 Héctor Carbajal-Contreras
Héctor Carbajal-Contreras Gerardo Gamba
Gerardo Gamba María Castañeda-Bueno
María Castañeda-Bueno
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 15 February 2023
Sec. Renal Physiology and Pathophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1100522
This article is part of the Research Topic 30th Anniversary of the Molecular Cloning and Identification of the Na-Cl Cotransporter, NCC View all 7 articles
The activity of the Na+-Cl- cotransporter (NCC) in the distal convoluted tubule (DCT) is finely tuned by phosphorylation networks involving serine/threonine kinases and phosphatases. While much attention has been paid to the With-No-lysine (K) kinase (WNK)- STE20-related Proline Alanine rich Kinase (SPAK)/Oxidative Stress Responsive kinase 1 (OSR1) signaling pathway, there remain many unanswered questions regarding phosphatase-mediated modulation of NCC and its interactors. The phosphatases shown to regulate NCC’s activity, directly or indirectly, are protein phosphatase 1 (PP1), protein phosphatase 2A (PP2A), calcineurin (CN), and protein phosphatase 4 (PP4). PP1 has been suggested to directly dephosphorylate WNK4, SPAK, and NCC. This phosphatase increases its abundance and activity when extracellular K+ is increased, which leads to distinct inhibitory mechanisms towards NCC. Inhibitor-1 (I1), oppositely, inhibits PP1 when phosphorylated by protein kinase A (PKA). CN inhibitors, like tacrolimus and cyclosporin A, increase NCC phosphorylation, giving an explanation to the Familial Hyperkalemic Hypertension-like syndrome that affects some patients treated with these drugs. CN inhibitors can prevent high K+-induced dephosphorylation of NCC. CN can also dephosphorylate and activate Kelch-like protein 3 (KLHL3), thus decreasing WNK abundance. PP2A and PP4 have been shown in in vitro models to regulate NCC or its upstream activators. However, no studies in native kidneys or tubules have been performed to test their physiological role in NCC regulation. This review focuses on these dephosphorylation mediators and the transduction mechanisms possibly involved in physiological states that require of the modulation of the dephosphorylation rate of NCC.
The Na+-Cl- cotransporter (NCC) is the most important apical route for Na+ reabsorption in the distal convoluted tubule (DCT) (Castañeda-Bueno and Ellison, 2022). The phosphorylation of its N-terminus by serine/threonine kinases positively modulates its activity in a critical manner, as shown by multiple in vitro, ex vivo and in vivo models (Pacheco-Alvarez et al., 2006; Yang et al., 2013; Cheng et al., 2015; Penton et al., 2016) and enables regulation of blood pressure and K+ homeostasis. This is most clear in the context of Familial Hyperkalemic Hypertension (FHHt), a monogenic disease in which the pathological increase in this transporter’s phosphorylation causes hypertension and hyperkalemia that are fully treatable with thiazides, the diuretics that inhibit NCC (Wilson et al., 2001).
Mutations in FHHt cause the gain-of-function of “With-No-lysine (K)” kinases 1 and 4 (WNK1 and WNK4) (Wilson et al., 2001), a pair of kinases that phosphorylate and activate the Ste20-related proline/alanine-rich kinase (SPAK) and oxidative stress responsive kinase 1 (OSR1) (Vitari et al., 2005), the enzymes that directly phosphorylate and activate NCC (Piechotta et al., 2002). WNK kinases are regulated by many mechanisms, including direct inhibitory Cl− binding (Piala et al., 2014; Bazúa-Valenti et al., 2015) and ubiquitylation by the KLHL3-CUL3 E3 ligase complex (Ohta et al., 2013; Shibata et al., 2013; Wakabayashi et al., 2013; Wu and Peng, 2013; McCormick et al., 2014). They are also modulated by other kinases, such as PKA, PKC (Castañeda-Bueno et al., 2017), SGK1 (Ring et al., 2007; Rozansky et al., 2009), and tyrosine kinases (Lin et al., 2015).
The phosphorylation of NCC is not only changed in FHHt, but also in response to homeostatic challenges. For instance, hormones such as adrenergic agonists (Mu et al., 2011; Terker et al., 2014; Poulsen et al., 2021), antidiuretic hormone (ADH) (Pedersen et al., 2010; Saritas et al., 2013), and parathyroid hormone (PTH) (Penton et al., 2019), as well as changes in the extracellular concentration of electrolytes such as K+ (Vallon et al., 2009; Sorensen et al., 2013; Terker et al., 2015, 2016) and Ca2+ (Bazúa-Valenti et al., 2018; Bahena-Lopez et al., 2022a) can modify the activity of NCC through changes in its phosphorylation. Much effort has been devoted to the understanding of the kinases involved in this signaling pathway, yet comparatively little is understood of the opposite processes mediated by protein phosphatases (PPs). These enzymes catalyze dephosphorylation and, thus, inactivation of the pathway. While their importance is very clear, something as fundamental as the bona fide substrates of the PPs involved remain uncertain, partly due to these enzymes’ catalytic subunits apparent promiscuity, and partly due to the conflicting results provided by different in vitro models and the use of pharmacological unspecific inhibitors of PPs in many studies.
Potential involvement in this signaling pathway has been suggested for Protein Phosphatase 1 (PP1), Protein Phosphatase 2A (PP2A), Calcineurin (CN, also known as Protein Phosphatase 3 or 2B, PP3 or PP2B), and Protein Phosphatase 4 (PP4). PPs are presumably central in the inhibition of NCC in response to high K+ intake, as their inhibition clearly blunts this phenomenon (Shoda et al., 2017; Shoda et al., 2020; Kortenoeven et al., 2021). PPs are also important players in cAMP signaling transduction (Penton et al., 2019), and possibly in other signaling pathways. This review aims to summarize the evidence available for the physiological roles played by PPs in the regulation of NCC and their modulation by different stimuli, along with controversies on the physiological relevance of said mechanisms.
PP1 is a ubiquitous enzyme in eukaryotic cells that has been estimated to catalyze up to one-third of serine/threonine dephosphorylation reactions (Cohen, 2002). Four human genes encode PP1 isoforms: PPP1CA (PP1α), PPP1CB (PP1β), PPP1CC (PP1γ1), and PPP1CCB (PP1γ2) (Zhang et al., 1993; Cohen, 2002). These enzymes associate with other proteins to produce PP1 holoenzymes. These regulatory subunits, of which there are dozens, confer catalytic regulation, specificity, and compartmentalization of the dephosphorylation processes mediated by PP1 (Ceulemans et al., 2002; Virshup and Shenolikar, 2009). The involvement of PP1 in the regulation of NCC and the WNK-SPAK/OSR1 signaling pathway has been studied by multiple groups.
NKCC1 and NKCC2, which are the most closely related transporters to NCC in our proteome, possess a conserved PP1-binding site (Darman et al., 2001; Gagnon and Delpire, 2010). This site has been validated in NKCC1. Mutations in this site can prevent inactivation of the transporter by precluding the binding of PP1 (Darman et al., 2001; Gagnon and Delpire, 2010). However, a putative PP1 binding site is absent in NCC, which begs the question: is PP1 involved in the direct dephosphorylation of NCC?
Evidence supporting that PP1 can directly dephosphorylate NCC includes PP1-NCC binding by yeast-two-hybrid assays, coimmunoprecipitation, and phosphatase assays of PP1 on the N-terminus of NCC (Picard et al., 2014). However, experiments from our group, performed in HEK293 cells, which allow for the dissection of individual components of this signaling system (albeit, through overexpression), have recently questioned such direct effect. In our hands, PP1 regulation of NCC cannot occur directly on SPAK or NCC in these cells, but strictly requires of WNK4 to occur (Carbajal-Contreras et al., 2022) (Figure 1). Thus, direct regulation of PP1 by NCC is still in question, and could either occur only through WNK4, or depend on a regulatory subunit that mediates interaction with NCC in vivo. Such seems to be the case for other reported substrates of SPAK, such as CFTR and NBCe1-b, which utilize the IP3 receptor binding protein released with IP3 (IRBIT) to allow PP1 binding (Yang et al., 2011; Vachel et al., 2018).
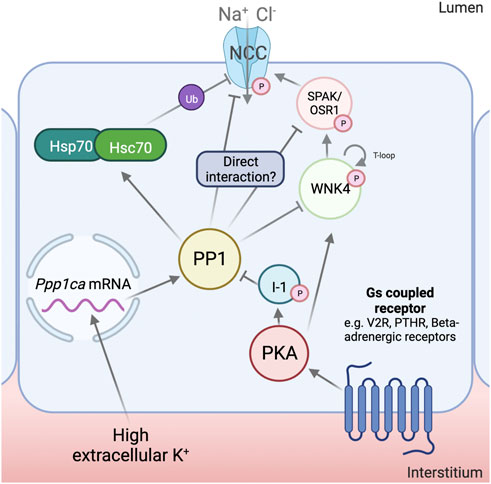
FIGURE 1. Mechanisms for NCC regulation by Protein Phosphatase 1. Protein phosphatase 1 (PP1) has been shown to increase its mRNA and protein abundance in response to high extracellular K+. Inhibitor-1 (I1), in response to cAMP increasing stimuli, is phosphorylated by Protein Kinase A (PKA). This, in turn, results in the inhibition of PP1 activity. PP1 has been shown to regulate With-No-Lysine (K) kinase (WNK4), Ste20-related Proline/Alanine rich Kinase (SPAK) and Na+-Cl- Cotransporter (NCC) phosphorylation, though it is at the moment uncertain if these interactions are physiological and the degree to which they contribute to NCC regulation. PP1 has also been shown to dephosphorylate and activate the 70 kDa Heat-Shock Protein (Hsp70) and Heat-Shock Cognate 70 (Hsc70), which have been proposed to play an important role in the ubiquitylation and degradation of NCC. Created with QmlvcmVuZGVyLmNvbQ==.
The par excellence homeostatic challenge associated with the dephosphorylation of NCC is an increase in extracellular K+ (Sorensen et al., 2013). This condition increases the transcript levels of Ppp1ca, but not of Ppp1cb or Ppp1cc, in mouse tubule suspensions (Kortenoeven et al., 2021). Protein abundance of PP1α also increases under these conditions, as well as in the kidney cortex of mice fed a high K+ diet for 4 days. Oppositely, low extracellular K+ decreases the abundance of this enzyme. This points towards a physiological role for PP1 upon changes in extracellular K+ in the renal tubules. It remains to be determined whether the reported changes occur in the DCT and play a role in NCC regulation by K+.
An unequivocal role for PP1 in the regulation of NCC was uncovered by mice lacking I1 (Picard et al., 2014), which have reduced levels of phosphorylated NCC and blood pressure. I1, encoded by PPP1R1A, is a regulatory subunit of PP1 that, when phosphorylated by PKA at T35, functions as a potent, specific inhibitor of PP1 (Nimmo and Cohen, 1978). The absence of I1 was then shown to reduce the response to cAMP increasing stimuli in the DCT. The main event regulating the increase in NCC phosphorylation under these conditions was proposed to be the direct downregulation of the transporter’s dephosphorylation rate, since the increase in pNCC with forskolin/IBMX was attenuated in I1−/− mice, and no changes in the phosphorylation of SPAK/OSR1 could be observed (Penton et al., 2019). However, this contrasts with findings by other groups using other cAMP-increasing stimuli, such as desmopressin (Saritas et al., 2013), where changes in SPAK/OSR1 phosphorylation could be observed. An attenuated response to beta-adrenergic agonists (which increase intracellular cAMP) has been reported in OSR1−/−, but not SPAK−/− mice, which further supports a role for kinase activation in this pathway (Terker et al., 2014). It is also important to note that I1 is a validated substrate of CN, which could serve as a link between PP1 and this other phosphatase (Mulkey et al., 1994). CN activation could also activate PP1 and, thus, inactivate NCC indirectly.
PP1 could also modulate the activity of NCC through the dephosphorylation of Hsp70 and Hsc70, which, in this state, could induce the ubiquitylation and degradation of NCC (Kortenoeven et al., 2021). This has been shown to occur under high extracellular K+ conditions in tubule suspensions, and to be prevented by the specific PP1 inhibitor, tautomycetin, and by the Hsp70 inhibitor, Ver155008, but not by the PP2A and CN inhibitor deltamethrin, the CN inhibitor FK506, or the PP4 inhibitor fostriecin (Kortenoeven et al., 2021). However, Hsp70 has many molecular targets, and its inhibition could have many unintended side-effects. Also, previous work has shown that NCC phosphorylation, which is known to be diminished by high extracellular K+, is inversely correlated with its stability and ubiquitylation (Yang et al., 2013; Rosenbaek et al., 2014).
PP1 may also play a role in the regulation of SPAK/OSR1. Results obtained in X. laevis oocytes have shown PP1-dependent inhibition of SPAK that depends on other scaffolding proteins, such as AATYK or NKCC1 itself (Gagnon et al., 2007; Gagnon and Delpire, 2010). SPAK has also been shown to coimmunoprecipitate with IRBIT and PP1 in other studies, though no regulation of dephosphorylation activity towards SPAK was analyzed (Yang et al., 2011; Vachel et al., 2018).
Regarding the upstream WNK kinases, there is also evidence for interaction with PP1. WNK1’s T-loop was shown to, in vitro, be dephosphorylated by PP1γ, which diminishes its catalytic activity (Zagórska et al., 2007). Another work, dedicated to screening and validating novel substrates of PP1α, found a positive hit with WNK1 (Hendrickx et al., 2009). In the DCT, this interaction might only be relevant for the kidney-specific WNK1 (KS-WNK1), as the catalytically active long isoform, L-WNK1, is thought to be physiologically irrelevant in these cells (Castañeda-Bueno et al., 2012; Thomson et al., 2020; Bahena-Lopez et al., 2022b).
Finally, WNK4, the master kinase regulating NCC, has a PP1-binding site in its C-terminus that has been validated in vitro (Murillo-de-Ozores et al., 2018). When this site is deleted or mutated, there is a noticeable gain of function of this kinase, accompanied by an increase in the phosphorylation of its T-loop. PP1 overexpression induces the dephosphorylation of wildtype WNK4, but not of kinase mutants lacking this binding site (Murillo-de-Ozores et al., 2018). Additionally, results from our group show that forskolin-induced I1 phosphorylation induces a dramatic increase in the phosphorylation of WNK4, which is blunted in the PP1 binding-deficient WNK4 mutant. Our results in HEK293 cells show that WNK4’s dephosphorylation, but not that of NCC or SPAK, can be regulated by I1 inhibition of PP1 (Carbajal-Contreras et al., 2022). This hints towards a central role of WNK4 as a mediator of stimuli that act through this phosphatase.
Despite the previously mentioned evidence, it must be noted that PP1-mediated regulation of the WNK4-SPAK/OSR1-NCC pathway remains polemical, as negative results have also been reported. For instance, mice fed a high K+ gavage showed no changes in the abundance of PP1, phosphorylated (inhibited) PP1, or I1 in their kidneys; and the administration of tautomycetin (PP1 inhibitor) to these mice did not prevent the dephosphorylation of NCC (Shoda et al., 2017). This suggested that, at least in an acute setting, PP1 plays a minor role in the regulation of pNCC by K+. Furthermore, at least two groups found that, in kidney slices, treatment with calyculin A can indeed increase the phosphorylation of NCC, but cannot prevent the transporter’s dephosphorylation under high K+ conditions (Penton et al., 2016; Mukherjee et al., 2021). As will be discussed later, an effect of PP1 inhibition in kidney slices during high-K+ conditions was only observed in mice genetically lacking CN activity in the DCT (Banki et al., 2021). Thus, PP1 may be involved in the dephosphorylation of the WNK4 SPAK/OSR1-NCC pathway under some, but not all situations.
CN inhibitors (CNIs) are widely used in the clinic as immunosuppressants. In some patients, the side effects of CNIs resemble the alterations observed in FHHt, including hypertension, hyperkalemia, metabolic acidosis and hypercalciuria. Thus, several groups have tested the effects of CNIs on the activity of NCC in rodents. Treatment with CNIs, including tacrolimus (or FK506) and cyclosporin, produces FHHt-like alterations and promotes NCC phosphorylation (Hoorn et al., 2011; Melnikov et al., 2011). Interestingly, the hypertensive effect of tacrolimus treatment was not observed in NCC knockout mice or in thiazide-treated mice, suggesting a central role for NCC modulation in the development of these side effects. The effects of tacrolimus in NCC phosphorylation, plasma K+, and blood pressure levels were attenuated in tubule specific FKBP12−/− mice (Lazelle et al., 2016). FKBP12 is the 12 kDa FK506-binding protein that inhibits CN when bound to tacrolimus. Thus, it was shown that the effects of tacrolimus on NCC are mediated by direct inhibition of CN in the renal epithelium.
These observations have been extended to patients treated with CNIs. Increased urinary vesicle abundance of NCC and pNCC in patients treated with CNIs has been observed (Tutakhel et al., 2017). This has also been shown to correlate with increased blood pressure (Rojas-Vega et al., 2015) and with thiazide responsiveness (Tutakhel et al., 2017). While a greater response to thiazides has been observed for patients treated with tacrolimus (Hoorn et al., 2011), an actual benefit for NCC blockade with thiazides compared to other antihypertensive drugs remains controversial (Moes et al., 2017), and further studies are required to test this hypothesis in the clinical setting.
The effects of CNIs on NCC in animal models are accompanied by changes in the expression or phosphorylation levels of known regulators of NCC activity. For instance, increased renal expression levels of WNK4 (Hoorn et al., 2011; Melnikov et al., 2011; Ishizawa et al., 2019), WNK1 (Ishizawa et al., 2019), SPAK (Hoorn et al., 2011), and increased phosphorylation of SPAK (Ishizawa et al., 2019) and KLHL3 (Ishizawa et al., 2019) have been observed. Changes in abundance of WNK kinases may be secondary to changes in phosphorylation of KLHL3 at S433, given that phosphorylation of this site has been shown to prevent binding to WNK kinases (Shibata et al., 2014). This phosphorylation was shown to be modulated by CN in in vitro experiments (Ishizawa et al., 2019). Moreover, KLHL3-S433 phosphorylation was shown to increase in mice on a low K+ diet (Ishizawa et al., 2016) and to decrease in response to high extracellular K+ in an in vitro cell model (Ishizawa et al., 2019). This latter effect was prevented by tacrolimus. Accordingly, it was proposed that high extracellular K+ may activate CN to promote KLHL3 dephosphorylation, leading to decreased WNK4 and WNK1 levels (Figure 2).
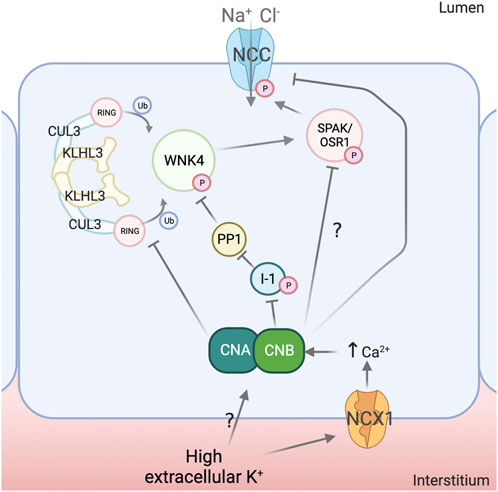
FIGURE 2. Mechanisms for NCC regulation by calcineurin. Calcineurin (CN) activity has been implicated in the dephosphorylation of the Na+-Cl- Cotransporter (NCC) in response to high extracellular K+. Modulation of the Na+-Ca2+ exchanger (NCX1) activity by K+ has been postulated as a mechanism to induce changes intracellular Ca2+ concentration and thus promote CN activation, although this mechanism requires confirmation and additional mechanisms may also participate. Evidence suggests that CN can directly dephosphorylate NCC and also affect upstream elements of the signaling pathway. For instance, CN appears to dephosphorylate Kelch-Like 3 (KLHL3), which leads to increased ubiquitylation of With-No-Lysine (K) kinase 4 (WNK4) and Kidney-Specific With-No-Lysine (K) kinase 1 (KS-WNK1) and, thus, decreased activation of Ste20-related Proline/Alanine rich Kinase (SPAK) and Oxidative Stress Responsive kinase 1 (OSR1). Inhibitor 1 (I1) has also been shown to be a substrate of CN and, consequently, this protein may function as a node linking CN activation to Protein phosphatase 1 (PP1) activation, with the implications that this may have in the DCT (see Figure 1). Created with QmlvcmVuZGVyLmNvbQ==.
There is also evidence of direct dephosphorylation of NCC by CN. Lazelle et al. showed that in cells expressing constitutively active SPAK, dephosphorylation of NCC induced by depolarization with BaCl2 was prevented with tacrolimus (Lazelle et al., 2016). Shoda et al. proposed that CN mediates direct dephosphorylation of NCC in response to acute increases in extracellular K+ (Shoda et al., 2017). In mice that were given an oral K+ load via gavage, rapid NCC dephosphorylation was completely prevented by pre-treatment with tacrolimus or with the calmodulin inhibitor W7. No changes in WNK4, SPAK or pSPAK were observed, suggesting a direct effect on NCC phosphorylation. However, this conclusion must be taken with reserve given that, in another study with a similar experimental setting, no measurable decrease in pSPAK/OSR1 levels was observed through Western blot of whole kidney lysates, but could be clearly noted in DCT cells by immunofluorescence (Mukherjee et al., 2021).
Given that CN is regulated by Ca2+, a link between high extracellular K+ and intracellular Ca2+ was proposed to be mediated by the Na+-Ca2+ exchanger NCX1, expressed in the basolateral membrane of the DCT. This antiporter might work in reverse mode and transport Ca2+ into the cell with high K+-induced depolarization of the DCT (Shoda et al., 2020). In HEK293 cells, Ca2+ and CN-dependent dephosphorylation of NCC in response to high extracellular K+ was observed. This was prevented by knockdown of NCX1 or by treatment with the NCX1 inhibitor SEA0400. Similarly, in mice, administration of SEA0400 1 hour prior to an acute oral K+ load prevented NCC dephosphorylation. In contrast to the results obtained in the aforementioned in vivo experiments, tacrolimus was unable to prevent NCC dephosphorylation in kidney slices incubated in high K+ solutions. It remains to be shown, however, if tacrolimus can effectively inhibit CN in this system, as low drug permeability may be a plausible explanation for these inconsistencies. Supporting this, it was shown that treatment with tacrolimus of kidney tubules in suspension results in a clear increase in NCC phosphorylation (Tutakhel et al., 2017). Moreover, genetic disruption of CN activity coupled to pharmacologic disruption of PP1 activity significantly prevented NCC dephosphorylation by high K+ in kidney slices (Banki et al., 2021).
Finally, the role of CN in NCC regulation was more recently addressed with an inducible, DCT-specific knockout mouse model for the B1 subunit of CN (Banki et al., 2021). CN normally functions as a heterodimer formed by an A subunit that provides catalytic activity and a calmodulin-binding site, and a Ca2+-binding regulatory B subunit. Both subunits are necessary for full phosphatase activity. Whereas the A subunit is encoded by three separate genes (PPP3CA, PPP3CB, and PPP3CC), the B subunit is encoded by two genes (PPP3R1 and PPP3R2). Only the B1 isoform (CaNB1, encoded by PPP3R1) appears to be normally expressed in the DCT (Chen et al., 2021). Thus, by knocking out CaNB1 in the DCT, CN activity was disrupted.
Surprisingly, 3 weeks after disruption of CaNB1 only hypomagnesemia and metabolic acidosis were observed, but no hypertension, hyperkalemia, hypercalciuria, or increased pNCC levels. Hypomagnesemia was associated with decreased expression of proteins involved in transcellular Mg2+ transport. Several DCT-enriched molecules were downregulated at the mRNA and protein levels, like NCC, TRPM6, KS-WNK1, and specially parvalbumin, whose mRNA and protein levels were drastically reduced. Interestingly, pNCC levels were similar to those of wild type animals, despite lower total NCC levels, suggesting that reduction of CN activity may indeed lead to increased NCC phosphorylation. Supporting this, tacrolimus treatment did not increase pNCC levels in the knockouts. It was proposed that the discrepancy between the effects observed with genetic disruption of CaNB1 and pharmacologic inhibition of CN could be due to either incomplete suppression of CN activity by drugs or to the length of the suppression of activity, as most previous pharmacologic studies were performed with treatment in shorter timeframes (Hoorn et al., 2011; Lazelle et al., 2016; Shoda et al., 2017; Ishizawa et al., 2019). It was concluded that disruption of CN activity strongly affects the transcriptome and the proteome of the DCT.
PP2A was proposed to regulate the activity of NKCC1 through work done in Calu-3 cells (Liedtke et al., 2005). This has not been explored in further works.
PP4 overexpression in X. laevis oocytes reduced NCC activity. The use of NCC phosphoablative or phosphomimetic mutants abolished this effect (Glover et al., 2010). It was not explored if this occurs directly or through upstream mediators. This work also showed enriched PP4 expression in the DCT through immunohistochemistry, although this is not evident at the transcript level (Chen et al., 2021). The physiological relevance of these findings has not been further tested.
The works reviewed here address the role of serine-threonine phosphatases in the signaling pathways implicated in NCC modulation and particularly highlight PP1 and CN as relevant modulators in these pathways. However, more physiologically accurate models ought to be studied to better understand the actual relevance of some of the findings. For instance, knock-in models for protein phosphatases’ binding sites of proposed interactors, or unbiased techniques with samples derived from DCT cells to study the true interactors of PPs involved in NCC regulation could allow further dissection of the pathways. In addition, it is very likely that the PPs reviewed here play other yet unknown roles in the cell biology of the DCT. These other roles are also likely to influence epithelial transport processes and thus, their characterization will be necessary to fully understand the impact of PPs activity in the function of the DCT.
HC-C, GG, and MC-B wrote the manuscript.
HC-C is a doctoral student from the “Programa de Estudios Combinados en Medicina (PECEM)”, at the Universidad Nacional Autónoma de México (UNAM)” and received a fellowship from CONACyT. Research performed by GG is supported by CONACyT Mexico, grant no. A1-S-8290. Research performed by MC-B is supported by CONACyT Mexico, grant no. 101720.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor DE declared past co-authorships with the authors.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bahena-Lopez J. P., Gamba G., Castañeda-Bueno M. (2022a). WNK1 in the kidney. Curr. Opin. Nephrol. Hypertens. 31 (5), 471–478. doi:10.1097/MNH.0000000000000820
Bahena-Lopez J. P., Rojas-Vega L., Chávez-Canales M., Bazua-Valenti S., Bautista-Pérez R., Lee J.-H., et al. (2022b). Glucose/fructose delivery to the distal nephron activates the sodium-chloride cotransporter via the calcium-sensing receptor. J. Am. Soc. Nephrol. 34, 55–72. doi:10.1681/ASN.2021121544
Banki E., Fisi V., Moser S., Wengi A., Carrel M., Loffing-Cueni D., et al. (2021). Specific disruption of calcineurin-signaling in the distal convoluted tubule impacts the transcriptome and proteome, and causes hypomagnesemia and metabolic acidosis. Kidney Int. 100 (4), 850–869. doi:10.1016/J.KINT.2021.06.030
Bazúa-Valenti S., Chávez-Canales M., Rojas-Vega L., González-Rodríguez X., Vázquez N., Rodríguez-Gama A., et al. (2015). The effect of WNK4 on the Na+-Cl-cotransporter is modulated by intracellular chloride. J. Am. Soc. Nephrol. 26 (8), 1781–1786. doi:10.1681/ASN.2014050470
Bazúa-Valenti S., Rojas-Vega L., Castañeda-Bueno M., Barrera-Chimal J., Bautista R., Cervantes-Pérez L. G., et al. (2018). The calcium-sensing receptor increases activity of the renal NCC through the WNK4-SPAK pathway. J. Am. Soc. Nephrol. 29 (7), 1838–1848. doi:10.1681/ASN.2017111155
Carbajal-Contreras H., Murillo-de-Ozores A. R., Magaña G. R., Bourqui L., Vázquez N. H., Loffing J., et al. (2022). PKA Regulates the WNK-SPAK-NCC/NKCC2 Signaling Pathway Through Phosphorylation of WNK4 and I-1 [Abstract]. J Am Soc Nephrol 33, 136–137.
Castañeda-Bueno M., Arroyo J. P., Zhang J., Puthumana J., Yarborough O., Shibata S., et al. (2017). Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. U. S. A. 114 (5), E879–E886. doi:10.1073/PNAS.1620315114
Castañeda-Bueno M., Cervantes-Peŕez L. G., Vaźquez N., Uribe N., Kantesaria S., Morla L., et al. (2012). Activation of the renal Na+:Cl-cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. U. S. A. 109 (20), 7929–7934. doi:10.1073/PNAS.1200947109
Castañeda-Bueno M., Ellison D. H. (2022). “Blood pressure effects of sodium transport along the distal nephron,” in Kidney international (Netherlands: Elsevier B.V). doi:10.1016/j.kint.2022.09.009
Ceulemans H., Stalmans W., Bollen M. (2002). Regulator-driven functional diversification of protein phosphatase-1 in eukaryotic evolution. BioEssays News Rev. Mol. Cell. Dev. Biol. 24 (4), 371–381. doi:10.1002/BIES.10069
Chen L., Chou C.-L., Knepper M. A. (2021). A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J. Am. Soc. Nephrol. JASN 32 (4), 897–912. doi:10.1681/ASN.2020101406
Cheng L., Wu Q., Kortenoeven M. L. A., Pisitkun T., Fenton R. A. (2015). A systems level analysis of vasopressin-mediated signaling networks in kidney distal convoluted tubule cells. Sci. Rep. 5, 12829. doi:10.1038/SREP12829
Cohen P. T. W. (2002). Protein phosphatase 1--targeted in many directions. J. Cell Sci. 115 (2), 241–256. doi:10.1242/JCS.115.2.241
Darman R. B., Flemmer A., Forbush B. (2001). Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J. Biol. Chem. 276 (37), 34359–34362. doi:10.1074/JBC.C100368200
Gagnon K. B., Delpire E. (2010). Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: The N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for ste20-related proline/alanine-rich kinase (SPAK) and PP1. J. Biol. Chem. 285 (19), 14115–14121. doi:10.1074/JBC.M110.112672
Gagnon K. B. E., England R., Diehl L., Delpire E. (2007). Apoptosis-associated tyrosine kinase scaffolding of protein phosphatase 1 and SPAK reveals a novel pathway for Na-K-2C1 cotransporter regulation. Am. J. Physiology - Cell Physiology 292 (5), C1809–C1815. doi:10.1152/ajpcell.00580.2006
Glover M., Zuber A. M., Figg N., O’Shaughnessy K. M. (2010). The activity of the thiazide-sensitive Na(+)-Cl(-) cotransporter is regulated by protein phosphatase PP4. Can. J. Physiology Pharmacol. 88 (10), 986–995. doi:10.1139/Y10-080
Hendrickx A., Beullens M., Ceulemans H., den Abt T., van Eynde A., Nicolaescu E., et al. (2009). Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol. 16 (4), 365–371. doi:10.1016/J.CHEMBIOL.2009.02.012
Hoorn E. J., Walsh S. B., McCormick J. A., Fürstenberg A., Yang C. L., Roeschel T., et al. (2011). The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat. Med. 17 (10), 1304–1309. doi:10.1038/NM.2497
Ishizawa K., Wang Q., Li J., Yamazaki O., Tamura Y., Fujigaki Y., et al. (2019). Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc. Natl. Acad. Sci. U. S. A. 116 (8), 3155–3160. doi:10.1073/PNAS.1817281116
Ishizawa K., Xu N., Loffing J., Lifton R. P., Fujita T., Uchida S., et al. (2016). Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem. Biophysical Res. Commun. 480 (4), 745–751. doi:10.1016/J.BBRC.2016.10.127
Kortenoeven M. L. A., Esteva-Font C., Dimke H., Poulsen S. B., Murali S. K., Fenton R. A. (2021). High dietary potassium causes ubiquitin-dependent degradation of the kidney sodium-chloride cotransporter. J. Biol. Chem. 297 (2), 100915. doi:10.1016/J.JBC.2021.100915
Lazelle R. A., McCully B. H., Terker A. S., Himmerkus N., Blankenstein K. I., Mutig K., et al. (2016). Renal deletion of 12 kDa FK506-binding protein attenuates tacrolimus-induced hypertension. J. Am. Soc. Nephrol. 27 (5), 1456–1464. doi:10.1681/ASN.2015040466
Liedtke C. M., Wang X., Smallwood N. D. (2005). Role for protein phosphatase 2A in the regulation of Calu-3 epithelial Na+-K+-2Cl-type 1 co-transport function. J. Biol. Chem. 280 (27), 25491–25498. doi:10.1074/JBC.M504473200
Lin D. H., Yue P., Yarborough O., Scholl U. I., Giebisch G., Lifton R. P., et al. (2015). Src-family protein tyrosine kinase phosphorylates WNK4 and modulates its inhibitory effect on KCNJ1 (ROMK). Proc. Natl. Acad. Sci. U. S. A. 112 (14), 4495–4500. doi:10.1073/pnas.1503437112
McCormick J. A., Yang C. L., Zhang C., Davidge B., Blankenstein K. I., Terker A. S., et al. (2014). Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J. Clin. Investigation 124 (11), 4723–4736. doi:10.1172/JCI76126
Melnikov S., Mayan H., Uchida S., Holtzman E. J., Farfel Z. (2011). Cyclosporine metabolic side effects: Association with the WNK4 system. Eur. J. Clin. Investigation 41 (10), 1113–1120. doi:10.1111/J.1365-2362.2011.02517.X
Moes A. D., Hesselink D. A., van den Meiracker A. H., Zietse R., Hoorn E. J. (2017). Chlorthalidone versus amlodipine for hypertension in kidney transplant recipients treated with tacrolimus: A randomized crossover trial. J. Natl. Kidney Found. 69 (6), 796–804. doi:10.1053/J.AJKD.2016.12.017
Mu S. Y., Shimosawa T., Ogura S., Wang H., Uetake Y., Kawakami-Mori F., et al. (2011). Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat. Med. 17 (5), 573–580. doi:10.1038/NM.2337
Mukherjee A., Yang C. L., McCormick J. A., Martz K., Sharma A., Ellison D. H. (2021). Roles of WNK4 and SPAK in K+-mediated dephosphorylation of the NaCl cotransporter. Am. J. Physiology. Ren. Physiology 320 (5), F719–F733. doi:10.1152/AJPRENAL.00459.2020
Mulkey R. M., Endo S., Shenolikar S., Malenka R. C. (1994). Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369 (6480), 486–488. doi:10.1038/369486A0
Murillo-de-Ozores A. R., Rodríguez-Gama A., Bazúa-Valenti S., Leyva-Ríos K., Vázquez N., Pacheco-Álvarez D., et al. (2018). C-terminally truncated, kidney-specific variants of the WNK4 kinase lack several sites that regulate its activity. J. Biol. Chem. 293 (31), 12209–12221. doi:10.1074/JBC.RA118.003037
Nimmo G. A., Cohen P. (1978). The regulation of glycogen metabolism. Phosphorylation of inhibitor-1 from rabbit skeletal muscle, and its interaction with protein phosphatases-III and -II. Eur. J. Biochem. 87 (2), 353–365. doi:10.1111/J.1432-1033.1978.TB12384.X
Ohta A., Schumacher F. R., Mehellou Y., Johnson C., Knebel A., Macartney T. J., et al. (2013). The CUL3–KLHL3 E3 ligase complex mutated in gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 451 (1), 111–122. doi:10.1042/BJ20121903
Pacheco-Alvarez D., San Cristóbal P., Meade P., Moreno E., Vazquez N., Muñoz E., et al. (2006). The Na+:Cl-cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281 (39), 28755–28763. doi:10.1074/jbc.M603773200
Pedersen N. B., Hofmeister M. v., Rosenbaek L. L., Nielsen J., Fenton R. A. (2010). Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 78 (2), 160–169. doi:10.1038/KI.2010.130
Penton D., Czogalla J., Wengi A., Himmerkus N., Loffing-Cueni D., Carrel M., et al. (2016). Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl- -dependent and independent mechanisms. J. Physiology 594 (21), 6319–6331. doi:10.1113/JP272504
Penton D., Moser S., Wengi A., Czogalla J., Rosenbaek L. L., Rigendinger F., et al. (2019). Protein phosphatase 1 inhibitor–1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J. Am. Soc. Nephrol. 30 (5), 737–750. doi:10.1681/ASN.2018050540
Piala A. T., Moon T. M., Akella R., He H., Cobb M. H., Goldsmith E. J. (2014). Chloride sensing by WNK1 kinase involves inhibition of autophosphorylation. Sci. Signal 7 (324), 1–21. doi:10.1126/scisignal.2005050.Chloride
Picard N., Trompf K., Yang C. L., Miller R. L., Carrel M., Loffing-Cueni D., et al. (2014). Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J. Am. Soc. Nephrol. 25 (3), 511–522. doi:10.1681/ASN.2012121202
Piechotta K., Lu J., Delpire E. (2002). Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 277 (52), 50812–50819. doi:10.1074/jbc.M208108200
Poulsen S. B., Cheng L., Penton D., Kortenoeven M. L. A., Matchkov V. v., Loffing J., et al. (2021). Activation of the kidney sodium chloride cotransporter by the β2-adrenergic receptor agonist salbutamol increases blood pressure. Kidney Int. 100 (2), 321–335. doi:10.1016/J.KINT.2021.04.021
Ring A. M., Leng Q., Rinehart J., Wilson F. H., Kahle K. T., Hebert S. C., et al. (2007). An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc. Natl. Acad. Sci. U. S. A. 104 (10), 4025–4029. doi:10.1073/PNAS.0611728104
Rojas-Vega L., Jiménez-Vega A. R., Bazúa-Valenti S., Arroyo-Garza I., Jiménez J. V., Gómez-Ocádiz R., et al. (2015). Increased phosphorylation of the renal Na+-Cl-cotransporter in male kidney transplant recipient patients with hypertension: A prospective cohort. Am. J. Physiology. Ren. Physiology 309 (10), F836–F842. doi:10.1152/AJPRENAL.00326.2015
Rosenbaek L. L., Kortenoeven M. L. A., Aroankins T. S., Fenton R. A. (2014). Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J. Biol. Chem. 289 (19), 13347–13361. doi:10.1074/JBC.M113.543710
Rozansky D. J., Cornwall T., Subramanya A. R., Rogers S., Yang Y. F., David L. L., et al. (2009). Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J. Clin. Investigation 119 (9), 2601–2612. doi:10.1172/JCI38323
Saritas T., Borschewski A., McCormick J. A., Paliege A., Dathe C., Uchida S., et al. (2013). SPAK differentially mediates vasopressin effects on sodium cotransporters. J. Am. Soc. Nephrol. 24 (3), 407–418. doi:10.1681/ASN.2012040404
Shibata S., Arroyo J. P., Castañeda-Bueno M., Puthumana J., Zhang J., Uchida S., et al. (2014). Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc. Natl. Acad. Sci. U. S. A. 111 (43), 15556–15561. doi:10.1073/PNAS.1418342111
Shibata S., Zhang J., Puthumana J., Stone K. L., Lifton R. P. (2013). Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. U. S. A. 110 (19), 7838–7843. doi:10.1073/PNAS.1304592110/-/DCSUPPLEMENTAL/PNAS.201304592SI
Shoda W., Nomura N., Ando F., Mori Y., Mori T., Sohara E., et al. (2017). Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int. 91 (2), 402–411. doi:10.1016/J.KINT.2016.09.001
Shoda W., Nomura N., Ando F., Tagashira H., Iwamoto T., Ohta A., et al. (2020). Sodium-calcium exchanger 1 is the key molecule for urinary potassium excretion against acute hyperkalemia. PloS One 15 (6), e0235360. doi:10.1371/JOURNAL.PONE.0235360
Sorensen M. v., Grossmann S., Roesinger M., Gresko N., Todkar A. P., Barmettler G., et al. (2013). Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 83 (5), 811–824. doi:10.1038/KI.2013.14
Terker A. S., Yang C. L., McCormick J. A., Meermeier N. P., Rogers S. L., Grossmann S., et al. (2014). Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertens. (Dallas, Tex 64 (1), 178–184. doi:10.1161/HYPERTENSIONAHA.114.03335
Terker A. S., Zhang C., Erspamer K. J., Gamba G., Yang C. L., Ellison D. H. (2016). Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 89 (1), 127–134. doi:10.1038/KI.2015.289
Terker A. S., Zhang C., McCormick J. A., Lazelle R. A., Zhang C., Meermeier N. P., et al. (2015). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21 (1), 39–50. doi:10.1016/J.CMET.2014.12.006
Thomson M. N., Cuevas C. A., Bewarder T. M., Dittmayer C., Miller L. N., Si J., et al. (2020). WNK bodies cluster WNK4 and SPAK/OSR1 to promote NCC activation in hypokalemia. Am. J. Physiology. Ren. Physiology 318 (1), F216–F228. doi:10.1152/AJPRENAL.00232.2019
Tutakhel O. A. Z., Moes A. D., Valdez-Flores M. A., Kortenoeven M. L. A., Vrie M. v. d., Jelen S., et al. (2017). NaCl cotransporter abundance in urinary vesicles is increased by calcineurin inhibitors and predicts thiazide sensitivity. PLoS ONE 12 (4), e0176220. doi:10.1371/JOURNAL.PONE.0176220
Vachel L., Shcheynikov N., Yamazaki O., Fremder M., Ohana E., Son A., et al. (2018). Modulation of Cl-signaling and ion transport by recruitment of kinases and phosphatases mediated by the regulatory protein IRBIT. Sci. Signal. 11 (554), eaat5018. doi:10.1126/SCISIGNAL.AAT5018
Vallon V., Schroth J., Lang F., Kuhl D., Uchida S. (2009). Expression and phosphorylation of the Na+-Cl-cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiology. Ren. Physiology 297 (3), F704–F712. doi:10.1152/AJPRENAL.00030.2009
Virshup D. M., Shenolikar S. (2009). From promiscuity to precision: Protein phosphatases get a makeover. Mol. Cell 33 (5), 537–545. doi:10.1016/J.MOLCEL.2009.02.015
Vitari A. C., Deak M., Morrice N. A., Alessi D. R. (2005). The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391 (1), 17–24. doi:10.1042/BJ20051180
Wakabayashi M., Mori T., Isobe K., Sohara E., Susa K., Araki Y., et al. (2013). Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 3 (3), 858–868. doi:10.1016/J.CELREP.2013.02.024
Wilson F. H., Disse-Nicodè S., Choate K. A., Ishikawa K., Nelson-Williams C., Desitter I., et al. (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112. doi:10.1126/science.1062844
Wu G., Peng J. (2013). Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett. 587 (12), 1717–1722. doi:10.1016/J.FEBSLET.2013.04.032
Yang D., Li Q., So I., Huang C. L., Ando H., Mizutani A., et al. (2011). IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J. Clin. Investigation 121 (3), 956–965. doi:10.1172/JCI43475
Yang S., Fang Y. W., Tseng M. H., Chu P. Y., Yu I. S., Wu H. C., et al. (2013). Phosphorylation regulates NCC stability and transporter activity in vivo. J. Am. Soc. Nephrol. 24 (10), 1587–1597. doi:10.1681/ASN.2012070742
Zagórska A., Pozo-Guisado E., Boudeau J., Vitari A. C., Rafiqi F. H., Thastrup J., et al. (2007). Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 176 (1), 89–100. doi:10.1083/JCB.200605093
Keywords: calcineurin (CaN), protein phosphatase (PP) 1, protein phophatase, with No lysine kinase (WNK), distal convoluted tubule, inhibitor 1 of protein phosphatase 1, tacrolimus, cyclosporin
Citation: Carbajal-Contreras H, Gamba G and Castañeda-Bueno M (2023) The serine-threonine protein phosphatases that regulate the thiazide-sensitive NaCl cotransporter. Front. Physiol. 14:1100522. doi: 10.3389/fphys.2023.1100522
Received: 16 November 2022; Accepted: 17 January 2023;
Published: 15 February 2023.
Edited by:
David Ellison, Oregon Health and Science University, United StatesReviewed by:
Robert A. Fenton, Aarhus University, DenmarkCopyright © 2023 Carbajal-Contreras, Gamba and Castañeda-Bueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Castañeda-Bueno, bWFyaWEuY2FzdGFuZWRhYkBpbmNtbnN6Lm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.