
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 15 August 2022
Sec. Integrative Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.962769
This article is part of the Research TopicSirtuins and Brain HomeostasisView all 6 articles
 Mateusz Watroba*
Mateusz Watroba* Dariusz Szukiewicz
Dariusz SzukiewiczBoth basic pathomechanisms underlying Alzheimer’s disease and some premises for stipulating a possible preventive role of some sirtuins, especially SIRT1 and SIRT3, protective against Alzheimer’s disease-related pathology, are discussed in this article. Sirtuins can inhibit some processes that underlie Alzheimer’s disease-related molecular pathology (e.g., neuroinflammation, neuroinflammation-related oxidative stress, Aβ aggregate deposition, and neurofibrillary tangle formation), thus preventing many of those pathologic alterations at relatively early stages of their development. Subsequently, the authors discuss in details which mechanisms of sirtuin action may prevent the development of Alzheimer’s disease, thus promoting brain homeostasis in the course of aging. In addition, a rationale for boosting sirtuin activity, both with allosteric activators and with NAD+ precursors, has been presented.
Alzheimer’s disease (AD) is a neurodegenerative disorder, clinically manifesting with a progressive loss of memory and cognitive functions (Scheltens et al., 2016; Fernando and Wijayasinghe, 2021). Histopathologic findings in AD patients’ brains usually occur much earlier than clinical manifestations of the disease; they include β-amyloid deposition in the interneural space and accumulation of abnormal, hyperphosphorylated tau proteins within neurons (Sperling et al., 2011; Kumar et al., 2015; Hanseeuw et al., 2019).
β-amyloid deposits, referred to as Aβ aggregates, are produced during degradation of amyloid-precursor protein (APP) which is quite large transmembrane glycoprotein, cleaved by β- and γ-secretases to about 40-aminoacid peptides called Aβ monomers (Zheng and Koo, 2011; Chen et al., 2017). APP protein itself is quite abundant in the brain, playing a signaling role in neuronal development, maintenance of synapses, and neuronal homeostasis (van der Kant and Goldstein, 2015). Some Aβ monomers tend to condensate into insoluble oligomers, in the form of fibrils or plaques. Senile plaques, very characteristic for AD, contain mainly fibrillary proteins referred to as Aβ1-42. Aβ neurotoxicity is attributed mainly to its soluble oligomeric form, which is capable to disrupt intraneuronal calcium homeostasis through causing an excessive calcium influx into the neurons, with a subsequent mitochondrial damage and neuronal death (Arispe et al., 1993; Zhao et al., 2012; Colvin et al., 2016; Wälti et al., 2016) (Supplementary Data Sheet S1). In some mouse models of AD, progressive deposition of Aβ aggregates is found mainly in hippocampus and cerebral cortex (Zhao et al., 2012). Intraneuronal deposition of abnormal tau proteins can usually be observed within a few years after the onset of Aβ deposition in the interneural space (Musiek and Holtzman, 2015; Sasaguri et al., 2017). Tau protein itself is a microtubule-associated protein, stabilizing the microtubules and thus significant for axonal transport. Abnormal phosphorylation of tau proteins results in their dissociation from microtubules and formation of fibrillary structures called neurofibrillary tangles (NFTs) (Iqbal et al., 2010; Wang and Mandelkow, 2016). Intraneuronal accumulation of NFTs results in neuronal malfunction, followed by neuronal death.
In addition, abnormalities in cerebral metabolism of cholesterol have been found in Alzheimer’s disease (Feringa and van der Kant, 2021). Accumulation of cholesterol within neurons promotes APP interactions with β- and γ-secretases, resulting in the production of aforementioned Aβ aggregates (Di Paolo and Kim, 2011). Because apolipoprotein E regulates cholesterol transport to the brain and lipid rafts function in astrocytes, a positive correlation can be found between AD risk and possessing certain alleles of apolipoprotein E-encoding gene (Corder et al., 1993; Wang et al., 2021).
In addition to accumulation of abnormal protein aggregates, neuroinflammation—i.e., inflammation within the central nervous system (CNS) also plays a role in the pathogenesis of AD (Heneka et al., 2015; Heppner et al., 2015; Calsolaro and Edison, 2016; Hampel et al., 2020). Neuroinflammatory response is an element of innate immunity, dependent on many types of cells, including microglial cells, astrocytes, cerebral vascular endothelial cells, mast cells and leukocytes reaching the cerebrospinal fluid through abnormally permeable blood-brain barrier (Das Sarma, 2014; 't Hart and den Dunnen, 2013). However, in the further part of this work, we focus mainly on microglial cells and astrocytes (Colombo and Farina, 2016; Ransohoff, 2016). The problem with neuroinflammation is that it can be potentially neurotoxic in its chronic form, despite being useful and neuroprotective in its acute form, through removal of pathogens from the brain (Kinney et al., 2018). Pro-inflammatory activity of microglial cells tends to increase with age, resulting in the increased production of pro-inflammatory mediators, inducing neuroinflammation, and increased permeability of blood-brain barrier (Schuitemaker et al., 2012; Elahy et al., 2015). Microglial cells obtained from old people show abnormalities in their morphology and function, impairing phagocytosis, proteostasis, and cell capability for migration (Mosher and Wyss-Coray, 2014). Furthermore, neuroinflammation can be additionally aggravated by the presence of abnormal protein aggregates, such as Aβ aggregates (Zenaro et al., 2017). In AD patients, a positive correlation is observed between the abundance of Aβ aggregates and intraneuronal deposits of tau proteins, and the extent of pro-inflammatory phenotype induction in microglial cells and blood-brain barrier permeability (Parbo et al., 2017; Dani et al., 2018; Nordengen et al., 2019). Microglial cells which have transited from their homeostatic phenotype to pro-inflammatory phenotype are located mainly in the vicinity of senile plaques (Heneka et al., 2015; Navarro et al., 2018). Aβ aggregates are responsible for such phenotypic transition of microglial cells, which results in the induction of many pro-inflammatory mediators promoting neuronal deaths, such as IL-1β, IL-6, TNF-α, chemokines, nitric oxide and prostaglandins (Glass et al., 2010). In addition, it is supposed that neuroinflammation can promote NFT formation in AD patients (Kitazawa et al., 2004; Kinney et al., 2018). Moreover, elevated concentrations of circulating pro-inflammatory cytokines (IL-1, IL-6, TNF-α) have been found in people suffering from dementia (Koyama et al., 2013; Lai et al., 2017; Darweesh et al., 2018; Shen et al., 2019).
Microglial cells comprise a component of innate immunity and are basically derived from macrophages (Ginhoux et al., 2010). Main functions of their homeostatic, phenotypically quiescent forms in healthy brain include elimination of pathogens, repairing tissue damage, immune surveillance, and homeostatic functions (maintenance of neurogenesis, neuronal plasticity, and synaptic well-being, thus promoting proper cognitive skills) (Nimmerjahn et al., 2005; Ji et al., 2013; Parkhurst et al., 2013). Phenotype of microglial cells can be switched from homeostatic to pro-inflammatory by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), such as lipopolysaccharides of bacterial walls (LPS), misfolded proteins, or even some pesticides and air pollutants (Lull and Block, 2010). Such phenotypically switched microglial cells change their morphology, as well as activate phagocytosis and inflammation-associated signaling pathways (ElAli and Rivest, 2016; Minter et al., 2016; Salter and Stevens, 2017). In addition, the extent of this kind of phenotypic alteration of microglial cells increases with age, along with other innate immunity associated pro-inflammatory phenomena, such as toll-like receptor (TLR) signaling and inflammasome activation (Sierra et al., 2007; Cribbs et al., 2012). Several kinds of receptors mediate microglial cell phenotypic transition from homeostatic to pro-inflammatory, including TLRs, nucleotide-binding oligomerization domain-like receptors (NLRs), receptors for advanced glycation products, formyl peptide receptors, scavenger receptors, and receptors for immunoglobulin Fc fragments and complement components (Doens and Fernández, 2014; Fiebich et al., 2018). In the course of Alzheimer’s disease, microglial cells interact with Aβ aggregates through such receptors as TLR2, TLR4, TLR6, TLR9, scavenger receptors such as CD36, CD37, and scavenger receptor A1 (SR-A1), as well as receptors for advanced glycation products and complement components, like complement receptor 3 (CR3) (Doens and Fernández, 2014; Fiebich et al., 2018). In the course of aging, as well as in metabolic syndrome-associated systemic inflammation, microglial cells can be abnormally recruited to induce neuroinflammation (Perry and Teeling, 2013; Niraula et al., 2017). Both metabolic syndrome-associated systemic inflammation and aging-associated systemic inflammation are characterized by increased plasma concentrations of IL-6 and TNF-α, which is positively correlated with cognitive impairment and AD-resembling symptoms. This may suggest that such symptoms are mediated by phenotypic switching of microglial cells from their homeostatic/surveillant phenotype to pro-inflammatory phenotype, due to increased systemic concentration of pro-inflammatory cytokines (Holmes et al., 2011).
Studies on mouse models of AD indicate that LPS-stimulated microglial cells produce increased amounts of IL-1β which in turn stimulates astrocytes to produce chemokines, such as chemokine C-C motif ligand 2 (CCL2), chemokine C-X-C motif ligand 1 (CXCL1) and chemokine C-X-C motif ligand 10 (CXCL10) (Lopez-Rodriguez et al., 2021). Results of those studies suggest that microglia-astrocyte interactions in response to microglial cell acquiring a pro-inflammatory phenotype may aggravate neuroinflammation, which can sometimes result in the impairment of cognitive functions (González-Reyes et al., 2017). Microglial cells which have acquired a pro-inflammatory phenotype in Aβ-dependent manner may adhere to the sites of Aβ deposition as disease-associated microglia (DAM) (Keren-Shaul et al., 2017; Shahidehpour et al., 2021). In early stages of AD, DAM cells can be useful, removing Aβ aggregates in triggering receptor expressed on myeloid cells 2 (TREM2)-dependent manner (Keren-Shaul et al., 2017; Ulland and Colonna, 2018). Possessing some rarely occurring alleles of TREM2 encoding gene is a risk factor of late-onset Alzheimer’s disease (Gratuze et al., 2018). In the course of aging, replicative stress imposed on microglial cells can hinder their efficacy in Aβ clearance, which may promote Aβ deposits growing larger (Hu et al., 2021). This may in turn functionally overload microglial cells, decreasing the effectiveness of phagocytosis because of reduced expression of Aβ-binding proteins, such as SR-A1, CD36 and receptor for advanced glycation products (RAGE), as well as reduced expression of Aβ-degrading enzymes (Hickman et al., 2008). Overaccumulation of Aβ aggregates in DAM cells may also promote neuroinflammation through stimulating the expression of pro-inflammatory mediators (IL-1, IL-6, TNF-alfa) as well as other neurotoxic substances that can promote the progress of AD (e.g., nitric oxide and superoxide anion) (Block et al., 2007; Hickman et al., 2008; Smith et al., 2012; Deczkowska et al., 2018). Furthermore, intracellular accumulation of Aβ aggregates may result in microglial cell necrosis, with a subsequent release of Aβ aggregates back to the extracellular space, which can further promote Aβ deposits enlargement (Baik et al., 2016). Phagocytic efficacy of microglial cells can be restored by reducing Aβ burden (Krabbe et al., 2013). Since chronic and excessive imposing of pro-inflammatory phenotype on microglia promotes formation of neurofibrillary tangles (intraneuronal deposits of tau proteins), moderating this kind of microglial cell phenotypic transition is considered to be a potentially useful strategy in the prevention and treatment of Alzheimer’s disease (Kitazawa et al., 2004).
Aβ binding to TLR receptors on microglial cells activates the same signaling pathways as are generally used for pathogen destruction. Directly, it can activate myeloid differentiation primary response (Myd88) transcription factor, which can transactivate other transcription factors, including nuclear factor kappa B (NF-κB) (Kawai and Akira, 2007). Active NF-κB may in turn promote the production of pro-IL-1β and NLR family pyrin domain containing 3 (NLRP3) cytoplasmic receptor (Bauernfeind et al., 2009). IL-1β is the main pro-inflammatory cytokine associated with neuroinflammation in the course of Alzheimer’s disease (Shaftel et al., 2008). In addition, it has been found that increased IL-1β expression in human microglial cells in the course of aging is underlied by a selective hypomethylation of IL-1β gene proximal promoter (Cho et al., 2015). However, IL-1β is synthesized in the form of inactive precursor—pro-IL-1β which can be transformed to IL-1β in the presence of active caspase 1, an intracellular pro-inflammatory caspase (Halle et al., 2008). Caspase 1 is also produced in the form of its inactive precursor—procaspase 1, and transforming of procaspase 1 to caspase 1 requires its proteolytic processing in inflammasomes, the most important being NLRP3 inflammasome. NLRP3 inflammasome consists of NLRP3 receptor, apoptosis-associated speck-like protein containing a CARD (ASC protein), and caspase 1 protease. Inflammasomes are responsible for detection of potential tissue insults and inducing an inflammatory response if such insults are indeed detected. NLRP3 inflammasome can be stimulated by several factors at the level of inflammasome assembly activation. Those factors include potassium efflux from intracellular fluids, reactive oxygen species (ROS), mitochondrial and phagolysosomal damage, as well as pathogens, such as bacteria, viruses, fungi and parasites (He et al., 2016; Zheng et al., 2020). Overexpression of active caspase 1 in microglial cells has been found in patients suffering from mild cognitive impairment or AD (Heneka et al., 2013). In addition, NLRP3 receptor expression is also transcriptionally controlled by NF-κB (Bauernfeind et al., 2009). It has been recently found that fibrillary Aβ aggregates can directly activate NLRP3 inflammasomes in microglial cells, which promotes caspase 1 activation (Nakanishi et al., 2018; Lučiūnaitė et al., 2020). Moreover, it has been confirmed that NLRP3 inflammasome indeed contributes to Aβ deposits formation in mice (Venegas and Heneka, 2019). In physiology, IL-1β can increase core body temperature through stimulation of thermoregulation center in the hypothalamus. In addition, IL-1β can promote sleep and sickness behavior in response to infections. While small amounts of IL-1β can promote long term potentiation (LTP) and thus acquisition of cognitive skills, large amounts of IL-1β are thought to be detrimental in the course of AD, mainly through suppression of hippocampal neurogenesis (Hevett et al., 2012).
Inflammasome activation in mouse microglial cells has been found to promote formation of neurofibrillary tangles (Ising et al., 2019). This phenomenon is mediated by increased phosphorylation of tau proteins by p38 kinase, stimulated by IL-1 (Li et al., 2003). In addition, pro-inflammatory stimuli, such as lipopolysaccharide (LPS) have also been found to promote hyperphosphorylation of tau proteins by cyclin-dependent kinase 5 (CDK-5), which can in turn be stimulated by IL-6 (Quintanilla et al., 2004; Kitazawa et al., 2005).
Mechanisms of microglial cell contribution to neuroinflammation and cognitive impairment in the course of Alzheimer’s disease are graphically illustrated in Figure 1
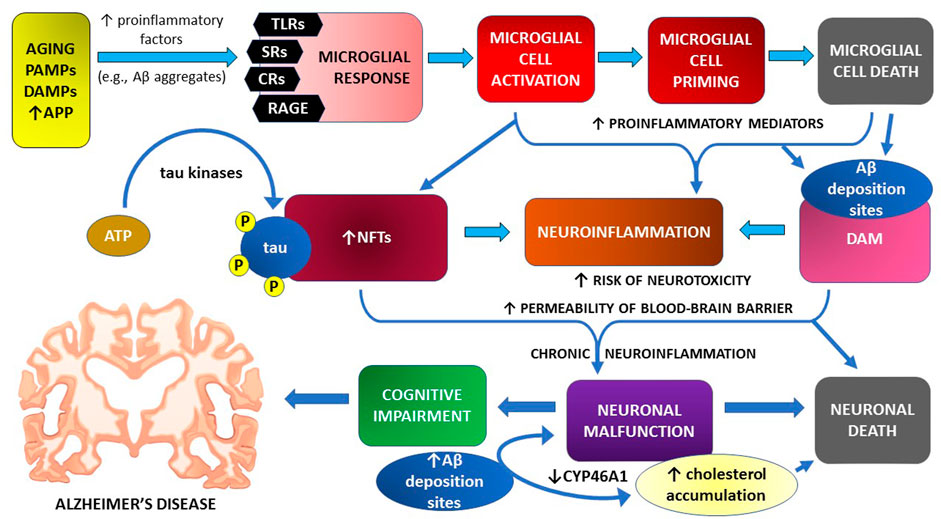
FIGURE 1. Microglial cell response and neuroinflammation in the pathomechanism of cognitive impairment—the main symptom of Alzheimer’s disease.
Sirtuins comprise a family of evolutionarily conserved enzymes performing NAD+ dependent protein deacetylation/deacylation (North and Verdin, 2004). Sirtuins have been initially discovered as transcription silencing factors in yeast, extending yeast replicative lifespan through histone deacetylation, resulting in heterochromatin formation and silencing mating-associated genes (Kaeberlein et al., 1999; Imai et al., 2000). More recently, sirtuins have been found to extend lifespan, or at least healthspan, in flatworms, fruit flies and mice (Imai and Guarente, 2016). In addition, sirtuin induction has been associated with caloric restriction-dependent lifespan extension in mammals (Bishop and Guarente, 2007; Guarente, 2013). Seven sirtuins have been identified in mammals so far; they can possess various enzymatic activity profiles and different subcellular location, but all of them share evolutionarily conserved catalytic core, consisting of NAD+ binding domain and zinc binding domain. Sirtuin domains other than aforementioned catalytic core seem to take part in substrate recognition and activity regulation (Feldman et al., 2012). Sirtuins can deacetylate both histone and non-histone substrates, including transcription factors, manganese superoxide dismutase (MnSOD) and tubulin. Mammalian sirtuins (SIRT1—7) have different profiles of action, substrate affinity, and subcellular compartmentation. Yet, all of them share a similar catalytic domain and use NAD+ as a co-substrate. Although initially identified as deacetylases, sirtuins are now known to have much more kinds of enzymatic activity, including deacylase and O-ADP-ribosylase activity (Michan and Sinclair, 2007). SIRT3, SIRT4, and SIRT5 are mitochondrial proteins, while SIRT1, SIRT6 and SIRT7 are nuclear enzymes, and—as such—can take part in the epigenetic regulation of cell phenotype, especially that they target histones and transcription factors. SIRT2 can be shuttled between nucleus and cytoplasm, depending on the phase of the cell cycle. Through exerting posttranslational regulatory modifications (PTRMs) on their target substrates, sirtuins can regulate a plethora of intracellular processes, such as energy expenditure, metabolic pattern, ROS concentration, DNA conservation, DNA damage repair, and cellular aging (Michan and Sinclair, 2007; Haigis and Sinclair, 2010; Houtkooper et al., 2012).
Sirtuins are quite abundantly expressed in the brain (Sidorova-Darmos et al., 2014; Jayasena et al., 2016). There is much evidence that various sirtuins are produced in different regions of the brain, while their activity can change with age. Furthermore, sometimes sirtuins’ enzymatic activity becomes reduced with age, despite their concentration increasing, which has been confirmed in mice in reference to SIRT1 and is generally attributed to falling concentration of NAD+ within cells (Braidy et al., 2015). However, in some circumstances, e.g., in rat hippocampal cells, SIRT1 expression becomes also reduced with age (Yan et al., 2019). The same problem may exist in reference to mitochondrial sirtuins (SIRT3-5) (Braidy et al., 2015). In neurons and glial cells cultured in vitro, the most expressed sirtuins include SIRT1-3 (Jayasena et al., 2016). Moreover, the levels of SIRT1 and SIRT3 in AD patients brains are reduced (Lutz et al., 2014; Lee et al., 2018; Yin et al., 2018). In addition, even in the plasma obtained from old mammals, SIRT1 and SIRT3 concentrations are decreased, which is correlated with general frailty (Kumar et al., 2014). In AD patients serum levels of SIRT1 are reduced, while SIRT6 levels are also reduced—both in the CNS and in the plasma, both in AD patients and in mouse models of AD (Kumar et al., 2013; Jung et al., 2016; Kaluski et al., 2017).
Sirtuins play an important role in the maintenance of neuronal well-being during aging (Herskovits and Guarente, 2014). In addition, they regulate many AD-associated processes, including APP processing, tau protein processing, mitochondrial functions, oxidative stress level, and neuroinflammation (Lalla and Donmez, 2013; Jęśko et al., 2017; Lee et al., 2018; Mohamad Nasir et al., 2018; Rizzi and Roriz-Cruz, 2018).
SIRT1 inhibits Aβ aggregate production through activating a disintegrin and metalloproteinase domain-containing protein 10 (ADAM-10), and thus stimulating APP processing to non-amyloidogenic, soluble metabolites, called soluble APPα (sAPPα) (Qin et al., 2006; Lee et al., 2014; Zhang et al., 2020). Furthermore, SIRT1 facilitates Aβ peptide degradation by upregulating lysosome number in primary astrocytes (Li et al., 2018).
SIRT1 levels in cerebral cortex of AD patients are reduced, and decreased SIRT1 concentration and activity are positively correlated with Aβ deposits formation in the extracellular space and NFT formation inside neurons (Julien et al., 2009). Furthermore, caloric restriction as a classic SIRT1 inducer alleviates Aβ-dependent pathology on animal models of AD (Wang et al., 2005; Qin et al., 2006). Aβ aggregates can reduce the expression of SIRT6 which is important for DNA damage repair and maintenance of genomic stability (Kugel and Mostoslavsky, 2014; Jung et al., 2016). Increased expression of SIRT6 may protect hippocampal neurons from Aβ-dependent DNA damage (Jung et al., 2016). Main mechanisms underlying inhibitory actions of SIRT1 towards Aβ aggregate deposition and related pathology are graphically illustrated in Figure 2.
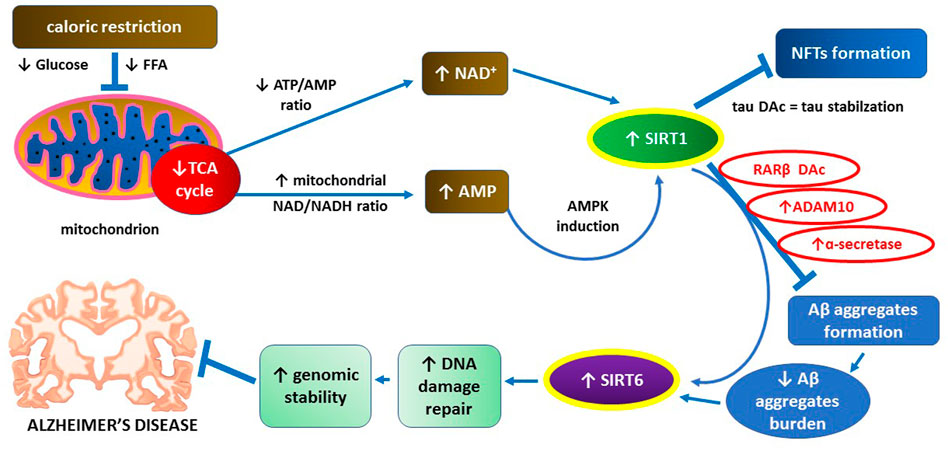
FIGURE 2. The key role of SIRT1 in supporting neuroprotective action of SIRT6 through preventing Aβ aggregates formation.
NFT formation is usually preceded by increased posttranslational regulatory modifications of tau proteins, such as phosphorylation and acetylation (Wang and Mandelkow, 2016; Guo et al., 2017). Acetylation of tau proteins inhibits their degradation, especially in reference to their phosphorylated forms, which promotes tau accumulation and neurotoxicity (Min et al., 2010; Cohen et al., 2011; Min et al., 2015; Tracy et al., 2016). In mouse models of tauopathy, SIRT1 overexpression or activation counteracts tau acetylation, which alleviates tau-related neurotoxicity (Min et al., 2018). It has been also shown that tau acetylation in mice can be promoted by Aβ aggregates through inhibition of SIRT3 expression (Yin et al., 2018). In mouse hippocampal neurons, SIRT3 activity induction reduces the extent of tau acetylation, while SIRT3 inhibition has the opposite effect (Li et al., 2019). Phosphorylation of tau proteins may be prevented by SIRT6 which inhibits glycogen synthase kinase 3 (GSK3) as a tau-phosphorylating enzyme (Kaluski et al., 2017).
The key actions of sirtuins, preventing NFT formation, are presented graphically in Figure 3.
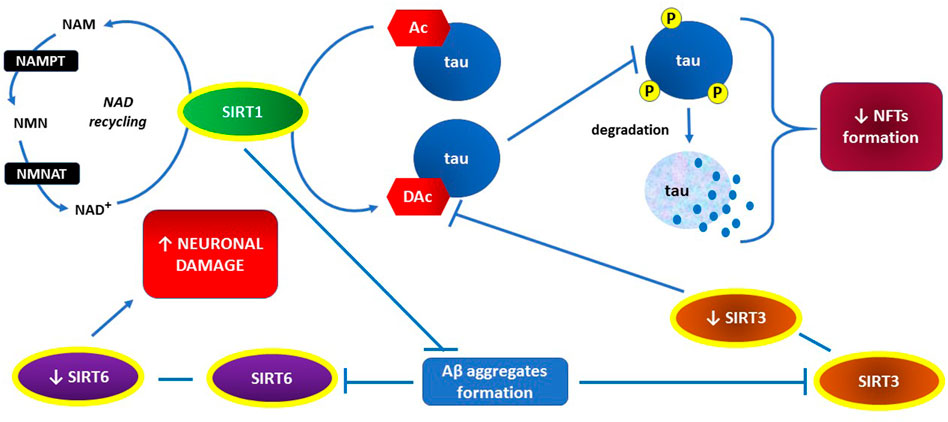
FIGURE 3. The key role of SIRT1 in both, inhibiting NFTs formation through preventing hyperphosphorylation of tau proteins and recovering activities of SIRT3 and SIRT6 through preventing Aβ aggregates formation.
NF-κB activation may occur through the canonical or non-canonical pathway. Still, in standard conditions, the canonical pathway is blocked by default due to IkB proteins, which sequestrate NF-κB in the cytoplasm. However, pro-inflammatory stimuli may activate IkB kinase (IKK), which promotes IkB degradation through inhibitory phosphorylation, and thus relocation of NF-κB to the cell nucleus. Sirtuins may inhibit NF-κB both directly and indirectly. Firstly—SIRT1 and SIRT2 can deacetylate NF-κB’s p65 subunit at lysine 310, which directly inhibits NF-κB activity (Yeung et al., 2004). Furthermore, such acetylation impedes methylation of adjacent lysine residues (K314 and K315), promoting ubiquitination and degradation of p65 (Rothgiesser et al., 2010; Yang et al., 2010). Secondly—SIRT1 can inhibit NF-κB through inhibitory phosphorylation of its transcriptional activators, such as PARP-1 and p300 histone acetyltransferase (Bouras et al., 2005; Rajamohan et al., 2009). Thirdly—SIRT1 and SIRT6 may inhibit the expression of NF-κB target genes due to transcriptional silencing through H3K9 DAC (Kawahara et al., 2009). In this way, SIRT1 exerts anti-inflammatory actions, counteracting neuroinflammation. Aβ interactions with microglial cells promote p65 subunit acetylation, while SIRT1 activation or overexpression prevents this effect. Therefore, SIRT1 protects CNS from Aβ neurotoxicity through inhibiting NF-κB dependent pro-inflammatory signaling pathway (Chen et al., 2005; Yang et al., 2012).
SIRT6 can induce the production of IkB at the level of transcription, which exerts an anti-inflammatory effect because IkB blocks the canonical pathway of NF-κB activation by default (Kawahara et al., 2009). In addition, SIRT6 may both desensitize cells to TNF-alpha, an upstream inducer of NF-κB, and inhibit TNF-alpha secretion. SIRT1 and SIRT6 actions described above are primarily responsible for their anti-inflammatory effects.
In addition, SIRT1 inhibits the production of IL-1β, a pro-inflammatory cytokine. This effect is dependent on activatory deacetylation of DNA (cytosine-5)-methyltransferase 1 (DNMT1)—an enzyme that inhibits biosynthesis of IL-1β at the level of transcription, through DNA methylation at IL-1β proximal promoter (Peng et al., 2011; Cho et al., 2015; Heo et al., 2017). If SIRT1 activity is reduced with age, the extent of DNA methylation at IL-1β proximal promoter is also reduced, which can facilitate IL-1β biosynthesis at the level of transcription, thus aggravating neuroinflammation. SIRT1 activators, such as resveratrol, can prevent this effect (Yan et al., 2019).
SIRT2 can also inhibit neuroinflammation through direct deacetylation of p65 at lysine 310 (Rothgiesser et al., 2010; Pais et al., 2013). SIRT2 inhibition may promote transition of microglial cells from homeostatic/quiescent phenotype to pro-inflammatory phenotype on mouse model of traumatic brain damage, through reactivation of NF-κB dependent pro-inflammatory signaling pathway (Yuan et al., 2016). It has been also found that SIRT2 overexpression in rats reduces neuroinflammation exactly through p65 deacetylation (Zhang and Chi, 2018). On the other hand, results of other research studies reveal some potentially pro-inflammatory actions of SIRT2. Inhibition of SIRT2 blocks NF-κB molecule translocation to cell nucleus, thus abrogating TNF-α and IL-6 expression in mouse microglial cells exposed to LPS. Thus, SIRT2 seems to be necessary to induce LPS-dependent neuroinflammation (Wang et al., 2016). Pharmacologic inhibition of SIRT2 reduces TNF-α and nitric oxide production in LPS-exposed microglial cells (Harrison et al., 2018). Furthermore, SIRT2 inhibition attenuates α-synuclein neurotoxicity on mouse models of Parkinson’s disease (Outeiro et al., 2007; Chen et al., 2015). Similarly, SIRT2 inhibition in mice alleviates cognitive deficits on mouse models of Alzheimer’s disease, through inhibition of Aβ formation (Biella et al., 2016). Although TNF-α signaling dependent on TNF-R1 receptor is thought to be pro-inflammatory and thus deleterious in the course of AD, TNF-α may also exert some neuroprotective effects through acting on TNF-R2 receptors. Since neuroprotective actions of TNF-α may include protection against demyelination, excitotoxicity and cerebral ischemia (Probert, 2015), this may—at least in part—explain why SIRT2 inhibition can be neuroprotective in some circumstances. Therefore, further research studies are needed to verify overall effect of SIRT2 and its inhibitors towards neuroinflammation in the course of AD, although inhibitors of TNF-α dependent signaling usually improve the cognitive performance of AD patients (He et al., 2007). In general, the outcome of NF-κB activation depends very much on the cell type and the stimuli present, since it determines which signaling pathway becomes activated. This may account for some discrepancies related to SIRT2 activation/inhibition effects towards inflammatory response.
Unlike TNF-α, IL-6 seems to have mainly deleterious effects towards aging brain, through promoting gliosis and inflammation, inhibiting LTP in hippocampal neurons, enhancing the neurotoxic properties of NMDA, as well as reducing adult neurogenesis in the hippocampal dentate gyrus (Godbout and Johnson, 2004). Furthermore, severity of dementia in the course of AD is positively correlated with IL-6 concentration in serum (Kalman et al., 1997). When having taken into consideration that IL-6 production is stimulated by NF-κB and its upstream inducers, both SIRT1 and SIRT6, which inactivate NF-κB, may exert their beneficial effects on the brain exactly through possessing this property.
Main preventive actions of sirtuins against both neuroinflammation and neuroinflammation-related oxidative stress are presented graphically in Figure 4.
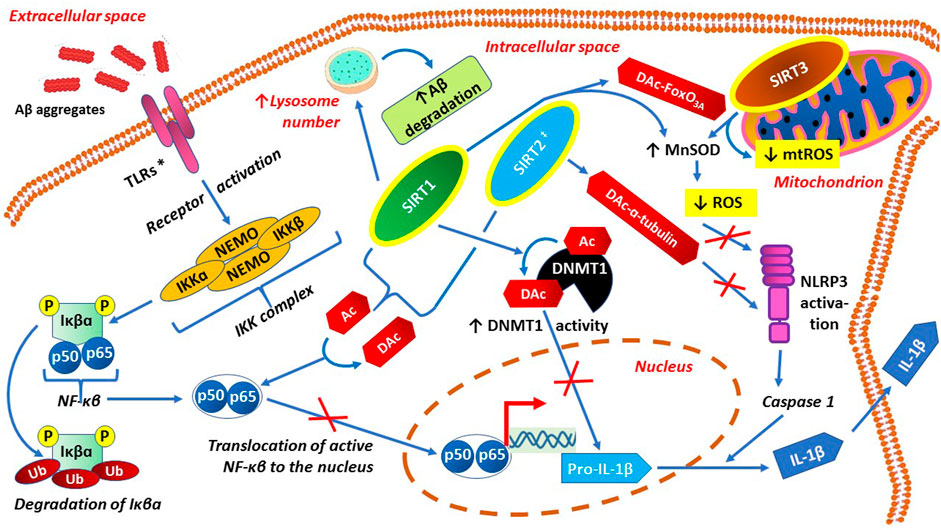
FIGURE 4. Anti-neuroinflammatory actions of sirtuins, through inactivation of p65 subunit of NF-κB, activation of DNMT1 and anti-oxidative effects.
SIRT3, a mitochondrial sirtuin, is quite significant for counteracting oxidative stress, since it both optimizes the action of respiratory chain enzymes and activates MnSOD. Therefore, SIRT3 inhibits ROS production and facilitates ROS inactivation (Ansari et al., 2017; Meng et al., 2019). SIRT3 activity falls during neuroinflammation in LPS-exposed microglial cells, while SIRT3 activation can prevent both LPS-induced neuroinflammation and mitochondrial dysfunction resulting in microglial cell death (Zhou and Jiang, 2019). Microglia-derived pro-inflammatory cytokines may induce apoptosis of neural tissue stem cells, as well as inhibit their proliferation. Using co-cultures of microglial cells and neural tissue stem cells, it has been found that Aβ-induced microglial cell transition to pro-inflammatory phenotype results in neural tissue stem cell necrosis through cytokine-dependent inhibition of SIRT3 and MnSOD, with a subsequent rise in intracellular ROS concentration. SIRT3 activation or overexpression protects the cells from such cytokine-dependent oxidative stress (Jiang et al., 2017). SIRT3 also protects mice from cognitive deficits induced by surgery/anesthesia brain injury. In old mice with cognitive impairments, loss of function of both SIRT3 and MnSOD has been found in hippocampal cells (Liu et al., 2021).
SIRT1 can also counteract oxidative stress through forkhead box O3A (FoxO3A) deacetylation, resulting in MnSOD activation by deacetylated FoxO3A (Brunet et al., 2004). As to SIRT3, it may promote both FoxO3A deacetylation and direct activation of MnSOD, also through deacetylation (Tao et al., 2014; Rangarajan et al., 2015).
Both ROS and mitochondrial degradation products can exert pro-inflammatory actions through activating NLRP3 inflammasome (Zhou et al., 2011; Wilkins et al., 2017). In this context, hyperactivation of inflammasomes as innate immunity components may promote neuroinflammation in the course of AD, while inflammasome activity inhibition can prevent neuroinflammation (Venegas and Heneka, 2019). This is why both SIRT1 and SIRT3 can prevent neuroinflammation through their mitochondria-protective and antioxidative effects (Zhang et al., 2017; Zou et al., 2018). SIRT2 may also inhibit NLRP3 inflammasome through deacetylation of α-tubulin, which is necessary in its acetylated form for inflammasome activation (Misawa et al., 2015). SIRT2 may also directly deacetylate pyrin domains significant for inflammasome activation (He et al., 2020).
Growing evidence suggests that mitochondrial dysfunction within CNS cells, as well as the resulting oxidative stress, are strongly associated with Alzheimer’s disease (Manoharan et al., 2016; Kausar et al., 2018; Llanos-González et al., 2020). In this context, activation of SIRT1 and SIRT3 can prevent AD through boosting their antioxidative and mitochondria-protective actions (Woodbury et al., 2013; Ye et al., 2019).
Aβ aggregates can also activate NLRP3 inflammasome through inducing phagolysomal damage in microglial cells, followed by leakage of lysosomal proteases and cathepsin B into the cytoplasm (Halle et al., 2008; Heid et al., 2013; Wu et al., 2013; Campden and Zhang, 2019; Kelley et al., 2019). Since SIRT1 inhibits Aβ aggregate formation, increasing its activity in the brain may causally prevent AD-associated pathology (Gay et al., 2020).
Conclusion: boosting sirtuins activity, especially in reference to SIRT1 and SIRT3, both through allosteric activation and through NAD+ replenishment, can be regarded as very promising strategy of promoting brain homeostasis and AD prevention, especially if the applied boosters are well-tolerated, safe, and easily crossing the blood-brain barrier.
Some pathomechanisms of Alzheimer’s disease have not been addressed in this article. Those mechanisms not addressed include: pathogenic role of some bacteria, such as P. gingivalis, in the induction of neuroinflammation (Dominy et al., 2019), as well as potentially pathogenic role of some metals—especially aluminum—in promoting Aβ aggregate oligomerization (Zhang et al., 2019). Similarly, we have not discussed the detailed mechanisms of neurotoxicity of misfolded proteins, such as tau proteins. In spite of that, it can be assumed that neuroinflammation etiology does not take part in the mechanisms of neuroinflammation-alleviating action of sirtuins. In other words, sirtuins can alleviate neuroinflammation regardless of its cause, since they inhibit an essential pro-inflammatory signaling pathway dependent on NF-κB. Although eradication of pro-neuroinflammatory bacterial infections is useful and desired, detailed microbiology of those infections is not a topic of this paper, while AD-promoting effect of such infections may occur either through stimulated production of pro-inflammatory cytokines in some regions adjacent to the CNS, so that those pro-inflammatory cytokines can act on the CNS in a paracrine manner, or through penetration of some bacterial toxins through the blood-brain barrier, thus exerting a direct neurotoxic effect.
As to the role of aluminum in the pathogenesis of Alzheimer’s disease, it seems to basically consist in promoting the oligomerization of peptides produced from APP by β- and γ-secretases, which raises the risk of Aβ aggregate formation. Since SIRT1 induction stimulates α-secretases, thus reducing the risk of APP processing by β- and γ-secretases, it can neutralize aluminum influence on the CNS, because aluminum excess seems to be harmful only if there is an already existing excess of β- and γ-secretase products.
Another matter is de facto lack of empirical, measurable effects of applying sirtuin boosters discovered so far on the course of Alzheimer’s disease in hitherto performed clinical trials. However, it should be taken into consideration that sirtuin allosteric activators discovered so far do not cross blood-brain barrier with 100% efficacy, while sirtuin activity boosters in the form of close NAD+ precursors are not widely available in pharmacy retail trade—either as medications or as dietary supplements, which limits their use. In addition, it is worth remembering that beneficial effects of sirtuins towards the course of Alzheimer’s disease, discussed in this paper, are mainly preventive, which means that empirical and measurable confirmation of the sirtuins’ actions assumed may require introducing the treatment with sirtuin activity boosters 15–20 years prior to the onset of AD clinical symptoms, in case of detection of AD risk factors (e.g., Aβ deposits or Aβ-associated alterations in neuroimaging). Moreover, hitherto known allosteric activators of sirtuins, such as resveratrol (SIRT1 activator), honokiol (SIRT3 activator), or SRT1720 may require chemical modifications to improve their crossing through blood-brain barrier, while sirtuin activity boosters in the form of close NAD+ precursors require introducing to the pharmaceutical retail market to provide their broad availability for people who would like to use them within the frames of AD prevention. Summing up: even if sirtuin boosters as a possible method of AD prevention were introduced today, their beneficial effects might be observed in a time interval equivalent to the time amount usually required for a progression of AD from its initial pathological and molecular manifestations to its clinical stage.
Someone could ask whether a preventive action of sirtuin activation refers to all sirtuins, or merely to those widely described as neuroprotective. The answer is: probably such a beneficial effects refer to all sirtuins, with only SIRT2 being a possible exception, although even this is uncertain, since there are some results of research studies indicating anti-inflammatory actions of SIRT2 (Rothgiesser et al., 2010; Pais et al., 2013; Yuan et al., 2016).
Another possible question is whether sirtuins are the only enzymes using NAD+ as a coenzyme/co-substrate. According to current knowledge, the answer is “no”, because there are strong premises to claim that beneficial effects of NAD+ replenishment are strictly correlated exactly with boosting the activity of sirtuins (Imai and Guarente, 2016), while focusing on all enzymes using NAD+ as a coenzyme would largely exceed the scope of this paper.
The question related to the previous one is whether using close metabolic precursors of NAD+ affects the activity of enzymes other than sirtuins. The answer is “yes”, and thus it cannot be excluded that at least some beneficial effects of NAD+ replenishment strategies are mediated by affecting enzymes other than sirtuins (e.g., poly-ADP-ribosyltransferases, glycohydrolases, mitochondrial enzymes coupling TCA reactions with oxidative phosphorylation and ATP biosynthesis). In other words, it is possible that NAD+ replenishment exerts its beneficial effects through affecting the activity of enzymes other than sirtuins. Although there are some premises that NAD+ precursors exert their beneficial effects indeed through sirtuin activation (Guarente, 2013; Imai and Guarente, 2016), additional research studies should be made to pinpoint the mechanisms of action of NAD+ replenishment strategies by comparing the phenotypic beneficial effects related to NAD+ replenishment with the effects achieved through selective overexpression of particular sirtuins. This kind of research studies may be necessary to verify whether NAD+ replenishment effects are sirtuin-specific or not.
DS and MW contributed to conception of the article. MW wrote a draft of the manuscript, while DS designed the figures. Both authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.962769/full#supplementary-material
't Hart B. A., den Dunnen W. F. (2013). Commentary on special issue: CNS diseases and the immune system. J. Neuroimmune Pharmacol. 8 (4), 757–759. doi:10.1007/s11481-013-9486-0
Ansari A., Rahman M. S., Saha S. K., Saikot F. K., Deep A., Kim K.-H., et al. (2017). Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 16, 4–16. doi:10.1111/acel.12538
Arispe N., Rojas E., Pollard H., January B. (1993). Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U. S. A. 90 (2), 567–571. doi:10.1073/pnas.90.2.567
Baik S. H., Kang S., Son S. M., Mook-Jung I. (2016). Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 64, 2274–2290. doi:10.1002/glia.23074
Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi:10.4049/jimmunol.0901363
Biella G., Fusco F., Nardo E., Bernocchi O., Colombo A., Lichtenthaler S. F., et al. (2016). Sirtuin 2 inhibition improves cognitive performance and acts on amyloid-β protein precursor processing in two Alzheimer’s disease mouse models. J. Alzheimers Dis. 53, 1193–1207. doi:10.3233/JAD-151135
Bishop N. A., Guarente L. (2007). Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat. Rev. Genet. 8, 835–844. doi:10.1038/nrg2188
Block M. L., Zecca L., Hong J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi:10.1038/nrn2038
Bouras T., Fu M., Sauve A. A., Wang F., Quong A. A., Perkins N. D., et al. (2005). SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280 (11), 10264–10276. doi:10.1074/jbc.M408748200
Braidy N., Poljak A., Grant R., Jayasena T., Mansour H., Chan-Ling T., et al. (2015). Differential expression of sirtuins in the aging rat brain. Front. Cell. Neurosci. 9, 167. doi:10.3389/fncel.2015.00167
Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., et al. (2004). Stress-Dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. doi:10.1126/science.1094637
Calsolaro V., Edison P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi:10.1016/j.jalz.2016.02.010
Campden R. I., Zhang Y. (2019). The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 670, 32–42. doi:10.1016/j.abb.2019.02.015
Chen G., Xu T., Yan Y., Zhou Y., Jiang Y., Melcher K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi:10.1038/aps.2017.28
Chen J., Zhou Y., Mueller-Steiner S., Chen L.-F., Kwon H., Yi S., et al. (2005). SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 280, 40364–40374. doi:10.1074/jbc.M509329200
Chen X., Wales P., Quinti L., Zuo F., Moniot S., Herisson F., et al. (2015). The Sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS One 10, e0116919. doi:10.1371/journal.pone.0116919
Cho S.-H., Chen J. A., Sayed F., Ward M. E., Gao F., Nguyen T. A., et al. (2015). SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 35, 807–818. doi:10.1523/JNEUROSCI.2939-14.2015
Cohen T. J., Guo J. L., Hurtado D. E., Kwong L. K., Mills I. P., Trojanowski J. Q., et al. (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2, 252. doi:10.1038/ncomms1255
Colombo E., Farina C. (2016). Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620. doi:10.1016/j.it.2016.06.006
Colvin M. T., Silvers R., Ni Q. Z., Can T. V., Sergeyev I., Rosay M., et al. (2016). Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674. doi:10.1021/jacs.6b05129
Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi:10.1126/science.8346443
Cribbs D. H., Berchtold N. C., Perreau V., Coleman P. D., Rogers J., Tenner A. J., et al. (2012). Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflammation 9, 179. doi:10.1186/1742-2094-9-179
Dani M., Wood M., Mizoguchi R., Fan Z., Walker Z., Morgan R., et al. (2018). Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. doi:10.1093/brain/awy188
Darweesh S. K. L., Wolters F. J., Ikram M. A., de Wolf F., Bos D., Hofman A., et al. (2018). Inflammatory markers and the risk of dementia and Alzheimer’s disease: a meta-analysis. Alzheimers Dement. 14, 1450–1459. doi:10.1016/j.jalz.2018.02.014
Das Sarma J. (2014). Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 20 (2), 122–136. doi:10.1007/s13365-013-0188-4
Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I., et al. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi:10.1016/j.cell.2018.05.003
Di Paolo G., Kim T.-W. (2011). Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296. doi:10.1038/nrn3012
Doens D., Fernández P. L. (2014). Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 11, 48. doi:10.1186/1742-2094-11-48
Dominy S. S., Lynch C., Ermini F., Benedyk M., Marczyk A., Konradi A., et al. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small molecule inhibitors. Sci. Adv. 5, eaau3333. eaau 3333. doi:10.1126/sciadv.aau3333
Elahy M., Jackaman C., Mamo J. C. L., Lam V., Dhaliwal S. S., Giles C., et al. (2015). Blood–brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 12, 2. doi:10.1186/s12979-015-0029-9
ElAli A., Rivest S. (2016). Microglia in Alzheimer’s disease: a multifaceted relationship. Brain Behav. Immun. 55, 138–150. doi:10.1016/j.bbi.2015.07.021
Feldman J. L., Dittenhafer-Reed K. E., Denu J. M. (2012). Sirtuin catalysis and regulation. J. Biol. Chem. 287, 42419–42427. doi:10.1074/jbc.R112.378877
Feringa F. M., van der Kant R. (2021). Cholesterol and Alzheimer’s disease; from risk genes to pathological effects. Front. Aging Neurosci. 13, 690372. doi:10.3389/fnagi.2021.690372
Fernando K. K. M., Wijayasinghe Y. S. (2021). Sirtuins as potential therapeutic targets for mitigating neuroinflammation associated with Alzheimer’s disease. Front. Cell. Neurosci. 15, 746631. doi:10.3389/fncel2021.746631
Fiebich B. L., Batista C. R. A., Saliba S. W., Yousif N. M., de Oliveira A. C. P. (2018). Role of microglia TLRs in neurodegeneration. Front. Cell. Neurosci. 12, 329. doi:10.3389/fncel.2018.00329
Gay N. H., Suwanjang W., Ruankham W., Songtawee N., Wongchitrat P., Prachayasittikul V., et al. (2020). Butein, isoliquiritigenin, and scopoletin attenuate neurodegeneration via antioxidant enzymes and SIRT1/ADAM10 signaling pathway. RSC Adv. 10, 16593–16606. doi:10.1039/C9RA06056A
Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi:10.1126/science.1194637
Glass C. K., Saijo K., Winner B., Marchetto M. C., Gage F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi:10.1016/j.cell.2010.02.016
Godbout W. E., Johnson R. W. (2004). Interleukin 6 in the aging brain. J. Neuroimmunol. 147, 141–144. doi:10.1016/j.jneuroim.2003.10.031
González-Reyes R. E., Nava-Mesa M. O., Vargas-Sánchez K., Ariza-Salamanca D., Mora-Muñoz L. (2017). Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 10, 427. doi:10.3389/fnmol.2017.00427
Gratuze M., Leyns C. E. G., Holtzman D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13, 66. doi:10.1186/s13024-018-0298-9
Guarente L. (2013). Calorie restriction and sirtuins revisited. Genes Dev. 27, 2072–2085. doi:10.1101/gad.227439.113
Guo T., Noble W., Hanger D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi:10.1007/s00401-017-1707-9
Haigis M. C., Sinclair D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295. doi:10.1146/annurev.pathol.4.110807.092250
Halle A., Hornung V., Petzold G. C., Stewart C. R., Monks B. G., Reinheckel T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. doi:10.1038/ni.1636
Hampel H., Caraci F., Cuello A. C., Caruso G., Nisticò R., Corbo M., et al. (2020). A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front. Immunol. 11, 456. doi:10.3389/fimmu.2020.00456
Hanseeuw B. J., Betensky R. A., Jacobs H. I. L., Schultz A. P., Sepulcre J., Becker J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. doi:10.1001/jamaneurol.2019.1424
Harrison I. F., Smith A. D., Dexter D. T. (2018). Pathological histone acetylation in Parkinson’s disease: neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 666, 48–57. doi:10.1016/j.neulet.2017.12.037
He M., Chiang H.-H., Luo H., Zheng Z., Qiao Q., Wang L., et al. (2020). An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 31, 580–591. e5. doi:10.1016/j.cmet.2020.01.009
He P., Zhong Z., Lindholm K., Berning L., Lee W., Lemere C., et al. (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841. doi:10.1083/jcb.200705042
He Y., Hara H., Núñez G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends biochem. Sci. 41, 1012–1021. doi:10.1016/j.tibs.2016.09.002
Heid M. E., Keyel P. A., Kamga C., Shiva S., Watkins S. C., Salter R. D., et al. (2013). Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 191, 5230–5238. doi:10.4049/jimmunol.1301490
Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 14, 388–405. doi:10.1016/S1474-4422(15)70016-5
Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi:10.1038/nature11729
Heo J., Lim J., Lee S., Jeong J., Kang H., Kim Y., et al. (2017). Sirt1 regulates DNA methylation and differentiation potential of embryonic stem cells by antagonizing Dnmt3l. Cell Rep. 18, 1930–1945. doi:10.1016/j.celrep.2017.01.074
Heppner F. L., Ransohoff R. M., Becher B. (2015). Immune attack: the role of inflammation in alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi:10.1038/nrn3880
Herskovits A. Z., Guarente L. (2014). SIRT1 in neurodevelopment and brain senescence. Neuron 81, 471–483. doi:10.1016/j.neuron.2014.01.028
Hevett S. J., Jackman N. A., Claycomb R. J. (2012). Interleukin-1β in central nervous system injury and repair. Eur. J. Neurodegener. Dis. 1 (2), 195–211.
Hickman S. E., Allison E. K., El Khoury J. (2008). Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi:10.1523/JNEUROSCI.0616-08.2008
Holmes C., Cunningham C., Zotova E., Culliford D., Perry V. H. (2011). Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 77, 212–218. doi:10.1212/WNL.0b013e318225ae07
Houtkooper R. H., Pirinen E., Auwerx J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi:10.1038/nrm3293
Hu Y., Fryatt G. L., Ghorbani M., Obst J., Menassa D. A., Martin-Estebane M., et al. (2021). Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 35, 109228. doi:10.1016/j.celrep.2021.109228
Imai S.-I., Guarente L. (2016). It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2, 16017. doi:10.1038/npjamd.2016.17
Imai S., Armstrong C. M., Kaeberlein M., Guarente L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. doi:10.1038/35001622
Iqbal K., Liu F., Gong C.-X., Grundke-Iqbal I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi:10.2174/156720510793611592
Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S. V., Vieira-Saecker A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi:10.1038/s41586-019-1769-z
Jayasena T., Poljak A., Braidy N., Zhong L., Rowlands B., Muenchhoff J., et al. (2016). Application of targeted mass spectrometry for the quantification of sirtuins in the central nervous system. Sci. Rep. 6, 35391. doi:10.1038/srep35391
Jęśko H., Wencel P., Strosznajder R. P., Strosznajder J. B. (2017). Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem. Res. 42, 876–890. doi:10.1007/s11064-016-2110-y
Ji K., Akgul G., Wollmuth L. P., Tsirka S. E. (2013). Microglia actively regulate the number of functional synapses. PLoS One 8, e56293. doi:10.1371/journal.pone.0056293
Jiang D.-Q., Wang Y., Li M.-X., Ma Y.-J., Wang Y. (2017). SIRT3 in neural stem cells attenuates microglia activation-induced oxidative stress injury through mitochondrial pathway. Front. Cell. Neurosci. 11, 7. doi:10.3389/fncel.2017.00007
Julien C., Tremblay C., Émond V., Lebbadi M., Salem N., Bennett D. A., et al. (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58. doi:10.1097/NEN.0b013e3181922348
Jung E. S., Choi H., Song H., Hwang Y. J., Kim A., Ryu H., et al. (2016). p53-dependent SIRT6 expression protects Aβ42-induced DNA damage. Sci. Rep. 6, 25628. doi:10.1038/srep25628
Kaeberlein M., McVey M., Guarente L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580. doi:10.1101/gad.13.19.2570
Kalman J., Juhasz A., Laird G., Dickens P., Jardanhazy T., Rimanoczy A., et al. (1997). Serum interleukin-6 levels correlate with the severity of dementia in Down syndrome and in Alzheimer’s disease. Acta Neurol. Scand. 96, 236–240. doi:10.1111/j.1600-0404.1997.tb00275.x
Kaluski S., Portillo M., Besnard A., Stein D., Einav M., Zhong L., et al. (2017). Neuroprotective functions for the histone deacetylase SIRT6. Cell Rep. 18, 3052–3062. doi:10.1016/j.celrep.2017.03.008
Kausar S., Wang F., Cui H. (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 7, 274. doi:10.3390/cells7120274
Kawahara T. L., Michishita E., Adler A. S., Damian M., Berber E., Lin M., et al. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB- dependent gene expression and organismal life span. Cell 136 (1), 62–74. doi:10.1016/j.cell.2008.10.052
Kawai T., Akira S. (2007). Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med. 13, 460–469. doi:10.1016/j.molmed.2007.09.002
Kelley N., Jeltema D., Duan Y., He Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328. doi:10.3390/ijms20133328
Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290. e17. doi:10.1016/j.cell.2017.05.018
Kinney J. W., Bemiller S. M., Murtishaw A. S., Leisgang A. M., Salazar A. M., Lamb B. T., et al. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590. doi:10.1016/j.trci.2018.06.014
Kitazawa M., Oddo S., Yamasaki T. R., Green K. N., LaFerla F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853. doi:10.1523/JNEUROSCI.2868-05.2005
Kitazawa M., Yamasaki T., Laferla F. M. (2004). Microglia as a potential bridge between the amyloid β-peptide and tau. Ann. N. Y. Acad. Sci. 1035, 85–103. doi:10.1196/annals.1332.006
Koyama A., O’Brien J., Weuve J., Blacker D., Metti A. L., Yaffe K., et al. (2013). The role of peripheral inflammatory markers in dementia and Alzheimer’s disease: a meta-analysis. J. Gerontol. A Biol. Sci. Med. Sci. 68, 433–440. doi:10.1093/gerona/gls187
Krabbe G., Halle A., Matyash V., Rinnenthal J. L., Eom G. D., Bernhardt U., et al. (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8, e60921. doi:10.1371/journal.pone.0060921
Kugel S., Mostoslavsky R. (2014). Chromatin and beyond: the multitasking roles for SIRT6. Trends biochem. Sci. 39, 72–81. doi:10.1016/j.tibs.2013.12.002
Kumar A., Singh A., Ekavali (2015). A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 67, 195–203. doi:10.1016/j.pharep.2014.09.004
Kumar R., Chaterjee P., Sharma P. K., Singh A. K., Gupta A., Gill K., et al. (2013). Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PLoS One 8, e61560. doi:10.1371/journal.pone.0061560
Kumar R., Mohan N., Upadhyay A. D., Singh A. P., Sahu V., Dwivedi S., et al. (2014). Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell 13, 975–980. doi:10.1111/acel.12260
Lai K. S. P., Liu C. S., Rau A., Lanctôt K. L., Köhler C. A., Pakosh M., et al. (2017). Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88, 876–882. doi:10.1136/jnnp-2017-316201
Lalla R., Donmez G. (2013). The role of sirtuins in Alzheimer’s disease. Front. Aging Neurosci. 5, 16. doi:10.3389/fnagi.2013.00016
Lee H. R., Shin H. K., Park S. Y., Kim H. Y., Lee W. S., Rhim B. Y., et al. (2014). Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 92, 1581–1590. doi:10.1002/jnr.23421
Lee J., Kim Y., Liu T., Hwang Y. J., Hyeon S. J., Im H., et al. (2018). SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 17, e12679. doi:10.1111/acel.12679
Li M. Z., Zheng L. J., Shen J., Li X. Y., Zhang Q., Bai X., et al. (2018). SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 13 (11), 2005–2013. doi:10.4103/1673-5374.239449
Li S., Yin J., Nielsen M., Beach T. G., Guo L., Shi J., et al. (2019). Sirtuin 3 mediates tau deacetylation. J. Alzheimers Dis. 69, 355–362. doi:10.3233/JAD-190014
Li Y., Liu L., Barger S. W., Griffin W. S. T. (2003). Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611. doi:10.1523/JNEUROSCI.23-05-01605.2003
Liu Q., Sun Y.-M., Huang H., Chen C., Wan J., Ma L.-H., et al. (2021). Sirtuin 3 protects against anesthesia/surgery-induced cognitive decline in aged mice by suppressing hippocampal neuroinflammation. J. Neuroinflammation 18, 41. doi:10.1186/s12974-021-02089-z
Llanos-González E., Henares-Chavarino Á. A., Pedrero-Prieto C. M., García-Carpintero S., Frontiñán-Rubio J., Sancho-Bielsa F. J., et al. (2020). Interplay between mitochondrial oxidative disorders and proteostasis in Alzheimer’s disease. Front. Neurosci. 13, 1444. doi:10.3389/fnins.2019.01444
Lopez-Rodriguez A. B., Hennessy E., Murray C. L., Nazmi A., Delaney H. J., Healy D., et al. (2021). Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 17, 1735–1755. doi:10.1002/alz.12341
Lučiūnaitė A., McManus R. M., Jankunec M., Rácz I., Dansokho C., Dalgëdienė I., et al. (2020). Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 155, 650–661. doi:10.1111/jnc.14945
Lull M. E., Block M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. doi:10.1016/j.nurt.2010.05.014
Lutz M. I., Milenkovic I., Regelsberger G., Kovacs G. G. (2014). Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromolecular Med. 16, 405–414. doi:10.1007/s12017-014-8288-8
Manoharan S., Guillemin G. J., Abiramasundari R. S., Essa M. M., Akbar M., Akbar M. D., et al. (2016). The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and huntington’s disease: a mini review. Oxid. Med. Cell. Longev. 2016, 8590578. doi:10.1155/2016/8590578
Meng H., Yan W.-Y., Lei Y.-H., Wan Z., Hou Y.-Y., Sun L.-K., et al. (2019). SIRT3 regulation of mitochondrial quality control in neurodegenerative diseases. Front. Aging Neurosci. 11, 313. doi:10.3389/fnagi.2019.00313
Michan S., Sinclair D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13. doi:10.1042/BJ20070140
Min S.-W., Chen X., Tracy T. E., Li Y., Zhou Y., Wang C., et al. (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162. doi:10.1038/nm.3951
Min S.-W., Cho S.-H., Zhou Y., Schroeder S., Haroutunian V., Seeley W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi:10.1016/j.neuron.2010.08.044
Min S.-W., Sohn P. D., Li Y., Devidze N., Johnson J. R., Krogan N. J., et al. (2018). SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J. Neurosci. 38, 3680–3688. doi:10.1523/JNEUROSCI.2369-17.2018
Minter M. R., Taylor J. M., Crack P. J. (2016). The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 136, 457–474. doi:10.1111/jnc.13411
Misawa T., Saitoh T., Kozaki T., Park S., Takahama M., Akira S., et al. (2015). Resveratrol inhibits the acetylated α-tubulin-mediated assembly of the NLRP3-inflammasome. Int. Immunol. 27, 425–434. doi:10.1093/intimm/dxv018
Mohamad Nasir N. F., Zainuddin A., Shamsuddin S. (2018). Emerging roles of sirtuin 6 in Alzheimer’s disease. J. Mol. Neurosci. 64, 157–161. doi:10.1007/s12031-017-1005-y
Mosher K. I., Wyss-Coray T. (2014). Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 88, 594–604. doi:10.1016/j.bcp.2014.01.008
Musiek E. S., Holtzman D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and “wingmen”. Nat. Neurosci. 18, 800–806. doi:10.1038/nn.4018
Nakanishi A., Kaneko N., Takeda H., Sawasaki T., Morikawa S., Zhou W., et al. (2018). Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm. Regen. 38, 27. doi:10.1186/s41232-018-0085-6
Navarro V., Sanchez-Mejias E., Jimenez S., Muñoz-Castro C., Sanchez-Varo R., Davila J. C., et al. (2018). Microglia in Alzheimer’s disease: activated: dysfunctional or degenerative. Front. Aging Neurosci. 10, 140. doi:10.3389/fnagi.2018.00140
Nimmerjahn A., Kirchhoff F., Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi:10.1126/science.1110647
Niraula A., Sheridan J. F., Godbout J. P. (2017). Microglia priming with aging and stress. Neuropsychopharmacology 42, 318–333. doi:10.1038/npp.2016.185
Nordengen K., Kirsebom B.-E., Henjum K., Selnes P., Gísladóttir B., Wettergreen M., et al. (2019). Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflammation 16, 46. doi:10.1186/s12974-019-1399-2
North B. J., Verdin E. (2004). Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5, 224. doi:10.1186/gb-2004-5-5-224
Outeiro T. F., Kontopoulos E., Altman S., Kufareva I., Strathearn K. E., Amore A. M., et al. (2007). Sirtuin 2 inhibitors rescue α-Synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519. doi:10.1126/science.1143780
Pais T. F., Szegő É. M., Marques O., Miller-Fleming L., Antas P., Guerreiro P., et al. (2013). The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 32, 2603–2616. doi:10.1038/emboj.2013.200
Parbo P., Ismail R., Hansen K. V., Amidi A., Mårup F. H., Gottrup H., et al. (2017). Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain 140, 2002–2011. doi:10.1093/brain/awx120.2011
Parkhurst C. N., Yang G., Ninan I., Savas J. N., Yates J. R., Lafaille J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi:10.1016/j.cell.2013.11.030
Peng L., Yuan Z., Ling H., Fukasawa K., Robertson K., Olashaw N., et al. (2011). SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell. Biol. 31, 4720–4734. doi:10.1128/MCB.06147-11
Perry V. H., Teeling J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. doi:10.1007/s00281-013-0382-8
Probert L. (2015). TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 302, 2–22. doi:10.1016/j.neuroscience.2015.06.038
Qin W., Yang T., Ho L., Zhao Z., Wang J., Chen L., et al. (2006). Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction ∗. J. Biol. Chem. 281, 21745–21754. doi:10.1074/jbc.M602909200
Quintanilla R. A., Orellana D. I., González-Billault C., Maccioni R. B. (2004). Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 295, 245–257. doi:10.1016/j.yexcr.2004.01.002
Rajamohan S. B., Pillai V. B., Gupta M., Sundaresan N. R., Birukov K. G., Samant S., et al. (2009). SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell. Biol. 29 (15), 4116–4129. doi:10.1128/MCB.00121-09
Rangarajan P., Karthikeyan A., Lu J., Ling E.-A., Dheen S. T. (2015). Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 311, 398–414. doi:10.1016/j.neuroscience.2015.10.048
Ransohoff R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi:10.1126/science.aag2590
Rizzi L., Roriz-Cruz M. (2018). Sirtuin 1 and Alzheimer’s disease: an up-to-date review. Neuropeptides 71, 54–60. doi:10.1016/j.npep.2018.07.001
Rothgiesser K. M., Erener S., Waibel S., Lüscher B., Hottiger M. O. (2010). SIRT2 regulates NF-κB-dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 123, 4251–4258. doi:10.1242/jcs.073783
Salter M. W., Stevens B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi:10.1038/nm.4397
Sasaguri H., Nilsson P., Hashimoto S., Nagata K., Saito T., De Strooper B., et al. (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36, 2473–2487. doi:10.15252/embj.201797397
Scheltens P., Blennow K., Breteler M. M. B., de Strooper B., Frisoni G. B., Salloway S., et al. (2016). Alzheimer’s disease. Lancet (London, Engl. 388, 505–517. doi:10.1016/S0140-6736(15)01124-1
Schuitemaker A., van der Doef T. F., Boellaard R., van der Flier W. M., Yaqub M., Windhorst A. D., et al. (2012). Microglial activation in healthy aging. Neurobiol. Aging 33, 1067–1072. doi:10.1016/j.neurobiolaging.2010.09.016
Shaftel S. S., Griffin W. S. T., O’Banion M. K. (2008). The role of interleukin-1 in neuroinflammation and alzheimer disease: an evolving perspective. J. Neuroinflammation 5, 7. doi:10.1186/1742-2094-5-7
Shahidehpour R. K., Higdon R. E., Crawford N. G., Neltner J. H., Ighodaro E. T., Patel E., et al. (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 99, 19–27. doi:10.1016/j.neurobiolaging.2020.12.003
Shen X.-N., Niu L.-D., Wang Y.-J., Cao X.-P., Liu Q., Tan L., et al. (2019). Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 90, 590–598. doi:10.1136/jnnp-2018-319148
Sidorova-Darmos E., Wither R. G., Shulyakova N., Fisher C., Ratnam M., Aarts M., et al. (2014). Differential expression of sirtuin family members in the developing, adult, and aged rat brain. Front. Aging Neurosci. 6, 333. doi:10.3389/fnagi.2014.00333
Sierra A., Gottfried-Blackmore A. C., McEwen B. S., Bulloch K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi:10.1002/glia.20468
Smith J. A., Das A., Ray S. K., Banik N. L. (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. doi:10.1016/j.brainresbull.2011.10.004
Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292. doi:10.1016/j.jalz.2011.03.003
Tao R., Vassilopoulos A., Parisiadou L., Yan Y., Gius D. (2014). Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal. 20, 1646–1654. doi:10.1089/ars.2013.5482
Tracy T. E., Sohn P. D., Minami S. S., Wang C., Min S.-W., Li Y., et al. (2016). Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90, 245–260. doi:10.1016/j.neuron.2016.03.005
Ulland T. K., Colonna M. (2018). TREM2 — a key player in microglial biology and alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi:10.1038/s41582-018-0072-1
van der Kant R., Goldstein L. S. B. (2015). Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 32, 502–515. doi:10.1016/j.devcel.2015.01.022
Venegas C., Heneka M. T. (2019). Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 33, 13075–13084. doi:10.1096/fj.201900439
Wälti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., et al. (2016). Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 113, E4976–E4984. doi:10.1073/pnas.1600749113
Wang B., Zhang Y., Cao W., Wei X., Chen J., Ying W., et al. (2016). SIRT2 plays significant roles in lipopolysaccharides-induced neuroinflammation and brain injury in mice. Neurochem. Res. 41, 2490–2500. doi:10.1007/s11064-016-1981-2
Wang H., Kulas J. A., Wang C., Holtzman D. M., Ferris H. A., Hansen S. B., et al. (2021). Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. U. S. A. 118, e2102191118. doi:10.1073/pnas.2102191118
Wang J., Ho L., Qin W., Rocher A. B., Seror I., Humala N., et al. (2005). Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 19, 659–661. doi:10.1096/fj.04-3182fje
Wang Y., Mandelkow E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi:10.1038/nrn.2015.1
Wilkins H. M., Weidling I. W., Ji Y., Swerdlow R. H. (2017). Mitochondria-Derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 8, 508. doi:10.3389/fimmu.2017.00508
Woodbury A., Yu S. P., Wei L., García P. (2013). Neuro-modulating effects of honokiol: a review. Front. Neurol. 4, 130. doi:10.3389/fneur.2013.00130
Wu Z., Sun L., Hashioka S., Yu S., Schwab C., Okada R., et al. (2013). Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiol. Aging 34, 2715–2725. doi:10.1016/j.neurobiolaging.2013.05.018
Yan J., Luo A., Gao J., Tang X., Zhao Y., Zhou B., et al. (2019). The role of SIRT1 in neuroinflammation and cognitive dysfunction in aged rats after anesthesia and surgery. Am. J. Transl. Res. 11, 1555–1568.
Yang H., Zhang W., Pan H., Feldser H. G., Lainez E., Miller C., et al. (2012). SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One 7, e46364. doi:10.1371/journal.pone.0046364
Yang X. D., Tajkhorshid E., Chen L. F. (2010). Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol. Cell. Biol. 30 (9), 2170–2180. doi:10.1128/MCB.01343-09
Ye J.-S., Chen L., Lu Y.-Y., Lei S.-Q., Peng M., Xia Z.-Y., et al. (2019). SIRT3 activator honokiol ameliorates surgery/anesthesia-induced cognitive decline in mice through anti-oxidative stress and anti-inflammatory in hippocampus. CNS Neurosci. Ther. 25, 355–366. doi:10.1111/cns.13053
Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., et al. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. doi:10.1038/sj.emboj.7600244
Yin J., Han P., Song M., Nielsen M., Beach T. G., Serrano G. E., et al. (2018). Amyloid-β increases tau by mediating sirtuin 3 in Alzheimer’s disease. Mol. Neurobiol. 55, 8592–8601. doi:10.1007/s12035-018-0977-0
Yuan F., Xu Z.-M., Lu L.-Y., Nie H., Ding J., Ying W.-H., et al. (2016). SIRT2 inhibition exacerbates neuroinflammation and blood–brain barrier disruption in experimental traumatic brain injury by enhancing NF-κB p65 acetylation and activation. J. Neurochem. 136, 581–593. doi:10.1111/jnc.13423
Zenaro E., Piacentino G., Constantin G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi:10.1016/j.nbd.2016.07.007
Zhang Q., Zhang F., Ni Y., Kokot S. (2019). Effects of aluminum on amyloid-beta aggregation in the context of Alzheimer’s disease. Arabian J. Chem. 12 (8), 2897–2904. doi:10.1016/j.arabjc.2015.06.019
Zhang X., Wu Q., Zhang Q., Lu Y., Liu J., Li W., et al. (2017). Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Front. Neurosci. 11, 611. doi:10.3389/fnins.2017.00611
Zhang Y., Chi D. (2018). Overexpression of SIRT2 alleviates neuropathic pain and neuroinflammation through deacetylation of transcription factor nuclear factor-kappa B. Inflammation 41, 569–578. doi:10.1007/s10753-017-0713-3
Zhang Z., Shen Q., Wu X., Zhang D., Xing D. (2020). Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Aβ levels in Alzheimer’s disease models. Aging Cell 19, e13054. doi:10.1111/acel.13054
Zhao L. N., Long H., Mu Y., Chew L. Y. (2012). The toxicity of amyloid β oligomers. Int. J. Mol. Sci. 13 (6), 7303–7327. doi:10.3390/ijms13067303
Zheng D., Liwinski T., Elinav E. (2020). Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 6, 36. doi:10.1038/s41421-020-0167-x
Zheng H., Koo E. H. (2011). Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 6, 27. doi:10.1186/1750-1326-6-27
Zhou D., Jiang Y. (2019). Sirtuin 3 attenuates neuroinflammation-induced apoptosis in BV-2 microglia. Aging 11, 9075–9089. doi:10.18632/aging.102375
Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi:10.1038/nature09663
Keywords: sirtuins (SIRTs), brain homeostasis, neuroinf lammation, alzheimer’s disease, aging
Citation: Watroba M and Szukiewicz D (2022) Sirtuins promote brain homeostasis, preventing Alzheimer’s disease through targeting neuroinflammation. Front. Physiol. 13:962769. doi: 10.3389/fphys.2022.962769
Received: 06 June 2022; Accepted: 18 July 2022;
Published: 15 August 2022.
Edited by:
Geoffrey A. Head, Baker Heart and Diabetes Institute, AustraliaReviewed by:
Tiina Kauppinen, University of Manitoba, CanadaCopyright © 2022 Watroba and Szukiewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mateusz Watroba, bWF0cGF0NzFAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.