 Marion Blayac
Marion Blayac Patrice Coll2
Patrice Coll2 Valérie Urbach
Valérie Urbach Pascale Fanen
Pascale Fanen Ralph Epaud
Ralph Epaud Sophie Lanone
Sophie Lanone
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 02 June 2022
Sec. Respiratory Physiology and Pathophysiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.908230
Cystic fibrosis (CF) is a lethal and widespread autosomal recessive disorder affecting over 80,000 people worldwide. It is caused by mutations of the CFTR gene, which encodes an epithelial anion channel. CF is characterized by a great phenotypic variability which is currently not fully understood. Although CF is genetically determined, the course of the disease might also depend on multiple other factors. Air pollution, whose effects on health and contribution to respiratory diseases are well established, is one environmental factor suspected to modulate the disease severity and influence the lung phenotype of CF patients. This is of particular interest as pulmonary failure is the primary cause of death in CF. The present review discusses current knowledge on the impact of air pollution on CF pathogenesis and aims to explore the underlying cellular and biological mechanisms involved in these effects.
Cystic Fibrosis (CF) is a monogenic disease caused by pathogenic mutations in the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) gene which encodes the CFTR protein, an anion channel responsible for chloride and bicarbonate ion transport across epithelial cells. For a more complete description of CF disease please, refer to Shteinberg et al. (2021). CFTR also regulates many other mechanisms in epithelial physiology and has been reported to inhibit the epithelial Na+ channel (ENaC) activity in the airways. Dysfunctional CFTR results in dehydration of airway surface liquid (ASL) and is associated with persistent airway infection and sustained inflammation, leading to obstructive lung disease and progressive structural damage (Boucher, 2007). While CF is a systemic disease characterized by altered lung mucociliary clearance, exocrine pancreatic insufficiency, intestinal obstruction and infertility, the bronchopulmonary disease remains the main driver of the high morbidity and early mortality of CF patients (Elborn, 2016). The progression of the disease mainly depends on pulmonary exacerbations defined as acute deterioration of respiratory symptoms which ultimately impair lung function and quality of life (Ferkol et al., 2006; Stanford et al., 2021). These exacerbations’ episodes, most frequently caused by lung bacterial infections, include increased cough, increased sputum production, increased use of antibiotics, dyspnea, and decreased lung function. Repeated exacerbation events, chronic infection and persistent inflammation lead to progressive and irreversible lung damage and respiratory failure (Sanders et al., 2010, 2011; de Boer et al., 2011; Waters et al., 2012).
CF patients present a great genotypic variability with more than 2,000 CFTR gene mutations identified so far (Rommens, 2011). These mutations are classified into six classes based on their effect on CFTR synthesis and function (Elborn, 2016). Each of these mutations occurs at a variable frequency (Lao et al., 2003; De Boeck et al., 2014) with a heterogenous geographical distribution (Mirtajani, 2017). Class I–III mutations are usually associated with a more severe CF disease prognosis as CFTR protein function is totally suppressed. Class IV–VI mutations are characterized by a residual CFTR function and usually cause a disease of more moderate severity (Elborn, 2016). Interestingly, while CFTR genotype correlates well globally with the pancreatic phenotype, it mostly fails to predict pulmonary-disease severity (Burke et al., 1992; The Cystic Fibrosis Genotype-Phenotype Consortium, 1993; Kerem and Kerem, 1996; Duguépéroux and De Braekeleer, 2004; Cutting, 2015; Raynal and Corvol, 2020). Twin and sibling studies have shown a smaller difference in lung function between monozygotic twins than between dizygotic twins or siblings (Collaco et al., 2010). Such difference appears to increase when twins and siblings start living apart and stop sharing common environment. This phenotypic variability suggests the involvement of other contributors especially to the CF airway disease, unrelated to CFTR itself. Several genes (i.e., NOS, TGFB1, MBL2, MUC4/MUC20, SLC9A3, SLC6A14 or HLA Class II) have been identified as possible gene modifiers of CF lung disease (Corvol et al., 2015; Sepahzad et al., 2021). The splicing machinery could also modify CF severity (Nissim-Rafinia and Kerem, 2005). Beside these genetic and epigenetic modifiers, non-genetic factors also appear as relevant modifying contributors to CF pathophysiology; environmental factors (Szczesniak et al., 2020) such as tobacco smoke (Campbell et al., 1992; Kopp et al., 2016), air pollution (Brugha et al., 2018), climate and seasonal changes (Ramsay et al., 2016) or socioeconomic status (Oates and Schechter, 2016). This review particularly focuses on the contribution of air pollution.
Outdoor air pollutants are numerous (several hundreds to several thousands of chemical species), for most of them at trace levels (from few ppb—parts per billion, to ppm—parts per million), and are gaseous (nitrogen dioxide—NO2, carbon dioxide—CO2, methane—CH4, etc.) but also under particular phases: these atmospheric particles, or aerosols—also referred to as particulate matter—are a suspension of liquid or solid in the gas phase and are categorized according to their size—coarse particles (PM10), fine particles (PM2.5) and ultrafine particles (PM0.1). Pollutants can be divided into two groups related to their origin: 1) primary pollutants which are air pollutants emitted directly from a source—as an illustration nitrogen oxides (NOX) or soot particles mainly emitted from traffic; 2) secondary pollutants that are not directly emitted, but are formed when other pollutants react in the lower atmosphere—as an illustration the production of the oxidant ozone (O3) resulting from reactions between nitrogen oxides (NOx) and volatile organic compounds (VOCs) (Figure 1A) (Walker, 1977). These numerous chemical compounds, gases and aerosols, react continuously in the atmosphere under solar irradiation, and are also stirred and transported in the atmosphere (Holton et al., 1995). Thus, the composition of the atmosphere varies according to time and space (in the order of few seconds and few meters for OH., and of 10 years and 1.000 km for CH4; illustrations and more extended descriptions may be found in (Wayne, 2000; Ravishankara, 2003). These well-known atmospheric composition and evolution are illustrated by Figures 1A,B. Much more details may be found in (Seinfeld and Pandis, 2016).
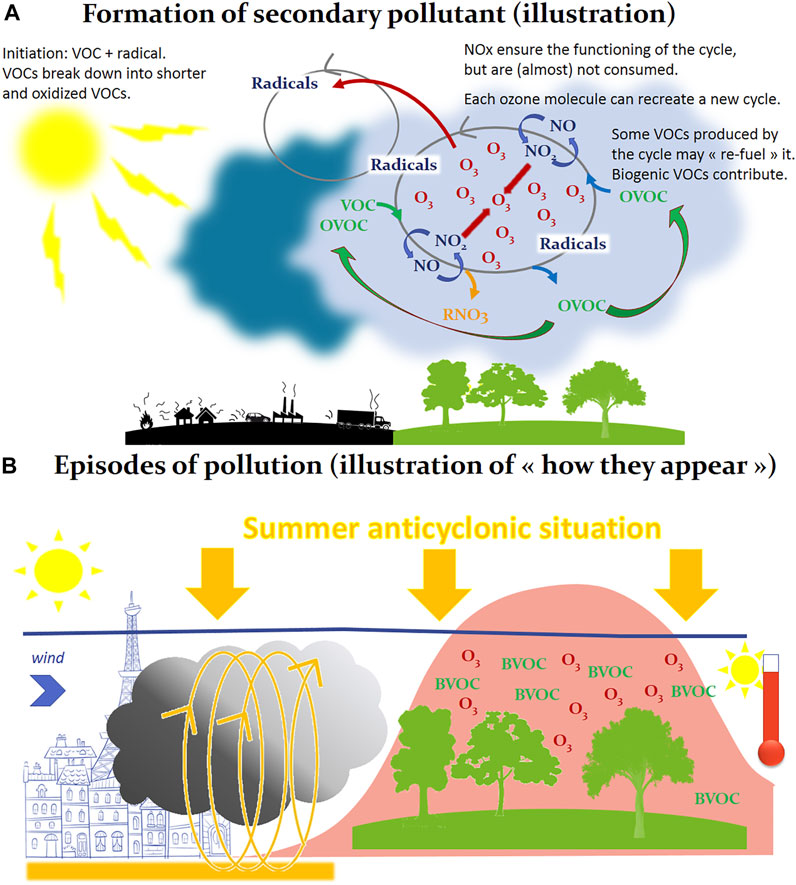
FIGURE 1. Formation of secondary pollutants (A) and episodes of pollution (B). In addition to primary pollutants (gases or particles, emitted naturally or by anthropogenic activity), many so-called “secondary” pollutants are not emitted but formed from other pollutants, as it is the case in Panel 1A in this illustration of the production of ozone (O3) in the gas phase. Here the formation of ozone results from reactions cycles between nitrogen oxides (NO/NO2) and VOCs, in the chemical reactor constituted by the atmosphere, these chemical reactions using solar energy as energy source. Atmospheric radicals are the intermediary species involved in these chemical cycles. All this explains how, with favorable conditions, very large quantities of ozone can be formed in a few hours of sunshine, downwind of cities. An illustration of such phenomenon is reported in Panel 1B, where we are facing a summer anticyclonic situation, the pollutants emitted for example in the city (left section of 1B) react in the urban atmosphere under the sunlight. Under the effect of the wind, this primary pollution can migrate to peri-urban or rural areas, cross other chemical compounds (as Biogenic Volatile Organic Compounds, BVOC, emitted by trees) and generate a secondary pollution, symbolized in Panel 1B by a production of ozone (O3, pollution peak). We are in the case of a secondary pollution at the regional scale, spatially extended, which will present a maximum in the afternoon because the sunshine irradiation, and consequently the intensity of the photochemistry, are maximum.
Public health concerns, with raising questions about the impact of air pollution on our health, has driven to monitor and predict air quality (as in the case of meteorology). Atmospheric measurements are carried out throughout the world, in a more or less intense way depending on the level of development of the countries. The number of measuring stations, and their distribution on the territories, answer to rules or laws (as an example in France, rules are stated at the European level). These rules are essential to guarantee a quality of measurement that is comparable from one region or country to another (Kracht et al., 2018). For example, in the Paris region, different types of measurements and analysis stations are spread over a 100 km radius around Paris and measure the quality of the air breathed by the population (more than 12 million inhabitants in the whole region), regardless of their environment, near or far from the sources of pollution. The network in charge of these measurements has about 70 measuring stations, including more than 50 permanent stations, acting as mini static laboratories (Rodriguez et al., 2019).
Automatic stations (measuring for example O3, NOx, sulfur dioxide—SO2, carbon monoxide—CO, etc.) ensure a continuous measurement every hour usually, and allow a real time follow-up of the air quality. The choice of the location of the automatic stations and of the types of the pollutants measured in these stations is primarily based on public health concerns. There are several types of stations: background stations (far from pollution sources, in particular traffic routes), near traffic stations (located near a road infrastructure as area representative of the maximum level of exposure for the population), urban stations (monitoring the average exposure of the population to air pollution phenomena known as “background” in urban centers), peri-urban stations (monitoring the photochemical pollution, in particular O3 and its precursors, and monitoring the average level of exposure of the population to “background” air pollution phenomena on the outskirts of the urban center), rural stations (monitoring of the exposure of ecosystems and of the population in rural areas to “background” air pollution, in particular photochemical pollution, at a regional scale) and industrial site stations (information on concentrations measured in areas representative of the maximum level to which the population near a stationary source is likely to be exposed by plume or accumulation phenomena). Such continuous monitoring allows to measure the level of exposure of individuals over different times scales, either acutely (referring to an exposure lasting from 1 day to a few days as during an air pollution episode) or chronically (referring to a continuous or repeated exposure over a long period of time, from months to years). In addition, some pollutants are not measured automatically and in real time by the stations but are measured by sampling followed by delayed analysis techniques in specific laboratories, or even by dedicated measurement field campaigns. Finally, automatic station network can be reinforced by measurements using diffusion tubes. This method guarantees a regular but not continuous monitoring and allows to calculate an annual average and to estimate the situation with regard to the regulatory values.
Numerical modeling (Chemistry Transport Models) is a complementary tool to pollutant emission measurements and inventories. It allows to estimate the concentrations of various pollutants at any point of a given geographical area, for past, present, or future periods. These numerical tools are based on the representation of the phenomena involved in the formation and fate of air pollution: chemical transformations, transport, dispersion, emissions, and depositions. These different numerical models operate at various temporal and geographical scales (from the street scale to the continental scale) and their use requires to characterize the parameters influencing the concentrations of pollutants (for example meteorology). Overall, the use of this mathematical tool supports air quality monitoring, as it allows to characterize the air pollution at any point of the represented geographical domain.
The challenge of measuring reactive trace species in the atmosphere (at ppt—parts per trillion to ppm levels), ideally on a continuous basis, and of integrating these data into the Chemistry Transport Models in order to characterize the exposure of populations at a given location over a given period of time, is therefore a very high-level challenge. This is the main barrier to linking the effects of air pollution to the observed health impacts on populations.
Amongst these hundreds of pollutants, some so-called “indicators” (such as O3, NO2, SO2, PM2.5, and PM10) are under official monitoring in most countries and are expected to fit air quality guidelines. World Health Organization (WHO) estimates that 99% of people worldwide breath polluted air, with effects on health that are clearly established (World Health Organization, 2021). Accounting for 4.2 million premature deaths yearly worldwide (World Health Organization, 2021), ambient air pollution currently constitutes the major environmental threat for human health and ranks as the fourth leading risk factor for premature deaths (Health Effects Institute, 2020). Regarding respiratory effects, it is well documented that air pollution reduces lung function (Paulin and Hansel, 2016), worsens respiratory condition of patients with pre-existing pathologies such as asthma and chronic obstructive pulmonary disease (COPD) (Hoek et al., 2013; Thurston et al., 2020) and increases lung cancer incidence (Raaschou-Nielsen et al., 2013). Since 2013, air pollution has been classified as carcinogenic to humans by the International Agency for Research Cancer (IARC) (International Agency for Research on Cancer, 2013). In a recent paper, the loss of life expectancy from ambient air pollution was shown to be 2.9 years (Lelieveld et al., 2020). In addition, it is estimated that the mean life expectancy would increase by 1.7 years if all anthropogenic emissions were removed. For children, air pollution is associated with increased preterm birth and low birth weight (Stieb et al., 2012; Dadvand et al., 2013; Fleischer et al., 2014), reduced pulmonary growth and lung function (Gauderman et al., 2007; Hwang et al., 2015; Milanzi et al., 2018) together with an increased risk of developing respiratory diseases (Goldizen et al., 2016).
With known effects for healthy people as well as for people with pre-existing condition(s), air pollution could constitute an important contributing factor for CF disease. Moreover, in a context where air pollution is often above WHO’s recommended values and where CF patients live older (while being diagnosed at birth in most cases), it is of particular interest to consider air pollution and its possible harmful effects on the course of the disease. Strikingly, studies dealing with specific implication of air pollution on the course of CF airway disease are scarce and only quite recent. Our goal here is to review the actual knowledge on the role of air pollution in the phenotypic variability observed in CF as well as to highlight recent advances in the underlying mechanisms.
Studies of interest were identified by searching PubMed from 1990 to present with the term “cystic fibrosis” or “CFTR” combined with the specific terms “air pollution” or “particulate matter,” “ozone,” “nitrogen dioxide,” “sulfur dioxide,” “diesel” or “exhaust.” Key words search was limited to title and abstract. Human as well as mouse studies were considered. Among the 62 studies retrieved after this search, 14 were chosen based on their title and abstract. Studies were included if they assessed health outcomes in people with CF or in CF experimental models in relation with air pollution exposure measurement data or experimental exposure. Though conducted in non-CF airway cells and non-CF mouse model, two more studies were included because they investigated direct effects of pollutants on CFTR expression and/or function and airway fluid secretion. Finally, one additional study was identified when reviewing references of the articles initially selected. A total of 17 studies were overall included in this review (Tables 1, 2, 3).
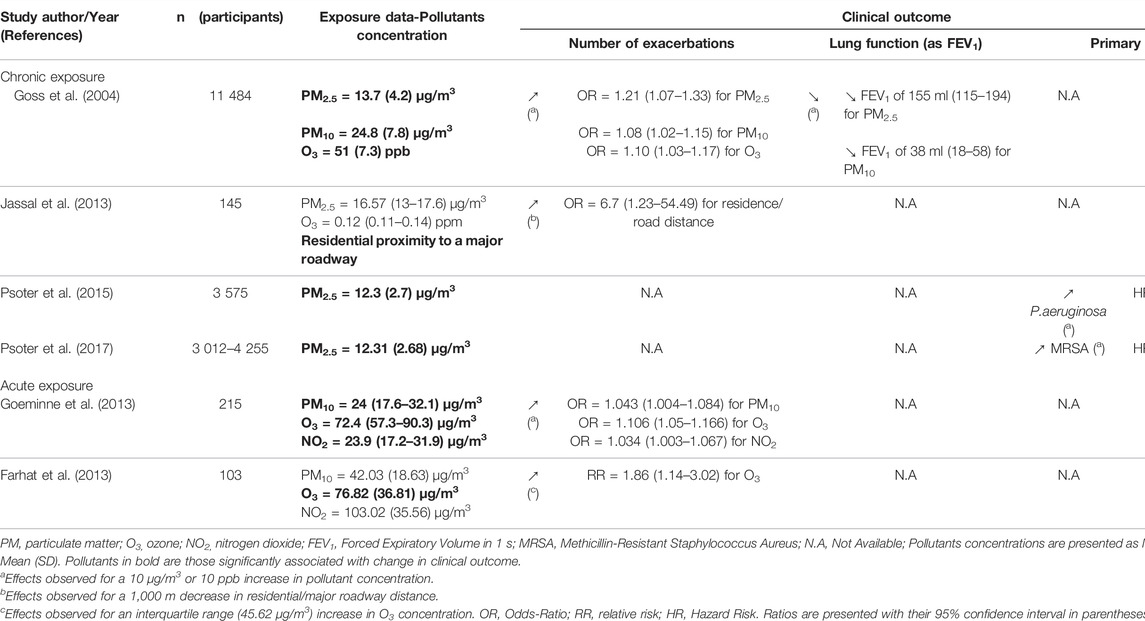
TABLE 1. Summary and comparison of results obtained by epidemiological studies.
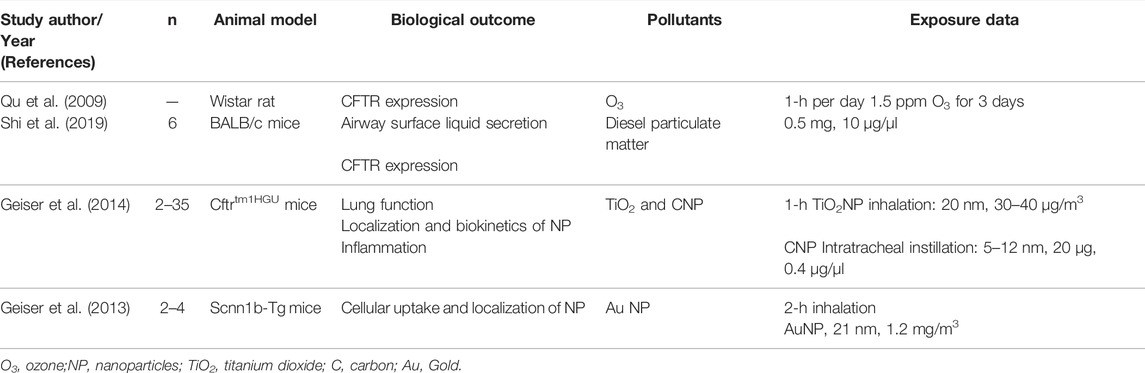
TABLE 2. In vivo experimental studies.
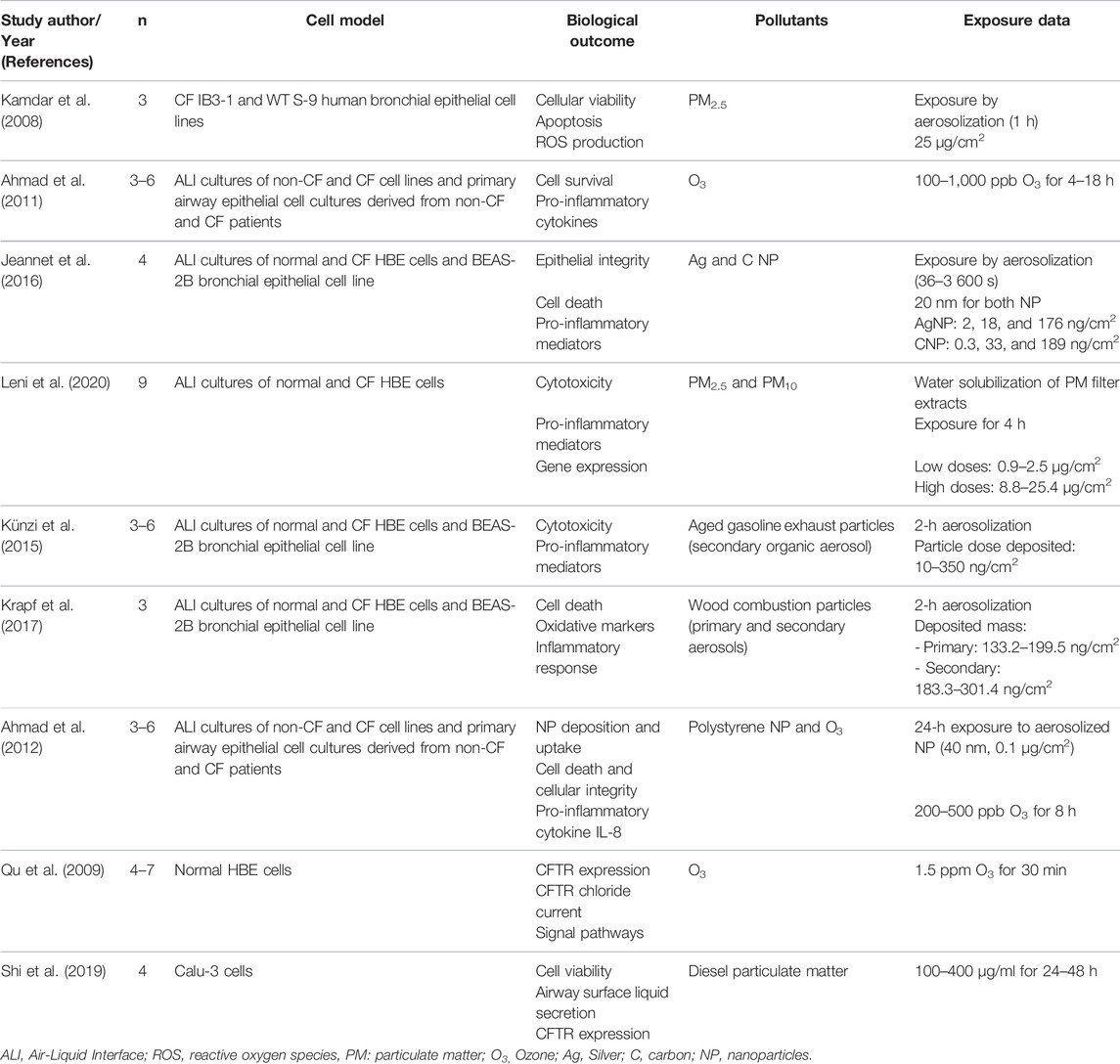
TABLE 3. In vitro experimental studies.
The first studies describing health outcomes of air pollution exposure in CF patients have focused on chronic exposures. There is only four of them so far. The very first study to address this issue has been published by Goss et al. (2004), in 2004, who surveyed a cohort of 11,484 patients during a 2 year-interval. In this study, the mean annual values for O3, NO2, SO2, CO, and particulate matter (PM) were considered. Authors linked the exposure data to pulmonary function, exacerbations and mortality data. Pulmonary function was estimated with forced expiratory volume in 1 s (FEV1) and exacerbations were defined as a “condition requiring hospitalization” or as “the at home use of intravenous antibiotics.” A negative linear association was found between lung function and particulate pollution (PM2.5 and PM10); for a 10 μg/m3 increase in PM2.5 and PM10, a significant decrease in FEV1 was observed (Table 1). No association was found with the other pollutants (O3, NO2, SO2, and CO). As for exacerbations, patients were divided into two groups according to the number of exacerbations experienced during the study period: none or one exacerbation versus two or more. The characterization of groups showed that the patients with more than two exacerbations were globally older, had a reduced lung function, a worse nutritional status and were more likely to be infected with the bacteria Pseudomonas aeruginosa (P.aeruginosa) and Burkholderia cepacia complex (B.cepacia) compared to those who suffer less than two exacerbations. Patients with two or more exacerbations also lived in regions with higher levels of PM2.5, PM10, and O3. Authors showed that, for every 10 μg/m3 increase in PM2.5 and PM10 concentration, there was respectively a 21% and 8% higher risk of presenting more than two exacerbations (Table 1). Moreover, a 10-ppb increase in O3 concentration resulted in 10% increase in that same risk. No difference in the mean annual concentrations of NO2, SO2, and CO was observed between the two groups and no significant association with the exacerbation number was noted for these pollutants. Finally, although the result was not significant, a 10 μg/m3 increase in PM2.5 exposure substantially increased the mortality risk (32% higher risk).
In another study, Jassal et al. (2013) took in consideration the participant’s home distance to a major road or freeway in addition to O3 and PM2.5. The goal here was to use the proximity to roadways as a proxy for long-term traffic pollution, as concentrations of particles are elevated near roads due to emissions by vehicles exhaust. This 5-year retrospective study, dedicated to study the effects of air pollution on pulmonary exacerbation frequency, was conducted in 145 CF patients. Exacerbation and patient’s categorization were defined as in Goss’s study. No significant difference in O3 and PM2.5 levels could be found between the two exacerbation groups. Residential proximity to a roadway was however higher for individuals who experienced more than two exacerbations (Table 1). While the pollutant levels were similar between studies, the failure to replicate O3 and PM2.5 effects observed by Goss is probably due to a lack of statistical power in Jassal’s study. This latter study is however the first to describe the influence of proximity to traffic on the clinical condition of CF patients as opposed to classical quantitative measures of pollutants. Of note, proximity to traffic implies the consideration of pollutants other than the ones usually measured. Eventually, these results suggest that residential proximity to roadway could increase the susceptibility for acute exacerbation events in CF patients.
Bacterial colonization constitutes a central element of exacerbations and a key determinant of the severity of exacerbation episodes in CF. Psoter et al. (2015) formulated the hypothesis that exposure to air pollution could increase the risk of primary infection by P.aeruginosa. Although this micro-organism is the major respiratory pathogen in CF patients with an initial colonization usually occurring within the first 10 years of life, earlier P.aeruginosa infection, particularly before the age of five is strongly associated with a more severe CF lung disease later (Pittman et al., 2011). Psoter et al. (2017) therefore followed children under the age of 6 (3,575) and recorded the time to initial acquisition of P.aeruginosa, defined as the apparition of a first positive respiratory culture while on study. For exposure evaluation, the mean yearly PM2.5 concentration during the year preceding birth of each child was considered. During a mean follow-up period of 1.8 years, 1,711 (48%) children acquired P.aeruginosa with a median age of acquisition of 15 months. Exposure to PM2.5 was found to be slightly higher among children who acquired P.aeruginosa during the follow-up period compared to those who remained uninfected. In addition, a 10 μg/m3 PM2.5 concentration increase was significantly associated with a 24% increase in the risk of primary colonization by P.aeruginosa (Table 1). In a companion study based on the same cohort and experimental design, Psoter and colleagues evaluated if exposure to PM2.5 could be a risk factor for the initial acquisition of other respiratory pathogens in young CF children. They looked at the methicillin-sensitive (MSSA) and -resistant (MRSA) forms of Staphylococcus aureus, Stenotrophomonas maltophilia and Achromobacter xylosoxidans. Similar to the initial study, children who declared colonization during the follow-up period had a greater PM2.5 exposure compared to those who remained free of pathogens, independently of the pathogen considered. A 10 μg/m3 increase in PM2.5 exposure was associated with a 68% increased risk of MRSA acquisition but was not associated with the acquisition of other respiratory pathogens (Table 1).
Only two studies so far have focused on the effects of acute exposure to pollution peaks on the course of CF disease. Goeminne et al. (2013) led a case-crossover retrospective study over a period of 12 years (Table 1). The goal was to explore the link between the short-term increase in PM10, O3, and NO2 and the onset of pulmonary exacerbations. An exacerbation was defined by the use of oral or intravenous antibiotics for respiratory event. Pollution levels were calculated for the last 2 days before and on the day of the exacerbation event. Data analysis showed an increase in the number of exacerbations on days with higher air pollution (Table 1). The authors found a significant association for a 10 μg/m3 increase in all three pollutants and same-day exacerbation onset. An overall increase in risk was also observed throughout the three measured days suggesting pollution peaks could have delayed effects. The strength of this study is its case-crossover design in which every subject is its own control, thus reducing the influence of confounding factors.
In a more size-limited study, Farhat et al. (2013) studied the effects of short-term variations in the concentrations of air pollutants. The study was conducted in the metropole of Sao Paulo on the occurrence of exacerbations in 103 CF children and teenagers for 1 year (Table 1). Exacerbation was defined as the presence of at least three of the following symptoms: fever, increase in sputum production or cough intensity, change in sputum color, worsening dyspnea, loss of appetite, >10% decrease of FEV1, or weight loss. The occurrence of exacerbation was then compared to daily mean concentrations of O3, SO2, NO2, PM10, and CO obtained from monitoring stations spread across the city. The authors reported a higher relative risk of exacerbation when O3 concentration increased by an interquartile range (45.62 μg/m3) (Table 1). A significant association was observed for an O3 increase 2 days before the onset of exacerbation and a tendency was observed for O3 exposure the day prior and the same day as the exacerbation. No statistically significant association was found between other pollutants and the occurrence of exacerbation in patients with CF. Indeed, O3 was the only pollutant exceeding national Brazilian standards as well as the WHO air quality guidelines during the study, hence reaching unhealthy levels while other pollutants levels were considered acceptable. However, this study raises several questions as many key elements are not described: the spatial variability of the concentrations of pollutants that allow to differentiate patients; the study of correlations between different pollutants (why are PM excluded from impacting pollutants when they are correlated with NO2?); the lack of temporal visibility of pollutant concentrations and the ability to predict the impact on disease on fine time scales; the insufficiently explicit use of clinical data and their very patchy aspect. Also, the physiological importance in defining exacerbation severity differs between criteria (fever vs. >10% decrease of FEV1). Overall, a clarification of the positive criteria related to an exacerbation would have provided the necessary information. The discrepancy with Goeminne’s study regarding the impact of PM10 and NO2 levels may be explained by the methods used for exposure data estimation. Interestingly, levels of PM10 and NO2 reported by Farhat were higher than Goeminne’s with respective mean concentrations of 42 μg/m3 and 103 μg/m3 against median concentrations of 24 μg/m3 for the two pollutants (Table 1) (Goeminne et al., 2013). These results are consistent with the fact that Farhat’s study was conducted in a highly polluted metropole. However, both studies came to comparable O3 concentration. More likely, the absence of association between the increase of PM10 and NO2 concentration and the occurrence of respiratory exacerbation in the Brazilian study might be explained by the smaller size of the panel but also by the difference in exacerbation definition and inclusion criteria. Goeminne’s definition is restrictive to severe exacerbations because a lot of exacerbations are treated with only oral and/or aerosolized antibiotics. Also, patients who had no exacerbation or no registered use of antibiotic were excluded. Exacerbation definition is less stringent in Farhat’s study which covers a larger panel of CF patients, including patients who did not undertake antibiotic therapy. This may have accounted for a possible not significant impact of other pollutants in Farhat’s study and could suggest that NO2 and PM10 mainly target airways and favor infection while O3 would have a wider impact on other organ function. Finally, patients’ status at the time of the study could also explain the difference observed. Indeed, in Farhat’s study, 70% of CF patients did not present a significant degree of bronchial obstruction and their median Shwachman-Kulczycki score (assessment of CF disease severity based on general activity, physical examination, nutrition and radiological findings) was 75 corresponding to a “Good” clinical condition, while no details on disease severity was provided in Goeminne’s study. Overall, both studies bring additional elements to consider acute exposure to air pollution as negatively impacting the course of CF disease.
Overall, epidemiological studies on CF patients have highlighted an increased risk of exacerbations, primary infections and reduced lung function associated with air pollution exposure. If these studies identify a negative impact of global air pollution on the course of CF, they however leave remaining questions about how these effects originate. The following chapter is dedicated to present experimental studies designed to answer these questions.
As central contributors to CF airway pathophysiology, the expression of CFTR and airway secretions have been investigated in response to air pollution. A markedly reduced immunofluorescence staining of CFTR was observed in tracheal respiratory epithelium of rats exposed to O3 (Qu et al., 2009). A decrease of about 25% in mRNA and protein expression of CFTR and aquaporins 1 (AQP1) and 5 (AQP5) was observed in normal BALB/c mice exposed to diesel particulate matter (Shi et al., 2019). The underlying mechanisms of action of such reduced CFTR expression have not been studied in vivo, nor the effect of diesel particulate matter on CFTR channel function. Importantly, the reduced expression of CFTR and aquaporins could result in a decreased cell permeability to water and ions and thus ASL secretion, two key features of CF. Shi et al. (2019) also described an increased Muc5AC mucin expression in BALF in the same mice. Interestingly, although not in the context of CF, O3, SO2, and particles were found to induce mucous cell metaplasia (Harkema et al., 1999; Sueyoshi et al., 2004), increase mucin expression (Jany and Basbaum, 1991; Cho et al., 1999; Huang et al., 2017) and impair mucociliary clearance and ciliary beating frequency (Pedersen, 1990; Saldiva et al., 1992; Bayram et al., 1998) in rat airway epithelium and human airway epithelial cells. These events would be interesting to consider since they are altered in CF but not yet studied in relation to air pollution exposure in CF models. Unfortunately, no dose-response relationship could be established, as only one dose for O3 and diesel particulate matter was used in these animal studies.
In vivo studies investigating the effects of air pollution on CF models remain sparse (Table 2), compared to those performed in other models of bronchial disease, such as asthma or COPD. Carbon nanoparticles (CNP), particularly abundant in urban air pollution, has been instilled in Cftrtm1HGU and WT mice by intratracheal route to evaluate their lung inflammatory response (Geiser et al., 2014). The Cftrtm1HGU model is produced by a targeted insertion mutagenesis in exon 10 of the Cftr gene (Dorin et al., 1992). It presents a markedly reduced but residual CFTR expression (about 10% of normal mRNA levels) resulting in a mild disease phenotype (Dorin et al., 1994). Of note, this mild airway phenotype is also influenced by genetic background and species differences as compared to humans. CNP instillation provoked, in WT and mutated mice, an acute and transient inflammatory response dominated by neutrophils. This response was maximal the day following exposure. Privileged interaction of NP with airway epithelial cells in mutants due to a reduced clearance of such NP could explain the induction of this inflammatory response. Indeed, Geiser et al. (2014) reported an alteration of titanium dioxide manufactured nanoparticles (TiO2NP) cellular uptake in Cftrtm1HGU mice. In WT mice, the cellular uptake of TiO2NP is entirely achieved by macrophages while in Cftrtm1HGu mice, 26% of TiO2NP are internalized by alveolar epithelial cells—the remaining 74% NP are internalized by macrophages. This suggests a reduced macrophagic clearance of NP in this CF model. These results corroborate those obtained by the same group on inhaled gold NP uptake and cellular localization in transgenic Scnn1b mice: compared to WT mice, Scnn1b transgenic mice showed a reduced macrophage uptake of gold NP that were mainly localized at the epithelium surface (Geiser et al., 2013). This is of particular interest because this mouse model—characterized by an airway-targeted overexpression of the ENaC β subunit—constitutes presently the most complete murine model reproducing human CF pulmonary phenotype (Mall et al., 2004; Zhou et al., 2011). Finally, a reduced airway macrophage phagocytosis of carbonaceous PM has also been described in CF children (Liu et al., 2020), adding to the notion of an overall altered clearance of particulate material in CF disease.
An important feature of CF airway pathogenesis is the excessive and inefficient inflammatory process that could be explained by an altered resolution of inflammation (Roesch et al., 2018; Briottet et al., 2020). The finding that resolution of inflammation differs between Cftrtm1Hgu and WT mice is consistent with this aspect of the clinical description of the CF airway disease. Indeed, at the third day post-instillation, neutrophil count in bronchoalveolar lavages fluid (BALF) is decreased by 70% in WT mice while only by 25% in Cftrtm1HGU mice. BALF pro-inflammatory cytokines levels were also higher in Cftrtm1HGU mice compared to WT for most cytokines tested. As a striking example, G-CSF concentration was 13 times higher in Cftr mutant mice, consistent with the slower decline of BALF neutrophils and suggesting a delayed or impaired resolution of inflammation in Cftr-deficient mice. The specialized pro-resolving mediators (SPM) which are bioactive lipids triggering the inflammation resolution phase, are thus of particular interest in this context. Interestingly, the levels of 15-hydroxyecosatetraenoic (15-HETE), a metabolite of the lipoxygenase pathway showing anti-inflammatory properties, was significantly lower in Cftr mutant mice exposed to CNP (Karp et al., 2004; Ringholz et al., 2014), which is in line with the compromised resolution of CNP-induced airway inflammation in Cftr mutants (Geiser et al., 2014). Furthermore, the SPM lipoxin A4 (LXA4) was found to be decreased in C57Bl/6 mice in response to cigarette and wood smoke (Awji et al., 2015). SPM also have a therapeutic potential in reversing or preventing pollution-induced inflammation (Kilburg-Basnyat et al., 2018; Lu et al., 2018; Thatcher et al., 2019). Finally, in the airway of CF patients, the biosynthesis of LXA4 and Resolvin D1 (RvD1) SPM are reduced (Karp et al., 2004; Ringholz et al., 2014; Eickmeier et al., 2017; Briottet et al., 2020; Isopi et al., 2020) and related to the sustained levels of pro-inflammatory cytokines and bacterial infection, the altered airway surface hydration and pulmonary function (Verrière et al., 2012; Eickmeier et al., 2017; Codagnone et al., 2018; Ringholz et al., 2018; Isopi et al., 2020). Taken together, these data highlight a possible particular pulmonary susceptibility of CF patients exposed to particulate air pollution; under these conditions, insufficient inflammation resolution could favor the onset of a chronic inflammatory condition and accelerated progression of the CF airway disease.
In non-CF HBE cells, it has been shown that O3 decreased CFTR mRNA, protein expression and activity (Qu et al., 2009). This modulation of CFTR expression is dependent of the JAK2/STAT1 pathway which is itself activated by reactive oxygen species (ROS) (Qu et al., 2009). Indeed, ROS inhibitors prevented O3-induced CFTR expression decrease by reducing STAT1 phosphorylation. As supporting evidence, a direct decrease of CFTR expression and activity due to oxidative stress has been previously described (Bebok et al., 2002; Cantin et al., 2006a). CFTR internalization or retrograde trafficking to the endoplasmic reticulum as induced by cigarette smoke exposure (Clunes et al., 2012; Marklew et al., 2019) could also be considered to explain the reduced CFTR expression and activity induced by O3. In Calu-3 cells, diesel particulate matter (DPM), which contains oxidants, dose-dependently reduced CFTR expression as well as its binding to the plasma membrane (Shi et al., 2019). Although not directly investigated, the authors suggest that previously described mechanisms involving JAK2/STAT1 in reducing CFTR expression could be similarly involved in these effects. Interestingly, the expression of AQP1 and AQP5 expression was also significantly decreased after DPM exposure, while an increased cAMP intracellular content could be detected, potentially promoting CFTR activity. It must be noted that the inhibiting effects of such exposure on CFTR expression and membrane localization were stronger. In another study in Calu-3 cells, CFTR function was found to be reduced by DPM. However, this acquired CFTR dysfunction could be rescued by CFTR potentiator VX-770 (Nguyen et al., 2021). Interestingly, polystyrene NP activated CFTR ion channel in Calu-3 cells (McCarthy et al., 2011). Although not yet studied in CF cells, the modification of CFTR expression after exposure to air pollutants is thus important to consider because the existing low CFTR expression and altered airway fluid secretion in CF patients could be deepen. In class IV–VI CF patients, with residual CFTR function mutations, this could lead to a potential total suppression of CFTR function which would certainly worsen their pathophysiological state.
Studies published on PM2.5/PM10, O3, and NP exposure effects on CF cells (Table 3) have highlighted three common cellular mechanisms—cell death, oxidative stress, and inflammation (Figure 2). Such effects are not surprising as they are classically described and known to participate in CF pathophysiology (Rosenfeld et al., 2001; Rottner et al., 2007, 2011; Sagel et al., 2012; Voisin et al., 2014; Ziady and Hansen, 2014; Antus, 2016; Roesch et al., 2018) and induced by air pollution exposure (Dagher et al., 2006; Lee et al., 2018; de Oliveira Alves et al., 2020; Gangwar et al., 2020; Li et al., 2020).
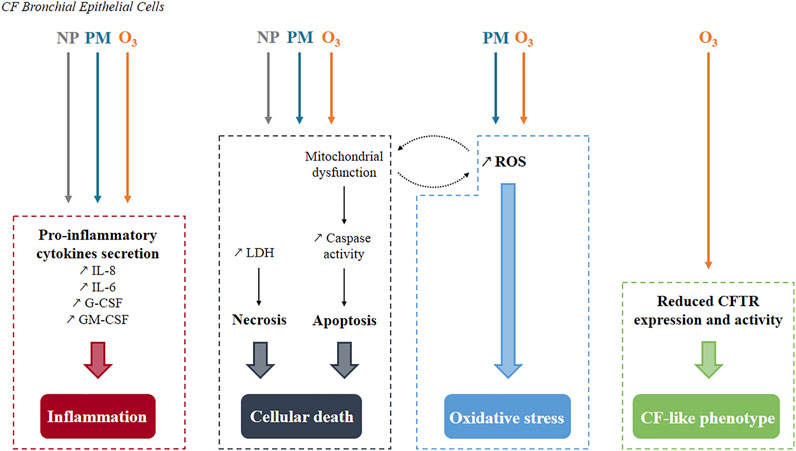
FIGURE 2. Summary of pollutants’ effects known so far in human CF bronchial epithelial cells. Three common cellular mechanisms have been identified. Inflammation and cell death occur after exposure to either nanoparticles, particulate matter or ozone while oxidative stress is generated only by particulate matter and ozone. Mitochondrial dysfunction and reactive oxygen species production are interconnected as they stimulate each other. Specifically to ozone, a CF-like phenotype is produced with decreased CFTR expression and activity. NP, Nanoparticles; PM, Particulate Matter; O3, Ozone; ROS, Reactive Oxygen Species.
Cell death occurs either by apoptosis or necrosis or even by both processes in the case of exposure to PM2.5 (Kamdar et al., 2008; Leni et al., 2020). Exposure of transformed human CF bronchial epithelial cells (CF HBE cells) or wild-type ones (non-CF HBE cells) to PM2.5 (Kamdar et al., 2008) or O3 (Ahmad et al., 2011) induced an increase in DNA fragmentation, propidium iodide staining, caspase 3/7 and 9 activity and an increased expression of pro-apoptotic mediators (Figures 2, 3). Of note, this increase was greater in CF HBE cells suggesting that these cells are more sensitive to PM2.5 and O3 than non-CF ones. While the level of anti-apoptotic proteins remained unchanged with exposure, the overexpression of the anti-apoptotic Bcl-xL contributes to significantly reduce apoptosis in CF HBE cells exposed to PM2.5. Mitochondrial dysfunction seems to be at the origin of such apoptosis (Kamdar et al., 2008; Ahmad et al., 2011). Indeed, a decrease of mitochondria membrane potential, of cytochrome C release and pro-apoptotic Bcl-2 associated X (Bax) translocation to mitochondria were reported upon exposure to PM2.5 and O3. Mitochondria-dependent generation of reactive oxygen species (ROS) was induced by exposure to PM2.5 only (Kamdar et al., 2008). Likewise, baseline production of ROS was higher in CF HBE cells revealing increased oxidative stress in CF cells. By promoting further mitochondrial dysfunction, ROS generation maintains oxidative stress, which could be responsible for the higher baseline apoptosis observed in CF cells. Furthermore, as a marker of cytotoxicity, increase lactate dehydrogenase (LDH) release from damaged cells has been linked to necrosis in CF cells exposed to silver (AgNP) and carbon manufactured NP (Jeannet et al., 2016). Also, a dose-dependent increase in LDH release was observed after exposure of normal and CF HBE cells to PM2.5 and PM10 (Leni et al., 2020), aged gasoline exhaust particles (Künzi et al., 2015) or wood combustion particles (Krapf et al., 2017). A loss of transepithelial resistance was showed in CF cells exposed to O3 suggesting the disruption of tight junctions (Ahmad et al., 2011). However, these NP did not interfere with epithelial integrity as intact apical junctional complexes were reported after such exposure (Ahmad et al., 2012; Jeannet et al., 2016). The observed modification of tight junction properties after O3 but not NP exposure indicates a more pronounced susceptibility of CF cells to O3 than to NP. Yet, CF HBE cells sensitivity to NP could be potentiated in presence of O3. Indeed, after cell surface attachment, NP are mainly internalized in cytosol but this localization was partly switched to nucleus when cells were co-exposed to O3 thus giving NP a potential to exert further harmful effects (Ahmad et al., 2012).
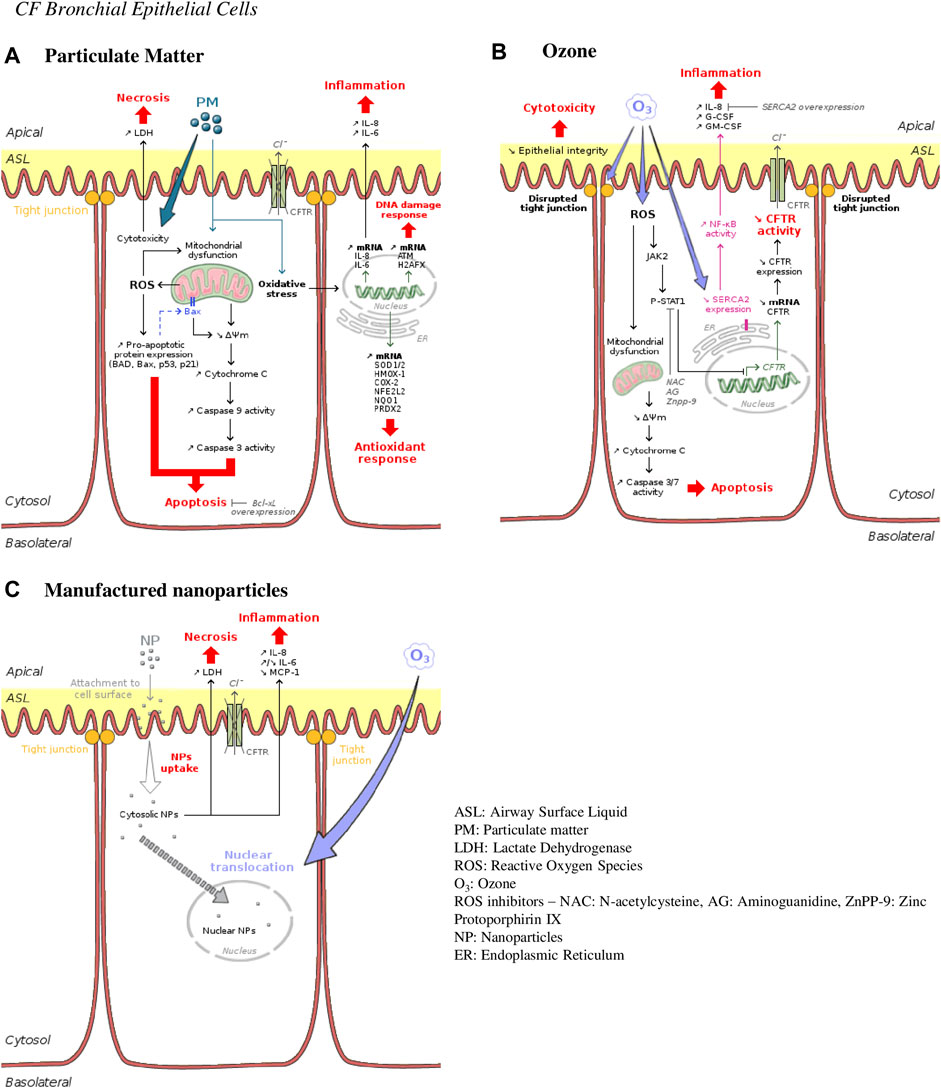
FIGURE 3. Ambient air pollution mediated effects and mechanisms induced in human CF bronchial epithelial cells. (A) Particulate matter effects. Particles are responsible for cellular death occurring either by necrosis or apoptosis. They are also responsible for inflammation, DNA damage and oxidative stress by inducing expression of typical genes involved in such responses—interleukin 8 and 6 (IL-8 and IL-6) for inflammation; histone H2A family member X (H2AFX) for DNA damage repair; superoxide dismutase (SOD1/2), heme oxygenase 1(HMOX-1), nuclear factor erythroid-derived 2-like 2 (NFE2L2) or peroxiredoxin (PRDX2) for antioxidant response. (B) Ozone effects. Ozone is directly cytotoxic for bronchial epithelial cells by disrupting tight junctions and epithelial integrity and by causing apoptosis. Promoting the JAK2-STAT1 pathway, ozone leads to pro-inflammatory cytokines secretion through NF-κB activation. CFTR expression is also downregulated leading to CFTR activity decrease and a CF-like phenotype. (C) Manufactured nanoparticles effects. Nanoparticles are internalized into bronchial epithelial cells provoking necrosis and inflammation. Co-exposure with ozone causes increase in intranuclear delivery of these nanoparticles.
Oxidative stress response has been shown to be stronger in CF HBE cells primary cultures compared to non-CF HBE cells. Wood combustion particles upregulated the expression of the oxidative stress markers heme oxygenase 1 (HMOX-1) and cyclooxygenase 2 (COX-2) (Krapf et al., 2017). PM2.5 and PM10 also upregulated genes involved in oxidative stress (superoxide dismutase one and 2—SOD1 and 2), antioxidant defense (HMOX-1, nuclear factor erythroid-derived 2-like 2—NFE2L2), oxidative protection (NADP(H) quinone dehydrogenase—NQO1, peroxiredoxin—PRDX2) as well as genes involved in DNA damage repair (ataxia telangiectasia mutated kinase—ATM, histone H2A family member H2AFX) (Leni et al., 2020). This oxidative/anti-oxidative imbalance could yield to an inflammation cascade. Indeed, IL-8, G-CSF, and GM-CSF levels were found to be markedly increased following O3 exposure in CFBE41o- cell line (Ahmad et al., 2011). O3-mediated inflammation is dependent on the NF-κB signaling pathway modulated by SERCA2 (Sarcoendoplasmic Reticulum Calcium ATPase). SERCA2 is downregulated by O3 and the specific downregulation of SERCA2 activates NF-κB pathway and the subsequent IL-8 release (Ahmad et al., 2011). The O3-induced IL-8 secretion was reversed by SERCA2 overexpression. Interestingly, SERCA2 expression is constitutively decreased in CF airway epithelia, which could potentially explain the basal inflammation observed in these cells (Ahmad et al., 2009). Indeed, basal IL-8, G-CSF, and GM-CSF levels were greater in CF airway epithelial cell lines and CF primary cultures than in non-CF cells (Ahmad et al., 2011) as well as IL-6 and MCP-1 levels which are, in CF cells, constitutively largely exceeding those of non-CF cells (Künzi et al., 2015; Jeannet et al., 2016). Such higher pro-inflammatory cytokines levels could also participate in turn to ROS generation. As regard to cytokine secretion, the effects of particle exposure are more controversial. Indeed, in both non-CF and CF HBE cells IL-8 levels increased after exposure to PM (Leni et al., 2020), AgNP and CNP (Jeannet et al., 2016) while polystyrene NP did not induce modification of IL-8 release (Ahmad et al., 2012). IL-6 levels either increased in response to PM, AgNP, and CNP in normal and CF HBE cells but decreased after exposure to aged gasoline particles in CF HBE cells. Upon exposure to PM and NP, MCP-1 levels increased in normal cells while they decreased in CF HBE cells (Künzi et al., 2015; Jeannet et al., 2016; Leni et al., 2020). Wood combustion particles did not significantly alter any cytokine release in any of the cell model considered (Krapf et al., 2017).
Unfortunately, there is no epidemiological data on CF prior to the industrial revolution that could bring to light the influence of air pollution composition on CFTR or on the CF airway disease. Indeed, anthropological studies suggests that CFTR mutation arose in the early Bronze Age and that the mutations spread from west to southeast Europe during ancient migrations via healthy carriers (CF is an autosomal recessive disease—people with CF inherit one mutated copy of the CFTR gene from each parent, referred to as a carrier). However, the first description of the CF disease dates only back to the 1930s (Andersen, 1938). In the 1950s, the median life expectancy for patients with CF was a few months, not allowing any conclusion to be drawn regarding the impact of air pollution. Finally, the causative CFTR gene was only discovered 30 years ago (Riordan et al., 1989). There are geographic differences in life expectancy of CF patients which could be related to different air pollution, but these discrepancies are mostly related to the status of developing country or not, where the weight of accessibility to treatments is bigger than that of air pollution. Substantial increased number of adults with CF in developed countries has resulted from delivery of care in well-organized, multidisciplinary CF centers and the use of effective drugs to treat infection and improve mucociliary clearance (Castellani et al., 2009; McCormick et al., 2010). In contrast, in countries with less well-resourced health-care systems, median life expectancy can be in the second decade of life because of the lack of access to treatment.
Epidemiological studies provide information on the global effect of air pollution on the course of CF disease. However, individual exposure is difficult to follow and only few pollutants are currently explored. Exposure estimation is tricky. Except one study which used spatial interpolation model (Goeminne et al., 2013), all the others estimated individual exposure on average measures obtained by the closest oriented-population monitoring air stations rather than direct monitoring at the patient’s residence. As a consequence, possible exposure misclassification could occur. Monitoring stations are mainly found in urban areas. How accurately assess exposure in rural areas? In addition, only few pollutants are monitored, but are they representative of the atmospheric mixture? Actual individual exposure also depends on factors like time spent outdoors, distance from pollution sources and weather patterns. For example, meteorological conditions are of primary importance because of their pivotal link to both CF and pollution: they have effects on CF clinical condition—as shown for temperature (Collaco et al., 2011) and season (Psoter et al., 2013) on P.aeruginosa lung infection—and they also influence on pollution chemical composition and evolution. Therefore, the inclusion of meteorological variables should be considered. A correct exposure estimation is also in relation with an accurate determination of possible confounders. In most epidemiological studies reviewed here, adjustment was made on socio-economic status. People of low socio-economic status tend to live in areas with higher ambient air pollution, have lower income and thus limited access to health care which could explain poorer health outcomes. Adjustment on socio-economic status may have reduced residual confounding due to these variables. However, these studies reveal the lack of inclusion of other confounding exposures such as tobacco smoke. Of note, cigarette smoking exposure has been associated with worse clinical prognosis in CF—decreased growth and weight (Rubin, 1990; Kovesi et al., 1993; Kopp et al., 2015), smaller lung function (Kovesi et al., 1993; Smyth et al., 1994; Collaco et al., 2008), increased colonization by MRSA (Kopp et al., 2015), longer periods of intravenous antibiotic treatment (Gilljam et al., 1990), airway surface dehydration (Clunes et al., 2012) and CFTR dysfunction (Cantin et al., 2006b; Raju et al., 2013; Marklew et al., 2019; Rasmussen et al., 2020). In a very recent study, tobacco smoke has also been shown to limit the therapeutic benefit of tezacaftor/ivacaftor therapy in pediatric CF patients (Baker et al., 2021). Similar limiting effects of air pollution on such treatments could be considered in CF patients. On the opposite, the effects of cigarette smoke exposure were reversed with the use of the CFTR potentiator ivacaftor (VX-770) in non-CF HBE cells (Sloane et al., 2012; Raju et al., 2017). One could thus also hypothesize the potential prevention of air pollution effects by CFTR modulators as shown with DPM-induced CFTR dysfunction rescue by VX-770 (Nguyen et al., 2021). Tobacco smoke and air pollution co-exposure must be considered in the context of worsening exacerbations and lung function. The important point here is that air pollution is part of a wider concept called exposome. Exposome corresponds to the sum of all environmental exposures an organism experiences throughout his/her life, from conception to death (Wild, 2012). This concept emphasizes that it is impossible to know to what extent air pollution exposure only influences CF phenotype as one individual is never exposed to air pollution alone. Environmental exposures more likely interact with each other and have cumulative effects. That is why, to be as realistic and accurate as possible, air pollution should be considered as part of a complete and complex exposome to reduce potential residual confounding. In addition to the complexity of this external exposome—all the external exposures—comes the complexity of an internal exposome—all the biological responses to exposures—that takes its origin, in the context of CF, in the great diversity of CF mutations and their respective consequences on the pleiotropic functions of CFTR itself. Keeping in mind this notion of exposome is of the utmost importance in developing new experimental concepts and lessen the misinterpretation of data. This is true for epidemiological studies as well as for experimental ones. Indeed, studying environmental exposures on animals maintained in strictly controlled conditions—i.e., same exposome—can only provide approximative indications on the potential biological responses in human populations who are genetically heterogenous, live in different environments, have different occupations, etc. and thus have complex and divergent internal and external exposomes (Bolon and Haschek, 2020).
Experimental studies use animal and cellular models. CF animal models are numerous and different based on the nature of the mutation in the Cftr locus (Wilke et al., 2011). In mice, most of the knock-out models present a severe mortality due to intestinal obstruction which limits the possibility to study the disease at adult age. F508del mutation models have a good survival until maturity and have the advantage to reproduce the most common mutation found in patients. However, they mainly reproduce the intestinal aspect of the disease with no basal specific lung phenotype. On the opposite, the Scnn1b transgenic model presents a CF-like lung disease but has no intestinal defect. In addition, this model is not mutated in the Cftr gene thus diverting the origin of CF disease and limiting other possible effects resulting from Cftr mutations. Apart from mice, other CF animal models developing a “spontaneaous” lung disease have emerged such as pigs, ferrets or rats (Semaniakou et al., 2019). As for CF animal models, there is a wide range of existing CF epithelial cell models (Castellani et al., 2018). The airway epithelium is characterized by a wide diversity of cell types: basal cells, ciliated cells, goblet cells, club cells, ionocytes and more (Montoro et al., 2018). These different cell types could have different susceptibility to air pollution. In the specific context of CF, ionocytes—cells with a high level of CFTR expression (Plasschaert et al., 2018; Scudieri et al., 2020)—could be particularly interesting to examine. Yet, this cellular complexity of the respiratory epithelium is not always considered. Indeed, transformed or immortalized cell lines constitute a simplistic single barely differentiated cell type model. On the other hand, fully differentiated primary HBE cells cultured at air-liquid interface, despite subjected to donors’ variability, more closely mimic the in vivo lung environment and therefore relevant exposure conditions. These differences in model construction could explain why responses might be very different from one study to another.
Beside the limits of the different animal and cellular models of CF exposed above, another major limitation of the experimental studies reviewed here resides in pollutants consideration. Several challenges remain to be tackled. As mentioned earlier, air pollution consists of a complex reactive mixture evolving in space and time. Yet only few pollutants—used as indicators and representing a tiny part of the atmospheric mixture (O3, NO2, SO2, PM2.5, and PM10)—are currently monitored. As an example, ultrafine particles (PM0.1) are not measured directly nor subjected to guideline values while causing deleterious health effects (MacNee and Donaldson, 2003; Schraufnagel, 2020). Moreover, many toxicological studies have been based on the study of individual pollutants. As a primary approach, studying individual pollutants is interesting for mechanistic insight and to give a first glimpse of their respective toxicity. However, it is limited when numerous pollutants interact together. One isolated pollutant will not necessarily have the same effects alone or in combination with others. The relevance of existing experimental studies can therefore be largely challenged based on the absence of consideration of such synergism and reactivity between pollutants making it thus difficult to extrapolate results to “real life.” In this context of considering pollutants’ dynamic, few studies exist on pulmonary effects of secondary or aged aerosols (Delfino et al., 2010; Gaschen et al., 2010; Rager et al., 2011; Diaz et al., 2012; Künzi et al., 2013; Künzi et al., 2015; Krapf et al., 2017). Ahmad et al. are to our knowledge the only ones to have combined NP and O3 exposition, a first step toward consideration of multi-pollutant interactions (Ahmad et al., 2012). A disruptive approach consists to reproduce multiphasic chemical processes in the laboratory—i.e., continuously produce environments representative of urban atmospheres—in order to study the impact of air pollution in particular in the multi-pollutant synergy and multiphasic dimensions (Coll et al., 2018). For example, the authors have developed a platform, named PolluRisk, which allows to reproduce for days the atmospheric phases (gases and aerosols) similar to those of cities like Paris or Beijing for example. It is resulting from the coupling of an atmospheric simulation chamber with exposure devices able to host preclinical models. The experimental protocol consists in the continuous injection of primary pollutants (NOx, organic compounds mix simulating anthropogenic emissions, SO2, soot, inorganic salts and potentially mineral dust particles if needed) at low concentrations (ppb levels in air) in the chamber. Due to the residence time in the atmospheric simulation chamber—around 4 h to represent air masses at a regional scale—the mixture is exposed to an artificial solar irradiation, allowing secondary pollutants such as O3, NO2, formaldehyde, and peroxyacetyl nitrate (PANs) as well as complex polyfunctional organics including Secondary Organic Aerosol (SOA) to be formed. This way, they can reach their chemical steady state. Exposure devices are then “fed” with a constant flow of such a mixture. A challenge of such chambers is to reach realistic pollutants doses, a key point as in vivo and in vitro studies usually expose animal models and cells to non-realistic elevated pollutants concentrations. For example, in the studies reviewed here (Tables 2, 3), an O3 exposure from 100 ppb to 1.5 ppm corresponds to 195–3 000 μg/m3, levels far from the WHO guidelines values—100 μg/m3 8-h daily maximum and 60 μg/m3 8-h mean (World Health Organization, 2021)—and not reached in real life. For particulate exposure, doses are usually more realistic but mostly match a high-exposure scenario.
A complex relationship exists between gene and environment into determining human disease phenotypes. If CF is definitely caused by a genetic defect, environmental aggressions most probably participate to the progression of CF airway disease. Epidemiological studies provide some data for a possible contribution of air pollution to CF airway severity and morbidity. In addition and although scarce, the existing experimental literature supports this possible contribution of air pollution to CF airway disease and gives an insight for possible mechanisms involved. However, it also underlies limitations, both conceptual—missing the complexity of exposome considerations—and experimental—linked to the use/evaluation of one pollutant at a time. For sure these questions need to be addressed in future research.
Conceptualization, MB and SL; methodology, MB; formal analysis, MB and VU; writing—original draft preparation, MB, PC, and SL; writing—review and editing, MB, VU, PC, PF, RE, and SL. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We want to thank the DIM Qi2 with the support of the Ile-de-France Region for funding MB. We also thank EU project #874753 REMEDIA which currently support the authors’ activity on the effects of exposome on the course of Cystic Fibrosis.
Ahmad S., Ahmad A., Dremina E. S., Sharov V. S., Guo X., Jones T. N., et al. (2009). Bcl-2 Suppresses Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase Expression in Cystic Fibrosis Airways. Am. J. Respir. Crit. Care Med. 179, 816–826. doi:10.1164/rccm.200807-1104OC
Ahmad S., Nichols D. P., Strand M., Rancourt R. C., Randell S. H., White C. W., et al. (2011). SERCA2 Regulates Non-CF and CF Airway Epithelial Cell Response to Ozone. PLOS ONE 6, e27451. doi:10.1371/journal.pone.0027451
Ahmad S., Raemy D. O., Loader J. E., Kailey J. M., Neeves K. B., White C. W., et al. (2012). Interaction and Localization of Synthetic Nanoparticles in Healthy and Cystic Fibrosis Airway Epithelial Cells: Effect of Ozone Exposure. J. Aerosol Med. Pulm. Drug Deliv. 25, 7–15. doi:10.1089/jamp.2011.0889
Andersen D. H. (1938). Cystic Fibrosis of the Pancreas and its Relation to Celiac Disease. Am. J. Dis. Child. 56, 344–399. doi:10.1001/archpedi.1938.01980140114013
Antus B. (2016). Oxidative Stress Markers in Sputum. Oxid. Med. Cell Longev. 2016, 2930434. doi:10.1155/2016/2930434
Awji E. G., Chand H., Bruse S., Smith K. R., Colby J. K., Mebratu Y., et al. (2015). Wood Smoke Enhances Cigarette Smoke-Induced Inflammation by Inducing the Aryl Hydrocarbon Receptor Repressor in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 52, 377–386. doi:10.1165/rcmb.2014-0142OC
Baker E., Harris W. T., Rowe S. M., Rutland S. B., Oates G. R. (2021). Tobacco Smoke Exposure Limits the Therapeutic Benefit of Tezacaftor/ivacaftor in Pediatric Patients with Cystic Fibrosis. J. Cyst. Fibros. 20, 612–617. doi:10.1016/j.jcf.2020.09.011
Bayram H., Devalia J. L., Sapsford R. J., Ohtoshi T., Miyabara Y., Sagai M., et al. (1998). The Effect of Diesel Exhaust Particles on Cell Function and Release of Inflammatory Mediators from Human Bronchial Epithelial CellsIn Vitro. Am. J. Respir. Cell Mol. Biol. 18, 441–448. doi:10.1165/ajrcmb.18.3.2882
Bebok Z., Varga K., Hicks J. K., Venglarik C. J., Kovacs T., Chen L., et al. (2002). Reactive Oxygen Nitrogen Species Decrease Cystic Fibrosis Transmembrane Conductance Regulator Expression and cAMP-Mediated Cl− Secretion in Airway Epithelia. J. Biol. Chem. 277, 43041–43049. doi:10.1074/jbc.M203154200
Bolon B., Haschek W. M. (2020). The Exposome in Toxicologic Pathology. Toxicol. Pathol. 48, 718–720. doi:10.1177/0192623320912403
Boucher R. C. (2007). Airway Surface Dehydration in Cystic Fibrosis: Pathogenesis and Therapy. Annu. Rev. Med. 58, 157–170. doi:10.1146/annurev.med.58.071905.105316
Briottet M., Shum M., Urbach V. (2020). The Role of Specialized Pro-resolving Mediators in Cystic Fibrosis Airways Disease. Front. Pharmacol. 11, 1290. doi:10.3389/fphar.2020.01290
Brugha R., Edmondson C., Davies J. C. (2018). Outdoor Air Pollution and Cystic Fibrosis. Paediatr. Respir. Rev. 28, 80–86. doi:10.1016/j.prrv.2018.03.005
Burke W., Aitken M. L., Chen S.-H., Scott C. R. (1992). Variable Severity of Pulmonary Disease in Adults with Identical Cystic Fibrosis Mutations. Chest 102, 506–509. doi:10.1378/chest.102.2.506
Campbell P. W., Parker R. A., Roberts B. T., Krishnamani M. R. S., Phillips J. A. (1992). Association of Poor Clinical Status and Heavy Exposure to Tobacco Smoke in Patients with Cystic Fibrosis Who Are Homozygous for the F508 Deletion. J. Pediatr. 120, 261–264. doi:10.1016/s0022-3476(05)80438-x
Cantin A. M., Bilodeau G., Ouellet C., Liao J., Hanrahan J. W. (2006a). Oxidant Stress Suppresses CFTR Expression. Am. J. Physiology-Cell Physiology 290, C262–C270. doi:10.1152/ajpcell.00070.2005
Cantin A. M., Hanrahan J. W., Bilodeau G., Ellis L., Dupuis A., Liao J., et al. (2006b). Cystic Fibrosis Transmembrane Conductance Regulator Function Is Suppressed in Cigarette Smokers. Am. J. Respir. Crit. Care Med. 173, 1139–1144. doi:10.1164/rccm.200508-1330OC
Castellani C., Southern K. W., Brownlee K., Dankert Roelse J., Duff A., Farrell M., et al. (2009). European Best Practice Guidelines for Cystic Fibrosis Neonatal Screening. J. Cyst. Fibros. 8, 153–173. doi:10.1016/j.jcf.2009.01.004
Castellani S., Di Gioia S., di Toma L., Conese M. (2018). Human Cellular Models for the Investigation of Lung Inflammation and Mucus Production in Cystic Fibrosis. Anal. Cell. Pathol. 2018, 1–15. doi:10.1155/2018/3839803
Cho H., Hotchkiss J. A., Harkema J. R. (1999). Inflammatory and Epithelial Responses during the Development of Ozone- Induced Mucous Cell Metaplasia in the Nasal Epithelium of Rats. Toxicol. Sci. 51, 135–145. doi:10.1093/toxsci/51.1.135
Clunes L. A., Davies C. M., Coakley R. D., Aleksandrov A. A., Henderson A. G., Zeman K. L., et al. (2012). Cigarette Smoke Exposure Induces CFTR Internalization and Insolubility, Leading to Airway Surface Liquid Dehydration. FASEB J. 26, 533–545. doi:10.1096/fj.11-192377
Codagnone M., Cianci E., Lamolinara A., Mari V. C., Nespoli A., Isopi E., et al. (2018). Resolvin D1 Enhances the Resolution of Lung Inflammation Caused by Long-Term Pseudomonas aeruginosa Infection. Mucosal Immunol. 11, 35–49. doi:10.1038/mi.2017.36
Coll P., Cazaunau M., Boczkowski J., Zysman M., Doussin J.-F., Gratien A., et al. (2018). Pollurisk: an Innovative Experimental Platform to Investigate Health Impacts of Air Quality. WIT Trans. Ecol. Environ. 230, 557–565. doi:10.2495/AIR180521
Collaco J. M., Blackman S. M., McGready J., Naughton K. M., Cutting G. R. (2010). Quantification of the Relative Contribution of Environmental and Genetic Factors to Variation in Cystic Fibrosis Lung Function. J. Pediatr. 157, 802–807. e3. doi:10.1016/j.jpeds.2010.05.018
Collaco J. M., McGready J., Green D. M., Naughton K. M., Watson C. P., Shields T., et al. (2011). Effect of Temperature on Cystic Fibrosis Lung Disease and Infections: A Replicated Cohort Study. PLOS ONE 6, e27784. doi:10.1371/journal.pone.0027784
Collaco J. M., Vanscoy L., Bremer L., McDougal K., Blackman S. M., Bowers A., et al. (2008). Interactions between Secondhand Smoke and Genes that Affect Cystic Fibrosis Lung Disease. JAMA 299, 417–424. doi:10.1001/jama.299.4.417
Corvol H., Blackman S. M., Boëlle P.-Y., Gallins P. J., Pace R. G., Stonebraker J. R., et al. (2015). Genome-wide Association Meta-Analysis Identifies Five Modifier Loci of Lung Disease Severity in Cystic Fibrosis. Nat. Commun. 6, 8382. doi:10.1038/ncomms9382
Cutting G. R. (2015). Cystic Fibrosis Genetics: from Molecular Understanding to Clinical Application. Nat. Rev. Genet. 16, 45–56. doi:10.1038/nrg3849
Dadvand P., Parker J., Bell M. L., Bonzini M., Brauer M., Darrow L. A., et al. (2013). Maternal Exposure to Particulate Air Pollution and Term Birth Weight: A Multi-Country Evaluation of Effect and Heterogeneity. Environ. Health Perspect. 121, 267–373. doi:10.1289/ehp.1205575
Dagher Z., Garçon G., Billet S., Gosset P., Ledoux F., Courcot D., et al. (2006). Activation of Different Pathways of Apoptosis by Air Pollution Particulate Matter (PM2.5) in Human Epithelial Lung Cells (L132) in Culture. Toxicology 225, 12–24. doi:10.1016/j.tox.2006.04.038
De Boeck K., Zolin A., Cuppens H., Olesen H. V., Viviani L. (2014). The Relative Frequency of CFTR Mutation Classes in European Patients with Cystic Fibrosis. J. Cyst. Fibros. 13, 403–409. doi:10.1016/j.jcf.2013.12.003
de Boer K., Vandemheen K. L., Tullis E., Doucette S., Fergusson D., Freitag A., et al. (2011). Exacerbation Frequency and Clinical Outcomes in Adult Patients with Cystic Fibrosis. Thorax 66, 680–685. doi:10.1136/thx.2011.161117
de Oliveira Alves N., Martins Pereira G., Di Domenico M., Costanzo G., Benevenuto S., de Oliveira Fonoff A. M., et al. (2020). Inflammation Response, Oxidative Stress and DNA Damage Caused by Urban Air Pollution Exposure Increase in the Lack of DNA Repair XPC Protein. Environ. Int. 145, 106150. doi:10.1016/j.envint.2020.106150
Delfino R. J., Staimer N., Tjoa T., Arhami M., Polidori A., Gillen D. L., et al. (2010). Associations of Primary and Secondary Organic Aerosols with Airway and Systemic Inflammation in an Elderly Panel Cohort. Epidemiology 21, 892–902. doi:10.1097/EDE.0b013e3181f20e6c
Diaz E. A., Chung Y., Papapostolou V., Lawrence J., Long M. S., Hatakeyama V., et al. (2012). Effects of Fresh and Aged Vehicular Exhaust Emissions on Breathing Pattern and Cellular Responses - Pilot Single Vehicle Study. Inhal. Toxicol. 24, 288–295. doi:10.3109/08958378.2012.668572
Dorin J. R., Dickinson P., Alton E. W. F. W., Smith S. N., Geddes D. M., Stevenson B. J., et al. (1992). Cystic Fibrosis in the Mouse by Targeted Insertional Mutagenesis. Nature 359, 211–215. doi:10.1038/359211a0
Dorin J. R., Stevenson B. J., Fleming S., Alton E. W. F. W., Dickinson P., Porteous D. J. (1994). Long-term Survival of the Exon 10 Insertional Cystic Fibrosis Mutant Mouse Is a Consequence of Low Level Residual Wild-type Cftr Gene Expression. Mamm. Genome 5, 465–472. doi:10.1007/bf00369314
Duguépéroux I., De Braekeleer M. (2004). De Braekeleer, M., and Participating Centres to the French National Cystic Fibrosis RegistryGenotype-Phenotype Relationship for Five CFTR Mutations Frequently Identified in Western France. J. Cyst. Fibros. 3, 259–263. doi:10.1016/j.jcf.2004.07.004
Eickmeier O., Fussbroich D., Mueller K., Serve F., Smaczny C., Zielen S., et al. (2017). Pro-resolving Lipid Mediator Resolvin D1 Serves as a Marker of Lung Disease in Cystic Fibrosis. PLoS One 12, e0171249. doi:10.1371/journal.pone.0171249
Farhat S. C. L., Almeida M. B., Silva-Filho L. V. R. F., Farhat J., Rodrigues J. C., Braga A. L. F. (2013). Ozone Is Associated with an Increased Risk of Respiratory Exacerbations in Patients with Cystic Fibrosis. Chest 144, 1186–1192. doi:10.1378/chest.12-2414
Ferkol T., Rosenfeld M., Milla C. E. (2006). Cystic Fibrosis Pulmonary Exacerbations. J. Pediatr. 148, 259–264. doi:10.1016/j.jpeds.2005.10.019
Fleischer N. L., Merialdi M., van Donkelaar A., Vadillo-Ortega F., Martin R. V., Betran A. P., et al. (2014). Betran Ana Pilar, et alOutdoor Air Pollution, Preterm Birth, and Low Birth Weight: Analysis of the World Health Organization Global Survey on Maternal and Perinatal Health. Environ. Health Perspect. 122, 425–430. doi:10.1289/ehp.1306837
Gangwar R. S., Bevan G. H., Palanivel R., Das L., Rajagopalan S. (2020). Oxidative Stress Pathways of Air Pollution Mediated Toxicity: Recent Insights. Redox Biol. 34, 101545. doi:10.1016/j.redox.2020.101545
Gaschen A., Lang D., Kalberer M., Savi M., Geiser T., Gazdhar A., et al. (2010). Cellular Responses after Exposure of Lung Cell Cultures to Secondary Organic Aerosol Particles. Environ. Sci. Technol. 44, 1424–1430. doi:10.1021/es902261m
Gauderman W. J., Vora H., McConnell R., Berhane K., Gilliland F., Thomas D., et al. (2007). Effect of Exposure to Traffic on Lung Development from 10 to 18 Years of Age: a Cohort Study. Lancet 369, 571–577. doi:10.1016/S0140-6736(07)60037-3
Geiser M., Quaile O., Wenk A., Wigge C., Eigeldinger-Berthou S., Hirn S., et al. (2013). Cellular Uptake and Localization of Inhaled Gold Nanoparticles in Lungs of Mice with Chronic Obstructive Pulmonary Disease. Part. Fibre Toxicol. 10, 19. doi:10.1186/1743-8977-10-19
Geiser M., Stoeger T., Casaulta M., Chen S., Semmler-Behnke M., Bolle I., et al. (2014). Biokinetics of Nanoparticles and Susceptibility to Particulate Exposure in a Murine Model of Cystic Fibrosis. Part. Fibre Toxicol. 11, 19. doi:10.1186/1743-8977-11-19
Gilljam H., Stenlund C., Ericsson-Hollsing A., Strandvik B. (1990). Passive Smoking in Cystic Fibrosis. Respir. Med. 84, 289–291. doi:10.1016/s0954-6111(08)80055-8
Goeminne P. C., Kiciński M., Vermeulen F., Fierens F., De Boeck K., Nemery B., et al. (2013). Impact of Air Pollution on Cystic Fibrosis Pulmonary Exacerbations. Chest 143, 946–954. doi:10.1378/chest.12-1005
Goldizen F. C., Sly P. D., Knibbs L. D. (2016). Respiratory Effects of Air Pollution on Children. Pediatr. Pulmonol. 51, 94–108. doi:10.1002/ppul.23262
Goss C. H., Newsom S. A., Schildcrout J. S., Sheppard L., Kaufman J. D. (2004). Effect of Ambient Air Pollution on Pulmonary Exacerbations and Lung Function in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 169, 816–821. doi:10.1164/rccm.200306-779OC
Harkema J. R., Hotchkiss J. A., Barr E. B., Bennett C. B., Gallup M., Lee J. K., et al. (1999). Long-Lasting Effects of Chronic Ozone Exposure on Rat Nasal Epithelium. Am. J. Respir. Cell Mol. Biol. 20, 517–529. doi:10.1165/ajrcmb.20.3.3227
Hoek G., Krishnan R. M., Beelen R., Peters A., Ostro B., Brunekreef B., et al. (2013). Long-term Air Pollution Exposure and Cardio- Respiratory Mortality: a Review. Environ. Health 12, 43. doi:10.1186/1476-069X-12-43
Holton J. R., Haynes P. H., McIntyre M. E., Douglass A. R., Rood R. B., Pfister L. (1995). Stratosphere-troposphere Exchange. Rev. Geophys. 33, 403–439. doi:10.1029/95rg02097
Huang L., Pu J., He F., Liao B., Hao B., Hong W., et al. (2017). Positive Feedback of the Amphiregulin-EGFR-ERK Pathway Mediates PM2.5 from Wood Smoke-Induced MUC5AC Expression in Epithelial Cells. Sci. Rep. 7, 11084. doi:10.1038/s41598-017-11541-1
Hwang B.-F., Chen Y.-H., Lin Y.-T., Wu X.-T., Leo Lee Y. (2015). Relationship between Exposure to Fine Particulates and Ozone and Reduced Lung Function in Children. Environ. Res. 137, 382–390. doi:10.1016/j.envres.2015.01.009
International Agency for Research on Cancer (2013). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 109. Outdoor air pollution.
Isopi E., Mattoscio D., Codagnone M., Mari V. C., Lamolinara A., Patruno S., et al. (2020). Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 11, 581. doi:10.3389/fimmu.2020.00581
Jany B., Basbaum C. B. (1991). Modification of Mucin Gene Expression in Airway Disease. Am. Rev. Respir. Dis. 144, S38–S41. doi:10.1164/ajrccm/144.3_pt_2.S38
Jassal M. S., Yu A. M., Bhatia R., Keens T. G., Davidson Ward S. L. (2013). Effect of Residential Proximity to Major Roadways on Cystic Fibrosis Exacerbations. Int. J. Environ. Health Res. 23, 119–131. doi:10.1080/09603123.2012.708917
Jeannet N., Fierz M., Schneider S., Künzi L., Baumlin N., Salathe M., et al. (2016). Acute Toxicity of Silver and Carbon Nanoaerosols to Normal and Cystic Fibrosis Human Bronchial Epithelial Cells. Nanotoxicology 10, 279–291. doi:10.3109/17435390.2015.1049233
Kamdar O., Le W., Zhang J., Ghio A. J., Rosen G. D., Upadhyay D. (2008). Air Pollution Induces Enhanced Mitochondrial Oxidative Stress in Cystic Fibrosis Airway Epithelium. FEBS Lett. 582, 3601–3606. doi:10.1016/j.febslet.2008.09.030
Karp C. L., Flick L. M., Park K. W., Softic S., Greer T. M., Keledjian R., et al. (2004). Defective Lipoxin-Mediated Anti-inflammatory Activity in the Cystic Fibrosis Airway. Nat. Immunol. 5, 388–392. doi:10.1038/ni1056
Kerem E., Kerem B. (1996). Genotype-phenotype Correlations in Cystic Fibrosis. Pediatr. Pulmonol. 22, 387–395. doi:10.1002/(sici)1099-0496(199612)22:6<387::aid-ppul7>3.0.co;2-g
Kilburg-Basnyat B., Reece S. W., Crouch M. J., Luo B., Boone A. D., Yaeger M., et al. (2018). Specialized Pro-resolving Lipid Mediators Regulate Ozone-Induced Pulmonary and Systemic Inflammation. Toxicol. Sci. 163, 466–477. doi:10.1093/toxsci/kfy040
Kopp B., Ortega-García J., Sadreameli S., Wellmerling J., Cormet-Boyaka E., Thompson R., et al. (2016). The Impact of Secondhand Smoke Exposure on Children with Cystic Fibrosis: A Review. Ijerph 13, 1003. doi:10.3390/ijerph13101003
Kopp B. T., Sarzynski L., Khalfoun S., Hayes D., Thompson R., Nicholson L., et al. (2015). Detrimental Effects of Secondhand Smoke Exposure on Infants with Cystic Fibrosis. Pediatr. Pulmonol. 50, 25–34. doi:10.1002/ppul.23016
Kovesi T., Corey M., Levison H. (1993). Passive Smoking and Lung Function in Cystic Fibrosis. Am. Rev. Respir. Dis. 148, 1266–1271. doi:10.1164/ajrccm/148.5.1266
Kracht O., Santiago J., Martin F., Piersanti A., Cremona G., Righini G., et al. (2018). Spatial Representativeness of Air Quality Monitoring Sites: Outcomes of the FAIRMODE/AQUILA Intercomparison Exercise. Luxembourg: EUR 28987 EN, Publications Office of the European Union. doi:10.2760/60611
Krapf M., Künzi L., Allenbach S., Bruns E. A., Gavarini I., El-Haddad I., et al. (2017). Wood Combustion Particles Induce Adverse Effects to Normal and Diseased Airway Epithelia. Environ. Sci. Process. Impacts 19, 538–548. doi:10.1039/C6EM00586A
Künzi L., Krapf M., Daher N., Dommen J., Jeannet N., Schneider S., et al. (2015). Toxicity of Aged Gasoline Exhaust Particles to Normal and Diseased Airway Epithelia. Sci. Rep. 5, 1–10. doi:10.1038/srep11801
Künzi L., Mertes P., Schneider S., Jeannet N., Menzi C., Dommen J., et al. (2013). Responses of Lung Cells to Realistic Exposure of Primary and Aged Carbonaceous Aerosols. Atmos. Environ. 68, 143–150. doi:10.1016/j.atmosenv.2012.11.055
Lao O., Andrés A. M., Mateu E., Bertranpetit J., Calafell F. (2003). Spatial Patterns of Cystic Fibrosis Mutation Spectra in European Populations. Eur. J. Hum. Genet. 11, 385–394. doi:10.1038/sj.ejhg.5200970
Lee H., Myung W., Jeong B.-H., Choi H., Jhun B. W., Kim H. (2018). Short- and Long-Term Exposure to Ambient Air Pollution and Circulating Biomarkers of Inflammation in Non-smokers: A Hospital-Based Cohort Study in South Korea. Environ. Int. 119, 264–273. doi:10.1016/j.envint.2018.06.041
Lelieveld J., Pozzer A., Pöschl U., Fnais M., Haines A., Münzel T. (2020). Loss of Life Expectancy from Air Pollution Compared to Other Risk Factors: a Worldwide Perspective. Cardiovasc. Res. 116, 1910–1917. doi:10.1093/cvr/cvaa025
Leni Z., Cassagnes L. E., Daellenbach K. R., El Haddad I., Vlachou A., Uzu G., et al. (2020). Oxidative Stress-Induced Inflammation in Susceptible Airways by Anthropogenic Aerosol. PLoS ONE 15, e0233425. doi:10.1371/journal.pone.0233425
Li Z., Liu Q., Xu Z., Guo X., Wu S. (2020). Association between Short-Term Exposure to Ambient Particulate Air Pollution and Biomarkers of Oxidative Stress: A Meta-Analysis. Environ. Res. 191, 110105. doi:10.1016/j.envres.2020.110105
Liu N. M., Miyashita L., Sanak M., Barratt B., Grigg J. (2021). Prostaglandin E2 and Phagocytosis of Inhaled Particulate Matter by Airway Macrophages in Cystic Fibrosis. J. Cyst. Fibros. 20 (4), 673–677. doi:10.1016/j.jcf.2020.11.010
Lu X., Fu H., Han F., Fang Y., Xu J., Zhang L., et al. (2018). Lipoxin A4 Regulates PM2.5-induced Severe Allergic Asthma in Mice via the Th1/Th2 Balance of Group 2 Innate Lymphoid Cells. J. Thorac. Dis. 10, 1449–1459. doi:10.21037/jtd.2018.03.02
MacNee W., Donaldson K. (2003). Mechanism of Lung Injury Caused by PM10 and Ultrafine Particles with Special Reference to COPD. Eur. Respir. J. 21, 47s–51s. doi:10.1183/09031936.03.00403203
Mall M., Grubb B. R., Harkema J. R., O'Neal W. K., Boucher R. C. (2004). Increased Airway Epithelial Na+ Absorption Produces Cystic Fibrosis-like Lung Disease in Mice. Nat. Med. 10, 487–493. doi:10.1038/nm1028
Marklew A. J., Patel W., Moore P. J., Tan C. D., Smith A. J., Sassano M. F., et al. (2019). Cigarette Smoke Exposure Induces Retrograde Trafficking of CFTR to the Endoplasmic Reticulum. Sci. Rep. 9, 13655. doi:10.1038/s41598-019-49544-9
McCarthy J., Gong X., Nahirney D., Duszyk M., Radomski M. (2011). Polystyrene Nanoparticles Activate Ion Transport in Human Airway Epithelial Cells. Ijn 6, 1343–1356. doi:10.2147/IJN.S21145
McCormick J., Mehta G., Olesen H. V., Viviani L., Macek M., Mehta A., et al. (2010). Comparative Demographics of the European Cystic Fibrosis Population: a Cross-Sectional Database Analysis. Lancet 375, 1007–1013. doi:10.1016/S0140-6736(09)62161-9
Milanzi E. B., Koppelman G. H., Smit H. A., Wijga A. H., Oldenwening M., Vonk J. M., et al. (2018). Air Pollution Exposure and Lung Function until Age 16 years: the PIAMA Birth Cohort Study. Eur. Respir. J. 52, 1800218. doi:10.1183/13993003.00218-2018
Mirtajani S. B., Farnia P., Hassanzad M., Ghanavi J. (2017). Geographical Distribution of Cystic Fibrosis. past 70 years data Anal. 1, 105–112. doi:10.4103/bbrj.bbrj_81_17
Montoro D. T., Haber A. L., Biton M., Vinarsky V., Lin B., Birket S. E., et al. (2018). A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature 560, 319–324. doi:10.1038/s41586-018-0393-7
Nguyen J. P., Huff R. D., Cao Q. T., Tiessen N., Carlsten C., Hirota J. A. (2021). Effects of Environmental Air Pollutants on CFTR Expression and Function in Human Airway Epithelial Cells. Toxicol. Vitro 77, 105253. doi:10.1016/j.tiv.2021.105253
Nissim-Rafinia M., Kerem B. (2005). The Splicing Machinery Is a Genetic Modifier of Disease Severity. Trends Genet. 21, 480–483. doi:10.1016/j.tig.2005.07.005
Oates G. R., Schechter M. S. (2016). Socioeconomic Status and Health Outcomes: Cystic Fibrosis as a Model. Expert Rev. Respir. Med. 10, 967–977. doi:10.1080/17476348.2016.1196140
Paulin L., Hansel N. (2016). Particulate Air Pollution and Impaired Lung Function. F1000Res 5, 201. doi:10.12688/f1000research.7108.1
Pittman J. E., Calloway E. H., Kiser M., Yeatts J., Davis S. D., Drumm M. L., et al. (2011). Age of Pseudomonas aeruginosa Acquisition and Subsequent Severity of Cystic Fibrosis Lung Disease. Pediatr. Pulmonol. 46, 497–504. doi:10.1002/ppul.21397
Plasschaert L. W., Žilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., et al. (2018). A Single-Cell Atlas of the Airway Epithelium Reveals the CFTR-Rich Pulmonary Ionocyte. Nature 560, 377–381. doi:10.1038/s41586-018-0394-6
Psoter K. J., De Roos A. J., Mayer J. D., Kaufman J. D., Wakefield J., Rosenfeld M. (2015). Fine Particulate Matter Exposure and InitialPseudomonas aeruginosaAcquisition in Cystic Fibrosis. Ann. ATS 12, 385–391. doi:10.1513/AnnalsATS.201408-400OC
Psoter K. J., De Roos A. J., Wakefield J., Mayer J. D., Rosenfeld M. (2017). Air Pollution Exposure Is Associated with MRSA Acquisition in Young U.S. Children with Cystic Fibrosis. BMC Pulm. Med. 17, 106. doi:10.1186/s12890-017-0449-8
Psoter K. J., De Roos A. J., Wakefield J., Mayer J., Rosenfeld M. (2013). Season Is Associated with Pseudomonas aeruginosa Acquisition in Young Children with Cystic Fibrosis. Clin. Microbiol. Infect. 19, E483–E489. doi:10.1111/1469-0691.12272
Qu F., Qin X.-Q., Cui Y.-R., Xiang Y., Tan Y.-R., Liu H.-J., et al. (2009). Ozone Stress Down-Regulates the Expression of Cystic Fibrosis Transmembrane Conductance Regulator in Human Bronchial Epithelial Cells. Chemico-Biological Interact. 179, 219–226. doi:10.1016/j.cbi.2008.10.059
Raaschou-Nielsen O., Andersen Z. J., Beelen R., Samoli E., Stafoggia M., Weinmayr G., et al. (2013). Air Pollution and Lung Cancer Incidence in 17 European Cohorts: Prospective Analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 14, 813–822. doi:10.1016/S1470-2045(13)70279-1
Rager J. E., Lichtveld K., Ebersviller S., Smeester L., Jaspers I., Sexton K. G., et al. (2011). A Toxicogenomic Comparison of Primary and Photochemically Altered Air Pollutant Mixtures. Environ. Health Perspect. 119, 1583–1589. doi:10.1289/ehp.1003323
Raju S. V., Jackson P. L., Courville C. A., McNicholas C. M., Sloane P. A., Sabbatini G., et al. (2013). Cigarette Smoke Induces Systemic Defects in Cystic Fibrosis Transmembrane Conductance Regulator Function. Am. J. Respir. Crit. Care Med. 188, 1321–1330. doi:10.1164/rccm.201304-0733OC
Raju S. V., Lin V. Y., Liu L., McNicholas C. M., Karki S., Sloane P. A., et al. (2017). The Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor Augments Mucociliary Clearance Abrogating Cystic Fibrosis Transmembrane Conductance Regulator Inhibition by Cigarette Smoke. Am. J. Respir. Cell Mol. Biol. 56, 99–108. doi:10.1165/rcmb.2016-0226OC
Ramsay K., Stockwell R., Bell S., Kidd T. (2016). Infection in Cystic Fibrosis: Impact of the Environment and Climate. Expert Rev. Respir. Med. 10, 505–519. doi:10.1586/17476348.2016.1162715
Rasmussen L. W., Stanford D., Patel K., Raju S. V. (2020). Evaluation of Secondhand Smoke Effects on CFTR Function In Vivo. Respir. Res. 21, 70. doi:10.1186/s12931-020-1324-3
Ravishankara A. R. (2003). Introduction: Atmospheric ChemistryLong-Term Issues. Chem. Rev. 103, 4505–4508. doi:10.1021/cr020463i
Raynal C., Corvol H. (2020). Variant Classifications, Databases and Genotype-Phenotype Correlations. Arch. Pédiatrie 27, eS13–eS18. doi:10.1016/S0929-693X(20)30045-2
Ringholz F. C., Buchanan P. J., Clarke D. T., Millar R. G., McDermott M., Linnane B., et al. (2014). Reduced 15-lipoxygenase 2 and Lipoxin A4/leukotriene B4 Ratio in Children with Cystic Fibrosis. Eur. Respir. J. 44, 394–404. doi:10.1183/09031936.00106013
Ringholz F. C., Higgins G., Hatton A., Sassi A., Moukachar A., Fustero-Torre C., et al. (2018). Resolvin D1 Regulates Epithelial Ion Transport and Inflammation in Cystic Fibrosis Airways. J. Cyst. Fibros. 17, 607–615. doi:10.1016/j.jcf.2017.11.017
Riordan J. R., Rommens J. M., Kerem B.-S., Alon N., Rozmahel R., Grzelczak Z., et al. (1989). Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 245, 1066–1073. doi:10.1126/science.2475911
Rodriguez D., Valari M., Payan S., Eymard L. (2019). On the Spatial Representativeness of NOX and PM10 Monitoring-Sites in Paris, France. Atmos. Environ. X 1, 100010. doi:10.1016/j.aeaoa.2019.100010
Roesch E. A., Nichols D. P., Chmiel J. F. (2018). Inflammation in Cystic Fibrosis: An Update. Pediatr. Pulmonol. 53, S30–S50. doi:10.1002/ppul.24129
Rommens J. M. (2011). Cystic Fibrosis Mutation Database. Available at: http://www.genet.sickkids.on.ca/StatisticsPage.html (Accessed January 30, 2021).
Rosenfeld M., Gibson R. L., McNamara S., Emerson J., Burns J. L., Castile R., et al. (2001). Early Pulmonary Infection, Inflammation, and Clinical Outcomes in Infants with Cystic Fibrosis. Pediatr. Pulmonol. 32, 356–366. doi:10.1002/ppul.1144
Rottner M., Kunzelmann C., Mergey M., Freyssinet J. M., Martínez M. C. (2007). Exaggerated Apoptosis andNF‐KBactivation in Pancreatic and Tracheal Cystic Fibrosis Cells. FASEB J. 21, 2939–2948. doi:10.1096/fj.06-7614com
Rottner M., Tual-Chalot S., Mostefai H. A., Andriantsitohaina R., Freyssinet J.-M., Martínez M. C. (2011). Increased Oxidative Stress Induces Apoptosis in Human Cystic Fibrosis Cells. PLOS ONE 6, e24880. doi:10.1371/journal.pone.0024880
Rubin B. K. (1990). Exposure of Children with Cystic Fibrosis to Environmental Tobacco Smoke. N. Engl. J. Med. 323, 782–788. doi:10.1056/NEJM199009203231203
Sagel S. D., Wagner B. D., Anthony M. M., Emmett P., Zemanick E. T. (2012). Sputum Biomarkers of Inflammation and Lung Function Decline in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 186, 857–865. doi:10.1164/rccm.201203-0507OC
Saldiva P. H. N., King M., Delmonte V. L. C., Macchione M., Parada M. A. C., Daliberto M. L., et al. (1992). Respiratory Alterations Due to Urban Air Pollution: an Experimental Study in Rats. Environ. Res. 57, 19–33. doi:10.1016/s0013-9351(05)80016-7
Sanders D. B., Bittner R. C. L., Rosenfeld M., Hoffman L. R., Redding G. J., Goss C. H. (2010). Failure to Recover to Baseline Pulmonary Function after Cystic Fibrosis Pulmonary Exacerbation. Am. J. Respir. Crit. Care Med. 182, 627–632. doi:10.1164/rccm.200909-1421OC
Sanders D. B., Bittner R. C., Rosenfeld M., Redding G. J., Goss C. H. (2011). Pulmonary Exacerbations Are Associated with Subsequent FEV1 Decline in Both Adults and Children with Cystic Fibrosis. Pediatr. Pulmonol. 46, 393–400. doi:10.1002/ppul.21374
Schraufnagel D. E. (2020). The Health Effects of Ultrafine Particles. Exp. Mol. Med. 52, 311–317. doi:10.1038/s12276-020-0403-3
Scudieri P., Musante I., Venturini A., Guidone D., Genovese M., Cresta F., et al. (2020). Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 9, 2090. doi:10.3390/cells9092090
Seinfeld J. H., Pandis S. N. (2016). Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. 3rd Edition. New-York: John Wiley & Sons.
Semaniakou A., Croll R. P., Chappe V. (2019). Animal Models in the Pathophysiology of Cystic Fibrosis. Front. Pharmacol. 9, 1475. doi:10.3389/fphar.2018.01475
Sepahzad A., Morris-Rosendahl D. J., Davies J. C. (2021). Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes 12, 562. doi:10.3390/genes12040562
Shi R., Su W.-W., Zhu Z.-T., Guan M.-Y., Cheng K.-L., Fan W.-Y., et al. (2019). Regulation Effects of Naringin on Diesel Particulate Matter-Induced Abnormal Airway Surface Liquid Secretion. Phytomedicine 63, 153004. doi:10.1016/j.phymed.2019.153004
Shteinberg M., Haq I. J., Polineni D., Davies J. C. (2021). Cystic Fibrosis. Lancet 397, 2195–2211. doi:10.1016/S0140-6736(20)32542-3
Sloane P. A., Shastry S., Wilhelm A., Courville C., Tang L. P., Backer K., et al. (2012). A Pharmacologic Approach to Acquired Cystic Fibrosis Transmembrane Conductance Regulator Dysfunction in Smoking Related Lung Disease. PLoS One 7, e39809. doi:10.1371/journal.pone.0039809
Smyth A., O'Hea U., Williams G., Smyth R., Heaf D. (1994). Passive Smoking and Impaired Lung Function in Cystic Fibrosis. Archives Dis. Child. 71, 353–354. doi:10.1136/adc.71.4.353
Stanford G. E., Dave K., Simmonds N. J. (2021). Pulmonary Exacerbations in Adults with Cystic Fibrosis. CHEST 159, 93–102. doi:10.1016/j.chest.2020.09.084
Stieb D. M., Chen L., Eshoul M., Judek S. (2012). Ambient Air Pollution, Birth Weight and Preterm Birth: a Systematic Review and Meta-Analysis. Environ. Res. 117, 100–111. doi:10.1016/j.envres.2012.05.007
Sueyoshi S., Miyata Y., Masumoto Y., Ishibashi Y., Matsuzawa S., Harano N., et al. (2004). Reduced Airway Inflammation and Remodeling in Parallel with Mucin 5AC Protein Expression Decreased by S-Carboxymethylcysteine, a Mucoregulant, in the Airways of Rats Exposed to Sulfur Dioxide. Int. Arch. Allergy Immunol. 134, 273–280. doi:10.1159/000079164
Szczesniak R., Rice J. L., Brokamp C., Ryan P., Pestian T., Ni Y., et al. (2020). Influences of Environmental Exposures on Individuals Living with Cystic Fibrosis. Expert Rev. Respir. Med. 14, 737–748. doi:10.1080/17476348.2020.1753507
Thatcher T. H., Woeller C. F., McCarthy C. E., Sime P. J. (2019). Quenching the Fires: Pro-resolving Mediators, Air Pollution, and Smoking. Pharmacol. Ther. 197, 212–224. doi:10.1016/j.pharmthera.2019.02.001
The Cystic Fibrosis Genotype-Phenotype Consortium (1993). Correlation between Genotype and Phenotype in Patients with Cystic Fibrosis. N. Engl. J. Med. 329, 1308–1313. doi:10.1056/NEJM199310283291804
Thurston G. D., Balmes J. R., Garcia E., Gilliland F. D., Rice M. B., Schikowski T., et al. (2020). Outdoor Air Pollution and New-Onset Airway Disease. An Official American Thoracic Society Workshop Report. Ann. ATS 17, 387–398. doi:10.1513/AnnalsATS.202001-046ST
Verrière V., Higgins G., Al-Alawi M., Costello R. W., McNally P., Chiron R., et al. (2012). Lipoxin A4 Stimulates Calcium-Activated Chloride Currents and Increases Airway Surface Liquid Height in Normal and Cystic Fibrosis Airway Epithelia. PLoS One 7, e37746. doi:10.1371/journal.pone.0037746
Voisin G., Bouvet G. F., Legendre P., Dagenais A., Massé C., Berthiaume Y. (2014). Oxidative Stress Modulates the Expression of Genes Involved in Cell Survival in ΔF508 Cystic Fibrosis Airway Epithelial Cells. Physiol. Genomics 46, 634–646. doi:10.1152/physiolgenomics.00003.2014
Waters V., Stanojevic S., Atenafu E. G., Lu A., Yau Y., Tullis E., et al. (2012). Effect of Pulmonary Exacerbations on Long-Term Lung Function Decline in Cystic Fibrosis. Eur. Respir. J. 40, 61–66. doi:10.1183/09031936.00159111
Wayne R. P. (2000). Chemistry of Atmospheres: An Introduction to the Chemistry of the Atmospheres of Earth, the Planets, and Their Satellites. Third Edition. Oxford, New York: Oxford University Press.
Wild C. P. (2012). The Exposome: from Concept to Utility. Int. J. Epidemiol. 41, 24–32. doi:10.1093/ije/dyr236
Wilke M., Buijs-Offerman R. M., Aarbiou J., Colledge W. H., Sheppard D. N., Touqui L., et al. (2011). Mouse Models of Cystic Fibrosis: Phenotypic Analysis and Research Applications. J. Cyst. Fibros. 10 (Suppl. 2), S152–S171. doi:10.1016/S1569-1993(11)60020-9
World Health Organization (2021). Ambient (Outdoor) Air Pollution. Available at: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health.
Zhou Z., Duerr J., Johannesson B., Schubert S. C., Treis D., Harm M., et al. (2011). The ENaC-Overexpressing Mouse as a Model of Cystic Fibrosis Lung Disease. J. Cyst. Fibros. 10 (Suppl. 2), S172–S182. doi:10.1016/S1569-1993(11)60021-0
Keywords: cystic fibrosis, air pollution, environmental factors, acute exposure, chronic exposure, CF models
Citation: Blayac M, Coll P, Urbach V, Fanen P, Epaud R and Lanone S (2022) The Impact of Air Pollution on the Course of Cystic Fibrosis: A Review. Front. Physiol. 13:908230. doi: 10.3389/fphys.2022.908230
Received: 30 March 2022; Accepted: 13 May 2022;
Published: 02 June 2022.
Edited by:
Amy L. Ryan, The University of Iowa, United StatesReviewed by:
Steven Molinski, University of Toronto, CanadaCopyright © 2022 Blayac, Coll, Urbach, Fanen, Epaud and Lanone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Lanone, c29waGllLmxhbm9uZUBpbnNlcm0uZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.