 Patricia P. Centeno
Patricia P. Centeno Lenah S. Binmahfouz1,2
Lenah S. Binmahfouz1,2 Khaleda Alghamdi
Khaleda Alghamdi Donald T. Ward
Donald T. Ward
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 31 March 2023
Sec. Membrane Physiology and Membrane Biophysics
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1154374
This article is part of the Research TopicThe Calcium-Sensing Receptor: from Physiology to PharmacologyView all 7 articles
As both a sensor of extracellular calcium (Ca2+o) concentration and a key controller of Ca2+o homeostasis, one of the most interesting properties of the calcium-sensing receptor (CaR) is its sensitivity to, and modulation by, ions and small ligands other than Ca2+. There is emerging evidence that extracellular phosphate can act as a partial, non-competitive CaR antagonist to modulate parathyroid hormone (PTH) secretion, thus permitting the CaR to integrate mineral homeostasis more broadly. Interestingly, phosphorylation of certain intracellular CaR residues can also inhibit CaR responsiveness. Thus, negatively charged phosphate can decrease CaR activity both extracellularly (via association with arginine) and intracellularly (via covalent phosphorylation).
Ca2+ is the fifth most abundant element both in the human body and the Earth’s crust. Of the environmentally available ions, Ca2+ is the most reactive and thus has been selected as a signalling molecule that can control millisecond duration intracellular events, for example, heartbeat, memory and neuro- and hormone secretion and locomotion (Williams, 2006). Outside of the cell, Ca2+ serves more of a structural role, both in tight junctions but most notably as a core component of bone hydroxyapatite. As a result of these differential functions, the 104 M extracellular versus intracellular Ca2+ (Ca2+i) concentration gradient must be restored after each intracellular Ca2+ (Ca2+i) signal has had its effect. Otherwise, sustained rises in Ca2+i concentration will drive Ca2+ overload and cell death (Duchen, 2000).
The other major component of hydroxyapatite is inorganic phosphate (PO4). Phosphorous is the sixth most abundant element in the body, though is much rarer in the terrestrial environment than calcium. PO4 is the most abundant intracellular anion and exists in millimolar concentrations inside the cell both as a free ion (involved in pH buffering) and as a critical component of the energy-releasing molecules phosphocreatine and adenosine triphosphate (ATP) as well as the other nucleotides. The protein kinase-mediated addition of negatively-charged PO4 groups to the sidechains of certain tyrosine and serine/threonine residues can effect local conformational changes in protein tertiary structure that can produce profound functional changes in those proteins (Hunter, 2012). Indeed, protein phosphorylation and Ca2+i mobilisation together represent two of the most fundamental mechanisms of intracellular signalling in nature. Therefore, any protein that plays a key role in controlling the total availability of both Ca2+ and PO4 in the body is of critical importance for both extracellular structure and intracellular signalling.
The CaR is a homodimeric G protein-coupled receptor (GPCR; family C) expressed with greatest abundance in organs involved in mineral ion homeostasis, specifically the parathyroid gland, kidneys, C cells of the thyroid and bone (Leach et al., 2020). Though capable of coupling to multiple heterotrimeric G proteins, most in vitro studies of CaR activity, measure Gq/11-mediated changes in either inositol phosphate metabolism, Ca2+i mobilisation or extracellular signal-regulated (ERK) activation, which have helped define CaR pharmacology.
While the CaR has long been known to sense and respond to Ca2+o, there is now emerging evidence that CaR is sensitive to, and inhibited by, PO4 as well (Centeno et al., 2019). Therefore, it is possible that the homeostasis of these two ions may be specifically coordinated. It should be noted that the free, ionised Ca2+ concentration in plasma is ∼1.2 mM while the concentration of PO4 tends to be a little lower at ∼0.8 mM, though with a wider normal range. Interestingly, this is a similar ratio to that seen for Ca2+ and PO4 abundance in hydroxyapatite (Ca5(PO4)3OH) and therefore coordinated regulation of their relative plasma concentrations might conceivably be obligatory.
In addition to the proposed inhibitory effect of extracellular PO4 ions on CaR activation, in the cytosol, the terminal PO4 of ATP can also inhibit CaR signalling, via covalent attachment to Ser-875 and Thr-888 residues (i.e., phosphorylation) in the receptor’s intracellular domain (ICD) (McCormick et al., 2010; Binmahfouz et al., 2019). While the dual but distinct roles of extracellular (structural) and intracellular (signalling/metabolic) Ca2+ and PO4 may be purely an example of energetic efficiency (i.e., multi-tasking with the same, simple elementary chemicals), this nevertheless represents an interesting biological parallel. And more so that for CaR, Ca2+o is stimulatory while PO4 is inhibitory.
The CaR exhibits promiscuous pharmacology sensing a broad range of ligands in addition to Ca2+ (Leach et al., 2020). Some CaR ligands act as orthosteric agonists, e.g., Mg2+ and spermine. Other CaR ligands act as positive allosteric modulators (PAMs; e.g., L-amino acids and calcimimetics) or negative allosteric modulators (NAMs; e.g., calcilytics) (Leach et al., 2020). Structurally, PAMs act by stabilising the CaR’s active conformation, while NAMs stabilise the inactive conformation (Leach et al., 2015). Interestingly, H+ and Na+ ions, at least at high concentrations are, in effect, NAMs of the CaR. But we have recently shown that PO4 ions are also inhibitory for CaR, acting potentially as non-competitive partial CaR antagonists. Here we set out the structural evidence for this (Geng et al., 2016; Zhang et al., 2016) followed by the functional (Centeno et al., 2019; Goodman et al., 2022).
Two groups have now generated four crystal models of the CaR’s extracellular domain (ECD) (Geng et al., 2016; Zhang et al., 2016) and more recently these structures have been largely confirmed by Cryo-EM (Gao et al., 2021). Three of these crystal models were obtained in the active conformation, while the fourth was obtained in the CaR’s inactive conformation (Geng et al., 2016). Sequence alignment followed by structural superpositioning of the CaR ECD crystal structures (active conformation) confirm the similarity of the two groups’ models, despite differences in their crystallisation environments (Figure 1). That said, the two models still exhibit significant differences in their predicted ligand binding sites and occupancy.
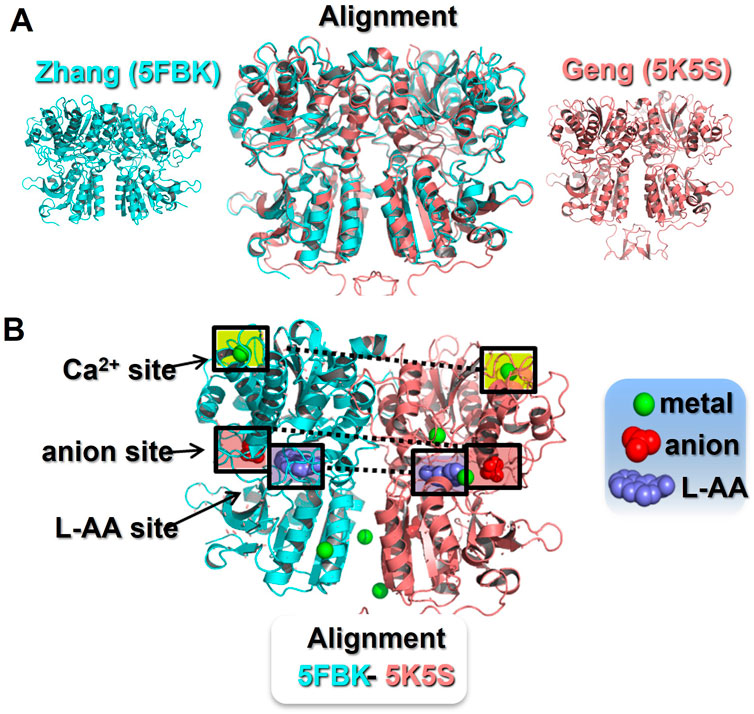
FIGURE 1. Identification of ionic binding sites in the CaR extracellular domain. Panel (A) Sequence alignment and structural superposition of the currently available CaR ECD models in the active conformation. The Zhang et al. (2016) model (5FBK) is shown in blue (left) and the Geng et al. (2016) model (5K5S) is shown in pink (right). In the middle, an alignment of both models reveals an almost identical CaR ECD structure with minor differences. Pymol root mean square deviation (RMSD) 0.5, after 5 cycles of iteration. Panel (B) Active conformation of the CaR ECD with reported ligand binding sites. The Zhang model (5FBH) is shown as the left monomer, with the Geng model (5K5S) on the right. The Zhang model describes three Ca2+-binding sites, one in the upper domain and two in the lower domain facing the interface between monomers. At the same locations, the Geng model describes two Ca2+-binding sites, but two additional Ca2+-binding sites in the cleft between the upper and lower domains. Both models identified a common L-amino acid binding site and a common anion binding site, both located at the cleft between the upper and lower domains. In addition, the Geng model includes an anion binding site located in the lower domain. The ligand binding sites highlighted in boxes are those found in both crystal models. L-AA, L-amino acid.
For example, Zhang et al. (2016) reported three metal binding sites (occupied by Mg2+ and Gd3+), one orthosteric L-aromatic amino acid binding site (occupied by L-1,2,3,4-tetrahydronorharman-3-carboxylic acid (TNCA), an L-Trp derivate), and one potential anion binding site (occupied by bicarbonate). In contrast, the CaR active conformation reported by Geng et al. (2016) suggested four metal binding sites (occupied by Ca2+), one orthosteric L-aromatic amino acid binding site (occupied by L-Trp itself), and two anion binding sites (occupied by PO4; see Figure 1). In addition, Geng’s inactive model revealed two additional anion binding sites (here occupied by SO4) (Geng et al., 2016). These discrepancies could be explained, in part, by the different crystallisation environments. That is, Zhang et al (2016) employed Mg2+ and Gd3+ in abundance, but with less Ca2+ and no L-aromatic amino acids, whereas Geng et al. (2016) included Ca2+ and L-Trp in abundance in their crystallisation buffer, and also PO4 and SO4. Given the greater potency and abundance of Ca2+ over Mg2+ in vivo, the metal binding sites appear more likely to be physiological Ca2+-binding sites. Also, based on previous literature, L-Trp and other aromatic amino acids are presumed to be the more likely ligands for the orthosteric L-aromatic amino acid-binding site, rather than TNCA (Mun et al., 2004; Conigrave and Hampson, 2006), though this remains to be confirmed.
The two crystal models identified four anion binding sites in the CaR’s ECD for the first time. However, it is the anion-binding sites present in the inactive conformation of the Geng et al (2016) model that we believe to be of particular interest. This is because the two, closely-associated anion binding sites appear to play an important role in the stabilisation of the inactive conformation. The first site mainly involves Arg-62 and Tyr-63, while the second involves Arg-66, Arg-69, Thr-412, and Arg-415. When unbound, Arg-62 and Arg-66 may mediate interactions that stabilise the closure of the Venus flytrap (VFT) domain. These interactions, a hydrogen bond (R66-S301) and a salt bridge (R62-E277), are directly breakable by anion binding to the two sites, which would then reduce the free energy needed for the VFT to open and to change to an inactive conformation (Geng et al., 2016).
Serum PO4 levels vary between 0.8 and 1.2 mM in healthy adults, which includes diurnal variation (Kemp et al., 1992). PO4 homeostasis is largely determined by the kidney because of PTH- and/or FGF23-mediated phosphaturia (Agoro and White, 2023), though PTH also increases 1α-hydroxylation of 25(OH)-vitamin D thus affecting intestinal PO4 absorption (Bergwitz and Juppner, 2010; Komaba and Fukagawa, 2016; Ide et al., 2018). Together, PTH/PTH1R and FGF23/Klotho pathways coordinate to maintain PO4 homeostasis (Shimada et al., 2004; Quinn et al., 2013; Kawakami et al., 2017; Fan et al., 2018; Ide et al., 2018). In chronic kidney disease (CKD) however, decreased phosphaturia commonly results in hyperphosphataemia as well as accumulation of calciprotein particles (CPPs), which are associated with soft-tissue calcification, especially vascular calcification and increased risk of death (Block et al., 2004; Tentori et al., 2008; Streja et al., 2014). Crucially, how mammals sense changes in their serum PO4 concentration remains unclear (Komaba and Fukagawa, 2016) though the presence of one or more PO4 sensors in bone and parathyroid cells, to regulate PTH and FGF23 release, appears likely.
The stimulatory effect of high PO4 concentration on PTH secretion has been demonstrated repeatedly in vivo and in vitro (Almaden et al., 1996; Nielsen et al., 1996; Slatopolsky et al., 1996; Almaden et al., 2003; Rodriguez et al., 2005), but without a clear linking mechanism. Interestingly, when studied ex vivo, the PO4 effect on PTH secretion was only observed in intact parathyroid tissue preparations but not in dispersed cells, where CaR expression becomes quickly reduced (Nielsen et al., 1996). Patients with secondary hyperparathyroidism (SHPT) show a left-shift in their PTH-Ca2+ curve, indicating that higher levels of serum Ca2+ are needed to activate CaR-mediated inhibition of PTH secretion (Rodriguez et al., 2005). Our hypothesis for this left-shift in the PTH-Ca2+ curve is that the hyperphosphataemia of CKD will promote the direct binding of PO4 to the CaR stabilising its inactive conformation and thus permitting increased PTH secretion. Indeed, we have shown that over its pathophysiological concentration range for CKD, PO4 inhibits CaR signalling in transfected human embryonic kidney (HEK-293) cells (Centeno et al., 2019). More specifically, PO4 lowers the efficacy of Ca2+o at the CaR (i.e., Emax) as opposed to altering the CaR’s sensitivity to Ca2+o (EC50). As such, the PO4 appears to act as a non-competitive CaR (partial) antagonist. Furthermore, raising buffer PO4 concentrations rapidly induced PTH secretion from primary human parathyroid cells and from murine parathyroid glands ex vivo. The rapid and reversible nature of this PO4 effect is indicative of a receptor-mediated event. Mutation of CaR residue Arg-62 (expressed in HEK-293 cells), overcame the inhibitory effect of the added PO4 suggesting that the Arg-62 residue may be the PO4 binding site, or at least a critical part of it. As mentioned earlier, CaRR62 was reported by Geng et al. (2016) to be a PO4 binding site present in the inactive conformation of the ECD but not its active conformation. Thus, the current functional data supports this structural prediction so far.
Following publication of the idea that the CaR itself serves as a parathyroid PO4 sensor, the clinical trials that had previously demonstrated efficacy for Cinacalcet and Etelcalcetide (used to lower PTH levels in end-stage renal disease) were reanalysed with regards to the prevailing serum PO4 concentrations in the patients (Goodman et al., 2022). Subjects were grouped according to whether their serum PO4 concentrations were above or below one of three different serum PO4 thresholds and these designations were dynamic over time, depending on whether their serum PO4 had risen above or dropped below the given threshold in the intervening time. By analysing the calcimimetic responses this way, it was found that calcimimetic-induced decreases in serum PTH levels were attenuated in subjects with higher serum PO4 concentrations. The inhibitory effect of high PO4 was quite modest for Etelcalcetide though more marked for Cinacalcet, especially at higher PO4 concentrations (Goodman et al., 2022). We would argue that, teleologically, it would make sense for PO4 to be able to moderate Ca2+-induced CaR activity/suppression of PTH secretion but not to be able to ablate it. For example, if one experienced simultaneous hypercalcaemia and hyperphosphataemia then without an inhibitory input from PO4, the high Ca2+o could maximally suppress PTH secretion abrogating the phosphaturia needed to resolve the hyperphosphataemia. By blunting this Ca2+o-induced suppression of PTH secretion, the mineral homeostatic system can resolve both issues, albeit potentially less quickly with regards to Ca2+. However, if high PO4 concentration could suppress CaR activity completely, thus acting effectively as a potent calcilytic, then the additional PTH-induced phosphaturia would resolve the hyperphosphataemia but would worsen the hypercalcaemia, perhaps even dangerously so. Therefore, it might be that as a non-competitive partial antagonist, elevated PO4 concentrations could integrate with Ca2+ to achieve the optimal PTH secretion for both minerals and not just for Ca2+. Furthermore, pathophysiological PO4 concentration also partially attenuated the effect of spermine, an endogenous polyamine and CaR agonist (Centeno et al., 2019). Thus, by disrupting the maintenance of VFT closure, PO4 may represent a general attenuator of positive CaR modulation, by acting as a non-competitive CaR antagonist.
Although CaR may couple to a broad range of heterotrimeric G protein families (Leach et al., 2020), it is CaR-induced Gαq/11 activation that has been most studied and is likely of the greatest importance for its biological functions. In mice, ablation of Gαq/11 results in a phenotype closely resembling that of the CaR knockout (Wettschureck et al., 2007). Similarly, in humans, some gain-of-function Gα11 mutations result in autosomal dominant hypocalcaemia (ADH) type-2 while some loss-of-function Gα11 mutations produce a familial hypocalciuric hypercalcemia (FHH)-like condition (Nesbit et al., 2013).
Initially, five putative protein kinase C (PKC) phosphorylation sites were identified in CaR, two in the first and third intracellular loops (Thr-646 and Ser-794) and three in the intracellular tail (Thr-888, Ser-895 and Ser-915) (Bai et al., 1998). Mutation of the two intracellular loop residues to non-phosphorylatable residues had no detectable effect on CaR activation, whereas mutation of CaRT888, CaRS895 and CaRS915 increased CaR responsiveness, but with the greatest effect resulting from CaRT888 mutation (Bai et al., 1998). Indeed, multiple studies have identified CaRT888 as the key PKC phosphorylation site in the CaR (Bai et al., 1998; Davies et al., 2007; Young et al., 2014). Furthermore, the identification of a family with ADH having a novel missense mutation in the PKC phosphorylation site Thr-888 (CaRT888M) provides evidence for the physiological importance of CaRT888 in humans (Lazarus et al., 2011).
Oscillatory Ca2+i signalling is usually ascribed to the IP3 receptor (IP3R) and how it becomes inhibited by the very rise in Ca2+i concentration that it mediates. As the cytosolic Ca2+ is returned to the Ca2+i stores by sarco/endoplasmic reticulum calcium ATPase (SERCA), the IP3R then reopens permitting the next cycle of Ca2+i release (Woll and Van Petegem, 2022). However, CaR-induced Ca2+i oscillations may also be explained by the existence of cycles of PKC-dependent GPCR phosphorylation and dephosphorylation. This idea was first proposed for metabotropic glutamate receptor 5 (mGluR5)-induced Ca2+i oscillations (Nakahara et al., 1997; Bradley et al., 2009). Since CaR shares significant structural and sequence homology with mGluR5, it was then proposed that CaR might also share similarities in terms of Ca2+i signalling (Young et al., 2014). In agreement with this, different studies have shown that agonist-induced PKC activation leads to CaR phosphorylation, mostly at CaRT888, which uncouples the receptor from its Gq/11/PLCβ signalling and thus inhibits Ca2+i release (McCormick et al., 2010; Ward and Riccardi, 2012). In contrast, inhibition of PKC results in decreased levels of CaRT888 phosphorylation resulting in CaR reactivation and a subsequent rise in Ca2+i mobilisation. Overall, these alternating cycles of CaRT888 phosphorylation and dephosphorylation might underlie CaR-induced Ca2+i oscillations (Conigrave and Ward, 2013) integrated in some way with the action of the IP3R.
The residue believed most likely to be responsible for the PKC-mediated inhibition of mGluR5 is Ser-839 (Kim et al., 2005) since mGluR5S839A does not exhibit Ca2+i oscillations whereas wild-type mGluR5 does (Kim et al., 2005). Interestingly, mGluR5S839 aligns not with CaRT888 but with CaRS875, which was not initially considered a likely PKC site. However, mutation of CaRS875 to alanine increased CaR sensitivity to Ca2+, while its mutation to aspartate (a phospho-mimetic site) decreased CaR sensitivity to Ca2+, suggesting that CaRS875 is another phosphorylation site with an inhibitory action on CaR signalling (Binmahfouz et al., 2019) (Figure 2).
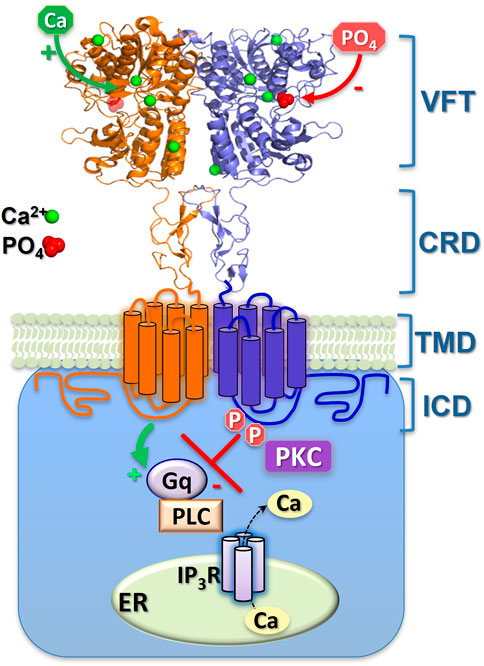
FIGURE 2. Schematic representation of CaR inhibition by extracellular phosphate ions and by intracellular, covalent phosphorylation. The binding-sites of the (activating) Ca2+ and (inhibitory) phosphate (PO4) ions shown here are approximate, though their 4:1 ratio is consistent with both the crystal models and functional Hill coefficients. Sustained phosphorylation (P) of ICD residues CaRS875 and CaRT888 supresses Ca2+i mobilisation. Episodic dephosphorylation of CaRT888 (at least) permits Ca2+i oscillations, while continuous dephosphorylation of these sites elicits enhanced, sustained Ca2+i mobilisation. The two monomers of the CaR homodimer are shown either as orange or blue. Gq, G protein-q/11; PLC, phospholipase C; IP3R, IP3 receptor; ER, Ca2+ stores of the endoplasmic reticulum; PKC, protein kinase C. The CaR domains shown are the venus flytrap (VFT), cysteine-rich domain (CRD), transmembrane domain (TMD) and intracellular domain (ICD).
In fact, human CaR has 54 serine and threonine residues in either its intracellular domain (ICD) or intracellular loops (Garrett et al., 1995). Depending on the phosphosite prediction software used, at least 17 of these could be phosphorylation sites, though the total number could be as high as 40. The most likely phosphorylation sites are those serine/threonine residues present in the juxtamembrane region of the ICD, as opposed to those in the carboxyl-terminus. While CaRT888A exhibits enhanced, less oscillatory signalling than for wild-type CaR, cotreatment with a PKC inhibitor can produce a further enhancement of signalling indicating that CaRT888 is not the sole target of PKC action (Garrett et al., 1995; Binmahfouz et al., 2019). In contrast, the CaRS875A/T888A double mutation elicits completely non-oscillatory signalling, the same as occurs with PKC inhibition, suggesting that dephosphorylation of both sites is required to abolish the inhibitory effect of PKC (Binmahfouz et al., 2019). Indeed, it has been suggested that the precise pattern of phosphorylation on any given GPCR could vary depending on the cellular context. The so-called “phospho-barcode” hypothesis posits that different phosphorylation “barcode” patterns could elicit distinct downstream signalling outcomes (Tobin et al., 2008; Yang et al., 2017).
In this review, we have set out how PO4 may inhibit CaR activity both as an extracellular bound anion, but also as an intracellular moiety resulting from phosphorylation. However, there is also recent evidence that raises fascinating questions about the evolutionary purpose of the CaR’s ECD. Synthetic calcimimetics bind within the CaR’s transmembrane domain (TMD), as demonstrated by the observation that their effect is maintained in “headless” CaR mutants lacking the ECD (Hauache et al., 2000; Ray and Northup, 2002; Mun et al., 2004; Rey et al., 2005). Interestingly, Ca2+ sensitivity is also retained in these headless mutants, indicating the presence of additional orthosteric Ca2+ binding sites in the TMD (Petrel et al., 2004). Indeed, in a headless CaR that also contained the mutation CaRT888A (i.e., where an inhibitory phospho-site was replaced by a non-phosphorylatable alanine), the Ca2+o sensitivity was restored to levels not dissimilar to those seen in wild-type CaR (Binmahfouz et al., 2019). This suggests that the CaR ECD is not essential for Ca2+o sensitivity per se, and thus it might be hypothesised that the ECD a) fine-tunes Ca2+o-sensitivity to make CaR more physiologically optimal for animal Ca2+o homeostasis and/or b) provides for regulation by a range of other physiological modulators (e.g., PO4, L-amino acids) making for more sophisticated mineral homeostatic integration. Therefore, further work will be needed to decipher the precise role(s) of the CaR’s ECD.
The CaR is not merely a Ca2+o-sensor and Ca2+o homeostasis controller but can sense other endogenous ligands, such as phosphate, thus permitting it to integrate mineral homeostasis broadly.
DW wrote the review making use of portions of text from PC and LB PhD thesis. DW modified the figures made originally by PC and KA provided additional text, insights and all co-authors proofread the review.
The authors acknowledge the support of the Marie Sklodowska-Curie Actions of the European Union’s Horizon 2020 program (675228, CaSR Biomedicine network) as well as Kidney Research UK (RP_005_20220704). Some subsections and figure components were based on text originally submitted for PhD examination by Patricia Centeno and Lenah Binmahfouz but not published elsewhere.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agoro, R., and White, K. E. (2023). Regulation of FGF23 production and phosphate metabolism by bone-kidney interactions. Nat. Rev. Nephrol. Jan. 9, 185–193. doi:10.1038/s41581-022-00665-x
Almaden, Y., Canalejo, A., Hernandez, A., Ballesteros, E., Garcia-Navarro, S., Torres, A., et al. (1996). Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J. Bone Min. Res. 11, 970–976. doi:10.1002/jbmr.5650110714
Almaden, Y., Felsenfeld, A. J., Rodriguez, M., Canadillas, S., Luque, F., Bas, A., et al. (2003). Proliferation in hyperplastic human and normal rat parathyroid glands: Role of phosphate, calcitriol, and gender. Kidney Int. 64, 2311–2317. doi:10.1046/j.1523-1755.2003.00331.x
Bai, M., Trivedi, S., Lane, C. R., Yang, Y., Quinn, S. J., Brown, E. M., et al. (1988). Protein kinase C phosphorylation of threonine at position 888 in Ca2+-sensing receptor (CaR) inhibits coupling to Ca2+ store release. J. Biol. Chem. 273, 21267–21275. doi:10.1074/jbc.273.33.21267
Bergwitz, C., and Juppner, H. (2010). Regulation of phosphate homeostasis by PTH, vitamin D, and Fgf23. Ann. Rev. Med. 61, 91–104. doi:10.1146/annurev.med.051308.111339
Binmahfouz, L. S., Centeno, P. P., Conigrave, A. D., and Ward, D. T. (2019). Identification of serine-875 as an inhibitory phosphorylation site in the calcium-sensing receptor. Mol. Pharmacol. 96, 204–211. doi:10.1124/mol.119.116178
Block, G. A., Martin, K. J., de Francisco, A. L., Turner, S. A., Avram, M. M., Suranyi, M. G., et al. (2004). Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 350, 1516–1525. doi:10.1056/NEJMoa031633
Bradley, S. J., Watson, J. M., and Challiss, R. A. J. (2009). Effects of positive allosteric modulators on single-cell oscillatory Ca2+ signaling initiated by the type 5 metabotropic glutamate receptor. Mol. Pharmacol. 76, 1302–1313. doi:10.1124/mol.109.059170
Centeno, P. P., Herberger, A., Mun, H-C., Tu, C., Nemeth, E. F., Chang, W., et al. (2019). Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Comm. 10, 4693. doi:10.1038/s41467-019-12399-9
Conigrave, A. D., and Hampson, D. R. (2006). Broad-spectrum L-amino acid sensing by class 3 G-protein-coupled receptors. Trends Endocrinol. Metab. 17, 398–407. doi:10.1016/j.tem.2006.10.012
Conigrave, A. D., and Ward, D. T. (2013). Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best Prac. Res. Clin. Endocrinol. Metab. 27, 315–331. doi:10.1016/j.beem.2013.05.010
Davies, S. L., Ozawa, A., McCormick, W. D., Dvorak, M. M., and Ward, D. T. (2007). Protein kinase C-mediated phosphorylation of the calcium-sensing receptor is stimulated by receptor activation and attenuated by calyculin-sensitive phosphatase activity. J. Biol. Chem. 282, 15048–15056. doi:10.1074/jbc.M607469200
Duchen, M. R. (2000). Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell. Calcium 28, 339–348. doi:10.1054/ceca.2000.0170
Fan, Y., Liu, W., Bi, R., Densmore, M. J., Sato, T., Mannstadt, M., et al. (2018). Interrelated role of Klotho and calcium-sensing receptor in parathyroid hormone synthesis and parathyroid hyperplasia. Proc. Natl. Acad. Sci. U.S.A. 115, E3749–E3758. doi:10.1073/pnas.1717754115
Gao, Y., Robertson, M. J., Rahman, S. N., Seven, A. B., Zhang, C., Meyerowitz, J. G., et al. (2021). Asymmetric activation of the calcium-sensing receptor homodimer. Nature 595, 455–459. doi:10.1038/s41586-021-03691-0
Garrett, J. E., Capuano, I. V., Hammerland, L. G., Hung, B. C., Brown, E. M., Hebert, S. C., et al. (1995). Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J. Biol. Chem. 270, 12919–12925. doi:10.1074/jbc.270.21.12919
Geng, Y., Mosyak, L., Kurinov, I., Zuo, H., Sturchler, E., Cheng, T. C., et al. (2016). Structural mechanism of ligand activation in human calcium-sensing receptor. Elife 5, e13662. doi:10.7554/eLife.13662
Goodman, W. G., Ward, D. T., Martin, K. J., Drayer, D., Moore, C., Xu, J., et al. (2022). Activation of the calcium receptor by calcimimetic agents is preserved despite modest attenuating effects of hyperphosphatemia. J. Am. Soc. Nephrol. 33, 201–212. doi:10.1681/ASN.2021060825
Hauache, O. M., Hu, J., Ray, K., and Spiegel, A. M. (2000). Functional interactions between the extracellular domain and the seven-transmembrane domain in Ca2+ receptor activation. Endocrine 13, 63–70. doi:10.1385/ENDO:13:1:63
Hunter, T. (2012). Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2513–2516. doi:10.1098/rstb.2012.0013
Ide, N., Ye, R., Courbebaisse, M., Olauson, H., Densmore, M. J., Larsson, T. E., et al. (2018). In vivo evidence for an interplay of FGF23/Klotho/PTH axis on the phosphate handling in renal proximal tubules. Am. J. Physiol. - Ren. Physiol. 315, F1261–F1270. doi:10.1152/ajprenal.00650.2017
Kawakami, K., Takeshita, A., Furushima, K., Miyajima, M., Hatamura, I., Kuro, O. M., et al. (2017). Persistent fibroblast growth factor 23 signalling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci. Rep. 7, 40534. doi:10.1038/srep40534
Kemp, G. J., Blumsohn, A., and Morris, B. W. (1992). Circadian changes in plasma phosphate concentration, urinary phosphate excretion, and cellular phosphate shifts. Clin. Chem. 38, 400–402. doi:10.1093/clinchem/38.3.400
Kim, C. H., Braud, S., Isaac, J. T. R., and Roche, K. W. (2005). Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on Serine 839 regulates Ca2+ oscillations. J. Biol. Chem. 280, 25409–25415. doi:10.1074/jbc.M502644200
Komaba, H., and Fukagawa, M. (2016). Phosphate. A poison for humans? Kid. Int. 90, 753–763. doi:10.1016/j.kint.2016.03.039
Lazarus, S., Pretorius, C., Khafagi, F., Campion, K. L., Brennan, S. C., Conigrave, A. D., et al. (2011). A novel mutation of the primary protein kinase C phosphorylation site in the calcium-sensing receptor causes autosomal dominant hypocalcemia. Eur. J. Endocrinol. 164, 429–435. doi:10.1530/EJE-10-0907
Leach, K., Conigrave, A. D., Sexton, P. M., and Christopoulos, A. (2015). Towards tissue-specific pharmacology: Insights from the calcium-sensing receptor as a paradigm for GPCR (Patho)physiological bias. Trends Pharmacol. Sci. 36, 215–225. doi:10.1016/j.tips.2015.02.004
Leach, K., Hannan, F. M., Josephs, T. M., Keller, A. N., Møller, T. C., Ward, D. T., et al. (2020). International union of basic and clinical pharmacology: Calcium-sensing receptor nomenclature, pharmacology, and function. Pharmacol. Rev. 72, 558–604. doi:10.1124/pr.119.018531
McCormick, W. D., Atkinson-Dell, R., Campion, K. L., Mun, H-C., Conigrave, A. D., and Ward, D. T. (2010). Increased receptor stimulation elicits differential calcium-sensing ReceptorT888 dephosphorylation. J. Biol. Chem. 285, 14170–14177. doi:10.1074/jbc.M109.071084
Mun, H. C., Franks, A. H., Culverston, E. L., Krapcho, K., Nemeth, E. F., and Conigrave, A. D. (2004). The venus fly trap domain of the extracellular Ca2+ -sensing receptor is required for L-amino acid sensing. J. Biol. Chem. 279, 51739–51744. doi:10.1074/jbc.M406164/200
Nakahara, K., Okada, M., and Nakanishi, S. (1997). The metabotropic glutamate receptor mGluR5 induces calcium oscillations in cultured astrocytes via protein kinase C phosphorylation. J. Neurochem. 69, 1467–1475. doi:10.1046/j.1471-4159.1997.69041467.x
Nesbit, M. A., Hannan, F. M., Howles, S. A., Babinsky, V. N., Head, R. A., Cranston, T., et al. (2013). Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N. Eng. J. Med. 368, 2476–2486. doi:10.1056/NEJMoa1300253
Nielsen, P. K., Feldt-Rasmussen, U., and Olgaard, K. (1996). A direct effect in vitro of phosphate on PTH release from bovine parathyroid tissue slices but not from dispersed parathyroid cells. Nephrol. Dial. Transpl. 11, 1762–1768. doi:10.1093/oxfordjournals.ndt.a027665
Petrel, C., Kessler, A., Dauban, P., Dodd, R. H., Rognan, D., and Ruat, M. (2004). Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J. Biol. Chem. 279, 18990–18997. doi:10.1074/jbc.M400724200
Quinn, S. J., Thomsen, A. R., Pang, J. L., Kantham, L., Brauner-Osborne, H., Pollak, M., et al. (2013). Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am. J. Physiol. Endocrinol. Metab. 304, E310–E320. doi:10.1152/ajpendo.00460.2012
Ray, K., and Northup, J. (2002). Evidence for distinct cation and calcimimetic compound (NPS 568) recognition domains in the transmembrane regions of the human Ca2+ receptor. J. Biol. Chem. 277, 18908–18913. doi:10.1074/jbc.M202113200
Rey, O., Young, S. H., Yuan, J., Slice, L., and Rozengurt, E. (2005). Amino acid-stimulated Ca2+ oscillations produced by the Ca2+-sensing receptor are mediated by a phospholipase C/inositol 1,4,5-trisphosphate-independent pathway that requires G12, rho, filamin-A, and the actin cytoskeleton. J. Biol. Chem. 280, 22875–22882. doi:10.1074/jbc.M503455200
Rodriguez, M., Nemeth, E., and Martin, D. (2005). The calcium-sensing receptor: A key factor in the pathogenesis of secondary hyperparathyroidism. Am. J. Physiol. Ren. Physiol. 288, F253–F264. doi:10.1152/ajprenal.00302.2004
Shimada, T., Kakitani, M., Yamazaki, Y., Hasegawa, H., Takeuchi, Y., Fujita, T., et al. (2004). Targeted ablation of Fgf23 demonstrates an essential physiological role of Fgf23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568. doi:10.1172/JCI19081
Slatopolsky, E., Finch, J., Denda, M., Ritter, C., Zhong, M., Dusso, A., et al. (1996). Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion in vitro. J. Clin. Invest. 97, 2534–2540. doi:10.1172/JCI118701
Streja, E., Wang, H., Lau, W. L., Molnar, M. Z., Kovesdy, C. P., Kalantar-Zadeh, K., et al. (2014). Mortality of combined serum phosphorus and parathyroid hormone concentrations and their changes over time in hemodialysis patients. Bone 61, 201–207. doi:10.1016/j.bone.2014.01.016
Tentori, F., Blayney, M. J., Albert, J. M., Gillespie, B. W., Kerr, P. G., Bommer, J., et al. (2008). Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: The dialysis outcomes and practice patterns study (DOPPS). Am. J. Kidney Dis. 52, 519–530. doi:10.1053/j.ajkd.2008.03.020
Tobin, A. B., Butcher, A. J., and Kong, K. C. (2008). Location, location, location.site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling, site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29, 413–420. doi:10.1016/j.tips.2008.05.006
Ward, D. T., and Riccardi, D. (2012). New concepts in calcium-sensing receptor pharmacology and signalling. Br. J. Pharmacol. 165, 35–48. doi:10.1111/j.1476-5381.2011.01511.x
Wettschureck, N., Lee, E., Libutti, S. K., Offermanns, S., Robey, P. G., Spiegel, A. M., et al. (2007). Parathyroid-specific double knockout of Gq and G11 α-subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+-sensing receptor. Mol. Endocrinol. 21, 274–280. doi:10.1210/me.2006-0110
Williams, R. J. P. (2006). The evolution of calcium biochemistry. Biochim. Biophys. Acta. 1763, 1139–1146. doi:10.1016/j.bbamcr.2006.08.042
Woll, K. A., and Van Petegem, F. (2022). Calcium-release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 102, 209–268. doi:10.1152/physrev.00033.2020
Yang, Z., Yang, F., Zhang, D., Liu, Z., Lin, A., Liu, C., et al. (2017). Phosphorylation of G protein-coupled receptors: From the barcode hypothesis to the flute model. Mol. Pharmacol. 92, 201–210. doi:10.1124/mol.116.107839
Young, S. H., Rey, O., Sinnett-Smith, J., and Rozengurt, E. (2014). Intracellular Ca2+ oscillations generated via the Ca2+-sensing receptor are mediated by negative feedback by PKCα at Thr888. Am. J. Physiol. - Cell. Physiol. 306, C298–C306. doi:10.1152/ajpcell.00194.2013
Keywords: calcium-sensing receptor, parathyroid hormone, phosphate-sensing, secondary hyperparathyroidism, receptor phosphorylation
Citation: Centeno PP, Binmahfouz LS, Alghamdi K and Ward DT (2023) Inhibition of the calcium-sensing receptor by extracellular phosphate ions and by intracellular phosphorylation. Front. Physiol. 14:1154374. doi: 10.3389/fphys.2023.1154374
Received: 30 January 2023; Accepted: 20 March 2023;
Published: 31 March 2023.
Edited by:
Daniela Riccardi, Cardiff University, United KingdomReviewed by:
Mariano Rodriguez, University of Cordoba, SpainCopyright © 2023 Centeno, Binmahfouz, Alghamdi and Ward. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald T. Ward, ZC53YXJkQG1hbmNoZXN0ZXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.