 Jialin Li
Jialin Li Sigen Huang
Sigen Huang Shengyin Liu1,2
Shengyin Liu1,2 Quanliang Liu
Quanliang Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 25 May 2023
Sec. Renal Physiology and Pathophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1118342
The solute-linked carrier 26 (SLC26) protein family is comprised of multifunctional transporters of substrates that include oxalate, sulphate, and chloride. Disorders of oxalate homeostasis cause hyperoxalemia and hyperoxaluria, leading to urinary calcium oxalate precipitation and urolithogenesis. SLC26 proteins are aberrantly expressed during kidney stone formation, and consequently may present therapeutic targets. SLC26 protein inhibitors are in preclinical development. In this review, we integrate the findings of recent reports with clinical data to highlight the role of SLC26 proteins in oxalate metabolism during urolithogenesis, and discuss limitations of current studies and potential directions for future research.
Kidney Stone, also known as urolithiasis, is a common disease that has long threatened human health. Its current prevalence of 14.8% (RULE et al., 2020) is increasing worldwide, with a 5-year recurrence rate of up to 50% (URIBARRI et al., 1989). Kidney stones are a direct cause of substantial morbidity and economic burdens, and may be complicated by hydronephrosis, urinary tract infections, and neoplasia.
The etiology of urolithiasis is complex and multifactorial. Urolithogenesis often results from abnormal urine chemistry (HOWLES and THAKKER, 2020). Predispositions include high urinary concentrations of calcium, oxalate, and uric acid (THONGPRAYOON et al., 2020). When the kidney is affected by intrinsic factors (including genetics, nutrition, metabolic abnormalities, urinary tract obstruction, or infection) and extrinsic factors (including natural and socio-economic environmental stressors), the dynamic balance between urine constituents and stone-inhibiting molecules (citrate or magnesium) is disturbed, leading to the precipitation of solutes, cellular debris, bacteria, and other components (MULAY et al., 2017; HAN et al., 2019).
Transporter proteins play important roles in ion homeostasis that are partially understood, and that deserve further study. SLC26 proteins are categorized as solute carriers, the second largest group of human membrane proteins, and affect various functions (MOUNT and ROMERO, 2004). These multifunctional proteins carry various substrates; primarily chloride, bicarbonate, oxalate, sulphate, formate, and other anions; and thereby regulate ion homeostasis (ALPER and SHARMA, 2013).
The SLC26 family of anion transporters plays important roles in the etiology of urolithiasis, mediating the transport of several key molecules essential to stone formation, including oxalate (CRIVELLI et al., 2020; WANG et al., 2021). For example, hyperoxaluria-induced injury of the renal tubular epithelium stimulates expression and secretion of macromolecules such as cadherin, osteoprotegrin, and collagen, thereby promoting the attachment of nascent crystals to the epithelial surface and consequent crystal nucleation, aggregation, and growth (JOSHI et al., 2015; HOWLES and THAKKER, 2020; WITTING et al., 2021). Here, we review the available evidence of the SLC26 protein family’s relationship to the risk of urolithiasis and detail the studies of its function.
The chemical composition of kidney stones is variable. Calcium, oxalate, and phosphate account for 80% of stones, while the remainder are composed of uric acid, struvite, cystine, insoluble drug molecules (such as the HIV protease inhibitor indinavir) and metabolites (KHAN et al., 2017). As one of the most prevalent disorders of ion metabolism in patients with urolithiasis, oxalate dysregulation has long served as a research priority. Sources of oxalate are classified as exogenous (e.g., dietary intake) and endogenous (e.g., hepatic synthesis).
The chronic dietary intake of foods rich in oxalate, especially in its soluble form (e.g., vegetables, nuts, and some cereals), predisposes to hyperoxaluria (MITCHELL et al., 2019) (CHEN et al., 2001). Gastrointestinal diseases and bariatric surgery may increase enteric epithelial permeability (DUFFEY et al., 2010) (TARPLIN et al., 2015), thus causing oxalate hyperabsorption that leads to hyperoxalemia and consequent hyperoxaluria. In addition, gut dysbiosis has been associated with urolithiasis, and particularly with calcium oxalate (CaOx) stones (MILLER et al., 2019). The gut microbiome communicates with enterocytes to perform various biological processes that may impact oxalate metabolism. For example, Oxalobacter formingenes stimulates enteric oxalate excretion/secretion and also consumes oxalate as a carbon source and thereby reduces oxaluria (HATCH et al., 2006; ARVANS et al., 2017).
Hepatic synthesis is the primary endogenous source of oxalate. Several autosomal recessive disorders, such as mutations of alanine/glyoxylate amino (AGT) transferase (WOOD et al., 2019), glyoxylate/hydroxypyruvate reductase (HOPPE et al., 2009), and the liver-specific mitochondrial enzyme 4-hydroxy-2-ketoglutaratealdolase-1 (4-hydroxy-2-oxoglutaratealdolase, HOGA1) (MONICO et al., 2011) increase hepatic oxalic acid production and raise the risk of urolithiasis.
The SLC26 family is an evolutionarily conserved class of transporter proteins encoded by the APC gene superfamily (ALPER and SHARMA, 2013) in taxonomically diverse organisms. Examples include the bacterial protein SLC26/SulP and the plant/yeast-associated protein SLC26/Sultr. Ten isoforms have been identified in humans (among them, SLC26A10 is a transcribed pseudogene), which are expressed across multiple organ systems (Figure 1). SLC26 proteins operate as anion exchangers that transport an anion that is coupled to another anion gradient, but there are a few exceptions. For example, SLC26A5 (prestin) acts as an electrical stress-sensitive motor protein responsible for the electrokinetic behavior of outer hair cells in an anion-dependent manner (BAVI et al., 2021).
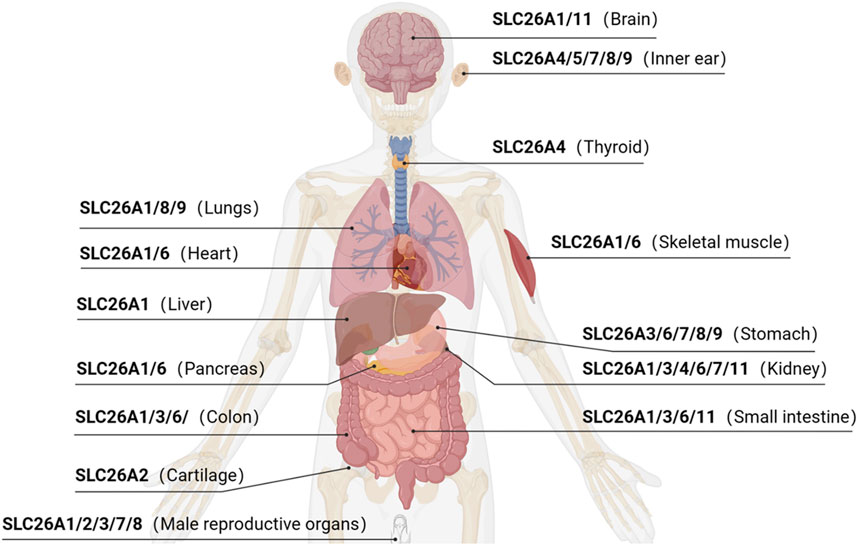
FIGURE 1. Anatomic distribution of SLC26 protein expressions.
SLC26 family members share similar structures, including an N-terminal structural domain, a segment consisting of 10–14 transmembrane segments, a C-terminal sulfate transporter and anti-sigma factor antagonist structural domain (STAS), and a PDZ structural domain (CHANG et al., 2019) (Figure 2). Remarkably, the highest variability and most functionally significant variations of STAS structural domains are located in a defined region designated as the intervening sequence or variable loop found between the first α-helix and the third β-sheet. This loop interacts with the cystic fibrosis transmembrane conductance regulator (R structural domain in the cystic fibrosis transmembrane regulator) and mediates epithelial chloride and bicarbonate transport (MOUNT and ROMERO, 2004). This function may be cross-regulated by SLC26 and related transporters. SLC26 proteins feature monomeric structures, but often act by forming structural and functional dimers in lipid membranes (TOURé, 2019), which may cause structural-functional alterations of SLC26 in vivo.
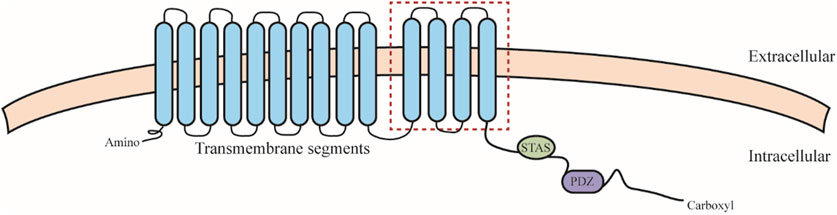
FIGURE 2. SLC26 protein structure. SLC26 including an N-terminal structural domain, a segment consisting of 10–14 transmembrane segments (the red square indicates differences in hydrophobic span that may exist between members), a C-terminal sulfate transporter and anti-sigma factor antagonist structural domain (STAS), and a PDZ structural domain.
Pathogenic mutations have been associated with SLC26A1 (CaOx stones) (WHITTAMORE et al., 2019); SLC26A2 (skeletal deformities and abnormal cartilage development) (ZHENG et al., 2019); SLC26A3 (chloride diarrhea) (KUMAR et al., 2021); SLC26A4 (Pendred syndrome, deafness, enlarged vestibular aqueduct syndrome, and thyroid lesions) (YUAN et al., 2020; TREPICCIONE et al., 2021); SLC26A5 (deafness) (BAVI et al., 2021); SLC26A6 (bicarbonate ion metabolism related diseases) (SONG et al., 2012); SLC26A7 (hypothyroidism and gastric neuroendocrine tumors) (CANGUL et al., 2018; ISHII et al., 2019); SLC26A8 (asthenozoospermia) (GAO et al., 2022); and SLC26A9 (cystic fibrosis) (PINTO et al., 2021). No pathogenic SLC26A11 mutants have been reported (Table 1). Accordingly, all mutations in the SLC26 gene family have been characterized through their corresponding models. In summary, the study of SLC26-mediated molecular pathogenesis can elucidate mechanisms of disease and facilitate the discovery of therapeutic targets.
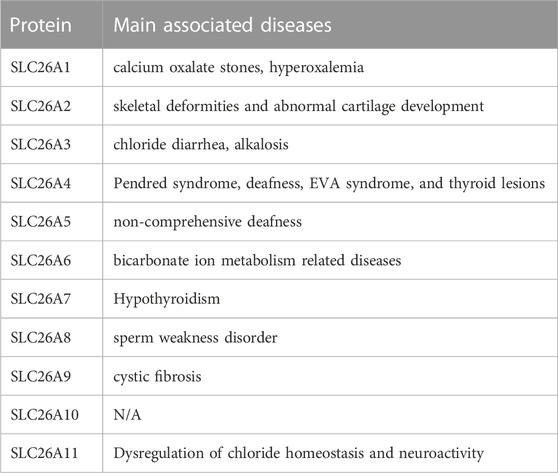
TABLE1. SLC26 family member-related diseases.
The major SLC26 proteins expressed in human renal tissue are SLC26A1, SLC26A3, and SLC26A6. Molecular, cellular, and in vivo studies have confirmed the regulatory role of SLC26 proteins in urolithiasis, as detailed below.
SLC26A1, also known as sulfate anion transporter protein-1 (SAT1), is an epithelial transporter that maintains oxalate and sulfate homeostasis. It is expressed primarily in the kidney, liver, and intestine. SLC26A1 exhibits high homology (78% amino acid identity) and similar tissue distribution between humans and mice (LEE et al., 2005; SEIDLER and NIKOLOVSKA, 2019), suggests the usefulness of animal models as preclinical models for studying SLC26 family proteins. A rat model of CaOx nephrolithiasis showed that SLC26A1 was expressed primarily on the basolateral membranes of renal proximal tubular and enteric epithelia, thus affecting oxalate absorption and excretion (GEE et al., 2016). SLC26A1 deletion alters the dynamic balance between oxalate and sulfate, causing hyperoxalemia and consequent hyperoxaluria, renal calcium deposits, and CaOx urolithiasis, and may also predispose to acetaminophen-induced hepatotoxicity (DAWSON et al., 2010). In addition, intense leukocytic infiltration was detected in the perivascular renal cortex of SLC26A1-deficient mice, suggesting the involvement of inflammatory pathways in this process. Meanwhile, sulfate homeostasis, another important factor in lithogenesis, is also disturbed, shown by hyposulfatemia and hypersulfaturia, which together with disturbances in oxalate metabolism increased the risk of stone formation.
The introduction of gene profiling into clinical practice enabled the identification of SLC26A1abnormalities in patients with kidney stones. Twenty-one clinically significant missense mutations of SLC26A1 were identified in patients with nephrolithiasis (ZIYADOV et al., 2021). Detection of SLC26A1 variant expression and comparison with the NCBI database showed that loss of function was associated with decreased protein stability (no significant difference in the number of transcripts). Notably, sequencing of a specimen from a patient with severe renal calcinosis revealed a substitution of a non-conserved amino acid (M132T) in the transmembrane structural domain of SLC26A1 (DAWSON et al., 2013), thus altering the structure of the hydrophobic region of the third segment, suggesting that the loss of SLC26A1 protein function may lead to hyperoxaluria and urolithiasis. The above evidence suggests that understanding the function of SLC26A1 at the genetic level may be a useful strategy.
SLC26A3 mediates chloride and bicarbonate exchange and regulates the balance between oxalate secretion and reabsorption. It is expressed primarily in the intestine, kidney, and adrenal glands. Generally, all human intestinal segments engage in a dynamic balance of oxalate absorption and secretion to avoid hyperoxalemia. SLC26A3-KO mice showed a 66% reduction in daily urinary oxalate excretion compared to wild-type mice, indicating the importance of the SLC26A3 transporter in intestinal oxalate uptake. Furthermore, in the same study, SLC26A6 levels were increased 3-fold in the duodenum and jejunum, suggesting a possible complementarity between SLC26A3 and SLC26A6 expressions (FREEL et al., 2013). However, another study showed that SLC26A3 overexpression in the murine intestine did not increase oxaluria and also reduced renal CaOx deposition, suggesting a role in preventing rather than promoting urolithiasis (LIU et al., 2021). These lines of evidence suggests that abnormal SLC26A3 expression, a risk factor for urolithiasis, is related to other mechanisms of urolithogenesis in addition to the regulation of oxalate metabolism.
Over the past decade, researchers have suggested that SLC26A3 may serve as a therapeutic target to treat hyperoxaluria and prevent CaOx urolithiasis. CIL et al. (2022) demonstrated that DRAinh-A270 selectively inhibits SLC26A3-mediated chloride/bicarbonate exchange. Notably, the same study reported that the reduced function of SLC26A3 mutants, which may decrease male fertility, may be caused by a weakened interaction between the specific structural domain STAS and the cystic fibrosis transmembrane conductance-regulated channel, thereby affecting chloride transport. To date, no studies have confirmed the role of SLC26A3 in renal physiology, but its role in the regulation of enteric anion exchange suggests that targeting SLC26A3 may represent a novel strategy to prevent CaOx urolithiasis, as it regulates oxalic acid metabolism in the body to reduce the risk of kidney stones caused by hyperoxaluria. However, further studies are needed to confirm this hypothesis.
SLC26A6, also known as PAT-1, is a chloride/oxalate transporter that is highly expressed in the kidney, pancreas, and intestine, and plays an important role in urolithogenesis. MONICO et al. (2008) first identified and characterized a SLC26A6 variant arising from a mutation in the STAS structural domain (D23H/D673N) in hyperoxaluric patients. This change impairs SLC26A6 expression and function, and disrupts the dynamic balance between citrate and oxalate, thereby promoting urolithogenesis (MONICO et al., 2008). This missense mutation is often complicated by hypertension and cystic fibrosis (SHIMSHILASHVILI et al., 2020). These findings have led to a growing interest in the relationship between SLC26A6 and kidney stones. Furthermore, JIANG et al. (2018) showed that renal but not systemic SLC26A6 hyperexpression increased oxalate secretion by renal tubular epithelial cells. In vitro experiments showed that elevated oxalate concentrations stimulated the intracellular production of reactive oxygen species and pro-inflammatory factors, leading to further cellular injury and crystal nucleation (JIANG et al., 2018). This finding also corroborates the above-mentioned involvement of the SLC26 family in inflammatory signaling.
Citrate prevents pathological mineralization of CaOx by inhibiting early nucleation, during the early stages of stone formation, there is often a dysregulation of citrate metabolism. Urinary citrate is reabsorbed mainly through NaDC-1, a Na+-dicarboxylic acid cotransporter in the proximal tubule. Increased renal NaDC-1 activity enhances tubular reabsorption of citrate and facilitates CaOx crystallization (JIANG et al., 2018). OHANA et al. (2013) found that the STAS structural domain of SLC26A6 interacted with the first intracellular loop (ICL1), a functional structural domain of NaDC-1, to inhibit NaDC-1 activity, thereby limiting citrate uptake, suggesting that the STAS-ICL1 interaction underlies the dynamic regulation of citrate/oxalate exchange by SCL26A6/NaDC-1, and that feedback between proteins the two balances their expression and function. This interaction is partially mediated by the scaffolding protein IRBIT through stimulation of the succinate receptor SUCNR-1 (KHAMAYSI et al., 2019).
SLC26A6 expression is regulated by a variety of factors, such as inflammation and miRNA. Protein kinases A (PKA) may positively regulate SLC26A6 levels and alter its intrinsic activity by increasing its expressions on the apical membranes of renal and intestinal epithelia. At the initiation of stone formation, PKA activation not only upregulates SLC26A6 expression, but also enhances the transporter activity of SLC26A2 and SLC26A6 (ARVANS et al., 2020). Similarly, in obesity-induced systemic inflammation, high levels of pro-inflammatory factors can significantly reduce SLC26A6 levels, reversing oxalate transport from net secretion to net absorption, leading to hyperoxalemia/uria, thus promoting urolithogenesis (AMIN et al., 2018). Notably, obesity alters the gut microbiome (MARUVADA et al., 2017). Furthermore, the upregulation of SLC26A6 by short-chain fatty acids (acetate, propionate, and butyrate) produced by gut microbes (LIU et al., 2021) reduced renal CaOx crystals in a rat model; this study suggests a close association between enteric microflora and oxalate metabolism, and also suggests a role of short-chain fatty acid food supplements in the prevention of CaOx urolithiasis.
In vivo studies have shown that SLC26A6 is modulated by molecules other than inflammatory mediators. Urinary glycine concentrations were significantly lower in patients with hyperoxaluria and were associated with downregulated SLC26A6 and NaDC-1 post-transcriptional levels via miRNA-411-3p linking the 3′ end untranslated regions of both mRNAs, thus decreasing urinary oxalate/citrate ratios and ultimately reducing CAOx crystallization in the rat kidney (JUNG et al., 2018). Moreover, SLC26A6 expression may be endocrine-dependent, as it may be regulated by parathyroid hormone (TSENG et al., 2020). In summary, SLC26A6 regulates oxalate metabolism, and loss of function variants promote the formation of kidney stones.
Other proteins of the SLC26 family are closely associated with kidney stones. For example, HIRATA et al. (2012) identified the role of SLC26A5 in CaOx urolithiasis by using a Drosophila model that may facilitate genetic studies regarding renal tubular ion transport and CaOx crystallization. Another study simulated the human physiology of SLC26A5 in the Drosophila stone model, and showed that in addition to transporting oxalate, it also regulates sulphate levels through competitive transport, thereby decreasing urinary calcium salt saturation and reducing CaOx precipitation (LANDRY et al., 2016).
Recent research has elucidated the role of SLC26 proteins in CaOx urolithiasis. These proteins have been increasingly recognized as important transporters that regulate the homeostasis of ions associated with stone development and have become a focus for drug development. SLC26 family proteins are widely expressed in multiple tissues of the gastrointestinal, urinary, skeletal, and reproductive systems. The expressions of SLC26 family members and their physiologic roles vary considerably between species, suggesting different pathogenic mechanisms of SLC26 mutants between humans and animal models (SOLEIMANI, 2013). Abnormalities of urinary ion concentrations, such as hyperoxaluria, are important pathogenic factors (SIENER and Hesse, 2021). The SLC26 family is centrally involved in oxalate and sulfate ion transport in urolithogenesis, and the STAS structural domain is an important site of action.
The study of SLC26 transporter proteins is still incomplete. For example, SLC26A1 is normally expressed in the intestine and kidney and regulates oxalate absorption and secretion; however, a study that demonstrated renal tubular deposition of calcium oxalate in SLC26A1-KO mice could not associate this finding directly to SLC26A1 deficiency (KO et al., 2012). Concomitant urinary pH and osmolarity may be altered through SLC26-mediated transport; however, no studies have been conducted to test this hypothesis. In addition, SLC26 proteins may reduce the risk of hyperoxaluria and stones to a greater degree in female compared to male rats, suggesting sex-based differences in SLC26 family protein expression and activity (ALPER and SHARMA, 2013). Unfortunately, most animal studies of SLC26 have used male mice. On the other hand, the expression patterns of most SLC26 family proteins are still unclear, which may be due to bias caused by poor reproducibility in animal models and low specificity of corresponding antibodies, resulting in divergent results (DAWSON et al., 2010; KO et al., 2012; GEE et al., 2016; WHITTAMORE et al., 2019). However, it is noteworthy that genetic studies of patients and animal models have revealed multiple variants of specific SLC26 proteins, which may serve as drug targets for the prevention of urolithiasis. The development of SLC26 protein-targeting drugs has made some progress, with the identification of small-molecule drug candidates, such as DRAinh-A270, which exhibits protein inhibition in vitro. However, potential side effects and clinical utility are still unknown, and are currently being explored. Future work will advance the understanding of SLC26 proteins and the development of therapeutic strategies based on their multiple physiologic roles.
JL: study design, literature search, and manuscript preparation; SH, SL, XL, and SY: literature search and manuscript preparation; QL: supervision. All authors contributed to the article and approved the submitted version.
We thank Medjaden Inc. for scientific editing of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alper, S. L., and Sharma, A. K. (2013). The SLC26 gene family of anion transporters and channels. Mol. aspects Med. 34 (2-3), 494–515. doi:10.1016/j.mam.2012.07.009
Amin, R., Asplin, J., Jung, D., Bashir, M., Alshaikh, A., Ratakonda, S., et al. (2018). Reduced active transcellular intestinal oxalate secretion contributes to the pathogenesis of obesity-associated hyperoxaluria. Kidney Int. 93 (5), 1098–1107. doi:10.1016/j.kint.2017.11.011
Arvans, D., Alshaikh, A., Bashir, M., Weber, C., and Hassan, H. (2020). Activation of the PKA signaling pathway stimulates oxalate transport by human intestinal Caco2-BBE cells. Am. J. physiology Cell physiology 318 (2), C372–C379. doi:10.1152/ajpcell.00135.2019
Arvans, D., Jung, Y. C., Antonopoulos, D., Koval, J., Granja, I., Bashir, M., et al. (2017). Oxalobacter formigenes-derived bioactive factors stimulate oxalate transport by intestinal epithelial cells. J. Am. Soc. Nephrol. JASN 28 (3), 876–887. doi:10.1681/ASN.2016020132
Bavi, N., Clark, M. D., Contreras, G. F., Shen, R., Reddy, B. G., Milewski, W., et al. (2021). The conformational cycle of prestin underlies outer-hair cell electromotility. Nature 600 (7889), 553–558. doi:10.1038/s41586-021-04152-4
Cangul, H., Liao, X. H., Schoenmakers, E., Kero, J., Barone, S., Srichomkwun, P., et al. (2018). Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI insight 3 (20), e99631. doi:10.1172/jci.insight.99631
Chang, Y. N., Jaumann, E. A., Reichel, K., Hartmann, J., Oliver, D., Hummer, G., et al. (2019). Structural basis for functional interactions in dimers of SLC26 transporters. Nat. Commun. 10 (1), 2032. doi:10.1038/s41467-019-10001-w
Chen, C. L., Fang, H. C., Chou, K. J., Wang, J. S., and Chung, H. M. (2001). Acute oxalate nephropathy after ingestion of star fruit. Am. J. kidney Dis. 37 (2), 418–422. doi:10.1053/ajkd.2001.21333
Cil, O., Chu, T., Lee, S., Haggie, P. M., and Verkman, A. S. (2022). Small-molecule inhibitor of intestinal anion exchanger SLC26A3 for treatment of hyperoxaluria and nephrolithiasis. JCI insight 7 (13), e153359. doi:10.1172/jci.insight.153359
Crivelli, J. J., Mitchell, T., Knight, J., Wood, K. D., Assimos, D. G., Holmes, R. P., et al. (2020). Contribution of dietary oxalate and oxalate precursors to urinary oxalate excretion. Nutr. 13 (1), 62. doi:10.3390/nu13010062
Dawson, P. A., Russell, C. S., Lee, S., McLeay, S. C., van Dongen, J. M., Cowley, D. M., et al. (2010). Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J. Clin. investigation 120 (3), 706–712. doi:10.1172/JCI31474
Dawson, P. A., Sim, P., Mudge, D. W., and Cowley, D. (2013). Human SLC26A1 gene variants: A pilot study. TheScientificWorldJournal 2013, 541710. doi:10.1155/2013/541710
Duffey, B. G., Alanee, S., Pedro, R. N., Hinck, B., Kriedberg, C., Ikramuddin, S., et al. (2010). Hyperoxaluria is a long-term consequence of roux-en-Y gastric bypass: A 2-year prospective longitudinal study. J. Am. Coll. Surg. 211 (1), 8–15. doi:10.1016/j.jamcollsurg.2010.03.007
Freel, R. W., Whittamore, J. M., and Hatch, M. (2013). Transcellular oxalate and Cl-absorption in mouse intestine is mediated by the DRA anion exchanger Slc26a3, and DRA deletion decreases urinary oxalate. Am. J. physiology Gastrointest. liver physiology 305 (7), G520–G527. doi:10.1152/ajpgi.00167.2013
Gao, Y., Wu, H., Xu, Y., Shen, Q., Xu, C., Geng, H., et al. (2022). Novel biallelic mutations in SLC26A8 cause severe asthenozoospermia in humans owing to midpiece defects: Insights into a putative dominant genetic disease. Hum. Mutat. 43 (3), 434–443. doi:10.1002/humu.24322
Gee, H. Y., Jun, I., Braun, D. A., Lawson, J. A., Halbritter, J., Shril, S., et al. (2016). Mutations in SLC26A1 cause nephrolithiasis. Am. J. Hum. Genet. 98 (6), 1228–1234. doi:10.1016/j.ajhg.2016.03.026
Han, S., Zhao, C., Pokhrel, G., Sun, X., Chen, Z., and Xu, H. (2019). Hydroxycitric acid tripotassium inhibits calcium oxalate crystal formation in the Drosophila melanogaster model of hyperoxaluria. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 25, 3662–3667. doi:10.12659/MSM.913637
Hatch, M., Cornelius, J., Allison, M., Sidhu, H., Peck, A., and Freel, R. W. (2006). Oxalobacter sp. reduces urinary oxalate excretion by promoting enteric oxalate secretion. Kidney Int. 69 (4), 691–698. doi:10.1038/sj.ki.5000162
Hirata, T., Cabrero, P., Berkholz, D. S., Bondeson, D. P., Ritman, E. L., Thompson, J. R., et al. (2012). In vivo Drosophilia genetic model for calcium oxalate nephrolithiasis. Am. J. physiology Ren. physiology 303 (11), F1555–F1562. doi:10.1152/ajprenal.00074.2012
Hoppe, B., Beck, B. B., and Milliner, D. S. (2009). The primary hyperoxalurias. Kidney Int. 75 (12), 1264–1271. doi:10.1038/ki.2009.32
Howles, S. A., and Thakker, R. V. (2020). Genetics of kidney stone disease. Nat. Rev. Urol. 17 (7), 407–421. doi:10.1038/s41585-020-0332-x
Ishii, J., Suzuki, A., Kimura, T., Tateyama, M., Tanaka, T., Yazawa, T., et al. (2019). Congenital goitrous hypothyroidism is caused by dysfunction of the iodide transporter SLC26A7. Commun. Biol. 2, 270. doi:10.1038/s42003-019-0503-6
Jiang, H., Pokhrel, G., Chen, Y., Wang, T., Yin, C., Liu, J., et al. (2018). High expression of SLC26A6 in the kidney may contribute to renal calcification via an SLC26A6-dependent mechanism. PeerJ 6, e5192. doi:10.7717/peerj.5192
Joshi, S., Clapp, W. L., Wang, W., and Khan, S. R. (2015). Osteogenic changes in kidneys of hyperoxaluric rats. Biochimica biophysica acta 1852 (9), 2000–2012. doi:10.1016/j.bbadis.2015.06.020
Jung, D., Alshaikh, A., Ratakonda, S., Bashir, M., Amin, R., Jeon, S., et al. (2018). Adenosinergic signaling inhibits oxalate transport by human intestinal Caco2-BBE cells through the A2B adenosine receptor. Am. J. physiology Cell physiology 315 (5), C687–C698. doi:10.1152/ajpcell.00024.2017
Khamaysi, A., Anbtawee-Jomaa, S., Fremder, M., Eini-Rider, H., Shimshilashvili, L., Aharon, S., et al. (2019). Systemic succinate homeostasis and local succinate signaling affect blood pressure and modify risks for calcium oxalate lithogenesis. J. Am. Soc. Nephrol. JASN 30 (3), 381–392. doi:10.1681/ASN.2018030277
Khan, S. R., Pearle, M. S., Robertson, W. G., Gambaro, G., Canales, B. K., Doizi, S., et al. (2017). Kidney stones. Nat. Rev. Dis. Prim. 3, 17001. doi:10.1038/nrdp.2017.1
Ko, N., Knauf, F., Jiang, Z., Markovich, D., and Aronson, P. S. (2012). Sat1 is dispensable for active oxalate secretion in mouse duodenum. Am. J. physiology Cell physiology 303 (1), C52–C57. doi:10.1152/ajpcell.00385.2011
Kumar, A., Priyamvada, S., Ge, Y., Jayawardena, D., Singhal, M., Anbazhagan, A. N., et al. (2021). A novel role of SLC26A3 in the maintenance of intestinal epithelial barrier integrity. Gastroenterology 160 (4), 1240–1255.e3. doi:10.1053/j.gastro.2020.11.008
Landry, G. M., Hirata, T., Anderson, J. B., Cabrero, P., Gallo, C. J. R., Dow, J. A. T., et al. (2016). Sulfate and thiosulfate inhibit oxalate transport via a dPrestin (Slc26a6)-dependent mechanism in an insect model of calcium oxalate nephrolithiasis. Am. J. physiology Ren. physiology 310 (2), F152–F159. doi:10.1152/ajprenal.00406.2015
Lee, A., Dawson, P. A., and Markovich, D. (2005). NaSi-1 and sat-1: Structure, function and transcriptional regulation of two genes encoding renal proximal tubular sulfate transporters. Int. J. Biochem. Cell Biol. 37 (7), 1350–1356. doi:10.1016/j.biocel.2005.02.013
Liu, Y., Jin, X., Ma, Y., Jian, Z., Wei, Z., Xiang, L., et al. (2021). Short-chain fatty acids reduced renal calcium oxalate stones by regulating the expression of intestinal oxalate transporter SLC26A6. mSystems 6 (6), e0104521. doi:10.1128/mSystems.01045-21
Maruvada, P., Leone, V., Kaplan, L. M., and Chang, E. B. (2017). The human microbiome and obesity: Moving beyond associations. Cell host microbe 22 (5), 589–599. doi:10.1016/j.chom.2017.10.005
Miller, A. W., Choy, D., Penniston, K. L., and Lange, D. (2019). Inhibition of urinary stone disease by a multi-species bacterial network ensures healthy oxalate homeostasis. Kidney Int. 96 (1), 180–188. doi:10.1016/j.kint.2019.02.012
Mitchell, T., Kumar, P., Reddy, T., Wood, K. D., Knight, J., Assimos, D. G., et al. (2019). Dietary oxalate and kidney stone formation. Am. J. physiology Ren. physiology 316 (3), F409–F413. doi:10.1152/ajprenal.00373.2018
Monico, C. G., Rossetti, S., Belostotsky, R., Cogal, A. G., Herges, R. M., Seide, B. M., et al. (2011). Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin. J. Am. Soc. Nephrol. CJASN 6 (9), 2289–2295. doi:10.2215/CJN.02760311
Monico, C. G., Weinstein, A., Jiang, Z., Rohlinger, A. L., Cogal, A. G., Bjornson, B. B., et al. (2008). Phenotypic and functional analysis of human SLC26A6 variants in patients with familial hyperoxaluria and calcium oxalate nephrolithiasis. Am. J. kidney Dis. 52 (6), 1096–1103. doi:10.1053/j.ajkd.2008.07.041
Mount, D. B., and Romero, M. F. (2004). The SLC26 gene family of multifunctional anion exchangers. Pflugers Archiv Eur. J. physiology 447 (5), 710–721. doi:10.1007/s00424-003-1090-3
Mulay, S. R., Eberhard, J. N., Desai, J., Marschner, J. A., Kumar, S. V. R., Weidenbusch, M., et al. (2017). Hyperoxaluria requires TNF receptors to initiate crystal adhesion and kidney stone disease. J. Am. Soc. Nephrol. JASN 28 (3), 761–768. doi:10.1681/ASN.2016040486
Ohana, E., Shcheynikov, N., Moe, O. W., and Muallem, S. (2013). SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J. Am. Soc. Nephrol. JASN 24 (10), 1617–1626. doi:10.1681/ASN.2013010080
Pinto, M. C., Quaresma, M. C., Silva, I. A. L., Railean, V., Ramalho, S. S., and Amaral, M. D. (2021). Synergy in cystic fibrosis therapies: Targeting SLC26A9. Int. J. Mol. Sci. 22 (23), 13064. doi:10.3390/ijms222313064
Rule, A. D., Lieske, J. C., and Paisjr, V. M. (2020). Management of kidney stones in 2020. Jama 323 (19), 1961–1962. doi:10.1001/jama.2020.0662
Seidler, U., and Nikolovska, K. (2019). Slc26 family of anion transporters in the gastrointestinal tract: Expression, function, regulation, and role in disease. Compr. Physiol. 9 (2), 839–872. doi:10.1002/cphy.c180027
Shimshilashvili, L., Aharon, S., Moe, O. W., and Ohana, E. (2020). Novel human polymorphisms define a key role for the slc26a6-STAS domain in protection from Ca2+-oxalate lithogenesis. Front. Pharmacol. 11, 405. doi:10.3389/fphar.2020.00405
Siener, R., and Hesse, A. (2021). Effect of black tea consumption on urinary risk factors for kidney stone formation. Nutrients 13 (6), 4434. doi:10.3390/nu13124434
Soleimani, M. (2013). SLC26 Cl-/HCO3- exchangers in the kidney: Roles in health and disease. Kidney Int. 84 (4), 657–666. doi:10.1038/ki.2013.138
Song, Y., Yamamoto, A., Steward, M. C., Ko, S. B. H., Stewart, A. K., Soleimani, M., et al. (2012). Deletion of SLC26A6 alters the stoichiometry of apical Cl-/HCO-3 exchange in mouse pancreatic duct. Am. J. physiology Cell physiology 303 (8), C815–C824. doi:10.1152/ajpcell.00151.2012
Tarplin, S., Ganesan, V., and Monga, M. (2015). Stone formation and management after bariatric surgery. Nat. Rev. Urol. 12 (5), 263–270. doi:10.1038/nrurol.2015.67
Thongprayoon, C., Krambeck, A. E., and Rule, A. D. (2020). Determining the true burden of kidney stone disease. Nat. Rev. Nephrol. 16 (12), 736–746. doi:10.1038/s41581-020-0320-7
Touré, A. (2019). Importance of SLC26 transmembrane anion exchangers in sperm post-testicular maturation and fertilization potential. Front. Cell Dev. Biol. 7, 230. doi:10.3389/fcell.2019.00230
Trepiccione, F., Suzumoto, Y., Perna, A., and Capasso, G. (2021). Pure Gitelman-like syndrome secondary to SLC26A4 (pendrin) mutation. Kidney Int. 100 (4), 947–948. doi:10.1016/j.kint.2021.06.020
Tseng, Y. S., Wu, W. B., Chen, Y., Lo Yang, F., and Ma, M. C. (2020). Small intestine resection increases oxalate and citrate transporter expression and calcium oxalate crystal formation in rat hyperoxaluric kidneys. Clin. Sci. 134 (19), 2565–2580. London, England : 1979. doi:10.1042/CS20200973
Uribarri, J., Oh, M. S., and Carroll, H. J. (1989). The first kidney stone. Ann. Intern. Med. 111 (12), 1006–1009. doi:10.7326/0003-4819-111-12-1006
Wang, Z., Zhang, Y., Zhang, J., Deng, Q., and Liang, H. (2021). Recent advances on the mechanisms of kidney stone formation (Review). Int. J. Mol. Med. 48 (2), 149. doi:10.3892/ijmm.2021.4982
Whittamore, J. M., Stephens, C. E., and Hatch, M. (2019). Absence of the sulfate transporter SAT-1 has no impact on oxalate handling by mouse intestine and does not cause hyperoxaluria or hyperoxalemia. Am. J. physiology Gastrointest. liver physiology 316 (1), G82–g94. doi:10.1152/ajpgi.00299.2018
Witting, C., Langman, C. B., Assimos, D., Baum, M. A., Kausz, A., Milliner, D., et al. (2021). Pathophysiology and treatment of enteric hyperoxaluria. Clin. J. Am. Soc. Nephrol. CJASN 16 (3), 487–495. doi:10.2215/CJN.08000520
Wood, K. D., Holmes, R. P., Erbe, D., Liebow, A., Fargue, S., and Knight, J. (2019). Reduction in urinary oxalate excretion in mouse models of Primary Hyperoxaluria by RNA interference inhibition of liver lactate dehydrogenase activity. Biochimica biophysica acta Mol. basis Dis. 1865 (9), 2203–2209. doi:10.1016/j.bbadis.2019.04.017
Yuan, J., Song, Y., Pan, W., Li, Y., Xu, Y., Xie, M., et al. (2020). LncRNA SLC26A4-AS1 suppresses the MRN complex-mediated DNA repair signaling and thyroid cancer metastasis by destabilizing DDX5. Oncogene 39 (43), 6664–6676. doi:10.1038/s41388-020-01460-3
Zheng, C., Lin, X., Xu, X., Wang, C., Zhou, J., Gao, B., et al. (2019). Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. EBioMedicine 40, 695–709. doi:10.1016/j.ebiom.2019.01.010
Keywords: SLC26, kidney stone, oxalate, hyperoxaluria, oxalate metabolism
Citation: Li J, Huang S, Liu S, Liao X, Yan S and Liu Q (2023) SLC26 family: a new insight for kidney stone disease. Front. Physiol. 14:1118342. doi: 10.3389/fphys.2023.1118342
Received: 07 December 2022; Accepted: 18 May 2023;
Published: 25 May 2023.
Edited by:
Carolyn Mary Ecelbarger, Georgetown University, United StatesReviewed by:
Linto Thomas, University of South Florida, United StatesCopyright © 2023 Li, Huang, Liu, Liao, Yan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanliang Liu, bGl1cXVhbmxpYW5nMjAwOEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.