
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 13 June 2022
Sec. Renal Physiology and Pathophysiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.886779
This article is part of the Research Topic Inflammation in Hypertensive Disorders View all 5 articles
 Sarika Chaudhari†
Sarika Chaudhari† Grace S. Pham†
Grace S. Pham† Calvin D. Brooks
Calvin D. Brooks Viet Q. Dinh
Viet Q. Dinh Cassandra M. Young-Stubbs
Cassandra M. Young-Stubbs Caroline G. Shimoura
Caroline G. Shimoura Keisa W. Mathis*
Keisa W. Mathis*Despite extensive research and a plethora of therapeutic options, hypertension continues to be a global burden. Understanding of the pathological roles of known and underexplored cellular and molecular pathways in the development and maintenance of hypertension is critical to advance the field. Immune system overactivation and inflammation in the kidneys are proposed alternative mechanisms of hypertension, and resistant hypertension. Consideration of the pathophysiology of hypertension in chronic inflammatory conditions such as autoimmune diseases, in which patients present with autoimmune-mediated kidney inflammation as well as hypertension, may reveal possible contributors and novel therapeutic targets. In this review, we 1) summarize current therapies used to control blood pressure and their known effects on inflammation; 2) provide evidence on the need to target renal inflammation, specifically, and especially when first-line and combinatory treatment efforts fail; and 3) discuss the efficacy of therapies used to treat autoimmune diseases with a hypertension/renal component. We aim to elucidate the potential of targeting renal inflammation in certain subsets of patients resistant to current therapies.
Hypertension remains the leading modifiable risk factor for cardiovascular morbidity, kidney disease, stroke, and premature death. In the last 3 decades, the global economic burden of hypertension has doubled not only due to a growing and aging population (Zhou et al., 2021), but also due to stress and a societal shift towards unhealthy lifestyles and diets. The International Society of Hypertension released new global practice guidelines in 2020, providing an updated definition of hypertension (Unger et al., 2020). The Society defined hypertension as a provider-measured blood pressure greater than 140/90 mm Hg, but recognized that 24-h ambulatory blood pressure monitoring in these individuals may show an average of greater than 130/80 mm Hg (Unger et al., 2020). The World Health Organization’s Guideline for the Pharmacological Treatment of Hypertension in Adults stresses the importance of having multiple blood pressure measurements, preferably across multiple visits, to accurately diagnose hypertension, but they recognize these conditions are not feasible in all settings (WHO, 2021). The United States continues to use hypertension guidelines set forth by the American Heart Association and American College of Cardiology in 2017 (Carey et al., 2018b). These guidelines define hypertension as blood pressure equal to or greater than 130/80 mm Hg, so that more efforts can be directed to control elevations in blood pressure before the disease progresses (Carey et al., 2018b). In the United States itself, half of the population has hypertension and only one out of four adults with hypertension have their blood pressure controlled (CDC, 2019b). Hypertension as a primary or contributing cause resulted in over 500,000 deaths in the United States in 2019 (CDC, 2019a).
Despite several advances in science, the etiology of hypertension still eludes physiologists (Ferdinand and Nasser, 2017). There are several mechanisms in place in the body to ensure blood pressure remains within physiological norms. Baroreceptors sense changes in blood pressure and then correct these changes through neural and hormonal pathways. These pathways augment heart rate, stroke volume, and total peripheral resistance, as well as control body fluid and electrolyte homeostasis (Pappano, 2010). The kidneys also have an undeniably important role in the fluid-electrolyte balance in the body through their intrinsic regulation of glomerular filtration rate and changes in electrolyte handling caused by neuroendocrine factors such as antidiuretic hormone, aldosterone, angiotensin II (ANGII), atrial natriuretic peptide, and brain natriuretic peptide (Oparil et al., 2018). Any disturbance of fluid-electrolyte homeostasis can contribute to hypertension (Coleman et al., 1975). As such, targeting water and electrolyte homeostatic systems through the kidney has been a cornerstone in the treatment of hypertension, although with limited success in some subpopulations of hypertensive patients.
It is well accepted that inflammation plays an important role in the pathogenesis of hypertension (Drummond et al., 2019). Although the role of the immune cells in hypertension has been known for decades (Okuda and Grollman, 1967; Svendsen, 1976), the direct role of inflammatory processes in the pathogenesis of hypertension was only recently appreciated. The causal role of lymph nodes and the thymus in hypertension in rodents was demonstrated in the 1960-70s (Okuda and Grollman, 1967; Svendsen, 1976). It has been confirmed that adaptive immunity with T lymphocytes [e.g., T helper (Th) cells] is an important player in modulating the inflammatory cytokine/chemokine production and hence in the immune-mediated pathogenesis of experimental hypertension in rodents (Guzik et al., 2007). Furthermore, knockout models such as B cell activating factor (BAFF) receptor knockout (BAFF-R−/−) mice or depletion of B and/or T cell activity using treatments like rituximab or mycophenolate mofetil blunt experimental hypertension in rodents (Herrera et al., 2006; Zheng et al., 2010; Chan et al., 2015; Taylor and Ryan, 2017; Taylor et al., 2018). The potential of mycophenolate mofetil to reduce blood pressure in chronic inflammatory settings has also been realized in hypertensive humans with psoriasis and rheumatoid arthritis (Herrera et al., 2006).
Cells of the innate immune system, including monocytes, macrophages, natural killer cells and dendritic cells (DCs) also contribute to hypertension (Lu and Crowley, 2020). Innate immunity is a non-specific response to antigens and causes inflammation directly through the activation of pattern recognition receptors such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors. Activation of these receptors on innate immune cells leads to increased oxidative stress while also increasing the release of various inflammatory cytokines that play important role in the development of hypertension (Mian et al., 2014; Van Beusecum et al., 2021). Indirectly they also cause activation of the adaptive immunity via the Th1 response or the humoral/antibody Th2 response, triggering further secretion of potent pro-inflammatory cytokines like interleukin (IL)-6, interferon (IFN)-γ, and IL-17, in turn contributing to the hypertension pathology (Mikolajczyk and Guzik, 2019).
Interestingly, several pharmacologic agents that reduce blood pressure have general anti-inflammatory effects as well (Silva et al., 2019). Below we discuss common therapies for hypertension and how hypertensive drugs from different classes are often used in combination to reach the target blood pressure reduction in situations where blood pressure is more difficult to control (Tsioufis and Thomopoulos, 2017). A complete list of pharmacologic anti-hypertensive therapies are summarized in Table 1. We will elaborate on those showing effects on the inflammation specifically.
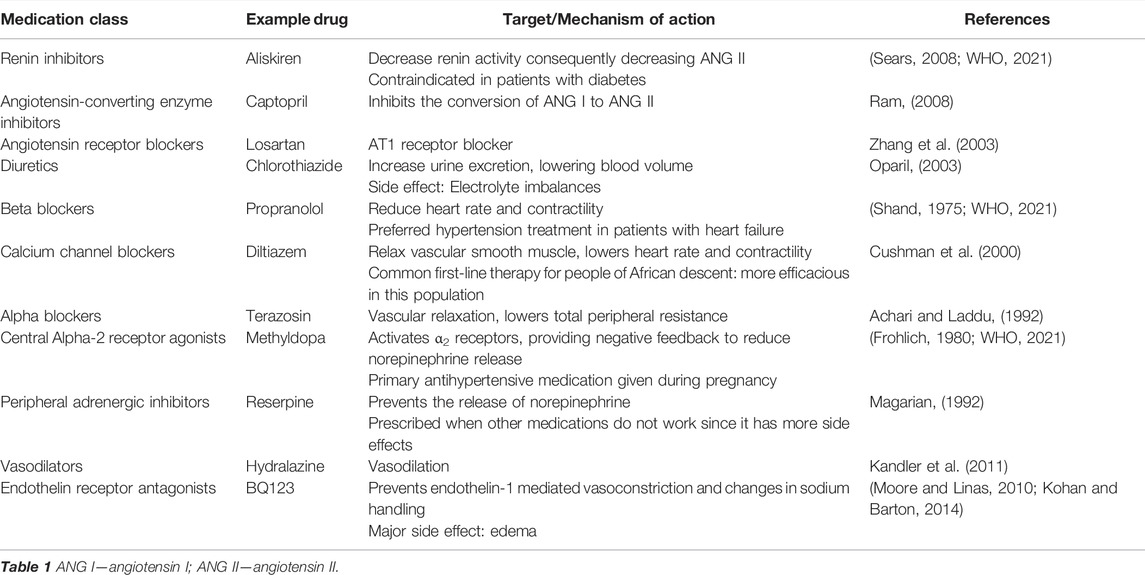
TABLE 1. Common hypertensive drugs and their mechanisms of action.
The renin angiotensin system (RAS) is a target for common first-line therapies to treat hypertension (WHO, 2021). Renin is a peptide released by juxtaglomerular (JG) cells, modified smooth muscle cells of the afferent arteriole of the kidney nephron, when 1) perfusion pressure within the afferent arteriole decreases; 2) renal sympathetic nerves activate the smooth muscle in the afferent arterioles; or 3) the macula densa senses a decrease in NaCl concentration in the ultrafiltrate. Angiotensinogen, a peptide produced by the liver, is cleaved by renin to produce angiotensin I (ANG I). Angiotensin-converting enzyme (ACE), mostly located on pulmonary vascular endothelial cells, then further converts ANG I into the active form, ANG II (Pappano, 2010). ANG II acts on cells in the adrenal cortex and posterior pituitary to stimulate the release of aldosterone and ADH, respectively. These hormones, in concert with ANG II, promote vasoconstriction and increase sodium and water reabsorption in the kidney, effects that combine to increase blood pressure (Stanton and Koeppen, 2010). ANG II has also been implicated as a crucial mediator of immune and inflammatory processes in hypertension (Ruiz-Ortega et al., 2000; Benigni et al., 2010) and hence ANG II infusion is a popular and consistent in vivo animal model for inducing hypertension with an inflammatory component in preclinical research (Schiffrin and Touyz, 2003). Upon binding to its ANG II-type 1 (AT1) receptor, ANG II increases vascular permeability and increases chemokine expression, which aids the inflammatory process and promotes immune cell infiltration (Suzuki et al., 2003). AT1 activation also promotes the proliferation and activation of Th1 and Th17 cells, which are both pro-inflammatory (Liu et al., 2009; Platten et al., 2009). On the other hand, ANG II binding to its AT2 receptor may have anti-inflammatory effects (Benndorf et al., 2009).
Interventions blocking components of the RAS are effective in decreasing blood pressure since they act on several targets including the kidneys, vasculature, brain, and immune system. Inhibition of renin and ACE prevents downstream production and action of ANG II, limiting rises in blood pressure. Aliskiren is a renin inhibitor that was approved for the treatment of essential hypertension (Sears, 2008), but the benefits of this drug may span beyond blood pressure control as aliskiren also decreases levels of systemic inflammatory cytokines like CD45 and CD3 T cells as well as F4/80-positive macrophages in hypertensive mice (Yen et al., 2013). Captopril is an ACE inhibitor that effectively lowers blood pressure in humans and animals, but also has an anti-inflammatory effect (Fukuzawa et al., 1997; De Albuquerque et al., 2004; Sepehri et al., 2016; Gan et al., 2018; Mitchell et al., 2021). Another ACE inhibitor, lisinopril, was shown to reduce autoimmune-induced inflammation in a mouse model of multiple sclerosis (Platten et al., 2009). ANG II receptor blockers (ARB), such as losartan, prevent the binding of ANG II to the AT1 receptor, the receptor primarily responsible for the vasoconstrictor and pro-inflammatory effects of ANG II (Brasier et al., 2002; Zhang et al., 2003; Gonçalves et al., 2004). ARBs have been shown to be as effective as ACE inhibitors in reducing cardiovascular risks (Li et al., 2014) while also reducing systemic inflammation in various disease states (Fairbrass et al., 2021; Gamboa et al., 2012).
While agents targeting RAS are first-line drugs to defend against hypertension, there are other drugs that target different mechanisms. Diuretics like thiazides, potassium sparing diuretics, and loop diuretics are often used in combination with other antihypertensive drugs; they reduce blood pressure by increasing excretion of sodium and water. The loop diuretic, furosemide, reduced the concentrations of proinflammatory cytokines TNF-α, IL-6 and IL-1β and activated polarization of macrophages from proinflammatory M1 type to anti-inflammatory M2 type. (Yuengsrigul et al., 1999; Xu et al., 2006; Wang et al., 2020; Tuttolomondo et al., 2021). In addition, hydrochlorothiazide from the thiazide diuretic group inhibits the secretion of proinflammatory cytokines like TNF-α and IFN-γ (Luo et al., 2011; Aloud et al., 2020). Interestingly, the last decade has also emphasized the anti-inflammatory properties of calcium channel blockers in addition to their antihypertensive effects due to reduced entry of calcium in the cardiac cells and smooth muscle cells of the vasculature in turn decreasing the contractile force. The calcium channel blocker lercanidipine lowers the number of polymorphonuclear leukocytes and C reactive protein in patients of essential hypertension while nicardipine inhibits the Th2-mediated airway inflammation and IFN-γ-induced neuro-inflammation of the microglial cells. (Gomes et al., 2007; Farah et al., 2013; Huang et al., 2014; Saddala et al., 2020). Beta-blockers inhibit the action of norepinephrine and epinephrine on β-adrenergic receptors. These are generally used to reduce the work output of the heart, promote relaxation of the vasculature and lower the blood pressure (Shand, 1975). In addition, β-blockers inhibit renin release from the kidneys and have demonstrated anti-inflammatory cytokine profile after their use (Ohtsuka et al., 2001; Gage et al., 2004; Hagiwara et al., 2009; Jachs et al., 2021). Methyldopa is a centrally-acting α2-adrenergic receptor agonist (Frohlich, 1980) and analog of DOPA (3,4-hydroxyphenylanine) that inhibits the adrenergic neuronal outflow and vasoconstriction response reducing the blood pressure. Methyldopa blocked the antigen presentation and inflammatory T cell responses in type 1 diabetes patients, suggesting its potential to combat autoimmunity (Ostrov et al., 2018; Bogacz et al., 2021). The vasodilator class of anti-hypertensive drugs, for example hydralazine, is beneficial as an antioxidant and anti-inflammatory agent in rodent models of sepsis, renal and myocardial ischemia-reperfusion injury, and the spontaneously hypertensive rat (Rodrigues et al., 2008; Li et al., 2020; Leu et al., 2021; Santos et al., 2021). Endothelin-1 (ET-1) is a potent vasoconstrictor and regulator of sodium excretion that induces inflammation and oxidative stress, so endothelin receptor antagonists have been investigated as a treatment for hypertension (Kohan and Barton, 2014). These drugs have not had wide attraction or success in treating hypertension due to adverse side effects like significant edema (Moore and Linas, 2010; Kohan and Barton, 2014). It is not yet known if drugs from other classes of antihypertensives like peripheral α-1 adrenergic inhibitors have anti-inflammatory properties.
These studies linking the anti-inflammatory properties to anti-hypertensive medications do not necessarily indicate a causal role of inflammation in the pathogenesis of hypertension. An interesting study by Marvar et al. actually demonstrated that hydralazine lowered blood pressure and, in turn, indirectly prevented the ANG II-induced T cell activation and vascular infiltration of leukocytes (Marvar et al., 2010). The association between inflammation and antihypertensive drugs may vary depending on the type of the inflammatory marker studied, the disease model under investigation, and the stage of the disease. It is highly likely that inflammation and blood pressure modulation may influence each other in a complex way. For example the drug Etanercept, which blocks TNF-α, reduced blood pressure in ANG II-induced hypertension (Guzik et al., 2007) and an autoimmune model of chronic inflammation (Venegas-Pont et al., 2010), but not in salt-dependent hypertension (Elmarakby et al., 2008). The recent Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) demonstrated that the IL-1β inhibition with canakinumab reduced major adverse cardiovascular event rates and C reactive protein, without effecting blood pressure (Dixon et al., 2020). Nevertheless, these studies emphasize the importance of identifying subpopulations of patients that may respond to particular anti-inflammatory therapy, while another cohort may not respond to that same therapy.
The American College of Cardiology stresses the importance of lifestyle modification in addition to or in place of pharmacological agents (Goetsch et al., 2021). The World Health Organization suggests initial treatment with one drug targeting the RAS combined with a thiazide-like diuretic, along with a drug from a third category like calcium channel blockers if necessary (WHO, 2021). However, some medications may be preferred or contraindicated based on the patient’s situation, e.g., β-blockers are a preferred hypertension treatment for heart failure patients due to their beneficial effects on the heart, and certain RAS inhibitors are contraindicated for people of African descent, making calcium channel blockers a common first-line therapy for these patients (Unger et al., 2020). The most commonly used initial combination of therapies include a long-acting calcium channel blocker (CCB), a blocker of RAS (ACE inhibitor or ARB), and a diuretic. In many cases, it is still difficult to control blood pressure even with a combination of drugs. The American College of Cardiology and American Heart Association defines “resistant hypertension” as occurring when blood pressure remains above the target blood pressure of 130/80 mmHg despite concurrent use of three antihypertensive agents of different classes, or when four or more antihypertensive agents are needed to control blood pressure at the optimum level (Carey et al., 2018a). True resistant hypertension can be differentiated from the pseudo- or apparent resistant hypertension, in which the latter is due to measurement error, the “white coat effect”, or nonadherence to treatment. Use of proper blood pressure measurement techniques by trained staff, using out–of-office, ambulatory and self-monitoring of the blood pressure, monitoring prescription refills and achieving a good doctor-patient relationship are some of the ways to avoid pseudo-resistant hypertension (Carey et al., 2018a). The cause of resistant hypertension is typically further investigated for 1) suboptimal lifestyle factors, e.g., obesity, dietary sodium intake, and physical inactivity; 2) other drugs interfering with the action of the antihypertensive therapy; and/or 3) other conditions that lead to hypertension such as endocrine disorders, renal disease, renal artery stenosis, and coarctation of aorta (Carey et al., 2018a). The fact that resistant hypertension cannot be controlled easily by the current anti-hypertensive regimens calls for new cellular and molecular targets.
Many investigators have demonstrated the causal role of renal inflammation in the form of immune cell (both innate and adaptive) infiltration and a pro-inflammatory milieu in the pathogenesis of hypertension (Vanegas et al., 2005; Franco et al., 2006). In fact, recent studies demonstrate that the immune cell infiltration in the kidneys may alter the renal vascular function and the fluid electrolyte balance in addition to causing renal injury (Franco et al., 2013; Rucker et al., 2018). Infiltrating T cells in the kidneys accentuate the Dahl salt-sensitive phenotype by increasing intrarenal ANG II and oxidative stress (Mattson, 2014). Proinflammatory cytokines like IL-17A and IFN-γ can induce hypertension by altering the reabsorption of sodium through its transporters: sodium-hydrogen exchanger-3 (NHE3), sodium-potassium-chloride cotransporter (NKCC) and sodium-chloride cotransporter (NCC) on the renal tubules (Kamat et al., 2015). Additionally, the polarization of macrophages to the pro-inflammatory M1 phenotype in the kidney can promote macrophage recruitment and added release of various inflammatory cytokines leading to renal damage and altered fluid-electrolyte balance (Fehrenbach and Mattson, 2020). Studies show that stimulation of dendritic cells (DCs) in the kidneys results in oxidative stress, fluid retention, and increased blood pressure (Lu et al., 2020). The importance of DCs in hypertension was demonstrated by adoptive transfer experiments of splenic DCs from ANG II hypertensive mice that was able to activate T-cells in the recipient mice. Furthermore, adoptive transfer of DCs from animals that underwent renal denervation led to a decrease in total leukocytes, CD3+, CD4+ and CD8+ T cells in response to low dose of ANG II. This suggests an association of the renal sympathetic nerves and renal inflammation in the development of hypertension (Xiao et al., 2015), which has been confirmed by many others (Xiao et al., 2015; Banek et al., 2018; Banek et al., 2019; Osborn et al., 2021). Besides renal nerves, sympathoexcitation of other nerves also contributes to hypertension and the contribution of the sympathetic vasomotor outflow seems to be dependent on the hypertension model and the stage of the disease. For example, in ANG II-induced hypertension, denervation of the splanchnic nerve, but not the renal or lumbar nerve, was able to blunt hypertension (Osborn and Fink, 2010). On the other hand, both renal and splanchnic nerve contributes to hypertension in the Genetically Hypertensive Schlager (BPH/2J) mice (Asirvatham-Jeyaraj et al., 2021). Renal denervation was unable to reduce blood pressure in hypertensive mice with late-stage systemic lupus erythematosus (Mathis et al., 2013). Thus, sympathetic activation has an important, but complicated role in the development and maintenance of hypertension and renal inflammation and needs further investigation.
Autoimmune diseases are characteristic of overactivation of immune cells, and chronic inflammation. Coincidently, autoimmune diseases with a renal component are associated with hypertension and cardiovascular morbidities (Boesen and Kakalij, 2021). Examples include systemic lupus erythematosus (SLE), Goodpasture syndrome, idiopathic membranous nephropathy, immunoglobulin A nephropathy, anti-neutrophil cytoplasmic antibody-associated glomerulonephritis, idiopathic thrombocytopenic purpura, rheumatoid arthritis and psoriasis (Panoulas et al., 2007; Coumbe et al., 2014; Giannelou and Mavragani, 2017; Liu and Kaplan, 2018; Tumurkhuu et al., 2019; Boesen and Kakalij, 2021). The renal pathological mechanisms involved in autoimmune-induced hypertension are still not completely understood; however, we do know that the loss of self-tolerance leads to autoantibody production and these autoantibodies form complexes, deposit into tissues like the kidneys and promote activation of other immune cells and the complement system. The resultant secretion of inflammatory mediators locally promotes chronic renal inflammation and oxidative stress (Choi et al., 2012; Small et al., 2018), which may increase fluid and electrolyte imbalance in the kidneys and/or cause renal vascular dysfunction leading to hypertension (Makino et al., 2002; Liu et al., 2012; Cowley et al., 2015; Gonzalez-Vicente et al., 2019).
SLE is the prototypical autoimmune disease with a renal inflammatory component usually referred to as lupus nephritis. Lupus nephritis develops following the production of diverse complex-forming nuclear autoantibodies that attack various self-antigens, including double-stranded DNA (dsDNA), histones, cardiolipins, phospholipids, Smith antigen and ribonucleoproteins (sm/RNP), and complement 1q (C1q) (Artim-Esen et al., 2014; Dema and Charles, 2016; Chen and Tsokos, 2021). Lupus nephritis is prevalent in more than half of SLE patients and it causes severe damage to glomerular, tubular and/or renal vascular structures (Najafi et al., 2001; Hsieh et al., 2011; Tsumiyama et al., 2013; Suarez-Fueyo et al., 2017; Ding et al., 2018). Lupus nephritis precedes hypertension in both female human SLE and the well-accepted female mouse model of SLE, the NZBWF1 mouse, so this enables the investigation of contributory mechanisms of hypertension in the setting of chronic renal inflammation. In addition, therapies that suppress lupus nephritis warrant identification and introduction to the hypertension field so that the scope of use of these drugs in patients with hard-to-control hypertension with a renal inflammatory component (e.g., resistant hypertension) can be considered.
Currently the prevalence of SLE in the United States is 72.8 per 100,000, with a threefold increase in incidence due to improved screening measures (Izmirly et al., 2021), and 9 out of 10 SLE patients are women of reproductive age. There are extreme disparities in SLE, since Black, Hispanic, Asian and Native American women are most commonly affected for unknown reasons. Several studies have confirmed a high prevalence of hypertension in SLE patients, ranging from 40% to as high as 74% in some cohorts (Sabio et al., 2011). Hypertension is a prominent baseline risk factor for severe ischemic stroke and cardiovascular disease in SLE patients (Mikdashi et al., 2007; Sabio et al., 2011; Munguia-Realpozo et al., 2019). Since cardiovascular disease is the leading cause of mortality among SLE patients, it is critical that we understand the pathogenesis of hypertension in SLE.
Current guidelines disregard the specific management of hypertension in SLE patients and focus on the treatment of lupus nephritis, skin manifestations, and pulmonary hypertension instead. Tselios et al. does recommend, however, a reduction of blood pressure to ≤130/80 for SLE patients using ACE inhibitors and ARBs, a similar goal and regimen recommended by the American Society of Cardiology for hypertensive persons (Tselios et al., 2020). The use of ACE inhibitors like captopril may be beneficial since it reduces renal injury and inflammation in spontaneously hypertensive rats (SHR) while reducing blood pressure (Gan et al., 2018). Captopril also provides renal protection when administered to diabetic patients and Albuquerque et al. confirmed the renoprotective effects of captopril in a mouse model of autoimmunity (De Albuquerque et al., 2004). The same group discovered that ACE inhibition decreases the renal expression of the pro-inflammatory mediator transforming growth factor (TGF)-β. These studies stress captopril’s ability to improve renal inflammatory processes, but more studies are needed to determine the efficacy in SLE.
Evidence of the effect of first-line SLE therapies on blood pressure is limited. However, we do know that traditional therapies that target lupus nephritis possess various adverse effects (see Table 2). Corticosteroids, the most widely-used agent for all autoimmune diseases, cause systemic side effects such as psychosis, Cushing’s syndrome, and importantly, hypertension in SLE patients (Petri et al., 2014; Kassel and Odum, 2015), while the NZBWF1 mice develop osteopenia and adrenal gland atrophy following chronic corticosteroid therapy (Jia et al., 2018). Novel immunomodulatory drugs are an exciting option for limiting immune system activation in SLE, but they often dampen the immune system excessively. For example, belimumab, a drug approved for SLE to inhibit B lymphocyte stimulator (BLyS) to induce apoptosis of autoreactive B cells (Weide et al., 2003; Petri, 2004; Pisoni et al., 2005; Chugh and Kalra, 2013), unfortunately causes progressive multifocal leukoencephalopathy in elderly SLE patients (Fredericks et al., 2014). Previous studies have shown that B cell or plasma cell depletion can prevent the development of hypertension if administered before the onset of SLE (Mathis et al., 2014; Taylor et al., 2018). Similar treatments exist for human SLE patients; rituximab binds to CD20 to deplete B cell activity, while mycophenolate mofetil and cyclophosphamide inhibit B cell activation and plasma cell synthesis. All have been efficacious in treating human SLE hypertension (Jiang et al., 2020; Lee et al., 2021). Mycophenolate mofetil also potentially reduces renal lymphocyte and macrophage infiltration while combating renal inflammation (Bravo et al., 2007). The most recent FDA-approved drug for SLE, anifrolumab, targets type 1 IFN by binding to its receptor IFNAR1 and inhibiting downstream inflammatory and immunological processes (Tanaka and Tummala, 2021). Anifrolumab caused adverse reactions such as nasopharyngitis, upper respiratory tract infection, and bronchitis (Mode and Stockholm, 2021), but its effect on renal inflammation and hypertension in SLE has not been investigated. Because of these unwanted and serious side effects, new and better therapies are still warranted to maintain the quality of life in SLE patients.

TABLE 2. A summary of drugs for SLE hypertension/lupus nephritis.
If we can target renal inflammation effectively, then there will likely be an improvement in blood pressure control in SLE given that majority of patients with lupus nephritis present with hypertension (Shaharir et al., 2015). Coincidently, SLE patients have significantly higher risks of resistant hypertension compared to non-SLE patients and this resistant hypertension is associated with lower renal function, and increased circulation inflammatory markers (erythrocyte sedimentation rate and C-reactive protein) (Gandelman et al., 2020).
Many studies have revealed mechanistic pathways regulating renal inflammation in SLE hypertension (Figure 1) (Ryan, 2009). Because SLE is a female-dominant disease, we often turn to the contributions of estrogen. Estrogen stimulates autoreactive B cells to increase autoantibody production, resulting in the rise of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-6, and C-reactive protein. Pharmacological blockade of estrogen early in life reduces renal inflammation by decreasing the population of B cells and immune complex deposition in NZBWF1 mice (Wu et al., 2000; Sthoeger et al., 2003). This suggests a mechanistic role of estrogen in contributing to renal inflammatory processes early in life in SLE. Another mechanism involves the increased production of endothelin-1, along with hyperactivation of the RAS increasing oxidative stress, which has been known to promote SLE hypertension (Kuryliszyn-Moskal et al., 2008; Mathis et al., 2012; Shah et al., 2014; Munguia-Realpozo et al., 2019). Since endothelin receptor A (ETA) blockade ameliorates renal inflammation and albuminuria in the diabetic rats (Saleh et al., 2011) it may be capable of doing the same in SLE hypertension.
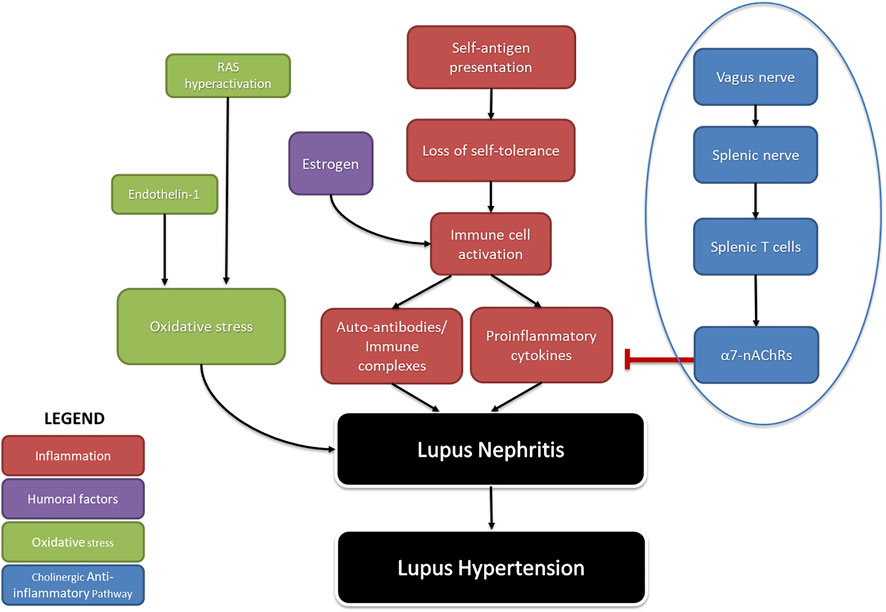
FIGURE 1. The pathophysiology of SLE-induced hypertension: A summary of factors that contribute to hypertension in SLE. Inflammation due to autoimmunity (red), as well as humoral factors that increase it (purple), and aberrant activity causing oxidative stress (green) comprise endogenous causes of lupus hypertension. The cholinergic anti-inflammatory pathway (blue) represents a neurogenic cause of lupus hypertension. Arrows (→) indicate stimulation; line with flathead (--|) indicates inhibition (RAS- renin angiotensin system; α7nAchRs-alpha seven nicotine acetyl choline receptors).
Less discussed are the possible neurogenic influences on renal inflammation in SLE hypertension. Basal activity of the hypothalamic-pituitary-adrenal (HPA) axis contributes to the suppression of peripheral inflammation by the release of corticosterone. Additionally, decreased parasympathetic (vagal) tone, as indicated by decreased heart rate variability, has been consistently identified in clinical studies involving SLE patients since 1997 (Laversuch et al., 1997; Maule et al., 1997; Thanou et al., 2016; Matusik et al., 2018; Zinglersen et al., 2021). An impaired HPA axis is present in SLE and may explain why renal inflammation is not resolved in SLE (Rovenský et al., 1998; Pham and Mathis, 2018). Tracey et al. established the significance of the inflammatory reflex—specifically the efferent arm of this reflex, known as the cholinergic anti-inflammatory pathway—a neural circuit that attenuates excessive inflammation (Huston et al., 2007; Tracey, 2021). This circuit runs from the vagus nerve to the splenic nerve and causes the release of acetylcholine from splenic T cells. This acetylcholine activates the alpha seven subunit of the nicotinic acetylcholine receptor (α7-nAChR) on splenic immune cells, ultimately resulting in the blunted release of proinflammatory cytokines and overall suppression of systemic inflammation and tissue damage (Pavlov et al., 2009; Martelli et al., 2014a); (Figure 2). Both the cholinergic anti-inflammatory pathway and the HPA axis rely on an active vagus nerve; therefore, decreased vagal nerve activity in SLE likely contributes to decreased activity of both of these anti-inflammatory pathways (Stein et al., 1996; Thanou, 2014; Mathis, 2015).
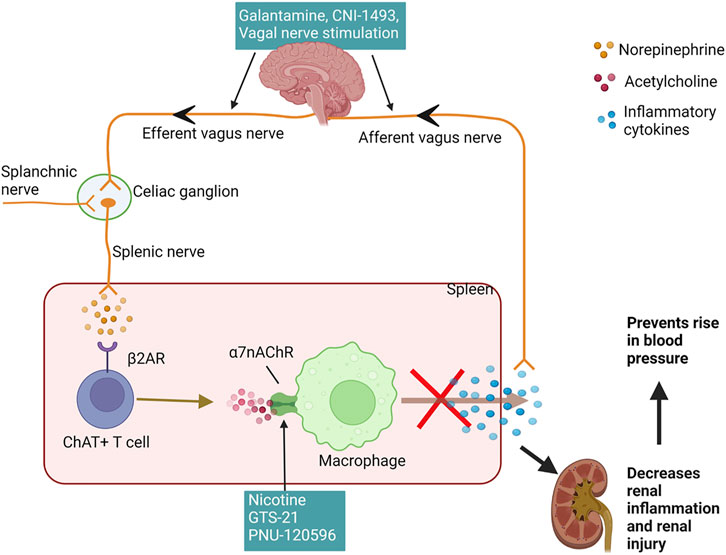
FIGURE 2. Cholinergic anti-inflammatory pathway reduces renal inflammation and hypertension: The afferent vagus nerve detects inflammatory cytokines and relays this information centrally to increase the efferent vagus, which synapses on the celiac ganglion and activates the splenic nerve. Another possible mechanism for activation of splenic nerve is via the splanchnic nerve. The splenic nerve has sympathetic fibers and stimulates splenic ChAT + T cells to synthesize and secrete acetylcholine, which acts upon various immune cells in the spleen, including macrophages, to inhibit the production and release of inflammatory cytokines. A reduction in splenic inflammation and proinflammatory cytokines in circulation decreases renal inflammation and renal injury preventing the rise in the blood pressure. The efferent vagus nerve can be stimulated using pharmacological agents galantamine and CNI-1493 or direct electrical stimulation. GTS-21 and nicotine, agonists of the α7nAchR and PNU-120596, positive allosteric modulator for this receptor leads to activation of this receptor to inhibit the cytokine release from immune cells. (β2AR—beta 2 adrenergic receptor; ChAT + T cells—choline acetyltransferase positive T cells; α7nAchRs—alpha seven nicotine acetylcholine receptors). Created with Biorender.com.
Studies from our lab suggest that stimulation of this cholinergic anti-inflammatory pathway can attenuate renal inflammation and the progression of hypertension in SLE, presenting the nerve as a novel, potential target for intervention for SLE hypertension (Fairley and Mathis, 2017). Galantamine is a centrally acting acetylcholinesterase inhibitor that enhances efferent vagus nerve activity and further enhances the cholinergic anti-inflammatory pathway (Pavlov et al., 2009), (Figure 2). Chronic systemic administration of galantamine in SLE mice successfully protects from hypertension by attenuating renal and splenic inflammation, while also decreasing the levels of autoantibodies (Pham et al., 2018). Similarly, the action of semapimod hydrochloride (CNI-1493), a tetravalent guanylhydrazone molecule, which inhibits the synthesis of proinflammatory cytokines, is mediated in part by the activation of efferent vagal nerve fibers (Borovikova et al., 2000; Bernik et al., 2002). When treated with systemic CNI-1493, SLE mice showed attenuated hypertension, decreased albuminuria, improved renal blood flow and decreased renal vascular resistance (Maloy et al., 2016). Whether long-term activation of the vagus nerve attenuates renal inflammation and hypertension in SLE patients is a prospective study to consider. Already, a randomized controlled pilot trial subjecting human SLE patients to 4 days of transcutaneous auricular vagus nerve stimulation successfully reduced pain and fatigue in these patients (Aranow et al., 2021), but the effect on renal inflammation and hypertension were not considered. Taken together, vagus nerve stimulation is promising in animal studies and humans so additional studies are needed to support these findings and to understand the detailed molecular mechanisms for its effect in the pathogenesis of lupus nephritis and SLE hypertension.
However, we must consider that the cholinergic anti-inflammatory pathway features several communication nodes, namely the vagus nerve, celiac ganglion, splenic nerve, and spleen (Tracey, 2007; Rosas-Ballina and Tracey, 2009; Olofsson et al., 2012; Pavlov and Tracey, 2017). Yet, this pathway and the proposed connections are controversial (Martelli et al., 2014a; Martelli et al., 2014c). Specifically, the neuroimmune mechanism is thought to be mediated by communication between the vagus and splenic nerves via the celiac ganglion. Several studies have shown the importance of these nodes of transmission in the protection offered by vagus nerve stimulation (Borovikova et al., 2000; Huston et al., 2008; Rosas-Ballina et al., 2008); however, morphological and anatomical studies suggest there may be no such connection between the two nerves (Martelli et al., 2014c), that the source of catecholamines may paradoxically be the vagus itself (Verlinden et al., 2016), and/or that other nerves like the splanchnic nerve may be the source of activation instead (Figure 2; Martelli et al., 2014b; Komegae et al., 2018). A recent study from our lab showed that unilateral vagotomy decrease renal inflammation and blunt hypertension in SLE mice, contrary from our initial hypothesis that vagotomy would worsen SLE hypertension and inflammation due to the disruption of the anticholinergic anti-inflammatory pathway. These data suggest the existence of compensatory neuroimmune mechanisms that prevail even after unilateral vagotomy that need further attention and investigation (Pham et al., 2020).
Attempts at reducing renal inflammation and attenuating SLE hypertension at other nodes within the cholinergic anti-inflammatory pathway were less successful in our hands. While systemic administration of nicotine, an agonist of α7-nAChR (Figure 2), reduced splenic and renal cortical expression of proinflammatory cytokines and blood pressure (Fairley and Mathis, 2017), partial agonist, GTS-21, and PNU-120596, a positive allosteric modulator (PAM) did not significantly reduce inflammation or blood pressure in SLE mice (Morales et al., 2021). However, it is possible that the lack of beneficial effect of GTS-21 and PNU-120596 was due to the mice being treated at an advanced stage of SLE, and future studies that use an earlier timeline for the administration of these α7 ligands may prove to be more successful. Additionally, the absence of a coexisting endogenous agonist (i.e., acetylcholine) due to decreased vagal tone in SLE may lead to ineffectiveness of PAM action. Interestingly, the importance of cholinergic anti-inflammatory pathway in the treatment of resistant hypertension has recently been recognized. In their study, Hilderman et al. collected whole blood samples at different time points from patients with resistant hypertension treated with renal denervation. These samples, when further treated with GTS-21, showed long-term suppression of LPS-induced TNF compared to the short-term suppression in the vehicle group. Although the authors could not study the effects on renal inflammation, their study and ours suggest a role of compromised anti-inflammatory pathways in the development of renal inflammation in SLE hypertension (Hilderman et al., 2019).
The antimalarial drug hydroxychloroquine, commonly used in SLE, suppresses the activity of lysosomal enzymes, thereby preventing major histocompatibility complex (MHC) class II-mediated autoantigen presentation. This drug also binds to dsDNA and inhibits toll-like receptor (TLR) signaling and/or TLR binding in order to reduce the production of proinflammatory cytokines (Rainsford et al., 2015). Gomez-Gutman et al. showed that hydroxychloroquine decreases renal inflammation, blood pressure, and renal injury, but does not decrease levels of anti-dsDNA antibodies in the NZBWF1 mouse model (Gómez-Guzmán et al., 2014). However, hydroxychloroquine was administered to the NZBWF1 mice at 30 weeks of age, when autoimmune-mediated hypertension and renal injury have already developed. It is likely that the timing of hydroxychloroquine administration is crucial to attenuate the extent of the autoantibodies production, reflected by the fact that hydroxychloroquine is the first-line drug for mild, early SLE in humans (Gies et al., 2020).
Finally, other drugs with unique mechanisms are also beneficial in reducing renal inflammation and blood pressure in SLE, introducing other potential mechanisms. Rosiglitazone, a peroxisome proliferator activator receptor gamma (PPARγ) agonist, targets adipose tissue and has been shown to reduce endothelin-1 and attenuate hypertension as well as renal inflammation and injury in SLE mice model (Venegas-Pont et al., 2009). The TNF-α inhibitor, etanercept, decreases mean arterial pressure as well as renal injury measured by glomerulosclerosis and albuminuria, and also decreases renal monocyte infiltration and oxidative stress (Venegas-Pont et al., 2010). Etanercept has been approved in many autoimmune conditions like rheumatoid arthritis, psoriasis, and systemic sclerosis to alleviate TNF-α-mediated immune responses and inflammation. It also decreases blood pressure in AngII-induced hypertension (Guzik et al., 2007) and pulmonary hypertension (Zhang et al., 2016); however, its safety and role in human SLE hypertension needs further investigation. Another promising drug for treating lupus nephritis is calcineurin inhibitors, drugs that block T cell activation through the suppression of the calcium/calcimodulin-dependent phosphatase. The novel calcineurin inhibitor, voclosporin acts by reducing the activation of T cells, causing stabilization of podocytes and reduction of proteinuria (Mok, 2017; Mejía-Vilet and Romero-Díaz, 2021). Voclosporin has recently been approved as induction therapy in lupus nephritis in combination with myocophenolate mofetil and a glucocorticoid. Its effects on blood pressure in lupus have not been demonstrated (Rovin et al., 2021).
In summary, several immune and molecular pathways play a role in executing the control of renal inflammation. Future mechanistic studies exploring the possible pathways that contribute to the renal inflammation in SLE-induced hypertension may help to modify the treatment strategies for hypertension in the setting of chronic inflammation or resistant hypertension.
There have been tremendous advances in treating hypertension. However, there are still subpopulations of hypertensive patients that do not respond to current combinations of antihypertensive agents indicating that alternative mechanisms are at play. In this review, we have provided an overview of what is currently known of the association between inflammation and hypertension, and evidence of the need to target renal inflammation in some hypertension patients. We provide intriguing examples that suggest that hypertension therapy could be more efficacious in those with underlying renal inflammation if the therapy was combined with an agent that combats renal inflammatory processes. There is a high prevalence of hypertension in SLE, so this presents an important disease model to study the control of renal inflammation in hypertension. Resistant hypertension is highly prevalent in SLE patients, so the link between the two also warrants further investigation. Some common SLE therapies have been successful in reducing both renal inflammation and blood pressure, while the efficacies of others have not been determined in humans or animal models of SLE. Studies determining the ability of other common SLE drugs to reduce blood pressure in more conventional animal models of hypertension and in hypertensive humans are warranted. Indeed, McCarthy et al. has already demonstrated the beneficial effects of chloroquine in spontaneously hypertensive rat (McCarthy et al., 2016) and chloroquine is well known to decrease vascular resistance and blood pressure in humans (Anigbogu et al., 1993). Development of diagnostic tools that identify the cohorts of hypertensive patients responding to a particular anti-inflammatory therapy combating renal inflammation is a step further towards personalized medicine. Whether drugs used to treat SLE have a beneficial effect on hypertension in the vulnerable population of resistant hypertensive patients is intriguing and warrants investigation.
All authors listed made a substantial and intellectual contribution to this review, and approved it for publication. GSP and KWM conceived the idea, SC and GSP prepared the first draft, and all authors made revisions to the subsequent drafts. The manuscript was finalized by KWM.
Funded by R01HL153703, K01HL139859 and a seed grant from the UNTHSC, Department of Physiology to KWM; T32AG020494 to CDB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Achari R., Laddu A. (1992). Terazosin: a New Alpha Adrenoceptor Blocking Drug. J. Clin. Pharmacol. 32 (6), 520–523. doi:10.1177/009127009203200605
Aloud B. M., Petkau J. C., Yu L., McCallum J., Kirby C., Netticadan T., et al. (2020). Effects of Cyanidin 3-O-Glucoside and Hydrochlorothiazide on T-Cell Phenotypes and Function in Spontaneously Hypertensive Rats. Food Funct. 11 (10), 8560–8572. doi:10.1039/d0fo01778g
Anigbogu C. N., Adigun S. A., Inyang I., Adegunloye B. J. (1993). Chloroquine Reduces Blood Pressure and Forearm Vascular Resistance and Increases Forearm Blood Flow in Healthy Young Adults. Clin. Physiol. 13 (2), 209–216. doi:10.1111/j.1475-097x.1993.tb00381.x
Aranow C., Atish-Fregoso Y., Lesser M., Mackay M., Anderson E., Chavan S., et al. (2021). Transcutaneous Auricular Vagus Nerve Stimulation Reduces Pain and Fatigue in Patients with Systemic Lupus Erythematosus: a Randomised, Double-Blind, Sham-Controlled Pilot Trial. Ann. Rheum. Dis. 80 (2), 203–208. doi:10.1136/annrheumdis-2020-217872
Artim-Esen B., Çene E., Şahinkaya Y., Ertan S., Pehlivan Ö., Kamali S., et al. (2014). Cluster Analysis of Autoantibodies in 852 Patients with Systemic Lupus Erythematosus from a Single Center. J. Rheumatol. 41 (7), 1304–1310. doi:10.3899/jrheum.130984
Asirvatham-Jeyaraj N., Gauthier M. M., Banek C. T., Ramesh A., Garver H., Fink G. D., et al. (2021). Renal Denervation and Celiac Ganglionectomy Decrease Mean Arterial Pressure Similarly in Genetically Hypertensive Schlager (BPH/2J) Mice. Hypertension 77 (2), 519–528. doi:10.1161/HYPERTENSIONAHA.119.14069
Banek C. T., Gauthier M. M., Baumann D. C., Van Helden D., Asirvatham-Jeyaraj N., Panoskaltsis-Mortari A., et al. (2018). Targeted Afferent Renal Denervation Reduces Arterial Pressure but Not Renal Inflammation in Established DOCA-Salt Hypertension in the Rat. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 314 (6), R883–R891. doi:10.1152/ajpregu.00416.2017
Banek C. T., Gauthier M. M., Van Helden D. A., Fink G. D., Osborn J. W. (2019). Renal Inflammation in DOCA-Salt Hypertension. Hypertension 73 (5), 1079–1086. doi:10.1161/HYPERTENSIONAHA.119.12762
Benigni A., Cassis P., Remuzzi G. (2010). Angiotensin II Revisited: New Roles in Inflammation, Immunology and Aging. EMBO Mol. Med. 2 (7), 247–257. doi:10.1002/emmm.201000080
Benndorf R. A., Krebs C., Hirsch-Hoffmann B., Schwedhelm E., Cieslar G., Schmidt-Haupt R., et al. (2009). Angiotensin II Type 2 Receptor Deficiency Aggravates Renal Injury and Reduces Survival in Chronic Kidney Disease in Mice. Kidney Int. 75 (10), 1039–1049. doi:10.1038/ki.2009.2
Bernik T. R., Friedman S. G., Ochani M., DiRaimo R., Ulloa L., Yang H., et al. (2002). Pharmacological Stimulation of the Cholinergic Antiinflammatory Pathway. J. Exp. Med. 195 (6), 781–788. doi:10.1084/jem.20011714
Boesen E. I., Kakalij R. M. (2021). Autoimmune-mediated Renal Disease and Hypertension. Clin. Sci. 135 (17), 2165–2196. doi:10.1042/cs20200955
Bogacz A., Mikołajczak P. Ł., Wolek M., Górska A., Szulc M., Ożarowski M., et al. (2021). Combined Effects of Methyldopa and Flavonoids on the Expression of Selected Factors Related to Inflammatory Processes and Vascular Diseases in Human Placenta Cells-An In Vitro Study. Molecules 26 (5), 1259. doi:10.3390/molecules26051259
Borovikova L. V., Ivanova S., Nardi D., Zhang M., Yang H., Ombrellino M., et al. (2000). Role of Vagus Nerve Signaling in CNI-1493-Mediated Suppression of Acute Inflammation. Auton. Neurosci. 85 (1-3), 141–147. doi:10.1016/S1566-0702(00)00233-2
Brasier A. R., Recinos A., Eledrisi M. S. (2002). Vascular Inflammation and the Renin-Angiotensin System. Arteriosclerosis, Thrombosis, Vasc. Biol. 22 (8), 1257–1266. doi:10.1161/01.atv.0000021412.56621.a2
Bravo Y., Quiroz Y., Ferrebuz A., Vaziri N. D., Rodríguez-Iturbe B. (2007). Mycophenolate Mofetil Administration Reduces Renal Inflammation, Oxidative Stress, and Arterial Pressure in Rats with Lead-Induced Hypertension. Am. J. Physiology-Renal Physiology 293 (2), F616–F623. doi:10.1152/ajprenal.00507.2006
Carey R. M., Calhoun D. A., Bakris G. L., Brook R. D., Daugherty S. L., Dennison-Himmelfarb C. R., et al. (2018a). Resistant Hypertension: Detection, Evaluation, and Management: a Scientific Statement from the American Heart Association. Hypertension 72 (5), e53–e90. doi:10.1161/HYP.0000000000000084
Carey R. M., Whelton P. K., Committee* A. A. H. G. W. (2018b). Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Synopsis of the 2017 American College of Cardiology/American Heart Association Hypertension Guideline. Ann. Intern Med. 168 (5), 351–358. doi:10.7326/m17-3203
CDC (2019a). About Multiple Cause of Death, 1999–2019. CDC WONDER Online Database Website. Atlanta, GA: Centers for Disease Control and Prevention.
CDC (2019b). Hypertension Cascade: Hypertension Prevalence, Treatment and Control Estimates Among US Adults Aged 18 Years and Older Applying the Criteria from the American College of Cardiology and American Heart Association's 2017 Hypertension Guideline—NHANES 2013–2016. Atlanta, GA: US Department of Health and Human Services.
Chan C. T., Sobey C. G., Lieu M., Ferens D., Kett M. M., Diep H., et al. (2015). Obligatory Role for B Cells in the Development of Angiotensin II-dependent Hypertension. Hypertension 66 (5), 1023–1033. doi:10.1161/hypertensionaha.115.05779
Chen P.-M., Tsokos G. C. (2021). T Cell Abnormalities in the Pathogenesis of Systemic Lupus Erythematosus: an Update. Curr. Rheumatol. Rep. 23 (2), 1–9. doi:10.1007/s11926-020-00978-5
Choi J., Kim S. T., Craft J. (2012). The Pathogenesis of Systemic Lupus Erythematosus-An Update. Curr. Opin. Immunol. 24 (6), 651–657. doi:10.1016/j.coi.2012.10.004
Chugh P. K., Kalra B. S. (2013). Belimumab: Targeted Therapy for Lupus. Int. J. Rheum. Dis. 16 (1), 4–13. doi:10.1111/1756-185x.12002
Coleman T. G., Guyton A. C., Young D. B., DeClue J. W., Norman R. A., Manning R. D., et al. (1975). The Role of the Kidney in Essential Hypertension. Clin. Exp. Pharmacol. Physiol. 2 (6), 571–581. doi:10.1111/j.1440-1681.1975.tb01862.x
Coumbe A. G., Pritzker M. R., Duprez D. A. (2014). Cardiovascular Risk and Psoriasis: beyond the Traditional Risk Factors. Am. J. Med. 127 (1), 12–18. doi:10.1016/j.amjmed.2013.08.013
Cowley A. W., Abe M., Mori T., O'Connor P. M., Ohsaki Y., Zheleznova N. N. (2015). Reactive Oxygen Species as Important Determinants of Medullary Flow, Sodium Excretion, and Hypertension. Am. J. Physiology-Renal Physiology 308 (3), F179–F197. doi:10.1152/ajprenal.00455.2014
Cushman W. C., Reda D. J., Perry H. M., Williams D., Abdellatif M., Materson B. J., et al. (2000). Regional and Racial Differences in Response to Antihypertensive Medication Use in a Randomized Controlled Trial of Men with Hypertension in the United States. Arch. Intern Med. 160 (6), 825–831. doi:10.1001/archinte.160.6.825
De Albuquerque D. A., Saxena V., Adams D. E., Boivin G. P., Brunner H. I., Witte D. P., et al. (2004). An ACE Inhibitor Reduces Th2 Cytokines and TGF-Β1 and TGF-Β2 Isoforms in Murine Lupus Nephritis. Kidney Int. 65 (3), 846–859. doi:10.1111/j.1523-1755.2004.00462.x
de Bourcy C. F. A., Dekker C. L., Davis M. M., Nicolls M. R., Quake S. R. (2017). Dynamics of the Human Antibody Repertoire after B Cell Depletion in Systemic Sclerosis. Sci. Immunol. 2 (15), eaan8289. doi:10.1126/sciimmunol.aan8289
Dema B., Charles N. (2016). Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies 5 (1), 2. doi:10.3390/antib5010002
Ding Y., Nie L.-M., Pang Y., Wu W.-J., Tan Y., Yu F., et al. (2018). Composite Urinary Biomarkers to Predict Pathological Tubulointerstitial Lesions in Lupus Nephritis. Lupus 27 (11), 1778–1789. doi:10.1177/0961203318788167
Dixon D. L., Wohlford IV G. F., Abbate A. (2020). Inflammation and Hypertension: Causal or Not? Hypertension 75, 297–298. Am Heart Assoc. doi:10.1161/HYPERTENSIONAHA.119.14195
Drummond G. R., Vinh A., Guzik T. J., Sobey C. G. (2019). Immune Mechanisms of Hypertension. Nat. Rev. Immunol. 19 (8), 517–532. doi:10.1038/s41577-019-0160-5
Eickenberg S., Mickholz E., Jung E., Nofer J. R., Pavenstädt H. J., Jacobi A. M. (2012). Mycophenolic Acid Counteracts B Cell Proliferation and Plasmablast Formation in Patients with Systemic Lupus Erythematosus. Arthritis Res. Ther. 14 (3), R110–R114. doi:10.1186/ar3835
Elmarakby A. A., Quigley J. E., Imig J. D., Pollock J. S., Pollock D. M. (2008). TNF-α Inhibition Reduces Renal Injury in DOCA-Salt Hypertensive Rats. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 294 (1), R76–R83. doi:10.1152/ajpregu.00466.2007
Fairbrass K. M., Hoshen D., Gracie D. J., Ford A. C. (2021). Effect of ACE Inhibitors and Angiotensin II Receptor Blockers on Disease Outcomes in Inflammatory Bowel Disease. Gut 70 (1), 218–219. doi:10.1136/gutjnl-2020-321186
Fairley A. S., Mathis K. W. (2017). Cholinergic Agonists Reduce Blood Pressure in a Mouse Model of Systemic Lupus Erythematosus. Physiol. Rep. 5 (7), e13213. doi:10.14814/phy2.13213
Farah R., Khamisy-Farah R., Shurtz-Swirski R. (2013). Calcium Channel Blocker Effect on Insulin Resistance and Inflammatory Markers in Essential Hypertension Patients. Int. Angiol. 32 (1), 85–93. https://www.minervamedica.it/en/journals/international-angiology/article.php?cod=R34Y2013N01A0085.
Fava A., Petri M. (2019). Systemic Lupus Erythematosus: Diagnosis and Clinical Management. J. Autoimmun. 96, 1–13. doi:10.1016/j.jaut.2018.11.001
Fehrenbach D. J., Mattson D. L. (2020). Inflammatory Macrophages in the Kidney Contribute to Salt-Sensitive Hypertension. Am. J. Physiology-Renal Physiology 318 (3), F544–F548. doi:10.1152/ajprenal.00454.2019
Ferdinand K. C., Nasser S. A. (2017). Management of Essential Hypertension. Cardiol. Clin. 35 (2), 231–246. doi:10.1016/j.ccl.2016.12.005
Franco M., Martínez F., Rodríguez-Iturbe B., Johnson R. J., Santamaría J., Montoya A., et al. (2006). Angiotensin II, Interstitial Inflammation, and the Pathogenesis of Salt-Sensitive Hypertension. Am. J. Physiology-Renal Physiology 291 (6), F1281–F1287. doi:10.1152/ajprenal.00221.2006
Franco M., Tapia E., Bautista R., Pacheco U., Santamaria J., Quiroz Y., et al. (2013). Impaired Pressure Natriuresis Resulting in Salt-Sensitive Hypertension Is Caused by Tubulointerstitial Immune Cell Infiltration in the Kidney. Am. J. Physiology-Renal Physiology 304 (7), F982–F990. doi:10.1152/ajprenal.00463.2012
Fredericks C., Kvam K., Bear J., Crabtree G., Josephson S. (2014). A Case of Progressive Multifocal Leukoencephalopathy in a Lupus Patient Treated with Belimumab. Lupus 23 (7), 711–713. doi:10.1177/0961203314524292
Frohlich E. D. (1980). Methyldopa. Mechanisms and Treatment 25 Years Later. Archives Intern. Med. 140 (7), 954–959. doi:10.1001/archinte.140.7.954
Fukuzawa M., Satoh J., Sagara M., Muto G., Muto Y., Nishimura S., et al. (1997). Angiotensin Converting Enzyme Inhibitors Suppress Production of Tumor Necrosis Factor-α In Vitro and In Vivo. Immunopharmacology 36 (1), 49–55. doi:10.1016/s0162-3109(96)00160-9
Gage J. R., Fonarow G., Hamilton M., Widawski M., Martínez-Maza O., Vredevoe D. L. (2004). Beta Blocker and Angiotensin-Converting Enzyme Inhibitor Therapy Is Associated with Decreased Th1/Th2 Cytokine Ratios and Inflammatory Cytokine Production in Patients with Chronic Heart Failure. Neuroimmunomodulation 11 (3), 173–180. doi:10.1159/000076766
Gan Z., Huang D., Jiang J., Li Y., Li H., Ke Y. (2018). Captopril Alleviates Hypertension-Induced Renal Damage, Inflammation, and NF-Κb Activation. Braz J. Med. Biol. Res. 51, e7338. doi:10.1590/1414-431X20187338
Gandelman J. S., Khan O. A., Shuey M. M., Neal J. E., McNeer E., Dickson A., et al. (2020). Increased Incidence of Resistant Hypertension in Patients with Systemic Lupus Erythematosus: a Retrospective Cohort Study. Arthritis Care Res. 72 (4), 534–543. doi:10.1002/acr.23880
Gamboa J. L., Pretorius M., Todd-Tzanetos D. R., Luther J. M., Yu C., Ikizler T. A., et al. (2012) Comparative Effects of Angiotensin-Converting Enzyme Inhibition and Angiotensin-Receptor Blockade on Inflammation During Hemodialysis. J. Am. Soc. Nephrol. 23 (2), 334–342. doi:10.1681/ASN.2011030287
Giannelou M., Mavragani C. P. (2017). Cardiovascular Disease in Systemic Lupus Erythematosus: a Comprehensive Update. J. Autoimmun. 82, 1–12. doi:10.1016/j.jaut.2017.05.008
Gies V., Bekaddour N., Dieudonné Y., Guffroy A., Frenger Q., Gros F., et al. (2020). Beyond Anti-viral Effects of Chloroquine/hydroxychloroquine. Front. Immunol. 11, 1409. doi:10.3389/fimmu.2020.01409
Goetsch M. R., Tumarkin E., Blumenthal R. S., Whelton S. P. (2021). New Guidance on Blood Pressure Management in Low-Risk Adults with Stage 1 Hypertension. American College of Cardiology.
Gomes B., Cabral M. D., Gallard A., Savignac M., Paulet P., Druet P., et al. (2007). Calcium Channel Blocker Prevents T Helper Type 2 Cell-Mediated Airway Inflammation. Am. J. Respir. Crit. Care Med. 175 (11), 1117–1124. doi:10.1164/rccm.200607-1026oc
Gómez-Guzmán M., Jiménez R., Romero M., Sánchez M., Zarzuelo M. J., Gómez-Morales M., et al. (2014). Chronic Hydroxychloroquine Improves Endothelial Dysfunction and Protects Kidney in a Mouse Model of Systemic Lupus Erythematosus. Hypertension 64 (2), 330–337. doi:10.1161/hypertensionaha.114.03587
Gonçalves A. R. R., Fujihara C. K., Mattar A. L., Malheiros D. M. A. C., Noronha I. L., De Nucci G., et al. (2004). Renal Expression of COX-2, ANG II, and AT1receptor in Remnant Kidney: Strong Renoprotection by Therapy with Losartan and a Nonsteroidal Anti-inflammatory. Am. J. Physiology-Renal Physiology 286 (5), F945–F954. doi:10.1152/ajprenal.00238.2003
Gonzalez-Vicente A., Hong N., Garvin J. L. (2019). Effects of Reactive Oxygen Species on Renal Tubular Transport. Am. J. Physiology-Renal Physiology 317 (2), F444–F455. doi:10.1152/ajprenal.00604.2018
Guzik T. J., Hoch N. E., Brown K. A., McCann L. A., Rahman A., Dikalov S., et al. (2007). Role of the T Cell in the Genesis of Angiotensin II-Induced Hypertension and Vascular Dysfunction. J. Exp. Med. 204 (10), 2449–2460. doi:10.1084/jem.20070657
Hagiwara S., Iwasaka H., Maeda H., Noguchi T. (2009). LANDIOLOL, AN ULTRASHORT-ACTING β1-ADRENOCEPTOR ANTAGONIST, HAS PROTECTIVE EFFECTS IN AN LPS-INDUCED SYSTEMIC INFLAMMATION MODEL. Shock 31 (5), 515–520. doi:10.1097/shk.0b013e3181863689
Herrera J., Ferrebuz A., MacGregor E. G., Rodriguez-Iturbe B. (2006). Mycophenolate Mofetil Treatment Improves Hypertension in Patients with Psoriasis and Rheumatoid Arthritis. J. Am. Soc. Nephrol. 17 (12 Suppl. 3), S218–S225. doi:10.1681/ASN.2006080918
Hilderman M., Qureshi A. R., Abtahi F., Witt N., Jägren C., Olbers J., et al. (2019). The Cholinergic Anti-inflammatory Pathway in Resistant Hypertension Treated with Renal Denervation. Mol. Med. 25 (1), 39–10. doi:10.1186/s10020-019-0097-y
Hsieh C., Chang A., Brandt D., Guttikonda R., Utset T. O., Clark M. R. (2011). Predicting Outcomes of Lupus Nephritis with Tubulointerstitial Inflammation and Scarring. Arthritis Care Res. 63 (6), 865–874. doi:10.1002/acr.20441
Huang B.-R., Chang P.-C., Yeh W.-L., Lee C.-H., Tsai C.-F., Lin C., et al. (2014). Anti-neuroinflammatory Effects of the Calcium Channel Blocker Nicardipine on Microglial Cells: Implications for Neuroprotection. PloS one 9 (3), e91167. doi:10.1371/journal.pone.0091167
Huston J. M., Gallowitsch-Puerta M., Ochani M., Ochani K., Yuan R., Rosas-Ballina M., et al. (2007). Transcutaneous Vagus Nerve Stimulation Reduces Serum High Mobility Group Box 1 Levels and Improves Survival in Murine Sepsis *. Crit. care Med. 35 (12), 2762–2768. doi:10.1097/00003246-200712000-00014
Huston J. M., Wang H., Ochani M., Ochani K., Rosas-Ballina M., Gallowitsch-Puerta M., et al. (2008). Splenectomy Protects Against Sepsis Lethality and Reduces Serum HMGB1 Levels. J. Immunol. 181, 3535–3539.
Izmirly P. M., Parton H., Wang L., McCune W. J., Lim S. S., Drenkard C., et al. (2021). Prevalence of Systemic Lupus Erythematosus in the United States: Estimates from a Meta‐Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol. 73 (6), 991–996. doi:10.1002/art.41632
Jachs M., Hartl L., Schaufler D., Desbalmes C., Simbrunner B., Eigenbauer E., et al. (2021). Amelioration of Systemic Inflammation in Advanced Chronic Liver Disease upon Beta-Blocker Therapy Translates into Improved Clinical Outcomes. Gut 70 (9), 1758–1767. doi:10.1136/gutjnl-2020-322712
Jia Z., Wang X., Wei X., Zhao G., Foster K. W., Qiu F., et al. (2018). Micelle-forming Dexamethasone Prodrug Attenuates Nephritis in Lupus-Prone Mice without Apparent Glucocorticoid Side Effects. ACS Nano 12 (8), 7663–7681. doi:10.1021/acsnano.8b01249
Jiang Y.-P., Zhao X.-X., Chen R.-R., Xu Z.-H., Wen C.-P., Yu J. (2020). Comparative Efficacy and Safety of Mycophenolate Mofetil and Cyclophosphamide in the Induction Treatment of Lupus Nephritis. Med. Baltim. 99 (38), e22328. doi:10.1097/MD.0000000000022328
Kamat N. V., Thabet S. R., Xiao L., Saleh M. A., Kirabo A., Madhur M. S., et al. (2015). Renal Transporter Activation during Angiotensin-II Hypertension Is Blunted in Interferon-γ −/− and Interleukin-17A −/− Mice. Hypertension 65 (3), 569–576. doi:10.1161/hypertensionaha.114.04975
Kandler M. R., Mah G. T., Tejani A. M., Stabler S. N., Salzwedel D. M. (2011). Hydralazine for Essential Hypertension. Cochrane Database Syst. Rev. 11, CD004934. doi:10.1002/14651858.CD004934.pub4
Kassel L. E., Odum L. E. (2015). Our Own Worst Enemy: Pharmacologic Mechanisms of Hypertension. Adv. chronic kidney Dis. 22 (3), 245–252. doi:10.1053/j.ackd.2014.10.002
Kohan D. E., Barton M. (2014). Endothelin and Endothelin Antagonists in Chronic Kidney Disease. Kidney Int. 86 (5), 896–904. doi:10.1038/ki.2014.143
Komegae E. N., Farmer D. G. S., Brooks V. L., McKinley M. J., McAllen R. M., Martelli D. (2018). Vagal Afferent Activation Suppresses Systemic Inflammation via the Splanchnic Anti-Inflammatory Pathway. Brain Behav. Immun. 73, 441–449.
Kuryliszyn-Moskal A., Klimiuk P. A., Ciolkiewicz M., Sierakowski S. (2008). Clinical Significance of Selected Endothelial Activation Markers in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 35 (7), 1307–1313. https://www.jrheum.org/content/jrheum/35/7/1307.full.pdf.
Laversuch C. J., Seo H., Modarres H., Collins D. A., McKenna W., Bourke B. E. (1997). Reduction in Heart Rate Variability in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 24 (8), 1540–1544. https://pubmed.ncbi.nlm.nih.gov/9263148/.
Lee D. S. W., Rojas O. L., Gommerman J. L. (2021). B Cell Depletion Therapies in Autoimmune Disease: Advances and Mechanistic Insights. Nat. Rev. Drug Discov. 20 (3), 179–199. doi:10.1038/s41573-020-00092-2
Leu J.-G., Su W.-H., Chen Y.-C., Liang Y.-J. (2021). Hydralazine Attenuates Renal Inflammation in Diabetic Rats with Ischemia/reperfusion Acute Kidney Injury. Eur. J. Pharmacol. 910, 174468. doi:10.1016/j.ejphar.2021.174468
Li E. C., Heran B. S., Wright J. M. (2014). Angiotensin Converting Enzyme (ACE) Inhibitors versus Angiotensin Receptor Blockers for Primary Hypertension. Cochrane Database Syst. Rev. (8), 1–49. doi:10.1002/14651858.CD009096.pub2
Li C., Su Z., Ge L., Chen Y., Chen X., Li Y. (2020). Cardioprotection of Hydralazine against Myocardial Ischemia/reperfusion Injury in Rats. Eur. J. Pharmacol. 869, 172850. doi:10.1016/j.ejphar.2019.172850
Liu X., Zhu X., Wang A., Fan H., Yuan H. (2009). Effects of Angiotensin-II Receptor Blockers on Experimental Autoimmune Myocarditis. Int. J. Cardiol. 137 (3), 282–288. doi:10.1016/j.ijcard.2009.09.540
Liu J., Kennedy D. J., Yan Y., Shapiro J. I. (2012). Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. Int. J. Nephrol. 2012, 1–14. doi:10.1155/2012/381320
Liu Y., Kaplan M. J. (2018). Cardiovascular Disease in Systemic Lupus Erythematosus: an Update. Curr. Opin. rheumatology 30 (5), 441–448. doi:10.1097/bor.0000000000000528
Lu X., Crowley S. D. (2020). Actions of Immune Cells in the Hypertensive Kidney. Curr. Opin. Nephrol. Hypertens. 29 (5), 515–522. doi:10.1097/mnh.0000000000000635
Lu X., Rudemiller N. P., Privratsky J. R., Ren J., Wen Y., Griffiths R., et al. (2020). Classical Dendritic Cells Mediate Hypertension by Promoting Renal Oxidative Stress and Fluid Retention. Hypertension 75 (1), 131–138. doi:10.1161/hypertensionaha.119.13667
Luo J., Gao X., Peng L., Sun H., Dai G. (2011). Effects of Hydrochlorothiazide on Cardiac Remodeling in a Rat Model of Myocardial Infarction-Induced Congestive Heart Failure. Eur. J. Pharmacol. 667 (1-3), 314–321. doi:10.1016/j.ejphar.2011.06.012
Magarian G. J. (1992). Reserpine: A Relic from the Past or a Neglected Drug of the Present for Achieving Cost Containment in Treating Hypertension? J. Gen. Intern Med. 7 (1), 124. doi:10.1007/bf02599123
Makino A., Skelton M. M., Zou A.-P., Roman R. J., Cowley A. W. (2002). Increased Renal Medullary Oxidative Stress Produces Hypertension. Hypertension 39 (2), 667–672. doi:10.1161/hy0202.103469
Maloy C. I., Fairley A. S., Pham G. S., Mathis K. W. (2016). Improvement in Blood Pressure and Renal Injury Following Vagal Nerve Stimulation. FASEB J. 30, 964–966. https://faseb.onlinelibrary.wiley.com/doi/abs/10.1096/fasebj.30.1_supplement.964.6.
Martelli D., McKinley M. J., McAllen R. M. (2014a). The Cholinergic Anti-inflammatory Pathway: a Critical Review. Aut. Neurosci. 182, 65–69. doi:10.1016/j.autneu.2013.12.007
Martelli D., Yao S., McKinley M. J., McAllen R. M. (2014b). Reflex Control of Inflammation by Sympathetic Nerves, not the Vagus. J. Physiol. 592 (7), 1677–1686.
Martelli D., Yao S., Mancera J., McKinley M., McAllen R. (2014c). Reflex Control of Inflammation by the Splanchnic Anti-Inflammatory Pathway is Sustained and Independent of Anesthesia. Am. J. Physiol. 307 (9), R1085–R1091.
Marvar P. J., Thabet S. R., Guzik T. J., Lob H. E., McCann L. A., Weyand C., et al. (2010). Central and Peripheral Mechanisms of T-Lymphocyte Activation and Vascular Inflammation Produced by Angiotensin II-Induced Hypertension. Circulation Res. 107 (2), 263–270. doi:10.1161/circresaha.110.217299
Mathis K. W. (2015). An Impaired Neuroimmune Pathway Promotes the Development of Hypertension in Systemic Lupus Erythematosus. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 309 (9), R1074–R1077. doi:10.1152/ajpregu.00143.2015
Mathis K. W., Venegas-Pont M., Masterson C. W., Stewart N. J., Wasson K. L., Ryan M. J. (2012). Oxidative Stress Promotes Hypertension and Albuminuria during the Autoimmune Disease Systemic Lupus Erythematosus. Hypertension 59 (3), 673–679. doi:10.1161/hypertensionaha.111.190009
Mathis K. W., Venegas-Pont M., Flynn E. R., Williams J. M., Maric-Bilkan C., Dwyer T. M., et al. (2013). Hypertension in an Experimental Model of Systemic Lupus Erythematosus Occurs Independently of the Renal Nerves. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 305 (7), R711–R719. doi:10.1152/ajpregu.00602.2012
Mathis K. W., Wallace K., Flynn E. R., Maric-Bilkan C., LaMarca B., Ryan M. J. (2014). Preventing Autoimmunity Protects against the Development of Hypertension and Renal Injury. Hypertension 64 (4), 792–800. doi:10.1161/hypertensionaha.114.04006
Mattson D. L. (2014). Infiltrating Immune Cells in the Kidney in Salt-Sensitive Hypertension and Renal Injury. Am. J. Physiology-Renal Physiology 307 (5), F499–F508. doi:10.1152/ajprenal.00258.2014
Matusik P. S., Matusik P. T., Stein P. K. (2018). Heart Rate Variability in Patients with Systemic Lupus Erythematosus: a Systematic Review and Methodological Considerations. Lupus 27 (8), 1225–1239. doi:10.1177/0961203318771502
Maule S., Quadri R., Mirante D., Pellerito R. A., Marucco E., Marinone C., et al. (1997). Autonomic Nervous Dysfunction in Systemic Lupus Erythematosus (SLE) and Rheumatoid Arthritis (RA): Possible Pathogenic Role of Autoantibodies to Autonomic Nervous Structures. Clin. Exp. Immunol. 110 (3), 423–427. doi:10.1046/j.1365-2249.1997.4501466.x
McCarthy C. G., Wenceslau C. F., Goulopoulou S., Ogbi S., Matsumoto T., Webb R. C. (2016). Autoimmune Therapeutic Chloroquine Lowers Blood Pressure and Improves Endothelial Function in Spontaneously Hypertensive Rats. Pharmacol. Res. 113, 384–394. doi:10.1016/j.phrs.2016.09.008
Mejía-Vilet J. M., Romero-Díaz J. (2021). Voclosporin: a Novel Calcineurin Inhibitor for the Management of Lupus Nephritis. Expert Rev. Clin. Immunol. 17 (9), 937–945. doi:10.1080/1744666x.2021.1967747
Mian M. O. R., Paradis P., Schiffrin E. L. (2014). Innate Immunity in Hypertension. Curr. Hypertens. Rep. 16 (2), 413. doi:10.1007/s11906-013-0413-9
Mikdashi J., Handwerger B., Langenberg P., Miller M., Kittner S. (2007). Baseline Disease Activity, Hyperlipidemia, and Hypertension Are Predictive Factors for Ischemic Stroke and Stroke Severity in Systemic Lupus Erythematosus. Stroke 38 (2), 281–285. doi:10.1161/01.str.0000254476.05620.14
Mikolajczyk T. P., Guzik T. J. (2019). Adaptive Immunity in Hypertension. Curr. Hypertens. Rep. 21 (9), 68–12. doi:10.1007/s11906-019-0971-6
Mitchell C. S., Premaratna S. D., Bennett G., Lambrou M., Stahl L. A., Jois M., et al. (2021). Inhibition of the Renin-Angiotensin System Reduces Gene Expression of Inflammatory Mediators in Adipose Tissue Independent of Energy Balance. Front. Endocrinol. (Lausanne) 12, 682726. doi:10.3389/fendo.2021.682726
Mode D., Stockholm L. C. (2021). AstraZeneca: Saphnelo (Anifrolumab) Approved in the US for Moderate to Severe Systemic Lupus Erythematosus.
Mok C. C. (2017). Calcineurin Inhibitors in Systemic Lupus Erythematosus. Best Pract. Res. Clin. Rheumatology 31 (3), 429–438. doi:10.1016/j.berh.2017.09.010
Moore R., Linas S. (2010). Endothelin Antagonists and Resistant Hypertension in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 19 (5), 432–436. doi:10.1097/mnh.0b013e32833a7a25
Morales J. Y., Young-Stubbs C. M., Shimoura C. G., Kem W. R., Uteshev V. V., Mathis K. W. (2021). Systemic Administration of α7-Nicotinic Acetylcholine Receptor Ligands Does Not Improve Renal Injury or Behavior in Mice with Advanced Systemic Lupus Erythematosus. Front. Med. (Lausanne) 8, 642960. doi:10.3389/fmed.2021.642960
Munguia-Realpozo P., Mendoza-Pinto C., Sierra Benito C., Escarcega R. O., Garcia-Carrasco M., Mendez Martinez S., et al. (2019). Systemic Lupus Erythematosus and Hypertension. Autoimmun. Rev. 18 (10), 102371. doi:10.1016/j.autrev.2019.102371
Najafi C. C., Korbet S. M., Lewis E. J., Schwartz M. M., Reichlin M., Evans J. (2001). Significance of Histologic Patterns of Glomerular Injury upon Long-Term Prognosis in Severe Lupus Glomerulonephritis. Kidney Int. 59 (6), 2156–2163. doi:10.1046/j.1523-1755.2001.00730.x
Ohtsuka T., Hamada M., Hiasa G., Sasaki O., Suzuki M., Hara Y., et al. (2001). Effect of Beta-Blockers on Circulating Levels of Inflammatory and Anti-inflammatory Cytokines in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 37 (2), 412–417. doi:10.1016/s0735-1097(00)01121-9
Okuda T., Grollman A. (1967). Passive Transfer of Autoimmune Induced Hypertension in the Rat by Lymph Node Cells. Tex Rep. Biol. Med. 25 (2), 257–264.
Olofsson P. S., Rosas-Ballina M., Levine Y. A., Tracey K. J. (2012). Rethinking Inflammation: Neural Circuits in the Regulation of Immunity. Immunol. Rev. 248 (1),188–204.
Oparil S., Acelajado M. C., Bakris G. L., Berlowitz D. R., Cífková R., Dominiczak A. F., et al. (2018). Hypertension. Nat. Rev. Dis. Prim. 4, 18014. doi:10.1038/nrdp.2018.14
Oparil S. (2003). Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) Practical Implications. Hypertension 41, 1006–1009. Am Heart Assoc. doi:10.1161/01.hyp.0000070905.09395.f6
Osborn J. W., Fink G. D. (2010). Region-specific Changes in Sympathetic Nerve Activity in Angiotensin II-Salt Hypertension in the Rat. Exp. Physiol. 95 (1), 61–68. doi:10.1113/expphysiol.2008.046326
Osborn J. W., Tyshynsky R., Vulchanova L. (2021). Function of Renal Nerves in Kidney Physiology and Pathophysiology. Annu. Rev. Physiol. 83, 429–450. doi:10.1146/annurev-physiol-031620-091656
Ostrov D. A., Alkanani A., McDaniel K. A., Case S., Baschal E. E., Pyle L., et al. (2018). Methyldopa Blocks MHC Class II Binding to Disease-specific Antigens in Autoimmune Diabetes. J. Clin. investigation 128 (5), 1888–1902. doi:10.1172/jci97739
Panoulas V. F., Douglas K. M. J., Milionis H. J., Stavropoulos-Kalinglou A., Nightingale P., Kita M. D., et al. (2007). Prevalence and Associations of Hypertension and its Control in Patients with Rheumatoid Arthritis. Rheumatology 46 (9), 1477–1482. doi:10.1093/rheumatology/kem169
Pappano A. J. (2010). “Regulation of the Heart and Vasculature,” in Berne and Levy Physiology. sixth editionupdated ed (Philadelphia: Elsevier), 370–392.
Pavlov V. A., Parrish W. R., Rosas-Ballina M., Ochani M., Puerta M., Ochani K., et al. (2009). Brain Acetylcholinesterase Activity Controls Systemic Cytokine Levels through the Cholinergic Anti-inflammatory Pathway. Brain, Behav. Immun. 23 (1), 41–45. doi:10.1016/j.bbi.2008.06.011
Pavlov V., Tracey K. (2017). Neural Regulation of Immunity: Molecular Mechanisms and Clinical Translation. Nat. Neurosci. 20 (2), 156–166.
Petri M., Bechtel B., Dennis G., Shah M., McLaughlin T., Kan H., et al. (2014). Burden of Corticosteroid Use in Patients with Systemic Lupus Erythematosus: Results from a Delphi Panel. Lupus 23 (10), 1006–1013. doi:10.1177/0961203314532699
Petri M. (2004). Cyclophosphamide: New Approaches for Systemic Lupus Erythematosus. Lupus 13 (5), 366–371. doi:10.1191/0961203303lu1028oa
Pham G., Mathis K. (2018). Lipopolysaccharide Challenge Reveals Hypothalamic-Pituitary-Adrenal axis Dysfunction in Murine Systemic Lupus Erythematosus. Brain Sci. 8 (10), 184. doi:10.3390/brainsci8100184
Pham G. S., Wang L. A., Mathis K. W. (2018). Pharmacological Potentiation of the Efferent Vagus Nerve Attenuates Blood Pressure and Renal Injury in a Murine Model of Systemic Lupus Erythematosus. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 315 (6), R1261–R1271. doi:10.1152/ajpregu.00362.2017
Pham G. S., Shimoura C. G., Chaudhari S., Kulp D. V., Mathis K. W. (2020) Chronic Unilateral Cervical Vagotomy Reduces Renal Inflammation, Blood Pressure, and Renal Injury in a Mouse Model of Lupus. Am. J. Physiol. Renal. Physiol. 31 (2), F155–F161.
Pisoni C., Karim Y., Cuadrado M. (2005). Mycophenolate Mofetil and Systemic Lupus Erythematosus: an Overview. Lupus 14 (3_Suppl. l), 9–11. doi:10.1191/0961203305lu2111oa
Platten M., Youssef S., Hur E. M., Ho P. P., Han M. H., Lanz T. V., et al. (2009). Blocking Angiotensin-Converting Enzyme Induces Potent Regulatory T Cells and Modulates TH1- and TH17-Mediated Autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 106 (35), 14948–14953. doi:10.1073/pnas.0903958106
Rainsford K. D., Parke A. L., Clifford-Rashotte M., Kean W. F. (2015). Therapy and Pharmacological Properties of Hydroxychloroquine and Chloroquine in Treatment of Systemic Lupus Erythematosus, Rheumatoid Arthritis and Related Diseases. Inflammopharmacol 23 (5), 231–269. doi:10.1007/s10787-015-0239-y
Ram C. V. S. (2008). Angiotensin Receptor Blockers: Current Status and Future Prospects. Am. J. Med. 121 (8), 656–663. doi:10.1016/j.amjmed.2008.02.038
Rodrigues S. F., de Oliveira M. A., dos Santos R. A., Soares A. G., de Cássia Tostes R., Carvalho M. H., et al. (2008). Hydralazine Reduces Leukocyte Migration through Different Mechanisms in Spontaneously Hypertensive and Normotensive Rats. Eur. J. Pharmacol. 589 (1-3), 206–214. doi:10.1016/j.ejphar.2008.05.003
Rosas-Ballina M., Ochani M., Parrish W. R., Ochani K., Harris Y. T., Huston J. M., et al. (2008). Splenic Nerve is Required for Cholienrgic Antiinflammatory Pathway Control of TNF in Endotoxemia. Proc. Natl. Acad. Sci. 105 (31), 11008–11013.
Rovenský J., Blažíĉková S., Rauovaá L., Ježová D., Koŝka J., Lukáĉ J., et al. (1998). The Hypothalamic-Pituitary Response in SLE. Regulation of Prolactin, Growth Hormone and Cortisol Release. Lupus 7 (6), 409–413. doi:10.1191/096120398678920325
Rovin B. H., Teng Y. K. O., Ginzler E. M., Arriens C., Caster D. J., Romero-Diaz J., et al. (2021). Efficacy and Safety of Voclosporin versus Placebo for Lupus Nephritis (AURORA 1): a Double-Blind, Randomised, Multicentre, Placebo-Controlled, Phase 3 Trial. Lancet 397 (10289), 2070–2080. doi:10.1016/s0140-6736(21)00578-x
Rucker A. J., Rudemiller N. P., Crowley S. D. (2018). Salt, Hypertension, and Immunity. Annu. Rev. Physiol. 80, 283–307. doi:10.1146/annurev-physiol-021317-121134
Ruiz-Ortega M., Lorenzo O., Ruperez M., Egido J. (2000). ACE Inhibitors and AT(1) Receptor Antagonists-Beyond the Haemodynamic Effect. Nephrol. Dial. Transpl. 15 (5), 561–565. doi:10.1093/ndt/15.5.561
Ryan M. J. (2009). Young Investigator Award Lecture of the APS Water and Electrolyte Homeostasis Section, 2008: The Pathophysiology of Hypertension in Systemic Lupus Erythematosus. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 296 (4), R1258–R1267. doi:10.1152/ajpregu.90864.2008
Sabio J. M., Vargas-Hitos J. A., Navarrete-Navarrete N., Mediavilla J. D., Jiménez-jáimez J., Díaz-chamorro A., et al. (2011). Prevalence of and Factors Associated with Hypertension in Young and Old Women with Systemic Lupus Erythematosus. J. Rheumatol. 38 (6), 1026–1032. doi:10.3899/jrheum.101132
Saddala M. S., Lennikov A., Mukwaya A., Yang Y., Hill M. A., Lagali N., et al. (2020). Discovery of Novel L-type Voltage-Gated Calcium Channel Blockers and Application for the Prevention of Inflammation and Angiogenesis. J. Neuroinflammation 17 (132), 1–23. doi:10.1186/s12974-020-01801-9
Saleh M. A., Pollock J. S., Pollock D. M. (2011). Distinct Actions of Endothelin A-Selective versus Combined Endothelin A/B Receptor Antagonists in Early Diabetic Kidney Disease. J. Pharmacol. Exp. Ther. 338 (1), 263–270. doi:10.1124/jpet.111.178988
Santos D. M. d., Da Silva E. A. P., Oliveira J. Y. S., Marinho Y. Y. d. M., Santana I. R. d., Heimfarth L., et al. (2021). The Therapeutic Value of Hydralazine in Reducing Inflammatory Response, Oxidative Stress, and Mortality in Animal Sepsis: Involvement of the PI3K/AKT Pathway. Shock Inj. Inflamm. Sepsis Laboratory Clin. Approaches 56 (5), 782–792. doi:10.1097/shk.0000000000001746
Schiffrin E. L., Touyz R. M. (2003). Multiple Actions of Angiotensin II in Hypertension. J. Am. Coll. Cardiol. 42 (5), 911–913. doi:10.1016/s0735-1097(03)00845-3
Schrezenmeier E., Dörner T. (2020). Mechanisms of Action of Hydroxychloroquine and Chloroquine: Implications for Rheumatology. Nat. Rev. Rheumatol. 16 (3), 155–166. doi:10.1038/s41584-020-0372-x
Sears E. (2008). “New Drugs Approved in 2007,” in Baylor University Medical Center Proceedings (Taylor & Francis), 186–191.
Segarra A., Arredondo K. V., Jaramillo J., Jatem E., Salcedo M. T., Agraz I., et al. (2020). Efficacy and Safety of Bortezomib in Refractory Lupus Nephritis: a Single-Center Experience. Lupus 29 (2), 118–125. doi:10.1177/0961203319896018
Sepehri Z., Masoumi M., Ebrahimi N., Kiani Z., Nasiri A. A., Kohan F., et al. (2016). Atorvastatin, Losartan and Captopril Lead to Upregulation of TGF-β, and Downregulation of IL-6 in Coronary Artery Disease and Hypertension. PLoS One 11 (12), e0168312. doi:10.1371/journal.pone.0168312
Shah D., Mahajan N., Sah S., Nath S. K., Paudyal B. (2014). Oxidative Stress and its Biomarkers in Systemic Lupus Erythematosus. J. Biomed. Sci. 21 (1), 23–13. doi:10.1186/1423-0127-21-23
Shaharir S. S., Mustafar R., Mohd R., Mohd Said M. S., A. Gafor H. (2015). Persistent Hypertension in Lupus Nephritis and the Associated Risk Factors. Clin. Rheumatol. 34 (1), 93–97. doi:10.1007/s10067-014-2802-0
Silva I. V. G., de Figueiredo R. C., Rios D. R. A. (2019). Effect of Different Classes of Antihypertensive Drugs on Endothelial Function and Inflammation. Int. J. Mol. Sci. 20 (14), 3458. doi:10.3390/ijms20143458
Small H. Y., Migliarino S., Czesnikiewicz-Guzik M., Guzik T. J. (2018). Hypertension: Focus on Autoimmunity and Oxidative Stress. Free Radic. Biol. Med. 125, 104–115. doi:10.1016/j.freeradbiomed.2018.05.085
Stanton B. A., Koeppen B. M. (2010). “Control of Body Fluid Osmolality and Volume,” in Berne and Levy Physiology (Pliladelphia Elsevier), 594–618.
Stein K., McFarlane I., Goldberg N., Ginzler E. (1996). Heart Rate Variability in Patients with Systemic Lupus Erythematosus. Lupus 5 (1), 44–48. doi:10.1177/096120339600500109
Sthoeger Z. M., Zinger H., Mozes E. (2003). Beneficial Effects of the Anti-oestrogen Tamoxifen on Systemic Lupus Erythematosus of (NZBxNZW)F1 Female Mice Are Associated with Specific Reduction of IgG3 Autoantibodies. Ann. rheumatic Dis. 62 (4), 341–346. doi:10.1136/ard.62.4.341
Suárez-Fueyo A., Bradley S. J., Klatzmann D., Tsokos G. C. (2017). T Cells and Autoimmune Kidney Disease. Nat. Rev. Nephrol. 13 (6), 329–343. doi:10.1038/nrneph.2017.34
Suzuki Y., Ruiz-Ortega M., Lorenzo O., Ruperez M., Esteban V., Egido J. (2003). Inflammation and Angiotensin II. Int. J. Biochem. Cell Biol. 35 (6), 881–900. doi:10.1016/s1357-2725(02)00271-6
Svendsen U. G. (1976). Evidence for an Initial, Thymus Independent and a Chronic, Thymus Dependent Phase of DOCA and Salt Hypertension in Mice. Acta Pathol. Microbiol. Scand. A 84 (6), 523–528. doi:10.1111/j.1699-0463.1976.tb00150.x
Tanaka Y., Tummala R. (2021). Anifrolumab, a Monoclonal Antibody to the Type I Interferon Receptor Subunit 1, for the Treatment of Systemic Lupus Erythematosus: An Overview from Clinical Trials. Mod. Rheumatol. 31 (1), 1–12. doi:10.1080/14397595.2020.1812201
Taylor E. B., Barati M. T., Powell D. W., Turbeville H. R., Ryan M. J. (2018). Plasma Cell Depletion Attenuates Hypertension in an Experimental Model of Autoimmune Disease. Hypertension 71 (4), 719–728. doi:10.1161/hypertensionaha.117.10473
Taylor E. B., Ryan M. J. (2017). Immunosuppression with Mycophenolate Mofetil Attenuates Hypertension in an Experimental Model of Autoimmune Disease. J. Am. Heart Assoc. 6 (3). doi:10.1161/JAHA.116.005394
Thanou A., Stavrakis S., Dyer J. W., Munroe M. E., James J. A., Merrill J. T. (2016). Impact of Heart Rate Variability, a Marker for Cardiac Health, on Lupus Disease Activity. Arthritis Res. Ther. 18, 197. doi:10.1186/s13075-016-1087-x
Thanou K. (2014). “Heart Rate Variabilty: New Window in Lupus Disease Activity,” in 14th Annual Meeting Of the American College Of Rheumatology.
Tracey K. J. (2021). Hacking the Inflammatory Reflex. Lancet Rheumatology 3 (4), e237–e239. doi:10.1016/s2665-9913(20)30448-3
Tracey K. J. (2007) Physiology and Immunology of the Cholinergic Antiinflammatory Pathway. J. Clin. Invest. 117 (2), 289–296.
Tselios K., Gladman D. D., Su J., Urowitz M. (2020). Impact of the New American College of Cardiology/American Heart Association Definition of Hypertension on Atherosclerotic Vascular Events in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 79 (5), 612–617. doi:10.1136/annrheumdis-2019-216764
Tsioufis C., Thomopoulos C. (2017). Combination Drug Treatment in Hypertension. Pharmacol. Res. 125 (Pt B), 266–271. doi:10.1016/j.phrs.2017.09.011
Tsumiyama K., Hashiramoto A., Takimoto M., Tsuji-Kawahara S., Miyazawa M., Shiozawa S. (2013). IFN-γ-Producing Effector CD8 T Lymphocytes Cause Immune Glomerular Injury by Recognizing Antigen Presented as Immune Complex on Target Tissue. J. Immunol. 191 (1), 91–96. doi:10.4049/jimmunol.1203217
Tumurkhuu G., Montano E., Jefferies C. (2019). Innate Immune Dysregulation in the Development of Cardiovascular Disease in Lupus. Curr. Rheumatol. Rep. 21 (9), 46–10. doi:10.1007/s11926-019-0842-9
Tuttolomondo A., Maida C., Casuccio A., Di Raimondo D., Fonte R., Vassallo V., et al. (2021). Effects of Intravenous Furosemide Plus Small‐volume Hypertonic Saline Solutions on Markers of Heart Failure. Esc. Heart Fail. 8 (5), 4174–4186. doi:10.1002/ehf2.13511
Unger T., Borghi C., Charchar F., Khan N. A., Poulter N. R., Prabhakaran D., et al. (2020). 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 75, 1334–1357. doi:10.1161/hypertensionaha.120.15026
Van Beusecum J. P., Moreno H., Harrison D. G. (2021). Innate Immunity and Clinical Hypertension. J. Hum. Hypertens., 1–7. doi:10.1038/s41371-021-00627-z
Vanegas V., Ferrebuz A., Quiroz Y., Rodríguez-Iturbe B. (2005). Hypertension in Page (Cellophane-wrapped) Kidney Is Due to Interstitial Nephritis. Kidney Int. 68 (3), 1161–1170. doi:10.1111/j.1523-1755.2005.00508.x
Venegas-Pont M., Sartori-Valinotti J. C., Maric C., Racusen L. C., Glover P. H., McLemore G. R., et al. (2009). Rosiglitazone Decreases Blood Pressure and Renal Injury in a Female Mouse Model of Systemic Lupus Erythematosus. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 296 (4), R1282–R1289. doi:10.1152/ajpregu.90992.2008
Venegas-Pont M., Manigrasso M. B., Grifoni S. C., LaMarca B. B., Maric C., Racusen L. C., et al. (2010). Tumor Necrosis Factor-α Antagonist Etanercept Decreases Blood Pressure and Protects the Kidney in a Mouse Model of Systemic Lupus Erythematosus. Hypertension 56 (4), 643–649. doi:10.1161/hypertensionaha.110.157685
Verlinden T., Rijkers K., Hoogland G., Herrler A. (2016). Morphology of the Human Cervical Vagus Nerve: Implications for Vagus Nerve Stimulation Treatment. Acta. Neurol. Scand. 133, 173–182.
Wang Z., Vilekar P., Huang J., Weaver D. F. (2020). Furosemide as a Probe Molecule for the Treatment of Neuroinflammation in Alzheimer's Disease. ACS Chem. Neurosci. 11 (24), 4152–4168. doi:10.1021/acschemneuro.0c00445
Weide R., Heymanns J., Pandorf A., Köppler H. (2003). Successful Long-Term Treatment of Systemic Lupus Erythematosus with Rituximab Maintenance Therapy. Lupus 12 (10), 779–782. doi:10.1191/0961203303lu449cr
WHO (2021). Guideline for the Pharmacological Treatment of Hypertension in Adults: Web Annex A: Summary of Evidence. Geneva: World Health Organization.
Wu W.-M., Lin B.-F., Su Y.-C., Suen J.-L., Chiang B.-L. (2000). Tamoxifen Decreases Renal Inflammation and Alleviates Disease Severity in Autoimmune NZB/W F1 Mice. Scand. J. Immunol. 52 (4), 393–400. doi:10.1046/j.1365-3083.2000.00789.x
Xiao L., Kirabo A., Wu J., Saleh M. A., Zhu L., Wang F., et al. (2015). Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ. Res. 117 (6), 547–557. doi:10.1161/CIRCRESAHA.115.306010
Xu B., Makris A., Thornton C., Ogle R., Horvath J. S., Hennessy A. (2006). Antihypertensive Drugs Clonidine, Diazoxide, Hydralazine and Furosemide Regulate the Production of Cytokines by Placentas and Peripheral Blood Mononuclear Cells in Normal Pregnancy. J. Hypertens. 24 (5), 915–922. doi:10.1097/01.hjh.0000222762.84605.03
Yen T.-H., Yang H.-Y., Yeh Y.-H., Chu P.-H., Wen C.-J., Fu J.-F., et al. (2013). Aliskiren Attenuates Proteinuria in Mice with Lupus Nephritis by a Blood Pressure-independent Mechanism. Lupus 22 (2), 180–189. doi:10.1177/0961203312471871
Yuengsrigul A., Chin T. W., Nussbaum E. (1999). Immunosuppressive and Cytotoxic Effects of Furosemide on Human Peripheral Blood Mononuclear Cells. Ann. Allergy, Asthma & Immunol. 83 (6), 559–566. doi:10.1016/s1081-1206(10)62870-0
Zhang C., Hein T. W., Wang W., Kuo L. (2003). Divergent Roles of Angiotensin II at 1 and at 2 Receptors in Modulating Coronary Microvascular Function. Circulation Res. 92 (3), 322–329. doi:10.1161/01.res.0000056759.53828.2c
Zhang L.-l., Lu J., Li M.-t., Wang Q., Zeng X.-f. (2016). Preventive and Remedial Application of Etanercept Attenuate Monocrotaline-Induced Pulmonary Arterial Hypertension. Int. J. Rheum. Dis. 19 (2), 192–198. doi:10.1111/1756-185x.12304
Zheng Y., Li M., Zhang Y., Shi X., Li L., Jin M. (2010). The Effects and Mechanisms of Mycophenolate Mofetil on Pulmonary Arterial Hypertension in Rats. Rheumatol. Int. 30 (3), 341–348. doi:10.1007/s00296-009-0966-8
Zhou B., Carrillo-Larco R. M., Danaei G., Riley L. M., Paciorek C. J., Stevens G. A., et al. (2021). Worldwide Trends in Hypertension Prevalence and Progress in Treatment and Control from 1990 to 2019: a Pooled Analysis of 1201 Population-Representative Studies with 104 Million Participants. Lancet 398 (10304), 957–980. doi:10.1016/S0140-6736(21)01330-1
Keywords: resistant hypertension, blood pressure, immune cells, kidney, autoimmunity, systemic lupus erythematosus, lupus
Citation: Chaudhari S, Pham GS, Brooks CD, Dinh VQ, Young-Stubbs CM, Shimoura CG and Mathis KW (2022) Should Renal Inflammation Be Targeted While Treating Hypertension?. Front. Physiol. 13:886779. doi: 10.3389/fphys.2022.886779
Received: 28 February 2022; Accepted: 17 May 2022;
Published: 13 June 2022.
Edited by:
Hana A. Itani, American University of Beirut, LebanonReviewed by:
Adam J. Case, Texas A&M University, United StatesCopyright © 2022 Chaudhari, Pham, Brooks, Dinh, Young-Stubbs, Shimoura and Mathis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keisa W. Mathis, a2Vpc2EubWF0aGlzQHVudGhzYy5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.