 Sahil Zaveri
Sahil Zaveri Ujala Srivastava1
Ujala Srivastava1 Yongxia Sarah Qu
Yongxia Sarah Qu Mohamed Chahine
Mohamed Chahine Mohamed Boutjdir
Mohamed Boutjdir
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 21 March 2023
Sec. Cardiac Electrophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1144069
This article is part of the Research TopicThe Roles of Ion Channels in Disease Occurrence and DevelopmentView all 5 articles
Ca2+ plays a crucial role in excitation-contraction coupling in cardiac myocytes. Dysfunctional Ca2+ regulation alters the force of contraction and causes cardiac arrhythmias. Ca2+ entry into cardiomyocytes is mediated mainly through L-type Ca2+ channels, leading to the subsequent Ca2+ release from the sarcoplasmic reticulum. L-type Ca2+ channels are composed of the conventional Cav1.2, ubiquitously expressed in all heart chambers, and the developmentally regulated Cav1.3, exclusively expressed in the atria, sinoatrial node, and atrioventricular node in the adult heart. As such, Cav1.3 is implicated in the pathogenesis of sinoatrial and atrioventricular node dysfunction as well as atrial fibrillation. More recently, Cav1.3 de novo expression was suggested in heart failure. Here, we review the functional role, expression levels, and regulation of Cav1.3 in the heart, including in the context of cardiac diseases. We believe that the elucidation of the functional and molecular pathways regulating Cav1.3 in the heart will assist in developing novel targeted therapeutic interventions for the aforementioned arrhythmias.
Cardiac excitation-contraction coupling is a process where electrical excitation of the cardiomyocyte leads to a muscular contraction in the heart (Bers, 2002). L-type Ca2+ channels play an essential role in excitation-contraction coupling by mediating Ca2+ influx and membrane excitability (Bodi et al., 2005; Striessnig et al., 2014; Catterall et al., 2020). These Ca2+ channels are modulated by small concentrations of different chemical classes of Ca2+ antagonists, including dihydropyridines (Berjukow et al., 2000; Tang et al., 2016). There are four types of L-type Ca2+ channels: Cav1.1, Cav1.2, Cav1.3, and Cav1.4 (Berjukow et al., 2000; Berger and Bartsch, 2014; Striessnig et al., 2014). The Cav1.1 and Cav1.4 channels are restricted to the skeletal muscle and retina/immune cells, respectively. However, Cav1.2 is more widely expressed in the heart/smooth muscle, neurons (somatodendritic), and endocrine cells, while Cav1.3 is expressed in the heart, neurons (somatodendritic), endocrine cells, and sensory cells (Striessnig et al., 2014; Mesirca et al., 2015).
Cav1.3, the focus of this review, was initially thought to be of neuroendocrine origin (Qu et al., 2005b; Zhang et al., 2005; Lu et al., 2015). However, it was subsequently discovered in the adult heart with distinct expression exclusively in the supraventricular tissues (atria, sinoatrial node, atrioventricular node) and not in the ventricles (Mangoni et al., 2003; Qu et al., 2005a). Genetic deletion of Cav1.3 in mice (Cav1.3−/−) causes congenital deafness, sinus bradycardia, and various degrees of atrioventricular (AV) block consistent with region-specific expression (Mangoni et al., 2003; Hu et al., 2004; Qu et al., 2005a). Furthermore, Cav1.3−/− mice display impaired Ca2+ homeostasis associated with atrial fibrillation (AF) (Figure 1) (Mancarella et al., 2008). Interestingly, loss of Cav1.3 function in humans was associated with sinoatrial node dysfunction and deafness (SANDD) syndrome with a cardiac and auditory phenotype similar to Cav1.3−/− mice (Baig et al., 2011; Liaqat et al., 2019; Torrente et al., 2020).
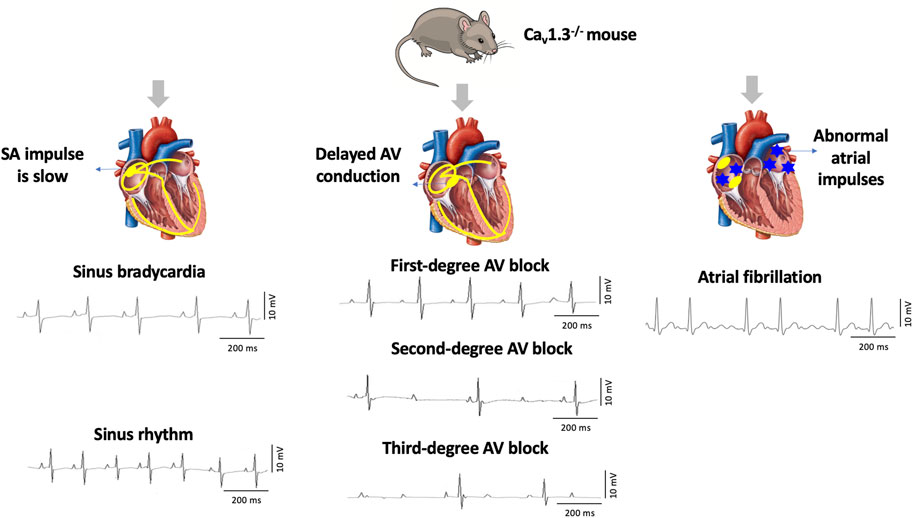
FIGURE 1. Deletion of Cav1.3 in mice leads to electrographic abnormalities. The absence of Cav1.3 in mice leads to the development of sinoatrial (SA) and atrioventricular (AV) node dysfunction leading to sinus bradycardia and first-degree, second-degree, and third-degree AV block as well as atrial fibrillation (AF).
Numerous neurotransmitters regulate Cav1.3 in the heart. Phosphorylation of the channel by cAMP-dependent protein kinase A (PKA) is at serine residues located at positions 1743 and 1816 of the C-terminus (Mitterdorfer et al., 1996). Protein kinase C (PKC) also plays a vital role in regulating Cav1.3 in an isozyme-specific manner, with the regulation site being a serine residue located at position 81 of the N-terminal domain (Baroudi et al., 2006). When calmodulin-dependent protein kinase II (CaMKII) is co-expressed with densin, which binds to Cav1.3, it facilitates the increase of Ca2+ current (ICaL) as a result of high-frequency stimulation (Maier and Bers, 2002; Jenkins et al., 2010; Tokumitsu and Sakagami, 2022). This provides another mechanism for Cav1.3 regulation (Jenkins et al., 2010; Tokumitsu and Sakagami, 2022).
Alternative splicing in the Cav1.3 C-terminus affects its electrophysiological properties by reducing Ca2+-dependent inactivation of the Cav1.3 channels (Tan et al., 2011). A recent study by Lu et al. showed that the C-terminus of Cav1.3 undergoes cleavage and translocation to the nucleus, where it acts as a transcription factor that affects the function of Ca2+-activated K+ channels in atrial cardiomyocytes (Lu et al., 2015).
This review summarizes the functional role and regulation of Cav1.3 in healthy and diseased hearts. Specifically, we provide current knowledge on Cav1.3 regulation across different cardiac conditions and the resulting implications for diseases and potential novel therapies.
In cardiac musculature, Cav1.3 is involved in pacemaking and AV conduction of the heart (Mesirca et al., 2016b). Cav1.3−/− mice are deaf and exhibit bradycardia and arrhythmia resulting from sinoatrial (SA) node dysfunction (Platzer et al., 2000; Ortner, 2023). This is likely because of the crucial role that Cav1.3 channels play in the diastolic depolarization of SA node pacemaker cells (Mangoni et al., 2003). In this regard, action potentials recorded from the SA nodes in Cav1.3−/− mice show a significant reduction in beating frequency and diastolic depolarization rate compared with Cav1.3+/− or wild-type littermates, suggesting that this decrease is intrinsic to the SA node (Zhang et al., 2002).
Another study reported that Cav1.3 deficiency impaired intracellular Ca2+ ([Ca2+]i) dynamics by decreasing the frequency of local [Ca2+]i release, eventually leading to dysfunctional synchronization (Torrente et al., 2016). Cav1.3 appeared to stimulate and synchronize ryanodine receptor (RyR)-dependent [Ca2+]i release during regular SA node pacemaker activity. Thus, Cav1.3 plays dual roles by mediating inward ICaL and stimulating RyR-dependent [Ca2+]i release. This provides an additional pathophysiological mechanism for congenital SA node dysfunction and heart block linked to the loss of Cav1.3 function in humans (Boutjdir, 2000; Torrente et al., 2016). Cav1.3 was implicated as an essential molecular component of the voltage-dependent, dihydropyridine-sensitive Na+ current (Ist), essential in SA node automaticity. Hence, Ist and ICaL share Cav1.3 as a common molecular determinant in the SA node, despite the relatively unknown molecular nature of Ist (Toyoda et al., 2017).
Cav1.3 is generally less abundant than Cav1.2, the predominant L-type Ca2+ channel in the heart and brain (Berger and Bartsch, 2014). The expression and localization of Cav1.3 are developmentally regulated. Two forms of Cav1.3 (250 kD and 190 kD) were observed, with the full-length (250 kD) channel protein predominant in the prenatal stages. Cav1.3 channel protein was expressed in both atria and ventricles at fetal and neonatal stages but was absent in adult ventricles. The short form of Cav1.3 is only expressed in the adult and is restricted to the atria (Qu et al., 2011).
The 190 kD form of Cav1.3 represents the channel with a truncated C-terminus (Qu et al., 2011). Interestingly, this truncation of Cav1.3 has been shown to translocate to the nucleus, functioning as a transcriptional regulator to alter the function of KCa2 in atrial myocytes. Nuclear translocation of the C-terminal domain of Cav1.3 is modulated by [Ca2+]i. This results in a decrease in protein expression of myosin light chain 2, which interacts with and increases the membrane localization of KCa2 channels (Lu et al., 2015). Another study reported that the total and membrane expression of Cav1.3 were significantly impaired by overexpression of the protein Snapin, resulting in the ubiquitin-proteasomal degradation of the channel and a consequent reduction of the total ICaL densities (Sun et al., 2017).
In embryonic atrial cardiomyocytes, elevated Cav1.3 expression was reported upon truncation and subsequent inhibition of Cav1.2 in murine models. Western blot analysis indicated an increase of Cav1.3 protein in the atrium, likely compensating for the functional loss of the truncated Cav1.2 channel in these murine embryonic atrial cardiomyocytes by upregulating the Cav1.3 channel (Ding et al., 2013).
The C-terminal part of the Cav1.3 channel is encoded by exons 39 to 49 and it is the subject of intensive alternative splicing events that affect its function (Figure 2) (Singh et al., 2008; Scharinger et al., 2015; Hofer et al., 2021). Several splicing variants have been reported in the nervous system and their role in heart is not yet well elucidated (Hofer et al., 2021). The C-terminus is a strong target for alternative splicing due to the C-terminus gating modulator’s ability to prevent Ca2+ inactivation of the channels (Singh et al., 2008; Scharinger et al., 2015; Hofer et al., 2021). The long isoform (Cav1.342L) possesses all the regulatory domains, whereas two short splicing isoforms (Cav1.342A and Cav1.343S) are characterized by the absence of the distal C-terminal regulatory domain or both proximal and distal C-terminal regulatory domains (Singh et al., 2008; Tan et al., 2011; Scharinger et al., 2015). Alternative splicing in the C-terminus of Cav1.3 modulates its electrophysiological properties (Singh et al., 2008; Scharinger et al., 2015; Hofer et al., 2021). Activation of ICaL through Cav1.342A channels increased at negative voltages, and inactivation was faster due to enhanced Ca2+-dependent inactivation (Singh et al., 2008). Furthermore, the C-terminal modulator domain in the Cav1.342 isoforms competed with calmodulin (CaM) in regards to binding to the IQ domain (Kuzmenkina et al., 2019).
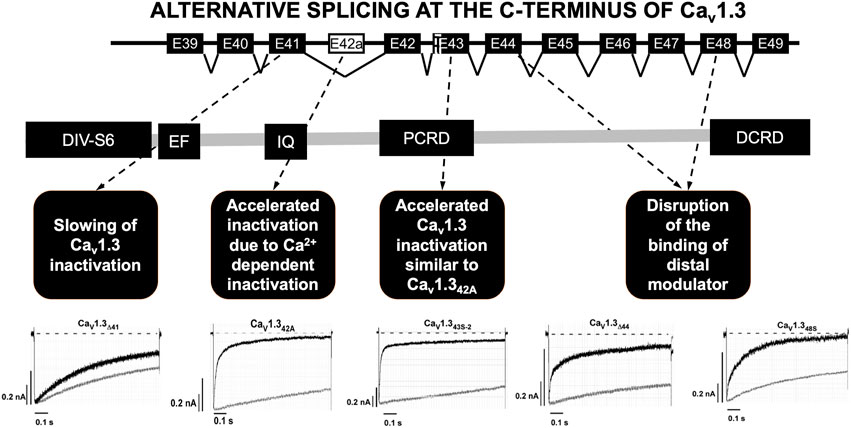
FIGURE 2. Alternative splicing at the C-terminus of Cav1.3. Alternative splicing of exon 41 that removes the IQ motif resulted in a truncated Cav1.3 protein with diminished inactivation. Splicing of exon 43 which causes a frameshift variant and is susceptible to accelerating the inactivation similar to Cav1.342A. Ca2+ current through Cav1.342A channels increased at negative voltages, and inactivation was faster because of Ca2+-dependent inactivation. Splicing of exons 44 and 48 was an in-frame variant and caused disruption of the binding of distal modulator to the IQ domain. PCRD (proximal C-terminal regulatory domain), DCRD (distal C-terminal regulatory domain). Current tracing are from reference Tan et al., 2011.
Alternative splicing was identified at four other different loci in the C-terminus of Cav1.3. The splicing of exon 41 removes the IQ motif resulting in a truncated Cav1.3 protein with diminished inactivation. Secondly, splicing of exon 43 results in a frameshift variant and is susceptible to increased inactivation similar to Cav1.342A. Lastly, the splicing of exons 44 and 48 in-frame causes disruption of the distal modulator binding to the IQ domain (Tan et al., 2011).
Cav1.3 is upregulated through the PKA-cAMP pathway (Figure 3A) (Qu et al., 2005b; Ramadan et al., 2009). Specifically, Ramadan et al. showed 3 PKA consensus sites phosphorylated on the proximal C-terminus of the Cav1.3 α1-subunit at serines 1743, 1816 and 1964 using mass spectrometry (Ramadan et al., 2009). Additional site-directed mutagenesis followed by patch clamp studies demonstrated that serines 1743 and 1816 were major functional PKA consensus sites while the phosphorylation of serine 1964 was not functionally relevant. The resulting PKA phosphorylation of Cav1.3 increased channel activity in the SA node and atria (Qu et al., 2005b). The upregulation Cav1.3 activity may account for as much as a 25% increase in total ICaL (Qu et al., 2005b; Mahapatra et al., 2012; Vandael et al., 2013). On the other hand, decrease in PKA activity and subsequent downregulation of Cav1.3 was reported in mice with a frameshift variant in the natriuretic peptide precursor A gene linked to AF (Menon et al., 2019). Collectively, these findings show that Cav1.3 is a target for sympathetic control of heart rhythm via PKA.
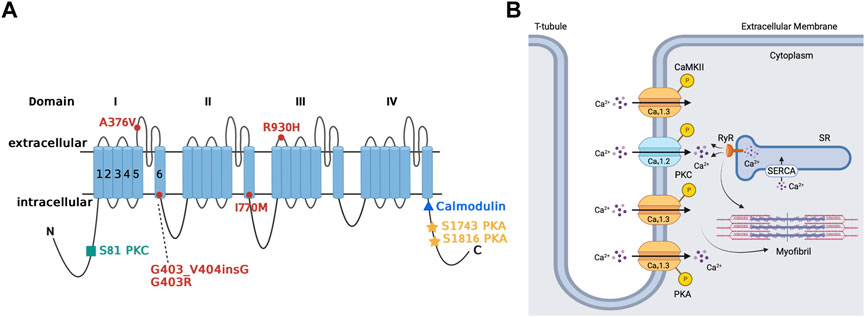
FIGURE 3. Regulation of Cav1.3 L-type Ca2+ channel by protein kinase A, protein kinase C, and calmodulin-dependent protein kinase II. Panel (A) Schematic representation of the four homologous domains (I-IV) of the Cav1.3 α1-subunit with 6 transmembrane segments (S1-S6) and N- and C- termini. Phosphorylation of the channel by PKA is at serine residues located at positions 1743 and 1816 of the C-terminus. PKC phosphorylates at the N-terminal domain at serine residue located at position 81. Calmodulin binding site is on the proximal C-terminus. Missense variant A376V and the founder variant G403_V404insG, as well as heterozygous non-synonymous variant R930H in CACNA1D gene have been associated with sinoatrial node dysfunction (Liaqat et al., 2019; Rinné et al., 2022). The missense variants G403R and I770M has been found in patients with atrioventricular node dysfunction (Scholl et al., 2013). Panel (B) The sketch summarizes the regulation of atrial Cav1.3 channel by the different kinases, including PKA, PKC and CaMKII. Ca2+ entry through Cav1.3 channel and subsequent Ca2+ release from RyR contributes to pacemaking, while Ca2+ entry through Cav1.2 contributes to excitation-contraction coupling. PKA (protein kinase A), PKC (protein kinase C), SR (sarcoplasmic reticulum), SERCA (sarcoendoplasmic reticulum calcium ATPase), RyR (ryanodine receptor), P (phosphorylation site), CaMKII (calmodulin-dependent protein kinase II).
There is limited available information about the regulation of Cav1.3 by PKC in the heart. We showed that Cav1.3 is inhibited through PKC activation by phosphorylation of its N-terminal domain (Figure 3A) (Baroudi et al., 2006). PKC activation reduces Cav1.3 ICaL by up to 50% by reducing the probability of Cav1.3 to remain in an open state while increasing the likelihood and time spent in the closed state (Baroudi et al., 2006; Chahine et al., 2008). Interestingly, βIIPKC and εPKC are key PKC isozymes implicated in this regulatory mechanism, and serine 81 represents an essential site for PKC-mediated phosphorylation of Cav1.3 (Baroudi et al., 2006). βIIV5-3 and εV1-2 peptides, which inhibit βIIPKC and εPKC, respectively, prevent the downregulation of Cav1.3 by PKC. This further supports the importance of isozyme specific PKC in the regulation of Cav1.3 and the essential consensus site at serine 81 in the downregulation of Cav1.3 (Baroudi et al., 2006; Ferreira et al., 2012).
The prevailing understanding of CaM modulation of Cav1.3 appears not to be limited to the binding of CaM to the C-terminus of the channel (Figure 3A) (Maier and Bers, 2002; Ben Johny et al., 2013). Johny et al. demonstrated that CaM might also bind to alternative sites on Cav1.3, and subsequent binding of Ca2+ leads to alternative configurations of the CaM-Cav1.3 complex, resulting in alternative forms of Ca2+-dependent inactivation (Ben Johny et al., 2013). Thus, in addition to the previously assumed association of CaM to the C-terminal IQ domain, the N-lobe may bind to the proximal Ca2+-inactivating region. Furthermore, if Ca2+ binds to the N-lobe of CaM, then it will bind onto the NSCaTE region of the Cav1.3 channel (Dick et al., 2008; Ben Johny et al., 2013). Banerjee et al. reinforce this idea by demonstrating that CaM must bind to Cav1.3 on both of its lobes to modulate Cav1.3 to exit its preinhibited configuration (Banerjee et al., 2018). If CaM only binds on one of its sites to the Cav1.3 channel, the channel will fail to be inhibited and operate as if CaM is not present. The C-terminal modulator domain in the distal C-terminus of the Cav1.3 channel can interfere with CaM binding, resulting in subsequent inhibition of channel activity (Kuzmenkina et al., 2019). Cav1.3 may also be modulated by CaMKII for increased channel activity (Maier and Bers, 2002; Gao et al., 2006; Tokumitsu and Sakagami, 2022). CaMKII-mediated phosphorylation of Cav1.3 channel may result in the channels staying open for longer, and the increased channel activity manifests in an action potential that looks similar to an ascending staircase with multiple depolarizations from holding potential (Maier and Bers, 2002). The specific pathway of insulin growth factor 1 stimulates phospholipase C-ɣ to facilitate Ca2+ release from IP3-sensitive stores, thereby activating CaMKII and phosphorylating the Cav1.3 channel (Gao et al., 2006).
Autoimmune-associated congenital heart block (aCHB) is an electrophysiological abnormality affecting the SA and AV nodes of structurally healthy hearts in fetuses and neonates. Clinical symptoms of aCHB include a spectrum of variations of sinus bradycardia and AV block. Of these variations, third-degree AV block is the most critical and lethal manifestation, having the greatest mortality rate (Lazzerini et al., 2017). aCHB is presumably acquired passively when the mother transmits anti-Ro/SSA auto-antibodies through the placenta to the fetus (Boutjdir, 2000; Lazzerini et al., 2017). Anti-Ro/SSA antibodies are the most common antibodies associated with aCHB (Karnabi et al., 2010; Qu et al., 2019). Qu et al. showed that human Cav1.3 expression and associated electrophysiological activity were inhibited by anti-Ro/SSA positive IgG antibodies obtained from mothers that had children with aCHB (Qu et al., 2005a). This study also showed the ability of anti-Ro/SSA positive IgG to recognize the Cav1.3 channel protein, which suggests cross-reactivity between the anti-Ro/SSA antibodies with the Cav1.3 channel (Qu et al., 2005a; Qu and Boutjdir, 2012).
Autoimmune diseases provide an additional level of insight into the development of cardiovascular diseases since autoantibodies have been found to modulate cardiac electrophysiological activity (Qu et al., 2019). Voltage-gated L-type Ca2+ channels, specifically Cav1.3, play a key role in the pathophysiology of cardiac arrhythmias in the presence of autoimmune antibodies such as anti-Ro/SSA and anti-La/SSB (Qu et al., 2019; Lazzerini et al., 2021). In particular, the anti-Ro/SSA antibodies interact with Cav1.3 through a mechanism of direct channel inhibition (Qu et al., 2005a; Lazzerini et al., 2018; Qu et al., 2019). There are two types of anti-Ro/SSA antibodies: anti-52 kD and anti-60 kD, and they are formed as a result of an autoimmune response to the Ro/SSA antigen (Lazzerini et al., 2018; Qu et al., 2019). In experiments with immunized (Ro/SSA antigens) and non-immunized Cav1.3−/− mice, only immunized Cav1.3−/− mice displayed severe sinus bradycardia, significantly prolonged PR interval and significantly lower fetal parity when compared to non-immunized mice (Karnabi et al., 2011).
Additional experiments with pregnant mice that were injected with positive IgG from human mothers that had children with aCHB showed that the timing of immunization during gestation was important (Mazel et al., 1999). Although pups from the 8 days-, 11 days-, and 16 days-gestation groups showed electrocardiographic symptoms, pups that were at least 11 days along in gestation were more likely to develop a higher degree of sinus bradycardia and PR prolongation (Mazel et al., 1999; Boutjdir, 2000). Hu et al. showed that ICaL was reduced in rabbit SA node cells following superfusion of maternal anti-Ro/SSA positive IgG from mothers who had children with aCHB resulting in slow spontaneous action potentials consistent with sinus bradycardia (Hu et al., 2004). As a potential therapy for aCHB, genetic or drug-induced deactivation of the muscarinic-gated K+ channel in Cav1.3−/− mice has been proposed to allow a net inward current to be maintained to prevent dysfunction of SA node pacemaking activity (Mesirca et al., 2016a). The role of autoimmune cardiac channelopathies involving Cav1.3 in the development of cardiac arrhythmias represents an avenue for future investigation (Capecchi et al., 2019).
A hallmark of aCHB is complete AV block almost always being accompanied by first-, second- or third-degree AV block (Boutjdir, 2000). In prospective studies of pregnancies in anti-Ro/SSA-positive women with no previously affected children, the risk of aCHB was estimated to be 2%–5%, whereas the risk of recurrence in mothers with a previously affected child increases to approximately 15%–20% (Boutjdir, 2000; Qu and Boutjdir, 2012). Treatment for aCHB includes dexamethasone, plasmapheresis, sympathomimetic and in utero cardiac pacing therapy, but none of these are definitive in successfully treating AV block (Boutjdir, 2000).
AV block was successfully induced in isolated Langendorff perfused human fetal hearts by purified IgG fractions and anti-52 kD Ro/SSA antibodies from mothers of children with aCHB (Boutjdir et al., 1997). Perfusion of maternal anti-Ro/SSA positive IgG into rat hearts resulted in the development of bradycardia associated with second-degree AV block, which then degenerated into complete AV block. Experiments have shown that anti-Ro/SSA antibodies led to a reduction of Cav1.3 ICaL by 35% in naive cardiomyocytes (Qu et al., 2019). Additionally, 14% of anti-Ro/SSA antibody-positive IgG was reactive with domain I of the extracellular S5-S6 loop in the pore-forming subunit of Cav1.3 (Karnabi et al., 2010).
Altered electrophysiological activity was noted in mice and rabbits that were immunized with recombinant anti-Ro 52 antigen (Lazzerini et al., 2017; Lazzerini et al., 2019; Qu et al., 2019). Peak ICaL recorded in human fetal cardiomyocytes was significantly inhibited by maternal 52 kD anti-Ro/SSA antibodies. Prior work has also shown that 52 kD anti-Ro/SSA showed more incidence of second-and third-degree AV block. In rabbit AV node cells, action potentials recorded following perfusion of human positive IgG showed significant reduction in the beating heart (Boutjdir, 2000). To culminate these findings, the inclusion of anti-Ro/SSA antibodies in isolated multicellular AV node preparations and Langendorff-perfused whole hearts resulted in bradycardia and AV block (Restivo et al., 2001; Qu and Boutjdir, 2012; Qu et al., 2019). IgG antibodies from mothers with children having aCHB reacted with the sarcolemma of human fetal cardiomyocytes and recognized Cav1.3 subunits, as opposed to anti-Ro/SSA negative IgG antibodies from mothers that had healthy children (Qu et al., 2005a). Collectively, Cav1.3 plays a critical role in the pathogenesis of conduction abnormalities seen in aCHB and can be a preferential target for novel therapies.
Initially, no known human channelopathies were described for Cav1.3 channels or its associated CACNA1D gene (Striessnig et al., 2010). Rinné et al. conducted a study on a three-generation Turkish family where whole genome sequencing was used to identify a variant of CACNA1D associated with SA dysfunction (Figure 3A) (Rinné et al., 2022) Specifically, examination of exon 22 on the CACNA1D gene led to characterization of the p (Arg930His) variant of the CACNA1D gene, which induces the alteration of the Cav1.3 long isoform, thus resulting in loss of function of the channel which leads to SANDD. In this variant, there is a substitution of arginine for a histamine residue at position 930 of the extracellular linker between the S1 and S2 transmembrane segments of domain III on the Cav1.3 channel, which is associated with the channel’s gating properties, resulting in loss of function (Rinné et al., 2022). Later, Baig et al. showed that an alteration in CACNA1D resulted in a glycine residue insertion near the Cav1.3 pore, thus reducing Ca2+ entry, which became an identifying feature of SANDD (Baig et al., 2011). The CACNA1D gene holds significance in Pakistani lineages, where variants have been discovered that lead to inhibited Cav1.3 function and possibly result in SA node dysfunction. Liaqat et al. identified and further characterized the founding variant p (G403_V404insG) and a new missense variant p (A376V), both of which exhibited a phenotype of SANDD (Liaqat et al., 2019).
The CACNA1D gene is also expressed in the AV node, meaning any variants have implication for Cav1.3-related channelopathies in the AV node. In this regard, AV block was reported in members of a Turkish family that expressed the p (Arg930His) variant of the CACNA1D gene (Figure 3A) (Rinné et al., 2022). Scholl et al. were able to show that variants of the CACNA1D gene resulted in altered glycine (G403R) and isoleucine residues (I770M) in the S6 of Cav1.3 domain I and II. This substitution increased channel activation and inhibited inactivation, leading to gain of function in Cav1.3 for patients with aldosteronism; the cardiac implications are yet to be characterized and further clinical studies are warranted (Scholl et al., 2013).
AF is the most common cardiac arrhythmia that contributes substantially to morbidity and mortality. The cellular mechanisms underlying AF are multifactorial. A reduction in ICaL density was initially reported in atrial myocytes from patients with AF (Van Wagoner et al., 1999). Subsequently, atrial samples from patients with AF also showed a significant decrease in Cav1.3 channel mRNA, pointing to a functional role for Cav1.3 in AF development (Gaborit et al., 2005). Cav1.3−/− mice showed atrial electrical dysfunction and predisposition to the development of AF (Mancarella et al., 2008). The electrical abnormalities in the Cav1.3−/− mice were associated with reduced total ICaL density, [Ca2+]i transient, and dysfunctional [Ca2+]i handling and atrial stimulation induced AF in Cav1.3−/− mice (Mancarella et al., 2008). Whole-cell ICaL in atrial myocytes from Cav1.3−/− mice showed a significant depolarizing shift in voltage-dependent activation (Mancarella et al., 2008). In contrast, there were no significant differences in the ICaL recorded from ventricular myocytes between wild-type and null Cav1.3−/− mice (Zhang et al., 2005). These data further support a potentially important role for Cav1.3 in the development of AF.
Studies of molecular mechanisms for the role of Cav1.3 in AF are still nascent. Reports show a reduction in ankyrin-B expression in the atria of patients with documented AF, suggesting that ankyrin-B was required for the membrane targeting and function of Cav1.3 in atrial myocytes (Mesirca et al., 2021). Ankyrin-B was shown to associate with Cav1.3 directly. Loss of ankyrin-B in atrial myocytes resulted in decreased Cav1.3 expression, membrane localization, and function, leading to shortened atrial action potentials and arrhythmias (Cunha et al., 2011; Wolf et al., 2013; Mesirca et al., 2021). In a subsequent study, reduced expression of Cav1.3 paralleled with enhanced expression of Snapin was seen in atrial samples from AF patients (Sun et al., 2017). Upon further investigation, it appeared that Snapin downregulated Cav1.3 membrane expression and promoted its degradation through the ubiquitin-proteasome pathway, thus functioning as a novel regulator for Cav1.3 protein trafficking in atrial myocytes. Further mechanistic insights came from a study from our group, which showed that Cav1.3 and the Ca2+ activated K+ channel SK4 were coupled in the atria and that Cav1.3 deletion led to decreased SK4 mRNA and brain natriuretic peptide secretion from the atria (Srivastava et al., 2017). Regulation of atrial endocrine secretion by Cav1.3 is a possible candidate pathway for generating cardiac arrhythmias such as AF. Particularly, cardiac Ca2+ (Cav1.2/Cav1.3) channel expression and ICaL, along with the action potential durations, were significantly reduced in mice with frameshift human natriuretic peptide precursor A genes, providing further evidence of the significant role of Cav1.3 in AF (Menon et al., 2019). Rose et al. showed that chronic iron overload reduced Cav1.3 mRNA and ICaL, thereby suppressing channel function (Rose et al., 2011). They suggested this mechanism as a possible contributor to the development of AF.
Heart failure (HF) is the heart’s inability to maintain adequate blood circulation to the body’s tissues or to pump out the venous blood returned to it by venous circulation (Dyck et al., 2022). There is substantial evidence that the contractility of failing human hearts is depressed (Houser and Margulies, 2003). As described above briefly, Ca2+ enters the cardiomyocytes via voltage-gated Ca2+ channels upon electrical excitation, which causes further Ca2+ release from the sarcoplasmic reticulum (Figure 3B). This raises free [Ca2+]i, thereby activating the contraction of cardiac tissue (Bers and Despa, 2006). Therefore, abnormalities in basal Ca2+ regulation and dysfunctional Ca2+ signaling cause contractile dysfunction and arrhythmias in pathophysiological conditions such as HF (Houser et al., 2000; Houser and Margulies, 2003).
However, the role of the L-type Ca2+ channels which provide Ca2+ entry to failing cardiomyocytes is unclear and controversial (Beuckelmann et al., 1992; Mukherjee et al., 1998; Benitah et al., 2010). Among L-type Ca2+ channels, Cav1.2 is highly expressed in the ventricles; therefore, research on HF focused on Cav1.2. No research except a recent study by Srivastava et al. has addressed the potential role of Cav1.3 in HF (Srivastava et al., 2020). Published results on Cav1.2 gene expression during HF are inconsistent and show either decreased or insignificant changes in mRNA levels (Takahashi et al., 1992; Schröder et al., 1998; Hong et al., 2012). Furthermore, there are contradictory reports where some studies observed an increase in ICaL during HF, while others reported no changes (Schröder et al., 1998; Chen et al., 2002; Mørk et al., 2007).
Several fetal genes, including the T-type Ca2+ channel Cav3.1, are re-expressed during ventricular remodeling following experimental myocardial infarction in rats (Gidh-Jain et al., 1998; Huang et al., 2000). Atrial natriuretic peptide, hyperpolarization activated cyclic nucleotide gated potassium channel 4, β-myosin heavy chain, skeletal α-actin, and smooth muscle 22α are other genes that are also re-expressed in HF (Kuwahara et al., 2012; Nandi and Mishra, 2015; Sano et al., 2021). Therefore, Cav1.3 was postulated as a possible candidate to be involved in ventricular remodeling in HF (Figure 4) (Nikolaidou et al., 2015; Menon et al., 2019; Zhang et al., 2020). In HF patients, we observed a 6.2-fold increase in Cav1.3 mRNA levels and a 14.9-fold decrease in Cav1.2 mRNA levels in failing hearts compared to healthy human left ventricular control tissue (Srivastava et al., 2020). This alteration was also reported for Cav1.3 protein with western blots in seven failing hearts, demonstrating high expression of Cav1.3 mRNA. A functional re-expression of Cav1.3 might serve as a novel compensatory mechanism for the ventricle to improve cardiac function in HF (Ding et al., 2013; Srivastava et al., 2020; Torrente et al., 2020). Single-cell RNA sequencing data from human dilated and hypertrophic cardiomyopathy demonstrated that Cav1.3 is highly expressed in activated fibroblasts (Chaffin et al., 2022). Thus, it cannot be excluded that the re-expression of Cav1.3 in HF could also originate from cardiac fibroblasts, a common feature of ventricular cardiac remodeling in HF. To elucidate the potential role of Cav1.3 in the ventricles of adult failing hearts, further investigations into its effect on ICaL and inotropy in the ventricle will be required. It will be crucial to delineate the role of Cav1.3 re-expression in HF for developing novel therapeutic interventions.
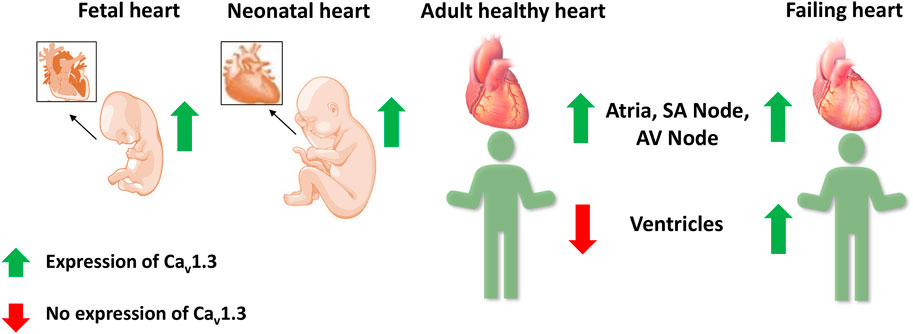
FIGURE 4. Expression of Cav1.3 L-type Ca2+ channel in the fetal, neonatal, adult, and failing heart. Cav1.3 is expressed in the supraventricular and ventricular tissue of the fetal and neonatal hearts. However in adult hearts, it is expressed only in the atria, sinoatrial (SA) node, and atrioventricular (AV) node, but not in the ventricles. Recent evidence suggests a Cav1.3 de novo expression in the ventricles of adult failing hearts.
The physiological role of L-type Ca2+ channels has been studied extensively, aided by generating gene knockout animal models. Given their crucial role in excitation-contraction coupling and maintaining a delicate balance of [Ca2+]i in cardiomyocytes, there is a need for further investigation into these channels in diseased states, particularly Cav1.3, as a therapeutic target. Table 1 reports published literature on the role of Cav1.3 in SA node dysfunction, AV node conduction defects, AF, HF, and autoimmune cardiac channelopathies. However, most studies have not elucidated the molecular mechanisms that underlie disease progression and management. Downregulation or upregulation of Cav1.3 observed in these various diseases will likely facilitate the maintenance of [Ca2+]i and generating and regulating pacemaking. Hence, detailed mechanistic insights into the role of Cav1.3 and its expression and function in the heart will assist in identifying new therapies targeted towards treating the aforementioned cardiovascular diseases.
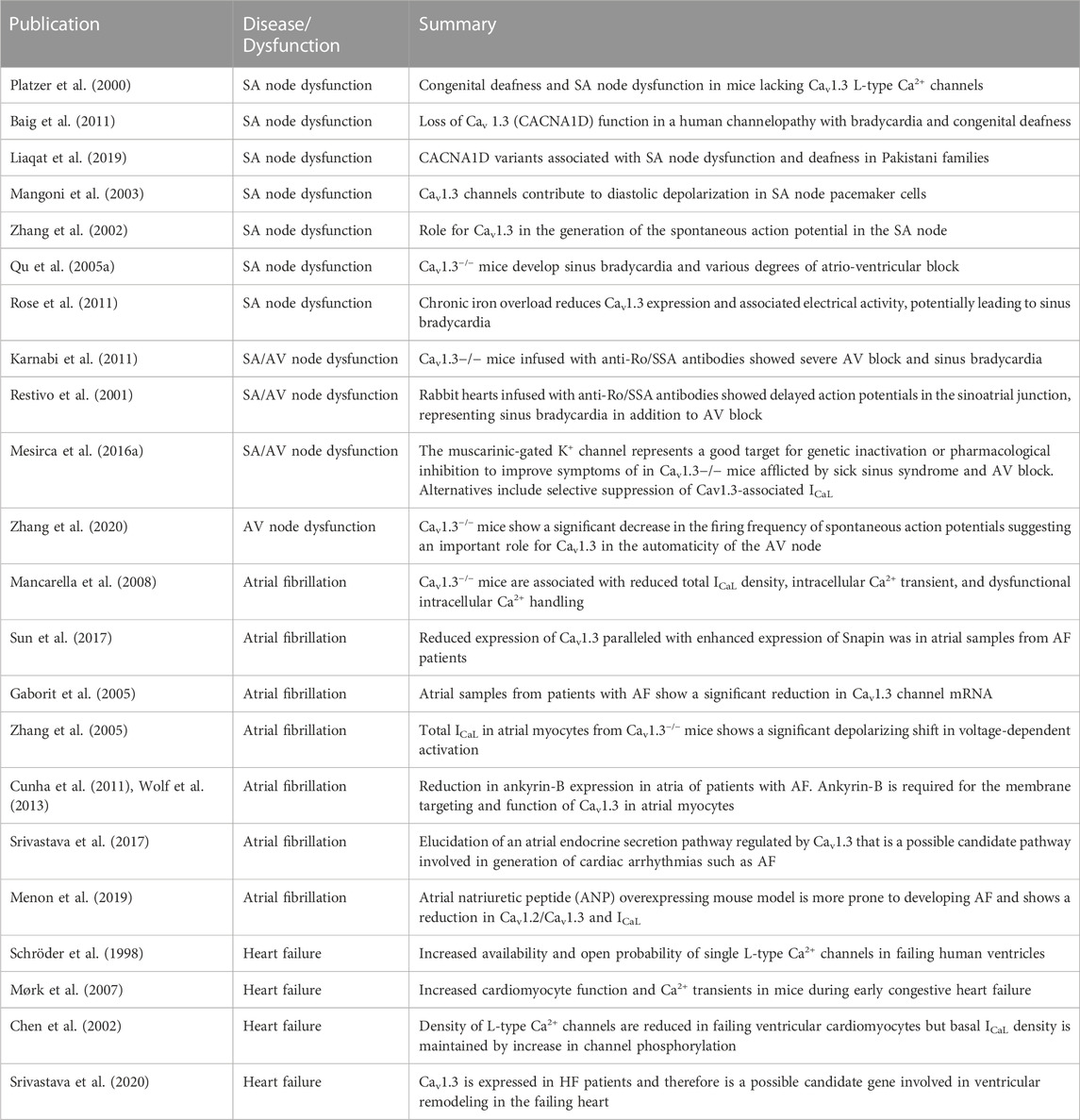
TABLE 1. Summary of published literature on Cav1.3 in SA/AV node dysfunction, atrial fibrillation, and heart failure.
SZ performed a secondary literature review and rewrote the initial draft. US performed the initial literature review and writing of the first draft of the manuscript. YQ reviewed and edited the manuscript with emphasis on the clinical and therapeutic aspects. MC and MB reviewed and rewrote all the manuscript versions. MC and MB contributed equally.
This work was supported by a Merit Review grant I01 BX002137 from Biomedical Laboratory Research & Development Service of Veterans Affairs Office of Research and Development to MB; National Heart, Lung, and Blood Institute 1R01HL164415-01 to MB; and US Department of Defense award number W81XWH-21-1-0424 to MB; and the Canadian Institutes of Health Research grant (MOP-130373) to MC; a US. Department of Defense grant (USAMRAA W81XWH-21-1-0426) to MC
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
aCHB, autoimmune-associated congenital heart block; AF, atrial fibrillation; AV, atrioventricular; CaM, calmodulin; CaMKII, calmodulin-dependent protein kinase II; HF, heart failure; PKA, protein kinase A; PKC, protein kinase C; SA, sinoatrial; SANDD, sinoatrial node dysfunction and deafness.
Baig, S. M., Koschak, A., Lieb, A., Gebhart, M., Dafinger, C., Nürnberg, G., et al. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14 (1), 77–84. doi:10.1038/nn.2694
Banerjee, R., Yoder, J. B., Yue, D. T., Amzel, L. M., Tomaselli, G. F., Gabelli, S. B., et al. (2018). Bilobal architecture is a requirement for calmodulin signaling to Ca(V)1.3 channels. Proc. Natl. Acad. Sci. U. S. A. 115 (13), E3026–e3035. doi:10.1073/pnas.1716381115
Baroudi, G., Qu, Y., Ramadan, O., Chahine, M., and Boutjdir, M. (2006). Protein kinase C activation inhibits Cav1.3 calcium channel at NH2-terminal serine 81 phosphorylation site. Am. J. Physiol. Heart Circ. Physiol. 291 (4), H1614–H1622. doi:10.1152/ajpheart.00095.2006
Ben Johny, M., Yang, P. S., Bazzazi, H., and Yue, D. T. (2013). Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun. 4, 1717. doi:10.1038/ncomms2727
Benitah, J. P., Alvarez, J. L., and Gómez, A. M. (2010). L-type Ca(2+) current in ventricular cardiomyocytes. J. Mol. Cell Cardiol. 48 (1), 26–36. doi:10.1016/j.yjmcc.2009.07.026
Berger, S. M., and Bartsch, D. (2014). The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 357 (2), 463–476. doi:10.1007/s00441-014-1936-3
Berjukow, S., Marksteiner, R., Gapp, F., Sinnegger, M. J., and Hering, S. (2000). Molecular mechanism of calcium channel block by isradipine. Role of a drug-induced inactivated channel conformation. J. Biol. Chem. 275 (29), 22114–22120. doi:10.1074/jbc.M908836199
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415 (6868), 198–205. doi:10.1038/415198a
Bers, D. M., and Despa, S. (2006). Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 100 (5), 315–322. doi:10.1254/jphs.cpj06001x
Beuckelmann, D. J., Näbauer, M., and Erdmann, E. (1992). Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85 (3), 1046–1055. doi:10.1161/01.cir.85.3.1046
Bodi, I., Mikala, G., Koch, S. E., Akhter, S. A., and Schwartz, A. (2005). The L-type calcium channel in the heart: The beat goes on. J. Clin. Invest 115 (12), 3306–3317. doi:10.1172/jci27167
Boutjdir, M., Chen, L., Zhang, Z. H., Tseng, C. E., DiDonato, F., Rashbaum, W., et al. (1997). Arrhythmogenicity of IgG and anti-52-kD SSA/Ro affinity-purified antibodies from mothers of children with congenital heart block. Circ. Res. 80 (3), 354–362. doi:10.1161/01.res.80.3.354
Boutjdir, M. (2000). Molecular and ionic basis of congenital complete heart block. Trends Cardiovasc Med. 10 (3), 114–122. doi:10.1016/s1050-1738(00)00059-1
Capecchi, P. L., Laghi-Pasini, F., El-Sherif, N., Qu, Y., Boutjdir, M., and Lazzerini, P. E. (2019). Autoimmune and inflammatory K(+) channelopathies in cardiac arrhythmias: Clinical evidence and molecular mechanisms. Heart rhythm. 16 (8), 1273–1280. doi:10.1016/j.hrthm.2019.02.017
Catterall, W. A., Lenaeus, M. J., and Gamal El-Din, T. M. (2020). Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154. doi:10.1146/annurev-pharmtox-010818-021757
Chaffin, M., Papangeli, I., Simonson, B., Akkad, A. D., Hill, M. C., Arduini, A., et al. (2022). Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature 608 (7921), 174–180. doi:10.1038/s41586-022-04817-8
Chahine, M., Qu, Y., Mancarella, S., and Boutjdir, M. (2008). Protein kinase C activation inhibits alpha1D L-type Ca channel: A single-channel analysis. Pflugers Arch. 455 (5), 913–919. doi:10.1007/s00424-007-0342-z
Chen, X., Piacentino, V., Furukawa, S., Goldman, B., Margulies, K. B., and Houser, S. R. (2002). L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91 (6), 517–524. doi:10.1161/01.res.0000033988.13062.7c
Cunha, S. R., Hund, T. J., Hashemi, S., Voigt, N., Li, N., Wright, P., et al. (2011). Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124 (11), 1212–1222. doi:10.1161/circulationaha.111.023986
Dick, I. E., Tadross, M. R., Liang, H., Tay, L. H., Yang, W., and Yue, D. T. (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451 (7180), 830–834. doi:10.1038/nature06529
Ding, J., Domes, K., Hofmann, F., and Wegener, J. W. (2013). Truncation of murine CaV1.2 at Asp 1904 increases CaV1.3 expression in embryonic atrial cardiomyocytes. Pflugers Arch. 465 (7), 955–964. doi:10.1007/s00424-012-1212-x
Dyck, J. R. B., Sossalla, S., Hamdani, N., Coronel, R., Weber, N. C., Light, P. E., et al. (2022). Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell Cardiol. 167, 17–31. doi:10.1016/j.yjmcc.2022.03.005
Ferreira, J. C., Mochly-Rosen, D., and Boutjdir, M. (2012). Regulation of cardiac excitability by protein kinase C isozymes. Front. Biosci. Sch. Ed. 4 (2), 532–546. doi:10.2741/283
Gaborit, N., Steenman, M., Lamirault, G., Le Meur, N., Le Bouter, S., Lande, G., et al. (2005). Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 112 (4), 471–481. doi:10.1161/circulationaha.104.506857
Gao, L., Blair, L. A., Salinas, G. D., Needleman, L. A., and Marshall, J. (2006). Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. J. Neurosci. 26 (23), 6259–6268. doi:10.1523/jneurosci.0481-06.2006
Gidh-Jain, M., Huang, B., Jain, P., Gick, G., and El-Sherif, N. (1998). Alterations in cardiac gene expression during ventricular remodeling following experimental myocardial infarction. J. Mol. Cell Cardiol. 30 (3), 627–637. doi:10.1006/jmcc.1997.0628
Hofer, N. T., Pinggera, A., Nikonishyna, Y. V., Tuluc, P., Fritz, E. M., Obermair, G. J., et al. (2021). Stabilization of negative activation voltages of Cav1.3 L-Type Ca(2+)-channels by alternative splicing. Channels (Austin) 15 (1), 38–52. doi:10.1080/19336950.2020.1859260
Hong, T. T., Smyth, J. W., Chu, K. Y., Vogan, J. M., Fong, T. S., Jensen, B. C., et al. (2012). BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart rhythm. 9 (5), 812–820. doi:10.1016/j.hrthm.2011.11.055
Houser, S. R., and Margulies, K. B. (2003). Is depressed myocyte contractility centrally involved in heart failure? Circ. Res. 92 (4), 350–358. doi:10.1161/01.Res.0000060027.40275.A6
Houser, S. R., Piacentino, V., and Weisser, J. (2000). Abnormalities of calcium cycling in the hypertrophied and failing heart. J. Mol. Cell Cardiol. 32 (9), 1595–1607. doi:10.1006/jmcc.2000.1206
Hu, K., Qu, Y., Yue, Y., and Boutjdir, M. (2004). Functional basis of sinus bradycardia in congenital heart block. Circ. Res. 94 (4), e32–e38. doi:10.1161/01.Res.0000121566.01778.06
Huang, B., Qin, D., Deng, L., Boutjdir, M., and N, E.-S. (2000). Reexpression of T-type Ca2+ channel gene and current in post-infarction remodeled rat left ventricle. Cardiovasc Res. 46 (3), 442–449. doi:10.1016/s0008-6363(00)00017-1
Jenkins, M. A., Christel, C. J., Jiao, Y., Abiria, S., Kim, K. Y., Usachev, Y. M., et al. (2010). Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30 (15), 5125–5135. doi:10.1523/jneurosci.4367-09.2010
Karnabi, E., Qu, Y., Mancarella, S., and Boutjdir, M. (2011). Rescue and worsening of congenital heart block-associated electrocardiographic abnormalities in two transgenic mice. J. Cardiovasc Electrophysiol. 22 (8), 922–930. doi:10.1111/j.1540-8167.2011.02032.x
Karnabi, E., Qu, Y., Wadgaonkar, R., Mancarella, S., Yue, Y., Chahine, M., et al. (2010). Congenital heart block: Identification of autoantibody binding site on the extracellular loop (domain I, S5-S6) of alpha(1D) L-type Ca channel. J. Autoimmun. 34 (2), 80–86. doi:10.1016/j.jaut.2009.06.005
Kuwahara, K., Nishikimi, T., and Nakao, K. (2012). Transcriptional regulation of the fetal cardiac gene program. J. Pharmacol. Sci. 119 (3), 198–203. doi:10.1254/jphs.12r04cp
Kuzmenkina, E., Novikova, E., Jangsangthong, W., Matthes, J., and Herzig, S. (2019). Single-Channel resolution of the interaction between C-terminal Ca(V)1.3 isoforms and calmodulin. Biophys. J. 116 (5), 836–846. doi:10.1016/j.bpj.2019.01.025
Lazzerini, P. E., Capecchi, P. L., El-Sherif, N., Laghi-Pasini, F., and Boutjdir, M. (2018). Emerging arrhythmic risk of autoimmune and inflammatory cardiac channelopathies. J. Am. Heart Assoc. 7 (22), e010595. doi:10.1161/jaha.118.010595
Lazzerini, P. E., Capecchi, P. L., Laghi-Pasini, F., and Boutjdir, M. (2017). Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat. Rev. Cardiol. 14 (9), 521–535. doi:10.1038/nrcardio.2017.61
Lazzerini, P. E., Laghi-Pasini, F., Boutjdir, M., and Capecchi, P. L. (2021). Anti-Ro/SSA antibodies and the autoimmune long-QT syndrome. Front. Med. (Lausanne) 8, 730161. doi:10.3389/fmed.2021.730161
Lazzerini, P. E., Laghi-Pasini, F., Boutjdir, M., and Capecchi, P. L. (2019). Cardioimmunology of arrhythmias: The role of autoimmune and inflammatory cardiac channelopathies. Nat. Rev. Immunol. 19 (1), 63–64. doi:10.1038/s41577-018-0098-z
Liaqat, K., Schrauwen, I., Raza, S. I., Lee, K., Hussain, S., Chakchouk, I., et al. (2019). Identification of CACNA1D variants associated with sinoatrial node dysfunction and deafness in additional Pakistani families reveals a clinical significance. J. Hum. Genet. 64 (2), 153–160. doi:10.1038/s10038-018-0542-8
Lu, L., Sirish, P., Zhang, Z., Woltz, R. L., Li, N., Timofeyev, V., et al. (2015). Regulation of gene transcription by voltage-gated L-type calcium channel, Cav1.3. J. Biol. Chem. 290 (8), 4663–4676. doi:10.1074/jbc.M114.586883
Mahapatra, S., Marcantoni, A., Zuccotti, A., Carabelli, V., and Carbone, E. (2012). Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J. Physiol. 590 (20), 5053–5073. doi:10.1113/jphysiol.2012.236729
Maier, L. S., and Bers, D. M. (2002). Calcium, calmodulin, and calcium-calmodulin kinase II: Heartbeat to heartbeat and beyond. J. Mol. Cell Cardiol. 34 (8), 919–939. doi:10.1006/jmcc.2002.2038
Mancarella, S., Yue, Y., Karnabi, E., Qu, Y., El-Sherif, N., and Boutjdir, M. (2008). Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am. J. Physiol. Heart Circ. Physiol. 295 (5), H2017–H2024. doi:10.1152/ajpheart.00537.2008
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. U. S. A. 100 (9), 5543–5548. doi:10.1073/pnas.0935295100
Mazel, J. A., El-Sherif, N., Buyon, J., and Boutjdir, M. (1999). Electrocardiographic abnormalities in a murine model injected with IgG from mothers of children with congenital heart block. Circulation 99 (14), 1914–1918. doi:10.1161/01.cir.99.14.1914
Menon, A., Hong, L., Savio-Galimberti, E., Sridhar, A., Youn, S. W., Zhang, M., et al. (2019). Electrophysiologic and molecular mechanisms of a frameshift NPPA mutation linked with familial atrial fibrillation. J. Mol. Cell Cardiol. 132, 24–35. doi:10.1016/j.yjmcc.2019.05.004
Mesirca, P., Bidaud, I., Briec, F., Evain, S., Torrente, A. G., Le Quang, K., et al. (2016a). G protein-gated IKACh channels as therapeutic targets for treatment of sick sinus syndrome and heart block. Proc. Natl. Acad. Sci. U. S. A. 113 (7), E932–E941. doi:10.1073/pnas.1517181113
Mesirca, P., Bidaud, I., and Mangoni, M. E. (2016b). Rescuing cardiac automaticity in L-type Cav1.3 channelopathies and beyond. J. Physiol. 594 (20), 5869–5879. doi:10.1113/jp270678
Mesirca, P., Fedorov, V. V., Hund, T. J., Torrente, A. G., Bidaud, I., Mohler, P. J., et al. (2021). Pharmacologic approach to sinoatrial node dysfunction. Annu. Rev. Pharmacol. Toxicol. 61, 757–778. doi:10.1146/annurev-pharmtox-031120-115815
Mesirca, P., Torrente, A. G., and Mangoni, M. E. (2015). Functional role of voltage gated Ca(2+) channels in heart automaticity. Front. Physiol. 6, 19. doi:10.3389/fphys.2015.00019
Mitterdorfer, J., Froschmayr, M., Grabner, M., Moebius, F. F., Glossmann, H., and Striessnig, J. (1996). Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel alpha 1 subunits. Biochemistry 35 (29), 9400–9406. doi:10.1021/bi960683o
Mørk, H. K., Sjaastad, I., Sande, J. B., Periasamy, M., Sejersted, O. M., and Louch, W. E. (2007). Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J. Mol. Cell Cardiol. 43 (2), 177–186. doi:10.1016/j.yjmcc.2007.05.004
Mukherjee, R., Hewett, K. W., Walker, J. D., Basler, C. G., and Spinale, F. G. (1998). Changes in L-type calcium channel abundance and function during the transition to pacing-induced congestive heart failure. Cardiovasc Res. 37 (2), 432–444. doi:10.1016/s0008-6363(97)00128-4
Nandi, S. S., and Mishra, P. K. (2015). Harnessing fetal and adult genetic reprograming for therapy of heart disease. J. Nat. Sci. 1 (4), e71.
Nikolaidou, T., Cai, X. J., Stephenson, R. S., Yanni, J., Lowe, T., Atkinson, A. J., et al. (2015). Congestive heart failure leads to prolongation of the PR interval and atrioventricular junction enlargement and ion channel remodelling in the rabbit. PLoS One 10 (10), e0141452. doi:10.1371/journal.pone.0141452
Ortner, N. J. (2023). CACNA1D-Related channelopathies: From hypertension to autism. Handb. Exp. Pharmacol. doi:10.1007/164_2022_626
Platzer, J., Engel, J., Schrott-Fischer, A., Stephan, K., Bova, S., Chen, H., et al. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102 (1), 89–97. doi:10.1016/s0092-8674(00)00013-1
Qu, Y., Baroudi, G., Yue, Y., and Boutjdir, M. (2005a). Novel molecular mechanism involving alpha1D (Cav1.3) L-type calcium channel in autoimmune-associated sinus bradycardia. Circulation 111 (23), 3034–3041. doi:10.1161/circulationaha.104.517326
Qu, Y., Baroudi, G., Yue, Y., El-Sherif, N., and Boutjdir, M. (2005b). Localization and modulation of {alpha}1D (Cav1.3) L-type Ca channel by protein kinase A. Am. J. Physiol. Heart Circ. Physiol. 288 (5), H2123–H2130. doi:10.1152/ajpheart.01023.2004
Qu, Y., and Boutjdir, M. (2012). Pathophysiology of autoimmune-associated congenital heart block. Appl. Cardiopulm. Pathophysiol. 16, 96–112.
Qu, Y., Karnabi, E., Ramadan, O., Yue, Y., Chahine, M., and Boutjdir, M. (2011). Perinatal and postnatal expression of Cav1.3 α1D Ca2⁺ channel in the rat heart. Pediatr. Res. 69 (6), 479–484. doi:10.1203/PDR.0b013e318217a0df
Qu, Y. S., Lazzerini, P. E., Capecchi, P. L., Laghi-Pasini, F., El Sherif, N., and Boutjdir, M. (2019). Autoimmune calcium channelopathies and cardiac electrical abnormalities. Front. Cardiovasc Med. 6, 54. doi:10.3389/fcvm.2019.00054
Ramadan, O., Qu, Y., Wadgaonkar, R., Baroudi, G., Karnabi, E., Chahine, M., et al. (2009). Phosphorylation of the consensus sites of protein kinase A on alpha1D L-type calcium channel. J. Biol. Chem. 284 (8), 5042–5049. doi:10.1074/jbc.M809132200
Restivo, M., Kozhevnikov, D. O., and Boutjdir, M. (2001). Optical mapping of activation patterns in an animal model of congenital heart block. Am. J. Physiol. Heart Circ. Physiol. 280 (4), H1889–H1895. doi:10.1152/ajpheart.2001.280.4.H1889
Rinné, S., Stallmeyer, B., Pinggera, A., Netter, M. F., Matschke, L. A., Dittmann, S., et al. (2022). Whole exome sequencing identifies a heterozygous variant in the Cav1.3 gene CACNA1D associated with familial sinus node dysfunction and focal idiopathic epilepsy. Int. J. Mol. Sci. 23 (22), 14215. doi:10.3390/ijms232214215
Rose, R. A., Sellan, M., Simpson, J. A., Izaddoustdar, F., Cifelli, C., Panama, B. K., et al. (2011). Iron overload decreases CaV1.3-dependent L-type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ. Arrhythm. Electrophysiol. 4 (5), 733–742. doi:10.1161/circep.110.960401
Sano, T., Yasuno, H., and Watanabe, T. (2021). Ion channel mRNA distribution and expression in the sinoatrial node and right atrium of dogs and monkeys. J. Toxicol. Pathol. 34 (3), 223–230. doi:10.1293/tox.2020-0089
Scharinger, A., Eckrich, S., Vandael, D. H., Schönig, K., Koschak, A., Hecker, D., et al. (2015). Cell-type-specific tuning of Cav1.3 Ca(2+)-channels by a C-terminal automodulatory domain. Front. Cell Neurosci. 9, 309. doi:10.3389/fncel.2015.00309
Scholl, U. I., Goh, G., Stölting, G., de Oliveira, R. C., Choi, M., Overton, J. D., et al. (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45 (9), 1050–1054. doi:10.1038/ng.2695
Schröder, F., Handrock, R., Beuckelmann, D. J., Hirt, S., Hullin, R., Priebe, L., et al. (1998). Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 98 (10), 969–976. doi:10.1161/01.cir.98.10.969
Singh, A., Gebhart, M., Fritsch, R., Sinnegger-Brauns, M. J., Poggiani, C., Hoda, J. C., et al. (2008). Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283 (30), 20733–20744. doi:10.1074/jbc.M802254200
Srivastava, U., Aromolaran, A. S., Fabris, F., Lazaro, D., Kassotis, J., Qu, Y., et al. (2017). Novel function of α(1D) L-type calcium channel in the atria. Biochem. Biophys. Res. Commun. 482 (4), 771–776. doi:10.1016/j.bbrc.2016.11.109
Srivastava, U., Jennings-Charles, R., Qu, Y. S., Sossalla, S., Chahine, M., and Boutjdir, M. (2020). Novel re-expression of L-type calcium channel Ca(v)1.3 in left ventricles of failing human heart. Heart rhythm. 17 (7), 1193–1197. doi:10.1016/j.hrthm.2020.02.025
Striessnig, J., Bolz, H. J., and Koschak, A. (2010). Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 460 (2), 361–374. doi:10.1007/s00424-010-0800-x
Striessnig, J., Pinggera, A., Kaur, G., Bock, G., and Tuluc, P. (2014). L-type Ca(2+) channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal 3 (2), 15–38. doi:10.1002/wmts.102
Sun, X. L., Yuan, J. F., Jin, T., Cheng, X. Q., Wang, Q., Guo, J., et al. (2017). Physical and functional interaction of Snapin with Cav1.3 calcium channel impacts channel protein trafficking in atrial myocytes. Cell Signal 30, 118–129. doi:10.1016/j.cellsig.2016.11.019
Takahashi, T., Allen, P. D., Lacro, R. V., Marks, A. R., Dennis, A. R., Schoen, F. J., et al. (1992). Expression of dihydropyridine receptor (Ca2+ channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J. Clin. Invest 90 (3), 927–935. doi:10.1172/jci115969
Tan, B. Z., Jiang, F., Tan, M. Y., Yu, D., Huang, H., Shen, Y., et al. (2011). Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J. Biol. Chem. 286 (49), 42725–42735. doi:10.1074/jbc.M111.265207
Tang, L., Gamal El-Din, T. M., Swanson, T. M., Pryde, D. C., Scheuer, T., Zheng, N., et al. (2016). Structural basis for inhibition of a voltage-gated Ca(2+) channel by Ca(2+) antagonist drugs. Nature 537 (7618), 117–121. doi:10.1038/nature19102
Tokumitsu, H., and Sakagami, H. (2022). Molecular mechanisms underlying Ca(2+)/calmodulin-dependent protein kinase kinase signal transduction. Int. J. Mol. Sci. 23 (19), 11025. doi:10.3390/ijms231911025
Torrente, A. G., Mesirca, P., Bidaud, I., and Mangoni, M. E. (2020). Channelopathies of voltage-gated L-type Cav1.3/α(1D) and T-type Cav3.1/α(1G) Ca(2+) channels in dysfunction of heart automaticity. Pflugers Arch. 472 (7), 817–830. doi:10.1007/s00424-020-02421-1
Torrente, A. G., Mesirca, P., Neco, P., Rizzetto, R., Dubel, S., Barrere, C., et al. (2016). L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc Res. 109 (3), 451–461. doi:10.1093/cvr/cvw006
Toyoda, F., Mesirca, P., Dubel, S., Ding, W. G., Striessnig, J., Mangoni, M. E., et al. (2017). Ca(V)1.3 L-type Ca(2+) channel contributes to the heartbeat by generating a dihydropyridine-sensitive persistent Na(+) current. Sci. Rep. 7 (1), 7869. doi:10.1038/s41598-017-08191-8
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., McCarthy, P. M., and Nerbonne, J. M. (1999). Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 85 (5), 428–436. doi:10.1161/01.res.85.5.428
Vandael, D. H., Mahapatra, S., Calorio, C., Marcantoni, A., and Carbone, E. (2013). Cav1.3 and Cav1.2 channels of adrenal chromaffin cells: Emerging views on cAMP/cGMP-mediated phosphorylation and role in pacemaking. Biochim. Biophys. Acta 1828 (7), 1608–1618. doi:10.1016/j.bbamem.2012.11.013
Wolf, R. M., Glynn, P., Hashemi, S., Zarei, K., Mitchell, C. C., Anderson, M. E., et al. (2013). Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: A computational analysis. Am. J. Physiol. Heart Circ. Physiol. 304 (9), H1253–H1266. doi:10.1152/ajpheart.00734.2012
Zhang, X., Li, Y., Zhang, X., Piacentino, V., Harris, D. M., Berretta, R., et al. (2020). A low voltage activated Ca(2+) current found in a subset of human ventricular myocytes. Channels (Austin) 14 (1), 231–245. doi:10.1080/19336950.2020.1794420
Zhang, Z., He, Y., Tuteja, D., Xu, D., Timofeyev, V., Zhang, Q., et al. (2005). Functional roles of cav1.3(alpha1D) calcium channels in atria: Insights gained from gene-targeted null mutant mice. Circulation 112 (13), 1936–1944. doi:10.1161/circulationaha.105.540070
Keywords: calcium channel, sinoatrial node dysfunction, atrial fibrillation, heart failure, protein kinase regulation
Citation: Zaveri S, Srivastava U, Qu YS, Chahine M and Boutjdir M (2023) Pathophysiology of Cav1.3 L-type calcium channels in the heart. Front. Physiol. 14:1144069. doi: 10.3389/fphys.2023.1144069
Received: 13 January 2023; Accepted: 07 March 2023;
Published: 21 March 2023.
Edited by:
Fabien Brette, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Pietro Mesirca, INSERM U1191 Institut de Génomique Fonctionnelle (IGF), FranceCopyright © 2023 Zaveri, Srivastava, Qu, Chahine and Boutjdir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Boutjdir, bW9oYW1lZC5ib3V0amRpckB2YS5nb3Y=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.