 Dora (Jun Ping) Xiong
Dora (Jun Ping) Xiong James G. Martin
James G. Martin Anne-Marie Lauzon
Anne-Marie Lauzon
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 05 October 2022
Sec. Clinical and Translational Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.993406
This article is part of the Research TopicSmooth Muscle in Health and DiseaseView all 4 articles
Known to have affected around 340 million people across the world in 2018, asthma is a prevalent chronic inflammatory disease of the airways. The symptoms such as wheezing, dyspnea, chest tightness, and cough reflect episodes of reversible airway obstruction. Asthma is a heterogeneous disease that varies in clinical presentation, severity, and pathobiology, but consistently features airway hyperresponsiveness (AHR)—excessive airway narrowing due to an exaggerated response of the airways to various stimuli. Airway smooth muscle (ASM) is the major effector of exaggerated airway narrowing and AHR and many factors may contribute to its altered function in asthma. These include genetic predispositions, early life exposure to viruses, pollutants and allergens that lead to chronic exposure to inflammatory cells and mediators, altered innervation, airway structural cell remodeling, and airway mechanical stress. Early studies aiming to address the dysfunctional nature of ASM in the etiology and pathogenesis of asthma have been inconclusive due to the methodological limitations in assessing the intrapulmonary airways, the site of asthma. The study of the trachealis, although convenient, has been misleading as it has shown no alterations in asthma and it is not as exposed to inflammatory cells as intrapulmonary ASM. Furthermore, the cartilage rings offer protection against stress and strain of repeated contractions. More recent strategies that allow for the isolation of viable intrapulmonary ASM tissue reveal significant mechanical differences between asthmatic and non-asthmatic tissues. This review will thus summarize the latest techniques used to study ASM mechanics within its environment and in isolation, identify the potential causes of the discrepancy between the ASM of the extra- and intrapulmonary airways, and address future directions that may lead to an improved understanding of ASM hypercontractility in asthma.
According to the 2018 Global Asthma Report, 339 million people are affected by asthma worldwide, leading to 1,000 deaths daily (Network, 2018). Asthma is an inflammatory disease characterized by reversible airway obstruction and airway hyperresponsiveness (AHR), an excessive narrowing of the airways in response to various stimuli. Bronchospasm in asthma has conventionally been alleviated with bronchodilators [β2-adrenergic receptor agonists that relax airway smooth muscle (ASM)] underscoring the significant role played by ASM in bronchoconstriction. However, in more severe cases anti-inflammatory drugs such as leukotriene modifiers (Patel and Shaw, 2015; Wang et al., 2015) and oral corticosteroids (Sy and Siracusa, 2016) are also used, as well as the newer agents that target more specific inflammatory pathways, namely antibodies against interleukin (IL)-5, IL-5 receptor α receptor, IL-13, IL-4 receptor α, IgE, the alarmins [thymic stromal lymphopoietin (TSLP), IL-25 and IL-33], and chemoattractant receptor-homologous molecule expressed on T helper type 2 cells (CRTH2) antagonists (Zhu et al., 2018). While the inflammatory cells and mediators create an environment with conditions for exacerbations, the exaggerated contraction of the ASM is the ultimate culprit of airway obstruction and AHR. Thus, is the ASM altered genetically in asthma, or is it altered due to allergenic, viral or pollution exposures, and is mechanical stress positively feeding more mechanical alterations? These are all questions that remain only partly answered.
AHR is defined as an increase in airway narrowing in response to various pharmacological, chemical, and physical stimuli that would otherwise not induce any effect in healthy subjects (Baroffio et al., 2009). The stimuli can be described either as direct or indirect, with direct stimuli acting on specific ASM receptors (e.g., acetylcholine, histamine, cysteinyl leukotrienes, and prostaglandins), or indirect stimuli that act through various intermediate pathways that often involve mediators released from inflammatory cells (e.g., exercise, hypertonic aerosols, and adenosine) (Cockcroft, 2010).
The severity of AHR correlates with the severity of asthma, thus measurements of airway responsiveness have been deemed useful for confirming the diagnosis of asthma and potentially for determining the intensity of treatment required to control symptoms (Juniper et al., 1981; O’Byrne and Inman, 2003). Airway challenge tests using inhaled histamine or methacholine are commonly used and their results are quantified using the volume of air exhaled within the first second of a forced expiration (FEV1). In this measurement, increasing doses of contractile agonists are administered to subjects until a 20% decrease in FEV1 is reached (Chai et al., 1975; Juniper et al., 1981; Cockcroft, 2010). The concentration used to attain this drop is defined as the provocative concentration (PC20) (Cockcroft, 2010). This value is generally lower in asthmatics (PC20 < 8 mg/ml) than non-asthmatics (PC20 > 16 ml), thus a lower PC20 is indicative of greater airway responsiveness (O’Byrne and Inman, 2003). Dosimeters are also employed and equivalence between the provocative doses and concentrations has been determined (Coates et al., 2017).
It is also possible to assess airway responsiveness by comparing agonist dose-response curves of asthmatics and non-asthmatics. In vitro ASM strips are known to produce a sigmoidal dose-response curve when exposed to different concentrations of histamine or methacholine (Brink et al., 1980). A similar curve can also be fitted to FEV1 responses of subjects exposed to inhaled agonists at various concentrations, spanning a non-response inducing concentration, through a quasi-linear increase, to a plateau where no further fall in the FEV1 is evoked (Woolcock et al., 1984). The position and slope of this curve can be used as an indication of the sensitivity and reactivity of the airways, respectively. Hyperresponsive airways are more sensitive and reactive to stimuli, thus asthmatic subjects tend to produce curves that are steeper and shifted leftwards compared to non-asthmatics (Woolcock et al., 1984). In only the mildest asthmatics is it possible to demonstrate a limit to the bronchoconstrictive response with increasing doses of agonist (Woolcock et al., 1984) (Figure 1).
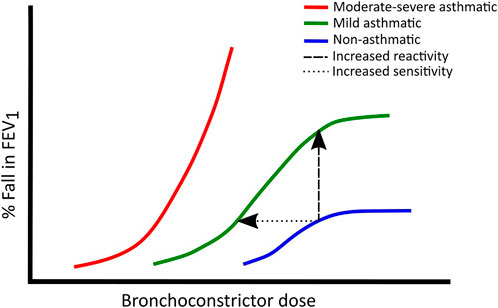
FIGURE 1. Typical dose-response curves in non-asthmatics (blue), mild asthmatics (green) and moderate to severe asthmatics (red). Leftward shifts show increased sensitivity whereas upward shifts show increased reactivity. Adapted from (Woolcock et al., 1984).
Nowadays, mechanical properties of the respiratory system can be measured by forced oscillometry (Dubois et al., 1955; Ram et al., 2019; King et al., 2020). Perturbations are applied during tidal breathing and thus the technique has the advantage of being non-invasive, versatile, and requiring minimal patient cooperation (Kim et al., 2001; Oostveen et al., 2003). Measurements are usually carried out with a stimulus being applied to the respiratory system through the mouth. The patient breathes through a mouthpiece while supporting their cheeks with their hands and blocking the nose with a noseclip. While breathing, a sound generator applies air flow oscillations of varying frequencies to the airstream and respiratory tract and the resulting pressure is measured (Ram et al., 2019). Using Fourier transform techniques, complex impedance is calculated which can be resolved into its resistive and reactive (elastic and inertial) components (Bates, 2009). The oscillations at low frequencies (<2 Hz) give information predominantly about the rheological properties of the lung tissues whereas the higher frequencies (4 Hz > f < 32 Hz) estimate mostly the properties of the proximal and distal airways (Bates, 2009; Ram et al., 2019). While these techniques have been very useful in clinical settings to evaluate patient’s asthma, they only provide indirect information about ASM mechanics.
It has long been hypothesized that during the development of asthma the ASM undergoes fundamental changes that make it hypercontractile. Bronchoprovocation studies using a wide variety of stimuli have shown that AHR is not specific to one agonist, indicating that the phenomenon is related to a general abnormal behaviour of ASM and not to upregulation of specific agonist receptors (Curry, 1946; Dubois and Dautrebande, 1958; Nadel et al., 1965; Mcneill and Ingram, 1966; Mathé et al., 1973; Peters-Golden and Henderson, 2007; Driessen et al., 2012; Pascoe et al., 2012; Hyrkäs-Palmu et al., 2018). Another observation that points towards abnormalities in ASM is that a deep inhalation does not cause bronchodilation in asthmatic subjects, and in severe disease may lead to bronchoconstriction (Fish et al., 1981). This finding highlights the importance of stretch as a bronchorelaxant under normal circumstances. Furthermore, when non-asthmatic subjects are prevented from taking deep breaths, they experience AHR similar to asthmatic patients (Skloot et al., 1995). Thus, ASM stretching via breathing decreases airway resistance in normal lungs but not in asthmatic patients (Fish et al., 1981; Skloot et al., 1995).
While AHR has become accepted as a defining feature of asthma, the question of how each airway tissue component contributes to this phenomenon remains unanswered. Airway responsiveness measurements performed in vivo are likely to assess a combination of factors that include ASM contraction, airway wall stiffness and airway-parenchymal interdependence. As the constrictor of the airways, exaggerated ASM contraction is directly linked to AHR. Changes to ASM contractile properties and increases in ASM mass are likely the major players in enhanced airway responsiveness by causing airways to contract more easily in response to stimuli, often leaving the airways in a chronically contracted state (Pascoe et al., 2012; Berair et al., 2013). Increases in airway resistance in response to inhaled histamine is sensitive to the lung volume at which the measurements are made (Balassy et al., 1995). Higher lung volume reduces airway responses whereas at low volume, airway narrowing is exaggerated. These observations highlight the importance of mechanical impedances such as lung elasticity in determining the degree of airway narrowing for any particular activation state of the ASM. Given all these factors that contribute to AHR, elucidating the precise pathophysiology underlying exaggerated ASM shortening in asthma has not been easy.
Due to limitations in accessing and working with viable ASM tissue from human asthmatic subjects, a variety of animal models have been used to understand the role of ASM in asthma. These include the Fisher and Lewis rat model of innate AHR (Eidelman et al., 1991; Tao et al., 1999; Tao et al., 2003; An et al., 2006; Gil et al., 2006; Tao et al., 2012), horses with heaves which serves as a model of naturally occurring inflammatory asthma (Lowell, 1990; Leclere et al., 2011; Matusovsky et al., 2016), the Brown Norway rat and canine models of induced AHR (Antonissen et al., 1979; Gunst, 1983; Shore et al., 1983; Jiang et al., 1992; Watanabe et al., 1995; Ramos-Barbón et al., 2005; Matusovsky et al., 2014), and AHR following challenge of naturally sensitized sheep (Soler et al., 1991).
The anatomy of the ASM differs based on its location in the airways (Suarez et al., 2012; Widmaier et al., 2019). Along the posterior end of the trachea and main bronchi, a transverse sheet of ASM connects the U-shaped cartilage rings of these segments. In the medium-sized bronchial generations, the ASM lies between the cartilage plaques and the epithelium. It extends to surround the entire airway so that its contraction leads to a decrease in airway lumen. In the next airway generations, the muscle strands become interwoven as a network of spirals that forms a mesh that lies between the cartilage and epithelium. The muscle strands become thinner as the caliber of the tubes becomes progressively smaller. In the bronchioles, the smooth muscle lies between the adventitial tissue and the epithelium but has become relatively large in proportion to the airway wall area, suggesting that it may have more functional importance in this airway generation (Macklin, 1929; Shore, 1984). Contraction of these spirally distributed ASM strands leads to a decrease in both airway diameter and length, which leads to a complex effect on airway resistance (Bates and Martin, 1990). Thus, contraction of ASM and the consequent bronchoconstriction compromises ventilation, although it may have a functional role in fine -tuning ventilation to perfusion ratios within the normal lung. While the role of ASM in healthy adult lungs remains unclear, recent findings have shown that ASM function extends beyond its ability to regulate bronchomotor tone (Camoretti-Mercado et al., 2021). For example, ASM cells can also have secretory or immunomodulatory functions (Ozier et al., 2011; Xia et al., 2013).
Airway remodeling in asthma describes the changes that occur to structural components of the airway wall including changes in number, composition, distribution, thickness, and mass or volume (Fehrenbach et al., 2017). These changes arise in response to either a disturbance in lung development or chronic injury and/or lung inflammation (Fehrenbach et al., 2017). Remodeling has been observed to occur in various airway components including the epithelium, nerve tissue, bronchial vasculature and smooth muscle (Fehrenbach et al., 2017). Physical changes to the ASM, such as hypertrophy (increase in ASM cell size) and/or hyperplasia (increase in ASM cell number), are common in airway remodeling and which lead to increased ASM mass and therefore increased airway wall thickness (Cockcroft and Davis, 2006). Increases in ASM mass have been found to be associated with a decrease in lung function in severe asthma (Pepe et al., 2005; Kaminska et al., 2009). However, while the quantity of ASM was found to serve as an important factor contributing to differences in airway responsiveness between Fisher and Lewis rats, it could not account for variability of AHR within strain, suggesting that other factors relating to the ASM itself may impact AHR (Eidelman et al., 1991).
The extent of airway remodeling can be clinically correlated to asthma severity, as well as AHR (Boulet et al., 1997). Although ASM cells are structural in nature, they have also been shown to exhibit immunomodulatory functions and may secrete cytokines and chemokines, and express cellular adhesion molecules, which can be important for modulating airway inflammation (Panettieri, 2002). While it is unclear whether remodeling is driven by inflammation or if the two processes occur simultaneously, there is evidence to suggest that ASM itself may interact with the inflammatory cells and mediators in its surrounding environment in a positive feedback loop that furthers airway remodeling throughout asthma pathogenesis (Panettieri, 2002).
ASM from bronchial biopsies of asthmatic subjects has been shown to express CXCL10 and ASM cells stimulated with pro-inflammatory cytokines secrete CX3CL1 (Sukkar et al., 2004; Brightling et al., 2012; Manuel Tunon-de-Lara Ousova et al., 2022). These molecules are chemo-attractants for mast cells that infiltrate ASM and have the potential to alter its responses (Sukkar et al., 2004; Brightling et al., 2012; Manuel Tunon-de-Lara Ousova et al., 2022). Activation and degranulation of mast cells are known to play a role in the extracellular deposition of inflammatory products which may enhance ASM mass (Bara et al., 2010). Additionally, ASM can release a range of extra-cellular matrix (ECM) proteins such as fibronectin, perlecan, elastin, laminin, thrombospondin, chondroitin sulfate, collagen I, III, IV, and V, versican, and decorin (Johnson et al., 2012). The increased deposition of ECM proteins around the ASM layer can lead to fibrosis, which contributes to the thickening of the airway wall. The ECM proteins may also directly alter ASM function by enhancing its proliferation, such as is the case for fibronectin (Hirst et al., 2012). Another consequence of an increased ECM is the release of active transforming growth factor (TGFβ), which is stored in the ECM and can be cleaved by matrix metalloproteinases such as MMP-9 (Bara et al., 2010). This profibrotic cytokine enhances α-smooth muscle actin, myosin light chain kinase (MLCK), and smooth muscle myosin heavy-chain contractile protein expression in human ASM cells, impairs β2-agonist induced relaxation (Ojiaku et al., 2019), and may also increase ASM force of contraction and shortening in response to contractile agonists (Goldsmith et al., 2006). Another fundamental feature of asthma is the development of a strong effector T lymphocyte helper type 2 (Th2) response by the adaptive immune system (Murdoch and Lloyd, 2010). Th2 differentiated CD4+ T cells have been observed to form direct contacts with ASM in ovalbumin-sensitized Brown Norway rats where they induce DNA synthesis, cell proliferation, reduced ASM cell apoptosis, and increased ASM mass after ovalbumin challenge (Ramos-Barbón et al., 2005). In the same model, direct contact with ASM also decreased apoptosis of CD4+ T cells, thus suggesting a reciprocal cycle of remodelling driven by both the T cells and ASMCs.
Increased ASM mass is a hallmark of airway remodeling in asthma (Bara et al., 2010). It is characterized by both hypertrophy and hyperplasia, although the degree to which each occurs is not consistent across asthmatics (Bentley and Hershenson, 2008). Hypertrophy was found to be present in some severe asthmatics, and is suggested to be activated through two potential pathways, either involving the mammalian target of rapamycin (mTOR) or an inhibition of glycogen synthase kinase (GSK)-3β with Akt, also known as protein kinase B (Bara et al., 2010). mTOR phosphorylates eukaryotic initiation factor-4E (eIF4E)-binding protein (4E-BP) to release eIF4E, which plays a key role in the initiation of translation (Zhou et al., 2005). Increased 4E-BP phosphorylation leads to greater rates of protein translation and cell hypertrophy (Zhou et al., 2005). GSK-3β inactivates eIF2B, which also catalyzes a key regulatory step in mRNA translation (Welsh et al., 1998). Thus, the inhibition of GSK-3β may induce hypertrophy in an eIF2B dependent manner. Hyperplasia is induced by growth factors such as TGFβ, epidermal growth factor (EGF), and platelet derived growth factor, as well as stimulation by contractile agonists such as histamine and leukotriene D4 (Doeing and Solway, 2013). Reactive oxygen species and mechanical stress have also been implicated as factors that induce ASM cell proliferation (Bara et al., 2010). Indeed, it has been shown that bronchoconstriction per se can lead to EGF receptor ligand release (Tschumperlin et al., 2004) and ASM remodeling.
Whether it be through increased ASM mass or airway wall thickness, airway remodeling tends to leave ASM in a shortened state. Due to its ability to adapt its optimal length, this allows for airways to contract with greater force at smaller diameters, and also reduces the effectiveness of breathing and deep inspiration for dilating the airways in asthmatics (Seow and Solway, 2011; Chin et al., 2012). A positive feedback cycle has also been recognized in which ASM that has become too great in mass or receives too little opposing force will stiffen and stretch less, which will in turn lead to further stiffening (Oliver et al., 2007).
ASM is innervated by the autonomic parasympathetic nervous system (Gosens et al., 2006). Activation of ASM is mediated by the neurotransmitter acetylcholine (ACh), which is released from nerve endings and binds to M3 muscarinic receptors, leading to an increase in cytoplasmic Ca2+ from the sarcoplasmic reticulum (SR) and the extracellular space, which is a key step in smooth muscle activation (Jude et al., 2008). Aside from activation through the nervous system, endogenous agonists that stimulate Ca2+ release include autocoids such as histamine and serotonin, lipid mediators such as cysteinyl leukotrienes C4, D4, and E4, prostanoids such as prostaglandins D2, F2α, and thromboxane A2 (Lam et al., 2019). Note that histamine also stimulates the release of Ach from nerve endings (Shore et al., 1983). Methacholine and carbachol are two drugs that mimic but are more stable in vivo than ACh so are used to study bronchoconstriction (Dubois and Dautrebande, 1958; Goldie et al., 1986; Gosens et al., 2006).
Under resting conditions, ASM cells maintain a low intracellular concentration of Ca2+. Contractile agonists stimulate G-protein-coupled receptors (GPCR) coupled to the Gα subunit and phospholipase C (PLC). Upon stimulation Ca2+ channels are opened throughout the cell to facilitate the influx of Ca2+ from the SR and the extracellular space (Jude et al., 2008; Perez-Zoghbi et al., 2009; Seow, 2021). The sarcolemma contains voltage-gated, Ca2+ store-operated, and receptor activated Ca2+ channels while the SR contains inositol 1,4,5-trisphosphate (IP3)-gated channels and ryanodine receptors (RyR’s) (Perez-Zoghbi et al., 2009; Seow, 2021). In addition, stimulation of the GPCR activates PLC which produces secondary messengers–inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG)—from the plasma membrane component phosphatidylinositol 4,5-bisphosphate (PIP2). IP3 releases Ca2+ from the SR, via the IP3-gated channels. Note that the Ca2+ increase in the cell is not monotonic but shows rhythmic oscillations (Perez-Zoghbi et al., 2009). These increases often start at one end of the cell and move throughout it as a wave, but the initiation site may fluctuate (Perez-Zoghbi et al., 2009).
In the cytoplasmic space, Ca2+ binds to the protein calmodulin (CaM). The Ca2+-CaM complex then binds MLCK and activates it so that it can phosphorylate the myosin regulatory light chain (LC20). Once LC20 is phosphorylated, the myosin head is able to interact with actin filaments to generate force i.e., muscle contraction (Jude et al., 2008; Seow, 2021). Myosin light chain phosphatase (MLCP) dephosphorylates LC20, and thus mediates muscle relaxation (Seow, 2021). However, two parallel pathways can inhibit this dephosphorylation. A RhoA/Rho-kinase pathway, where Rho kinase, a serine/threonine kinase, phosphorylates and therefore inhibits MLCP (Somlyo and Somlyo, 2003; Chiba and Misawa, 2004). A second mechanism involves the inhibition of MLCP by CPI-17 that is activated when phosphorylated through a DAG/protein kinase C mechanism (Kitazawa et al., 2000). These two mechanisms are Ca2+ independent but enhance the effect of Ca2+ and have therefore been named Ca2+ sensitization. These Ca2+ dependent and independent activation mechanisms are depicted in Figure 2.
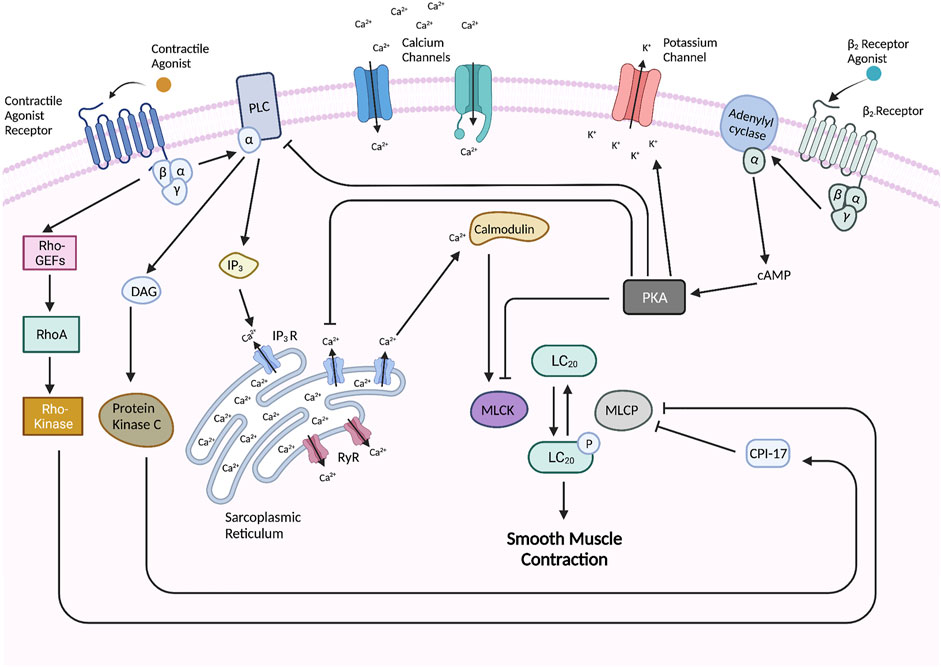
FIGURE 2. Molecular pathways underlying ASM contraction and relaxation. The activation of G-coupled protein receptors such as muscarinic and histamine receptors leads to subsequent interaction and activation of phospholipase C (PLC) which promotes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 interacts with IP3-gated channels on the SR to induce Ca2+ efflux into the cytoplasmic space. Ryanodine receptors (RyR’s) on the SR also become activated to release Ca2+. In the cytoplasmic space, Ca2+ binds to calmodulin (CaM) which activates it so that it may activate myosin light chain kinase (MLCK) which phosphorylates and activates the myosin regulatory light chain (LC20) of myosin to initiate cross-bridge cycling and thus muscle contraction. LC20 is dephosphorylated and thus inhibited by myosin light chain phosphatase (MLCP), but this dephosphorylation is inhibited by two pathways–including the Rho-kinase pathway which is stimulated by the activated GPCR and a CPI-17 pathway which is activated by protein kinase C. Muscle relaxation can be induced by stimulation of a β2-receptor which leads to the activation of adenylyl cyclase (AC) which catalyzes the formation of cyclic AMP (cAMP). cAMP binds and activates PKA which carries out various regulatory activities such as inhibiting MLCK, PLC, and IP3-gated channels, and activating Ca2+-gated potassium channels in the cellular membrane to release K+ which hyperpolarizes the cell. Created with BioRender.com.
After an excitatory event, Ca2+ is taken up into the SR stores or pumped back into the extracellular space. The main Ca2+ storage compartment in ASM is the SR (Sanders, 2001). It is surrounded by a non-Ca2+ permeable membrane which is lined with specialized active Ca2+ ATPases known as SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) pumps that maintain a 10,000-fold Ca2+ gradient between the SR lumen and the cytoplasm (Sanders, 2001). Mitochondria also play a role in Ca2+ uptake by way of a Ca2+ uniporter on the mitochondrial inner membrane that relies on an electrochemical gradient generated by the electron transport chain (Sanders, 2001). A major source of Ca2+ extrusion from the cytosol is the plasma membrane Ca2+-ATPase which uses ATP to transport Ca2+ back out of the cell in exchange for two protons. This exchange results in a net increase in protons within the cell which is compensated by transporters such as the Na+/H+ exchange (Sanders, 2001).
The pathways leading to ASM contraction are complex so there are several potential steps that can be altered and lead to hypercontractility. Neural control has been shown through animal models to be implicated in AHR (Shore et al., 1985; Wagner and Jacoby, 1999; Lourenço et al., 2020; Nie et al., 2020) but a similar connection has not been proven in humans (Sterk et al., 1985). Thus, the focus on possible mechanisms to explain abnormal ASM behaviour remains on post-synaptic processes such as the interaction of acetylcholine and M3 receptors at the neuromuscular junction (Ouedraogo and Roux, 2003) and following pathways that become activated downstream.
Abnormalities in one or more of the signalling or contractile proteins can occur genetically or by alterations at the transcriptional, translational, or post-translational levels. A greater expression of the [(+) insert] smooth muscle myosin heavy chain (SMMHC) isoform–which is more highly expressed in rapidly contracting phasic muscle–has been hypothesized to contribute to AHR (Gil et al., 2006). The trachealis of the hyperresponsive Fisher rats was reported to express greater mRNA levels of the [(+) insert] SMMHC isoform and greater protein content of this isoform relative to total SMMHC compared to the normoresponsive Lewis rats (Gil et al., 2006). The asthmatic horses were also found to express higher (+) insert SMMHC at the mRNA and protein levels in intrapulmonary and central airways, which was reduced after corticosteroid treatment or antigen avoidance (Boivin et al., 2014). The (+) insert SMMHC isoform was also shown to be increased at the mRNA level in human endobronchial biopsies of well characterized asthmatic patients compared to controls (Léguillette et al., 2009). However, such studies have yet to be repeated at the protein level. Increased ASM MLCK protein levels have been reported in allergen sensitized human airways (Ammit et al., 2012), and a heightened MLCK mRNA expression was noted in asthmatic ASM cells (Ma et al., 2002; Léguillette et al., 2009). Similar results have been obtained from biopsies in asthmatics with differing levels of asthma severity (Zweiman et al., 2003). A higher MLCK content should increase the extent of LC20 activation, which has been correlated with a potentiated rate and extent of ASM shortening in the canine model of allergic AHR (Jiang et al., 1995). Heightened LC20 activation was observed in ASM from Fisher rats and in tracheal tissue from an allergic dog model of asthma (Jiang et al., 1995; Gil et al., 2006).
Another possible culprit of ASM hypercontractility is the dysregulation of Ca2+ homeostasis. Studies performed with human tissues reported a decreased expression of SERCA in asthmatic ASM (Mahn et al., 2009) whereas an enhanced Ca2+ release in response to contractile agonists was reported in the ASM of hyperresponsive Fisher rats (Tao et al., 1999). When the expression of this Ca2+ reuptake protein in healthy ASM culture was inhibited with siRNA, the cells exhibited asthmatic ASM behaviour such as increased motility, secretion of pro-inflammatory chemokines, and decreased Ca2+ reuptake (Tao et al., 1999). Therapies targeting the Ca2+ sensitization pathways have also been investigated and have revealed several promising candidates (Koziol-White et al., 2016; Yoo, 2017; Yoo et al., 2017).
ASM may also become hypercontractile due to abnormalities in the relaxation pathway. ASM relaxation can be induced by stimulation of the β2-receptors, which are GPCRs classified under the Gs sub-family (Billington and Penn, 2003). Upon activation, the Gs protein binds to and activates adenylyl cyclase which then catalyzes the formation of cyclic adenosine 3′,5′-monophosphate (cAMP) from cytosolic ATP (Billington and Penn, 2003). cAMP then binds with high affinity to the regulatory subunits of protein kinase A (PKA) and causes these subunits to dissociate from the enzyme complex (Sassone-Corsi, 2012). The remaining catalytic subunits of PKA can then phosphorylate and regulate the activity of numerous downstream proteins (Billington and Penn, 2003). Of note, PKA phosphorylates myosin light chain kinase (MLCK) which prevents downstream phosphorylation of myosin light chain (LC20) and thus promotes ASM relaxation (Billington and Penn, 2003). Activated PKA also phosphorylates other specific GPCRs, phospholipase C (PLC), and inositol 1,4,5-triphosphate (IP3) receptor which leads to a reduction of calcium flux (Billington and Penn, 2003; Álvarez-Santos et al., 2020). Another important effector of PKA are Ca2+ gated potassium (Kca) channels which are densely distributed throughout the ASM cellular membrane (Kume et al., 1994). Upon phosphorylation, these channels open and allow K+ efflux which results in cell hyperpolarization. Figure 2 depicts the β2-mediated relaxation pathway.
Many inflammatory cytokines present in asthmatic airways decrease the relaxant response to isoproterenol in human ASM, including interleukin (IL)-13, IL-1β, tumour necrosis factor (TNF), and TGF-β through several different mechanisms (Shore, 2002). IL-13 decreases the β2-adrenergic response via an extracellular signal-regulated kinase (ERK) dependant pathway (Laporte et al., 2001) whereas IL-1β & TNF induce the expression of cyclooxygenase-2, resulting in prostanoid release (Laporte et al., 1998). IL-13 and TNF also augment Ca2+ release in response to contractile agonists, and IL-13 increases the expression of receptors for histamine and cysteinyl leukotrienes (Amrani et al., 1997; Deshpande et al., 2004; Manson et al., 2020). Additionally, IL-13 has been shown to increase ASM constriction in response to acetylcholine and histamine (Grunstein et al., 2002; Risse et al., 2011).
Excised airway segments from animal models or post-mortem human lungs have been used to study airway mechanics and to infer ASM mechanics. The side branches of the airway segments are sealed off to form a leak-free system, allowing measurements of ASM force generation through changes in pressure or volume (Gunst and Mitzner, 1981; Noble et al., 2007). When using airways from small animals, this can be done by measuring the pressure drop of a fluid perfused along an airway segment. When using airways from large animals ex vivo, it is possible to directly study the lumen of the airways either by endoscopy, anatomical optical coherence tomography, or ultrasound. While the excised segments retain many elements of the airways including the epithelial layer, nerves, and airway wall, they also provide an opportunity to perform more intrusive protocols such as electrical field stimulation and to compare differences between the central and peripheral airways (LaPrad et al., 2008; LaPrad et al., 2010; Noble et al., 2010; Ansell et al., 2013). The dynamics of stretch as a bronchodilator have been studied using excised airway segments (LaPrad et al., 2010; Ansell et al., 2013). It has been shown that applying strain rather than stress induces bronchodilation (Ansell et al., 2013). Furthermore, rapid stretches cause greater bronchodilation (Ansell et al., 2013) than slow ones (Ansell et al., 2013). One drawback to the approach of a fully integrated system in airway models is that the elucidation of cellular and molecular mechanisms is not possible, and thus studies using these models are often supplemented with in vitro experiments such as the use of human ASM cell culture.
In vitro experiments using human lungs are limited by the relative inaccessibility of smaller intrapulmonary airways, making it difficult to study the airways in the more peripheral regions (Lam et al., 2019). The early thick slices (Dandurand et al., 1993) have given way to thinner precision cut lung slices (PCLS) (Martin et al., 1996; Perez-Zoghbi et al., 2009; Wright et al., 2013). PCLS offer an approach to study ASM ex vivo in an integrated system (Martin et al., 1996; Wright et al., 2013; Lam et al., 2019). PCLS are prepared by filling intact lungs, lobes, or wedge preparations with liquid agarose that maintains tissue structure after solidifying (Martin et al., 1996; Lam et al., 2019; Liu et al., 2019). After the gel stiffens, it is then possible to generate lung slices that retain many features of the airway anatomy including the alveolar architecture, respiratory parenchyma, structural and immune cells, and connective tissue. PCLS can be maintained in culture medium for up to 14 days, where they maintain normal metabolic activity, tissue homeostasis, and structural integrity (Liu et al., 2019). As PCLS can be maintained for extended periods of time, it is also possible to study the impact of prolonged exposures to various stimuli (Cooper and Panettieri, 2008; Cooper et al., 2011). Innate immune responses have been observed in PCLS after lipopolysaccharide exposure, bacterial infections, or viral infections (Chakrabarty et al., 2007; Henjakovic et al., 2008; Wu et al., 2010). The presence of other airway cells in PCLS also allows the assessment of the effects of airway inflammation and remodelling on ASM contraction.
Bronchoconstriction in response to agonists and electric field stimulation (EFS) can be captured and measured in real-time with videomicroscopy and digital imaging (Martin et al., 1996). To perform this method, PCLS are held down on a cover slip by a nylon mesh which has a hole cut in the middle to expose the airway (Bai and Sanderson, 2009). A second cover slip is placed over the mesh and sealed with silicon grease to create a perfusion chamber, and the lung slice is perfused by a gravity driven perfusion system (Bai and Sanderson, 2009). PCLS retains the parenchymal load against the contracting ASM, despite a loss of the 3D structure (Dandurand et al., 1993; Wright et al., 2013; Lam et al., 2019) so that experiments can be carried out without setting and controlling an arbitrary ASM length and tension, reducing the complexity of the measurements. The early studies using lung slices to compare inbred strains of rat differing in airway responsiveness showed that the intraparenchymal airways of the hyperresponsive Fisher rats constricted faster and to a larger extent than those of the normoresponsive Lewis rats, as had previously been reported in vivo (Tao et al., 1999). The AHR was also manifest at the ASM cellular level; Ca2+ mobilization by an agonist was enhanced in the Fisher compared to the Lewis rat (Tao et al., 2003; Tao et al., 2012).
PCLS has been a valuable tool for the study of Ca2+ signalling while retaining the gross architecture. ASM can also be loaded with fluorescent dyes to detect Ca2+ flux and sensitivity (Liu et al., 2019). PCLS have also been used to study Ca2+ oscillations in ASM and towards understanding the contributions of various Ca2+ channels (Perez-Zoghbi et al., 2009). The PCLS technique has been used with disease models as well as with transgenic mice to dissect the contributions of highly specific phenotypes (Sanderson, 2011). Through a combination of mathematical modelling and experimental work involving Ca2+ channel inhibitors, store-operated Ca2+ channels were shown to play a prominent role in maintaining Ca2+ and thus revealed their critical importance for sustaining ASM contraction and tone (Boie et al., 2017; Chen and Sanderson, 2017). Although not having as significant a role in replenishing Ca2+ during ASM contraction, PCLS studies in mice also have shown that the level of RyR sensitization impacts Ca2+ oscillations. Agonist-induced contractions result in Ca2+ depletion of the SR which normally causes RyR inactivation (Croisier et al., 2015). However, inflammatory cytokines such as TNF-α and IL-13 have been reported to modify RyR, possibly causing them to remain open during SR depletion, increasing Ca2+ sensitivity (Deshpande et al., 2004; Sieck et al., 2008). Small increases in the sensitivity of cytosolic and/or luminal RyR led to low-frequency Ca2+ oscillations while moderate to high luminal RyR sensitization led to an elevated average cytosolic Ca2+ concentration upon agonist stimulation (Croisier et al., 2015).
One important point to consider when using PCLS is the loss of diffusion barriers that are normally present in the 3D structure of the airway because agonists or mediators added to the culture can access all compartments of the airways. Without the epithelial barrier, the ASM is now directly exposed to various stimuli (Wright et al., 2013). This makes it difficult to observe which cell type is being affected by the stimuli and determine the exact source of a secreted endogenous substance. PCLS will also lose certain cell populations during longer-term culture, which can contribute to decreased sensitivity in response to external stimuli (Liu et al., 2019). However, insulin has been shown to preserve myosin heavy chain expression in mice PCLS when maintained in culture for 2 weeks and retains IL-13-induced ASM hypercontractility (Li et al., 2020).
PCLS used with post-mortem human lungs is improving our understanding of AHR. With PCLS, a reduced airway relaxation to isoproterenol was observed in the presence of ASM β2 receptor phosphorylation (Tyr 141 and Tyr 350), an effect that is induced in asthma by growth factors released from activated mast cells (Chachi et al., 2018). The PCLS technique has also been used to demonstrate the dilation of small airways by PI3k inhibitors (Koziol-White et al., 2016) and to study the potentiation of ASM contractility by inflammatory cytokines such as IL-17 (Willis et al., 2015). However, thus far there are no reports showing intrinsic differences in agonist responses of ASM between human asthmatic and control PCLS.
Although there are many benefits to measuring ASM contraction in integrated systems, one drawback to models using airway segments and PCLS is that the ASM contractile properties cannot be isolated from the influence of the other tissue elements. The presence of multiple cell types and a complex mechanical environment impose potential modulating effects which may mask the behaviour of ASM (Gunst, 2012). Therefore, the practice of using isolated ASM strips or bronchial/tracheal ASM rings has been employed to fill this knowledge gap. ASM strips have been obtained from animal species such as mice, rats, guinea pigs, dogs, pigs, horse, and cows, but human ASM strips are also used and obviously, provide the most relevant information for understanding asthma (Gunst, 1983; Wang et al., 2000; Latourelle et al., 2002; Moir et al., 2003; Bullimore et al., 2011; Dekkers et al., 2012; Matusovsky et al., 2016). Using ASM from larger animals such as horses has been integral to obtaining initial data regarding the deeper generations of the airways (Matusovsky et al., 2016). Due to limited supply of asthmatic human ASM tissue, control human ASM has been sensitized through incubation with asthmatic serum, IgE, or cytokines including IL-13, IL-5, TNF, and IL-1β to mimic the asthmatic inflammatory milieu (Black et al., 1989; Ellis et al., 1994; Tunon de Lara et al., 1995; Barchasz et al., 1999; Hakonarson et al., 1999; Grunstein et al., 2002).
ASM can be dissected from isolated airways, leaving a tissue strip, with or without epithelium, with muscle fibres that are aligned along the longitudinal axis. The strips can be attached to two aluminum foil clips which are used to mount the ASM on a force transducer that senses changes in contractile force and a length-sensing lever (Chin et al., 2012). This system allows the control over two important variables that characterize ASM function–force and length. It is then possible to focus on each variable independently of one another by studying either isometric or isotonic contractions. Changes in tissue tension (i.e., force) are measured by exposing the strip to contractile or relaxing agonists, or an electric field stimulation (EFS) (Ijpma et al., 2015). Assessing tissue changes in tensile force in response to gradually increasing concentrations of a drug or agonist gives a dose-response curve that has been used to compare differences in ASM responsiveness (see above) (O’Byrne and Inman, 2003). Using the quick-release technique (described below), it is possible to determine the shortening velocity (Seow and Stephens, 1986; Bullimore et al., 2010; Ijpma et al., 2015).
Important parameters of muscle mechanics include force, stress, length, and shortening velocity. Smooth muscle differs from striated muscle in that it is plastic and adapts to changes in length. It has, as for striated muscle, an optimal length at which it produces maximal isometric force (Stephens and Van Niekerk, 1977). However, SM changes its internal configuration to adapt to changes in length quite quickly. It is therefore important to maintain a constant reference length throughout testing conditions. Thus, changes in ASM length during a contraction are expressed as a percentage change of a predefined reference length (Chin et al., 2012; Matusovsky et al., 2016; Luo et al., 2019).
The magnitude of force generated by a smooth muscle bundle depends on the number of activated cross-bridges (Seow, 2021). Thus, an important parameter is the maximal force, obtained with maximal stimulation, and which is designated as Fmax (Matusovsky et al., 2016; Seow, 2021). Under in vitro conditions, this is usually measured at a fixed length, as the maximal isometric force. Muscles and cell bundles with larger cross-sectional areas can generate more contractile force because they have more myosin molecules that work in parallel. This is important to note as an increased ASM mass is a common feature in asthmatic airways (Dunnill et al., 1969; Carroll et al., 1993). Note that this increased ASM mass has been examined by stereology and was believed to be overestimated due to matrix proteins between ASM cells (Thomson et al., 1996). However, further careful stereology studies did not confirm any difference in matrix proteins between asthmatic and control ASM (James et al., 2012). Thus, to study ASM responses that are otherwise influenced by ASM mass, muscle force is normalized to the cross-sectional area which is referred to as stress (Ijpma et al., 2015).
The velocity of muscle shortening describes the rate at which the length of a muscle decreases during a contraction (Chin et al., 2012; Seow, 2021). ASM shortening velocity is inversely related to the load against which a muscle contracts. Velocity is highest in the initial stage of contraction and decreases over time throughout muscle shortening. Thus, for comparative purposes, it is important to measure velocity at the same timepoint after muscle stimulation.
Force-velocity plots are constructed using the quick-release technique. Briefly, the muscle is first activated under isometric conditions until the force generated reaches a plateau (Seow, 2021). At this point, the contraction is switched from isometric to isotonic (at a given specific load), which allows muscle shortening from its initial length. Assuming no viscoelastic recoil, the muscle’s shortening velocity at this load is taken as the change in length per unit of time. Repeating this with progressively declining loads generates the data that form a force-velocity plot. In theory, the muscle would achieve its maximal velocity (Vmax) if the load were dropped to zero, thus Vmax is calculated by extrapolation to the zero load of this plot. Vmax is commonly used because it is a good representation of the actin-myosin cross-bridge cycle.
Historically, there has been ambiguity in the data addressing whether ASM function is abnormal in asthma. While some in vitro studies using asthmatic ASM have demonstrated increased force generation, shortening, and agonist sensitivity (Schellenberg and Foster, 1984; de Jongste et al., 1987; Bai, 1990; Bramley et al., 1994; Thomson et al., 1996), others have also shown no difference (Roberts et al., 1984; Goldie et al., 1986; Whicker et al., 1988; Cerrina et al., 1989; Bjorck et al., 1992). However, many of the earlier studies were performed with small sample sizes and with various methodological limitations such as the lack of normalization of force with respect to ASM cross-sectional area, or without the capacity of measuring ASM shortening and shortening velocity (Seow et al., 2012).
Later studies that collected more robust data reported significantly increased maximal contractile capacity and shortening velocity in response to EFS as well as an increased response from asthmatic ASM cells to contractile agonists, such as histamine and bradykinin (Ma et al., 2002; Matsumoto et al., 2007; Sutcliffe et al., 2012). More recent studies that compared the ASM function between asthmatics and normal controls, used isolated human ASM bundles from the trachealis and main bronchi and they did not report any difference in stress and Vmax (Chin et al., 2012; Ijpma et al., 2015). However, increased passive stress and altered response to length oscillations were seen (Chin et al., 2012; Ijpma et al., 2015). An equine model of spontaneous and severe asthma was then used to study the ASM mechanics of intrapulmonary (IP) airways. IP ASM of the asthmatic horses exhibited a greater Vmax than their own trachealis, and greater than IP ASM of control horses, whereas the IP ASM of horses under remission showed increased stress (Matusovsky et al., 2016). Following the optimization of the IP ASM dissection done in the horse, comparisons between human IP (i.e., Third-fifth generations) and tracheal segments were performed and significantly greater contractile stress and stiffness were reported for the asthmatic IP ASM strips (Ijpma et al., 2020).
Modelling and ventilation imaging approaches have indicated heterogeneous airway constriction during asthmatic exacerbations with an emphasis on the importance of small airway constriction (Tgavalekos et al., 2005; Tzeng et al., 2009). These site -specific changes in ASM intrinsic contractility are speculated to be influenced by increased interactions with an inflammatory environment in the IP airways (Lutchen, 2016). The severity of inflammation has been shown to progressively increase towards the periphery (Hamid et al., 1997; Balzar et al., 2002; De Magalhães Simões et al., 2005), therefore IP ASM will encounter a greater number of inflammatory cells/mediators compared to that of the trachea and main bronchi. Of note, the increase in Vmax seen from the equine model was dependent on the time since the last corticosteroid treatment, indicating that acute inflammation enhanced ASM mechanics (Matusovsky et al., 2016). Conversely, the increased IP ASM stress observed in the horses under remission is presumably due to long term ASM remodelling. The normal Vmax but increased stress reported in the human IP ASM by Ijpma et al. (2020) were obtained from asthmatic subjects who died of other causes than asthma, and so did not necessarily exhibit high levels of inflammation. Their ASM properties were therefore closer to those of the horses under remission. Taken together, these data suggest that mechanical alterations observed in asthmatic IP ASM, depend on the type and level of inflammation in the surrounding airway environment and on the chronicity of the exposure.
It is noteworthy that while we often think of a hypercontractile ASM as a stronger muscle, ASM that exhibits a greater Vmax could, conceivably, lead to AHR. It has been suggested that due to the stretching and relaxing effect of tidal breathing, an ASM that re-contracts rapidly between each breath would maintain asthmatic airways in a more constricted state because the ASM would have time to shorten significantly between each breath, thus counteracting the relaxing effect of tidal breathing (Gunst, 1983; Solway and Fredberg, 1997). Indeed, computed tomography and transfer impedance studies showed that both healthy and asthmatic airways dilate upon lung inflation, but that the asthmatic airways rapidly narrow back to their initial diameter (Brown et al., 2001; Jackson et al., 2004). In addition, a greater Vmax has been observed in sensitized human bronchi (Mitchell et al., 1994), in single ASM cells from asthmatic human airways (Ma et al., 2002), and in mouse, rat and canine models of AHR (Antonissen et al., 1979; Jiang and Stephens, 1990; Duguet et al., 2000; Blanc et al., 2003; Fan et al., 2012).
The development of primary human ASM cell culture has provided substantial information on the molecular pathways and cellular interactions that influence ASM contraction (Hirst, 1996). ASM cells can be cultured through two methods (Hirst, 1996). In the first, cells are enzymatically dissociated from a minced ASM preparation (Hall and Kotlikoff, 1995). With the second approach, cells are grown from explants of ASM in petri dishes or tissue-culture flasks and eventually harvested after growing to confluence (Hall et al., 1992; Marsh and Hill, 1992).
ASM cell contraction can be studied using various methods. One frequent, although indirect measurement, is that of the concentration of intracellular Ca2+, which increases when ASM cells are triggered to contract. To do so, cells are loaded with the fluorescent Ca2+ indicator Indo-1 and monitored fluorometrically with an Indo-1 probe (Berger et al., 2001). Ca2+ can also be quantified using the ratiometric fluorophore FURA2-AM. Measurements of downstream events including the phosphorylation of LC20 are also made using western blots (Hunter et al., 2003). For greater sensitivity in quantifying LC20 phosphorylation, western blots using Phos-tag SDS PAGE gels can increase protein solubility and extraction efficiency, and results are confirmed with phosphor-Ser19- LC20 antibodies (Takeya et al., 2008). Cell contraction can also be more directly measured using a collagen gel substrate embedded with ASM cells (Lee et al., 1995; Matsumoto et al., 2007; Han et al., 2020). When assessing contraction, the gels are released from the wells and a contractile stimulus is added. The decrease in gel area can then be taken as a measure of cell contraction. This method requires caution while interpreting results as the free-floating gel provides very little load opposing the ASM, thus allowing for non-contractile cytoskeletal re-organization to manifest as a contraction. Moreover, it is infeasible to “relax” the gel and obtain repeated measurements (Ceresa et al., 2009). Collagen gel contractions have shown that asthmatic ASM cells contract significantly more in response to histamine compared to control ASM cells (Matsumoto et al., 2007).
Cultured ASM cells stiffen when exposed to contractile agonists (An et al., 2002). The stiffening may be measured by optical magnetic twisting cytometry (Wang and Ingber, 1994; Fabry et al., 2001; An et al., 2006; Wright et al., 2013). To do this, ferromagnetic microbeads that are coated with a synthetic RGD (Arg-Gly-Asp)-containing peptide are bound to the cell cytoskeleton via integrin-receptors on the cell surface. The beads are first magnetized horizontally (parallel to the cell plating surface) with a strong homogenous pulse and then twisted vertically with the application of a weaker external homogeneous magnetic field that varies sinusoidally in time. This results in displacement of the bead which can be detected optically and can be used alongside the applied torque value to calculate the cell’s stiffness. Changes in stiffness can be detected upon subsequent additions of contractile or relaxant agonists and can be related back to the contractile force. Using this approach, An and co-workers showed that ASM cells from the hyperresponsive Fisher rat exhibited greater and faster stiffening responses, as well as greater contractile forces, than those of the Lewis rats (An et al., 2006). ASMCs from Fisher rats also demonstrated a faster rate of cytoskeletal remodeling, allowing for the ASM cell to adapt to progressively shorter working lengths which is thought to be a major factor in contributing to excessive narrowing (An et al., 2006). More recently, using this technique, An et al. also showed enhanced stiffness in cells harvested from asthmatic subjects (An et al., 2016).
Another technique to estimate ASM cell force production is traction force microscopy (TFM) (Wang and Li, 2009). TFM is primarily used to quantify forces that contribute to cell migration, but one can also obtain a scalar measure of contractile strength known as the net contractile moment (An et al., 2009). TFM employs an elastic polyacrylamide substrate that is coated with ECM proteins and embedded with fluorescent particles. ASM cells are plated on this substrate and displace the fluorescent particles during a period in which they spread and stabilize. The displacement of cells can then be microscopically tracked after the cells are removed via trypsinization (Wright et al., 2013). This technique has shown that asthmatic ASM cells exhibit increased cell traction forces at baseline and also demonstrate a higher net contractile moment compared to non-asthmatic cells (An et al., 2016).
Following the recognition that several asthma phenotypes exist and that they impact the response to treatments, there has been great interest in developing biological therapies to target specific immune cells and inflammatory mediators (Zhu et al., 2018). These include antibodies against IL-13, IL-4 receptor α, CRTHT2, TSLP, IL-25, IL-13, IL-17A receptor, and CXC chemokine receptor 2 (CSCR2/IL-8) (Zhu et al., 2018). Certain mediators such as IL-4, IL-13, IL-25, and IL-17A are known to cause ASM hyperplasia and AHR (Kudo et al., 2012; Willis et al., 2015; Robinson et al., 2017), but most novel biologics are focused on their effects on inflammatory pathways. Of interest, IL-13 has been shown to enhance ASM contraction and impair relaxation (Corren, 2013). Dupilumab–an anti-IL-4/IL-13 biologic–was described to attenuate human ASM remodelling and contraction as assessed by studies using small bronchi explants and cell culture (Manson et al., 2020) and demonstrated efficacy in phase 3 trials (Wenzel et al., 2016; Robinson et al., 2017). IL-17A is another mediator that directly increases the contractility of ASM as was demonstrated in an allergen sensitized mouse model (Kudo et al., 2012). To inhibit the effects of IL-17A, brodalumab–which binds to the IL-17 receptor A and thus blocks IL-17A–was tested in a clinical trial with patients who had uncontrolled moderate to severe asthma, but did not demonstrate significant improvement in the overall study population, although a nominal significance in Asthma Control Questionnaire scores was observed in one treatment subgroup (Busse et al., 2013a). Current Food and Drug Administration-approved biologics such as tezepelumab which blocks TSLP (Menzies-Gow et al., 2021), omalizumab which blocks immunoglobulin (Ig)E (Busse et al., 2013b; Hanania et al., 2013; Chipps et al., 2017), and mepolizumab and reslizumab which bind and neutralize IL- 5 (Nair et al., 2009; Castro et al., 2012) were found to improve various asthmatic symptoms such as reducing exacerbation rates, free IgE levels, blood and sputum eosinophils and improved FEV1. However, their effects on ASM and AHR have not been identified. Bronchial thermoplasty is another novel treatment for adults with severe asthma that shows promising results (Dombret et al., 2014). With this technique, thermal energy is used to reduce the presence of ASM in the airways. The procedure is described to be minimally invasive, and patients in a 3 month follow-up have reported more symptom-free days, fewer exacerbations, and improved expiratory flow and airway responsiveness (Cox et al., 2007; Cox et al., 2012). However, a temporary worsening of respiratory symptoms was observed after the initial treatment period so there are concerns that the application of heat in the airways may inflict further injury on the bronchial wall (Dombret et al., 2014). Further studies are still required to explore the long-term effects of the procedure. As it is difficult to isolate ASM effects in patients, the benefit of many novel treatments on ASM remains unknown. As such, a greater understanding of ASM mechanics and its role in the airways will be beneficial for the identification and development of more targeted therapeutics.
Asthma is a multifactorial disease with alterations at several levels including changes to ASM contractility. Such changes derive from factors including airway hyperresponsiveness (which affects ASM sensitivity and reactivity to contractile agonists and reduces β2-receptor mediated relaxation) and airway remodeling (which results in increases in ASM mass that worsens the effect of bronchoconstriction). Alterations in calcium flux and dysregulation in the expression of signalling or contractile proteins occur by alterations at the transcriptional, translational, or post-translational levels possibly caused by heightened interactions with inflammatory mediators in asthmatic airways. The role of ASM in asthma is depicted through the measurement of its intrinsic mechanical features and how they change throughout disease pathogenesis. Multiple approaches have been used to understand the abnormalities of ASM in asthma, with each presenting different advantages and limitations. Ex vivo airway segments and precision cut lung slices may provide measurements that account for the interactions between ASM and other airway components while the ASM strip models focus on resolving important parameters of ASM mechanics including stress, length, and shortening velocity. ASM cell mechanics illustrate changes at a cellular level and can also be useful for providing an understanding of the changes at a molecular level through demonstrating the modifications that occur to contractile proteins and receptors. Current treatments for asthma may alleviate symptoms but cannot serve as a cure. Due to asthma having multiple phenotypes where each presents different underlying drivers of disease, conventional treatments are still insufficient in many cases and only work for specific types of the disease. Notwithstanding the insights gained to date, the elucidation of the root cause of the abnormal in vivo behaviour of ASM remains an important goal because ASM is the downstream effector of airway obstruction and AHR. A better understanding of the adverse changes that happen to ASM and its role in asthmatic airways may allow the discovery of therapeutics that provide a permanent cure and improved quality of life for asthmatics.
DX wrote the manuscript, JM and A-ML edited the manuscript.
Funds received for this article came from the Canadian Institutes of Health Research (CIHR) (PJT 156369).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Álvarez-Santos M. D., Álvarez-González M., Estrada-Soto S., Bazán-Perkins B. (2020). Regulation of myosin light-chain phosphatase activity to generate airway smooth muscle hypercontractility. Front. Physiol. 0, 701. doi:10.3389/fphys.2020.00701
Ammit A. J., Armour C. L., Black J. L. (2012). Smooth-muscle myosin light-chain kinase content is increased in human sensitized airways. Am. J. Respir. Crit. Care Med. 161, 257–263. doi:10.1164/ajrccm.161.1.9901005
Amrani Y., Krymskaya V., Maki C., Panettieri R. A. (1997). Mechanisms underlying TNF-α effects on agonist-mediated calcium homeostasis in human airway smooth muscle cells. Am. J. Physiol. 273, L1020–L1028. doi:10.1152/ajplung.1997.273.5.L1020
An S. S., Fabry B., Trepat X., Wang N., Fredberg J. J. (2006). Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am. J. Respir. Cell. Mol. Biol. 35, 55–64. doi:10.1165/rcmb.2005-0453OC
An S. S., Kim J., Ahn K., Trepat X., Drake K. J., Kumar S., et al. (2009). Cell stiffness, contractile stress and the role of extracellular matrix. Biochem. Biophys. Res. Commun. 382, 697–703. doi:10.1016/j.bbrc.2009.03.118
An S. S., Laudadio R. E., Lai J., Rogers R. A., Fredberg J. J. (2002). Stiffness changes in cultured airway smooth muscle cells. Am. J. Physiol. Cell. Physiol. 283, 792–801. doi:10.1152/ajpcell.00425.2001
An S. S., Mitzner W., Tang W. Y., Ahn K., Yoon A. R., Huang J., et al. (2016). An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. J. Allergy Clin. Immunol. 138, 294–297. doi:10.1016/j.jaci.2015.12.1315
Ansell T. K., McFawn P. K., Mitchell H. W., Noble P. B. (2013). Bronchodilatory response to deep inspiration in bronchial segments: The effects of stress vs. strain. J. Appl. Physiol. 115, 505–513. doi:10.1152/japplphysiol.01286.2012
Antonissen L. A., Mitchell R. W., Kroeger E. A., Kepron W., Tse K. S., Stephens N. L. (1979). Mechanical alterations of airway smooth muscle in a canine asthmatic model. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 46, 681–687. doi:10.1152/jappl.1979.46.4.681
Bai T. R. (1990). Abnormalities in airway smooth muscle in fatal asthma. Am. Rev. Respir. Dis. 141, 552–557. doi:10.1164/ajrccm/141.3.552
Bai Y., Sanderson M. J. (2009). The contribution of Ca2+ signaling and Ca2+ sensitivity to the regulation of airway smooth muscle contraction is different in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, 947–958. doi:10.1152/ajplung.90288.2008
Balassy Z., Mishima M., Bates J. H. T. (1995). Changes in regional lung impedance after intravenous histamine bolus in dogs: Effects of lung volume. J. Appl. Physiol. 78, 875–880. doi:10.1152/jappl.1995.78.3.875
Balzar S., Wenzel S. E., Chu H. W. (2002). Transbronchial biopsy as a tool to evaluate small airways in asthma. Eur. Respir. J. 20, 254–259. doi:10.1183/09031936.02.00261102
Bara I., Ozier A., Tunon De Lara J.-M., Marthan R., Berger P. (2010). Pathophysiology of bronchial smooth muscle remodelling in asthma. Eur. Respir. J. 36, 1174–1184. doi:10.1183/09031936.00019810
Barchasz E., NalinE E., MoliMardM. , Moreau J., GeOrges O., Emonds-Alt X., et al. (1999). Interleukin-1beta-induced hyperresponsiveness to [Sar9, Met(O2)11]substance P in isolated human bronchi. Eur. J. Pharmacol. 379, 87–95. doi:10.1016/s0014-2999(99)00484-7
Baroffio M., Barisione G., Crimi E., Brusasco V. (2009). Noninflammatory mechanisms of airway hyper-responsiveness in bronchial asthma: An overview. Ther. Adv. Respir. Dis. 3, 163–174. doi:10.1177/1753465809343595
Bates J. H. T., Martin J. G. (1990). A theoretical study of the effect of airway smooth muscle orientation on bronchoconstriction. J. Appl. Physiol. (1985) 69, 995–1001. doi:10.1152/jappl.1990.69.3.995
Bentley J. K., Hershenson M. B. (2008). Airway smooth muscle growth in asthma: Proliferation, hypertrophy, and migration. Proc. Am. Thorac. Soc. 5, 89–96. doi:10.1513/pats.200705-063VS
Berair R., Hollins F., Brightling C., Rousseau E. (2013). Airway smooth muscle hypercontractility in asthma. J. Allergy 2013, 185971. doi:10.1155/2013/185971
Berger P., Tunon-De-Lara J. M., Savineau J. P., Marthan R. (2001). Selected contribution: Tryptase-induced PAR-2-mediated Ca2+ signaling in human airway smooth muscle cells. J. Appl. Physiol. 91, 995–1003. doi:10.1152/jappl.2001.91.2.995
Billington C. K., Penn R. B. (2003). Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir. Res. 4, 4. doi:10.1186/1465-9921-4-2
Bjorck T., Gustafsson L. E., Dahlen S. E. (1992). Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am. Rev. Respir. Dis. 145, 1087–1091. doi:10.1164/ajrccm/145.5.1087
Black J. L., Marthan R., Armour C. L., Johnson P. R. A. (1989). Sensitization alters contractile responses and calcium influx in human airway smooth muscle. J. Allergy Clin. Immunol. 84, 440–447. doi:10.1016/0091-6749(89)90356-4
Blanc F. X., Coirault C., Salmeron S., Chemla D., Lecarpentier Y. (2003). Mechanics and crossbridge kinetics of tracheal smooth muscle in two inbred rat strains. Eur. Respir. J. 22, 227–234. doi:10.1183/09031936.03.00064203
Boie S., Chen J., Sanderson M. J., Sneyd J. (2017). The relative contributions of store‐operated and voltage‐gated Ca2+ channels to the control of Ca2+ oscillations in airway smooth muscle. J. Physiol. 595, 3129–3141. doi:10.1113/JP272996
Boivin R., Vargas A., Lefebvre-Lavoie J., Lauzon A.-M., Lavoie J.-P. (2014). Inhaled corticosteroids modulate the (+)insert smooth muscle myosin heavy chain in the equine asthmatic airways. Thorax 69, 1113–1119. doi:10.1136/thoraxjnl-2014-205572
Boulet L. P., LavioletteM. , Turcotte H., Cartier A., DugasM. , Malo J. L., et al. (1997). Bronchial subepithelial fibrosis correlates with airway responsiveness to methacholine. Chest 112, 45–52. doi:10.1378/chest.112.1.45
Bramley A. M., Thomson R. J., Roberts C. R., Schellenberg R. R. (1994). Hypothesis: Excessive bronchoconstriction in asthma is due to decreased airway elastance. Eur. Respir. J. 7, 337–341. doi:10.1183/09031936.94.07020337
Brightling C. E., Ammit A. J., Kaur D., Black J. L., Wardlaw A. J., Hughes J. M., et al. (2012). The CXCL10/CXCR3 Axis mediates human lung mast cell migration to asthmatic airway smooth muscle. Am. J. Respir. Crit. Care Med. 171, 1103–1108. doi:10.1164/rccm.200409-1220OC
Brink C., Grimaud C., Guillot C., Orehek J. (1980). The interaction between indomethacin and contractile agents on human isolated airway muscle. Br. J. Pharmacol. 69, 383–388. doi:10.1111/j.1476-5381.1980.tb07026.x
Brown R. H., Scichilone N., Mudge B., Diemer F. B., Permutt S., Togias A. (2001). High-resolution computed tomographic evaluation of airway distensibility and the effects of lung inflation on airway caliber in healthy subjects and individuals with asthma. Am. J. Respir. Crit. Care Med. 163, 994–1001. doi:10.1164/ajrccm.163.4.2007119
Bullimore S. R., Saunders T. J., Herzog W., Macintosh B. R. (2010). Calculation of muscle maximal shortening velocity by extrapolation of the force-velocity relationship: Afterloaded versus isotonic release contractions. Can. J. Physiol. Pharmacol. 88, 937–948. doi:10.1139/y10-068
Bullimore S. R., Siddiqui S., Donovan G. M., Martin J. G., Sneyd J., Bates J. H. T., et al. (2011). Could an increase in airway smooth muscle shortening velocity cause airway hyperresponsiveness? Am. J. Physiol. Lung Cell. Mol. Physiol. 300, 121–131. doi:10.1152/ajplung.00228.2010
Busse W., Spector S., Rosén K., Wang Y., Alpan O. (2013). High eosinophil count: A potential biomarker for assessing successful omalizumab treatment effects. J. Allergy Clin. Immunol. 132, 485–486. doi:10.1016/j.jaci.2013.02.032
Busse W. W., Holgate S., Kerwin E., Chon Y., Feng J. Y., Lin J., et al. (2013). Randomized, double-blind, placebo-controlled study of brodalumab, a human anti–IL-17 receptor monoclonal antibody.Moderate Severe Asthma 188, 1294–1302. doi:10.1164/rccm.201212-2318OC
Camoretti-Mercado B., Lockey R. F., Fla T. (2021). Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 147, 1983–1995. doi:10.1016/j.jaci.2021.03.035
Carroll N., Elliot J., Morton A., James A. (1993). The structure of large and small airways in nonfatal and fatal asthma. Am. Rev. Respir. Dis. 147, 405–410. doi:10.1164/ajrccm/147.2.405
Castro M., Mathur S., Hargreave F., Boulet L. P., Xie F., Young J., et al. (2012). Reslizumab for poorly controlled, eosinophilic asthma: A randomized, placebo-controlled study. Am. J. Respir. Crit. Care Med. 184, 1125–1132. doi:10.1164/rccm.201103-0396OC
Ceresa C. C., Knox A. J., Johnson S. R. (2009). Use of a three-dimensional cell culture model to study airway smooth muscle-mast cell interactions in airway remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, 1059–1066. doi:10.1152/ajplung.90445.2008
Cerrina J., Labat C., Haye-Legrande I., Raffestin B., Benveniste J., Brink C. (1989). Human isolated bronchial muscle preparations from asthmatic patients: Effects of indomethacin and contractile agonists. Prostaglandins 37, 457–469. doi:10.1016/0090-6980(89)90095-6
Chachi L., AlzAhrAni A., Koziol-White C., BiddleM. , Bagadood R., Panettieri R. A., et al. (2018). Increased β2‐adrenoceptor phosphorylation in airway smooth muscle in severe asthma: Possible role of mast cell‐derived growth factors. Clin. Exp. Immunol. 194, 253–258. doi:10.1111/cei.13191
Chai H., Farr R. S., Froehlich L. A., Mathison D. A., McLean J. A., Rosenthal R. R., et al. (1975). Standardization of bronchial inhalation challenge procedures. J. Allergy Clin. Immunol. 56, 323–327. doi:10.1016/0091-6749(75)90107-4
Chakrabarty K., Wu W., Booth J. L., Duggan E. S., Nagle N. N., Coggeshall K. M., et al. (2007). Human lung innate immune response to Bacillus anthracis spore infection. Infect. Immun. 75, 3729–3738. doi:10.1128/IAI.00046-07
Chen J., Sanderson M. J. (2017). Store‐operated calcium entry is required for sustained contraction and Ca2+ oscillations of airway smooth muscle. J. Physiol. 595, 3203–3218. doi:10.1113/JP272694
Chiba Y., Misawa M. (2004). The role of RhoA-mediated Ca 2+ sensitization of bronchial smooth muscle contraction in airway hyperresponsiveness. J. Smooth Muscle Res. 40, 155–167. doi:10.1540/jsmr.40.155
Chin L. Y. M., Bosse Y., Pascoe C., Hackett T. L., Seow C. Y., Pare P. D. (2012). Mechanical properties of asthmatic airway smooth muscle. Eur. Respir. J. 40, 45–54. doi:10.1183/09031936.00065411
Chipps B. E., Lanier B., Milgrom H., Deschildre A., Hedlin G., Szefler S. J., et al. (2017). Omalizumab in children with uncontrolled allergic asthma: Review of clinical trial and real-world experience. J. Allergy Clin. Immunol. 139, 1431–1444. doi:10.1016/j.jaci.2017.03.002
Coates A. L., Wanger J., Cockcroft D. W., Culver B. H., Diamant Z., Gauvreau G., et al. (2017). ERS technical standard on bronchial challenge testing: General considerations and performance of methacholine challenge tests. Eur. Respir. J. 49, 1601526. doi:10.1183/13993003.01526-2016
Cockcroft D. W., Davis B. E. (2006). Mechanisms of airway hyperresponsiveness. J. Allergy Clin. Immunol., 118 551–559. doi:10.1016/j.jaci.2006.07.012
Cockcroft D. W. (2010). Direct challenge tests: Airway hyperresponsiveness in asthma: Its measurement and clinical significance. Chest 138, 18S–24S. doi:10.1378/chest.10-0088
Cooper P. R., Kurten R. C., Zhang J., Nicholls D. J., Dainty I. A., Panettieri R. A. (2011). Formoterol and salmeterol induce a similar degree of β2-adrenoceptor tolerance in human small airways but via different mechanisms. Br. J. Pharmacol. 163, 521–532. doi:10.1111/j.1476-5381.2011.01257.x
Cooper P. R., Panettieri R. A. (2008). Steroids completely reverse albuterol-induced beta(2)-adrenergic receptor tolerance in human small airways. J. Allergy Clin. Immunol. 122, 734–740. doi:10.1016/j.jaci.2008.07.040
Corren J. (2013). Role of interleukin-13 in asthma. Curr. Allergy Asthma Rep. 13, 415–420. doi:10.1007/s11882-013-0373-9
Cox G., Miller J. D., McWilliams A., FitzGerald J. M., Lam S. (2012). Bronchial thermoplasty for asthma. Am. J. Respir. Crit. Care Med. 173, 965–969. doi:10.1164/rccm.200507-1162oc
Cox G., Thomson N. C., Rubin A. S., Niven R. M., Corris P. A., Siersted H. C., et al. (2007). Asthma control during the year after bronchial thermoplasty. N. Engl. J. Med. 356, 1327–1337. doi:10.1056/NEJMoa064707
Croisier H., Tan X., Chen J., Sneyd J., Sanderson M. J., Brook B. S. (2015). Ryanodine receptor sensitization results in abnormal calcium signaling in airway smooth muscle cells. Am. J. Respir. Cell. Mol. Biol. 53, 703–711. doi:10.1165/rcmb.2014-0386OC
Curry J. J. (1946). The action of histamine on the respiratory tract in normal and asthmatic subjects. J. Clin. Invest. 25, 785–791. doi:10.1172/JCI101764
Dandurand R. J., Wang C. G., Phillips N. C., Eidelman D. H. (1993). Responsiveness of individual airways to methacholine in adult rat lung explants. J. Appl. Physiol. 75, 364–372. doi:10.1152/jappl.1993.75.1.364
de Jongste J., Mons H., Bonta I., Kerrebijn K. (1987). In vitro responses of airways from an asthmatic patient. Eur. J. Respir. Dis. 71, 23–29.
De Magalhães Simões S., dos Santos M. A., da Silva OliveiraM. , Fontes E. S., Fernezlian S., Garippo A. L., et al. (2005). Inflammatory cell mapping of the respiratory tract in fatal asthma. Clin. Exp. Allergy 35, 602–611. doi:10.1111/j.1365-2222.2005.02235.x
Dekkers B. G. J., Bos I. S. T., Gosens R., Halayko A. J., Zaagsma J., Meurs H. (2012). The integrin-blocking peptide RGDS inhibits airway smooth muscle remodeling in a Guinea pig model of allergic asthma. Am. J. Respir. Crit. Care Med. 181, 556–565. doi:10.1164/rccm.200907-1065oc
Deshpande D. A., Dogan S., Walseth T. F., Miller S. M., Amrani Y., Panettieri R. A., et al. (2004). Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: Role of CD38/cyclic adenosine diphosphate ribose pathway. Am. J. Respir. Cell. Mol. Biol. 31, 36–42. doi:10.1165/rcmb.2003-0313OC
Doeing D. C., Solway J. (2013). Airway smooth muscle in the pathophysiology and treatment of asthma. J. Appl. Physiol. 114, 834–843. doi:10.1152/japplphysiol.00950.2012
Dombret M. C., Alagha K., Boulet L. P., Brillet P. Y., Joos G., Laviolette M., et al. (2014). Bronchial thermoplasty: A new therapeutic option for the treatment of severe, uncontrolled asthma in adults. Eur. Respir. Rev. 23, 510–518. doi:10.1183/09059180.00005114
Driessen J. M., Van Der Palen J., Van Aalderen W. M., De Jongh F. H., Thio B. J. (2012). Inspiratory airflow limitation after exercise challenge in cold air in asthmatic children. Respir. Med. 106, 1362–1368. doi:10.1016/j.rmed.2012.06.017
Dubois A. B., Botelho S. Y., Comroe J. H. (1955). A new method for measuring airway resistance in man using a body plethysmograph: Values in normal subjects and in patients with respiratory disease. J. Clin. Invest. 35, 327–335. doi:10.1172/JCI103282
Dubois A. B., Dautrebande L. (1958). Acute effects of breathing inert dust particles and of carbachol aerosol on the mechanical characteristics of the lungs in man; changes in response after inhaling sympathomimetic aerosols. J. Clin. Invest. 37, 1746–1755. doi:10.1172/JCI103767
Duguet A., Biyah K., Minshall E., Gomes R., Wang C. G., Taoudi-Benchekroun M., et al. (2000). Bronchial responsiveness among inbred mouse strains. Role of airway smooth-muscle shortening velocity. Am. J. Respir. Crit. Care Med. 161, 839–848. doi:10.1164/ajrccm.161.3.9906054
Dunnill M. S., Massarella G. R., Anderson J. A. (1969). A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, in chronic bronchitis, and in emphysema. Thorax 24, 176–179. doi:10.1136/thx.24.2.176
Eidelman D. H., DiMaria G. U., Bellofiore S., Wang N. S., Guttmann R. D., Martin J. G. (1991). Strain-related differences in airway smooth muscle and airway responsiveness in the rat. Am. Rev. Respir. Dis. 144, 792–796. doi:10.1164/ajrccm/144.4.792
Ellis J. L., Hubbard W. C., Meeker S., Undem B. J. (1994). Ragweed antigen E and anti-IgE in human central versus peripheral isolated bronchi. Am. J. Respir. Crit. Care Med. 150, 717–723. doi:10.1164/ajrccm.150.3.8087342
Fabry B., Maksym G. N., Butler J. P., GlogauerM. , Navajas D., Fredberg J. J. (2001). Scaling the microrheology of living cells. Phys. Rev. Lett. 87, 1481021–1481024. doi:10.1103/PhysRevLett.87.148102
Fan T., Yang M., Halayko A., Mohapatra S. S., Stephens N. L. (2012). Airway responsiveness in two inbred strains of mouse disparate in IgE and IL-4 production. Am. J. Respir. Cell. Mol. Biol. 17, 156–163. doi:10.1165/ajrcmb.17.2.2628
Fehrenbach H., Wagner C., Wegmann M. (2017). Airway remodeling in asthma: What really matters. Cell. Tissue Res. 367, 551–569. doi:10.1007/s00441-016-2566-8
Fish J. E., Ankin M. G., Kelly J. F., Peterman V. I. (1981). Regulation of bronchomotor tone by lung inflation in asthmatic and nonasthmatic subjects. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 50, 1079–1086. doi:10.1152/jappl.1981.50.5.1079
Gil F. R., Zitouni N. B., Azoulay E., Maghni K., Lauzon A. M. (2006). Smooth muscle myosin isoform expression and LC20 phosphorylation in innate rat airway hyperresponsiveness. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, 932–940. doi:10.1152/ajplung.00339.2004
Goldie R., Spina D., Henry P., Lulich K., Paterson (1986). In vitro responsiveness of human asthmatic bronchus to carbachol, histamine, beta-adrenoceptor agonists and theophylline. Br. J. Clin. Pharmacol. 22, 669–676. doi:10.1111/j.1365-2125.1986.tb02956.x
Goldsmith A. M., Bentley J. K., Zhou L., Jia Y., Bitar K. N., Fingar D. C., et al. (2006). Transforming growth factor-β induces airway smooth muscle hypertrophy. Am. J. Respir. Cell. Mol. Biol. 34, 247–254. doi:10.1165/rcmb.2005-0166OC
Gosens R., Zaagsma J., Meurs H., Halayko A. J. (2006). Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir. Res. 71, 73–15. doi:10.1186/1465-9921-7-73
Grunstein M. M., Hakonarson H., Leiter J., ChenM. , Whelan R., Grunstein J. S., et al. (2002). IL-13-dependent autocrine signaling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, 520–528. doi:10.1152/ajplung.00343.2001
Gunst S. J. (2012). “Airway smooth muscle and asthma,” in Fundamental biology and mechanisms of disease, 1359–1369. doi:10.1016/B978-0-12-381510-1.00104-6
Gunst S. J. (1983). Contractile force of canine airway smooth muscle during cyclical length changes. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 55, 759–769. doi:10.1152/jappl.1983.55.3.759
Gunst S. J., Mitzner W. (1981). Mechanical properties of contracted canine bronchial segments in vitro. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 50, 1236–1247. doi:10.1152/jappl.1981.50.6.1236
Hakonarson H., Maskeri N., Carter C., Chuang S., Grunstein M. M. (1999). Autocrine interaction between IL-5 and IL-1beta mediates altered responsiveness of atopic asthmatic sensitized airway smooth muscle. J. Clin. Invest. 104, 657–667. doi:10.1172/JCI7137
Hall I. P., Kotlikoff M. (1995). Use of cultured airway myocytes for study of airway smooth muscle. Am. J. Physiol. 268, L1–L11. doi:10.1152/ajplung.1995.268.1.L1
Hall I. P., Widdop S., Townsend P., Daykin K. (1992). Control of cyclic AMP levels in primary cultures of human tracheal smooth muscle cells. Br. J. Pharmacol. 107, 422–428. doi:10.1111/j.1476-5381.1992.tb12762.x
Hamid Q., Song Y., Kotsimbos T. C., Minshall E., Bai T. R., Hegele R. G., et al. (1997). Inflammation of small airways in asthma. J. Allergy Clin. Immunol. 100, 44–51. doi:10.1016/s0091-6749(97)70193-3
Han S., Son M., Lee Y., Cheon S. H., Kim S. W. (2020). 4-Methoxyphenyl (E)-3-(Furan-3-yl) acrylate inhibits vascular smooth muscle cell proliferation. J. Cardiovasc. Pharmacol. 76, 106–111. doi:10.1097/FJC.0000000000000831
Hanania N. A., Wenzel S., Rosen K., Hsieh H. J., Mosesova S., Choy D. F., et al. (2013). Exploring the effects of omalizumab in allergic asthma: An analysis of biomarkers in the EXTRA study. Am. J. Respir. Crit. Care Med. 187, 804–811. doi:10.1164/rccm.201208-1414OC
Henjakovic M., Sewald K., Switalla S., Kaiser D., MullerM. , Veres T. Z., et al. (2008). Ex vivo testing of immune responses in precision-cut lung slices. Toxicol. Appl. Pharmacol. 231, 68–76. doi:10.1016/j.taap.2008.04.003
Hirst S. J. (1996). Airway smooth muscle cell culture: Application to studies of airway wall remodelling and phenotype plasticity in asthma. Eur. Respir. J. 9, 808–820. doi:10.1183/09031936.96.09040808
Hirst S. J., Twort C. H. C., Lee T. H. (2012). Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am. J. Respir. Cell. Mol. Biol. 23, 335–344. doi:10.1165/ajrcmb.23.3.3990
Hunter I., Cobban H. J., Vandenabeele P., Macewan D. J., Nixon G. F. (2003). Tumor necrosis factor-α–induced activation of RhoA in airway smooth muscle cells: Role in the Ca2+ sensitization of myosin light Chain20 phosphorylation. Mol. Pharmacol. 63, 714–721. doi:10.1124/mol.63.3.714
Hyrkäs-Palmu H., Ikaheimo T. M., Laatikainen T., Jousilahti P., Jaakkola M. S., Jaakkola J. J. K. (2018). Cold weather increases respiratory symptoms and functional disability especially among patients with asthma and allergic rhinitis. Sci. Rep. 8, 10131. doi:10.1038/s41598-018-28466-y
Ijpma G., Kachmar L., Matusovsky O. S., Bates J. H. T., Benedetti A., Martin J. G., et al. (2015). Human trachealis and main bronchi smooth muscle are normoresponsive in asthma. Am. J. Respir. Crit. Care Med. 191, 884–893. doi:10.1164/rccm.201407-1296OC
Ijpma G., Kachmar L., Panariti A., Matusovsky O. S., Torgerson D., Benedetti A., et al. (2020). Intrapulmonary airway smooth muscle is hyperreactive with a distinct proteome in asthma. Eur. Respir. J. 56, 1902178. doi:10.1183/13993003.02178-2019
Jackson A. C., Murphy M. M., Rassulo J., Celli B. R., Ingram R. H. (2004). Deep breath reversal and exponential return of methacholine-induced obstruction in asthmatic and nonasthmatic subjects. J. Appl. Physiol. 96, 137–142. doi:10.1152/japplphysiol.00504.2003
James A. L., Elliot J. G., Jones R. L., Carroll M. L., Mauad T., Bai T. R., et al. (2012). Airway smooth muscle hypertrophy and hyperplasia in asthma. Am. J. Respir. Crit. Care Med. 185, 1058–1064. doi:10.1164/rccm.201110-1849OC
Jiang H., Rao K., Halayko A. J., Kepron W., Stephens N. L. (1992). Bronchial smooth muscle mechanics of a canine model of allergic airway hyperresponsiveness. J. Appl. Physiol. 72, 39–45. doi:10.1152/jappl.1992.72.1.39
Jiang H., Rao K., Liu X., Liu G., Stephens N. L. (1995). Increased Ca2+ and myosin phosphorylation, but not calmodulin activity in sensitized airway smooth muscles. Am. J. Physiol. 268, L739–L746. doi:10.1152/ajplung.1995.268.5.L739
Jiang H., Stephens N. L. (1990). Contractile properties of bronchial smooth muscle with and without cartilage. J. Appl. Physiol. 69, 120–126. doi:10.1152/jappl.1990.69.1.120
Johnson P. R. A., Black J. L., Carlin S., Ge Q., Underwood P. A. (2012). The production of extracellular matrix proteins by human passively sensitized airway smooth-muscle cells in culture: The effect of beclomethasone. Am. J. Respir. Crit. Care Med. 162, 2145–2151. doi:10.1164/ajrccm.162.6.9909111
Jude J. A., Wylam M. E., Walseth T. F., Kannan M. S. (2008). Calcium signaling in airway smooth muscle. Proc. Am. Thorac. Soc. 5, 15–22. doi:10.1513/pats.200704-047VS
Juniper E. F., Frith P. A., Hargreave F. E. (1981). Airway responsiveness to histamine and methacholine: Relationship to minimum treatment to control symptoms of asthma. Thorax 36, 575–579. doi:10.1136/thx.36.8.575
Kaminska M., Foley S., Maghni K., Storness-Bliss C., Coxson H., Ghezzo H., et al. (2009). Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J. Allergy Clin. Immunol. 124, 45–51. doi:10.1016/j.jaci.2009.03.049
Kim C. W., Kim J. S., Park J. W., Hong C.-S. (2001). Clinical applications of forced oscillation techniques (FOT) in patients with bronchial asthma. Korean J. Intern. Med. 16, 80–86. doi:10.3904/kjim.2001.16.2.80
King G. G., Bates J., Berger K. I., Calverley P., de Melo P. L., Dellaca R. L., et al. (2020). Technical standards for respiratory oscillometry. Eur. Respir. J. 55, 1900753. doi:10.1183/13993003.00753-2019
Kitazawa T., Eto M., Woodsome T. P., Brautigan D. L. (2000). Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 275, 9897–9900. doi:10.1074/jbc.275.14.9897
Koziol-White C. J., Yoo E. J., Cao G., Zhang J., Papanikolaou E., Pushkarsky I., et al. (2016). Inhibition of PI3K promotes dilation of human small airways in a rho kinase‐dependent manner. Br. J. Pharmacol. 173, 2726–2738. doi:10.1111/bph.13542
Kudo M., Melton A. C., Chen C., Engler M. B., Huang K. E., Ren X., et al. (2012). IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 18, 547–554. doi:10.1038/nm.2684
Kume H., Hall I. P., Washabau R. J., Takagi K., Kotlikoff M. (1994). Beta-adrenergic agonists regulate KCa channels in airway smooth muscle by cAMP-dependent and -independent mechanisms. J. Clin. Invest. 93, 371–379. doi:10.1172/JCI116969
Lam M., Lamanna E., Bourke J. E. (2019). Regulation of airway smooth muscle contraction in health and disease. Adv. Exp. Med. Biol. 1124, 381–422. doi:10.1007/978-981-13-5895-1_16
Laporte J. C., Moore P. E., Baraldo S., Jouvin M. H., Church T. L., Schwartzman I. N., et al. (2001). Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am. J. Respir. Crit. Care Med. 164, 141–148. doi:10.1164/ajrccm.164.1.2008060
Laporte J. D., Moore P. E., Panettieri R. A., Moeller W., Heyder J., Shore S. A. (1998). Prostanoids mediate IL-1beta-induced beta-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am. J. Physiol. 275, 491–501. doi:10.1152/ajplung.1998.275.3.L491
LaPrad A. S., Szabo T. L., Suki B., Lutchen K. R. (2010). Tidal stretches do not modulate responsiveness of intact airways in vitro. J. Appl. Physiol. 109, 295–304. doi:10.1152/japplphysiol.00107.2010
LaPrad A. S., West A. R., Noble P. B., Lutchen K. R., Mitchell H. W. (2008). Maintenance of airway caliber in isolated airways by deep inspiration and tidal strains. J. Appl. Physiol. 105, 479–485. doi:10.1152/japplphysiol.01220.2007
Latourelle J., Fabry B., Fredberg J. J. (2002). Dynamic equilibration of airway smooth muscle contraction during physiological loading. J. Appl. Physiol. 92, 771–779. doi:10.1152/japplphysiol.01090.2000
Leclere M., Lavoie-Lamoureux A., Lavoie J. P. (2011). Heaves, an asthma-like disease of horses. Respirology 16, 1027–1046. doi:10.1111/j.1440-1843.2011.02033.x
Lee R. T., Berditchevski F., Cheng G. C., Hemler M. E. (1995). Integrin-mediated collagen matrix reorganization by cultured human vascular smooth muscle cells. Circ. Res. 76, 209–214. doi:10.1161/01.res.76.2.209
Léguillette R., Laviolette M., Bergeron C., Zitouni N., Kogut P., Solway J., et al. (2009). Myosin, transgelin, and myosin light chain kinase: Expression and function in asthma. Am. J. Respir. Crit. Care Med. 179, 194–204. doi:10.1164/rccm.200609-1367OC
Li G., Cohen J. A., Martines C., Ram-Mohan S., Brain J. D., Krishnan R., et al. (2020). Preserving airway smooth muscle contraction in precision-cut lung slices. Sci. Rep. 101, 6480–6489. doi:10.1038/s41598-020-63225-y
Liu G., Betts C., Cunoosamy D. M., Aberg P. M., Hornberg J. J., Sivars K. B., et al. (2019). Use of precision cut lung slices as a translational model for the study of lung biology. Respir. Res. 201, 162–214. doi:10.1186/s12931-019-1131-x
Lourenço L. O., Ramos Lopes A. C., Zavan B., Soncini R. (2020). Vagotomy influences the lung response to adrenergic agonists and muscarinic antagonists. Respir. Physiol. Neurobiol. 274, 103358. doi:10.1016/j.resp.2019.103358
Lowell F. C. (1990). Observations on heaves: An asthma-like syndrome in the horse. Allergy Proc. 11, 149–150. doi:10.2500/108854190778880240
Luo L., Wang L., Paré P. D., Seow C. Y., Chitano P. (2019). The huxley crossbridge model as the basic mechanism for airway smooth muscle contraction. Am. J. Physiol. Lung Cell. Mol. Physiol. 317, L235–L246. doi:10.1152/ajplung.00051.2019
Lutchen K. R. (2016). Invited Editorial on ‘Measurement of intraindividual airway tone heterogeneity and its importance in asthma’: How does an airway and subsequently the lung become hyperresponsive? J. Appl. Physiol. 121, 221–222. doi:10.1152/japplphysiol.00444.2016
Ma X., Cheng Z., Kong H., Wang Y., Unruh H., Stephens N. L., et al. (2002). Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L1181–L1189. doi:10.1152/ajplung.00389.2001
Macklin C. C. (1929). The musculature of the bronchi and lungs. Physiol. Rev. 9, 1–60. doi:10.1152/physrev.1929.9.1.1
Mahn K., Hirst S. J., Ying S., Holt M. R., Lavender P., Ojo O. O., et al. (2009). Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc. Natl. Acad. Sci. U. S. A. 106, 10775–10780. doi:10.1073/pnas.0902295106
Manson M. L., Safholm J., James A., Johnsson A. K., Bergman P., Al-Ameri M., et al. (2020). IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J. Allergy Clin. Immunol. 145, 808–817. doi:10.1016/j.jaci.2019.10.037
Manuel Tunon-de-Lara Ousova J., Fayon M., Vernejoux J.-M., Amr El-Shazly R., Berger P., Girodet P.-O. (2022). Recruitment in asthma muscle cells contributes to mast cell fraktalkine produced by airway smooth. J. Immunol. Ref. 176, 1860–1868.
Marsh K. A., Hill S. J. (1992). Bradykinin B2 receptor-mediated phosphoinositide hydrolysis in bovine cultured tracheal smooth muscle cells. Br. J. Pharmacol. 107, 443–447. doi:10.1111/j.1476-5381.1992.tb12765.x
Martin C., Uhlig S., Ullrich V. (1996). Videomicroscopy of methacholine-induced contraction of individual airways in precision-cut lung slices. Eur. Respir. J. 9, 2479–2487. doi:10.1183/09031936.96.09122479
Mathé A. A., Hedqvist P., Holmgren A., Svanborg N. (1973). Bronchial hyperreactivity to prostaglandin F2α and histamine in patients with asthma. Br. Med. J. 1, 193–196. doi:10.1136/bmj.1.5847.193
Matsumoto H., Moir L. M., Oliver B. G. G., Burgess J. K., Roth M., Black J. L., et al. (2007). Comparison of gel contraction mediated by airway smooth muscle cells from patients with and without asthma. Thorax 62, 848–854. doi:10.1136/thx.2006.070474
Matusovsky O. S., Kachmar L., Ijpma G., Bates G., Zitouni N., Benedetti A., et al. (2016). Peripheral airway smooth muscle, but not the trachealis, is hypercontractile in an equine model of asthma. Am. J. Respir. Cell. Mol. Biol. 54, 718–727. doi:10.1165/rcmb.2015-0180OC
Matusovsky O. S., Nakada E. M., Kachmar L., Fixman E. D., Lauzon A. M. (2014). CD4+ T cells enhance the unloaded shortening velocity of airway smooth muscle by altering the contractile protein expression. J. Physiol. 592, 2999–3012. doi:10.1113/jphysiol.2014.270843
Mcneill R. S., Ingram C. G. (1966). Effect of propranolol on ventilatory function. Am. J. Cardiol. 18, 473–475. doi:10.1016/0002-9149(66)90072-5
Menzies-Gow A., Corren J., Bourdin A., Chupp G., Israel E., Wechsler M. E., et al. (2021). Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N. Engl. J. Med. 384, 1800–1809. doi:10.1056/NEJMoa2034975
Mitchell R. W., Ruhlmann E., Magnussen H., Leff A. R., Rabe K. F. (1994). Passive sensitization of human bronchi augments smooth muscle shortening velocity and capacity. Am. J. Physiol. 267, L218–L222. doi:10.1152/ajplung.1994.267.2.L218
Moir L. M., Ward J. P. T., Hirst S. J. (2003). Contractility and phenotype of human bronchiole smooth muscle after prolonged fetal bovine serum exposure. Exp. Lung Res. 29, 339–359. doi:10.1080/01902140303758
Murdoch J. R., Lloyd C. M. (2010). Chronic inflammation and asthma. Mutat. Res. 690, 24–39. doi:10.1016/j.mrfmmm.2009.09.005
Nadel J. A., Salem H., Tamplin B., Tokiwa Y. (1965). Mechanism of bronchoconstriction during inhalation of sulfur dioxide. J. Appl. Physiol. 20, 164–167. doi:10.1152/jappl.1965.20.1.164
Nair P., Pizzichini M. M. M., Kjarsgaard M., Inman M. D., Efthimiadis A., Pizzichini E., et al. (2009). Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 360, 985–993. doi:10.1056/NEJMoa0805435
Nie Z., Maung J. N., Jacoby D. B., Fryer A. D. (2020). Lung eosinophils increase vagus nerve-mediated airway reflex bronchoconstriction in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 318, L242–L251. doi:10.1152/ajplung.00040.2019
Noble P. B., McFawn P. K., Mitchell H. W. (2007). Responsiveness of the isolated airway during simulated deep inspirations: Effect of airway smooth muscle stiffness and strain. J. Appl. Physiol. 103, 787–795. doi:10.1152/japplphysiol.00314.2007
Noble P. B., West A. R., McLaughlin R. A., Armstrong J. J., Becker S., McFawn P. K., et al. (2010). Airway narrowing assessed by anatomical optical coherence tomography in vitro: Dynamic airway wall morphology and function. J. Appl. Physiol. 108, 401–411. doi:10.1152/japplphysiol.00511.2009
O’Byrne P. M., Inman M. D. (2003). Airway hyperresponsiveness. Chest 123, 411S–416S. doi:10.1378/chest.123.3_suppl.411s
Ojiaku C. A., Chung E., Parikh V., Williams J. K., Schwab A., Fuentes A. L., et al. (2019). Transforming growth factor-β1 decreases β2-Agonist-induced relaxation in human airway smooth muscle. Am. J. Respir. Cell. Mol. Biol. 61, 209–218. doi:10.1165/rcmb.2018-0301OC
Oliver M. N., Fabry B., Marinkovic A., Mijailovich S. M., Butler J. P., Fredberg J. J. (2007). Airway hyperresponsiveness, remodeling, and smooth muscle mass: Right answer, wrong reason? Am. J. Respir. Cell. Mol. Biol. 37, 264–272. doi:10.1165/rcmb.2006-0418OC
Oostveen E., MacLeoD D., Lorino H., FaRRe R., Hantos Z., Desager K., et al. (2003). The forced oscillation technique in clinical practice: Methodology, recommendations and future developments. Eur. Respir. J. 22, 1026–1041. doi:10.1183/09031936.03.00089403
Ouedraogo N., Roux E. (2003). Physiology of airway smooth muscle contraction: An overview. J. Pulm. Respir. Med. 284, 221. doi:10.4172/2161-105x.1000221
Ozier A. A., Allard B., Bara I., Girodet P. O., Trian T., Marthan R., et al. (2011). The pivotal role of airway smooth muscle in asthma pathophysiology. J. Allergy 201120, 742710. doi:10.1155/2011/742710
Panettieri R. A. (2002). Airway smooth muscle: An immunomodulatory cell. J. Allergy Clin. Immunol. 110, S269–S274.
Pascoe C. D., Wang L., Syyong H. T., Paré P. D. (2012). A brief history of airway smooth muscle’s role in airway hyperresponsiveness. J. Allergy 1–8, 768982. doi:10.1155/2012/768982
Patel M., Shaw D. (2015). A review of standard pharmacological therapy for adult asthma - steps 1 to 5. Chron. Respir. Dis. 12, 165–176. doi:10.1177/1479972315573529
Pepe C., Foley S., Shannon J., Lemiere C., Olivenstein R., Ernst P., et al. (2005). Differences in airway remodeling between subjects with severe and moderate asthma. J. Allergy Clin. Immunol. 116, 544–549. doi:10.1016/j.jaci.2005.06.011
Perez-Zoghbi J. F., Karner C., Ito S., Shepherd M., Alrashdan Y., Sanderson M. J. (2009). Ion channel regulation of intracellular calcium and airway smooth muscle function. Pulm. Pharmacol. Ther. 22, 388–397. doi:10.1016/j.pupt.2008.09.006
Peters-Golden M., Henderson W. R. (2007). Leukotrienes. N. Engl. J. Med. 357, 1841–1854. doi:10.1056/NEJMra071371
Ram J., Pineda-Cely J., Calhoun W. J. (2019). Forced oscillometry: A new tool for assessing airway function-is it ready for prime time? J. Allergy Clin. Immunol. Pract. 7, 2861–2862. doi:10.1016/j.jaip.2019.07.037
Ramos-Barbón D., Presley J. F., Hamid Q. A., Fixman E. D., Martin J. G. (2005). Antigen-specific CD4+ T cells drive airway smooth muscle remodeling in experimental asthma. J. Clin. Invest. 115, 1580–1589. doi:10.1172/JCI19711
Risse P. A., Jo T., Suarez F., Hirota N., Tolloczko B., Ferraro P., et al. (2011). Interleukin-13 inhibits proliferation and enhances contractility of human airway smooth muscle cells without change in contractile phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 300, 958–966. doi:10.1152/ajplung.00247.2010
Roberts J. A., Raeburn D., Rodger I. W., Thomson N. C. (1984). Comparison of in vivo airway responsiveness and in vitro smooth muscle sensitivity to methacholine in man. Thorax 39, 837–843. doi:10.1136/thx.39.11.837
Robinson D., HuMbertM. , Buhl R., Cruz A. A., Inoue H., Korom S., et al. (2017). Revisiting type 2-high and type 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clin. Exp. Allergy 47, 161–175. doi:10.1111/cea.12880
Sanders K. M. (2001). Invited review: Mechanisms of calcium handling in smooth muscles. J. Appl. Physiol. 91, 1438–1449. doi:10.1152/jappl.2001.91.3.1438
Sanderson M. J. (2011). Exploring lung physiology in health and disease with lung slices. Pulm. Pharmacol. Ther. 24, 452–465. doi:10.1016/j.pupt.2011.05.001
Sassone-Corsi P. (2012). The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 4, a011148. doi:10.1101/cshperspect.a011148
Schellenberg R. R., Foster A. (1984). In vitro responses of human asthmatic airway and pulmonary vascular smooth muscle. Int. Arch. Allergy Appl. Immunol. 75, 237–241. doi:10.1159/000233622
Seow C. Y. (2021). An introduction to smooth muscle mechanics. Second. Newcastle Upon Tyne: Cambridge Scholars Publishing.
Seow C. Y., Schellenberg R. R., Pare P. D. (2012). Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am. J. Respir. Crit. Care Med. 158, 179–186. doi:10.1164/ajrccm.158.supplement_2.13tac160
Seow C. Y., Solway J. (2011). Mechanical and structural plasticity. Compr. Physiol. 1, 283–293. doi:10.1002/cphy.c100024
Seow C. Y., Stephens N. L. (1986). Force-velocity curves for smooth muscle: Analysis of internal factors reducing velocity. Am. J. Physiol. 251, 362–368. doi:10.1152/ajpcell.1986.251.3.C362
Shore S. A., Bai T. R., Wang C. G., Martin J. G. (1985). Central and local cholinergic components of histamine-induced bronchoconstriction in dogs. J. Appl. Physiol. (1985) 58, 443–451. doi:10.1152/jappl.1985.58.2.443
Shore S. A. (2002). Cytokine regulation of β-adrenergic responses in airway smooth muscle. J. Allergy Clin. Immunol. 110, S255–S260. doi:10.1067/mai.2002.129947
Shore S. A. (1984). Factors modulating histamine induced contraction of canine airway. McGill University.
Shore S., Irvin C. G., Shenkier T., Martin J. G. (1983). Mechanisms of histamine-induced contraction of canine airway smooth muscle. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 55, 22–26. doi:10.1152/jappl.1983.55.1.22
Sieck G. C., White T. A., Thompson M. A., Pabelick C. M., Wylam M. E., Prakash Y. S. (2008). Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, 378–385. doi:10.1152/ajplung.00394.2007
Skloot G., Permutt S., Togias A. (1995). Airway hyperresponsiveness in asthma: A problem of limited smooth muscle relaxation with inspiration. J. Clin. Invest. 96, 2393–2403. doi:10.1172/JCI118296
Soler M., Sielczak M., Abraham W. M. (1991). Separation of late bronchial responses from airway hyperresponsiveness in allergic sheep. J. Appl. Physiol. 70, 617–623. doi:10.1152/jappl.1991.70.2.617
Solway J., Fredberg J. J. (1997). Perhaps airway smooth muscle dysfunction contributes to asthmatic bronchial hyperresponsiveness after all. Am. J. Respir. Cell. Mol. Biol. 17 (2), 144–146. doi:10.1165/ajrcmb.17.2.f137
Somlyo A. P., Somlyo A. V. (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358. doi:10.1152/physrev.00023.2003
Stephens N. L., Van Niekerk W. (1977). Isometric and isotonic contractions in airway smooth muscle. Can. J. Physiol. Pharmacol. 55, 833–838. doi:10.1139/y77-112
Sterk J., Daniel E. E., Zamel N., Hargreave F. E. (1985). Limited maximal airway narrowing in nonasthmatic subjects. Role of neural control and prostaglandin release. Am. Rev. Respir. Dis. 132, 865–870. doi:10.1164/arrd.1985.132.4.865
Suarez C. J., Dintzis S. M., Frevert C. W. (2012). Respiratory. Comp. Anat. Histol., 121–134. doi:10.1016/B978-0-12-381361-9.00009-3
Sukkar M. B., Issa R., Xie S., Oltmanns U., Newton R., Chung K. F. (2004). Fractalkine/CX3CL1 production by human airway smooth muscle cells: Induction by IFN-γ and TNF-α and regulation by TGF-β and corticosteroids. Am. J. Physiol. 287, 1230–1240. doi:10.1152/ajplung.00014.2004
Sutcliffe A., Hollins F., Gomez E., Saunders R., Doe C., Cooke M., et al. (2012). Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am. J. Respir. Crit. Care Med. 185, 267–274. doi:10.1164/rccm.201107-1281OC
Sy C. B., Siracusa M. C. (2016). The therapeutic potential of targeting cytokine alarmins to treat allergic airway inflammation. Front. Physiol. 7, 214. doi:10.3389/fphys.2016.00214
Takeya K., Loutzenhiser K., Shiraishi M., Loutzenhiser R., Walsh M. P. (2008). A highly sensitive technique to measure myosin regulatory light chain phosphorylation: The first quantification in renal arterioles. Am. J. Physiol. Ren. Physiol. 294, 1487–1492. doi:10.1152/ajprenal.00060.2008
Tao F. C., Shah S., Pradhan A. A., Tolloczko B., Martin J. G. (2003). Enhanced calcium signaling to bradykinin in airway smooth muscle from hyperresponsive inbred rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 284, L90–L99. doi:10.1152/ajplung.00023.2002
Tao F. C., Tolloczko B., Eidelman D. H., Martin J. C. (1999). Enhanced Ca2+ mobilization in airway smooth muscle contributes to airway hyperresponsiveness in an inbred strain of rat. Am. J. Respir. Crit. Care Med. 160, 446–453. doi:10.1164/ajrccm.160.2.9811098
Tao F. C., Tolloczko B., Mitchell C. A., Powell W. S., Martin J. G. (2012). Inositol (1,4,5)Trisphosphate metabolism and enhanced calcium mobilization in airway smooth muscle of hyperresponsive rats. Am. J. Respir. Cell. Mol. Biol. 23, 514–520. doi:10.1165/ajrcmb.23.4.3966
Tgavalekos N. T., Tawhai M., Harris R. S., Musch G., Mush G., Vidal-Melo M., et al. (2005). Identifying airways responsible for heterogeneous ventilation and mechanical dysfunction in asthma: An image functional modeling approach. J. Appl. Physiol. 99, 2388–2397. doi:10.1152/japplphysiol.00391.2005
Thomson R. J., Bramley A. M., Schellenberg R. R. (1996). Airway muscle stereology: Implications for increased shortening in asthma. Am. J. Respir. Crit. Care Med. 154, 749–757. doi:10.1164/ajrccm.154.3.8810615
Tschumperlin D. J., Dai G., Maly I. V., Kikuchi T., Laiho L. H., McVittie A. K., et al. (2004). Mechanotransduction through growth-factor shedding into the extracellular space. Nature 429, 83–86. doi:10.1038/nature02543
Tunon de Lara J. M., Okayama Y., Savineau J. P., Marthan R. (1995). IgE-induced passive sensitization of human isolated bronchi and lung mast cells. Eur. Respir. J. 8, 1861–1865. doi:10.1183/09031936.95.08111861
Tzeng Y.-S., Lutchen K., Albert M. (2009). The difference in ventilation heterogeneity between asthmatic and healthy subjects quantified using hyperpolarized 3He MRI. J. Appl. Physiol. 106, 813–822. doi:10.1152/japplphysiol.01133.2007
Wagner E. M., Jacoby D. B. (1999). Methacholine causes reflex bronchoconstriction. J. Appl. Physiol. 86, 294–297. doi:10.1152/jappl.1999.86.1.294
Wang J. H. C., Li B. (2009). Application of cell traction force microscopy for cell biology research. Methods Mol. Biol. 586, 301–313. doi:10.1007/978-1-60761-376-3_17
Wang K., Tian P., Fan Y., Wang Y., Liu C. (2015). Assessment of second-line treatments for patients with uncontrolled moderate asthma. Int. J. Clin. Exp. Med. 8, 19476–19480.
Wang L., Paré P. D., Seow C. Y. (2000). Effects of length oscillation on the subsequent force development in swine tracheal smooth muscle. J. Appl. Physiol. 88, 2246–2250. doi:10.1152/jappl.2000.88.6.2246
Wang N., Ingber D. E. (1994). Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 66, 2181–2189. doi:10.1016/S0006-3495(94)81014-8
Watanabe A., Renzi P. M., Xu L. J., Guttmann R. D., Martin J. G. (1995). Adoptive transfer of allergic airway responses with sensitized lymphocytes in BN rats. Am. J. Respir. Crit. Care Med. 152, 64–70. doi:10.1164/ajrccm.152.1.7599864
Welsh G. I., Miller C. M., Loughlin A. J., Price N. T., Proud C. G. (1998). Regulation of eukaryotic initiation factor eIF2B: Glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 421, 125–130. doi:10.1016/s0014-5793(97)01548-2
Wenzel S., Castro M., Corren J., Maspero J., Wang L., Zhang B., et al. (2016). Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: A randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 388, 31–44. doi:10.1016/S0140-6736(16)30307-5
Whicker S. D., Armour C. L., Black J. L. (1988). Responsiveness of bronchial smooth muscle from asthmatic patients to relaxant and contractile agonists. Pulm. Pharmacol. 1, 25–31. doi:10.1016/0952-0600(88)90007-5
Widmaier E. P., Raff H., Strang K. T., Shoepe T. C. (2019). Vander’s human Physiology. McGraw-Hill Education.
Willis C. R., Siegel L., Leith A., Mohn D., Escobar S., Wannberg S., et al. (2015). IL-17RA signaling in airway inflammation and bronchial hyperreactivity in allergic asthma. Am. J. Respir. Cell. Mol. Biol. 53, 810–821. doi:10.1165/rcmb.2015-0038OC
Woolcock A. J., Salome C. M., Yan K. (1984). The shape of the dose-response curve to histamine in asthmatic and normal subjects. Am. Rev. Respir. Dis. 130, 71–75. doi:10.1164/arrd.1984.130.1.71
Wright D., Sharma P., Ryu M. H., Risse P. A., Ngo M., Maarsingh H., et al. (2013). Models to study airway smooth muscle contraction in vivo, ex vivo and in vitro: Implications in understanding asthma. Pulm. Pharmacol. Ther. 26, 24–36. doi:10.1016/j.pupt.2012.08.006
Wu W., Booth J. L., Duggan E. S., Patel K. B., Coggeshall K. M., Metcalf J. P. (2010). Human lung innate immune cytokine response to adenovirus type 7. J. Gen. Virol. 91, 1155–1163. doi:10.1099/vir.0.017905-0
Xia Y. C., Redhu N. S., Moir L. M., Koziol-White C., Ammit A. J., Al-Alwan L., et al. (2013). Pro-inflammatory and immunomodulatory functions of airway smooth muscle: Emerging concepts. Pulm. Pharmacol. Ther. 26, 64–74. doi:10.1016/j.pupt.2012.05.006
Yoo E. J., Cao G., Koziol-White C. J., Ojiaku C. A., Sunder K., Jude J. A., et al. (2017). Gα12 facilitates shortening in human airway smooth muscle by modulating phosphoinositide 3‐kinase‐mediated activation in a RhoA‐dependent manner. Br. J. Pharmacol. 174, 4383–4395. doi:10.1111/bph.14040
Yoo E. J. (2017). Targeting calcium sensitization mechanisms in human airway smooth muscle for novel bronchodilator therapy in asthma. ProQuest Diss. Theses.
Zhou L., Goldsmith A. M., Bentley J. K., Jia Y., Rodriguez M. L., Abe M. K., et al. (2005). 4E-Binding protein phosphorylation and eukaryotic initiation factor-4E release are required for airway smooth muscle hypertrophy. Am. J. Respir. Cell. Mol. Biol. 33, 195–202. doi:10.1165/rcmb.2004-0411OC
Zhu L., Ciaccio C. E., Casale T. B. (2018). Potential new targets for drug development in severe asthma. World Allergy Organ. J. 11, 30. doi:10.1186/s40413-018-0208-1
Keywords: airway smooth muscle, airway hyperresponsiveness, asthma, airway smooth muscle contraction, airway remodeling
Citation: Xiong D(JP), Martin JG and Lauzon A-M (2022) Airway smooth muscle function in asthma. Front. Physiol. 13:993406. doi: 10.3389/fphys.2022.993406
Received: 13 July 2022; Accepted: 14 September 2022;
Published: 05 October 2022.
Edited by:
M. Teresa Perez-Garcia, University of Valladolid, SpainReviewed by:
Mai ElMallah, Duke University, United StatesCopyright © 2022 Xiong, Martin and Lauzon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne-Marie Lauzon, YW5uZS1tYXJpZS5sYXV6b25AbWNnaWxsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.