
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 28 June 2022
Sec. Metabolic Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.913611
This article is part of the Research Topic Inflammation in Diabetes and its Secondary Complications View all 8 articles
 Nikki L. Farnsworth1,2
Nikki L. Farnsworth1,2 Robert A. Piscopio2,3Wolfgang E. Schleicher2,3David G. Ramirez2,3Jose G. Miranda2,3
Robert A. Piscopio2,3Wolfgang E. Schleicher2,3David G. Ramirez2,3Jose G. Miranda2,3 Richard K. P. Benninger2,3*
Richard K. P. Benninger2,3*In type 1 diabetes (T1D), islet dysfunction occurs prior to diabetes onset. Pro-inflammatory cytokines can disrupt insulin secretion and Ca2+ homeostasis. Connexin36 (Cx36) gap junctions electrically couple β-cells to coordinate glucose-stimulated Ca2+ and insulin secretion. Cx36 gap junction coupling can also protect against cytokine-induced apoptosis. Our goal was to determine how islet gap junction coupling and Ca2+ dynamics are altered in mouse models of T1D prior to diabetes. Glucose tolerance was assessed in NOD and immunodeficient NOD-RAG1KO mice at 6–12 weeks age. Glucose-stimulated insulin secretion, Ca2+ dynamics, and gap junction coupling were measured in islets isolated at each age. Gap junction coupling was also measured in islets from mice that underwent transfer of diabetogenic splenocytes and from chromograninA knockout NOD mice. Cell death was measured in islets isolated from wild-type, Cx36 knockout or Cx36 over-expression mice, each treated with a cocktail of pro-inflammatory cytokines and KATP or SERCA activators/inhibitors. NOD mice over-expressing Cx36 were also monitored for diabetes development, and islets assessed for insulitis and apoptosis. NOD and NOD-RAG1KO controls showed similar glucose tolerance at all ages. Ca2+ dynamics and gap junction coupling were disrupted in islets of NOD mice at 9 weeks, compared to controls. Transfer of diabetogenic splenocytes also decreased gap junction coupling. Islets from chromograninA knockout mice displayed normal coupling. Overexpression of Cx36 protected islets from cytokine-induced apoptosis. A knockout of Cx36 amplified cytokine-induced apoptosis, which was reversed by KATP activation or SERCA activation. Cx36 overexpression in NOD mice delayed diabetes development compared to NOD controls. However, apoptosis and insulitis were not improved. Decreases in islet gap junction coupling occur prior to T1D onset. Such decreases alter islet susceptibility to apoptosis due to altered Ca2+. Future studies will determine if increasing Cx36 gap junction coupling in combination with restoring Ca2+ homeostasis protects against islet decline in T1D.
Type 1 diabetes (T1D) is characterized by the progressive immune destruction of insulin producing β-cells in the pancreatic islets of Langerhans. Activated immune cells in the pancreas initiate β-cell death in part through secretion of high levels of Th1 cytokines, including tumor necrosis factor-α (TNF-α) from activated macrophages and interleukin-1β (IL-1β) and interferon-γ (IFN-γ) from activated T-cells (Cnop, Welch et al., 2005; Padgett, Broniowska et al., 2013). Early in the progression of T1D, disruptions to insulin secretion have been reported prior to loss of glucose homeostasis, indicating a potential role for islet dysfunction in the pathogenesis of T1D (Lo, Hawa et al., 1992; Ize-Ludlow, Lightfoot et al., 2011; Soleimanpour and Stoffers 2013). High levels of pro-inflammatory cytokines can also increase β-cell endoplasmic reticulum (ER) and oxidative stress early in T1D progression (Lu, Liu et al., 2020). In vitro, pro-inflammatory cytokines reduce insulin secretion (Wang, Guan et al., 2010) and alter intracellular free-calcium (Ca2+) and ER calcium dynamics (Ramadan, Steiner et al., 2011; Kono, Ahn et al., 2012). Our previous work demonstrated in vitro, that cytokines decreased connexin36 (Cx36) gap junction coupling and disrupted the coordination of Ca2+ dynamics (Farnsworth, Walter et al., 2016). However, changes in islet Cx36 gap junction coupling and Ca2+ signaling dynamics have not been studied in T1D.
Cx36 gap junctions form channels between β-cells that mediates electrical coupling and regulates islet function in a glucose-dependent manner (Serre-Beinier, Gurun et al., 2000; Serre-Beinier, Bosco et al., 2009). At high glucose, Cx36 gap junction coupling coordinates glucose-induced membrane depolarization, pulsatile intracellular Ca2+ signaling, and insulin secretion across the islet (Ravier, Guldenagel et al., 2005; Benninger, Zhang et al., 2008; Farnsworth and Benninger 2014). At low glucose, Cx36 gap junction coupling coordinates hyper-polarizing electrical currents across the islet to suppress intracellular Ca2+ and insulin secretion (Benninger, Head et al., 2011; Farnsworth and Benninger 2014). Loss of Cx36 gap junction coupling between β-cells results in a loss of first phase insulin secretion and reduced glucose tolerance in mice (Head, Orseth et al., 2012; Perez-Armendariz 2013), similar to what is observed in pre-diabetic individuals (Lin, Hsia et al., 2010; Cersosimo, Triplitt et al., 2018). While decreases in first-phase insulin secretion are also a hallmark of preclinical T1D (Daneman 2006; Sosenko, Skyler et al., 2013), it is unknown whether changes in Cx36 gap junction coupling and Ca2+ signaling dynamics also occur early in the progression of T1D, when islet dysfunction is observed.
The goal of this study was to determine if changes in islet Cx36 gap junction coupling and Ca2+ signaling dynamics occur early in the progression of T1D, prior to changes in glucose tolerance. We utilized the NOD mouse model of T1D to study changes in Cx36 prior to disease onset, as progression in this mouse model is similar to human disease (Pearson, Wong et al., 2016). In addition to regulating islet intracellular Ca2+ signaling dynamics, Cx36 gap junction coupling also protects against β-cell apoptosis (Klee, Allagnat et al., 2011; Allagnat, Klee et al., 2013; Cigliola, Populaire et al., 2016). Specifically, Cx36 protects against cytokine-induced β-cell death in vitro via downregulation of oxidative and ER stress (Calabrese, Zhang et al., 2003; Ravier, Guldenagel et al., 2005; Allagnat, Klee et al., 2013). In vivo, overexpression of Cx36 resulting in increased β-cell coupling protects against streptozotocin (STZ) induced β-cell death (Klee, Allagnat et al., 2011). Therefore, this study also sought to determine if modulation of Cx36 and intracellular Ca2+ could protect against β-cell death and delay the onset of diabetes in the NOD mouse model.
All experiments utilizing mice were approved by the University of Colorado Denver Institutional Animal Care and Use Committee [Protocol # B-95817(05)1D]. Mice were housed in a temperature-controlled facility with access to food and water ad libitum on a 12 h light/dark cycle. In NOD mice diabetes progression was monitored by weekly ad lib glucose measurements taken from the tail vain. Mice with a blood glucose level >250 mg/dl for three consecutive measurements were considered diabetic and were euthanized via CO2 and cervical dislocation to minimize distress. All animals were bred in house except where otherwise noted.
Cx36 knockout (KO) mice were generated as previously described (Degen, Meier et al., 2004). The rat insulin promoter driven Cx36 (RIPCx36) plasmid construct was a gift from Dr. Philippe Klee (Klee, Allagnat et al., 2011). The expression cassette was removed by enzymatic digestion, purified, and injected into C57BL/6 embryos at the University of Colorado Anschutz Medical Campus Transgenic and Gene Targeting Core. Mice carrying the transgene were identified by PCR using primers designed for the mutant Cx36. RIPCx36 mice were backcrossed onto the NOD background by identifying two transgenic founders and successive breeding with NOD mice from a colony maintained at the University of Colorado Anschutz Medical Campus for up to 10 generations. Mice from generations 1–10 were used to characterize the overexpression of Cx36. Mice from generation 2 were backcrossed onto the NOD background using a “speed congenic” technique. Within 4 generations of backcrossing, >99% NOD genetic background was achieved. Mice from generations 5–10 were monitored to determine the diabetic phenotype of the colony compared to the phenotype of the original NOD colony. Littermate mice negative for RIPCx36 served as controls (WT-NOD).
Mice were fasted with access to water ad lib for 16 h prior to performing the GTT. Fasting glucose was measured via the tail vein and 200 mg/kg glucose was given by intraperitoneal (IP) injection. Blood glucose was monitored over 2 h post glucose bolus.
Pancreata were isolated from female 6, 9, or 12-week old NOD and NOD-RAG1KO mice; male and female 12-week old WT-NOD, RIPCx36-NOD; or female 12-week old chromogranin A knockout NOD (ChGAKO-NOD) mice. Female NOD mice were selected for experiments investigating diabetes onset, as they more reliably progress to diabetes than their male counterparts (Matthews, Xue et al., 2015). Male and female RIPCx36-NOD mice were selected to determine differences in Cx36 gene expression in the presence and absence of diabetes. Briefly, animals were injected with 100 mg/kg ketamine and 8 mg/kg xylazine and euthanized via exsanguination prior to inflating the pancreas with a collagenase solution and removing the pancreas, per our approved animal protocol. Islets were isolated by enzymatic digest of the pancreas with 2.5 mg/ml collagenase (C9263, Millipore-Sigma, Saint Louis, MO) at 37°C. Islets were handpicked into 1640 RPMI Medium (Millipore-Sigma) with 10% FBS, 10,000 U/mL Penicillin and 10,000 μg/ml Streptomycin and incubated overnight at 37°C and 5% CO2. Islet from WT, Cx36 KO, and RIP Cx36 mice were also cultured with a cytokine cocktail containing 10 ng/ml tumor necrosis factor-α (TNF-α, 410-MT-010/CF, R&D Systems, ED50 = 8–50 pg/ml), 5 ng/ml interleukin-1 β (IL-1β, 401-ML-005/CF, R&D systems, ED50 = 2–10 pg/ml), and 100 ng/ml interferon-γ (IFN-γ, 485-MI-100/CF, R&D Systems, ED50 = 0.3–0.9 ng/ml) for 24 h in culture media.
Splenocytes were isolated from female NOD mice with ad lib blood glucose levels >250 mg/dl by manual dissociation in cold Hanks Balanced Salt Solution (HBSS). Leukocytes were counted and one dose of 20 × 106 leukocytes in HBSS was injected into each 12–14-week old NOD-scid (Jackson Laboratory, Bar Harbor, ME) adoptive transfer recipient. NOD-scid vehicle controls were injected with an equal volume of HBSS. Islets were isolated as previously described 2 weeks after adoptive transfer.
Isolated islets were incubated with 4 µM Fluo-4 AM (Thermo Fisher Scientific, Waltham, MA) in BMHH buffer containing 125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, 0.1% bovine serum albumin (BSA), and 2 mM glucose at room temperature for 2 h in the dark. Islets were transferred to MatTek glass-bottom imaging dishes (MatTek, Ashland, MA) with fresh BMHH with either 2 mM or 5 mM glucose prior to imaging. Islets were imaged on one of three microscopes: 1) a Nikon Eclipse-Ti wide field microscope (Nikon, Melville, NY) with a x20 0.75 NA Plan Apo objective, a 490/40 nm band-pass excitation filter and a 525/36 nm band-pass emission filter, 2) a Zeiss 780 laser scanning microscope with a x40 1.2 NA water c-apochromat objective using a 488 nm Ar+ laser line and a QUASAR spectral detector, 3) a Zeiss 800 laser scanning microscope with a x40 1.2 NA c-apochromat objective using a 488 nm Ar+ laser with a 525/20 nm emission filter. Islets were imaged at 37°C, where images were acquired every second for 2.5–5 min following incubation at 2 mM glucose for 2 h, 5 mM glucose for 2 h, 11 mM glucose for 10 min, or 20 mM glucose for 10 min. The fraction of islet area active at 2 mM or 5 mM glucose and the fraction of islet area synchronized at 11 mM or 20 mM glucose was determined as previously described (Hraha, Bernard et al., 2014) using MATLAB (Mathworks, Natick, MA).
FRAP measurement of Cx36 coupling was performed as described in Farnsworth et al. (2014). Immediately after isolation, islets were adhered to MatTek glass bottom dishes with Cell-Tak (Thermo Fisher Scientific) per the manufacturer’s instructions and cultured overnight in RPMI as previously described. Prior to imaging, islets were incubated with 12.5 µM rhodamine 123 in Hanks Balanced Salt Solution (HBSS) for 30 min at 37°C. Islets were washed with fresh HBSS and imaged on either a Zeiss 510 LSM with a 488 nm Ar+ laser, a 700/488 nm short pass dichroic beam splitter, a 490 nm long pass secondary beam splitter, and a 505 nm long pass emission filter or a Zeiss 800 LSM as previously described for Ca2+ imaging. As described in Farnsworth et al. (2014), half of each islet was photobleached and the fluorescence recovery in the bleached region was measured over time. The fluorescence recovery rate, which correlates with Cx36 gap junction coupling, was calculated from the reverse exponential fluorescence recovery curve.
Islets from WT or Cx36 KO mice were cultured for 24 h with and without the cytokine cocktail mentioned previously. Islets were also cultured for 24 h with and without: 250 µM of the KATP channel activator Diazoxide (Millipore-Sigma), 20 mM of the K channel inhibitor tetraethylammonium (TEA, Millipore-Sigma), 1 µM of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) activator Ochratoxin A (Millipore-Sigma), and 1 µM of the SERCA inhibitor thapsigargin (Millipore). After incubation, islets were stained with 0.5 mg/ml fluorescein diacetate (Sigma) and 0.1 mg/ml propidium iodide (Sigma) in PBS to identify live and dead cells respectively. Islets were imaged on a Zeiss 800 scanning laser confocal microscope where 3 optical sections per islets were obtained and quantified manually for live and dead cells using ImageJ for 5–10 islets per condition.
Islets were isolated from 8 to 12-week old male and female NOD or RIPCx36-NOD littermates as previously described. Islet mRNA was isolated using the RNeasy mini kit (Qiagen, Germantown, MD) per the manufacturer’s instructions and RNA was eluted in 30 µl DI water. RNA was converted to cDNA using the 1st Strand cDNA synthesis kit (OriGene, Rockville, MD) per the manufacturer’s instructions with 1 µg of RNA per reaction. Primers for Cx36 were purchased from OriGene (NM_010290, forward primer: GTGGTGCTCAATCTGGCTGAAC, reverse primer: GACTGAGTCCTGCCGAAATTGG) and primers for the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT1) are as follows: 5′-TGGATACAGGCCAGACTTTGTT-3′ and 3′-GGGAAAATACAGCCAACACTGC-5′. PCR reactions were created using Eppendorf Matercycler Gradient Thermocycer and run on a CFX96 Real-Time System Thermocycler real-time PCR machine with Pre-soak 95°C for 10 min/Denaturation 95°C for 15 s/Annealing 60°C for 30 s for 40 cycles cycle/temp/time. Cycle time (Ct) values were calculated using Bio-Rad CFX Manager Software software and the relative change in Cx36 gene expression was calculated using the ΔΔCt method, where wild type (WT) NOD islets are the control samples (Ctcontrol) and RIPCx36-NOD islets are the experimental samples (Ctexp). Relative changes in gene expression were calculated as follows:
1) ΔCtcontrol = CtWT-NOD—CtHPRT1
2) ΔCtexp = CtRIPCx36-NOD—CtHPRT1
3) ΔΔCt = 2-(ΔCtexp-AVG(ΔCtcontrol))
Pancreata from 12-week old NOD and RIPCx36-NOD female mice were isolated and fixed in 4% paraformaldehyde at 4°C overnight. Fixed pancreata were embedded in paraffin and sliced into 5 µm sections. Sections were imaged on a Nikon Eclipse Ti widefield microscope (Nikon, Melville, NY) with ANDOR color camera with a x40 objective. Insulitis was determined from pancreatic sections stained with hematoxylin and eosin. Islets with no insulitis were given a score of 0, islets with peri-insulitis were given a score of 1, islets with <50% infiltrated area were given a score of 2, and islets with >50% infiltrated area were given a score of 3. The insulitis scores were averaged for all islets on 6 pancreatic sections taken from different regions of the pancreas (∼100 µm apart).
Subsequent pancreas sections were deparaffinized, permeabilized with 0.25% Triton-X-100 (Sigma) and stained for TUNEL positive cells with the ClickiT Plus TUNEL Assay (ThermoFisher) per the manufacturer’s instructions. TUNEL staining was verified with DNase treated pancreatic tissue slices. Slides were then stained with guinea pig anti-insulin primary antibody (ab7842, Abcam) diluted 1:100 in PBS with 5% normal donkey serum (NDS) at 4°C overnight. Slides were rinsed in PBS, then stained with AlexaFluor 647 Donkey anti-guinea pig secondary antibody (706-605-148, Jackson Immuno Research) diluted 1:500 in PBS with 5% NDS at room temperature for 2 h. Slides were rinsed, then mounted in Fluoromount with DAPI (ThermoFisher) and sealed. Insulin antibody staining was verified on pancreatic tissue slices from NOD-RAG1KO mice. Slides were imaged on a Nikon Eclipse Ti widefield microscope with x20 0.75 NA Plan Apo objective. DAPI was imaged with a 402/50 nm band-pass excitation filter and a 455/50 nm band-pass emission filter, TUNEL positive cells labeled with AlexaFluor488 were imaged with a 490/20 nm band-pass excitation filter and a 525/36 nm band-pass emission filter, and AlexaFluor647 was imaged with a 555/25 nm band-pass excitation filter and a 605/52 band-pass emission filter. Images of the whole pancreas slice were obtained using a tile scan and image stitching with 15% image overlap. TUNEL positive and insulin positive cells were quantified using ImageJ and insulin positive pancreas area was determined using MATLAB (MathWorks, Natick, MA).
To determine statistical significance, analysis of variance (ANOVA) with Tukey’s post hoc analysis or a student’s t-test was used where indicated. In each figure panel, all genotypes and treatments are compared against one another; however, only differences that were found to be statistically significant with a p-value <0.05 were reported in all figures. To determine clustering of NOD coupling and Ca2+ data, k-means clustering was used in combination with a t-test to determine significance between identified clusters of data. To determine statistical differences in survival of the WT NOD and RIPCx36-NOD mouse colonies, Kaplan-Meier survival analysis with a Breslow test was used where indicated.
To determine the optimal time frame to study pre-symptomatic diabetes in the NOD mouse we assessed glucose homeostasis in female mice at 6, 9, and 12 weeks via GTT. No significant changes in glucose tolerance test (Figure 1A, Supplementary Figures S1A,B) or area-under-the-curve (AUC) from the GTT curves (Figure 1B) were found at 6, 9, or 12 weeks compared to age-matched NOD-RAG1KO controls. Islets isolated from NOD and NOD-RAG1KO mice secreted similar amounts of insulin at 2 mM (Supplementary Figure S1C) and 20 mM glucose (Figure 1C) and had similar insulin content (Supplementary Figure S1D) at all ages investigated.
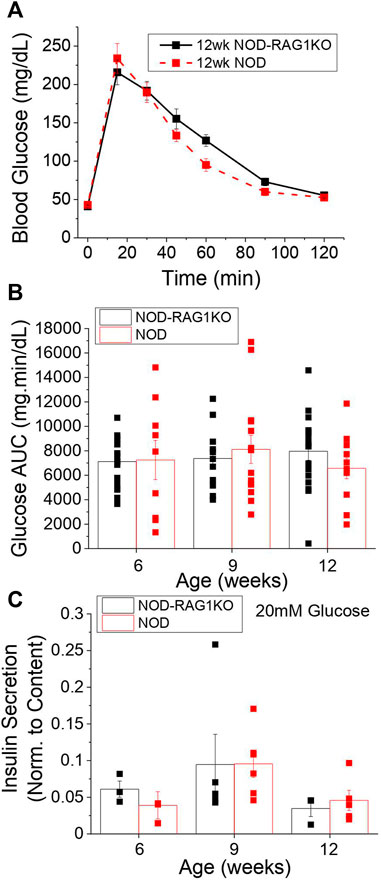
FIGURE 1. NOD mice are normoglycemic at 12 weeks of age. (A) Blood glucose levels in 12-week old NOD (n = 7–13) and NOD-RAG1KO (n = 8–15) mice following a glucose tolerance test starting at time 0. (B) Glucose area under the curve for 6, 9, and 12 week old NOD (red) and NOD-RAG1KO (black) mice calculated from blood glucose levels measured over 2 h following glucose challenge in as A. (C) Insulin secretion normalized to insulin content in isolated islets from 6, 9, and 12 week old NOD (n = 4–6) and NOD-RAG1KO (n = 3–5) mice at 20 mM glucose. Data in all panels represents the mean ± SEM. In B and C, data from individual mice are represented by black and red squares for NOD-RAG1KO and NOD mice respectively. All animals are female unless otherwise noted.
We next investigated changes in intracellular Ca2+ signaling dynamics and Cx36 gap junction coupling in islets from 6, 9, and 12-week old mice which were previously found to be normoglycemic. At 6 weeks no differences in Ca2+ signaling dynamics were observed between NOD and NOD-RAG1KO islets (Supplementary Figures S2A,B) at 11 mM glucose. At 9 and 12 weeks in the NOD mouse Ca2+ dynamics were visually disrupted (Figures 2A,B, Supplementary Figures S2C,D) with disrupted Ca2+ pulsatility and oscillation frequency compared to NOD-RAG1KO controls. We quantified the fraction of each islet that was active (i.e., pulsing intracellular Ca2+) and the fraction of the islet that showed coordinated Ca2+ oscillations. At 2, 11, and 20 mM glucose there were no significant differences in the fraction of the islet that was active between NOD and NOD-RAG1KO islets (Figures 2C,E, Supplementary Figure S2E) at any ages studied. At 5 mM glucose, there was no change in the fraction of the islet that was active in NOD islets compared with NOD-RAG1KO controls at any age studied (Figure 2D). At 11 mM glucose, there was a significant decrease in the fraction of the islet area with coordinated Ca2+ oscillations from 6 to 9 weeks in NOD mice (p = 0.025), compared to NOD-RAG1KO controls which were unchanged with age (Figure 2F). While the islet area with coordinated Ca2+ oscillations appears to decrease from 11 mM glucose to 20 mM glucose at 6 and 9 weeks of age (Figure 2F and Supplementary Figure S2F), this may be due to fast islet Ca2+ oscillations that could not be sampled at the 1 Hz imaging rate (Stozer, Klemen et al., 2021; Pohorec, Bombek et al., 2022). Overall, our results show that intracellular Ca2+ dynamics are disrupted in islets from NOD mice as early as 9 weeks of age.
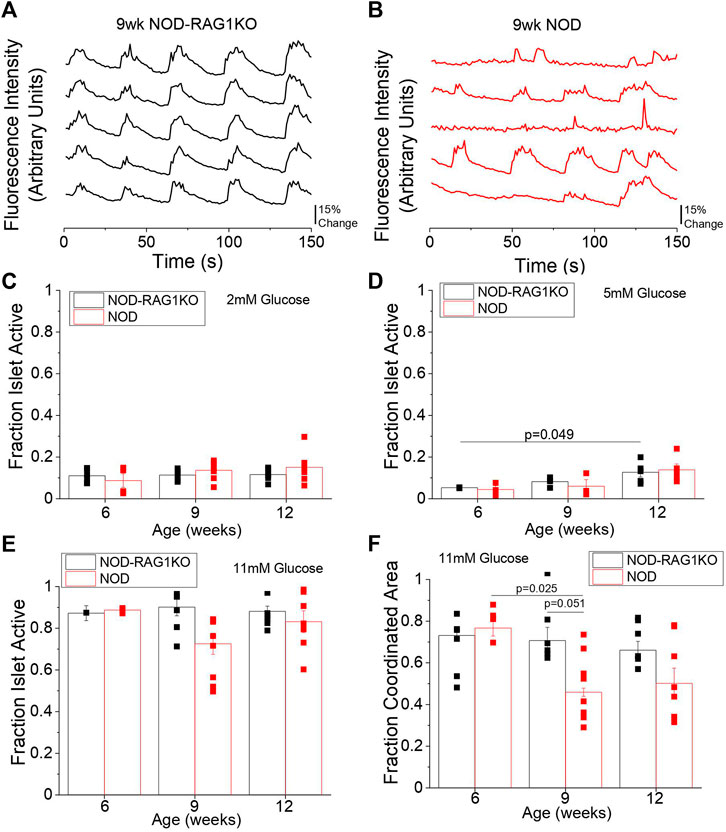
FIGURE 2. NOD mice have altered Ca2+ signaling as early as 9 weeks of age. Representative plots of intracellular Ca2+ as measured by fluorescence intensity at 11 mM glucose in 5 individual cells in the same islet over time in 9-week old (A) NOD-RAG1KO (black, n = 3–4) and (B) NOD (red, n = 3–5) mice. Quantification of the fraction of the islet area showing intracellular Ca2+ activity at (C) 2 mM glucose, (D) 5 mM glucose, and (E) 11 mM glucose in isolated islets from 6, 9, and 12-week old NOD and NOD-RAG1KO mice. (F) Quantification of the fraction of the islet area with coordinated Ca2+ oscillations at 11 mM glucose in isolated islets from 6, 9, and 12-week old NOD and NOD-RAG1KO mice. Data in C-F represents the mean ± SEM. Data from individual mice are represented by black and red squares for NOD-RAG1KO and NOD mice respectively (n = 3–6). p < 0.05 is significant as determined by ANOVA. All animals are female unless otherwise noted.
To determine if islet gap junction coupling was also altered in NOD mice, we used FRAP to measure changes in β-cell Cx36 gap junction coupling. As early as 9 weeks of age, we observed a 25% decrease in β-cell coupling (p = 0.008) in NOD mice compared to NOD-RAG1KO controls (Figure 3A). This decrease in coupling persisted to 12 weeks in NOD mice (p = 0.007). We next determined if changes in β-cell coupling were predictive of a future diabetic phenotype. We used K-means clustering analysis of β-cell coupling from Figure 3A plotted against the fraction of the islet area with coordinated Ca2+ oscillations at 11 mM glucose from Figure 2F in 9-week old NOD and NOD-RAG1KO mice. We found that a subset of NOD mice with similar gap junction coupling and high Ca2+ coordination (solid red squares) clustered with the NOD-RAG1KO control mice (Figure 3B). A second subset of NOD mice with similar gap junction coupling and lower Ca2+ coordination (open red squares) clustered separate from both the NOD-RAG1KO islets and the NOD islets with higher coupling and coordination (Figure 3B). No significant correlation was found between gap junction coupling rate and Ca2+ coordination (Figure 3B). When comparing the coordinated area for each cluster of NOD mice, we found a significant (p = 0.003) difference in Ca2+ coordination between cluster 1 (mean = 0.58 ± 0.05 SEM) and cluster 2 (mean = 0.32 ± 0.03 SEM) identified using K-means clustering (Figure 3B). These results show that populations of NOD mice can be identified that have reduced gap junction coupling and disrupted Ca2+ dynamics.

FIGURE 3. NOD mice have decreased β-cell coupling as early as 9 weeks of age and decreased β-cell coupling in the NOD mouse is mediated by autoreactive T-cells. (A) β-cell coupling as measured by fluorescence recovery rate in isolated islets from 6, 9, and 12-week old NOD (red, n = 6–9) and NOD-RAG1KO (black, n = 5–6) mice. Data represent the mean ± SEM. Data from individual mice are represented by black and red squares for NOD-RAG1KO and NOD mice respectively (n = 3–6). p < 0.05 is significant as determined by ANOVA. (B) K-means clustering analysis of the correlation between β-cell coupling and the fraction of the islet area with coordinated intracellular Ca2+ oscillations at 11 mM glucose in NOD and NOD-RAG1KO mice at 9 weeks of age. Red solid and dashed ovals indicate the clusters of animals identified by k-means analysis. (C) β-cell coupling as measured by fluorescence intensity recovery rate in isolated islets from 12-week old NOD (red), NOD-RAG1KO (black), and NOD chromogranin-A knockout (NOD-ChgA KO, green). Data from individual mice are represented by black, red, or green squares for NOD-RAG1KO, NOD, or NOD-ChgA KO mice respectively (n = 4–5). (D) β-cell coupling as measured by fluorescence intensity recovery rate in isolated islets from mice 2 weeks post-adoptive transfer with diabetogenic splenocytes or vehicle control. Data from individual mice are represented by solid or open squares for vehicle or adoptive transfer mice respectively (n = 3). Data in A and B represent the mean ± SEM. p < 0.05 is significant as determined by ANOVA in A and t-test in B. All animals are female unless otherwise noted.
We next determined if changes in β-cell gap junction coupling were mediated by activated immune cells, as changes in coupling occurred only in the NOD mice. To determine the role of activated T-cells in mediating disruptions to islet coupling, we utilized 12 week old NOD-RAG1KO mice, NOD mice, and NOD mice with a knockout of chromogranin A (NOD-ChgA KO). NOD-ChgA KO mice have a competent immune system, but do not develop T1D as they lack the primary antigen causing autoimmune diabetes in this strain of NOD mouse, ChromograninA (Baker, Bradley et al., 2016). NOD-ChgA KO mice showed significantly higher (p = 0.032) Cx36 gap junction coupling than age-matched NOD mice and showed similar levels of coupling as NOD-RAG1KO controls (Figure 3C). To further determine the role of diabetogenic T-cells in mediating decreases in β-cell coupling we transferred splenocytes from diabetic NOD mice into immunodeficient NOD-RAG1KO mice. Two weeks after splenocyte or vehicle transfer, islets from mice which received splenocytes had a 23% reduction (p = 0.027) in β-cell gap junction coupling compared to vehicle only control mice (Figure 3D). Overall, our results indicate that Cx36 coupling is reduced between β-cell by the presence of autoreactive T-cells.
Increases in Cx36 coupling have been shown to protect against cytokine-induced death in the islet (Allagnat, Klee et al., 2013). Similarly, the Ca2+ channel blocker Verapamil has been shown to protect against β-cell death in patients with T1D, suggesting a role for intracellular Ca2+ in mediating islet survival (Ovalle, Grimes et al., 2018). As we have shown that both Cx36 and intracellular Ca2+ signaling are disrupted in islets from pre-diabetic NOD mice, we next wanted to test if modulating Cx36 coupling and intracellular Ca2+ (outlined in Supplementary Table S1) impacts islet survival in the context of pro-inflammatory cytokine treatment. In islets overexpressing Cx36 (RIP-Cx36), levels of β-cell coupling as measured by FRAP are maintained over increasing cytokine concentration, with 0.01 and 0.1 relative cytokine concentration (RCC), as compared to a x1 solution of 10 ng/ml TNF-α, 5 ng/ml IL-1β, and 100 ng/ml IFN-γ as described in the methods section. In contrast, WT controls showed decreased β-cell gap junction coupling with increasing cytokine concentration (Figure 4A). RIP Cx36 islets were significantly protected against cytokine-induced death (p = 0.021); however, increasing intracellular Ca2+ by closing K+ channels, (including KATP channels) with TEA abolished this protective effect (Figure 4B). In contrast, Cx36 KO islets showed significantly increased cytokine-induced death (p = 0.015) compared to WT controls, and this effect was completely abolished by decreasing intracellular Ca2+ by activating KATP channels with diazoxide (Figure 4C). This difference between WT or Cx36 KO islets was maintained upon increasing intracellular Ca2+ with TEA (Figure 4D). Thus modulating cytosolic Ca2+ improved cell survival in islets with a loss of Cx36 function compared to WT controls

FIGURE 4. Modulation of Cx36 Coupling and Intracellular Ca2+ Mediate Cytokine-Induced Islet Death. (A) β-cell coupling rate as measured by FRAP in islets from WT and RIP Cx36 mice treated with 0, 0.01, or 0.1 relative cytokine concentration (RCC) for 24 h (n = 5). (B) Percent cell death islets from WT and RIP Cx36 mice treated with or without cytokines (1 RCC) and with or without the K+ channel inhibitor TEA for 24 h (n = 3–5). Percent cell death in islets from WT and Cx36 KO mice treated with or without (C) 250 nM of the KATP channel activator Diazoxide (n = 5) or (D) 20 mM of the K+ channel inhibitor TEA (n = 3) with or without a cytokine cocktail and for 24 h. Percent cell death in WT and Cx36 KO islets treated with or without (E) 1 µM of the SERCA activator Ochratoxin A (n = 5) or (F) 1 µM of the SERCA inhibitor thapsigargin (n = 5) with or without cytokines (1 RCC) for 24 h. Data represent the mean ± SEM. p < 0.05 is significant as determined by ANOVA. * Indicates p < 0.05 compared to untreated controls in A.
We next tested the effects of modulating ER Ca2+ on the exacerbation of cell death in Cx36 KO islets by applying the SERCA channel activator ochratoxin A and inhibitor thapsigargin. Increasing ER Ca2+ with ochratoxin A reversed the elevation in cytokine-induced death in Cx36 KO islets (Figure 4E). In contrast upon decreasing ER Ca2+ cell death was still elevated in Cx36 KO islets (Figure 4F). While treatment with thapsigargin for 24 h did not significantly increase cell death in WT or KO islets, previous studies in mouse islet suggest that 48 h treatment may be required for significant levels of cell death in mouse islets (Duprez and Jonas 2011).
Given that reduced Cx36 gap junction coupling can exacerbate cytokine-mediated β-cell death, we next examined the role of decreased Cx36 coupling in the pathogenesis of diabetes in the NOD mouse. To accomplish this, we backcrossed a β-cell specific Cx36 overexpression onto the NOD background (Klee, Allagnat et al., 2011). In 8–12-week old male NOD mice overexpressing Cx36 (RIPCx36-NOD) islets had ∼x13 higher Cx36 gene expression (p < 0.001, Figure 5A) and had 20% increased gap junction coupling compared to WT NOD islets (p = 0.014, Figure 5B). Islets from 12-week old female RIPCx36-NOD mice had ∼x6 higher Cx36 gene expression (p = 0.007, Figure 5C) and had ∼x1.3 higher gap junction coupling (p = 0.017, Figure 5D) compared to WT NOD controls. Of note, WT NOD islets had similar levels of Cx36 gap junction coupling compared to islets from the original NOD colony. These results confirm the successful development of a mouse model with increased Cx36 expression and gap junction coupling.
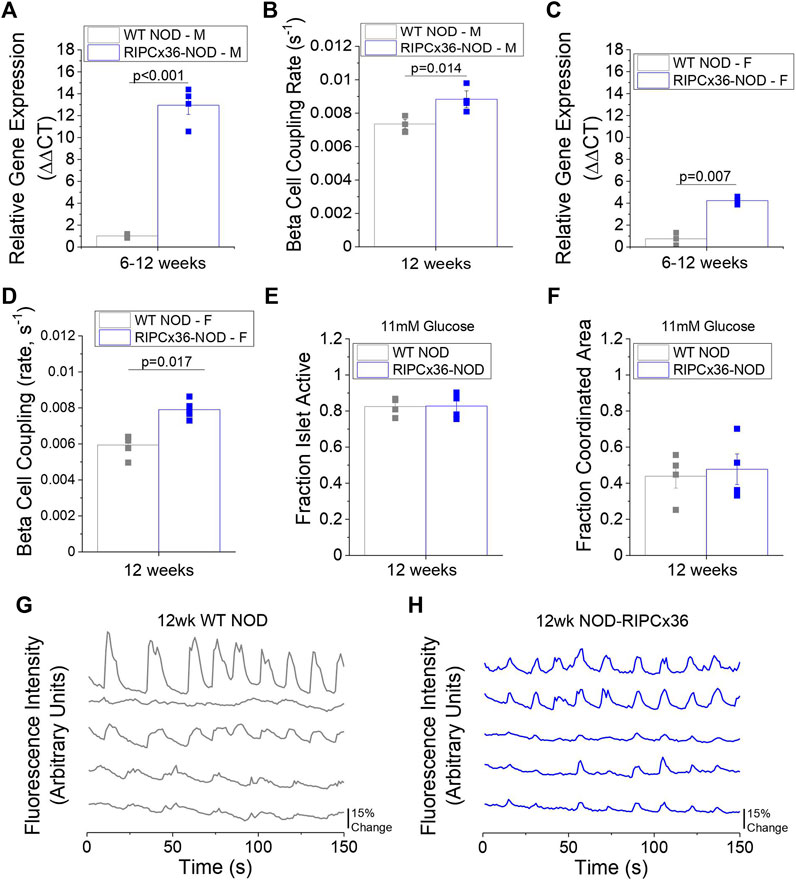
FIGURE 5. Overexpression of Cx36 in the NOD mouse. (A) Gene expression of Cx36 in 8–12-week old male WT NOD and RIPCx36-NOD mice normalized to HPRT1 housekeeping gene using the ΔΔCt method (n = 4). (B) β-cell coupling as measured by fluorescence recovery rate in isolated islets from 12-week old male WT NOD (grey) and RIPCx36-NOD (blue) mice (n = 4). (C) Gene expression of Cx36 in 12-week old female WT NOD and RIPCx36-NOD mice normalized to HPRT1 housekeeping gene using the ΔΔCt method (n = 3). (D) β-cell coupling as measured by fluorescence intensity recovery rate in isolated islets from WT NOD and RIPCx36-NOD mice (n = 4–5). Quantification of the fraction of the islet area showing intracellular Ca2+ activity (E) and the fraction of the islet area with coordinated Ca2+ oscillations (F) at 11 mM glucose in isolated islets from 12 week old WT NOD and RIPCx36-NOD mice (n = 3–4). Representative plots of intracellular Ca2+ as measured by fluorescence intensity at 11 mM glucose in 5 individual cells in the same islet over time in 12-week old (G) WT NOD (grey) and (H) RIPCx36-NOD (blue) mice. Data from individual mice are represented by grey or blue squares for WT NOD or RIPCx36-NOD mice respectively. Data in A-F represent the mean ± SEM. p < 0.05 is significant as determined by t-test in A-F. All animals are female unless otherwise noted.
To determine if increasing Cx36 coupling improved islet function in islets from NOD mice, we analyzed intracellular Ca2+ signaling dynamics and in vitro insulin secretion. No differences in the fraction of the islet active was observed at any glucose concentration studied in RIPCx36-NOD islets compared to WT NOD islets (Figure 5E, Supplementary Figures S3A,B). Similarly, while RIPCx36-NOD islets showed a trend to slightly higher coordinated Ca2+ oscillations, this was not significantly different compared to WT NOD islets (Figure 5F). Overall, intracellular Ca2+ dynamics at 11 mM glucose were similar in islets from 12-week old RIPCx36-NOD mice compared to WT NOD mice (Figures 5G,H). Insulin secretion at both 2 mM glucose and 20 mM glucose was similar in both WT NOD and RIPCx36-NOD islets (Supplementary Figure S3C). Overall, our data indicate that increased Cx36 coupling has little effect on islet function as measured by Ca2+ dynamics or in vitro insulin secretion.
While overexpression of Cx36 did not significantly protect against islet dysfunction, we next determined if increased Cx36 coupling protected against β-cell death and diabetes development. Female RIPCx36-NOD mice were significantly protected against developing diabetes at 12 weeks as determined by 95% confidence intervals compared to WT NOD controls (Figure 6A); however, the long-term survival of the RIPCx36-NOD mice as determined by Kaplan-Meier survival analysis was not significantly different than the WT NOD (p = 0.064, Figure 6A). Compared to the female mice in the original NOD colony, the female RIPCx36-NOD mice were significantly protected against diabetes onset as determined by 95% confidence intervals at 10, 11, 12, and 16 weeks (Figure 6B). The survival of the RIPCx36-NOD mice was also significantly higher than that of the NOD colony as determined by Kaplan-Meier survival analysis (p = 0.044, Figure 6B). There was no significant difference in survival between the WT NOD and NOD colony female mice (Supplementary Figure S3D). While the average age of diabetes onset decreased with each subsequent generation, there was no significant difference in the overall average age of diabetes onset between the WT NOD and original NOD colony (Supplementary Figure S3E).
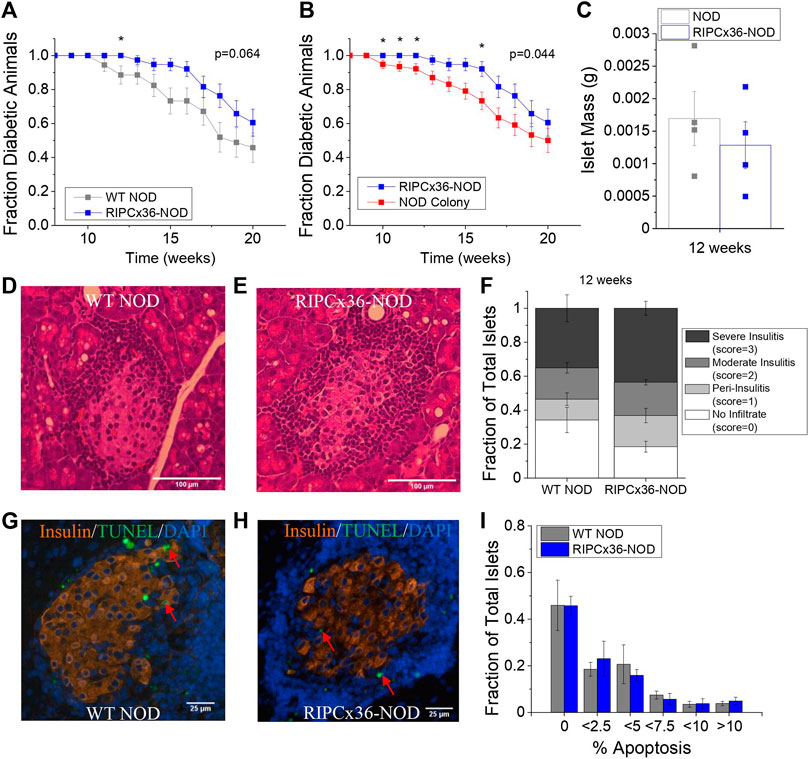
FIGURE 6. Overexpression of Cx36 protects against T1D onset between 10–16 weeks of age in the NOD mouse. (A) Kaplan-Meier survival analysis of diabetes onset in WT NOD (grey, n = 34) and RIPCx36-NOD (blue, n = 38) mice. p < 0.05 indicates a significant difference in survival probability as determined by the Breslow test. (B) Kaplan-Meier survival analysis of diabetes onset in NOD mice (NOD colony, red, n = 77) and RIPCx36-NOD (blue, n = 38) mice. p < 0.05 indicates a significant difference in survival probability as determined by the Breslow test. (C) Average islet mass per pancreas in 12-week old WT NOD (grey) and RIPCx36-NOD (blue) pancreata. Representative images of H&E staining in WT NOD (D) insulitis score = 2 and RIPCx36-NOD (E) insulitis score = 3 pancreas. (F) Fraction of total islets averaged over 6 slices per pancreas with an insulitis score of 0, 1, 2, or 3 in 12-week old WT NOD (n = 5) and RIPCx36-NOD (n = 6) mice as determined by IHC. Representative images of TUNEL and insulin staining in pancreas sections from WT NOD (G) and RIPCx36-NOD (H) mice. Red arrows indicate TUNEL+ insulin + cells. (I) Histogram of the average fraction of total islets with increasing percentage of apoptotic (TUNEL+) insulin+ cells per islet in WT NOD (grey, n = 4) and RIPCx36-NOD (blue, n = 4) islets. Data represents the mean ± SEM. All animals are female unless otherwise noted.
We next quantified cell death and immune cell infiltration in pancreas sections from WT NOD and RIPCx36-NOD female mice at 12 weeks where significant protection was observed (Figures 6D,E). No significant differences in immune infiltration (insulitis) or total islet mass as measured by insulin staining (Supplementary Figure S4A) were observed between pancreata from WT NOD and RIPCx36-NOD mice (Figure 6F). Similarly, we observed no difference in the percentage of apoptotic cells per islet as determine by TUNEL staining (Figures 6G,H, Supplementary Figure S4B) or the distribution of β-cell death per islet in islets from WT NOD and RIPCx36-NOD mice (Figure 6I). Overall, our data indicate that increased Cx36 coupling has a modest impact in protecting against diabetes progression but without detctable changes in insulitis or β-cell death.
The goal of this study was to determine the role of altered Cx36 coupling and Ca2+ signaling in the development of T1D in the NOD mouse. We found that euglycemic NOD mice had disrupted Ca2+ signaling dynamics and decreased Cx36-mediated gap junction coupling at 9 and 12 weeks age, compared to age-matched immunodeficient controls. These changes occurred prior to marked dysglycemia. Decreases in Cx36 gap junction coupling were mediated by autoreactive T-cells. Increases in Cx36 gap junction coupling and pharmacological agents that decreased cytosolic (Ca2+) and increased ER Ca2+ improved the survival of cytokine treated islets in vitro. While overexpression of Cx36 in the β-cells of NOD mice protected against early diabetes onset (at ∼12 weeks), increased Cx36 gap junction coupling did not significantly improve islet function or survival, resulting in only modest protection against T1D development. Our results suggest that while decreases in β-cell Cx36 gap junction coupling and altered Ca2+ signaling dynamics are associated with the development of T1D in the NOD mouse, increasing gap junction coupling alone is not sufficient to prevent the onset of disease.
Consistent with studies in prediabetic mouse models of type 2 diabetes (Carvalho, Oliviera et al., 2012; Gonzalez, Merino et al., 2013; Irles, Neco et al., 2015; Amaral, Kravets et al., 2020), we found that Cx36 gap junction coupling is decreased in islets from prediabetic NOD mice as early as 9 weeks of age compared to immunodeficient controls. Several studies have shown that glucose-induced Ca2+ oscillation coordination is mediated by electrical coupling between β-cells that coordinates membrane depolarization and subsequent Ca2+ influx across the islet (Ravier, Guldenagel et al., 2005; Benninger, Zhang et al., 2008; Perez-Armendariz 2013). This is supported by our data, where decreases in the coordination of glucose-induced Ca2+ oscillations directly correlated with decreases in Cx36 coupling. While we observed dysfunction to Cx36 coupling and Ca2+ oscillations in islets from NOD mice as early as 9 weeks of age, we did not observe dysfunction to insulin secretion at any of the time points studied. This is in line with previous studies where total KO of Cx36 in the islet disrupts insulin release pulsatility, but has a limited effect on overall insulin secretion levels (Head, Orseth et al., 2012). Our previous study has shown low levels of pro-inflammatory cytokines decrease Cx36 coupling and coordination of glucose-stimulated Ca2+ oscillations without causing dysfunction to levels of insulin secretion (Farnsworth, Walter et al., 2016). While decreased Cx36 coupling may lead to a shift in the range of glucose responsiveness observed in this study and our previous study, measurement of GSIS in isolated islets does not provide sufficient temporal resolution to determine shifts in glucose responsiveness and future study is required to determine any potential contributions to insulin secretion with loss of Cx36 coupling. In the early stages of T1D, levels of pro-inflammatory cytokines may be low enough to cause dysfunction to Cx36 gap junction coupling and Ca2+ dynamics with no significant effects on insulin secretion, as shown in this study. This is also supported by findings in type 2 diabetes, where low-grade inflammation caused impaired Ca2+ handling and islet dysfunction (Dula, Jecmenica et al., 2010). While measuring changes in Ca2+ dynamics in advanced stages of T1D in isolated mouse islets is technically challenging, future studies utilizing NOD pancreas slices from animals with advanced disease, coupled with immunocytochemical labeling of β-cells and T-cells, couldp provide insight into further alterations in Cx36 coupling and Ca2+ dynamics in T1D progression. Altogether, our results indicate a role for decreased Cx36 coupling in mediating islet dysfunction in early type 1 diabetes.
We also identified sub-populations of NOD mice with higher levels of dysfunction as determined by the decreased coordination of glucose-stimulated Ca2+ oscillations. This heterogeneity in islet function was not present in the immunodeficient control mice and therefore is likely associated with diabetes pathogenesis. However, we were unable to follow these mice to determine if this increased dysfunction correlated with earlier development of diabetes. We also noted that there was no correlation between gap junction coupling and Ca2+ oscillation coordination in islet from NOD mice, suggesting that changes in gap junction coupling alone are not representative of islet dysfunction. The identified sub-population of NOD mice with lower Ca2+ oscillation coordination did not have significantly different levels of coupling compared to the population of NOD mice with increased Ca2+ oscillation coordination. This suggests that other factors, such as KATP channel function or ER/mitochondrial Ca2+ buffering may also be impacted in diabetic animals; however, further study is required to determine how Ca2+ handling is altered. Previous studies in twins who were at risk for developing T1D have shown a positive correlation between decreases in first-phase insulin release and onset of T1D, supporting our hypothesis that greater islet dysfunction prior to changes in glucose homeostasis is associated with disease progression (Lo, Hawa et al., 1992). As altered first-phase insulin secretion occurs with a loss of Cx36 gap unction coupling in the islet, correlations between first-phase insulin secretion with levels of Cx36 coupling and altered Ca2+ signaling may predict disease onset.
Our results indicate that decreases in Cx36 coupling occur as a result of T1D onset, as levels of Cx36 are similar in NOD mice and immunodeficient controls at early ages but decrease starting at 9 weeks in the NOD mice. Deletion of chromogranin A in NOD mice, which lack autoreactive T-cells, preserves Cx36 coupling and transfer of diabetogenic splenocytes to immunodeficient mice decreases Cx36 coupling in islets (Baker, Bradley et al., 2015). Thus autoreactive immune cells are both sufficient and required to cause decreases in Cx36 gap junction coupling in the NOD mouse, which has not been previously shown. Islet dysfunction, specifically changes in islet electrophysiology, prior to diabetes onset has not been well characterized in T1D. This is partly due to the heterogeneity of T1D onset making it difficult to pinpoint the time when decreased islet function can be observed in the absence of significant cell death (Matthews, Xue et al., 2015). Additionally, techniques used to measure changes in islet electrophysiology frequently require destruction of the 3D islet architecture. Utilizing gap junction coupling and changes in Ca2+ dynamics as a measure of islet function and changes in electrophysiology provides an alternative technique to capture early decreases in function of islet in pre-diabetic NOD mice. Our results are in line with previous studies that showed islet dysfunction as measured by a reduction in first-phase insulin secretion in at-risk children who went on to develop T1D (Sherry, Tsai et al., 2005). Activated immune cells likely decrease Cx36 gap junction coupling by production of high levels of pro-inflammatory cytokines, which we have shown decrease Cx36 gap junction coupling and alter Ca2+ dynamics in vitro via activation of protein kinase C δ (Farnsworth, Walter et al., 2016).
Our results indicate that modulation of Cx36 gap junction coupling in the islet can mediate the effects of pro-inflammatory cytokines on islet survival. Gap junction coupling and Ca2+ regulation are tightly coupled, suppressing spontaneous elevations at low glucose levels and coordinating pulsatile Ca2+ at high glucose. Elevations in Ca2+ can also exacerbate apoptosis, thus suggesting that Cx36 gap junction coupling protects against cell death via Ca2+ regulation (Nicotera and Orrenius 1998). Both diazoxide activation of KATP channels, and OTA-activation of SERCA which will suppresses cytosolic Ca2+ elevations, counteracted the impact of a loss of Cx36 on cell death. The protection against cell death afforded by Cx36 overexpression was eliminated upon TEA inhibition of K+ channels, which will elevate cytosolic Ca2+ elevations. Together these findings indicate that a loss of Cx36 gap junction coupling exacerbates cytokine-mediated cell death via elevated spontaneous Ca2+ activity. This is supported by previous studies, where loss of gap junction coupling in islet β-cells resulted in increased intracellular Ca2+ at low glucose levels, likely due to heterogeneity in β-cell excitability in the islet (Benninger, Head et al., 2011; Benninger, Dorrell et al., 2018). This conclusion is supported by studies where cytokines cause apoptosis in β-cells by increasing intracellular Ca2+ (Chang, Cho et al., 2004). Additionally, reduced ER Ca2+ has been linked to ER stress induced apoptosis in β-cells, suggesting that increasing ER Ca2+ with the SERCA activator may protect against cell death (O'Neill, Lu et al., 2013). Further studies are required to determine if increasing ER Ca2+ can protect against cytokine-mediated cell death by reducing ER stress in islets with decreases gap junction coupling. Overall, our results support a role for elevated intracellular Ca2+ as a result of a loss of Cx36 coupling that exacerbates cytokine-induced islet cell death. However, the exact mechanisms by which Cx36-regulated Ca2+ impacts Cx36-mediated protection require further studies.
Our results indicate that decreases in Cx36 coupling are a result of T1D pathogenesis, therefore we also sought to determine if increasing Cx36 coupling could protect against the onset of disease in the NOD mouse. Overexpression of Cx36 did increase coupling at all time points studied; however, protection against disease onset was only observed early on from 10 to 12 weeks with a small difference in overall survival. We did not observe a significant improvement in islet Ca2+ dynamics or insulin secretion. These results are in contrast to our in vitro studies, as well as a previous study that used glibenclamide to increase Cx36 gap junction coupling and observed protection against T1D onset in the NOD mouse (Lamprianou, Gysemans et al., 2016). Glibenclamide also increases glucose-stimulated insulin secretion via KATP channel closure (Charpentier, Cancela et al., 2007; Ashcroft and Rorsman 2013), which may explain why improved insulin secretion was observed. While we found that overexpression of Cx36 increases β-cell gap junction coupling, dysfunction to intracellular Ca2+ dynamics remained. This suggests that glucose-stimulated Ca2+ signaling can be disrupted by changes in ER Ca2+ uptake in addition to decreased Cx36 coupling, as has been suggested by previous studies in vitro (Ramadan, Steiner et al., 2011; Kono, Ahn et al., 2012), and that increases in Cx36 gap junction coupling alone are insufficient to overcome altered intracellular Ca2+ handling.
Several studies have shown that Cx36 gap junction coupling between β-cells protects against cytokine-induced β-cell death in vitro (Klee, Allagnat et al., 2011; Allagnat, Klee et al., 2013) as well as streptozotocin-induced β-cell death in vivo (Klee, Allagnat et al., 2011). In contrast, we observed that increased Cx36 gap junction coupling provided little protection against β-cell apoptosis in NOD mice at 12 weeks, where mice were normoglycemic but significant insulitis was observed. This could be a result of the additional disruption to Ca2+ handling which was not recovered by increased Cx36 gap junction coupling. For example, recovery of ER Ca2+ homeostasis can protect against cytokine-mediated β-cell death (Clark, Kanekura et al., 2017), which follows with our results in vitro with KATP and SERCA modulators. The lack of protection against β-cell death with increased Cx36 gap junction coupling may also be due to β-cell death mediated by Ca2+-independent activation of the Fas ligand on the β-cell surface by activated immune cells (Lightfoot, Chen et al., 2012), as we did not observe differences in insulitis between NOD mice with normal or increased Cx36 gap junction coupling. While increases in Cx36 gap junction coupling alone is insufficient to protect against immune-mediated β-cell death in the NOD mouse, combination therapies which increase Cx36 gap junction coupling and recover ER Ca2+ homeostasis may show greater protection against β-cell death in T1D. Future studies that investigate changes in cytosolic and ER Ca2+ using genetically encoded and organelle targeted fluorescent Ca2+ probes may help to determine if modulating both intracellular Ca2+ and gap junction coupling can maintain ER Ca2+ homeostasis and normalize β-cell electrophysiology in islets from diabetic NOD mice.
In summary, we have shown that Cx36 gap junction coupling is reduced in the islets of NOD mice prior to changes in glucose homeostasis. This decrease in Cx36 gap junction coupling is accompanied by altered coordination of Ca2+ dynamics across the islet and is mediated by autoreactive immune cells in the NOD mouse. In vitro, loss of Cx36 gap junction coupling exacerbates cell death and Cx36 overexpression protects against cell death via regulating cytosolic Ca2+. While increasing Cx36 gap junction coupling in the NOD mouse protected against early onset of T1D; long term survival, islet function, and β-cell death were not improved. Thus while islet dysfunction occurs prior to changes in glucose homeostasis in T1D, increased β-cell gap junction coupling alone is insufficient to recover islet function or prevent diabetes onset. Future studies will determine if increasing Cx36 coupling in combination with therapies which restore Ca2+ homeostasis can protect against islet dysfunction and death in the NOD mouse.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee, University of Colorado Anschutz Medical Campus.
NLF contributed to study conception and design, data acquisition and analysis, drafting and revising the article, and final approval. RAP, WES, DGR, and JGM contributed to data acquisition and analysis, revising the article, and final approval. RKPB contributed to study conception and design, revising the article, and final approval.
This work was supported by Juvenile Diabetes Research Foundation (JDRF) Grants 5-CDA-2014-198-A-N (to RKPB) and 3-APF-2019-749-A-N (to NLF); National Institute of Health (NIH) grants R00 DK 085145, R01 DK102950, R01 DK106412 (to RKPB), and F32 DK102276 (to NLF). Microscopy experiments were performed in the Advanced Light Microscopy Core, supported in part by NIH grants P30 NS048154, P30 DK116073.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Kathryn Haskins for the generous donation of the ChGAKO mice for this study. We would like to thank the Barbara Davis Center Islet Isolation Core for facilitating islet isolations. We would also like to thank Laura Pyle for assistance with K-means clustering analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.913611/full#supplementary-material
Allagnat F., Klee P., Cardozo A. K., Meda P., Haefliger J. A. (2013). Connexin36 Contributes to INS-1E Cells Survival through Modulation of Cytokine-Induced Oxidative Stress, ER Stress and AMPK Activity. Cell Death Differ. 20, 1742–1752. doi:10.1038/cdd.2013.134
Ashcroft F. M., Rorsman P. (2013). KATP Channels and Islet Hormone Secretion: New Insights and Controversies. Nat. Rev. Endocrinol. 9, 660–669. doi:10.1038/nrendo.2013.166
Baker R. L., Bradley B., Wiles T. A., Lindsay R. S., Barbour G., Delong T., et al. (2015). Cutting Edge: Nonobese Diabetic Mice Deficient in Chromogranin A Are Protected from Autoimmune Diabetes. J. I. 196 (1), 39–43. doi:10.4049/jimmunol.1501190
Baker R. L., Bradley B., Wiles T. A., Lindsay R. S., Barbour G., Delong T., et al. (2016). Cutting Edge: Nonobese Diabetic Mice Deficient in Chromogranin A Are Protected from Autoimmune Diabetes. J. I. 196 (1), 39–43. doi:10.4049/jimmunol.1501190
Benninger R. K. P., Dorrell C., Hodson D. J., Rutter G. A. (2018). The Impact of Pancreatic Beta Cell Heterogeneity on Type 1 Diabetes Pathogenesis. Curr. Diab Rep. 18 (11), 112. doi:10.1007/s11892-018-1085-2
Benninger R. K. P., Head W. S., Zhang M., Satin L. S., Piston D. W. (2011). Gap Junctions and Other Mechanisms of Cell-Cell Communication Regulate Basal Insulin Secretion in the Pancreatic Islet. J. Physiology 589 (22), 5453–5466. doi:10.1113/jphysiol.2011.218909
Benninger R. K. P., Zhang M., Head W. S., Satin L. S., Piston D. W. (2008). Gap Junction Coupling and Calcium Waves in the Pancreatic Islet. Biophysical J. 95, 5048–5061. doi:10.1529/biophysj.108.140863
Calabrese A., Zhang M., Serre-Beinier V., Caton D., Mas C., Satin L. S., et al. (2003). Connexin 36 Controls Synchronization of Ca2+ Oscillations and Insulin Secretion in MIN6 Cells. Diabetes 52, 417–424. doi:10.2337/diabetes.52.2.417
Carvalho C. P. F., Oliveira R. B., Britan A., Santos-Silva J. C., Boschero A. C., Meda P., et al. (2012). Impaired β-cell-β-cell Coupling Mediated by Cx36 Gap Junctions in Prediabetic Mice. Am. J. Physiology-Endocrinology Metabolism 303, E144–E151. doi:10.1152/ajpendo.00489.2011
Cersosimo E., Triplitt C., Solis-Herrera C., Manadarino L. J., DeFronzo R. A. (2018). Pathogenesis of Type 2 Diabetes. MA: South DatmouthMDtext.
Chang I., Cho N., Kim S., Kim J. Y., Kim E., Woo J.-E., et al. (2004). Role of Calcium in Pancreatic Islet Cell Death by IFN-Γ/tnf-α. J. Immunol. 172 (11), 7008–7014. doi:10.4049/jimmunol.172.11.7008
Charpentier E., Cancela J., Meda P. (2007). Beta Cells Preferentially Exchange Cationic Molecules via Connexin 36 Gap Junction Channels. Diabetologia 50, 2332–2341.
Cigliola V., Populaire C., Pierri C. L., Deutsch S., Haefliger J. A., Fadista J., et al. (2016). A Variant of GJD2, Encoding for Connexin 36, Alters the Function of Insulin Producing β-Cells. PLOS One 11 (3), e0150880. doi:10.1371/journal.pone.0150880
Clark A. L., Kanekura K., Lavagnino Z., Spears L. D., Abreu D., Mahadevan J., et al. (2017). Targeting Cellular Calcium Homeostasis to Prevent Cytokine-Mediated Beta Cell Death. Sci. Rep. 7, 5611. doi:10.1038/s41598-017-05935-4
Cnop M., Welsh N., Jonas J.-C., Jörns A., Lenzen S., Eizirik D. L. (2005). Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Diabetes. Diabetes 54, S97–S107. doi:10.2337/diabetes.54.suppl_2.s97
Corezola do Amaral M. E., Kravets V., Dwulet J. M., Farnsworth N. L., Piscopio R., Miranda W. E. J. G., et al. (2020). Caloric Restriction Recovers Impaired β-cell-β-cell Gap Junction Coupling, Calcium Oscillation Coordination, and Insulin Secretion in Prediabetic Mice. Am. J. Physiology-Endocrinology Metabolism 319 (4), E709–E720. doi:10.1152/ajpendo.00132.2020
Degen J., Meier C., Van Der Giessen R. S., Söhl G., Petrasch-Parwez E., Urschel S., et al. (2004). Expression Pattern of lacZ Reporter Gene Representing Connexin36 in Transgenic Mice. J. Comp. Neurol. 473 (4), 511–525. doi:10.1002/cne.20085
Dula S. B., Jecmenica M., Wu R., Jahanshahi P., Verrilli G. M., Carter J. D., et al. (2010). Evidence that Low-Grade Systemic Inflammation Can Induce Islet Dysfunction as Measured by Impaired Calcium Handling. Cell Calcium 48 (2-3), 133–142. doi:10.1016/j.ceca.2010.07.007
Duprez J., Jonas J.-C. (2011). Role of Activating Transcription Factor 3 in Low Glucose- and Thapsigargin-Induced Apoptosis in Cultured Mouse Islets. Biochem. Biophysical Res. Commun. 415 (2), 294–299. doi:10.1016/j.bbrc.2011.10.048
Farnsworth N. L., Benninger R. K. P. (2014). New Insights into the Role of Connexins in Pancreatic Islet Function and Diabetes. FEBS Lett. 588 (8), 1278–1287. doi:10.1016/j.febslet.2014.02.035
Farnsworth N. L., Hemmati A., Pozzoli M., Benninger R. K. P. (2014). Fluorescence Recovery after Photobleaching Reveals Regulation and Distribution of Connexin36 Gap Junction Coupling within Mouse Islets of Langerhans. J. Physiol. 592 (20), 4431–4446. doi:10.1113/jphysiol.2014.276733
Farnsworth N. L., Walter R. L., Hemmati A., Westacott M. J., Benninger R. K. P. (2016). Low Level Pro-inflammatory Cytokines Decrease Connexin36 Gap Junction Coupling in Mouse and Human Islets through Nitric Oxide-Mediated Protein Kinase Cδ. J. Biol. Chem. 291 (7), 3184–3196. doi:10.1074/jbc.m115.679506
Gonzalez A., Merino B., Marroquí L., Ñeco P., Alonso-Magdalena P., Caballero-Garrido E., et al. (2013). Insulin Hypersecretion in Islets from Diet-Induced Hyperinsulinemic Obese Female Mice Is Associated with Several Functional Adaptations in Individual β-Cells. Endocrinology 154 (10), 3515–3524. doi:10.1210/en.2013-1424
Head W. S., Orseth M. L., Nunemaker C. S., Satin L. S., Piston D. W., Benninger R. K. P. (2012). Connexin-36 Gap Junctions Regulate In Vivo First- and Second-phase Insulin Secretion Dynamics and Glucose Tolerance in the Conscious Mouse. Diabetes 61 (7), 1700–1707. doi:10.2337/db11-1312
Hraha T. H., Bernard A. B., Nguyen L. M., Anseth K. S., Benninger R. K. P. (2014). Dimensionality and Size Scaling of Coordinated Ca2+ Dynamics in MIN6 β-cell Clusters. Biophysical J. 106 (1), 299–309. doi:10.1016/j.bpj.2013.11.026
Irles E., Ñeco P., Lluesma M., Villar-Pazos S., Santos-Silva J. C., Vettorazzi J. F., et al. (2015). Enhanced Glucose-Induced Intracellular Signaling Promotes Insulin Hypersecretion: Pancreatic Beta-Cell Functional Adaptations in a Model of Genetic Obesity and Prediabetes. Mol. Cell. Endocrinol. 404, 46–55. doi:10.1016/j.mce.2015.01.033
Ize-Ludlow D., Lightfoot Y. L., Parker M., Xue S., Wasserfall C., Haller M. J., et al. (2011). Progressive Erosion of β-Cell Function Precedes the Onset of Hyperglycemia in the NOD Mouse Model of Type 1 Diabetes. Diabetes 60 (8), 2086–2091. doi:10.2337/db11-0373
Klee P., Allagnat F., Pontes H., Cederroth M., Charollais A., Caille D., et al. (2011). Connexins Protect Mouse Pancreatic β Cells against Apoptosis. J. Clin. Invest. 121 (12), 4870–4879. doi:10.1172/jci40509
Kono T., Ahn G., Moss D. R., Gann L., Zarain-Herzberg A., Nishiki Y., et al. (2012). PPAR-γ Activation Restores Pancreatic Islet SERCA2 Levels and Prevents β-Cell Dysfunction under Conditions of Hyperglycemic and Cytokine Stress. Mol. Endocrinol. 26 (2), 257–271. doi:10.1210/me.2011-1181
Lamprianou S., Gysemans C., Bou Saab J., Pontes H., Mathieu C., Meda P. (2016). Glibenclamide Prevents Diabetes in NOD Mice. PLoS ONE 11 (12), e0168839. doi:10.1371/journal.pone.0168839
Lightfoot Y. L., Chen J., Mathews C. E. (2012). Immune-mediated β-cell Death in Type 1 Diabetes: Lessons from Human β-cell Lines. Eur. J. Clin. Investigation 42 (11), 1244–1251. doi:10.1111/j.1365-2362.2012.02711.x
Lin J.-D., Hsia T.-L., Wu C.-Z., Su C.-C., Ma W.-Y., Hsieh A.-T., et al. (2010). The First and Second Phase of Insulin Secretion in Naive Chinese Type 2 Diabetes Mellitus. Metabolism 59 (6), 780–786. doi:10.1016/j.metabol.2009.09.024
Lo S. S. S., Hawa M., Beer S. F., Pyke D. A., Leslie R. D. G. (1992). Altered Islet Beta-Cell Function before the Onset of Type 1 (Insulin-dependent) Diabetes Mellitus. Diabetologia 35, 277–282. doi:10.1007/bf00400930
Lu J., Liu J., Li L., Lan Y., Liang Y. (2020). Cytokines in Type 1 Diabetes: Mechanisms of Action and Immunotherapeutic Targets. Clin. Transl. Immunol. 9 (3), e1122. doi:10.1002/cti2.1122
Matthews C. E., Xue S., Posgai A., Lightfoot Y. L., Li X., Lin A., et al. (2015). Acute versus Progressive Onset of Diabetes in NOD Mice: Potential Implications for Therapeutic Interventions in Type 1 Diabetes. Diabetes 64 (11), 3885–3890. doi:10.2337/db15-0449
Nicotera P., Orrenius S. (1998). The Role of Calcium in Apoptosis. Cell Calcium 23 (2-3), 173–180. doi:10.1016/s0143-4160(98)90116-6
O'Neill C. M., Lu C., Corbin K. L., Sharma P. R., Dula S. B., Carter J. D., et al. (2013). Circulating Levels of IL-1B+IL-6 Cause ER Stress and Dysfunction in Islets from Prediabetic Male Mice. Endocrinology 154 (9), 3077–3088. doi:10.1210/en.2012-2138
Ovalle F., Grimes T., Xu G., Patel A. J., Grayson T. B., Thielen L. A., et al. (2018). Verapamil and Beta Cell Function in Adults with Recent-Onset Type 1 Diabetes. Nat. Med. 24, 1108–1112. doi:10.1038/s41591-018-0089-4
Padgett L. E., Broniowska K. A., Hansen P. A., Corbett J. A., Tse H. M. (2013). The Role of Reactive Oxygen Species and Proinflammatory Cytokines in Type 1 Diabetes Pathogenesis. Ann. N.Y. Acad. Sci. 1281, 16–35. doi:10.1111/j.1749-6632.2012.06826.x
Pearson J. A., Wong F. S., Wen L. (2016). The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J. Autoimmun. 66, 76–88. doi:10.1016/j.jaut.2015.08.019
Pérez-Armendariz E. M. (2013). Connexin 36, a Key Element in Pancreatic Beta Cell Function. Neuropharmacology 75, 557–566. doi:10.1016/j.neuropharm.2013.08.015
Pohorec V., Križančić Bombek L., Skelin Klemen M., Stožer J., Stozer A. (2022). Glucose-Stimulated Calcium Dynamics in Beta Cells from Male C57BL/6J, C57BL/6N, and NMRI Mice: A Comparison of Activation, Activity, and Deactivation Properties in Tissue Slices. Front. Endocrinol. 13, 867663. doi:10.3389/fendo.2022.867663
Ramadan J. W., Steiner S. R., O’Neill C. M., Nunemaker C. S. (2011). The Central Role of Calcium in the Effects of Cytokines on Beta-Cell Function: Implications for Type 1 and Type 2 Diabetes. Cell Calcium 50 (6), 481–490. doi:10.1016/j.ceca.2011.08.005
Ravier M. A., Güldenagel M., Charollais A., Gjinovci A., Caille D., Söhl G., et al. (2005). Loss of Connexin36 Channels Alters β-Cell Coupling, Islet Synchronization of Glucose-Induced Ca2+ and Insulin Oscillations, and Basal Insulin Release. Diabetes 54 (6), 1798–1807. doi:10.2337/diabetes.54.6.1798
Serre-Beinier V., Bosco D., Zulianello L., Charollais A., Caille D., Charpantier E., et al. (2009). Cx36 Makes Channels Coupling Human Pancreatic β-cells, and Correlates with Insulin Expression. Hum. Mol. Genet. 18 (3), 428–439. doi:10.1093/hmg/ddn370
Serre-Beinier V., Le Gurun S., Belluardo N., Trovato-Salinaro A., Charollais A., Haefliger J. A., et al. (2000). Cx36 Preferentially Connects Beta-Cells within Pancreatic Islets. Diabetes 49 (5), 727–734. doi:10.2337/diabetes.49.5.727
Sherry N. A., Tsai E. B., Herold K. C. (2005). Natural History of β-Cell Function in Type 1 Diabetes. Diabetes 54, S32–S39. doi:10.2337/diabetes.54.suppl_2.s32
Soleimanpour S. A., Stoffers D. A. (2013). The Pancreatic β Cell and Type 1 Diabetes: Innocent Bystander or Active Participant? Trends Endocrinol. Metabolism 24 (7), 324–331. doi:10.1016/j.tem.2013.03.005
Sosenko J. M., Skyler J. S., Beam C. A., Krischer J. P., Greenbaum C. J., Mahon J., et al. (2013). Acceleration of the Loss of the First-phase Insulin Response during the Progression to Type 1 Diabetes in Diabetes Prevention Trial-type 1 Participants. Diabetes 62 (12), 4179–4183. doi:10.2337/db13-0656
Stozer A., Klemen M. S., Gosak M., Bombek L. K., Pohorec V., Rupnik M. S., et al. (2021). Glucose-dependent Activation, Activity, and Deactivation of Beta Cell Networks in Acute Mouse Pancreas Tissue Slices. Am. J. Physiology - Endocrinol. Metabolism 321, E305–E323. doi:10.1152/ajpendo.00043.2021
Wang C., Guan Y., Yang J. (2010). Cytokines in the Progression of Pancreatic β-Cell Dysfunction. Int. J. Endocrinol. 2010, 515136. doi:10.1155/2010/515136
AUC Area under the curve
ANOVA Analysis of variance
Ca2+ Calcium
Ct Cycle time
ChGAKO-NOD Chromogranin A knockout NOD mouse
Cx36 Connexin36
FRAP Fluorescence recovery after photobleaching
HBSS Hanks balanced salt solution
HPRT1 hypoxanthine phosphoribosyltransferase 1
IFN-γ Interferon gamma
IL-1β Interleukin-1beta
RIP Rat insulin promoter
RCC Relative cytokine concentration
TNF-α Tumor necrosis factor alpha
WT Wild type
Keywords: calcium signaling, connexin36, cytokines, islet dysfunction, NOD mouse, type 1 diabetes
Citation: Farnsworth NL, Piscopio RA, Schleicher WE, Ramirez DG, Miranda JG and Benninger RKP (2022) Modulation of Gap Junction Coupling Within the Islet of Langerhans During the Development of Type 1 Diabetes. Front. Physiol. 13:913611. doi: 10.3389/fphys.2022.913611
Received: 05 April 2022; Accepted: 09 June 2022;
Published: 28 June 2022.
Edited by:
Paul Edward Squires, University of Lincoln, United KingdomReviewed by:
Laura Marroqui, Miguel Hernández University of Elche, SpainCopyright © 2022 Farnsworth, Piscopio, Schleicher, Ramirez, Miranda and Benninger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard K. P. Benninger, UmljaGFyZC5CZW5uaW5nZXJAY3VhbnNjaHV0ei5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.