The pathological changes in the brain of patients with Alzheimer’s disease are diffuse brain atrophy and are characterized by senile plaques (SP), neurofibrillary tangles (NFT) and neuronal reduction. The SP center is β-amyloid protein (Aβ), and the main component of NFT is a highly phosphorylated microtubule-associated protein, tau protein (Ju and Tam, 2022). Genetics is one of the major factors in the development of Alzheimer’s disease. Four genes have been identified to be associated with Alzheimer’s disease, namely, the amyloid precursor protein (APP) gene, the presenilin 1 (PSEN1) gene, the presenilin 2 (PSEN2) gene and the apolipoprotein E (ApoE) gene. Among them, mutations or polymorphisms in the first three genes are closely associated with early-onset familial Alzheimer’s disease, and ApoE is closely associated with sporadic Alzheimer’s disease (Riedel et al., 2016). The more recognized pathogenesis of Alzheimer’s disease suggests that imbalance of Aβ production and clearance is the initiating factor of neuronal degeneration and dementia development, which can induce a series of pathological processes such as tau protein hyperphosphorylation, inflammatory response, and neuronal death. Meanwhile, there is a wide range of neurotransmitter abnormalities in the brains of Alzheimer’s disease patients, including acetylcholine system, monoamine system, amino acids and neuropeptides (Ju and Tam, 2022).
In the clinical treatment of AD, exercise is considered to be an effective non-pharmacological treatment (Sujkowski et al., 2022), Second, prospective observational studies suggest that the benefits of regular exercise for AD are not limited to the treatment of AD, but rather to the prevention of AD, and that physical inactivity is often considered a predisposing factor for AD (Guure et al., 2017; Iso-Markku et al., 2022). Of course, many meta-analyses of randomized controlled clinical trials have also shown that exercise has a reversing effect on memory loss, decline in higher cognitive functions, AD-induced lifestyle changes, mood abnormalities, etc., In AD patients (Blondell et al., 2014; Cunningham et al., 2020; Yu et al., 2020). With the advances in molecular biology and medicine, the effects of exercise on AD have been studied in more depth, starting with the possible risk factors and their effects on the course of AD, and examining the benefits of exercise on AD in detail and at a microscopic level, such as the improvement of synaptic plasticity, the reduction of neuroinflammation, and the promotion of hippocampal neurogenesis in AD patients with increased physical activity (Barnes, 2015; Luan et al., 2019).
The scope of exercise type is extremely broad, including the type of exercise, exercise intensity, exercise frequency, exercise duration, etc. With the addition of these factors, different types of exercise show different characteristics, and the role of AD does not seem to be the same, some researchers believe that compared to high-intensity exercise, it seems that moderate intensity sustained aerobic exercise is more suitable for AD patients (Wang S. et al., 2021). In addition, the pathways through which exercise mediates improvement in the course of AD are also being explored and discussed by researchers. The growing number of hypotheses about potential therapeutic targets for AD and the advances in medical and biological technology have opened up new possibilities to explore the potential benefits of exercise for AD.
More and more researches tend to take a molecular biology perspective to reveal how exercise works on AD through the complex and large “network” inside the human body, using the coordination of tissues, organs and systems (Lourenco et al., 2019). Among many studies, the more concentrated area is the involvement of exercise in participating in complex biochemical reactions through signalling pathways, regulating or altering major lesion components in AD, abnormal protein expression, etc. Interestingly, in this basis, researchers have proposed a very explored concept: the “motor factor (Lee T. H.-Y. et al., 2019)”. That is, these biochemicals are produced or expressed in response to the stimulus of exercise; in other words, although they may be produced by different tissues, the involvement or non-involvement of exercise is crucial to their action in the body.
Therefore, the aim of this review is to systematically summarize, from a molecular biology perspective, the evidence from currently available studies regarding the pathways through which exercise may be mediated (signaling pathways) and which biomarkers (involvement of motor factors) are involved in the complex chemical reactions in vivo to alleviate AD. Figure 1. Interactions between motion and AD through signaling pathways.
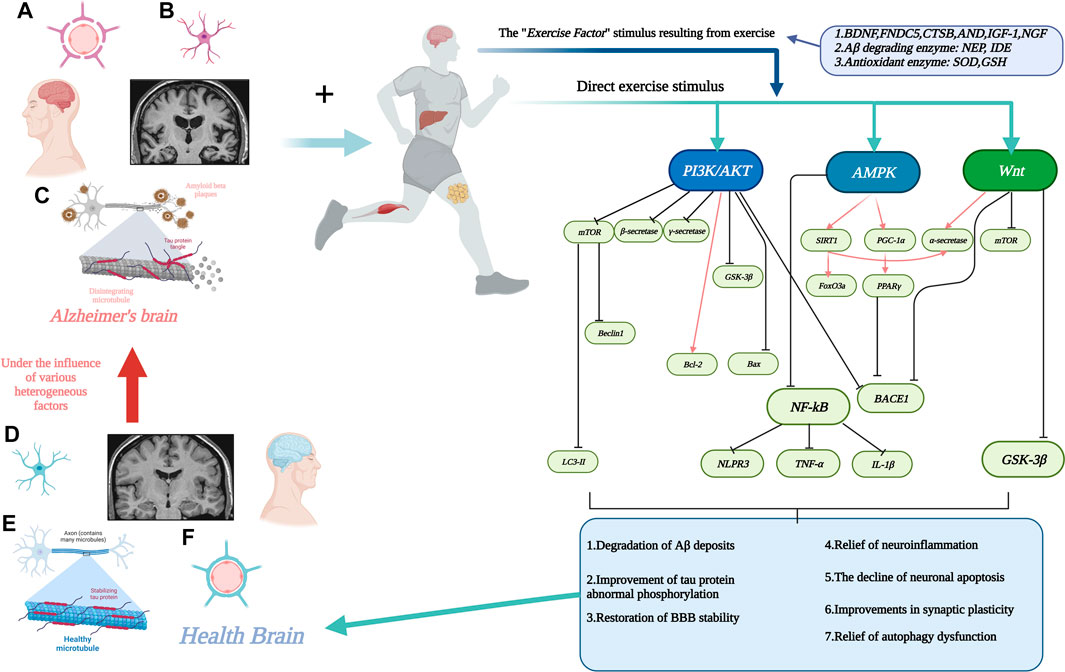
FIGURE 1. Interactions between motion and AD through signaling pathways. (A). Stabilization of the blood-brain barrier disrupted by AD with increased permeability. (B). Neuronal damage in the AD brain. (C). Aβ deposition around neurons and neuronal fiber tangle formation due to abnormal tau protein phosphorylation in the AD brain. (D). Neurons in healthy brain. (E). Healthy brain without Aβ deposition and neuronal fiber tangles. (F). Healthy brain with normal physiological function of the blood-brain barrier. The line segment with an arrow at the end represents facilitation and the line segment with a horizontal bar at the end represents inhibition. Created with BioRender.com.
Our study was based on the principles of the literature selection process suggested by PRISMA, with minor refinements, to identify the original research literature for the narrative review of this review (Page et al., 2021). The specific process is shown in the Figure 2.
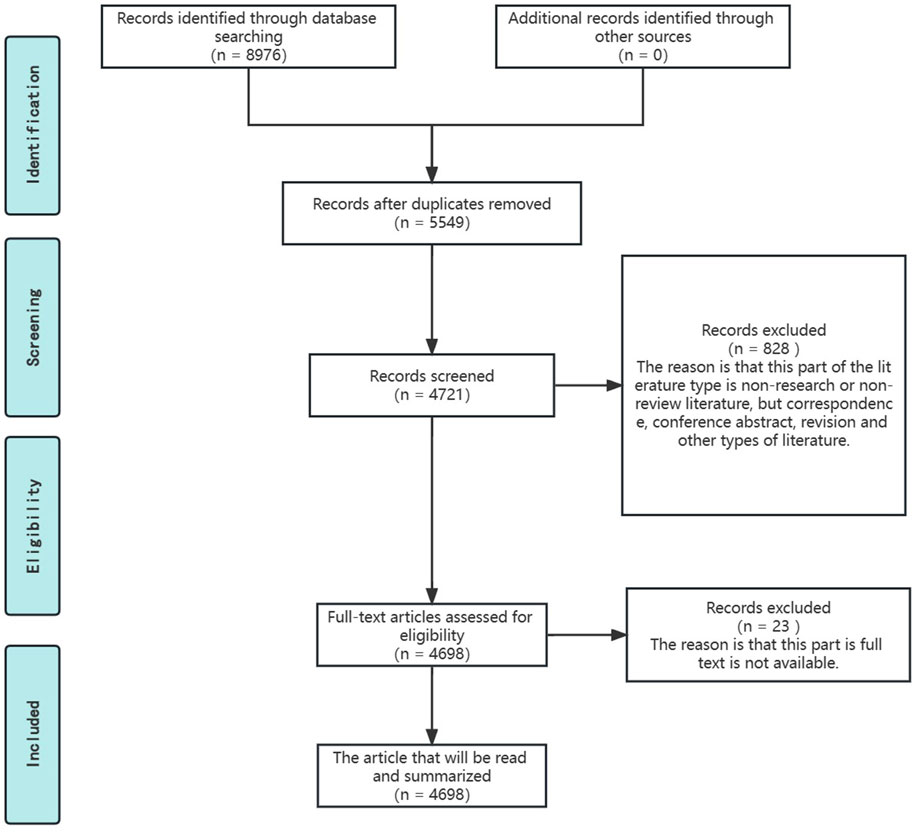
FIGURE 2. Literature reading process based on PRISMA recommendations.
Aβ is a normal product of body metabolism, which is produced by the hydrolysis of β-amyloid precursor protein (APP) by β-secretase and γ-secretase, and under normal conditions, Aβ maintains a stable state of production and elimination in the body (Jeong et al., 2022). In contrast, increased production of Aβ and decreased clearance of Aβ in the presence of abnormal APP metabolism caused by certain reasons can lead to abnormally large deposition of Aβ, which can have toxic effects on neurons. Although the mechanism of Aβ toxicity to neuronal cells is not clear, it has been pointed out that the molecular conformation and molecular state of Aβ are related to the neurotoxicity of Aβ, and the β-sheet structure can contribute to the aggregation of Aβ into insoluble fibers, which in turn form extremely insoluble deposits and consequently generate senile plaques (SPS), leading to the development of AD. In addition, the Aβ oligomer theory suggests that soluble Aβ oligomers are more neurotoxic than insoluble fibers, causing increased apoptosis or inducing glial cells to express nitric oxide synthase (I NOS) to produce large amounts of NO to kill neurons (Cioffi et al., 2019). Based on the available evidence that Aβ deposition plays a key role in neuronal cell death and brain atrophy, which results in the onset and progression of AD, mitigating Aβ deposition is a potentially promising target for the treatment of AD (Yoon and Jo, 2012).
Exercise mitigates Aβ deposition using the Wnt signaling pathway as a mediator: In several studies, it was found that the Wnt/β-catenin signaling pathway may be associated with the dissolution of Aβ deposition in AD, and activation of Wnt/β-catenin promoted α-secretase hydrolysis of APP (Jia et al., 2019). Interestingly, inhibition of Wnt/β-catenin activity increased the level of Aβ and also increased the ratio of Aβ42/Aβ40, in addition to the finding that Wnt/β-catenin also inhibited Aβ deposition by suppressing BACE1 transcription (Parr et al., 2015).
Exercise reduces Aβ deposition mediated by PI3K/AKT: A study confirmed that in AD mice, PI3K/AKT pathway was inhibited and Aβ42 was deposited in large amounts, making the memory ability impaired, while reactivation of PI3K/AKT and inhibition of β-catabolase and γ-catabolase activities not only promoted Aβ clearance but also inhibited Aβ deposition (Um et al., 2011). As a downstream target factor of PI3K/AKT signaling pathway, mTOR has a role in negatively regulating cellular autophagy. mTOR is over-activated in AD, which inhibits cellular autophagy and leads to Aβ deposition. In some researchers, it was pointed out that exercise can reduce Aβ deposition in mouse hippocampus by regulating PI3K/AKT signaling pathway. After 12 weeks of running-table exercise, the expression levels of phosphorylated PI3K (p-PI3K) and phosphorylated AKT (p-AKT) in the hippocampus of AD mice were found to be significantly increased, while the expression levels of mTOR returned to normal and the abnormal cellular autophagy was alleviated, while the expression of Beclin1 and LC3-II, which are associated with reduced Aβ deposition, increased and the Aβ content decreased, thus suggesting that the alleviating effect of exercise on AD may be achieved by activating the PI3K/AKT signaling pathway to alleviate the abnormal cellular autophagy in AD (Kang and Cho, 2015). As mentioned previously, it is possible that exercise may also act on SPS as an AD causative factor through the PI3K/AKT signaling pathway, and the possible mechanism is that exercise inhibits BACE1 expression, thereby suppressing Aβ production and reducing Aβ deposition.
Exercise alleviates Aβ deposition through the AMPK signaling pathway: AMPK is present in all mammalian cells as a fuel-sensing enzyme, and numerous studies have confirmed that an increase in AMP/ATP ratio leads to AMPK activation in skeletal muscle, while exercise is also considered to be the most significant physiological activator of AMPK (Jeon, 2016). Physical exercise is often considered a cheap and useful intervention for the treatment of some diseases, and its effects are also attributed to the activation of AMPK (Kjobsted et al., 2018). It is well known that PGC-1α and SIRT1 are downstream factors of AMPK and their expression is positively correlated with AMPK activation, while in AD, PGC-1α and SIRT1 are often considered as important targets for Aβ alleviation. After 35 sessions of moderate treadmill exercise, it was found that learning and memory deficits were improved in AD mice, and a reduction in Aβ deposition was observed. It is hypothesized that the possible mechanism is due to the activation of AMPK by long-term exercise which in turn upregulates the expression of PGC-1α, and the PPARγ activity regulated by it is similarly increased, both of which bind to the promoter of BACE1 and inhibit the transcription of BACE1, thus reducing Aβ42 and Aβ40 production, reducing Aβ deposition (Koo et al., 2017). A recent study suggested that after prolonged exercise activation of AMPK increases the expression of SIRT1, which is regulated by it, and subsequently shifts APP to the α-transferase pathway. Possible mechanisms include upregulation of SIRT1 to activate RARβ to further activate ADAM10, and in addition SIRT1 can deacetylate FoxO transcription factor 3a (FoxO3a) in the nucleus, causing ROCK1 is often phosphorylated at the Ser655 site of APP, and inhibition of its activity can reduce the β-catabolic pathway of APP and shift APP to the α-hydrolase pathway, thereby increasing the content of αAPPs and reducing Aβ deposition (Bedada et al., 2021).
It has also been suggested that Aβ deposition, in addition to the overexpression of BACE1, is associated with the ubiquitin-protease system (UPS), an enzyme system whose main function is to degrade abnormal intracellular or extracellular proteins, which is involved in restoring protein homeostasis and maintaining normal cellular function (Al Mamun et al., 2020a). In AD, a downregulation of UCHL-1 expression is often observed, while interestingly, in another study, UCHL-1 was found to be negatively correlated with BACE1. The possible mechanism is thought to be that the expression of UCHL-1, which is an E3 ligase, affects the function of UPS and thus the clearance of Aβ deposition. In contrast, exercise activates PI3K/AKT, which in turn upregulates the expression of UCHL-1 after activation, thereby improving Aβ clearance (Xu et al., 2022).
Tau protein, a highly soluble microtubule-associated protein (MAP), is abundant in neurons of the central nervous system (CNS). As a member of the MAPs family of proteins, tau protein mainly acts on the distal axon to maintain microtubule stability and flexibility, while other proteins such as MAP6 protein mainly acts on the proximal axon to fix microtubules, and MAP2 mainly maintains dendritic stability (Martin et al., 2013). Tau proteins interact with microtubule proteins (Tubulin) to stabilize microtubules while driving Tubulin assembly within microtubules. Tau proteins control microtubule stability through heterodimerization and phosphorylation (Ye et al., 2022). The phosphorylation of tau is regulated by a dynamic balance between tau kinase and phosphatase activities. Disruption of this balance is an important cause of abnormal tau phosphorylation, which causes tau protein aggregation and constitutes neuronal intracellular aggregates, thus causing neuronal fiber tangles (Al Mamun et al., 2020b).
The protein kinase of Tau, mainly PDPK, causes phosphorylation of tau through three pathways: A. Overexpression of GSK-3β induces abnormal phosphorylation of tau leading to neurodegeneration, resulting in a series of pathologies such as neuronal apoptosis, hippocampal neurodegeneration, memory deficits, increased neuroinflammatory Aβ deposition and reduced acetylcholine synthesis (Llorens-Martin et al., 2014). B. The most active form of CDK5, CSK5/p25 polymer, induces increased tau phosphorylation and neurodegeneration and induces neuronal apoptosis (Engmann and Giese, 2009). C. MAPK signaling pathway controlled by p38, Erk1/2, JNK1/2/3, where JNK activation is also involved in increased γ-secretase activity associated with increased production of Aβ (Ploia et al., 2011).
The activity of the Wnt signaling pathway is thought to be associated with hyperphosphorylation of tau in AD. In mice, dysfunction of Wnt was found to lead to phosphorylation of tau proteins, Thr231, Ser235 and AT8, resulting in hyperphosphorylation of tau, and reactivation of the Wnt signaling pathway revealed that hyperphosphorylation of tau was regulated, and in addition to this, GSK-3β activity was subsequently inhibited. It is hypothesized that in AD, inhibition of the Wnt signaling pathway leads to overexpression of GSK-3β, which induces hyperphosphorylation of tau and leads to neurogenic fiber tangles. In contrast, 12 weeks of aerobic exercise on a running platform upregulated Wnt-3 expression in the hippocampal tissue of MCI mice and inhibited GSK-3β expression, which somewhat alleviated Tau hyperphosphorylation and improved cognitive performance in mice.
As mentioned previously, a useful target for improving tau hyperphosphorylation and NFT deposition is thought to be GSK-3β. In addition to the Wnt pathway, the PI3K/AKT signaling pathway is also thought to be critical for reducing GSK-3β activity in AD. Exercise has been shown to have an effect on the activation of the PI3K/AKT signaling pathway, and exercise is beneficial in increasing the content and activity of PI3K in the hippocampus of AD brain, prompting the activation of Akt at Thr308 and Ser473, and the activated Akt inhibits GSK-3β activity by inhibiting GSK-3β phosphorylation at the Ser9 site, thus achieving the inhibition of abnormal Tau phosphorylation and improve NFT (Fang et al., 2018a). In addition, another study found that a single acute exercise session could also have the same effect, but with a time-limited effect, i.e., GSK-3β activity decreased immediately after exercise and then recovered (MacPherson et al., 2015), However, the effects of acute exercise on AD has remained controversial, with some suggesting that the oxidative stress that may be induced after acute exercise may be very detrimental to AD (Jin et al., 2015).
The AMPK signaling pathway is also often thought to be closely associated with AD. In this study, it was found that p-AMPK levels were positively correlated with p-AKT levels, and after prolonged exercise, p-AMPK levels were significantly elevated and phosphorylation of tau proteins at the Ser396 and Ser262 sites was inhibited. Similarly, mTORC1/P-70-S6K, which is regulated with AKT, was also activated and served to inhibit tau protein phosphorylation at the Ser396 site (Joa et al., 2017). In addition to this, some researchers have suggested that acetylation levels of tau are often associated with promoting tau hyperphosphorylation, and that aerobic exercise can reduce Ac-tau levels (Mankhong et al., 2020), sirt1, which is associated with changes in Ac-tau levels, was not found to be associated with exercise, i.e., it is inconclusive whether exercise can upregulate sirt1 and thus reduce Ac-tau levels. However, it is well established that increasing sirt1 levels can improve tau hyperphosphorylation in AD (Chen et al., 2022). Sports are widely considered to be sirt1 activators (Vargas-Ortiz et al., 2019), The aberrant phosphorylation of tau protein by exercise may be achieved through sirt1. Figure 3 visualises the content of 3.1 and 3.2.
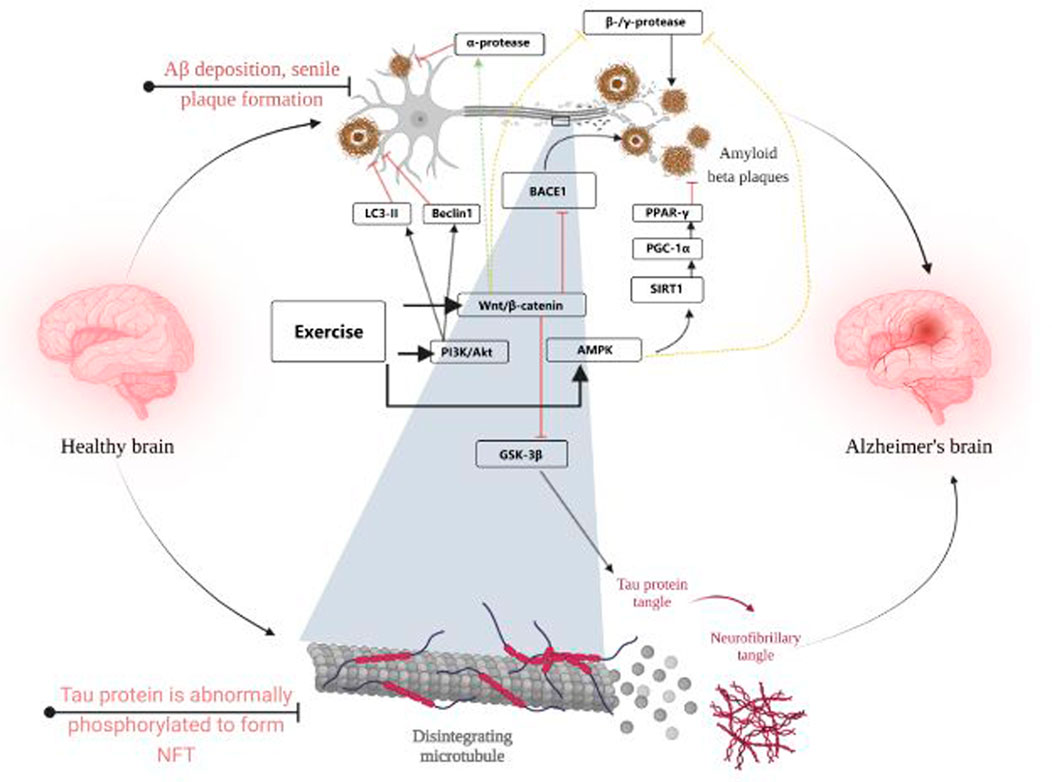
FIGURE 3. Visualises the content of 3.1 and 3.2. Created with BioRender.com.
Autophagy is an evolutionarily conserved and important process for the turnover of intracellular material in eukaryotes. In this process, some damaged proteins or organelles are wrapped in autophagic vesicles with a double membrane structure and sent to lysosomes for degradation and recycling to maintain cellular metabolic homeostasis and self-renewal (Nixon, 2013). In normal neurons, autophagic activity is active and efficient, but in AD, autophagy is impaired, and it has been suggested that dysfunction of autophagy and lysosomes precedes lesions in AD, and that this impairment has a facilitating effect on neuronal apoptosis and plaque formation (Zhang et al., 2022). Normal autophagy can help clear Aβ deposition and abnormally phosphorylated tau proteins. Downregulation of Beclin1 and LC3-II/I ratios was observed in AD mice (Lucin et al., 2013) and mTOR expression was upregulated (Liu et al., 2021), suggesting that normal autophagic activity is inhibited.
In their study, Liu et al. observed the activation of the AMPK/Beclin1 signaling pathway by aerobic exercise, thus serving to regulate abnormal autophagy in AD (Liu W. et al., 2019). Their further study found that aerobic exercise may activate the APN-AdipoR1/AMPK signaling pathway in mice to achieve the effect of regulating autophagy-lysosome pathway abnormalities in AD, thereby alleviating AD lesions. Adiponectin (APN) is often thought to have neuroprotective effects and is useful in alleviating cognitive impairment and memory loss (Jian et al., 2022). It is likely that exercise acts through APN and its receptor AdipoR1 for the recovery of neurodegenerative diseases.
The mTOR is an important regulator for autophagy, and overexpression of mTOR induces abnormal autophagic function. One of the upstream signaling pathways of mTOR is PI3K/AKT. In AD, it was found that inhibition of PI3K/AKT and upregulation of mTOR expression affect autophagy (Heras-Sandoval et al., 2014). According to Li et al., PI3K/AKT was activated in mice after high intensity interval training and medium intensity continuous training interventions, which inhibited mTOR expression and was helpful for the restoration of autophagic function. In addition to this, the expression of autophagy protein Beclin1 was also found to be upregulated after 12 weeks of running table exercise, which increased the autophagic activity that was also altered due to AD (Kang and Cho, 2015). Therefore, it is believed that the amelioration of abnormal autophagy function in AD by exercise could also exert its effect through the PI3K/AKT signaling pathway.
In AD, the neurotoxic effects of Aβ can affect the structural part of the postsynapse, particularly important for PSD-95, leading to inhibition of LTP (a major form of synaptic plasticity reflecting changes in physiological activities such as neuroelectricity in synaptic plasticity) and synaptic dysfunction, causing related changes in learning and memory abilities (Tu et al., 2014). The protein expression level of PSD-95 is also often used as a practical indicator of synaptic plasticity in the mouse hippocampus (Shao et al., 2011).
Also, Wnt activation can upregulate PSD-95 and then LTP, thus causing enhanced learning and memory capacity (Oliva et al., 2013). Ren et al. found that prolonged physical activity significantly inhibited DKK1, an inhibitor of Wnt, compared to sedentary patients, resulting in a decrease in DKK1 levels in patients (Ren et al., 2017). In contrast, overexpression of DKK1 significantly inhibits Wnt activity, which could explain the much higher levels of DKK1 found in the hippocampus of a subset of AD patients at autopsy and their synaptic degeneration (Ren et al., 2019). The mechanism by which long-term exercise improves synaptic plasticity via Wnt may be that exercise downregulates the abnormal upregulation of DKK1 expression in AD, reversing to some extent the abnormalities of Wnt and its regulated PSD-95 and LTP due to DKK1 overexpression.
In AD, in addition to the aforementioned changes in synaptic plasticity, synaptic damage is often observed, and this also causes neuronal loss (Fricker et al., 2018). As a key pathway controlling cell survival, many studies have found that the absence or inhibition of Wnt makes neurons more susceptible to apoptosis induced by Aβ deposition (De Ferrari and Moon, 2006; Rosso and Inestrosa, 2013; Pascual-Vargas and Salinas, 2021). Also, in AD, finding ways to reactivate Wnt could rescue this detrimental effect. Despite controversy, new evidence suggests that hippocampal neurogenesis in humans does exist in the aging brain and realizes a dramatic decline in the AD brain. The likely mechanism is the downregulation of Wnt proteins and messy proteins, while DKK1 expression is upregulated, combined with reduced levels of Wnt protein astrocytes due to aging, thereby disrupting adult hippocampal neurogenesis.
In AD, it is often accompanied by apoptosis of neuronal cells, and in the elderly, neuronal survival is important for the health of the organism. Among all apoptotic pathways, the Bcl-2 protein family is the downstream protein family of all pathways and is essential for apoptosis, especially Bcl-2, which plays an anti-apoptotic role, and Bax, which plays an apoptotic role. In normal brain, Bcl-2 is highly expressed and saves cells from apoptosis, while in AD brain, Bcl-2 expression is suppressed, Bax is overexpressed, and Bcl-2/Bax homeostasis is disrupted, triggering neuronal apoptosis. In the latest study, it was found that aerobic exercise improved neuronal apoptosis and was accompanied by activation of PI3K/AKT Further study revealed that it was the activation of PI3K/AKT that reversed the decrease of Bcl-2 expression and inhibited Bax expression, allowing the balance to be restored (Peng et al., 2022). Figure 4. Interaction between synaptic function, blood-brain barrier, neuroinflammation in AD.
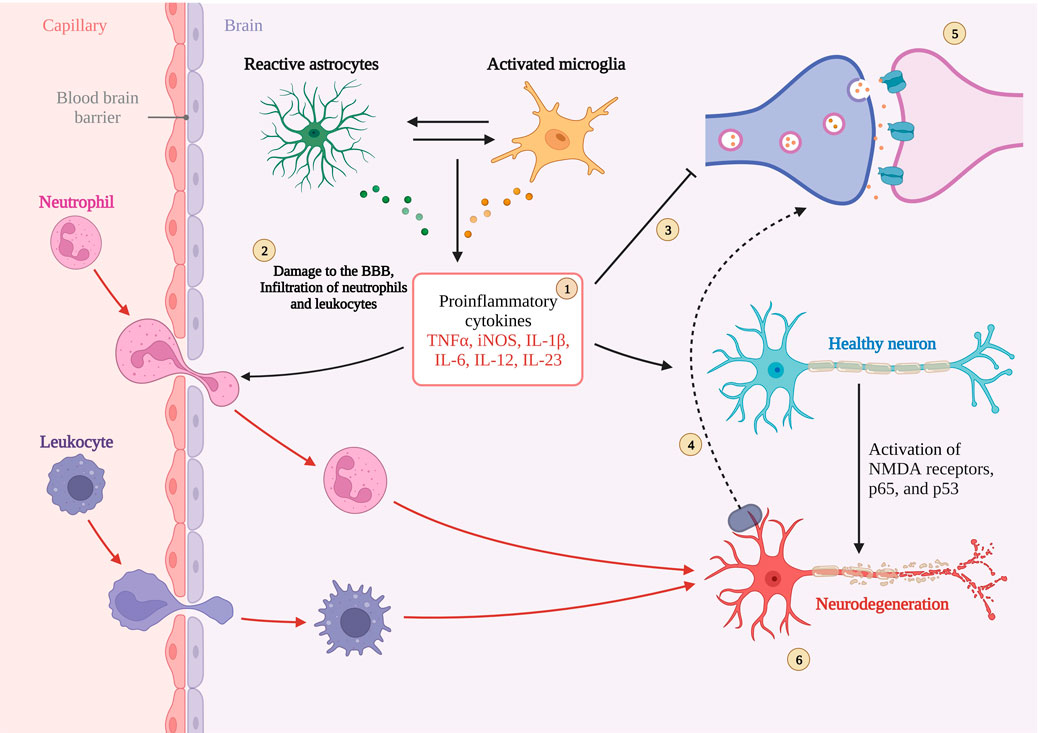
FIGURE 4. Interaction between synaptic function, blood-brain barrier, neuroinflammation in AD. 1. Sustained activation of microglia and astrocytes leading to cytokine overexpression. 2. Destabilization of the BBB leading to peripheral cytokines into the CNS.3 and 5 Inhibition and disruption of synaptic function by excess cytokines.6. Neuronal damage and death caused by cytokines.4. Magnified illustration of neuronal terminals. Created with BioRender.com.
In AD, although the exact mechanism of its pathogenesis is unknown, the more accepted hypotheses are the Aβ hypothesis and the NFT hypothesis due to Tau. In addition, more than 30 years ago, some researchers suggested that AD may also be associated with neuroinflammation, but there is no definite conclusion to prove whether neuroinflammation is the cause or effect of AD (Calsolaro and Edison, 2016). Under normal circumstances, moderate inflammation produced by the organism will subside on its own, but prolonged inflammation can lead to the development of chronic inflammation, i.e., the organism does not recover properly. Neuroinflammation, on the other hand, refers to the inflammatory response of the CNS with neuronal damage (Thawkar and Kaur, 2019). There is abundant evidence that such neuroinflammation is often observed in AD, in addition to the activation of astrocytes and microglia associated (Lyman et al., 2014; Calsolaro and Edison, 2016; Guo S. et al., 2022). These two types of cells are essential for the inflammatory response of the CNS. It is generally accepted that microglia are in a “quiescent” state under normal conditions, but can be in a state of monitoring of the brain and its environment, and are often considered to be neuroprotective cells with phagocytic functions and release of neurotrophic factors. Under abnormal conditions, microglia are activated, and although they do initially have a defensive role, if abnormal stimuli persist, chronic neuroinflammation occurs, and the release of cytokines (TNF-α, IL-1β, etc.) leads to a proinflammatory response, and the persistence of these cytokines continues to induce the production of APP in the AD brain, which itself has a disrupted Aβ homeostasis and produces more APP. Also, the AD brain, whose Aβ homeostasis is disrupted, produces more Aβ due to the increase of APP, forming a vicious circle (Kwon and Koh, 2020). In addition, Aβ can stimulate the NF-kB signaling pathway, which is a downstream target of pro-inflammatory cytokines (Sivandzade et al., 2019). Activated microglia also release substances such as free radicals, reactive oxygen species and neurotoxic substances that are detrimental to neuronal survival and normal synaptic function. The effects of neuroinflammation on the course of AD are not only accomplished by exacerbating Aβ deposition and NFT, but are also mixed with more complex factors. Therefore, the suppression of neuroinflammation may be a potential way to treat AD.
NF-kB, as an important pro-inflammatory signaling pathway in AD neuroinflammation, is involved in the expression of almost all inflammatory cytokines in AD. NF-kB is nucleated and overexpresses pro-inflammatory factors such as IL-1β and TNF-α to trigger neuroinflammation, while IL-1β and TNF-α can in turn act as agonists of NF-kB in reverse (Zhao et al., 2019). The activation of PI3K/AKT signaling pathway will play a role in inhibiting the activation of NF-kB. After 8 weeks of swimming training, the expression of p-PI3K and p-AKT was upregulated in the hippocampus of rats, the activity of NF-kB was inhibited, and the expression of TNF-α, NO, IL-1β and other pro-inflammatory factors regulated by them were downregulated (Wang et al., 2019).
The activation of AMPK signaling pathway by exercise is well documented, and AMPK is also considered as a “star signaling pathway” to improve some diseases, and some of the beneficial effects of AMPK are achieved through its regulation of downstream targets, such as SIRT1 and PGC-1α. Although SIRT1 has been shown in many studies to be useful in alleviating neuroinflammation in AD, it is unfortunately unknown whether the alleviating effect of exercise on neuroinflammation is mediated through the AMPK/SIRT1 axis, as SIRT1 can be activated by more than just AMPK. Exercise is important for SIRT1 expression, and in neuroinflammation in AD, SIRT1 directly deacetylates NF-kB at the p65 site, resulting in inhibition of NF-kB overactivation (Koo et al., 2017). In addition to IL-1β, TNF-α, pro-inflammatory factors that have been shown to be directly regulated by NF-kB, there is a pro-inflammatory factor NLRP3 that is also regulated by SIRT1. It is known that NLRP3 expression must be controlled by NF-kB, and overexpression of NLRP3 is often observed in insulin-resistant mice. Thus exercise appears to alleviate neuroinflammation through the SIRT1/NF-KB/NLPR3 axis (Zhao et al., 2023). According to previously described, microglia activation is also associated with NF-kB mediation, and protein expression of SIRT1 increases after exercise, thereby activating Nrf2 and PGC-1α to inhibit microglia activation and alleviate Aβ deposition and NFT formation (Fang et al., 2018b; Wu et al., 2018). Figure 5. Neuroinflammation with microglial activation in AD.
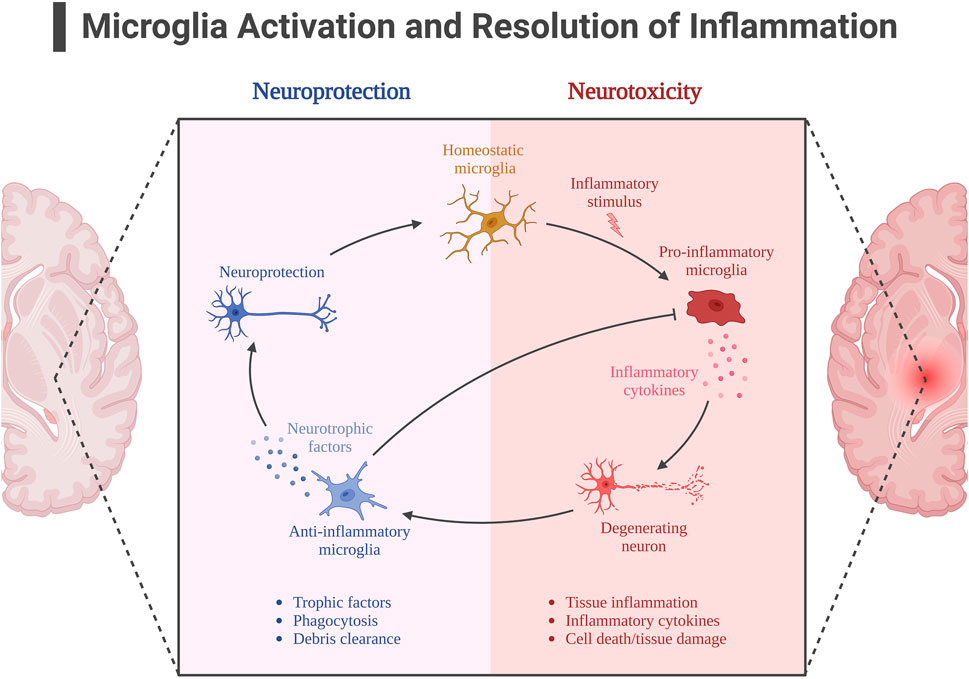
FIGURE 5. Neuroinflammation with microglial activation in AD. Created with BioRender.com.
Although controversial, recent studies suggest that the blood-brain barrier (BBB) is inextricably linked to AD, a functional unit that divides brain tissue from the peripheral environment, consisting of vascular endothelial cells tightly connected to each other by tight junction proteins (TJs) and adhesion-linked proteins, and pericytes and astrocytes covering the endothelial cells (Sweeney et al., 2019). It plays a role in limiting the entry of peripheral macromolecular proteins, cytotoxic substances, and peripheral inflammatory factors into the CNS, and maintaining the stability of the CNS internal environment and normal brain cell function (Liu X. et al., 2019).
Impairment of normal physiological function of BBB is often found in AD brain, such as crosstalk between neuroinflammation and reduced BBB stability, which is maintained in normal state, whereas in AD brain, the increased permeability of BBB allows the opportunity for peripheral inflammatory factors to enter the brain, which in turn is overexpressed by AD, possibly via NF-kB pathway (Vest and Wyss-Coray, 2022). This part of inflammatory factors enters the CNS, which activates glial cells and induces the differentiation of M1 phenotype, increasing the inflammatory state in AD brain, which in turn continues to impair the normal physiological function of the BBB and causes endothelial cell damage, forming a cycle. In addition, abnormal BBB function is often thought to be involved in the process of Aβ deposition. In normal brain, Aβ protein is transported from the brain to the brain via the BBB, whereas abnormal BBB function may lead to impairment of this process and abnormal clearance of Aβ transport thus causing Aβ deposition (Nelson et al., 2016; Montagne et al., 2017; Profaci et al., 2020).
In some studies on the alleviation of the AD process through physical activity it was found that exercise might possibly reverse the disruption of the BBB stability in the AD brain.
As previously mentioned, TJs are molecular substances that act as anchors to produce the BBB, and some proteins expressed by TJs are important for maintaining BBB stability, such as the claudin family and occludin. Downregulation of claudin-3 and -5 expression was observed in AD brain, while a protein that inhibits tight junctions in the brain, endothelial cell adhesion molecule 1 (PECAM -1) was upregulated. In contrast, regular physical exercise restored the expression of claudin-4 and occludin to basal levels in the brains of AD mice, while downregulating the expression of PECAM-1 for maintaining tight links in TJs (Malkiewicz et al., 2019). Interestingly, overexpression of GSK-3β may shorten the half-life and suppress the expression levels of ocludin and claudin-5, while physical exercise in turn suppresses the expression levels of GSK-3β in AD brains and indirectly restores TJs. In addition, in a study of AD induction by ALCL3 injection, it was found that physical exercise not only promoted ocludin and claudin-3 expression increases, but also decreases the downregulation of inflammation-related PHF-3 and TNF-α expression (chun et al., 2022).
It is the anti-inflammatory effect of exercise that may have a “blocking” effect on the vicious cycle between CNS neuroinflammation and BBB. For example, the secretion of the anti-inflammatory factors IL-6 and IL-10 is increased and the secretion of IL-1β and TNF-α is decreased. In addition, oxidative stress is often thought to be involved in the crosstalk between neuroinflammation and the BBB, with oxidative stress states causing disruption of the tight linkage state of TJs, while physical exercise can exert antioxidant effects via the AMPK/Nrf2 pathway to maintain TJs and protect neurons (Małkiewicz et al., 2019). It is also noteworthy that disruption of the BBB due to chronic inflammation has been observed not only in AD patients but also in patients with T2DM, dyslipidemia, obesity and insulin resistance, which are often associated with chronic inflammation and are often thought to be associated with an increased risk of AD. The role of exercise in AD risk prevention by eliminating the crosstalk between BBB and chronic inflammation cannot be ignored (Bertram et al., 2016; Brown et al., 2022). Figure 6. Alterations in blood-brain barrier function in AD.
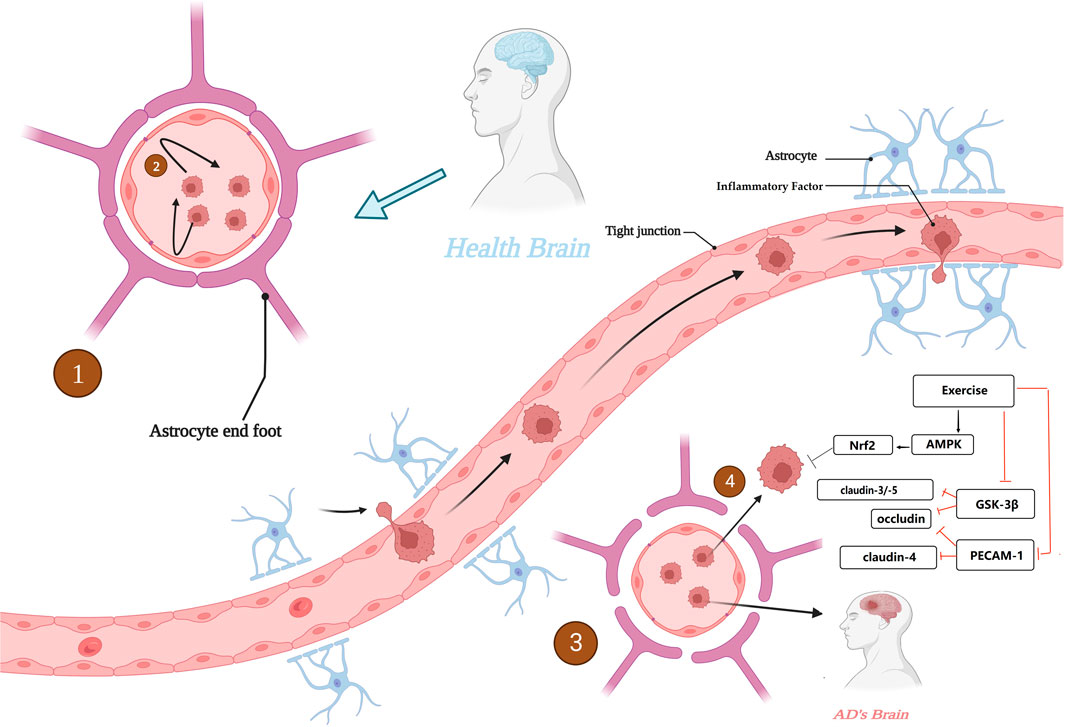
FIGURE 6. Alterations in blood-brain barrier function in AD. 1. BBB with normal physiological function in healthy brain. 2. BBB with normal physiological function to maintain the CNS from interference by peripheral harmful factors. 3. BBB with destabilization and increased permeability in AD brain. 4. The BBB, which is unable to function as a barrier, invades the CNS. Created with BioRender.com.
In conclusion, BBB plays an important role in alleviating the course of AD and preventing the risk of AD, and maintaining the stability of BBB may be a new target for the prevention and alleviation of AD, is a means that cannot be ignored
The blood-brain barrier is of great interest to researchers as an important defense mechanism in the human body. Based on this, it has been suggested that microorganisms in the gut and intestinal tract may interconnect with the CNS through the “brain-gut axis”, i.e., forming a bidirectional “microbe-gut-brain” pathway, in which the blood-brain barrier plays an important role. In AD brain, an increase in the abundance of harmful intestinal microorganisms was found (Goyal et al., 2021), and consequently, the type of short-chain fatty acids (SCFAs) produced by the intestinal microbiota changed, which is an important nutrient for maintaining the stability of the intestinal barrier and the BBB, and significant changes in SCFAs adversely affected the maintenance of BBB stability (Wang D. et al., 2021). For example, in mice treated with antibiotics, changes in intestinal flora species were found, with the consequent effect of suppressing SCFA homeostasis and expression levels of the claudin family of proteins and occludin proteins, two transmembrane proteins that are important for the maintenance of TJs, as discussed earlier (Wang, 2023). This highlights the relationship between maintaining a stable gut microbiota and BBB stability. In addition, chronic high-sugar and high-fat diets can also disrupt the normal gut microbiota, thereby inhibiting tight junction proteins. Thus, the gut microbiota may be an important target for improving AD and maintaining BBB stability, but current studies are controversial and therefore further research is needed to determine this (Bou Zerdan et al., 2022). Figure 7. Schematic diagram of the relationship between the “brain-gut axis” and the blood-brain barrier.
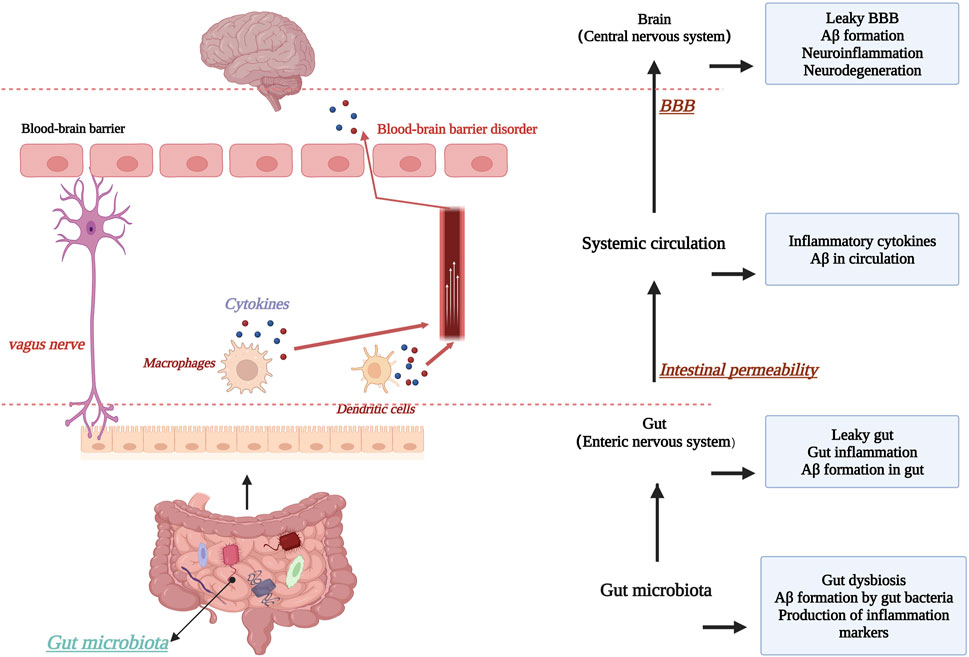
FIGURE 7. Schematic diagram of the relationship between the “brain-gut axis” and the blood-brain barrier. Created with BioRender.com.
There is a consensus that physical activity has beneficial effects on the body. The first half of this paper discusses the benefits of physical activity on AD in the context of signaling pathways, starting from AD and its possible causative factor. In addition, recent studies have focused on biomolecular substances called " Exerkines”, which are so named because they almost always act on the extracellular environment through autocrine, paracrine or endocrine action in response to exercise (Chow et al., 2022). Exerkines exert beneficial protective effects on CNS by crosstalking systems, tissues and organs, including but not limited to promoting neuronal regeneration, improving synaptic plasticity, and alleviating memory and higher cognitive decline.
BDNF is found in a wide range of areas such as the CNS, peripheral nervous system, endocrine system, bone and cartilage tissues, but is mainly expressed in the CNS, especially in the hippocampus and cortex. BDNF is the most abundant neurotrophic factor in the body and acts by binding to TrkB, an intracellular region with intrinsic tyrosine kinase activity, which is activated by BDNF binding to TrkB, causing enhanced phosphorylation of TrkB itself, which in turn activates the Ras-MAPK pathway and finally activates CREB at the final activation of CREB at the serine site of CAMP response element binding protein (CREB). CREB promotes neuronal cell survival, increases synaptic plasticity and neurogenesis by increasing the expression of BDNF gene and anti-apoptotic protein gene BCL-2 (Lu et al., 2013). The mode of action is specifically as follows: (1) Increases synaptic plasticity, which in turn affects LTP, the process underlying the learning process and the process of memory formation (second level memory). (2) Promoting neurogenesis especially in the hippocampus, Ernfors et al. found that heterozygous mice with BDNF knockout had much less neuropathy than wild-type mice. (3) Promoting cell survival, mainly by maintaining and promoting the developmental differentiation and growth regeneration of various neurons, especially 5-hydroxytryptamine (5-HT) and dopaminergic (DA) neurons (Allen et al., 2013; Gao et al., 2022). BDNF not only binds to the Trk family of receptors (TrkA, TrkB, TrkC) but also to the P75 neurotrophin receptor, a member of the tumor necrosis factor family with a molecular weight of 75 kD, exert the corresponding biological effects. P75 receptors are not required for the function of neurotrophins (NTs); the role of P75 may lie in enhancing the affinity of Trk receptors for NTs, facilitating nerve terminal uptake and retrograde transport (Eggert et al., 2022). In addition, BDNF inhibits the pro-inflammatory signaling pathway NF-kB, thereby inhibiting the secretion of pro-inflammatory factors activated by glial cells and exerting neuroprotective effects (Cai et al., 2014). Some scholars believe that neuronal damage caused by decreased DNA repair capacity due to DNA oxidation is often observed in AD, and that BDNF can counteract DNA damage and increase transcription factor activity by increasing protein expression of purine-free/pyrimidine-free nucleic acid endonuclease/oxidation reductase-1 (APE1/Ref-1) through the BDNF/CREB pathway (Yang et al., 2014). For aberrant phosphorylation of Tau, the possible signaling pathway of BDNF action is PI3K/AKT. In addition to this, increased BDNF can induce a shift in APP degradation toward the α-secretase pathway and prevent the hydrolysis of APP to Aβ by β-secretase.
The peroxisome protein, FNDC5, is a glycosylated type I membrane protein. FNDC5 and its small molecule, irisin, produced by protein hydrolysis, play a considerable beneficial role in the course of AD. FNDC5 is widely distributed throughout the body but is upregulated in skeletal muscle, serum and hippocampus after exercise stimulation, probably because its upstream regulator PGC-1α expression is upregulated under exercise conditions (Wrann et al., 2013). FNDC5/irisin is a biological target that regulates AD and its expression level is negatively correlated with AD-related symptoms, such as improving neuroinflammation and promoting increased synaptogenesis and plasticity (Lourenco et al., 2019), Another ability of irisin is its permeability to the BBB, and irisin crosses the BBB to upregulate BDNF expression (Guo P. et al., 2022). In AD patients, induction of hippocampal proliferation is one of the potential therapeutic tools, and irisin can promote hippocampal and neuronal proliferation and improve synaptic function by upregulating BDNF expression and subsequently STAT3 signaling (Rabiee et al., 2020). After moderate treadmill exercise, improved spatial learning and memory abilities were observed in mice, possibly because exercise blocked the binding of Aβ42 to neurons via AMPK/PGC-1α/FNDC5/irisin (Azimi et al., 2018). In addition to these effects, it was also observed that increased expression of irisin inhibited astrocyte phosphorylation and IkBα inhibition due to Aβ exposure, thereby suppressing NF-kB activity and alleviating neuroinflammation. Irisin is known as the “messenger goddess”, and its beneficial effects on AD cannot be separated from the regulation of FNDC5 and BDNF, and more importantly, from their activation by exercise.
CTSB, a cysteine protease, is a muscle " Exerkines” that is thought to be induced in large amounts by exercise and mediates an increase in BDNF and DCX, thereby causing an improvement in hippocampal function (Suzuki, 2016). In a clinical study, 26 weeks of treadmill training was found to increase CTSB expression and show beneficial cognitive changes in middle-aged and older AD patients (Gaitan et al., 2021). However, at the same time, some scholars have taken an opposing view, suggesting that CTSB may increase the activity of NLRP3 inflammatory vesicles and induce oxidative stress (Bai et al., 2018).
IL-6, a factor currently thought to be induced by exercise, seems to play a “paradoxical” role in the organism. In healthy individuals, IL-6 expression levels were found to increase dramatically after exercise and return to normal levels during rest periods, and IL-6 is thought to inhibit the expression of the pro-inflammatory factor TNF-α (Ng et al., 2018). At the same time, however, chronic elevation of IL-6 is thought to be detrimental to the normal physiological function of the CNS, and large amounts of IL-6 are found in aged plaques in AD brains. In addition, IL-6 can also cross the BBB, whose stability is destabilized by AD, and transmit peripheral inflammatory processes into the CNS (Silva et al., 2021). Whether the specific role of IL-6 in the AD population is pro-inflammatory or anti-inflammatory needs to be discussed and explained by more scientific studies in the future.
APN is an endogenous bioactive peptide or protein secreted by adipocytes that plays an effect on insulin sensitization. It has been found that APN is involved in and ameliorates some metabolic diseases that may induce AD, such as T2DM and insulin resistance. Exercise has considerable benefits in inducing improvements in the CNS involved in APN (Song and Lee, 2013). First, the effect of exercise on the induction of ADN is due to the activation of the AMPK signaling pathway stimulated by exercise. Activation of AMPK upregulates the expression of ADN, and the AdipoR1 (a receptor for ADN)/articulin, which is involved in the regulation of ADN, enhances hippocampal neurogenesis. The benefits of ADN for the hippocampus also include the promotion of its proliferation, relying on the AND/p38MAPK/GSK-3β/β-catenin axis to achieve this. In behavioral tests in mice, it was found that mice with normal ADN expression showed better cognitive function and LTP than knockout mice (Liu et al., 2020). As for neuroinflammation, perhaps two aspects can be discussed. A. Exercise induces the release of ADN, which in turn alleviates neuroinflammation via AdipoR1/AMPK/NF-kB/IL-1β. B. Acrp30/PPARγ, induces an anti-inflammatory M2 phenotype in microglia, secreting IL-4/IL-10, arignase1 and other anti-inflammatory factors. C. Mitigation of the disruption of BBB stability and permeability by neurotoxic substances of Aβ through Acrp30/AdipoR1/NF-kB axis. As previously described, the disruption of BBB has a facilitative effect on the development of neuroinflammation.
IGF-1 is a tissue growth factor secreted by the liver and is found to be widely expressed in the nervous system, especially in the CNS, and it was found that animal studies in which IGF-1 signaling was blocked revealed exacerbation of some AD conditions, such as increased Aβ deposition and abnormal phosphorylation of tau, whereas in human and animal studies it was found that concomitant with increased physical activity, IGF-1 expression in the upregulation of IGF-1 expression in the CNS and the remission of AD disorders were found in human and animal studies, where the binding of IGF-1 to its receptor led to phosphorylation of its own protein kinase and activation of downstream signaling molecules, such as insulin receptor substrates 1 and 2 (IRS-1/2) (Ostrowski et al., 2016), which activated the phosphatidylinositol 3-kinase (PI3K)-Akt pathway. In addition, activated receptor tyrosine kinase aggregates the bridging protein Shc, which subsequently activates mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) 1/2 via a cascade reaction. In addition, IGF-1 is useful for neural stem cell proliferation, possibly through PI3K/AKT signaling to induce differentiation of neural stem cells (Trueba-Saiz et al., 2017). In addition, IGF-1 has a regulatory effect on APP metabolism; IGF-1 promotes the α metabolic pathway of APP and facilitates the transfer of β/γ metabolic pathway to α metabolic pathway, and also inhibits the expression of BACE1 thereby suppressing Aβ deposition (Jiang et al., 2017).
Nerve growth factor (NGF) is the first identified neurotrophic factor that has the biological function of nourishing neurons and promoting protrusion growth, and has an important role in the development, differentiation, growth, and regeneration of central and peripheral neurons, and is widely expressed not only by neurons and glial cells, but also detected in peripheral cells, and reduces neuronal death in AD via the NGF/TrkA/PI3K pathway. A more consistent view is that in AD, NGF/TrkA mainly acts on non-amyloid proteins, and overexpression of NGF regulates TrkA binding to APP and promotes Golgi transport of APP, inhibits APP binding by BACE1, thus reducing Aβ production and eliminating imbalance, in addition to finding that NGF promotes APP transfer to the α-secretase pathway (Aloe et al., 2012). After exercise, NGF expression was increased, suggesting that exercise may have an important role in upregulating NGF expression.
In addition to NGF, another “Exerkines” widely distributed in various tissues and cells, VEGF, a highly specific pro-vascular endothelial cell growth factor, is also significantly regulated by exercise. VEGF has six structurally similar proteins, VEGF-A/B/C/D/E and placental growth factor. The VEGF family is known to promote increased vascular permeability, vascular endothelial cell migration, proliferation and angiogenesis (Melincovici et al., 2018). As mentioned above, changes in vascular function are often found in AD, which could also explain the decrease in VEGF expression levels found in AD. However, increasing physical activity in the course of AD upregulates VEGF expression, which is useful for hippocampal neurogenesis and improved synaptic plasticity, while the polymer of VEGF-C/VEGFR3 is useful for alleviating neuroinflammation, which not only induces M2-type polarization of microglia and promotes the production of anti-inflammatory factors, but also prevents neuronal apoptosis to some extent (Han et al., 2014). VEGF also appears to be reported to be associated with reduced Aβ deposition and improved abnormal phosphorylation of Tau proteins (Mahoney et al., 2021).
NEP, an enkephalinase, is a type II membrane metalloendopeptidase with a zinc-binding motif (HEXXH) at the active site in the extracellular carboxy-terminal structural domain. NEP is highly expressed throughout the body, and in the CNS, NEP is found mainly at the presynaptic neuron terminals. NEP expression is upregulated by increased physical activity and also increases its enzymatic activity, an effect found mainly in the hippocampus and cerebral cortex (Hui, 2007). In AD, hemizygous NEP deficiency is often found, and it is hypothesized that NEP deficiency is associated with impaired memory, astrocyte activation and increased Aβ deposition. NEP acts as an Aβ degrading enzyme, mainly acting on BACE1, and NEP expression upregulates consistent BACE1 production, thereby inhibiting Aβ deposition production and age plaque formation (McAllister et al., 2010); NEP may also alleviate Aβ toxicity through BBB penetration.
In addition to NEP, there is an enzyme that specifically targets β-constituents. Insulin-degrading enzyme (IDE) is a metalloproteinase, and although insulin is the primary substrate for its action, its catabolic cleavage for other peptides gives it the effect of degrading Aβ deposition. One study found that increased physical activity significantly upregulated IDE expression levels in the murine hippocampus, and it was in the presence of AD that IDE concentration and activity were significantly reduced in patients. It is hypothesized that exercise may upregulate IDE expression through the PPARγ/AMPK pathway so that the accumulation of Aβ42 is reversed (Li et al., 2009; Costes and Butler, 2014).
Oxidative stress is an important mechanism of the aging process of the body and may be harmful for CNS (Salim, 2017). Both free radicals and reactive oxygen species (ROS) “contribute” to the process of AD, such as increasing damage to synaptic plasticity and triggering neuroinflammation, but small amounts of ROS are irrelevant to AD, mainly because of the presence of oxidative stress defense systems consisting of antioxidant enzymes that can scavenge them, such as superoxide dismutase (SOD) and catalase. However, the expression of antioxidant enzymes in the body is not constant, and when the level of ROS and other substances exceeds and suppresses the antioxidant enzymes, the oxidative stress state in the body surges. As two major categories of pathological mechanisms in AD, abnormal phosphorylation of Tau and deposition of Aβ contribute to the production of ROS. Aβ may cause ROS production by causing mitochondrial dysfunction and lipid peroxidation of unsaturated fatty acids. An important risk factor biomarker in the course of AD is RCAN1, and upregulation of RCAN1 is thought to result from oxidative stress caused by a combination of Tau phosphorylation and Aβ (Greenough et al., 2013; Chen and Zhong, 2014; Kim et al., 2015).
SOD is a metalloenzyme whose defense against oxidative stress depends mainly on the scavenging of superoxide anion (O2-). There are three types of SODs, distinguished by the different metal ions they contain: SOD1 contains Cu2+ and Zn2+; SOD2 is an SOD containing Mn2+ mainly in mitochondria, and SOD3 is an SOD containing Cu2+ and the only secretory SOD. Physical exercise significantly increased SOD1 and SOD2 in the in vivo study. In AD mice, the Aβ42/Aβ40 ratio decreased with increased SOD2 and restored LTP (Ma et al., 2011). Overexpression of SOD1 prevents superoxide anion and oxidative stress induced by polymers of Cu and Aβ (Murakami et al., 2012).
Glutathione (GSH) is a gamma-amide-bonded and sulfhydryl-containing tripeptide composed of glutamate, cysteamine and glycine and is present in almost every cell of the body. It is commonly believed that oxidative stress-mediated AD is associated with reduced GSH expression. Studies in humans and animal models have shown that physical exercise is effective in increasing GSH content and GSH/GSH persulfide ratio in the CNS. GSH protects brain endothelial cells by inhibiting NO and ROS production, and in addition, upregulation of GSH expression activates the activity of the Nrf2 signaling pathway, and many downstream target factors under its control are often considered to be key antioxidants. GSH also has the potential to deplete and induce activation of pro-inflammatory signaling pathways in microglia and astrocytes, leading to the release of pro-inflammatory factors and exacerbating neuroinflammation. Similarly, for Aβ, a major pathological marker of AD, GSH deficiency and reduction contribute to Aβ formation, probably because GSH deficiency causes the accumulation of carboxy-terminal fragments of Aβ precursor proteins (Saharan and Mandal, 2014; Braidy et al., 2015; Mandal et al., 2019; Lin and Lane, 2021).
The issue of population aging is something that not only developed countries need to face, but also developing countries should consider. The changes in social life have amplified the harm of AD for individual members of society and the whole social aspect. The exact cause of AD is still unclear, and the treatment is relatively single, so how to prevent and treat it is a problem that every concerned person should consider. As an inexpensive and widely beneficial tool, the benefits of physical activity are cross-disease and cross-age, and increasing regular physical activity seems to be more effective for those with both AD and other diseases than the specificity of the effects of medications used to treat AD, which is true, as AD patients are often found to have other chronic diseases, and the greater benefit of physical activity is not curative but preventive (Suzuki, 2019).
This review takes three classical AD-related signaling pathways as the starting point and adds the perspective of “Exerkines” to summarize and discuss the current representative studies to reveal the molecular mechanisms of exercise to alleviate AD-related disorders. In short, exercise itself “Exerkines” released by motor stimulation act on three classical signaling pathways, AMPK, Wnt, and PI3K/AKT, or directly at the site of the disease to alleviate neuroinflammation, improve synaptic plasticity, promote neurogenesis, reduce Aβ deposition, decrease abnormal tau protein phosphorylation, maintain BBB stability, and improve autophagy, thereby reversing the effects of neuroinflammation, reduced synaptic plasticity, neuronal apoptosis, decreased memory capacity, and degeneration of higher cognitive abilities caused by AD.
Although exercise has been observed to be effective in the prevention and relief of AD in some human studies, and the same findings were reported in a summary meta-analysis (Farina et al., 2014; Hernández et al., 2015; Kivipelto et al., 2018), we believe that these studies are more focused on the discovery of “macroscopic phenomena”, although the rehabilitation of AD patients through exercise or the reduction of the risk of AD in healthy people through exercise is our ultimate goal. Although rehabilitating AD patients with exercise or reducing the risk of AD in healthy people with exercise is the ultimate goal, it seems that the complex molecular mechanisms of the interaction between AD and exercise need to be more finely “micro-explained” so that readers or researchers can consider the effect of exercise on AD prevention and treatment through both micro- and macro-validation when viewing the relevant articles. This is necessary for a scientific and critical study of the field of “exercise intervention in AD”. Therefore, although evidence from human studies is collected, the focus of this paper is more on the complex interactions between AD and exercise in vivo through molecular signaling pathways.
While it is true that some high quality reviews have focused on these issues, our study differs from previous studies in that it breaks down several topical conditions related to AD that currently exist, and under each topical condition, describes the signaling pathways through which exercise may play a beneficial role in AD, which is more clearly organized and may be easier for the reader to read.
In addition, we have systematically described “locomotor factors”, substances regulated by locomotion, which are secreted under the conditions of locomotion, and which have rarely been covered and discussed in previous reviews.
In some studies it is considered that AD is one of the concomitant causes of increased sarcopenia and therefore muscle mass in AD patients is one of the concerns (Yun et al., 2021), and it is precisely in this regard that the benefits of regular exercise are also highlighted, and after training, whether aerobic, resistance or mixed, AD patients get beneficial results both in terms of muscle genesis and muscle mass, improving their daily life capacity. In addition to this, the researchers suggest that there may be a link between the decrease in muscle mass and hippocampal genesis, and that there is a positive relationship between muscle mass and cognitive performance (Lee K. et al., 2019; Moon et al., 2019).
Interestingly, in summarizing the above literature, we found that the effect of exercise on stimulating signaling pathways to inhibit AD may also involve a cascade of responses between different signaling pathways, rather than a simple “one-way” unilinear development. In addition, many natural extracts of plants belonging to polyphenols may also have a mitigating effect on AD, such as epigallocatechin gallate, curcumin, resveratrol, etc. Moreover, the ameliorating effect of these substances on AD may also be produced through the stimulation of some of the above-mentioned signaling pathways, which is similar to the mechanism of the ameliorating effect of exercise on AD. Therefore, we hypothesize that the combination of polyphenol supplementation with increased physical activity may be a good approach, but unfortunately, there are few studies exploring the combined effect of exercise or polyphenols on AD compared to the effect of exercise or polyphenols on AD alone.
Although some studies have examined the effect of exercise on AD through the Wnt signaling pathway, there is a paucity of studies compared to “Wnt and AD” or “exercise stimulation of Wnt for other diseases”, and although this review also summarizes the current evidence based on exercise stimulation of Wnt to improve AD, there are still some elements that deserve further investigation by researchers to fill the gap.
Finally, recent research suggests that there may be an interaction between gut flora and the brain via the “brain-gut axis”, which appears to point to a new aspect of AD research, with the possibility that gut flora may be a new target for the treatment of AD. In summary, potential directions that still need to be developed in the field of exercise to improve AD include: A. Polyphenols combined with exercise for AD. B. Exercise through the brain-gut axis for AD. C. Exercise through the Wnt signaling pathway for AD.
ZH, KL, and PC contributed to conception and design of the study. ZH and LZ organized the database. ZH and KL wrote the first draft of the manuscript. ZH, KL, and LZ wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This research was funded by Shandong University Student Innovation and Entrepreneurship Training Program, grant number S202110439103.
We are very grateful to the BioRender website for providing a rich and powerful image library and to each author for adding to it.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1193031/full#supplementary-material
Al Mamun, A., Rahman, M. M., Zaman, S., Munira, S., Uddin, M. S., Rauf, A., et al. (2020a). Molecular insight into the crosstalk of UPS components and alzheimer’s disease. Curr. Protein Pept. Sci. 21, 1193–1201. doi:10.2174/1389203721666200923153406
Al Mamun, A., Uddin, M. S., Mathew, B., and Ashraf, G. M. (2020b). Toxic tau: Structural origins of tau aggregation in alzheimer’s disease. Neural Regen. Res. 15, 1417–1420. doi:10.4103/1673-5374.274329
Allen, S. J., Watson, J. J., Shoemark, D. K., Barua, N. U., and Patel, N. K. (2013). GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 138, 155–175. doi:10.1016/j.pharmthera.2013.01.004
Aloe, L., Rocco, M. L., Bianchi, P., and Manni, L. (2012). Nerve growth factor: From the early discoveries to the potential clinical use. J. Transl. Med. 10, 239. doi:10.1186/1479-5876-10-239
Andrews, S. J., Fulton-Howard, B., and Goate, A. (2020). Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 19, 326–335. doi:10.1016/S1474-4422(19)30435-1
Azimi, M., Gharakhanlou, R., Naghdi, N., Khodadadi, D., and Heysieattalab, S. (2018). Moderate treadmill exercise ameliorates amyloid-β-induced learning and memory impairment, possibly via increasing AMPK activity and up-regulation of the PGC-1α/FNDC5/BDNF pathway. Peptides 102, 78–88. doi:10.1016/j.peptides.2017.12.027
Bai, H., Yang, B., Yu, W., Xiao, Y., Yu, D., and Zhang, Q. (2018). Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 362, 180–187. doi:10.1016/j.yexcr.2017.11.015
Barnes, J. N. (2015). Exercise, cognitive function, and aging. Adv. Physiol. Educ. 39, 55–62. doi:10.1152/advan.00101.2014
Bedada, F. B., Ntekim, O. E., Nwulia, E. O., Fungwe, T. V., Nadarajah, S. R., and Obisesan, T. O. (2021). Exercise training-increased FBXO32 and FOXO1 in a gender-dependent manner in mild cognitively impaired african Americans: GEMS-1 study. Front. Aging Neurosci. 13, 641758. doi:10.3389/fnagi.2021.641758
Bertram, S., Brixius, K., and Brinkmann, C. (2016). Exercise for the diabetic brain: How physical training may help prevent dementia and alzheimer’s disease in T2DM patients. Endocrine 53, 350–363. doi:10.1007/s12020-016-0976-8
Blondell, S. J., Hammersley-Mather, R., and Veerman, J. L. (2014). Does physical activity prevent cognitive decline and dementia?: A systematic review and meta-analysis of longitudinal studies. BMC Public Health 14, 510. doi:10.1186/1471-2458-14-510
Bou Zerdan, M., Hebbo, E., Hijazi, A., El Gemayel, M., Nasr, J., Nasr, D., et al. (2022). The gut microbiome and alzheimer’s disease: A growing relationship. Curr. Alzheimer Res. doi:10.2174/1567205020666221227090125
Braidy, N., Zarka, M., Welch, J., and Bridge, W. (2015). Therapeutic approaches to modulating glutathione levels as a pharmacological strategy in alzheimer’s disease. Curr. Alzheimer Res. 12, 298–313. doi:10.2174/1567205012666150302160308
Brown, C., Pemberton, S., Babin, A., Abdulhameed, N., Noonan, C., Brown, M. B., et al. (2022). Insulin blood-brain barrier transport and interactions are greater following exercise in mice. J. Appl. Physiol. 132, 824–834. doi:10.1152/japplphysiol.00866.2021
Cai, Z., Zhang, X., Wang, G., Wang, H., Liu, Z., Guo, X., et al. (2014). BDNF attenuates IL-1β-induced F-actin remodeling by inhibiting NF-κB signaling in hippocampal neurons. Neuroendocrinol. Lett. 35, 13–19.
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers. Dement. 12, 719–732. doi:10.1016/j.jalz.2016.02.010
Chen, K., Zhang, X., and Du, L. (2022). Exercise enhances SIRT1 expression and improves alzheimer’s disease. Chin. J. Biochem. Mol. 38, 563–569. doi:10.13865/j.cnki.cjbmb.2021.07.1252
Chen, Z., and Zhong, C. (2014). Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 30, 271–281. doi:10.1007/s12264-013-1423-y
Chow, L. S., Gerszten, R. E., Taylor, J. M., Pedersen, B. K., van Praag, H., Trappe, S., et al. (2022). Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 18, 273–289. doi:10.1038/s41574-022-00641-2
Cioffi, F., Adam, R. H. I., and Broersen, K. (2019). Molecular mechanisms and genetics of oxidative stress in alzheimer’s disease. J. Alzheimers Dis. 72, 981–1017. doi:10.3233/JAD-190863
Costes, S., and Butler, P. C. (2014). Insulin-degrading enzyme inhibition, a novel therapy for type 2 diabetes? Cell Metab. 20, 201–203. doi:10.1016/j.cmet.2014.07.016
Cunningham, C., O’ Sullivan, R., Caserotti, P., and Tully, M. A. (2020). Consequences of physical inactivity in older adults: A systematic review of reviews and meta-analyses. Scand. J. Med. Sci. Sports 30, 816–827. doi:10.1111/sms.13616
De Ferrari, G. V., and Moon, R. T. (2006). The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553. doi:10.1038/sj.onc.1210064
Dubois, B., Villain, N., Frisoni, G. B., Rabinovici, G. D., Sabbagh, M., Cappa, S., et al. (2021). Clinical diagnosis of alzheimer’s disease: Recommendations of the international working group. Lancet Neurol. 20, 484–496. doi:10.1016/S1474-4422(21)00066-1
Eggert, S., Kins, S., Endres, K., and Brigadski, T. (2022). Brothers in arms: proBDNF/BDNF and sAPPα/Aβ-signaling and their common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in the progression of Alzheimer's disease. Biol. Chem. 403, 43–71. doi:10.1515/hsz-2021-0330
Engmann, O., and Giese, K. P. (2009). Crosstalk between Cdk5 and GSK3 beta: Implications for alzheimer’s disease. Front. Molec. Neurosci. 2, 2. doi:10.3389/neuro.02.002.2009
Fang, G., Zhang, D., Zhang, L., Zhao, J., Li, L., and Li, P. (2018a). Long-term aerobic exercise reduces tau phosphorylation by activating the pi3k/akt pathway in the Hippocampus of app/ps1 mice. J. Mech. Med. Biol. 18, 1840026. doi:10.1142/S0219519418400262
Fang, G., Zhao, J., Zhang, L., and Li, P. (2018b). The effect of aerobic exercise on keap1/nrf2 pathway in the cerebral cortex and hippocamus of APP/PS1 mice. Chin. J. Sports Med. 37, 839–846. doi:10.16038/j.1000-6710.2018.10.008
Farina, N., Rusted, J., and Tabet, N. (2014). The effect of exercise interventions on cognitive outcome in alzheimer’s disease: A systematic review. Int. Psychogeriatr. 26, 9–18. doi:10.1017/S1041610213001385
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi:10.1152/physrev.00011.2017
Gaitan, J. M., Moon, H. Y., Stremlau, M., Dubal, D. B., Cook, D. B., Okonkwo, O. C., et al. (2021). Effects of aerobic exercise training on systemic biomarkers and cognition in late middle-aged adults at risk for alzheimer’s disease. Front. Endocrinol. 12, 660181. doi:10.3389/fendo.2021.660181
Gao, L., Zhang, Y., Sterling, K., and Song, W. (2022). Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 11, 4. doi:10.1186/s40035-022-00279-0
Gaugler, J., James, B., Johnson, T., Reimer, J., Solis, M., Weuve, J., et al. (2022). 2022 Alzheimer’s disease facts and figures. Alzheimers. Dement. 18, 700–789. doi:10.1002/alz.12638
Goyal, D., Ali, S. A., and Singh, R. K. (2021). Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuro-Psychopharmacology Biol. Psychiatry 106, 110112. doi:10.1016/j.pnpbp.2020.110112
Greenough, M. A., Camakaris, J., and Bush, A. I. (2013). Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 62, 540–555. doi:10.1016/j.neuint.2012.08.014
Guo, P., Liu, L., Yang, X., Li, M., Zhao, Q., and Wu, H. (2022a). Irisin improves BBB dysfunction in SAP rats by inhibiting MMP-9 via the ERK/NF-Kappa B signaling pathway. Cell. Signal. 93, 110300. doi:10.1016/j.cellsig.2022.110300
Guo, S., Wang, H., and Yin, Y. (2022b). Microglia polarization from M1 to M2 in neurodegenerative diseases. Front. Aging Neurosci. 14, 815347. doi:10.3389/fnagi.2022.815347
Guure, C. B., Ibrahim, N. A., Adam, M. B., and Said, S. M. (2017). Impact of physical activity on cognitive decline, dementia, and its subtypes: Meta-analysis of prospective studies. Biomed. Res. Int. 2017, 9016924. doi:10.1155/2017/9016924
Han, J., Baker, K., Fournier, N., Calvo, C.-F., Duman, R., Eichmann, A., et al. (2014). VEGF-C/VEGFR3 signaling is required for maintenance and physical exercise-induced activity of adult hippocampal neural stem/progenitor cells. Angiogenesis 17, 312.
Heras-Sandoval, D., Perez-Rojas, J. M., Hernandez-Damian, J., and Pedraza-Chaverri, J. (2014). The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 26, 2694–2701. doi:10.1016/j.cellsig.2014.08.019
Hernández, S. S. S., Sandreschi, P. F., da Silva, F. C., Arancibia, B. A. V., da Silva, R., Gutierres, P. J. B., et al. (2015). What are the benefits of exercise for alzheimer’s disease? A systematic review of the past 10 years. J. Aging Phys. Act. 23, 659–668. doi:10.1123/japa.2014-0180
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi:10.1038/s41582-019-0244-7
Hui, K.-S. (2007). Brain-specific aminopeptidase: From enkephalinase to protector against neurodegeneration. Neurochem. Res. 32, 2062–2071. doi:10.1007/s11064-007-9356-3
Iso-Markku, P., Kujala, U. M., Knittle, K., Polet, J., Vuoksimaa, E., and Waller, K. (2022). Physical activity as a protective factor for dementia and alzheimer’s disease: Systematic review, meta-analysis and quality assessment of cohort and case-control studies. Br. J. Sports Med. 56, 701–709. doi:10.1136/bjsports-2021-104981
Jeon, S.-M. (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48, e245. doi:10.1038/emm.2016.81
Jeong, H., Shin, H., Hong, S., and Kim, Y. (2022). Physiological roles of monomeric amyloid-beta and implications for alzheimer’s disease therapeutics. Exp. Neurobiol. 31, 65–88. doi:10.5607/en22004
Jia, L., Pina-Crespo, J., and Li, Y. (2019). Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer's disease. Mol. Brain 12, 104. doi:10.1186/s13041-019-0525-5
Jian, Y., Yuan, S., Yang, J., Lei, Y., Li, X., and Liu, W. (2022). Aerobic exercise alleviates abnormal autophagy in brain cells of APP/PS1 mice by upregulating AdipoR1 levels. Int. J. Mol. Sci. 23, 9921. doi:10.3390/ijms23179921
Jiang, G., Wang, C., Zhang, J., and Liu, H. (2017). Mediation of insulin growth factor-1 in Alzheimer’s disease and the mechanism of PRNP genetic expression and the PI3K/Akt signaling pathway. Exp. Ther. Med. 13, 2763–2766. doi:10.3892/etm.2017.4320
Jin, C-H., Il-Young, P., Kwak, Y.-S., Jee, Y.-S., and Kim, J-Y. (2015). Exhaustive submaximal endurance and resistance exercises induce temporary immunosuppression via physical and oxidative stress. J. Exerc. Rehabilitation 11, 198–203. doi:10.12965/jer.150221
Joa, K.-L., Moon, S., Kim, J.-H., Shin, D. W., Lee, K.-H., Jung, S., et al. (2017). Effects of task-specific rehabilitation training on tau modification in rat with photothrombotic cortical ischemic damage. Neurochem. Int. 108, 309–317. doi:10.1016/j.neuint.2017.05.004
Ju, Y., and Tam, K. Y. (2022). Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 17, 543. doi:10.4103/1673-5374.320970
Kang, E.-B., and Cho, J. Y. (2015). Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. Phys. Activity Nutr. 19, 199–209. doi:10.5717/jenb.2015.15090806
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi:10.5607/en.2015.24.4.325
Kivipelto, M., Mangialasche, F., and Ngandu, T. (2018). Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 14, 653–666. doi:10.1038/s41582-018-0070-3
Kjobsted, R., Hingst, J. R., Fentz, J., Foretz, M., Sanz, M.-N., Pehmoller, C., et al. (2018). AMPK in skeletal muscle function and metabolism. Faseb J. 32, 1741–1777. doi:10.1096/fj.201700442R
ki, K., Jang, Y., Kyung, K. T., and Koo, J.-H. (2022). The effect of treadmill exercise and Sildenafil administration on tauopathy and blood-brain barrier function in Alzheimer’s disease mice model. 스포츠사이언스 40, 279–286.
Koo, J.-H., Kang, E.-B., Oh, Y.-S., Yang, D.-S., and Cho, J.-Y. (2017). Treadmill exercise decreases amyloid-beta burden possibly via activation of SIRT-1 signaling in a mouse model of Alzheimer’s disease. Exp. Neurol. 288, 142–152. doi:10.1016/j.expneurol.2016.11.014
Kwon, H. S., and Koh, S.-H. (2020). Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 9, 42. doi:10.1186/s40035-020-00221-2
Lee, K., Lee, Y. M., Park, J.-M., Lee, B.-D., Moon, E., Jeong, H.-J., et al. (2019a). Right hippocampus atrophy is independently associated with Alzheimer’s disease with psychosis. Psychogeriatrics 19, 105–110. doi:10.1111/psyg.12369
Lee, T. H.-Y., Formolo, D. A., Kong, T., Lau, S. W.-Y., Ho, C. S.-L., Leung, R. Y. H., et al. (2019b). “Potential exerkines for physical exercise-elicited pro-cognitive effects: Insight from clinical and animal research,” in Exercise on brain health. Editors S. Y. Yau, and K. F. So (San Diego: Elsevier Academic Press Inc), 361–395. doi:10.1016/bs.irn.2019.06.002
Li, J., Yan, Y., and Cai, Z. (2009). Research progress of insulin-degrading enzymes in the pathogenesis of Alzheimer’s disease. Chin. Bull. Life Sci. 21, 126–130.
Lin, C.-H., and Lane, H.-Y. (2021). Plasma glutathione levels decreased with cognitive decline among people with mild cognitive impairment (MCI): A two-year prospective study. Antioxidants 10, 1839. doi:10.3390/antiox10111839
Liu, B., Liu, J., Wang, J.-G., Liu, C.-L., and Yan, H.-J. (2020). AdipoRon improves cognitive dysfunction of Alzheimer's disease and rescues impaired neural stem cell proliferation through AdipoR1/AMPK pathway. Exp. Neurol. 327, 113249. doi:10.1016/j.expneurol.2020.113249
Liu, D.-X., Zhang, D., Hu, W.-M., Chang, Y.-F., Wang, X.-H., and Li, L. (2021). Geniposide protection against Aβ1-42 toxicity correlates with mTOR inhibition and enhancement of autophagy. J. Integr. Neurosci. 20, 67–75. doi:10.31083/j.jin.2021.01.242
Liu, W., Xia, Y., Kuang, H., Wang, Z., Liu, S., Tang, C., et al. (2019a). Proteomic profile of carbonylated proteins screen the regulation of calmodulin-dependent protein kinases-AMPK-beclin1 in aerobic exercise-induced autophagy in middle-aged rat Hippocampus. Gerontology 65, 620–633. doi:10.1159/000500742
Liu, X., Hou, D., Lin, A., Luo, J., Xie, J., Wang, Y., et al. (2019b). The role of neurovascular unit damage in the occurrence and development of Alzheimer’s disease. Rev. Neurosci. 30, 477–484. doi:10.1515/revneuro-2018-0056
Llorens-Martin, M., Jurado, J., Hernandez, F., and Avila, J. (2014). GSK-3 beta, a pivotal kinase in Alzheimer disease. Front. Molec. Neurosci. 7, 46. doi:10.3389/fnmol.2014.00046
Lourenco, M. V., Frozza, R. L., de Freitas, G. B., Zhang, H., Kincheski, G. C., Ribeiro, F. C., et al. (2019). Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 25, 165. doi:10.1038/s41591-018-0275-4
Lu, B., Nagappan, G., Guan, X., Nathan, P. J., and Wren, P. (2013). BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 14, 401–416. doi:10.1038/nrn3505
Luan, X., Tian, X., Zhang, H., Huang, R., Li, N., Chen, P., et al. (2019). Exercise as a prescription for patients with various diseases. J. Sport Health Sci. 8, 422–441. doi:10.1016/j.jshs.2019.04.002
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial Beclin 1 regulates retromer trafficking and phagocytosis and is impaired in alzheimer’s disease. Neuron 79, 873–886. doi:10.1016/j.neuron.2013.06.046
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: The role and consequences. Neurosci. Res. 79, 1–12. doi:10.1016/j.neures.2013.10.004
Ma, T., Hoeffer, C. A., Wong, H., Massaad, C. A., Zhou, P., Iadecola, C., et al. (2011). Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. 31, 5589–5595. doi:10.1523/JNEUROSCI.6566-10.2011
MacPherson, R. E. K., Baumeister, P., Peppler, W. T., Wright, D. C., and Little, J. P. (2015). Reduced cortical BACE1 content with one bout of exercise is accompanied by declines in AMPK, Akt, and MAPK signaling in obese, glucose-intolerant mice. J. Appl. Physiol. 119, 1097–1104. doi:10.1152/japplphysiol.00299.2015
Mahoney, E. R., Dumitrescu, L., Moore, A. M., Cambronero, F. E., De Jager, P. L., Koran, M. E., et al. (2021). Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatr. 26, 888–896. doi:10.1038/s41380-019-0458-5
Malkiewicz, M. A., Szarmach, A., Sabisz, A., Cubala, W. J., Szurowska, E., and Winklewski, P. J. (2019). Blood-brain barrier permeability and physical exercise. J. Neuroinflamm. 16, 15. doi:10.1186/s12974-019-1403-x
Mandal, P. K., Shukla, D., Tripathi, M., and Ersland, L. (2019). Cognitive improvement with glutathione supplement in alzheimer’s disease: A way forward. J. Alzheimers Dis. 68, 531–535. doi:10.3233/JAD-181054
Mankhong, S., Kim, S., Moon, S., Lee, K.-H., Jeon, H.-E., Hwang, B.-H., et al. (2020). Effects of aerobic exercise on tau and related proteins in rats with the middle cerebral artery Occlusion. Int. J. Mol. Sci. 21, 5842. doi:10.3390/ijms21165842
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M.-L., Yardin, C., et al. (2013). Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 12, 289–309. doi:10.1016/j.arr.2012.06.003
McAllister, C., Long, J., Bowers, A., Walker, A., Cao, P., Honda, S.-I., et al. (2010). Genetic targeting aromatase in male amyloid precursor protein transgenic mice down-regulates beta-secretase (BACE1) and prevents alzheimer-like pathology and cognitive impairment. J. Neurosci. 30, 7326–7334. doi:10.1523/JNEUROSCI.1180-10.2010
Melincovici, C. S., Bosca, A. B., Susman, S., Marginean, M., Mihu, C., Istrate, M., et al. (2018). Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 59, 455–467.
Montagne, A., Zhao, Z., and Zlokovic, B. V. (2017). Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 214, 3151–3169. doi:10.1084/jem.20171406
Moon, Y., Moon, W.-J., Kim, J. O., Kwon, K. J., and Han, S.-H. (2019). Muscle strength is independently related to brain atrophy in patients with alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 47, 306–314. doi:10.1159/000500718
Murakami, K., Murata, N., Noda, Y., Irie, K., Shirasawa, T., and Shimizu, T. (2012). Stimulation of the amyloidogenic pathway by cytoplasmic superoxide radicals in an alzheimer’s disease mouse model. Biosci. Biotechnol. Biochem. 76, 1098–1103. doi:10.1271/bbb.110934
Nelson, A. R., Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 1862, 887–900. doi:10.1016/j.bbadis.2015.12.016
Ng, A., Tam, W. W., Zhang, M. W., Ho, C. S., Husain, S. F., McIntyre, R. S., et al. (2018). IL-1 beta, IL-6, TNF-alpha and CRP in elderly patients with depression or alzheimer’s disease: Systematic review and meta-analysis. Sci. Rep. 8, 12050. doi:10.1038/s41598-018-30487-6
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi:10.1038/nm.3232
Oliva, C. A., Vargas, J. Y., and Inestrosa, N. C. (2013). Wnt signaling: Role in LTP, neural networks and memory. Ageing Res. Rev. 12, 786–800. doi:10.1016/j.arr.2013.03.006
Ostrowski, P. P., Barszczyk, A., Forstenpointner, J., Zheng, W., and Feng, Z.-P. (2016). Meta-analysis of serum insulin-like growth factor 1 in alzheimer’s disease. PLoS One 11, e0155733. doi:10.1371/journal.pone.0155733
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). Updating guidance for reporting systematic reviews: Development of the PRISMA 2020 statement. J. Clin. Epidemiol. 134, 103–112. doi:10.1016/j.jclinepi.2021.02.003
Parr, C., Mirzaei, N., Christian, M., and Sastre, M. (2015). Activation of the Wnt/β-catenin pathway represses the transcription of the β-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. Faseb J. 29, 623–635. doi:10.1096/fj.14-253211
Pascual-Vargas, P., and Salinas, P. C. (2021). A role for frizzled and their post-translational modifications in the mammalian central nervous system. Front. Cell. Dev. Biol. 9, 692888. doi:10.3389/fcell.2021.692888
Peng, Y., Chi, R., Liu, G., Tian, W., Zhang, J., and Zhang, R. (2022). Aerobic exercise regulates apoptosis through the PI3K/Akt/GSK-3 beta signaling pathway to improve cognitive impairment in alzheimer’s disease mice. Neural. Plast. 2022, 1500710. doi:10.1155/2022/1500710
Ploia, C., Antoniou, X., Sclipa, A., Grande, V., Cardinetti, D., Colombo, A., et al. (2011). JNK plays a key role in tau hyperphosphorylation in alzheimer’s disease models. J. Alzheimers Dis. 26, 315–329. doi:10.3233/JAD-2011-110320
Profaci, C. P., Munji, R. N., Pulido, R. S., and Daneman, R. (2020). The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 217, e20190062. doi:10.1084/jem.20190062
Rabiee, F., Lachinani, L., Ghaedi, S., Nasr-Esfahani, M. H., Megraw, T. L., and Ghaedi, K. (2020). New insights into the cellular activities of Fndc5/Irisin and its signaling pathways. Cell Biosci. 10, 51. doi:10.1186/s13578-020-00413-3
Ren, C., Gu, X., Li, H., Lei, S., Wang, Z., Wang, J., et al. (2019). The role of DKK1 in alzheimer’s disease: A potential intervention point of brain damage prevention? Pharmacol. Res. 144, 331–335. doi:10.1016/j.phrs.2019.04.033
Ren, H., Yang, L., Li, W., Yang, L., Che, Y., and Sun, J. (2017). Effects of exercise on body composition and serum Dickkpof1 levels in breast cancer patients. Chin. J. Gerontology 37, 5123–5124.
Riedel, B. C., Thompson, P. M., and Brinton, R. D. (2016). Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 160, 134–147. doi:10.1016/j.jsbmb.2016.03.012
Rosso, S. B., and Inestrosa, N. C. (2013). WNT signaling in neuronal maturation and synaptogenesis. Front. Cell. Neurosci. 7, 103. doi:10.3389/fncel.2013.00103
Rudnicka, E., Napierala, P., Podfigurna, A., Meczekalski, B., Smolarczyk, R., and Grymowicz, M. (2020). The World Health Organization (WHO) approach to healthy ageing. Maturitas 139, 6–11. doi:10.1016/j.maturitas.2020.05.018
Saharan, S., and Mandal, P. K. (2014). The emerging role of glutathione in alzheimer’s disease. J. Alzheimers Dis. 40, 519–529. doi:10.3233/JAD-132483
Salim, S. (2017). Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 360, 201–205. doi:10.1124/jpet.116.237503
Shao, C. Y., Mirra, S. S., Sait, H. B. R., Sacktor, T. C., and Sigurdsson, E. M. (2011). Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced A beta and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 122, 285–292. doi:10.1007/s00401-011-0843-x
Silva, N. M. L. e., Goncalves, R. A., Pascoal, T. A., Lima-Filho, R. A. S., Resende, E. de P. F., Vieira, E. L. M., et al. (2021). Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatr. 11, 251. doi:10.1038/s41398-021-01349-z
Sivandzade, F., Prasad, S., Bhalerao, A., and Cucullo, L. (2019). NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 21, 101059. doi:10.1016/j.redox.2018.11.017
Song, J., and Lee, J. E. (2013). Adiponectin as a new paradigm for approaching Alzheimer’s disease. Anat. Cell Biol. 46, 229–234. doi:10.5115/acb.2013.46.4.229
Sujkowski, A., Hong, L., Wessells, R. J., and Todi, S. (2022). The protective role of exercise against age-related neurodegeneration. Ageing Res. Rev. 74, 101543. doi:10.1016/j.arr.2021.101543
Suzuki, K. (2019). Chronic inflammation as an immunological abnormality and effectiveness of exercise. Biomolecules 9, 223. doi:10.3390/biom9060223
Suzuki, W. A. (2016). How body affects brain. Cell Metab. 24, 192–193. doi:10.1016/j.cmet.2016.07.022
Sweeney, M. D., Montagne, A., Sagare, A. P., Nation, D. A., Schneider, L. S., Chui, H. C., et al. (2019). Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers. Dement. 15, 158–167. doi:10.1016/j.jalz.2018.07.222
Thawkar, B. S., and Kaur, G. (2019). Inhibitors of NF-kappa B and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 326, 62–74. doi:10.1016/j.jneuroim.2018.11.010
Trueba-Saiz, A., Fernandez, A. M., Nishijima, T., Mecha, M., Santi, A., Munive, V., et al. (2017). Circulating insulin-like growth factor I regulates its receptor in the brain of male mice. Endocrinology 158, 349–355. doi:10.1210/en.2016-1468
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric A beta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9, 48. doi:10.1186/1750-1326-9-48
Um, H.-S., Kang, E.-B., Koo, J.-H., Kim, H.-T., Jin, L.-K., Kim, E.-J., et al. (2011). Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci. Res. 69, 161–173. doi:10.1016/j.neures.2010.10.004
Vargas-Ortiz, K., Perez-Vazquez, V., and Macias-Cervantes, M. H. (2019). Exercise and sirtuins: A way to mitochondrial health in skeletal muscle. Int. J. Mol. Sci. 20, 2717. doi:10.3390/ijms20112717
Vest, R. T., and Wyss-Coray, T. (2022). Molecular map of the human blood-brain barrier reveals links to Alzheimer’s disease. Nature. doi:10.1038/d41586-022-00350-w
Wang, D., Chen, F., Han, Z., Yin, Z., Ge, X., Lei, P., et al. (2021a). Neutralization of Hv1/HVCN1 with antibody enhances microglia/macrophages myelin clearance by promoting their migration in the brain. Front. Cell. Neurosci. 15, 768059. doi:10.3389/fncel.2021.768059
Wang, Q., Hu, J., Liu, Y., Li, J., Liu, B., Li, M., et al. (2019). Aerobic exercise improves synaptic-related proteins of diabetic rats by inhibiting FOXO1/NF-kappa B/NLRP3 inflammatory signaling pathway and ameliorating PI3K/Akt insulin signaling pathway. J. Mol. Neurosci. 69, 28–38. doi:10.1007/s12031-019-01302-2
Wang, S., Liu, H.-Y., Cheng, Y.-C., and Su, C.-H. (2021b). Exercise dosage in reducing the risk of dementia development: Mode, duration, and intensity-A narrative review. Int. J. Environ. Res. Public Health 18, 13331. doi:10.3390/ijerph182413331
Wang, Y. (2023). The role of the gut microbiota and microbial metabolites in the pathogenesis of alzheimer’s disease. CNS Neurol. Disord.-Drug Targets 22, 577–598. doi:10.2174/1871527321666220417005115
Wrann, C. D., White, J. P., Salogiannnis, J., Laznik-Bogoslavski, D., Wu, J., Ma, D., et al. (2013). Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 18, 649–659. doi:10.1016/j.cmet.2013.09.008
Wu, C., Yang, L., Tucker, D., Dong, Y., Zhu, L., Duan, R., et al. (2018). Beneficial effects of exercise pretreatment in a sporadic alzheimer’s rat model. Med. Sci. Sports Exerc. 50, 945–956. doi:10.1249/MSS.0000000000001519
Xu, L., Li, M., Wei, A., Yang, M., Li, C., Liu, R., et al. (2022). Treadmill exercise promotes E3 ubiquitin ligase to remove amyloid beta and P-tau and improve cognitive ability in APP/PS1 transgenic mice. J. Neuroinflamm. 19, 243. doi:10.1186/s12974-022-02607-7
Yang, J.-L., Lin, Y.-T., Chuang, P.-C., Bohr, V. A., and Mattson, M. P. (2014). BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromol. Med. 16, 161–174. doi:10.1007/s12017-013-8270-x
Ye, H., Han, Y., Li, P., Su, Z., and Huang, Y. (2022). The role of post-translational modifications on the structure and function of tau protein. J. Mol. Neurosci. 72, 1557–1571. doi:10.1007/s12031-022-02002-0
Yoon, S.-S., and Jo, S. A. (2012). Mechanisms of amyloid-beta peptide clearance: Potential therapeutic targets for alzheimer’s disease. Biomol. Ther. 20, 245–255. doi:10.4062/biomolther.2012.20.3.245
Yu, J.-T., Xu, W., Tan, C.-C., Andrieu, S., Suckling, J., Evangelou, E., et al. (2020). Evidence-based prevention of alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 91, 1201–1209. doi:10.1136/jnnp-2019-321913
Yun, J. H., Kim, D. H., and Chang, M. C. (2021). A simple bedside exercise method to enhance lower limb muscle strength in moderate alzheimer’s disease patients with sarcopenia. Healthcare 9, 680. doi:10.3390/healthcare9060680
Zhang, W., Xu, C., Sun, J., Shen, H.-M., Wang, J., and Yang, C. (2022). Impairment of the autophagy-lysosomal pathway in Alzheimer’s diseases: Pathogenic mechanisms and therapeutic potential. Acta Pharm. Sin. B 12, 1019–1040. doi:10.1016/j.apsb.2022.01.008
Zhao, J., Bi, W., Xiao, S., Lan, X., Cheng, X., Zhang, J., et al. (2019). Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 9, 5790. doi:10.1038/s41598-019-42286-8
Keywords: aging, Alzheimer’s disease, neurodegenerative diseases, exercise, nonpharmacological interventions
Citation: Hao Z, Liu K, Zhou L and Chen P (2023) Precious but convenient means of prevention and treatment: physiological molecular mechanisms of interaction between exercise and motor factors and Alzheimer’s disease. Front. Physiol. 14:1193031. doi: 10.3389/fphys.2023.1193031
Received: 24 March 2023; Accepted: 31 May 2023;
Published: 08 June 2023.
Edited by:
Gonul Babayigit Irez, Mugla Sitki Kocman University, TürkiyeReviewed by:
Lidia Castillo-Mariqueo, Universidad Católica de Temuco, ChileCopyright © 2023 Hao, Liu, Zhou and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Chen, Y3BAb3VjLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.