 Guangxin Peng
Guangxin Peng Xiaoling Tang1,2
Xiaoling Tang1,2 Yang Gui
Yang Gui Lifang Ye
Lifang Ye Liuyang Wu
Liuyang Wu Lihong Wang
Lihong Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 15 August 2022
Sec. Integrative Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.951980
This article is part of the Research Topic Transient Receptor Potential (TRP) Ion Channels in Non-Excitable Cells View all 6 articles
The transient receptor potential vanilloid subtype 1 (TRPV1), belonging to the TRPV channel family, is a non-selective, calcium-dependent, cation channel implicated in several pathophysiological processes. Collagen, an extracellular matrix component, can accumulate under pathological conditions and may lead to the destruction of tissue structure, organ dysfunction, and organ failure. Increasing evidence indicates that TRPV1 plays a role in the development and occurrence of fibrotic diseases, including myocardial, renal, pancreatic, and corneal fibrosis. However, the mechanism by which TRPV1 regulates fibrosis remains unclear. This review highlights the comprehensive role played by TRPV1 in regulating pro-fibrotic processes, the potential of TRPV1 as a therapeutic target in fibrotic diseases, as well as the different signaling pathways associated with TRPV1 and fibrosis.
Fibrosis is a pathophysiological feature primarily characterized by the accumulation of inflammatory cells; increased fibroblast proliferation, migration, and capacity to adhere and deposit extracellular matrix (ECM); and abnormal deposition of collagen-based ECM (Dees et al., 2020). The morbidity and mortality rates of fibrotic diseases remain high. Fibrosis, which is also an aging-related complication, is implicated in numerous chronic diseases; moreover, it can afflict almost any organ or tissue. Diseases associated with fibrosis include liver, lung, and renal diseases, myocardial fibrosis, systemic sclerosis, or any other disease in which, fibrotic remodeling leads to the destruction of organ structure and loss of function (Zhang and Zhang, 2020). Mounting evidence suggests that myofibroblasts play an instrumental role in ECM remodeling and fibrosis (Yazdani et al., 2017). Furthermore, fibroblasts are responsible for the production and early tensing of collagen and play key roles in fibrosis and wound healing. Smooth muscle actin (α-SMA), expressed in differentiated fibroblasts (i.e., myofibroblasts), increases the expression of type I and type III collagen, and inhibits the degradation of ECM macromolecules (Li et al., 2020). The onset and progression of fibrotic disease are associated with complex pathophysiological and biochemical processes regulated by a network of cytokines and intracellular signaling pathways (Henderson et al., 2020). However, identifying putative anti-fibrotic targets and formulating effective therapies for treating fibrotic diseases remain challenging. Transient receptor potential vanilloid (TRPV) channels are essential for various cellular functions and are involved in signaling pathways related to apoptosis, cell proliferation, cell migration, and invasion (Santoni et al., 2011). TRPV subtype 1 (TRPV1) is a member of the TRPV family, and mechanisms underlying its function are relatively well-understood. The TRPV1 receptor is highly permeable to Ca2+ and plays a critical role in maintaining Ca2+ levels in the cells (Zhai et al., 2020). In addition, calcium-dependent mechanisms are believed to play a crucial role in fibrogenesis via transient receptor potential channels (Inoue et al., 2019). Angiotensin II significantly increased the proliferation of cardiac fibroblasts, whereas activation of TRPV1 by capsaicin abolished the proliferation, differentiation and apoptosis of fibroblasts isolated from WT mice, but not in fibroblasts isolated from TRPV1-/- mice (Wang H.J et al., 2014). In recent years, a TRPV1 has been strong associated with fibrosis, and has been implicated in fibrotic diseases of various organs. For instance, TRPV1 is considered to regulate renal, lung, and cardiac disease and corneal fibrosis (Wang et al., 2008; Hutchinson et al., 2017; Inoue et al., 2019; Turan et al., 2021). Given the commonalities among fibrotic diseases, summarizing the direct or indirect relationship between TRPV1 and fibrosis as well as elucidating the novel role of TRPV1 in fibrosis will have far-reaching implications for future studies investigating fibrosis onset and development, pathogenesis, and potential novel therapies.
TRPV1, a capsaicin receptor first cloned in 1997 (Caterina et al., 1997), is a non-selective cation channel that allows the passage of cations such as H+, Na+, Ca2+, and Mg2+, with particularly high permeability for Ca2+ (Pedersen et al., 2005). TRPV1 is composed of 838 amino acids with a molecular weight of 95 kDa. The protein has a tetramer structure composed of six hydrophobic groups across the membrane and between the fifth and sixth transmembrane regions. The C- and N-termini of TRPV1 are located inside the cell membrane and regulate its functional activity (Liao et al., 2013). TRPV1 is a multimodal nociceptor that can be activated by a variety of stimuli, including capsaicin, lipopolysaccharide, low osmolarity, high temperature (>43 C), low pH (<6.0), and endogenous or synthetic ligands (Gavva et al., 2008; Gao et al., 2014). Although TRPV1 receptors are widely expressed throughout the nervous system, they are also found in other tissues and organs, including the heart, pancreas, lungs, trachea, kidneys, skin, intestines, and bladder (Faisy et al., 2007; Suri and Szallasi, 2008; Ditting et al., 2012; Thomas et al., 2012; Grundy et al., 2018; Cohen et al., 2019; Yoshie et al., 2020; Deng et al., 2021). At the cellular level, TRPV1 is expressed in lymphocytes, macrophages, dendritic cells, mast cells, endothelial cells, vascular smooth muscle cells, Epithelial cells, and fibroblasts (O'Connell et al., 2005; Kim et al., 2006; Razavi et al., 2006; Tóth et al., 2018; Otto et al., 2020; Luo et al., 2021; Shamsi et al., 2021). TRPV1 channels facilitate Ca2+ passage and thus, are involved in calcium signaling that regulates cellular processes related to proliferation, survival, migration, embryonic development, apoptosis, autophagy, and tumor cell metastasis (Waning et al., 2007; Takahashi et al., 2014; Malenczyk et al., 2017; Xu et al., 2018; Nakazawa et al., 2019; Chen et al., 2021; Güzel and Akpınar, 2021). Following TRPV1 channel activation, large amounts of calcium ions flow inward, while some potassium ions flow outward, thereby stimulating nerve endings to generate action potential and undergo depolarization. This activates a series of intracellular signals and the release of neurotransmitters such as calcitonin gene-related peptide (CGRP), substance P (SP), and somatostatin (SOM, a growth inhibitory hormone), thus, triggering multiple signaling cascades (Holzer, 1988). Most of the research studies investigating the functions of the TRPV1 channel have focused on screening TRPV1 agonists and antagonists and evaluating their role in TRPV1-related diseases and associated mechanisms. The exogenous agonists of TRPV1 include capsaicin, resin toxin, piperine, wogonin, norepinephrine, and cannabinol. The endogenous agonists of TRPV1 include cannabinoids, N-arachidonic ethanolamine, leukotriene B4, N-arachidonic dopamine, palmitoylethanolamide, and N-oleoyldopamine. The endogenous antagonists of TRPV1 are lysozyme D2 and norepinephrine, while the exogenous antagonists include capsazepine iodine resin toxin, caffeic acid, SB-366791, AMG-9810, and A-425619 (Li et al., 2021). Use of TRPV1 agonists and antagonists along with TRPV1 receptor knockout mice has revealed a strong connection between TRPV1 and the development of fibrotic diseases (Hutter et al., 2005; Ueda et al., 2008; Sumioka et al., 2014; Wang Q et al., 2014). Table 1 lists the modulators available for evaluating TRPV1 association with fibrosis.
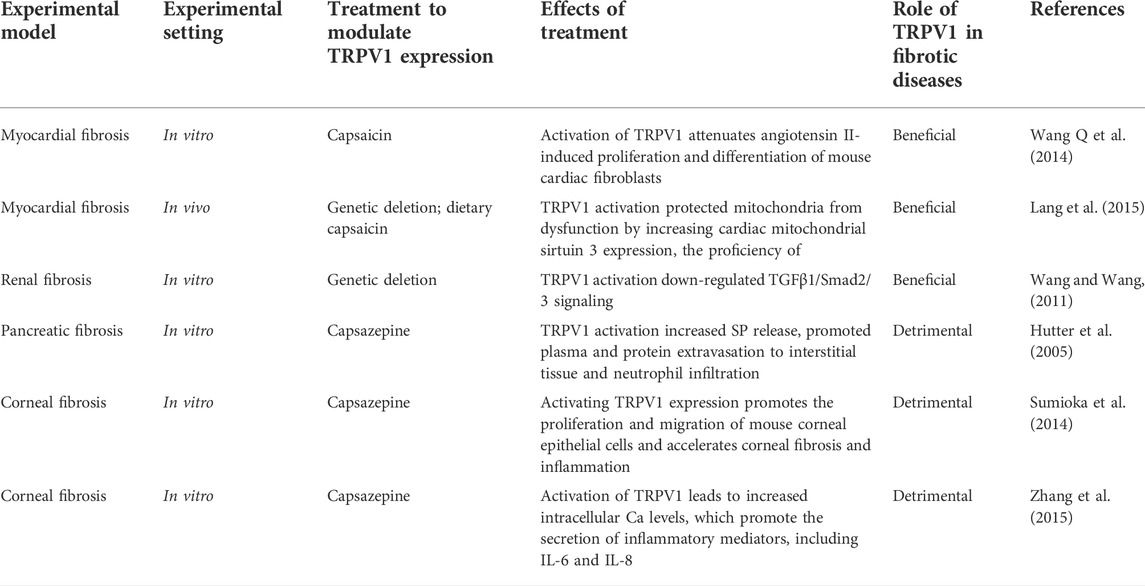
TABLE 1. Summary of the role played by TRPV1 in fibrotic disease.
Cardiac fibrosis is a pathological change observed in the later stage of various cardiovascular diseases, usually manifesting as cardiac hypertrophy, ECM deposition, and collagen synthesis, which eventually lead to irreversible heart structure remodeling and cardiac dysfunction (González et al., 2018). Pathological cardiac remodeling subjects the heart to increased pressure and volume overload and affects all cardiac cells, including myocytes, endothelial cells, stromal cells, and cardiac fibroblasts (Heusch et al., 2014). Calcium ions regulate various cellular functions, and abnormal intracellular calcium homeostasis can activate fibrosis-associated remodeling. Calcium plays an important role in regulating apoptosis, mast gene expression, and myocardial fibrosis, while TRPV1 mediates Ca2+ signal transduction in cells (Ramires et al., 1998). Cardiac fibroblasts treated with the TRPV1 agonist capsaicin exhibit activation of TRPV1 channels followed by a significant increase in intracellular Ca2+ concentration (Wang Q et al., 2014). Intracellular Ca2+ entry facilitates fibroblast function (Inoue et al., 2006); for instance, the occurrence of myocardial fibrosis depends on cardiac fibroblast proliferation (Krenning et al., 2010). Specifically, TRPV1 overexpression substantially reduces isoproterenol-induced proliferation and differentiation in mouse cardiac fibroblasts and prevents the development of myocardial fibrosis (Wang et al., 2016). Furthermore, a high-salt diet can cause cardiac hypertrophy and fibrosis in mice whereas long-term capsaicin consumption can substantially decrease cardiac hypertrophy and fibrosis. After long-term capsaicin intervention, WT mice show reduction in high-salt-induced cardiac insufficiency and cardiac hypertrophy; however, these symptoms are not improved in TRPV1-/- mice. Chronic dietary capsaicin intake can increase the proficiency of mitochondrial complex I OXPHOS, production of ATP, expression of cardiac mitochondrial sirtuin 3, and activity of complex I enzyme activity via activating TRPV1, leading to alleviation in cardiac mitochondrial dysfunction and prevention of cardiac hypertrophy induced by a high-salt diet (Lang et al., 2015). Ligation of the anterior descending branch of the left coronary artery is a common method to generate myocardial infarction models. This procedure damages myocardial contractility and aggravates concomitant development of left ventricular dilatation, myocardial fibrosis, and ventricular remodeling (Gao et al., 2010). Recently, capillary formation, myofibroblast deposition, and collagen expression in TRPV1-/- mice after ligation of the left anterior descending coronary artery were reported to be significantly higher than those in wild-type mice. This was accompanied by significant upregulation of vascular endothelial growth factors and matrix metalloproteinases, which exacerbated cardiac remodeling. Furthermore, TRPV1-/- mice displayed higher cardiac fibroblast infiltration, capillary density, and ECM collagen content. The affected processes markedly accelerated fibrosis and impaired myocardial contractility following myocardial infarction (Huang et al., 2010). Mice that received intrathecal resin toxin (TRPV1 agonist) showed reduced sympathetic nerve activity in an overactive heart and improved cardiac compliance. Resin toxin desensitizes cardiac afferent fibers by activating TRPV1 expression, thereby eliminating the sympathetic excitatory reflex of the heart, weakening cardiac remodeling, and alleviating cardiovascular dysfunction in rats with heart failure. Molecular evidence has demonstrated that resin toxin reduces cardiac hypertrophy, cardiac apoptosis, and fibrosis marker expression in rat models of chronic heart failure (Wang H.J et al., 2014; Wang et al., 2019). In addition, TRPV1 deficiency exacerbates myocardial hypertrophy and impairs diastolic function and myocardial interstitial fiber proliferation in a mouse model of transverse aortic constriction-induced de novo cardiac pressure overload (Zhong et al., 2018). After transverse aortic constriction (TAC) in TRPV1-/- mice, secretion of pro-inflammatory cytokines as well as infiltration by macrophages increase, and cardiac function is deteriorated. Moreover, neuropeptide CGRP, which regulates vascular tone and cardiovascular function, cannot be released in the absence of TRPV1. Therefore, TRPV1 contributes to the anti-inflammatory process by inhibiting pressure overload-induced myocardial hypertrophy and exerting cardioprotective effects. However, results obtained in other studies represent another point of view. In a study using TAC to simulate acute pressure overload, the cardiac function of TRPV1-/- mice was improved compared with that of wild type mice, and myocardial hypertrophy, fibrosis, and tissue remodeling were reduced (Buckley and Stokes, 2011). These findings suggest that TRPV1 knockout can preserve cardiac structure and function under simulated pressure overload. Further, the use of TRPV1 antagonists can prevent or improve myocardial hypertrophy and myocardial fibrosis and reduce the levels of apoptosis markers. Therefore, the TRPV1 receptor is a candidate therapeutic target for myocardial fibrosis. Notably, among other TRPV channels, knockout of the TRPV2 channel can effectively improve cardiac insufficiency and inhibit myocardial fibrosis (Entin-Meer et al., 2018). Activation of TRPV3 leads to interstitial fibrosis and aggravates cardiac dysfunction in pressure overloaded rats (Liu Y al., 2018) . TRPV4 gene deletion or siRNA-mediated downregulation is attenuated in vitro by TGF- β, and Trpv4-/- mice show preserved myocardial integrity and cardiac function as well as reduced cardiac fibrosis (Adapala et al., 2020).
Renal fibrosis is often associated with chronic kidney failure resulting from various causes, which eventually progresses to end-stage renal failure results in the destruction of kidney tissue structure and complete loss of kidney function (Humphreys, 2018). Hypertension-induced kidney injury elicits a fibrotic response via abnormal activation of renal fibroblasts and the overproduction of ECM proteins (Sun, 2019). Hypertension induced by deoxycorticosterone acetate salts (DOCA) causes glomerulosclerosis, increases the number of proliferating cell nuclear antigen (PCNA)-positive tubular cells, tubular interstitial fibrosis, and ECM over-deposition in both wild-type and TRPV1-/- mice; however, TRPV1-/- mice exhibit more severe symptoms than wild-type mice (Wang et al., 2008). TRPV1 knockdown exacerbates renal damage and fibrosis induced by DOCA salt-mediated hypertension; thus, TRPV1 may be a part of a protective mechanism against hypertension-induced organ damage. Consistently, TRPV1 activation reportedly protects against renal damage during hypertension as TRPV1 loss promotes the upregulation of fibronectin, expression of type I and III collagen, which can delay the progression of interstitial renal fibrosis. (Wang and Wang, 2011). Furthermore, DOCA salt induces upregulation of SMAD2/3 phosphorylation and increases TGF-β1 activity. TGF-β1 protein expression in the kidneys of DOCA salt-induced hypertensive TRPV1-/- mice is significantly higher than that in WT mice. These results indicate that excessive damage to renal function and structure is accompanied by increased ECM protein production and TGF-β1/SMAD2/3 signaling pathway activation. Thus, interfering with TGF-β1 production may help reduce ECM accumulation and delay progression of renal interstitial fibrosis (Wang and Wang, 2009). Further, increased production of pro-inflammatory cytokines/chemokines, including TNF-α, IL-6 and MCP-1, was reported in the TRPV1-/- mouse model of DOCA-induced acute salt-sensitive hypertension. Thus, in the absence of TRPV1, many pro-inflammatory cytokines harmful to the kidney are produced along with increased macrophage infiltration and tubulointerstitial injury. In addition, abnormal activation of the nuclear transcription factor NF-κB as well as multiple inflammation-related genes causes excessive inflammatory responses that exacerbate renal damage and pathogenesis. The absence of TRPV1 produces a large number of pro-inflammatory cytokines harmful to the kidney, accompanied by increased macrophage infiltration and tubulointerstitial injury. In DOCA salt-treated WT mice compared with that in TRPV1-/- mice, it was obvious that NF-κB activity in TRPV1-/- mice-increased further. These data suggest that NF-κB plays a central regulatory role in mediating the anti-inflammatory and protective effects of TRPV1. In addition, abnormal activation of the nuclear transcription factor NF-κB as well as multiple inflammation-related genes induces excessive inflammatory responses that exacerbate renal damage and pathogenesis. NF-κB activity was obviously increased in TRPV1-/- mice compared with that in DOCA salt-treated WT mice . These data suggest that NF-κB plays a central regulatory role in mediating the anti-inflammatory and protective effects of TRPV1 in renal tissues. Furthermore, available scientific evidence suggests that the TRPV1 receptor is expressed in human proximal tubular cells, mouse renal microvascular endothelial cells, canine renal tubular cells, and porcine renal tubular cells (Tsagogiorgas et al., 2012; Wang et al., 2017; Lu et al., 2020). In vitro studies have suggested that TRPV1 may be involved in renal fibrosis through the production of major pro-inflammatory mediators of apoptosis and can protect against renal fibrosis as a prospective regulator of inflammatory processes. Alternatively, in vivo studies using an ischemia/reperfusion model along with TRPV1 agonists (capsaicin and resin toxin) have shown that TRPV1 plays a role in the response to renal injury in mice. Moreover, TRPV1 agonists reportedly prevent ischemia/reperfusion-induced renal dysfunction and protect the kidneys from injury and fibrosis (Ueda et al., 2008; Chen et al., 2014). Thus, TRPV1 is involved in mediating inflammatory response and fibrosis in renal fibrosis, and its activation ameliorates both processes. The relationship between TRPV channels and renal fibrosis has received limited research attention. However, treatment with apigenin, an agonist of TRPV4, has been found to help alleviate the DOCA salt-induced damage to renal structure and function, reduce the expression of extracellular matrix proteins, and exert a beneficial effect against hypertension-induced renal fibrosis (Wei et al., 2017).
Chronic pancreatitis is an irreversible and progressive inflammation of the pancreatic parenchyma and pancreatic ducts. Its pathological features present as chronic inflammatory damage and interstitial fibrosis of the pancreatic parenchyma, calcification of the pancreatic parenchyma, dilatation of the pancreatic ducts, and changes such as pancreatic duct stones (Barry, 2018). Pancreatic fibrosis, a characteristic feature of chronic pancreatitis, presents with the fibrosis of the pancreatic parenchyma, atrophy of lung alveolar cells, and infiltration of inflammatory cells (Huang et al., 2021). In pancreatitis, TRPV1 is activated in pancreatic sensory nerves and spinal cord neurons, thereby inducing the release SP and CGRP, causing pain and inflammation (Wick et al., 2006). SP plays pro-inflammatory role in acute pancreatitis and pancreatitis-related lung injury by binding to neurokinin 1 receptor (NK1R) and acts on calcium channels in cell membranes via inositol triphosphate, causing depolarization of membrane potential and alteration of protein kinase activity (Bhatia et al., 1998). Furthermore, in a mouse model of cerulein-induced pancreatitis, the use of the TRPV1 antagonist capsazepine inhibited primary sensory neurons and reduced tissue inflammation, thereby inhibiting the release of SP and the subsequent NK1R activation, eventually alleviating the severity of fibrosis in the pancreas (Nathan et al., 2001; Hutter et al., 2005). Alternatively, in a rat model of pancreatitis, CGRP administration induced vasodilation and vascular injury, which aggravated pancreatitis symptoms. Moreover, CGRP removes active digestive enzymes and increases the levels of inflammatory mediators in pancreatic tissue to reduce pancreatic injury (Warzecha et al., 2001). The severity of pancreatitis may be determined by the aggregation and activation of inflammatory cells after alveolar cell injury and the production and release of cytokines and other inflammatory mediators (Liddle and Nathan, 2004). TRPV1 expression is upregulated in pancreatic dorsal root ganglion neurons during chronic pancreatitis, and a significant reversal of pancreatic inflammation and pain relief was reported with systemic administration of the TRPV1 antagonist SB-366791 (Xu et al., 2007). Furthermore, another study showed that pancreatic inflammation combined with significant changes in the activity and function of TRPV1 increased the excitability of pancreatic sensory neurons entering the spinal cord as well as the vagus nerve (Schwartz et al., 2011). Based on these observations, we conclude that TRPV1 plays a key role in neuroinflammation-driven pancreatic fibrosis and blocking neurogenic inflammation may improve disease outcomes. However, this needs to be investigated and validated in pre-clinical and clinical models. In other studies on TRPV channels and pancreatic fibrosis, the TRPV4 antagonist hc067047 has been found to effectively alleviate pain-related behavior in the rat chronic pancreatitis model induced by alcohol/high fat diet (AHF) and reduce hypersensitivity and fibrosis pathology (Zhang et al., 2015). TRPV6 is a novel susceptibility gene for chronic pancreatitis. Administration of cerulein to TRPV6 mut/mut mice leads to severe pancreatitis, characterized by elevated pancreatic enzyme levels, histological changes, and pancreatic fibrosis (Masamune et al., 2020).
Fibrosis of the corneal stroma, also known as scarring or progressive opacity, represents a disruption of corneal homeostasis following injury, resulting in abnormal tissue remodeling (Wilson et al., 2017). Excessive corneal fibrosis can form a scar in the center of the cornea, thereby reducing corneal transparency and causing visual impairments (Meek and Knupp, 2015). Recently, the role of TRPV1 in corneal fibrosis was explored in vitro and in vivo, and TRPV1 expression was observed in both corneal fibrotic tissue and epithelial cells. TRPV1 deletion reportedly inhibited ocular fibroblast differentiation as well as expression of TGF-β1 and other pro-inflammatory genes, while TRPV1 suppression inhibited inflammation and fibrosis onset during corneal healing in mouse model of corneal alkali-burn (Okada et al., 2011). Myofibroblast activation promotes fibrosis, and persistent induction of fibrotic response results in fibrosis observed in many types of corneal trauma (Wilson, 2020). Corneal wound-induced myofibroblast differentiation is greatly inhibited by capsazepine (a TRPV1 antagonist) via downregulating α-SMA expression (Yang et al., 2013a). Release of endocannabinoids such as 2-AG following injury-induced cannabinoid receptor 1 (CB1) activation downregulates TRPV1-mediated inflammation and corneal opacity (Yang et al., 2013b). This finding suggests that inhibition of TRPV1 activation reduces corneal fibrosis and inflammation and CB1 is a candidate drug target for regulating TRPV1-induced inflammation. Suppressing TRPV1 ion channel expression or using TRPV1 antagonists are potential therapeutic strategies to reduce inflammation and enhance the healing of fibrotic wounds. Furthermore, TRPV1 activity is closely associated with an inward flow of cytosolic calcium (Yang et al., 2013c). Treatment of human corneal epithelial cells with capsaicin increases intracellular Ca2+ levels and increases IL-6 and IL-8 release by 3.3- and 9-fold, respectively. Thus, TRPV1 activation induces the secretion and release of inflammatory mediators from the human corneal epithelium which subsequently leads to corneal damage and fibrosis (Zhang et al., 2007). Furthermore, capsaicin (a TRPV1 agonist) promotes the proliferation and migration of corneal epithelial cells, whereas SB366791 (a TRPV1 antagonist) delays corneal fibrosis in mice. These results indicate TRPV1 activation induces the upregulation of IL-6 and SP expression, thereby delaying the healing of the corneal epithelium in mice (Sumioka et al., 2014). Collectively, these findings suggest that TRPV1 is an important player in mediating inflammation following in vivo injury and should be further explored as a therapeutic target for corneal fibrosis. In line with this, TRPV4 activation contributes to the severe inflammatory fibrosis caused by alkali damage to the cornea of mice. TRPV4 antagonists can improve corneal function recovery during wound healing by reducing inflammatory fibrosis (Okada et al., 2016).
Pulmonary fibrosis is a chronic, interstitial lung disease characterized by thickening and scarring of the alveolar walls and is caused by long-term inflammation of the lungs, bacterial or viral infections, autoimmune reactions, and allergy and related tissue damage (Thannickal et al., 2004). Life expectancy after pulmonary fibrosis diagnosis is between 2 and 6 years (Selman et al., 2001). Pulmonary fibrosis is commonly characterized by coughing, difficulty in breathing, and fatigue (Swigris et al., 2005). However, few studies have examined the association between TRPV1 and pulmonary fibrosis. A recent study showed that bleomycin-induced pulmonary fibrosis is characterized by a positive correlation between neuropeptide SP, NK1R, and CGRP and cough sensitivity in animal models (Guan et al., 2021). Secretion of inflammatory factors and immune mediators indirectly activates the TRPV1 receptor. Furthermore, inflammation and the cough reflex pathway are markedly upregulated in pulmonary fibrosis, leading to increasing cough hypersensitivity and disease progression (Guo et al., 2019). Regarding bleomycin-induced pulmonary fibrosis, the symptoms of fibrosis are weaker in TRPV4-/- mice than in wild type mice, and gene deletion of TRPV4 inhibits myofibroblast differentiation (Rahaman et al., 2014).
Increasing evidence indicates that fibrotic diseases are associated with pathways such as those for inflammation, oxidative stress, the renin angiotensin system, and TGF- β/SMAD signaling (Chen et al., 2018; Otoupalova et al., 2020; Gupta et al., 2021, 2018). TGF-β1 receptor is one of the key elements associated with fibrosis and may play a critical role in infarct healing (Murphy-Ullrich and Suto, 2018). TGF-β1 is induced and activated in various fibrotic diseases, and it in turn induces its downstream mediators SMAD2 and SMAD3 to play their biological roles (Hu et al., 2018). In myocardial fibrosis, TRPV1-mediated angiotensin II-induced cardiac fibroblast differentiation inhibits ECM deposition via a downregulation-dependent TGF-β-SMAD signaling pathway (Huang et al., 2010; Wang H.J et al., 2014). Furthermore, TRPV1 activation upregulates TGF-β1 protein expression and can inhibit the TGF-β1 fibrogenic activity by downregulating SMAD2/3; these data suggest that TRPV1 inhibits the TGF- β/SMAD signaling pathway and plays a protective role in renal fibrosis induced by salt-sensitive hypertension (Wang and Wang, 2011). In pancreatic fibrosis, TRPV1 contributes to pain and fibrosis by regulating TGF-β expression via the SMAD pathway (Liu L et al., 2018). TRPV1 silencing in corneal fibroblasts promotes TGF-β-induced corneal myofibroblast differentiation via SMAD2-p38 signaling, which also induces oxidative stress and accelerates corneal fibrosis progression (Yang et al., 2013a). Recent studies have elucidated a new signal transduction pathway leading to corneal fibrosis. The p38-MAPK pathway is involved in the migration and proliferation of cells during corneal epithelial tissue wound healing in mice. TGF-β1-activated p38-MAPK is one of the most important signaling cascades involved in cell migration (Saika et al., 2004). The limited information available regarding the regulatory role played by TRPV1 in fibrosis suggests that TRPV1 mediates fibrosis mainly via TGF-β signaling. Research studies exploring the other TRPV channels have focused on elucidating their relationship with TGF-β signaling (Rahaman et al., 2014; Liu Y et al., 2018; Kato et al., 2022). TRPV4 also protects the heart from myocardial infarction-induced pathological myocardial remodeling by regulating cardiac fibroblast differentiation and myocardial fibrosis through the Rho/MRTF-A signaling pathway (Adapala et al., 2020).
In this review, we have comprehensively analyzed the role played by TRPV1 in fibrosis. Since TRPV1 channels are expressed in various organs, they play different roles in physiological and clinical conditions. TRPV1 mediates the entry of extracellular Ca2+ and promotes Ca2+ release from the endoplasmic reticulum into the cytoplasm, thereby regulating intracellular Ca2+ concentration and myofibroblast growth and migration. The neuropeptides CGRP and SP are released in conjunction with TRPV1 activation, causing neurogenic inflammation expressed as vasodilation, plasma extravasation, and podocyte enlargement. These neuropeptides promote leukocyte migration and regulate fibrosis progression in different organs. Furthermore, the TGF-β/SMAD signaling pathway is one of the promising targets to promote healing of fibrosis. Activation of TRPV1 favors renal fibrosis and inhibition of TRPV1 favors pancreatic fibrosis, corneal fibrosis and ocular corneal fibrosis. However, the contradictory role of TRPV1 in myocardial fibrosis makes it difficult to decide whether TRPV1 should be activated or inhibited when treating inflammation and fibrosis caused by organ damage. This phenomenon illuminates the complex role played by TRPV1 in pathophysiological conditions. Two experimental studies using TAC to simulate acute pressure overload reported contrasting results (Buckley and Stokes, 2011; Zhong et al., 2018). These contrasting results may be attributed to the different factors analyzed as well as different experimental conditions, such as different testing time points before and after cardiac echocardiography, differences in the size of the needles used to develop the TAC model, and different doses of antagonists used. The long-term efficacy and safety of the systemic interventions targeting TRPV1 remain unknown and may be influenced by post-inflammatory compensatory response. Although this makes understanding the role of TRPV1 and mechanisms underlying its function a challenge for developing anti-fibrotic therapy, TRPV1 is a potential therapeutic target to prevent fibrosis onset and development and ensuing organ damage. We previously showed that TRPV1 loss aggravates inflammation following myocardial infarction, impairs lesion healing, promotes myocardial fibrosis, and leads to poor ventricular remodeling (Wang and Wang, 2005). Nevertheless, TRPV1 may also protect the heart from injury by releasing the neurotransmitter SP, which acts as a cardiac anti-inflammatory molecule. Multiple studies have shown that TRPV1 activation can alleviate diabetes and cardiovascular diseases such as atherosclerosis and hypertension (Ma et al., 2011; Marshall et al., 2013). The neuropeptides released following TRPV1 activation exert multifaceted effects and regulate TRPV1 role in the inflammatory process at multiple levels. In conclusion, Ca2+, neuropeptides, and inflammation collectively modulate the role played by TRPV1 in fibrosis. Since little is known about the mechanisms underlying TRPV1-mediated fibrosis, further research to explore the role of TRPV1 in fibrosis is warranted. This will help in developing effective therapeutic interventions for fibrotic diseases in the future.
GP and XT: Designed the review and wrote the manuscript. YG, JY, and LY: Preparation, presentation of the published work, specifically writing the initial draft. LWu and YD: Revised the manuscript. LWa: Ensured that the descriptions are accurate and agreed by all authors.
This study was supported by the National Natural Science Foundation of China (Grant No. 81670447); the National Natural Science Foundation of Zhejiang Province (Grant No. LY15H020006); Zhejiang Province Key Subject of Medicine (Neurological Rehabilitation) and the Traditional Chinese Medicine Program of Zhejiang Provincial (Grant No. 2017ZZ001); the Zhejiang Provincial Health Commission Project (Grant No. 2017KY559); and the National Natural Science Foundation of Zhejiang Province (Grant No. LY19H070003). This research was supported by Zhejiang Provincial Natural Science Foundation of Chinaunder Grant No. LGF18H020007. LWa is sponsored by the Zhejiang Provincial Program for developing High-Level Innovative Health Talents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.951980/full#supplementary-material
Adapala R. K., Kanugula A. K., Paruchuri S., Chilian W. M., Thodeti C. K. (2020). TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res. Cardiol. 115, 14. doi:10.1007/s00395-020-0775-5
Bhatia M., Saluja A. K., Hofbauer B., Frossard J. L., Lee H. S., Castagliuolo I., et al. (1998). Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc. Natl. Acad. Sci. U. S. A. 95, 4760–4765. doi:10.1073/pnas.95.8.4760
Buckley C. L., Stokes A. J. (2011). Mice lacking functional TRPV1 are protected from pressure overload cardiac hypertrophy. Channels 5, 367–374. doi:10.4161/chan.5.4.17083
Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D., et al. (1997). The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824. doi:10.1038/39807
Chen L., Markó L., Kaßmann M., Zhu Y., Wu K., Gollasch M., et al. (2014). Role of TRPV1 channels in ischemia/reperfusion-induced acute kidney injury. PLoS One 9, e109842. doi:10.1371/journal.pone.0109842
Chen L., Yang T., Lu D. W., Zhao H., Feng Y. L., Chen H., et al. (2018). Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 101, 670–681. doi:10.1016/j.biopha.2018.02.090
Chen M., Dong X., Deng H., Ye F., Zhao Y., Cheng J., et al. (2021). Targeting TRPV1-mediated autophagy attenuates nitrogen mustard-induced dermal toxicity. Signal Transduct. Target. Ther. 6, 29. doi:10.1038/s41392-020-00389-z
Cohen J. A., Edwards T. N., Liu A. W., Hirai T., Jones M. R., Wu J., et al. (2019). Cutaneous TRPV1+ neurons trigger protective innate type 17 anticipatory immunity. Cell 178, 919–932. e14. doi:10.1016/j.cell.2019.06.022
Dees C., Pötter S., Zhang Y., Bergmann C., Zhou X., Luber M., et al. (2020). TGF-β–induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Invest. 130, 2347–2363. doi:10.1172/JCI122462
Deng F., Zhao B. C., Yang X., Lin Z. B., Sun Q. S., Wang Y. F., et al. (2021). The gut microbiota metabolite capsiate promotes Gpx4 expression by activating TRPV1 to inhibit intestinal ischemia reperfusion-induced ferroptosis. Gut Microbes 13, 1–21. doi:10.1080/19490976.2021.1902719
Ditting T., Freisinger W., Siegel K., Fiedler C., Small L., Neuhuber W., et al. (2012). Tonic postganglionic sympathetic inhibition induced by afferent renal nerves? Hypertension. Hypertension 59, 467–476. doi:10.1161/HYPERTENSIONAHA.111.185538
Entin-Meer M., Cohen L., Hertzberg-Bigelman E., Levy R., Ben-Shoshan J., Keren G., et al. (2018). TRPV2 knockout mice demonstrate an improved cardiac performance following myocardial infarction due to attenuated activity of peri-infarct macrophages. PLoS One 12, e0177132. doi:10.1371/journal.pone.0177132
Faisy C., Planquette B., Naline E., Risse P. A., Frossard N., Fagon J. Y., et al. (2007). Acid-induced modulation of airway basal tone and contractility: Role of acid-sensing ion channels (ASICs) and TRPV1 receptor. Life Sci. 81, 1094–1102. doi:10.1016/j.lfs.2007.08.026
Gao E., Lei Y. H., Shang X., Huang Z. M., Zuo L., Boucher M., et al. (2010). A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107 (12), 1445–1453. doi:10.1161/CIRCRESAHA.110.223925
Gao F., Liang Y., Wang X., Lu Z., Li L., Zhu S., et al. (2014). TRPV1 activation attenuates high-salt diet-induced cardiac hypertrophy and fibrosis through PPAR- δ upregulation. PPAR Res. 2014, 491963. doi:10.1155/2014/491963
Gavva N. R., Treanor J. J. S., Garami A., Fang L., Surapaneni S., Akrami A., et al. (2008). Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136, 202–210. doi:10.1016/j.pain.2008.01.024
González A., Schelbert E. B., Díez J., Butler J. (2018). Myocardial interstitial fibrosis in Heart Failure: Biological and translational perspectives. J. Am. Coll. Cardiol. 71, 1696–1706. doi:10.1016/j.jacc.2018.02.021
Grundy L., Daly D. M., Chapple C., Grundy D., Chess-Williams R. (2018). TRPV1 enhances the afferent response to P2X receptor activation in the mouse urinary bladder. Sci. Rep. 8, 197. doi:10.1038/s41598-017-18136-w
Guan M., Ying S., Wang Y. (2021). Increased expression of transient receptor potential channels and neurogenic factors associates with cough severity in a Guinea pig model. BMC Pulm. Med. 21, 187. doi:10.1186/s12890-021-01556-w
Guo Y., Ying S., Zhao X., Liu J., Wang Y. (2019). Increased expression of lung TRPV1/TRPA1 in a cough model of bleomycin-induced pulmonary fibrosis in Guinea pigs. BMC Pulm. Med. 19, 27. doi:10.1186/s12890-019-0792-z
Gupta D., Kumar A., Mandloi A., Shenoy V. (2021). Renin angiotensin aldosterone system in pulmonary fibrosis: Pathogenesis to therapeutic possibilities. Pharmacol. Res. 174, 105924. doi:10.1016/j.phrs.2021.105924
Güzel M., Akpınar O. (2021). Hydroxychloroquine attenuates acute inflammation (LPS)-induced apoptosis via inhibiting TRPV1 channel/ROS signaling pathways in human monocytes. Biology 10, 967. doi:10.3390/biology10100967
Henderson N. C., Rieder F., Wynn T. A. (2020). Fibrosis: From mechanisms to medicines. Nature 587, 555–566. doi:10.1038/s41586-020-2938-9
Heusch G., Libby P., Gersh B., Yellon D., Böhm M., Lopaschuk G., et al. (2014). Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383, 1933–1943. doi:10.1016/S0140-6736(14)60107-0
Holzer P. (1988). Local effector functions of capsaicin-sensitive sensory nerve endings: Involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience 24, 739–768. doi:10.1016/0306-4522(88)90064-4
Hu H. H., Chen D. Q., Wang Y. N., Feng Y. L., Cao G., Vaziri N. D., et al. (2018). New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 25, 76–83. doi:10.1016/j.cbi.2018.07.008
Huang C., Iovanna J., Santofimia-Castaño P. (2021). Targeting fibrosis: The bridge that connects pancreatitis and pancreatic cancer. Int. J. Mol. Sci. 22, 4970. doi:10.3390/ijms22094970
Huang W., Rubinstein J., Prieto A. R., Wang D. H. (2010). Enhanced postmyocardial infarction fibrosis via stimulation of the transforming growth factor-β-Smad2 signaling pathway: Role of transient receptor potential vanilloid type 1 channels. J. Hypertens. 28, 367–376. doi:10.1097/HJH.0b013e328333af48
Humphreys B. D. (2018). Mechanisms of renal fibrosis. Annu. Rev. Physiol. 80, 309–326. doi:10.1146/annurev-physiol-022516-034227
Hutchinson N., Gibbs A., Tonks A., Hope-Gill B. D. (2017). Airway expression of transient receptor potential (TRP) vanniloid-1 and ankyrin-1 channels is not increased in patients with idiopathic pulmonary fibrosis. PLoS One 12, e0187847. doi:10.1371/journal.pone.0187847
Hutter M. M., Wick E. C., Day A. L., Maa J., Zerega E. C., Richmond A. C., et al. (2005). Transient receptor potential vanilloid (TRPV-1) promotes neurogenic inflammation in the pancreas via activation of the neurokinin-1 receptor (NK-1R). Pancreas 30, 260–265. doi:10.1097/01.mpa.0000153616.63384.24
Inoue R., Jensen L. J., Shi J., Morita H., Nishida M., Honda A., et al. (2006). Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 99, 119–131. doi:10.1161/01.RES.0000233356.10630.8a
Inoue R., Kurahara L. H., Hiraishi K. (2019). TRP channels in cardiac and intestinal fibrosis. Semin. Cell Dev. Biol. 94, 40–49. doi:10.1016/j.semcdb.2018.11.002
Kato S., Shiozaki A., Kudou M., Shimizu H., Kosuga T., Ohashi T., et al. (2022). TRPV2 promotes cell migration and invasion in gastric cancer via the transforming growth factor-β signaling pathway. Ann. Surg. Oncol. 29, 2944–2956. doi:10.1245/s10434-021-11132-5
Kim S. J., Lee S. A., Yun S. J., Kim J. K., Park J.-S., Jeong H. S., et al. (2006). Expression of vanilloid receptor 1 in cultured fibroblast. Exp. Dermatol. 15, 362–367. doi:10.1111/j.0906-6705.2006.00418.x
Krenning G., Zeisberg E. M., Kalluri R. (2010). The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 225, 631–637. doi:10.1002/jcp.22322
Lang H., Li Q., Yu H., Li P., Lu Z., Xiong S., et al. (2015). Activation of TRPV1 attenuates high salt-induced cardiac hypertrophy through improvement of mitochondrial function. Br. J. Pharmacol. 172, 5548–5558. doi:10.1111/bph.12987
Li L., Chen C., Chiang C., Xiao T., Chen Y., Zhao Y., et al. (2021). The impact of TRPV1 on cancer pathogenesis and therapy: A systematic review. Int. J. Biol. Sci. 17, 2034–2049. doi:10.7150/ijbs.59918
Li X., Zhang W., Cao Q., Wang Z., Zhao M., Xu L., et al. (2020). Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 6, 80. doi:10.1038/s41420-020-00316-9
Liao M., Cao E., Julius D., Cheng Y. (2013). Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. doi:10.1038/nature12822
Liddle R. A., Nathan J. D. (2004). Neurogenic inflammation and pancreatitis. Pancreatology 4, 551–559. doi:10.1159/000082180
Liu L., Zhu Y., Noë M., Li Q., Pasricha P. J. (2018). Neuronal transforming growth factor beta signaling via SMAD3 contributes to pain in animal models of chronic pancreatitis. Gastroenterology 154, 2252–2265. e2. doi:10.1053/j.gastro.2018.02.030
Liu Y., Qi H., E M., Shi P., Zhang Q., Li S., et al. (2018). Transient receptor potential vanilloid-3 (TRPV3) activation plays a central role in cardiac fibrosis induced by pressure overload in rats via TGF-β1 pathway. Naunyn-Schmiedeberg's. Arch. Pharmacol. 391, 131–143. doi:10.1007/s00210-017-1443-7
Lu C. L., Liao C. H., Lu K. C., Ma M. C. (2020). TRPV1 hyperfunction involved in uremic toxin indoxyl sulfate-mediated renal tubular damage. Int. J. Mol. Sci. 21, 6212. doi:10.3390/ijms21176212
Luo X., Chen O., Wang Z., Bang S., Ji J., Lee S. H., et al. (2021). IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron 109, 2691–2706.e5. e5. doi:10.1016/j.neuron.2021.06.015
Ma L., Zhong J., Zhao Z., Luo Z., Ma S., Sun J., et al. (2011). Activation of TRPV1 reduces vascular lipid accumulation and attenuates atherosclerosis. Cardiovasc. Res. 92, 504–513. doi:10.1093/cvr/cvr245
Malenczyk K., Girach F., Szodorai E., Storm P., Segerstolpe Å., Tortoriello G., et al. (2017). A TRPV1‐to‐secretagogin regulatory axis controls pancreatic β‐cell survival by modulating protein turnover. EMBO J. 36, 2107–2125. doi:10.15252/embj.201695347
Marshall N. J., Liang L., Bodkin J., Dessapt-Baradez C., Nandi M., Collot-Teixeira S., et al. (2013). A role for TRPV1 in influencing the onset of cardiovascular disease in obesity. Hypertension 61, 246–252. doi:10.1161/HYPERTENSIONAHA.112.201434
Masamune A., Kotani H., Sörgel F. L., Chen J. M., Hamada S., Sakaguchi R., et al. (2020). Variants that affect function of calcium channel TRPV6 are associated with early-onset chronic pancreatitis. Gastroenterology 158, 1626–1641. e8. doi:10.1053/j.gastro.2020.01.005
Meek K. M., Knupp C. (2015). Corneal structure and transparency. Prog. Retin. Eye Res. 49, 1–16. doi:10.1016/j.preteyeres.2015.07.001
Murphy-Ullrich J. E., Suto M. J. (2018). Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 68-69, 28–43. doi:10.1016/j.matbio.2017.12.009
Nakazawa Y., Donaldson P. J., Petrova R. S. (2019). Verification and spatial mapping of TRPV1 and TRPV4 expression in the embryonic and adult mouse lens. Exp. Eye Res. 186, 107707. doi:10.1016/j.exer.2019.107707
Nathan J. D., Patel A. A., McVey D. C., Thomas J. E., Prpic V., Vigna S. R., et al. (2001). Capsaicin vanilloid receptor-1 mediates substance P release in experimental pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1322–G1328. doi:10.1152/ajpgi.2001.281.5.G1322
O’Connell P. J., Pingle S. C., Ahern G. P. (2005). Dendritic cells do not transduce inflammatory stimuli via the capsaicin receptor TRPV1. FEBS Lett. 579, 5135–5139. doi:10.1016/j.febslet.2005.08.023
Okada Y., Reinach P. S., Shirai K., Kitano A., Kao W. W. Y., Flanders K. C., et al. (2011). TRPV1 involvement in inflammatory tissue fibrosis in mice. Am. J. Pathol. 178, 2654–2664. doi:10.1016/j.ajpath.2011.02.043
Okada Y., Shirai K., Miyajima M., Reinach P. S., Yamanaka O., Sumioka T., et al. (2016). Loss of TRPV4 function suppresses inflammatory fibrosis induced by alkali-burning mouse corneas. PLoS One 11, e0167200. doi:10.1371/journal.pone.0167200
Otoupalova E., Smith S., Cheng G., Thannickal V. J. (2020). Oxidative stress in pulmonary fibrosis. Compr. Physiol. 12, 509–547. doi:10.1002/cphy.c190017
Otto M., Bucher C., Liu W., Müller M., Schmidt T., Kardell M., et al. (2020). 12(S)-HETE mediates diabetes-induced endothelial dysfunction by activating intracellular endothelial cell TRPV1. J. Clin. Invest. 130, 4999–5010. doi:10.1172/JCI136621
Pedersen S. F., Owsianik G., Nilius B. (2005). TRP channels: An overview. Cell Calcium 38, 233–252. doi:10.1016/j.ceca.2005.06.028
Rahaman S. O., Grove L. M., Paruchuri S., Southern B. D., Abraham S., Niese K. A., et al. (2014). TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Invest. 124, 5225–5238. doi:10.1172/JCI75331
Ramires F. J., Sun Y., Weber K. T. (1998). Myocardial fibrosis associated with aldosterone or angiotensin II administration: Attenuation by calcium channel blockade. J. Mol. Cell. Cardiol. 30, 475–483. doi:10.1006/jmcc.1997.0612
Razavi R., Chan Y., Afifiyan F. N., Liu X. J., Wan X., Yantha J., et al. (2006). TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell 127, 1123–1135. doi:10.1016/j.cell.2006.10.038
Saika S., Okada Y., Miyamoto T., Yamanaka O., Ohnishi Y., Ooshima A., et al. (2004). Role of p38 MAP kinase in regulation of cell migration and proliferation in healing corneal epithelium. Invest. Ophthalmol. Vis. Sci. 45, 100–109. doi:10.1167/iovs.03-0700
Santoni G., Farfariello V., Amantini C. (2011). TRPV channels in tumor growth and progression. Adv. Exp. Med. Biol. 704, 947–967. doi:10.1007/978-94-007-0265-3_49
Schwartz E. S., Christianson J. A., Chen X., La J., Davis B. M., Albers K. M., et al. (2011). Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology 140, 1283–1291. e2. doi:10.1053/j.gastro.2010.12.033
Selman M., King T. E., Pardo A. (2001). Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 134, 136–151. doi:10.7326/0003-4819-134-2-200101160-00015
Shamsi F., Piper M., Ho L. L., Huang T. L., Gupta A., Streets A., et al. (2021). Vascular smooth muscle-derived Trpv1+ progenitors are a source of cold-induced thermogenic adipocytes. Nat. Metab. 3, 485–495. doi:10.1038/s42255-021-00373-z
Sumioka T., Okada Y., Reinach P. S., Shirai K., Miyajima M., Yamanaka O., et al. (2014). Impairment of corneal epithelial wound healing in a TRPV1-deficient mouse. Invest. Ophthalmol. Vis. Sci. 55, 3295–3302. doi:10.1167/iovs.13-13077
Sun H. J. (2019). Current opinion for hypertension in renal fibrosis. Adv. Exp. Med. Biol. 1165, 37–47. doi:10.1007/978-981-13-8871-2_3
Suri A., Szallasi A. (2008). The emerging role of TRPV1 in diabetes and obesity. Trends Pharmacol. Sci. 29, 29–36. doi:10.1016/j.tips.2007.10.016
Swigris J. J., Stewart A. L., Gould M. K., Wilson S. R. (2005). Patients’ perspectives on how idiopathic pulmonary fibrosis affects the quality of their lives. Health Qual. Life Outcomes 3, 61. doi:10.1186/1477-7525-3-61
Takahashi N., Matsuda Y., Yamada H., Tabeta K., Nakajima T., Murakami S., et al. (2014). Epithelial TRPV1 signaling accelerates gingival epithelial cell proliferation. J. Dent. Res. 93, 1141–1147. doi:10.1177/0022034514552826
Thannickal V. J., Toews G. B., White E. S., Lynch J. P., Martinez F. J. (2004). Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 55, 395–417. doi:10.1146/annurev.med.55.091902.103810
Thomas K. C., Roberts J. K., Deering-Rice C. E., Romero E. G., Dull R. O., Lee J., et al. (2012). Contributions of TRPV1, endovanilloids, and endoplasmic reticulum stress in lung cell death in vitro and lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L111–L119. doi:10.1152/ajplung.00231.2011
Tóth E., Tornóczky T., Kneif J., Perkecz A., Katona K., Piski Z., et al. (2018). Upregulation of extraneuronal TRPV1 expression in chronic rhinosinusitis with nasal polyps. Rhinology 56, 245–254. doi:10.4193/Rhin17.108
Tsagogiorgas C., Wedel J., Hottenrott M., Schneider M. O., Binzen U., Greffrath W., et al. (2012). N-octanoyl-dopamine is an agonist at the capsaicin receptor TRPV1 and mitigates ischemia-induced [corrected] acute kidney injury in rat. PLoS One 7, e43525. doi:10.1371/journal.pone.0043525
Turan E., Valtink M., Reinach P. S., Skupin A., Luo H., Brockmann T., et al. (2021). L-carnitine suppresses transient receptor potential vanilloid type 1 activity and myofibroblast transdifferentiation in human corneal keratocytes. Lab. Invest. 101, 680–689. doi:10.1038/s41374-021-00538-0
Ueda K., Tsuji F., Hirata T., Takaoka M., Matsumura Y. (2008). Preventive effect of TRPV1 agonists capsaicin and resiniferatoxin on ischemia/reperfusion-induced renal injury in rats. J. Cardiovasc. Pharmacol. 51, 513–520. doi:10.1097/FJC.0b013e31816f6884
Wang D., Wu Y., Chen Y., Wang A., Lv K., Kong X., et al. (2019). Focal selective chemo-ablation of spinal cardiac afferent nerve by resiniferatoxin protects the heart from pressure overload-induced hypertrophy. Biomed. Pharmacother. 109, 377–385. doi:10.1016/j.biopha.2018.10.156
Wang H. J., Wang W., Cornish K. G., Rozanski G. J., Zucker I. H. (2014). Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 64, 745–755. doi:10.1161/HYPERTENSIONAHA.114.03699
Wang L., Wang D. H. (2005). TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 112, 3617–3623. doi:10.1161/CIRCULATIONAHA.105.556274
Wang Q., Ma S., Li D., Zhang Y., Tang B., Qiu C., et al. (2014). Dietary capsaicin ameliorates pressure overload-induced cardiac hypertrophy and fibrosis through the transient receptor potential vanilloid type 1. Am. J. Hypertens. 27, 1521–1529. doi:10.1093/ajh/hpu068
Wang Q., Zhang Y., Li D., Zhang Y., Tang B., Li G., et al. (2016). Transgenic overexpression of transient receptor potential vanilloid subtype 1 attenuates isoproterenol-induced myocardial fibrosis in mice. Int. J. Mol. Med. 38, 601–609. doi:10.3892/ijmm.2016.2648
Wang Y., Babánková D., Huang J., Swain G. M., Wang D. H. (2008). Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension 52, 264–270. doi:10.1161/hypertensionaha.108.110197
Wang Y., Cui L., Xu H., Liu S., Zhu F., Yan F., et al. (2017). TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 260, 13–19. doi:10.1016/j.atherosclerosis.2017.03.016
Wang Y., Wang D. H. (2009). Aggravated renal inflammatory responses in TRPV1 gene knockout mice subjected to DOCA-salt hypertension. Am. J. Physiol. Ren. Physiol. 297, F1550–F1559. doi:10.1152/ajprenal.00012.2009
Wang Y., Wang D. H. (2011). Protective effect of TRPV1 against renal fibrosis via inhibition of TGF-β/Smad signaling in DOCA-salt hypertension. Mol. Med. 17, 1204–1212. doi:10.2119/molmed.2011.00063
Waning J., Vriens J., Owsianik G., Stüwe L., Mally S., Fabian A., et al. (2007). A novel function of capsaicin-sensitive TRPV1 channels: Involvement in cell migration. Cell Calcium 42, 17–25. doi:10.1016/j.ceca.2006.11.005
Warzecha Z., Dembiński A., Ceranowicz P., Stachura J., Tomaszewska R., Konturek S. J., et al. (2001). Effect of sensory nerves and CGRP on the development of caerulein-induced pancreatitis and pancreatic recovery. J. Physiol. Pharmacol. 52, 679–704.
Wei X., Gao P., Pu Y., Li Q., Yang T., Zhang H., et al. (2017). Activation of TRPV4 by dietary apigenin antagonizes renal fibrosis in deoxycorticosterone acetate (DOCA)-salt-induced hypertension. Clin. Sci. 131, 567–581. doi:10.1042/CS20160780
Wick E. C., Hoge S. G., Grahn S. W., Kim E., Divino L. A., Grady E. F., et al. (2006). Transient receptor potential vanilloid 1, calcitonin gene-related peptide, and substance P mediate nociception in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G959–G969. doi:10.1152/ajpgi.00154.2005
Wilson S. E. (2020). Corneal myofibroblasts and fibrosis. Exp. Eye Res. 201, 108272. doi:10.1016/j.exer.2020.108272
Wilson S. E., Marino G. K., Torricelli A. A. M., Medeiros C. S. (2017). Injury and defective regeneration of the epithelial basement membrane in corneal fibrosis: A paradigm for fibrosis in other organs? Matrix Biol. 64, 17–26. doi:10.1016/j.matbio.2017.06.003
Xu G., Winston J. H., Shenoy M., Yin H., Pendyala S., Pasricha P. J., et al. (2007). Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology 133, 1282–1292. doi:10.1053/j.gastro.2007.06.015
Xu S., Zhang L., Cheng X., Yu H., Bao J., Lu R., et al. (2018). Capsaicin inhibits the metastasis of human papillary thyroid carcinoma BCPAP cells through the modulation of the TRPV1 channel. Food Funct. 9, 344–354. doi:10.1039/C7FO01295K
Yang Y., Wang Z., Yang H., Wang L., Gillespie S. R., Wolosin J. M., et al. (2013a). TRPV1 potentiates TGFβ-induction of corneal myofibroblast development through an oxidative stress-mediated p38-SMAD2 signaling loop. PLoS One 8, e77300. doi:10.1371/journal.pone.0077300
Yang Y., Yang H., Wang Z., Mergler S., Wolosin J. M., Reinach P. S., et al. (2013b). Functional TRPV1 expression in human corneal fibroblasts. Exp. Eye Res. 107, 121–129. doi:10.1016/j.exer.2012.11.004
Yang Y., Yang H., Wang Z., Varadaraj K., Kumari S. S., Mergler S., et al. (2013c). Cannabinoid receptor 1 suppresses transient receptor potential vanilloid 1-induced inflammatory responses to corneal injury. Cell. Signal. 25, 501–511. doi:10.1016/j.cellsig.2012.10.015
Yazdani S., Bansal R., Prakash J. (2017). Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv. Drug Deliv. Rev. 121, 101–116. doi:10.1016/j.addr.2017.07.010
Yoshie K., Rajendran P. S., Massoud L., Mistry J., Swid M. A., Wu X., et al. (2020). Cardiac TRPV1 afferent signaling promotes arrhythmogenic ventricular remodeling after myocardial infarction. JCI Insight 5, 124477. doi:10.1172/jci.insight.124477
Zhai K., Liskova A., Kubatka P., Büsselberg D. (2020). Calcium entry through TRPV1: A potential target for the regulation of proliferation and apoptosis in cancerous and healthy cells. Int. J. Mol. Sci. 21, 4177. doi:10.3390/ijms21114177
Zhang F., Yang H., Wang Z., Mergler S., Liu H., Kawakita T., et al. (2007). Transient receptor potential vanilloid 1 activation induces inflammatory cytokine release in corneal epithelium through MAPK signaling. J. Cell. Physiol. 213, 730–739. doi:10.1002/jcp.21141
Zhang L. P., Kline R. H. t., Deevska G., Ma F., Nikolova-Karakashian M., Westlund K. N., et al. (2015). Alcohol and high fat induced chronic pancreatitis: TRPV4 antagonist reduces hypersensitivity. Neuroscience 311, 166–179. doi:10.1016/j.neuroscience.2015.10.028
Zhang M., Zhang S. (2020). T cells in fibrosis and fibrotic diseases. Front. Immunol. 11, 1142. doi:10.3389/fimmu.2020.01142
Keywords: fibrotic diseases, transient receptor potential vanilloid type 1, signaling pathways, therapeutic target, neurogenic inflammation
Citation: Peng G, Tang X, Gui Y, Yang J, Ye L, Wu L, Ding Yh and Wang L (2022) Transient receptor potential vanilloid subtype 1: A potential therapeutic target for fibrotic diseases. Front. Physiol. 13:951980. doi: 10.3389/fphys.2022.951980
Received: 24 May 2022; Accepted: 11 July 2022;
Published: 15 August 2022.
Edited by:
Federico Davide Mussano, University of Turin, ItalyReviewed by:
Chandan Goswami, National Institute of Science Education and Research (NISER), IndiaCopyright © 2022 Peng, Tang, Gui, Yang, Ye, Wu, Ding and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lihong Wang, d2FuZ2xobmV3QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.