 Kensuke Ihara
Kensuke Ihara Tetsuo Sasano
Tetsuo Sasano
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 14 April 2022
Sec. Cardiac Electrophysiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.862164
This article is part of the Research Topic Interplay between the heart and the immune system: Focus on heart rhythm regulation View all 5 articles
Atrial fibrillation (AF) is one of the most common arrhythmias encountered in clinical practice. AF is a major risk factor for stroke, which is associated with high mortality and great disability and causes a significant burden on society. With the development of catheter ablation, AF has become a treatable disease, but its therapeutic outcome has been limited so far. In persistent and long-standing AF, the expanded AF substrate is difficult to treat only by ablation, and a better understanding of the mechanism of AF substrate formation will lead to the development of a new therapeutic strategy for AF. Inflammation is known to play an important role in the substrate formation of AF. Inflammation causes and accelerates the electrical and structural remodeling of the atria via pro-inflammatory cytokines and other inflammatory molecules, and enhances the AF substrate, leading to the maintenance of AF and further inflammation, which forms a vicious spiral, so-called “AF begets AF”. Breaking this vicious cycle is expected to be a key therapeutic intervention in AF. In this review, we will discuss the relationship between AF and inflammation, the inflammatory molecules included in the AF-related inflammatory process, and finally the potential of those molecules as a therapeutic target.
Atrial fibrillation (AF) is one of the most common arrhythmias encountered in clinical practice (Chugh et al., 2014) and is associated with severe life-threatening diseases such as cerebral infarction and heart failure resulting in a major social burden (Ball et al., 2013). Although some cases of AF are familial, AF is commonly thought to be a multifactorial disease that is caused by a combination of genetic factors, comorbidities, lifestyle, and aging (Said et al., 2018; Roselli et al., 2020). AF is initiated mainly by ectopic electrical excitation originating from the pulmonary veins (PV) (Haissaguerre et al., 1998), but if there is no arrhythmogenic substrate in the atria to maintain AF, AF does not persist. On the other hand, with the firm AF substrate, once AF starts, it does not stop spontaneously and persists for a long time. This is called persistent AF or long-standing AF (de Vos et al., 2010; Heijman et al., 2014).
In general, the therapeutic efficacy of antiarrhythmic drugs for AF is limited, and catheter ablation is effective, especially in symptomatic cases (Mark et al., 2019). Catheter ablation, the main therapeutic strategy of AF in the current medicine, is based on electrical PV isolation, which can eliminate the AF trigger (Calkins et al., 2017). Paroxysmal AF, which is an early stage of AF, can be treated with PV isolation alone with good outcomes. However persistent or long-standing AF in which AF substrate is firmly formed is known to have poor outcomes even with the repeated procedures (Arbelo et al., 2014) and the various ablation strategies (Verma et al., 2015). In the treatment of AF, the development of effective therapeutic strategies for AF substrate is a matter of the highest priority.
It is well known that inflammation is deeply involved in the formation of the AF substrate. Understanding its mechanism will lead to the development of new therapeutic targets and strategies. In addition, it is now known that not only inflammation contributes to the development of AF, but also that AF itself can induce inflammation, indicating that inflammation and AF form a vicious spiral. This review will discuss the interaction between inflammation and AF and the key molecules that contribute to it.
The incidence of AF increases in the presence of systemic inflammation. For example, it is known that patients with sepsis have a higher incidence of AF (Seguin et al., 2004; Christian et al., 2008). Meierhenrich et al. reported that the overall incidence of new AF in the patients admitted to the intensive care unit was 7.8%, while the incidence was much higher in septic patients (46%), and the elevation in plasma C-reactive protein (CRP) level was found before the onset of AF in AF group (Meierhenrich et al., 2010).
Patients with rheumatoid arthritis (RA) with active inflammation are also known to have a high incidence of AF (Lazzerini et al., 2017; Ungprasert et al., 2017). Although the patients with RA also have a high incidence of heart failure and ischemic heart disease, which are risk factors of AF development, the presence of systemic inflammation is an independent risk factor for AF development in them. Severe extra-articular manifestations and erythrocyte sedimentation rate are most associated with the development of AF in patients with RA (Bacani et al., 2015), suggesting that systemic inflammation contributes to the development of AF. Furthermore, patients with psoriasis, a chronic inflammatory disease, also have an increased risk of AF, and the risk of the development of AF is associated with its disease severity (Ahlehoff et al., 2012). Patients with inflammatory bowel disease, including Crohn’s disease and ulcerative colitis, which are also chronic inflammatory diseases, are reported to have an increased risk of developing AF, especially in Crohn’s disease (Choi et al., 2019). Common diseases such as hypertension and obesity are also known to be risk factors for AF (Kannel et al., 1998; Wang et al., 2004) and to cause systemic inflammation (Harrison et al., 2011; Nalliah et al., 2016). Finally, it is well known that high levels of inflammatory biomarkers are associated with the risk of developing AF even in the general population, suggesting that the presence of systemic and extracardiac inflammation may contribute to the development of AF.
Coronavirus disease-2019 (COVID-19) outbreak began at the end of 2019, and as of February 2022, the COVID-19 pandemic has reached 400 million infected people, 5.7 million dead, and no end in sight. AF is also a major clinical problem with COVID-19. In a meta-analysis of 21,582 hospitalized patients with COVID-19, AF was found in 11%, and the presence of AF was associated with an increase in all-cause mortality (OR 2.98) (Li et al., 2021). Also, in a recent report by Musikantow et al., the incidence of AF and atrial flutter (AFL) in hospitalized patients with COVID-19 was 10%, and new-onset AF/AFL was found in 4%. In this study, high levels of interleukin-6 (IL-6) and CRP in the blood were associated with the development of AF/AFL. When compared to patients with COVID-19, the incidence of AF/AFL was comparable in patients hospitalized for influenza, suggesting that the development of AF/AFL is not a specific issue to COVID19, but it was thought to be the result of systemic inflammation caused by severe viral infection (Musikantow et al., 2021).
Many studies investigated the involvement of inflammatory molecules in the initiation and progression of AF in the pathophysiological aspect and reported the utility of these molecules as a biomarker (Table 1).
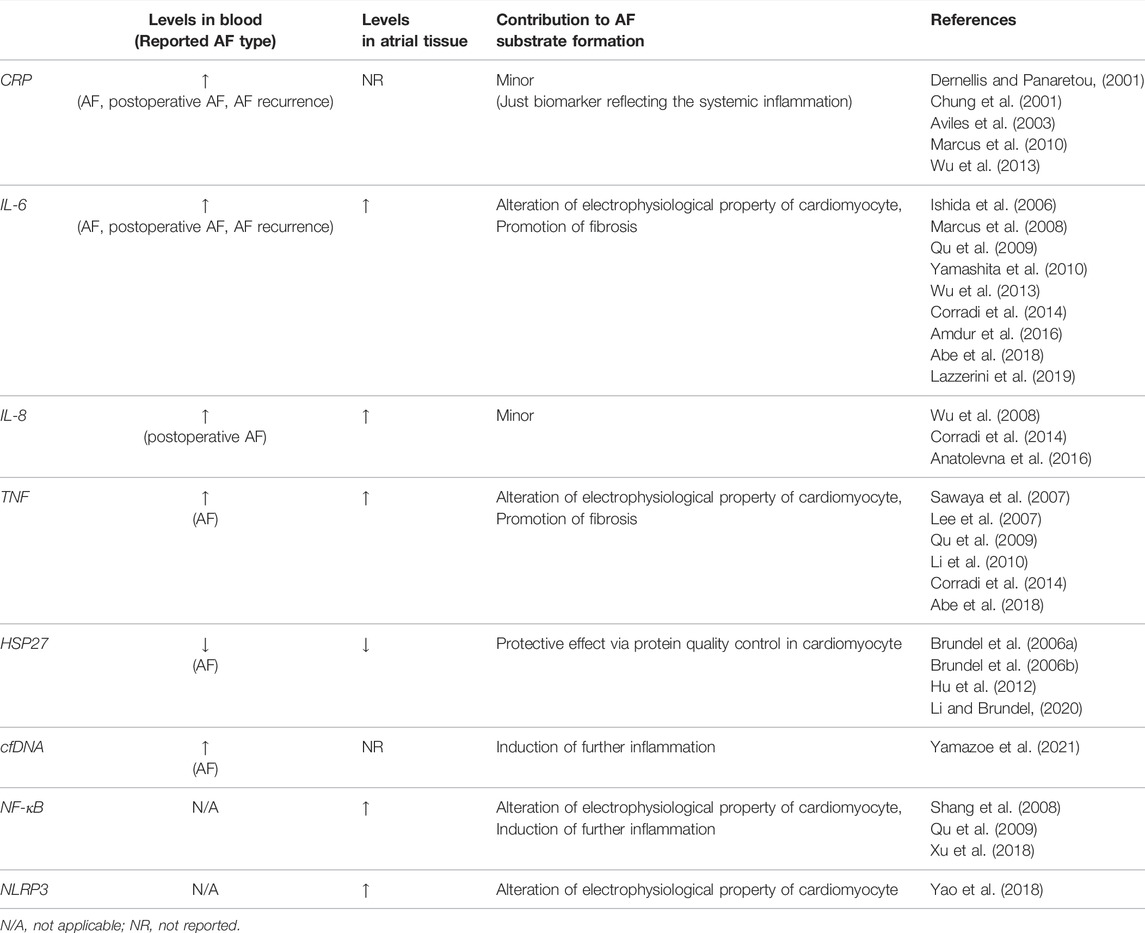
TABLE 1. Summary of the main inflammatory molecules in AF pathogenesis.
CRP, including high-sensitivity CRP (hs-CRP), is a biomarker most used in clinical practice to indicate the presence and severity of inflammation. Pro-inflammatory cytokines such as IL-6 and tumor necrosis factor (TNF, also known as TNFα) that are produced by monocytes and macrophages mainly act on hepatocytes, leading to the production of CRP. Many clinical studies reported the association between the level of CRP in the blood and AF. A high level of CRP has been observed in patients with paroxysmal AF (Dernellis and Panaretou, 2001), and even a higher level of CRP is observed in patients with persistent AF (Chung et al., 2001). In patients with paroxysmal AF, CRP level is higher during AF than during sinus rhythm (Marcus et al., 2010). Aviles et al. reported that CRP level was elevated in patients with AF, and that the undiagnosed patients with elevated CRP levels have a significantly higher incidence of AF during follow-up (Aviles et al., 2003). These findings of CRP indicate that the presence of inflammation contributes to the development of AF, however, they can also be interpreted as evidence that AF causes inflammation. A few reports are suggesting that CRP itself may contribute to the development of AF (Narducci et al., 2011; Chang et al., 2012).
IL-6 is a pro-inflammatory cytokine that is secreted by a wide range of cell types including endothelial cells, fibroblasts, myocytes, adipocytes, as well as macrophages and T cells (Hoene and Weigert, 2008). IL-6 has been reported to be useful as a biomarker predicting the development of AF in a limited group of patients, including patients with coronary artery disease and chronic kidney disease (Marcus et al., 2008; Wu et al., 2013; Amdur et al., 2016).
TNF is a pro-inflammatory cytokine that is mainly secreted by macrophages, however, it is also known to be secreted by a wide range of cell types including both immune and non-immune cells. Even cardiac fibroblasts and cardiomyocytes isolated from neonatal rats produce TNF upon lipopolysaccharide stimulation (Yokoyama et al., 1999). Concerning AF, serum TNF level is significantly higher in patients with AF especially in the patients with persistent or long-standing AF rather than paroxysmal AF (Li et al., 2010).
Heat shock proteins are important regulators of the inflammatory response (van Eden et al., 2005), and heat shock protein 27 (HSP27) induced by geranylgeranylacetone has been reported to prevent AF development in dogs (Brundel et al., 2006b). Brundel et al. also reported that in human atrial tissue, the expression levels of HSP27 were upregulated in patients with paroxysmal AF compared to that in control or in patients with persistent AF, and in murine atrial cardiomyocytes (HL-1 cells), induction of HSP27 had a protective effect on tachypacing-induced myolysis, indicating that elevated expression of HSP27 in human atrium might prevent AF substrate formation by its protective effect on atrial myolysis (Brundel et al., 2006a). HSP27 may not only prevent the development of AF controlling protein quality in cardiomyocytes, but also by suppression of inflammation (Li and Brundel, 2020). Hu et al. reported that serum HSP27 level was relatively low in patients with AF and inversely correlated with the atrial diameter and that HSP27 regulates TNF and IL-10 expression level, resulting in a low incidence of AF (Hu et al., 2012).
IL-8, also known as a neutrophil chemotactic factor, is produced by many cell types, including vascular endothelial cells as well as monocytes and macrophages (Liuba et al., 2008). A high level of IL-8 in the blood may be a predictor of the development of postoperative AF in the patients after coronary artery bypass graft (CABG) surgery (Wu et al., 2008; Anatolevna et al., 2016). In terms of postoperative AF, not only IL-8 but also Plasma IL-6 level is reported to be useful for predicting postoperative AF in the patients after CABG (Ishida et al., 2006). Other than the above, many inflammatory mediators, such as IL-1, IL-2, IL-10, IL-18, MCP1, and so on, are reported to be associated with AF (Hu et al., 2015).
Since AF is generally difficult to diagnose in its early stage with paroxysmal form, the application of these inflammation-related molecules as biomarkers is very useful to predict the onset of AF. Although all of these clinical findings suggest a strong association between AF and systemic inflammation, it is important to note that they did not prove their direct causal relationship. In addition, these studies about AF biomarkers may report different data depending on the study design and the study population, so the interpretation of the results should be considered carefully.
Although the various possible electrophysiological mechanisms for maintaining AF, such as multiple reentrant wavelets hypothesis and rotor hypothesis (Moe and Abildskov, 1959; Winfree, 1991; Jalife et al., 2002) are presented, these hypotheses are all premised on the alteration of electrophysiological properties and structure of the atrial tissue (electrical remodeling and structural remodeling), such as decreased conduction velocity, shortened action potential duration (APD), and their heterogeneity within the atrial tissue and enlargement of the atrium. The presence of inflammation in the atrial tissue is thought to be critical for these remodelings (Nattel et al., 2008). In fact, the infiltration of the inflammatory cells into the atrial tissue has been found in patients with AF (Chen et al., 2008), and especially the infiltration of CD68 positive macrophages has been observed (Smorodinova et al., 2017). Yamashita et al. reported that the production of IL-6 and transforming growth factor β (TGFβ) was increased in macrophages in atrial tissue of patients with AF (Yamashita et al., 2010). Furthermore, the expression of cytokines such as IL-6, IL-8, IL-10, and TNF is increased in the atrial tissue of patients with AF (Qu et al., 2009; Corradi et al., 2014), suggesting that the local production of these cytokines is enhanced by inflammatory cell infiltration.
The inflammatory molecules act directly on the atrial cardiomyocytes, altering their electrophysiological properties and contributing to generating atrial arrhythmogenesis.
Lazzeriniet al. reported that the parameters of the P wave on the body-surface 12-lead electrocardiogram (ECG) normalized along with the improvement of inflammatory biomarkers in the patients with inflammatory states, such as infection and autoimmune diseases (Lazzerini et al., 2019). In particular, serum IL-6 level correlated to P wave width and dispersion. They also demonstrated that the expression of both connexin40 (Cx40) and Cx43 in peripheral blood mononuclear cells strongly correlated with those in the atrial tissue, and that the expression of both Cx40 and Cx43 in peripheral blood was significantly decreased in patients with inflammatory diseases during the inflammatory phase compared with healthy subjects, and especially serum IL-6 level inversely correlated with the Cx43 expression level. In vitro experiments showed that direct exposure of IL-6 to HL-1 cells induced the rapid decrease of the expression of Cx40 and Cx43, and that the expression of them rapidly recovered upon cessation of IL-6 exposure (Lazzerini et al., 2019), suggesting that IL-6 regulates the expression of gap junction channels on cardiomyocytes, and contributes to forming the AF substrate. IL-6 is also known to act directly on murine or rat cardiomyocytes to alter their electrophysiological properties, such as enhancing ICaL and decreasing Sarco/endoplasmic reticulum calcium ATPase-2 (SERCA2) expression (Villegas et al., 2000; Tanaka et al., 2004; Hagiwara et al., 2007).
TNF has also been reported to have a direct effect on cardiomyocytes by altering their electrophysiological properties. In the mutant mice with cardiomyocyte-specific overexpression of TNF, atrial arrhythmias including AF were observed. In this paper, the decreased atrial conduction velocity was observed in the mutant mice accompanied by the decreased expression of Cx40 and the dyslocalization of Cx43 in the atrial cardiomyocytes (Sawaya et al., 2007). TNF is also reported to act directly on murine cardiomyocytes and to cause abnormal Ca handling (Saba et al., 2005; Kao et al., 2010; Zuo et al., 2018). Lee et al. reported that direct TNF treatment of the rabbit isolated PV cardiomyocytes resulted in decreased SERCA2 expression and abnormal Ca handling leading to increased susceptibility to delayed afterdepolarization (DAD) (Lee et al., 2007).
Nuclear factor-kappaB (NF-κB) is a transcription factor that regulates the transcription of inflammatory cytokines such as TNF, IL-1, IL-6, and IL-8, and is an important key regulator of inflammatory responses (Baeuerle and Henkel, 1994). NF-κB is expressed not only in immune cells but also in various cell types and is activated by various stress stimuli to induce inflammation. TLR4-MyD88 pathway is known to be one pathway that activates NF-κB (Byrd-Leifer et al., 2001). It has been reported that MyD88 protein and phosphorylated NF-κB are increased in atrial tissue of patients with AF (Xu et al., 2018), and NF-κB may also contribute to atrial inflammation in AF. Furthermore, NF-κB not only induces and exacerbates inflammation but also suppresses Scn5a gene expression in rat cardiomyocytes, which may contribute to electrical remodeling in AF (Shang et al., 2008). NF-κB also regulates the transcription of NLR family pyrin domain containing 3 (NLRP3) (He et al., 2016).
NLRP3 inflammasome consists of NLRP3, apoptosis-associated speck-like protein containing a CARD and caspase 1 and plays a central role in innate immunity. NLRP3 inflammasomes are also reported to be associated with atrial electrical remolding. Yao et al. reported that the activity of NLRP3 inflammasomes was increased in atrial cardiomyocytes in human atrial tissue, cardiomyocyte-specific NLRP3 transgenic mice, and a canine model of atrial tachy-pacing. Especially in cardiomyocyte-specific NLRP3 transgenic mice, the enhanced activity of NLRP3 inflammasomes in atrial cardiomyocytes was accompanied by the increased gene expression of Ryr2 and Kcna5 resulting in ectopic electrical excitation and shortening of the effective refractory period, and might contribute to the development of AF (Yao et al., 2018).
Not only inflammatory molecules but also inflammatory cells themselves interact directly with cardiomyocytes to alter their electrophysiological properties (Grune et al., 2021). It has been known that cardiac fibroblasts express connexins and interact with cardiomyocytes via gap junctions (Camelliti et al., 2004), and recently macrophages are also found to form gap junctions with cardiomyocytes (Hulsmans et al., 2017). Hulsmans et al. reported that the gap junction between cardiomyocytes and macrophages with Cx43 regulates APD and resting membrane potential of cardiomyocytes in the mouse atrioventricular (AV) node. In addition, AV block was induced in macrophage-specific Cx43 knockout and macrophage-deficient mice (Hulsmans et al., 2017). Currently, there is no evidence that the direct electrophysiological effects of noncardiomyocytes on cardiomyocytes contribute to the development of AF, and further investigation on their role in the formation of AF substrate is warranted.
Atrial fibrosis plays a major role in the formation of AF substrate by decreasing atrial conduction velocity and inducing its heterogeneity (Li et al., 1999). Cardiac fibrosis is caused by increased the production of extracellular matrix, which is induced by the activation of cardiac fibroblasts and their differentiation into myofibroblasts, and TGFβ is an important mediator of their activation (Nattel, 2017). Mechanical stretch also can activate rat cardiac fibroblasts in vitro, and enhance the promoter activity of alpha-smooth muscle actin, which expression is required for myofibroblast differentiation (Zhao et al., 2007). Recruitment of macrophages is critical for tissue fibrosis because they are an important source of IL-6 and TGFβ (Ma et al., 2012; Kong et al., 2014). Inflammatory cytokines such as TNF, IL-1β, and IL-6 can enhance the proliferation and activation of cardiac fibroblasts, and cardiac fibroblasts themselves secrete them and enhance the fibrosis (Travers et al., 2016).
Obesity is one of the most important risk factors for AF (Wang et al., 2004). Recently, epicardial adipose tissue (EAT) has been well studied as a source of inflammation and a risk factor for AF. The amount of EAT detected by the imaging modality, such as computed tomography and magnetic resonance imaging, is considered to be critical for developing AF (Zhou et al., 2020). Thanassoulis et al. reported that EAT is an independent risk factor for AF, whereas non-EAT fat (e.g. visceral fat) is not a risk factor for AF in a cohort (Thanassoulis et al., 2010). Al Chekakie et al. reported that the amount of EAT was significantly increased in patients with persistent AF with advanced left atrial remodeling compared with those with paroxysmal AF (Al Chekakie et al., 2010). Human EAT secretes cytokines such as IL-6, TNF, and TGFβ, which cause inflammation and subsequent fibrosis via a paracrine effect in adjacent atrial tissue (Mazurek et al., 2003; Iacobellis and Barbaro, 2008). Abe et al. investigated peri-atrial EAT in detail with the specimens of human left atrial appendage obtained from cardiac surgery patients and found that EAT invaded into the atrial tissue, and fibrosis was mainly observed at the boundary between EAT and atrial tissue (Abe et al., 2018). In this investigation, the amount of EAT and the severity of atrial fibrosis were associated with the presence of AF, and the expression of pro-inflammatory cytokines (IL-1β, IL-6, TNF, and so on) in the EAT was correlated with atrial fibrosis. The EAT is thought to induce atrial inflammation by increasing its amount and/or secreting pro-inflammatory cytokines as a response to systemic inflammation (Packer, 2018).
Local atrial inflammation can be induced by direct surgical invasion (Ishii et al., 2005; Maesen et al., 2012) or direct atrial pressure overload, such as heart failure (Verheule et al., 2003; Liao et al., 2010), and contributes to the development of AF. In our investigation, mechanical stretch to HL-1 cells induced the extracellular release of adenosine triphosphate (ATP) via pannexin-2 which is the main isoform of pannexin in the atrium, and subsequent macrophage migration. ATP released through pannexin-1 of apoptotic cells is well known to work as a find-me signal and recruit the macrophages (Chekeni et al., 2010), and ATP released through pannexin-2 is thought to work in the same manner. Furthermore, when pressure overload was applied to mouse heart by transverse aortic constriction, macrophages infiltrated into the atrial tissue, however, the administration of carbenoxolone, a pannexin-2 blocker, attenuated this macrophage infiltration, suggesting that atrial stretch in heart failure may cause the release of ATP from cardiomyocytes leading to atrial infiltration of macrophage and subsequent atrial inflammation (Oishi et al., 2012).
AF and inflammation are often observed together, and there is no doubt that they are deeply related. However, while many reports indicate that inflammation forms AF substrate, there are not many studies directly demonstrating that AF causes inflammation, and the mechanism by which AF induces inflammation remains unclear (Hu et al., 2015). In animal studies, atrial tachy-pacing in dogs was reported to cause the elevation of IL-6 and TNF in blood and atrial tissue (Zhao et al., 2018) and the activation of NLRP3 inflammasome in atrial cardiomyocytes (Yao et al., 2018) suggesting that atrial high-frequent contraction and excitation can cause inflammation. In the clinical study with the patients with persistent AF, defibrillation, and maintenance of sinus rhythm decrease plasma CRP level (Kallergis et al., 2008), suggesting that AF is the cause of inflammation.
Recently, we have researched cell-free DNA (cfDNA) in the blood to investigate the relationship between cfDNA and AF (Yamazoe et al., 2021). cfDNA is secreted by various types of cells and circulates in the blood. In the field of oncology, the mutation analysis of circulating tumor DNA released from cancer cells has been performed for early detection of cancer, prediction of sensitivity to anticancer drugs, and monitoring of recurrence after cancer treatment (Schwarzenbach et al., 2011). We quantified cfDNA from peripheral blood in patients with AF and found that the amount of cfDNA was higher in the elderly than in the young, in patients with AF than in healthy subjects, and in patients with persistent AF than in patients with paroxysmal AF. We also compared the copy numbers of nuclear-cfDNA (n-cfDNA) and mitochondrial-cfDNA (mt-cfDNA) and found that the copy number of mt-cfDNA correlated more with the presence of AF than that of n-cfDNA, suggesting that cfDNA, especially mt-cfDNA, may be useful as a biomarker for detecting AF. Furthermore, we found that the pacing of HL-1 cells in vitro increased cfDNA in the culture media and that the administration of the cfDNA to macrophages (J774.1 cells) increased the expression of IL-1β and IL-6. In particular, this increase in cytokine expression was significantly induced by mt-cfDNA but not by n-cfDNA, suggesting that unmethylated mt-cfDNA induces a TLR-9 mediated pre-immune response and that AF can induce systemic inflammation via cfDNA (Yamazoe et al., 2021).
Commonly referred to as “AF begets AF” (Wijffels et al., 1995), once AF occurs, electrical remodeling and structural remodeling is progressed and form further AF substrate, making AF easy to occur and sustain. As shown above, inflammation plays an important role in the vicious spiral through various inflammatory molecules, inflammatory cells, and fibrosis. Not only local but also systemic and extracardiac inflammation induces the generation of AF substrate and AF, in turn, induces local and systemic inflammation (Figure 1).
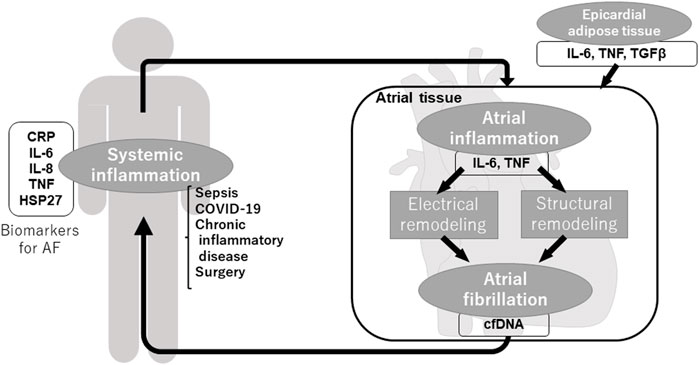
FIGURE 1. Inflammation and atrial fibrillation.
Inflammation is a major contributor to thrombus formation (Stark and Massberg, 2021). Cerebral embolism caused by thrombus in the left atrium (LA) is one of the most serious complications of AF, and inflammation is also involved in thrombus formation in AF. Stroke has been reported to be associated with the presence of systemic inflammation and increased inflammatory biomarkers in the blood, and plasma CRP level has been linked with the risk of stroke in patients with AF (Lip et al., 2007). Plasma IL-6 level is also important for predicting the complication of thromboembolism and the prognosis of vascular death in patients with AF (Aulin et al., 2015; Hijazi et al., 2016). The previous report has shown that high plasma CRP levels are associated with the presence of intracardiac thrombus and spontaneous echo contrast in the LA on transesophageal echocardiography in patients with AF (Cianfrocca et al., 2010). In addition, another report showed that high CRP levels in the blood correlate with blood flow velocity and shear rate in the LA appendage (Thambidorai et al., 2004), suggesting that the presence of inflammation contributes to dysfunction of LA appendage and thrombus formation. Lim et al. reported that in patients who underwent catheter ablation, AF caused increased platelet activation, thrombin generation, atrial endothelial dysfunction, and inflammation, whereas rapid pacing did not cause endothelial dysfunction and inflammation (Lim et al., 2013).
Inflammation in AF is important for AF development, but it also contributes to endothelial damage and dysfunction, platelet activation, and activation of the coagulation cascade, and, together with AF-induced loss of contractility of the LA, is thought to play a critical role in LA thrombus formation (Harada et al., 2015).
Anti-inflammatory drugs and drugs targeting inflammatory molecules have been investigated as potential drugs for the treatment of AF. Steroids are widely used as anti-inflammatory agents, and their therapeutic effects on AF have been extensively studied.
Steroids are commonly used for the treatment of severe infection, and it is reported that steroids reduced the incidence of AF in patients with sepsis (Launey et al., 2019). In patients with persistent AF, administration of steroids significantly reduced the plasma CRP level and the recurrence of AF (50–9.6%) during 2 years follow up after pharmacological or electrical defibrillation (Dernellis and Panaretou, 2004). Kim et al. reported that short-term steroid treatment can suppress the early recurrence of AF (48–23%) in patients with AF who underwent catheter ablation (Kim et al., 2015). Postoperative AF was also suppressed by steroid therapy in the patients who underwent CABG (Prasongsukarn et al., 2005). Although the benefit of steroids for AF was indicated by many studies, there are concerns about the side effect of steroids (Ho and Tan, 2009).
Colchicine is an anti-inflammatory drug and, for cardiovascular disease, is clinically used in the treatment of pericarditis. For treatment of AF, colchicine significantly reduced serum CRP and IL-6 level and prevented the recurrence of AF after catheter ablation for AF (Deftereos et al., 2012). It is also reported to be effective in the prevention of postoperative AF (Imazio et al., 2014), and administration of colchicine may be considered for patients postoperatively to reduce AF after cardiac surgery (class IIb indication) (January et al., 2014).
Although they are not so-called anti-inflammatory drugs, some antihypertensive drugs show anti-inflammatory effects, and they have also been studied for their therapeutic effects on AF. Angiotensin II induces inflammation by stimulation via AT1 receptors (Benigni et al., 2010). Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) are expected to prevent the formation of AF substrate and the development of AF via its anti-inflammatory effect, and many clinical studies have been conducted. In fact, ACEIs/ARBs suppress the inflammation in human patients (Takeda et al., 2003; Manabe et al., 2005), and some retrospective clinical researches demonstrated their effect for preventing the development of AF (Maggioni et al., 2005; Schmieder et al., 2008). However, subsequent large-scale prospective clinical trials have failed to demonstrate their efficacy (Disertori et al., 2009; Yusuf et al., 2011; Goette et al., 2012). Following those results, in the current clinical setting, ACEIs/ARBs are reasonable for primary prevention of new-onset AF only in patients with heart failure with reduced left ventricular ejection fraction (Class IIa indication) (January et al., 2014).
Statins, which are HMG-CoA reductase inhibitors and the drugs for the treatment of hypercholesterolemia, exert pleiotropic effects including anti-inflammatory action (Jain and Ridker, 2005). There are some reports to show the efficacy of statins for primary and secondary prevention of AF (Fang et al., 2012), and statin therapy may be reasonable for primary prevention of new-onset AF after coronary artery surgery (Class IIb indication) (Liakopoulos et al., 2009; January et al., 2014).
Drugs targeting inflammatory molecules have been clinically available for connective tissue diseases and other autoimmune diseases, and their application to cardiac diseases, including AF, is also underway (Su et al., 2021). Anti-IL-6 receptor antibody (tocilizumab) is widely used for the treatment of RA. Recently, in the clinical trial using anti-IL-6 receptor antibody in the patients with acute myocardial infarction, anti-IL-6 receptor antibody successfully suppressed the inflammatory response during myocardial infarction and ultimately reduced the infarct size (Kleveland et al., 2016; Broch et al., 2021). Similarly, the report using an anti-IL-1β antibody (canakinumab) reduced hospitalization for heart failure and heart failure-related mortality (Everett et al., 2019). In the observational study of the patients with psoriasis, the patients who were treated with TNF inhibitors (adalimumab, etanercept, and infliximab) had a lower risk of cardiovascular events compared to those treated with phototherapy patients (Wu et al., 2018). Although these drugs suppressed the inflammation and showed the therapeutic potential for treating cardiovascular diseases in those studies, their efficacy for the development of AF was not assessed, and further research is required.
AF and inflammation are deeply linked, forming a negative spiral in which the presence of AF exacerbates the inflammation and vice versa. Inflammation causes and accelerates the electrical and structural remodeling of the atria and enhances the AF substrate leading to the maintenance of AF and further inflammation. Inflammation is a central player in the concept of “AF begets AF” and breaking this vicious cycle is expected to be a key therapeutic intervention in AF. Currently, anti-inflammatory approaches for AF are considered in a limited patient population, but further elucidation of inflammatory mechanisms in AF and the development of novel anti-inflammatory agents will bring breakthroughs in AF treatment.
KI and TS wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe I., Teshima Y., Kondo H., Kaku H., Kira S., Ikebe Y., et al. (2018). Association of Fibrotic Remodeling and Cytokines/chemokines Content in Epicardial Adipose Tissue with Atrial Myocardial Fibrosis in Patients with Atrial Fibrillation. Heart Rhythm 15, 1717–1727. doi:10.1016/j.hrthm.2018.06.025
Ahlehoff O., Gislason G. H., Jørgensen C. H., Lindhardsen J., Charlot M., Olesen J. B., et al. (2012). Psoriasis and Risk of Atrial Fibrillation and Ischaemic Stroke: a Danish Nationwide Cohort Study. Eur. Heart J. 33, 2054–2064. doi:10.1093/eurheartj/ehr285
Al Chekakie M. O., Welles C. C., Metoyer R., Ibrahim A., Shapira A. R., Cytron J., et al. (2010). Pericardial Fat Is Independently Associated with Human Atrial Fibrillation. J. Am. Coll. Cardiol. 56, 784–788. doi:10.1016/j.jacc.2010.03.071
Amdur R. L., Mukherjee M., Go A., Barrows I. R., Ramezani A., Shoji J., et al. (2016). Interleukin-6 Is a Risk Factor for Atrial Fibrillation in Chronic Kidney Disease: Findings from the CRIC Study. PLoS One 11, e0148189. doi:10.1371/journal.pone.0148189
Anatolevna R. O., Veniaminovich F. O., Mikhaylovich K. S. (2016). Predictors of New-Onset Atrial Fibrillation in Elderly Patients with Coronary Artery Disease after Coronary Artery Bypass Graft. J. Geriatr. Cardiol. 13, 444–449.
Arbelo E., Brugada J., Hindricks G., Maggioni A. P., Tavazzi L., Vardas P., et al. (2014). The Atrial Fibrillation Ablation Pilot Study: a European Survey on Methodology and Results of Catheter Ablation for Atrial Fibrillation Conducted by the European Heart Rhythm Association. Eur. Heart J. 35, 1466–1478. doi:10.1093/eurheartj/ehu001
Aulin J., Siegbahn A., Hijazi Z., Ezekowitz M. D., Andersson U., Connolly S. J., et al. (2015). Interleukin-6 and C-Reactive Protein and Risk for Death and Cardiovascular Events in Patients with Atrial Fibrillation. Am. Heart J. 170, 1151–1160. doi:10.1016/j.ahj.2015.09.018
Aviles R. J., Martin D. O., Apperson-Hansen C., Houghtaling P. L., Rautaharju P., Kronmal R. A., et al. (2003). Inflammation as a Risk Factor for Atrial Fibrillation. Circulation 108, 3006–3010. doi:10.1161/01.cir.0000103131.70301.4f
Bacani A. K., Crowson C. S., Roger V. L., Gabriel S. E., Matteson E. L. (2015). Increased Incidence of Atrial Fibrillation in Patients with Rheumatoid Arthritis. Biomed. Res. Int. 2015, 809514. doi:10.1155/2015/809514
Baeuerle P. A., Henkel T. (1994). Function and Activation of NF-kappaB in the Immune System. Annu. Rev. Immunol. 12, 141–179. doi:10.1146/annurev.iy.12.040194.001041
Ball J., Carrington M. J., Mcmurray J. J. V., Stewart S. (2013). Atrial Fibrillation: Profile and burden of an Evolving Epidemic in the 21st century. Int. J. Cardiol. 167, 1807–1824. doi:10.1016/j.ijcard.2012.12.093
Benigni A., Cassis P., Remuzzi G. (2010). Angiotensin II Revisited: New Roles in Inflammation, Immunology and Aging. EMBO Mol. Med. 2, 247–257. doi:10.1002/emmm.201000080
Broch K., Anstensrud A. K., Woxholt S., Sharma K., Tøllefsen I. M., Bendz B., et al. (2021). Randomized Trial of Interleukin-6 Receptor Inhibition in Patients with Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 77, 1845–1855. doi:10.1016/j.jacc.2021.02.049
Brundel B. J. J. M., Henning R. H., Ke L., Van Gelder I. C., Crijns H. J. G. M., Kampinga H. H. (2006a). Heat Shock Protein Upregulation Protects against Pacing-Induced Myolysis in HL-1 Atrial Myocytes and in Human Atrial Fibrillation. J. Mol. Cell Cardiol. 41, 555–562. doi:10.1016/j.yjmcc.2006.06.068
Brundel B. J. J. M., Shiroshita-Takeshita A., Qi X., Yeh Y.-H., Chartier D., Van Gelder I. C., et al. (2006b). Induction of Heat Shock Response Protects the Heart against Atrial Fibrillation. Circ. Res. 99, 1394–1402. doi:10.1161/01.res.0000252323.83137.fe
Byrd-Leifer C. A., Block E. F., Takeda K., Akira S., Ding A. (2001). The Role of MyD88 and TLR4 in the LPS-Mimetic Activity of Taxol. Eur. J. Immunol. 31, 2448–2457. doi:10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n
Calkins H., Hindricks G., Cappato R., Kim Y.-H., Saad E. B., Aguinaga L., et al. (2017). 2017 HRS/EHRA/ECAS/APHRS/SOLAECE Expert Consensus Statement on Catheter and Surgical Ablation of Atrial Fibrillation. Heart Rhythm 14, e275–e444. doi:10.1016/j.hrthm.2017.05.012
Camelliti P., Green C. R., Legrice I., Kohl P. (2004). Fibroblast Network in Rabbit Sinoatrial Node. Circ. Res. 94, 828–835. doi:10.1161/01.res.0000122382.19400.14
Chang S.-N., Tsai C.-T., Wu C.-K., Lee J.-K., Lai L.-P., Huang S.-W., et al. (2012). A Functional Variant in the Promoter Region Regulates the C-Reactive Protein Gene and Is a Potential Candidate for Increased Risk of Atrial Fibrillation. J. Intern. Med. 272, 305–315. doi:10.1111/j.1365-2796.2012.02531.x
Chekeni F. B., Elliott M. R., Sandilos J. K., Walk S. F., Kinchen J. M., Lazarowski E. R., et al. (2010). Pannexin 1 Channels Mediate 'find-Me' Signal Release and Membrane Permeability during Apoptosis. Nature 467, 863–867. doi:10.1038/nature09413
Chen M.-C., Chang J.-P., Liu W.-H., Yang C.-H., Chen Y.-L., Tsai T.-H., et al. (2008). Increased Inflammatory Cell Infiltration in the Atrial Myocardium of Patients with Atrial Fibrillation. Am. J. Cardiol. 102, 861–865. doi:10.1016/j.amjcard.2008.05.038
Choi Y.-J., Choi E.-K., Han K.-D., Park J., Moon I., Lee E., et al. (2019). Increased Risk of Atrial Fibrillation in Patients with Inflammatory Bowel Disease: A Nationwide Population-Based Study. Wjg 25, 2788–2798. doi:10.3748/wjg.v25.i22.2788
Christian S.-A., Schorr C., Ferchau L., Jarbrink M. E., Parrillo J. E., Gerber D. R. (2008). Clinical Characteristics and Outcomes of Septic Patients with New-Onset Atrial Fibrillation. J. Crit. Care 23, 532–536. doi:10.1016/j.jcrc.2007.09.005
Chugh S. S., Havmoeller R., Narayanan K., Singh D., Rienstra M., Benjamin E. J., et al. (2014). Worldwide Epidemiology of Atrial Fibrillation. Circulation 129, 837–847. doi:10.1161/circulationaha.113.005119
Chung M. K., Martin D. O., Sprecher D., Wazni O., Kanderian A., Carnes C. A., et al. (2001). C-reactive Protein Elevation in Patients with Atrial Arrhythmias. Circulation 104, 2886–2891. doi:10.1161/hc4901.101760
Cianfrocca C., Loricchio M. L., Pelliccia F., Pasceri V., Auriti A., Bianconi L., et al. (2010). C-reactive Protein and Left Atrial Appendage Velocity Are Independent Determinants of the Risk of Thrombogenesis in Patients with Atrial Fibrillation. Int. J. Cardiol. 142, 22–28. doi:10.1016/j.ijcard.2008.12.052
Corradi D., Callegari S., Manotti L., Ferrara D., Goldoni M., Alinovi R., et al. (2014). Persistent Lone Atrial Fibrillation: Clinicopathologic Study of 19 Cases. Heart Rhythm 11, 1250–1258. doi:10.1016/j.hrthm.2014.02.008
de Vos C. B., Pisters R., Nieuwlaat R., Prins M. H., Tieleman R. G., Coelen R.-J. S., et al. (2010). Progression from Paroxysmal to Persistent Atrial Fibrillation. J. Am. Coll. Cardiol. 55, 725–731. doi:10.1016/j.jacc.2009.11.040
Deftereos S., Giannopoulos G., Kossyvakis C., Efremidis M., Panagopoulou V., Kaoukis A., et al. (2012). Colchicine for Prevention of Early Atrial Fibrillation Recurrence after Pulmonary Vein Isolation. J. Am. Coll. Cardiol. 60, 1790–1796. doi:10.1016/j.jacc.2012.07.031
Dernellis J., Panaretou M. (2001). C-reactive Protein and Paroxysmal Atrial Fibrillation. Acta Cardiologica 56, 375–380. doi:10.2143/ac.56.6.2005701
Dernellis J., Panaretou M. (2004). Relationship between C-Reactive Protein Concentrations during Glucocorticoid Therapy and Recurrent Atrial Fibrillation. Eur. Heart J. 25, 1100–1107. doi:10.1016/j.ehj.2004.04.025
Disertori M., Disertori M., Latini R., Barlera S., Franzosi M. G., Staszewsky L., et al. (2009). Valsartan for Prevention of Recurrent Atrial Fibrillation. N. Engl. J. Med. 360, 1606–1617. doi:10.1056/NEJMoa0805710
Everett B. M., Cornel J. H., Lainscak M., Anker S. D., Abbate A., Thuren T., et al. (2019). Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 139, 1289–1299. doi:10.1161/circulationaha.118.038010
Fang W.-t., Li H.-j., Zhang H., Jiang S. (2012). The Role of Statin Therapy in the Prevention of Atrial Fibrillation: a Meta-Analysis of Randomized Controlled Trials. Br. J. Clin. Pharmacol. 74, 744–756. doi:10.1111/j.1365-2125.2012.04258.x
Goette A., Schön N., Kirchhof P., Breithardt G., Fetsch T., Häusler K. G., et al. (2012). Angiotensin II-Antagonist in Paroxysmal Atrial Fibrillation (ANTIPAF) Trial. Circ. Arrhythmia Electrophysiol. 5, 43–51. doi:10.1161/circep.111.965178
Grune J., Yamazoe M., Nahrendorf M. (2021). Electroimmunology and Cardiac Arrhythmia. Nat. Rev. Cardiol. 18, 547–564. doi:10.1038/s41569-021-00520-9
Hagiwara Y., Miyoshi S., Fukuda K., Nishiyama N., Ikegami Y., Tanimoto K., et al. (2007). SHP2-mediated Signaling cascade through Gp130 Is Essential for LIF-dependent ICaL, [Ca2+]i Transient, and APD Increase in Cardiomyocytes. J. Mol. Cell Cardiol. 43, 710–716. doi:10.1016/j.yjmcc.2007.09.004
Haïssaguerre M., Jaïs P., Shah D. C., Takahashi A., Hocini M., Quiniou G., et al. (1998). Spontaneous Initiation of Atrial Fibrillation by Ectopic Beats Originating in the Pulmonary Veins. N. Engl. J. Med. 339, 659–666. doi:10.1056/nejm199809033391003
Harada M., Van Wagoner D. R., Nattel S. (2015). Role of Inflammation in Atrial Fibrillation Pathophysiology and Management. Circ. J. 79, 495–502. doi:10.1253/circj.cj-15-0138
Harrison D. G., Guzik T. J., Lob H. E., Madhur M. S., Marvar P. J., Thabet S. R., et al. (2011). Inflammation, Immunity, and Hypertension. Hypertension 57, 132–140. doi:10.1161/hypertensionaha.110.163576
He Y., Hara H., Núñez G. (2016). Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 41, 1012–1021. doi:10.1016/j.tibs.2016.09.002
Heijman J., Voigt N., Nattel S., Dobrev D. (2014). Cellular and Molecular Electrophysiology of Atrial Fibrillation Initiation, Maintenance, and Progression. Circ. Res. 114, 1483–1499. doi:10.1161/circresaha.114.302226
Hijazi Z., Aulin J., Andersson U., Alexander J. H., Gersh B., Granger C. B., et al. (2016). Biomarkers of Inflammation and Risk of Cardiovascular Events in Anticoagulated Patients with Atrial Fibrillation. Heart 102, 508–517. doi:10.1136/heartjnl-2015-308887
Ho K. M., Tan J. A. (2009). Benefits and Risks of Corticosteroid Prophylaxis in Adult Cardiac Surgery. Circulation 119, 1853–1866. doi:10.1161/circulationaha.108.848218
Hoene M., Weigert C. (2008). The Role of Interleukin-6 in Insulin Resistance, Body Fat Distribution and Energy Balance. Obes. Rev. 9, 20–29. doi:10.1111/j.1467-789X.2007.00410.x
Hu Y.-F., Chen Y.-J., Lin Y.-J., Chen S.-A. (2015). Inflammation and the Pathogenesis of Atrial Fibrillation. Nat. Rev. Cardiol. 12, 230–243. doi:10.1038/nrcardio.2015.2
Hu Y.-F., Yeh H.-I., Tsao H.-M., Tai C.-T., Lin Y.-J., Chang S.-L., et al. (2012). Electrophysiological Correlation and Prognostic Impact of Heat Shock Protein 27 in Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 5, 334–340. doi:10.1161/circep.111.965996
Hulsmans M., Clauss S., Xiao L., Aguirre A. D., King K. R., Hanley A., et al. (2017). Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522. doi:10.1016/j.cell.2017.03.050
Iacobellis G., Barbaro G. (2008). The Double Role of Epicardial Adipose Tissue as Pro- and Anti-inflammatory Organ. Horm. Metab. Res. 40, 442–445. doi:10.1055/s-2008-1062724
Imazio M., Brucato A., Ferrazzi P., Pullara A., Adler Y., Barosi A., et al. (2014). Colchicine for Prevention of Postpericardiotomy Syndrome and Postoperative Atrial Fibrillation. JAMA 312, 1016–1023. doi:10.1001/jama.2014.11026
Ishida K., Kimura F., Imamaki M., Ishida A., Shimura H., Kohno H., et al. (2006). Relation of Inflammatory Cytokines to Atrial Fibrillation after Off-Pump Coronary Artery Bypass Grafting. Eur. J. Cardio-Thoracic Surg. 29, 501–505. doi:10.1016/j.ejcts.2005.12.028
Ishii Y., Schuessler R. B., Gaynor S. L., Yamada K., Fu A. S., Boineau J. P., et al. (2005). Inflammation of Atrium after Cardiac Surgery Is Associated with Inhomogeneity of Atrial Conduction and Atrial Fibrillation. Circulation 111, 2881–2888. doi:10.1161/circulationaha.104.475194
Jain M. K., Ridker P. M. (2005). Anti-inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat. Rev. Drug Discov. 4, 977–987. doi:10.1038/nrd1901
Jalife J., Berenfeld O., Mansour M. (2002). Mother Rotors and Fibrillatory Conduction: a Mechanism of Atrial Fibrillation. Cardiovasc. Res. 54, 204–216. doi:10.1016/s0008-6363(02)00223-7
January C. T., Wann L. S., Alpert J. S., Calkins H., Cigarroa J. E., Cleveland J. C., et al. (2014). 2014 AHA/ACC/HRS Guideline for the Management of Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 64, e1–e76. doi:10.1016/j.jacc.2014.03.022
Kallergis E. M., Manios E. G., Kanoupakis E. M., Mavrakis H. E., Kolyvaki S. G., Lyrarakis G. M., et al. (2008). The Role of the post-cardioversion Time Course of Hs-CRP Levels in Clarifying the Relationship between Inflammation and Persistence of Atrial Fibrillation. Heart 94, 200–204. doi:10.1136/hrt.2006.108688
Kannel W. B., Wolf P. A., Benjamin E. J., Levy D. (1998). Prevalence, Incidence, Prognosis, and Predisposing Conditions for Atrial Fibrillation: Population-Based Estimates 11Reprints Are Not Available. Am. J. Cardiol. 82, 2N–9N. doi:10.1016/s0002-9149(98)00583-9
Kao Y.-H., Chen Y.-C., Cheng C.-C., Lee T.-I., Chen Y.-J., Chen S.-A. (2010). Tumor Necrosis Factor-α Decreases Sarcoplasmic Reticulum Ca2+-ATPase Expressions via the Promoter Methylation in Cardiomyocytes*. Crit. Care Med. 38, 217–222. doi:10.1097/ccm.0b013e3181b4a854
Kim Y. R., Nam G.-B., Han S., Kim S.-H., Kim K.-H., Lee S., et al. (2015). Effect of Short-Term Steroid Therapy on Early Recurrence during the Blanking Period after Catheter Ablation of Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 8, 1366–1372. doi:10.1161/circep.115.002957
Kleveland O., Kunszt G., Bratlie M., Ueland T., Broch K., Holte E., et al. (2016). Effect of a Single Dose of the Interleukin-6 Receptor Antagonist Tocilizumab on Inflammation and Troponin T Release in Patients with Non-ST-elevation Myocardial Infarction: a Double-Blind, Randomized, Placebo-Controlled Phase 2 Trial. Eur. Heart J. 37, 2406–2413. doi:10.1093/eurheartj/ehw171
Kong P., Christia P., Frangogiannis N. G. (2014). The Pathogenesis of Cardiac Fibrosis. Cell. Mol. Life Sci. 71, 549–574. doi:10.1007/s00018-013-1349-6
Launey Y., Lasocki S., Asehnoune K., Gaudriot B., Chassier C., Cinotti R., et al. (2019). Impact of Low-Dose Hydrocortisone on the Incidence of Atrial Fibrillation in Patients with Septic Shock: A Propensity Score-Inverse Probability of Treatment Weighting Cohort Study. J. Intensive Care Med. 34, 238–244. doi:10.1177/0885066617696847
Lazzerini P. E., Capecchi P. L., Laghi-Pasini F. (2017). Systemic Inflammation and Arrhythmic Risk: Lessons from Rheumatoid Arthritis. Eur. Heart J. 38, 1717–1727. doi:10.1093/eurheartj/ehw208
Lazzerini P. E., Laghi-Pasini F., Acampa M., Srivastava U., Bertolozzi I., Giabbani B., et al. (2019). Systemic Inflammation Rapidly Induces Reversible Atrial Electrical Remodeling: The Role of Interleukin-6-Mediated Changes in Connexin Expression. J. Am. Heart Assoc. 8, e011006. doi:10.1161/JAHA.118.011006
Lee S.-H., Chen Y.-C., Chen Y.-J., Chang S.-L., Tai C.-T., Wongcharoen W., et al. (2007). Tumor Necrosis Factor-α Alters Calcium Handling and Increases Arrhythmogenesis of Pulmonary Vein Cardiomyocytes. Life Sci. 80, 1806–1815. doi:10.1016/j.lfs.2007.02.029
Li D., Fareh S., Leung T. K., Nattel S. (1999). Promotion of Atrial Fibrillation by Heart Failure in Dogs. Circulation 100, 87–95. doi:10.1161/01.cir.100.1.87
Li J., Solus J., Chen Q., Rho Y. H., Milne G., Stein C. M., et al. (2010). Role of Inflammation and Oxidative Stress in Atrial Fibrillation. Heart Rhythm 7, 438–444. doi:10.1016/j.hrthm.2009.12.009
Li N., Brundel B. J. J. M. (2020). Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated with Atrial Fibrillation. Circ. Res. 127, 73–90. doi:10.1161/circresaha.119.316364
Li Z., Shao W., Zhang J., Ma J., Huang S., Yu P., et al. (2021). Prevalence of Atrial Fibrillation and Associated Mortality Among Hospitalized Patients with COVID-19: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 8, 720129. doi:10.3389/fcvm.2021.720129
Liakopoulos O. J., Choi Y.-H., Kuhn E. W., Wittwer T., Borys M., Madershahian N., et al. (2009). Statins for Prevention of Atrial Fibrillation after Cardiac Surgery: a Systematic Literature Review. J. Thorac. Cardiovasc. Surg. 138, 678–686. doi:10.1016/j.jtcvs.2009.03.054
Liao C.-h., Akazawa H., Tamagawa M., Ito K., Yasuda N., Kudo Y., et al. (2010). Cardiac Mast Cells Cause Atrial Fibrillation through PDGF-A-Mediated Fibrosis in Pressure-Overloaded Mouse Hearts. J. Clin. Invest. 120, 242–253. doi:10.1172/jci39942
Lim H. S., Willoughby S. R., Schultz C., Gan C., Alasady M., Lau D. H., et al. (2013). Effect of Atrial Fibrillation on Atrial Thrombogenesis in Humans: Impact of Rate and Rhythm. J. Am. Coll. Cardiol. 61, 852–860. doi:10.1016/j.jacc.2012.11.046
Lip G. Y. H., Patel J. V., Hughes E., Hart R. G. (2007). High-Sensitivity C-Reactive Protein and Soluble CD40 Ligand as Indices of Inflammation and Platelet Activation in 880 Patients with Nonvalvular Atrial Fibrillation. Stroke 38, 1229–1237. doi:10.1161/01.str.0000260090.90508.3e
Liuba I., Ahlmroth H., Jonasson L., Englund A., Jonsson A., Safstrom K., et al. (2008). Source of Inflammatory Markers in Patients with Atrial Fibrillation. Europace 10, 848–853. doi:10.1093/europace/eun111
Ma F., Li Y., Jia L., Han Y., Cheng J., Li H., et al. (2012). Macrophage-Stimulated Cardiac Fibroblast Production of IL-6 Is Essential for TGF β/Smad Activation and Cardiac Fibrosis Induced by Angiotensin II. PLoS One 7, e35144. doi:10.1371/journal.pone.0035144
Maesen B., Nijs J., Maessen J., Allessie M., Schotten U. (2012). Post-operative Atrial Fibrillation: a Maze of Mechanisms. Europace 14, 159–174. doi:10.1093/europace/eur208
Maggioni A. P., Latini R., Carson P. E., Singh S. N., Barlera S., Glazer R., et al. (2005). Valsartan Reduces the Incidence of Atrial Fibrillation in Patients with Heart Failure: Results from the Valsartan Heart Failure Trial (Val-HeFT). Am. Heart J. 149, 548–557. doi:10.1016/j.ahj.2004.09.033
Manabe S., Okura T., Watanabe S., Fukuoka T., Higaki J. (2005). Effects of Angiotensin II Receptor Blockade with Valsartan on Pro-inflammatory Cytokines in Patients with Essential Hypertension. J. Cardiovasc. Pharmacol. 46, 735–739. doi:10.1097/01.fjc.0000185783.00391.60
Marcus G. M., Smith L. M., Ordovas K., Scheinman M. M., Kim A. M., Badhwar N., et al. (2010). Intracardiac and Extracardiac Markers of Inflammation during Atrial Fibrillation. Heart Rhythm 7, 149–154. doi:10.1016/j.hrthm.2009.10.004
Marcus G. M., Whooley M. A., Glidden D. V., Pawlikowska L., Zaroff J. G., Olgin J. E. (2008). Interleukin-6 and Atrial Fibrillation in Patients with Coronary Artery Disease: Data from the Heart and Soul Study. Am. Heart J. 155, 303–309. doi:10.1016/j.ahj.2007.09.006
Mark D. B., Anstrom K. J., Sheng S., Piccini J. P., Baloch K. N., Monahan K. H., et al. (2019). Effect of Catheter Ablation vs Medical Therapy on Quality of Life Among Patients with Atrial Fibrillation. JAMA 321, 1275–1285. doi:10.1001/jama.2019.0692
Mazurek T., Zhang L., Zalewski A., Mannion J. D., Diehl J. T., Arafat H., et al. (2003). Human Epicardial Adipose Tissue Is a Source of Inflammatory Mediators. Circulation 108, 2460–2466. doi:10.1161/01.cir.0000099542.57313.c5
Meierhenrich R., Steinhilber E., Eggermann C., Weiss M., Voglic S., Bögelein D., et al. (2010). Incidence and Prognostic Impact of New-Onset Atrial Fibrillation in Patients with Septic Shock: a Prospective Observational Study. Crit. Care 14, R108. doi:10.1186/cc9057
Moe G. K., Abildskov J. A. (1959). Atrial Fibrillation as a Self-Sustaining Arrhythmia Independent of Focal Discharge. Am. Heart J. 58, 59–70. doi:10.1016/0002-8703(59)90274-1
Musikantow D. R., Turagam M. K., Sartori S., Chu E., Kawamura I., Shivamurthy P., et al. (2021). Atrial Fibrillation in Patients Hospitalized with COVID-19. JACC: Clin. Electrophysiol. 7, 1120–1130. doi:10.1016/j.jacep.2021.02.009
Nalliah C. J., Sanders P., Kottkamp H., Kalman J. M. (2016). The Role of Obesity in Atrial Fibrillation. Eur. Heart J. 37, 1565–1572. doi:10.1093/eurheartj/ehv486
Narducci M. L., Pelargonio G., Dello Russo A., Casella M., Biasucci L. M., La Torre G., et al. (2011). Role of Tissue C-Reactive Protein in Atrial Cardiomyocytes of Patients Undergoing Catheter Ablation of Atrial Fibrillation: Pathogenetic Implications. Europace 13, 1133–1140. doi:10.1093/europace/eur068
Nattel S., Burstein B., Dobrev D. (2008). Atrial Remodeling and Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 1, 62–73. doi:10.1161/circep.107.754564
Nattel S. (2017). Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC: Clin. Electrophysiol. 3, 425–435. doi:10.1016/j.jacep.2017.03.002
Oishi S., Sasano T., Tateishi Y., Tamura N., Isobe M., Furukawa T. (2012). Stretch of Atrial Myocytes Stimulates Recruitment of Macrophages via ATP Released through gap-junction Channels. J. Pharmacol. Sci. 120, 296–304. doi:10.1254/jphs.12202fp
Packer M. (2018). Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J. Am. Coll. Cardiol. 71, 2360–2372. doi:10.1016/j.jacc.2018.03.509
Prasongsukarn K., Abel J. G., Jamieson W. R. E., Cheung A., Russell J. A., Walley K. R., et al. (2005). The Effects of Steroids on the Occurrence of Postoperative Atrial Fibrillation after Coronary Artery Bypass Grafting Surgery: a Prospective Randomized Trial. J. Thorac. Cardiovasc. Surg. 130, 93–98. doi:10.1016/j.jtcvs.2004.09.014
Qu Y.-C., Du Y.-M., Wu S.-L., Chen Q.-X., Wu H.-L., Zhou S.-F. (2009). Activated Nuclear Factor-Κb and Increased Tumor Necrosis Factor-α in Atrial Tissue of Atrial Fibrillation. Scand. Cardiovasc. J. 43, 292–297. doi:10.1080/14017430802651803
Roselli C., Rienstra M., Ellinor P. T. (2020). Genetics of Atrial Fibrillation in 2020. Circ. Res. 127, 21–33. doi:10.1161/circresaha.120.316575
Saba S., Janczewski A. M., Baker L. C., Shusterman V., Gursoy E. C., Feldman A. M., et al. (2005). Atrial Contractile Dysfunction, Fibrosis, and Arrhythmias in a Mouse Model of Cardiomyopathy Secondary to Cardiac-specific Overexpression of Tumor Necrosis Factor-α. Am. J. Physiology-Heart Circulatory Physiol. 289, H1456–H1467. doi:10.1152/ajpheart.00733.2004
Said M. A., Verweij N., Van Der Harst P. (2018). Associations of Combined Genetic and Lifestyle Risks with Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol. 3, 693–702. doi:10.1001/jamacardio.2018.1717
Sawaya S. E., Rajawat Y. S., Rami T. G., Szalai G., Price R. L., Sivasubramanian N., et al. (2007). Downregulation of Connexin40 and Increased Prevalence of Atrial Arrhythmias in Transgenic Mice with Cardiac-Restricted Overexpression of Tumor Necrosis Factor. Am. J. Physiology-Heart Circulatory Physiol. 292, H1561–H1567. doi:10.1152/ajpheart.00285.2006
Schmieder R. E., Kjeldsen S. E., Julius S., Mcinnes G. T., Zanchetti A., Hua T. A. (2008). Reduced Incidence of New-Onset Atrial Fibrillation with Angiotensin II Receptor Blockade: the VALUE Trial. J. Hypertens. 26, 403–411. doi:10.1097/hjh.0b013e3282f35c67
Schwarzenbach H., Hoon D. S. B., Pantel K. (2011). Cell-free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 11, 426–437. doi:10.1038/nrc3066
Seguin P., Signouret T., Laviolle B., Branger B., Mallédant Y. (2004). Incidence and Risk Factors of Atrial Fibrillation in a Surgical Intensive Care Unit*. Crit. Care Med. 32, 722–726. doi:10.1097/01.ccm.0000114579.56430.e0
Shang L. L., Sanyal S., Pfahnl A. E., Jiao Z., Allen J., Liu H., et al. (2008). NF-κB-dependent Transcriptional Regulation of the Cardiac Scn5a Sodium Channel by Angiotensin II. Am. J. Physiology-Cell Physiol. 294, C372–C379. doi:10.1152/ajpcell.00186.2007
Smorodinova N., Bláha M., Melenovský V., Rozsívalová K., Přidal J., Ďurišová M., et al. (2017). Analysis of Immune Cell Populations in Atrial Myocardium of Patients with Atrial Fibrillation or Sinus Rhythm. PLoS One 12, e0172691. doi:10.1371/journal.pone.0172691
Stark K., Massberg S. (2021). Interplay between Inflammation and Thrombosis in Cardiovascular Pathology. Nat. Rev. Cardiol. 18, 666–682. doi:10.1038/s41569-021-00552-1
Su J.-H., Luo M.-Y., Liang N., Gong S.-X., Chen W., Huang W.-Q., et al. (2021). Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 12, 745061. doi:10.3389/fphar.2021.745061
Takeda T., Hoshida S., Nishino M., Tanouchi J., Otsu K., Hori M. (2003). Relationship between Effects of Statins, Aspirin and Angiotensin II Modulators on High-Sensitive C-Reactive Protein Levels. Atherosclerosis 169, 155–158. doi:10.1016/s0021-9150(03)00158-8
Tanaka T., Kanda T., Takahashi T., Saegusa S., Moriya J., Kurabayashi M. (2004). Interleukin-6-induced Reciprocal Expression of SERCA and Natriuretic Peptides mRNA in Cultured Rat Ventricular Myocytes. J. Int. Med. Res. 32, 57–61. doi:10.1177/147323000403200109
Thambidorai S. K., Parakh K., Martin D. O., Shah T. K., Wazni O., Jasper S. E., et al. (2004). Relation of C-Reactive Protein Correlates with Risk of Thromboembolism in Patients with Atrial Fibrillation. Am. J. Cardiol. 94, 805–807. doi:10.1016/j.amjcard.2004.06.011
Thanassoulis G., Massaro J. M., O'donnell C. J., Hoffmann U., Levy D., Ellinor P. T., et al. (2010). Pericardial Fat Is Associated with Prevalent Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 3, 345–350. doi:10.1161/circep.109.912055
Travers J. G., Kamal F. A., Robbins J., Yutzey K. E., Blaxall B. C. (2016). Cardiac Fibrosis. Circ. Res. 118, 1021–1040. doi:10.1161/circresaha.115.306565
Ungprasert P., Srivali N., Kittanamongkolchai W. (2017). Risk of Incident Atrial Fibrillation in Patients with Rheumatoid Arthritis: a Systematic Review and Meta-Analysis. Int. J. Rheum. Dis. 20, 434–441. doi:10.1111/1756-185x.12820
van Eden W., Van Der Zee R., Prakken B. (2005). Heat-shock Proteins Induce T-Cell Regulation of Chronic Inflammation. Nat. Rev. Immunol. 5, 318–330. doi:10.1038/nri1593
Verheule S., Wilson E., Everett T., Shanbhag S., Golden C., Olgin J. (2003). Alterations in Atrial Electrophysiology and Tissue Structure in a Canine Model of Chronic Atrial Dilatation Due to Mitral Regurgitation. Circulation 107, 2615–2622. doi:10.1161/01.cir.0000066915.15187.51
Verma A., Jiang C.-y., Betts T. R., Chen J., Deisenhofer I., Mantovan R., et al. (2015). Approaches to Catheter Ablation for Persistent Atrial Fibrillation. N. Engl. J. Med. 372, 1812–1822. doi:10.1056/nejmoa1408288
Villegas S., Villarreal F. J., Dillmann W. H. (2000). Leukemia Inhibitory Factor and Interleukin-6 Downregulate Sarcoplasmic Reticulum Ca 2+ ATPase (SERCA2) in Cardiac Myocytes. Basic Res. Cardiol. 95, 47–54. doi:10.1007/s003950050007
Wang T. J., Parise H., Levy D., D'agostino R. B., Sr , Wolf P. A., et al. (2004). Obesity and the Risk of New-Onset Atrial Fibrillation. JAMA 292, 2471–2477. doi:10.1001/jama.292.20.2471
Wijffels M. C. E. F., Kirchhof C. J. H. J., Dorland R., Allessie M. A. (1995). Atrial Fibrillation Begets Atrial Fibrillation. Circulation 92, 1954–1968. doi:10.1161/01.cir.92.7.1954
Winfree A. T. (1991). Varieties of Spiral Wave Behavior: An Experimentalist's Approach to the Theory of Excitable media. Chaos 1, 303–334. doi:10.1063/1.165844
Wu J. J., Sundaram M., Cloutier M., Gauthier-Loiselle M., Guérin A., Singh R., et al. (2018). The Risk of Cardiovascular Events in Psoriasis Patients Treated with Tumor Necrosis Factor-α Inhibitors versus Phototherapy: An Observational Cohort Study. J. Am. Acad. Dermatol. 79, 60–68. doi:10.1016/j.jaad.2018.02.050
Wu N., Xu B., Xiang Y., Wu L., Zhang Y., Ma X., et al. (2013). Association of Inflammatory Factors with Occurrence and Recurrence of Atrial Fibrillation: a Meta-Analysis. Int. J. Cardiol. 169, 62–72. doi:10.1016/j.ijcard.2013.08.078
Wu Z.-K., Laurikka J., Vikman S., Nieminen R., Moilanen E., Tarkka M. R. (2008). High Postoperative Interleukin-8 Levels Related to Atrial Fibrillation in Patients Undergoing Coronary Artery Bypass Surgery. World J. Surg. 32, 2643–2649. doi:10.1007/s00268-008-9758-7
Xu Q., Bo L., Hu J., Geng J., Chen Y., Li X., et al. (2018). High Mobility Group Box 1 Was Associated with Thrombosis in Patients with Atrial Fibrillation. Medicine (Baltimore) 97, e0132. doi:10.1097/md.0000000000010132
Yamashita T., Sekiguchi A., Iwasaki Y.-k., Date T., Sagara K., Tanabe H., et al. (2010). Recruitment of Immune Cells across Atrial Endocardium in Human Atrial Fibrillation. Circ. J. 74, 262–270. doi:10.1253/circj.cj-09-0644
Yamazoe M., Sasano T., Ihara K., Takahashi K., Nakamura W., Takahashi N., et al. (2021). Sparsely Methylated Mitochondrial Cell Free DNA Released from Cardiomyocytes Contributes to Systemic Inflammatory Response Accompanied by Atrial Fibrillation. Sci. Rep. 11, 5837. doi:10.1038/s41598-021-85204-7
Yao C., Veleva T., Scott L., Cao S., Li L., Chen G., et al. (2018). Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 138, 2227–2242. doi:10.1161/circulationaha.118.035202
Yokoyama T., Sekiguchi K., Tanaka T., Tomaru K., Arai M., Suzuki T., et al. (1999). Angiotensin II and Mechanical Stretch Induce Production of Tumor Necrosis Factor in Cardiac Fibroblasts. Am. J. Physiology-Heart Circulatory Physiol. 276, H1968–H1976. doi:10.1152/ajpheart.1999.276.6.h1968
Yusuf S., Yusuf S., Healey J. S., Pogue J., Chrolavicius S., Flather M., et al. (2011). Irbesartan in Patients with Atrial Fibrillation. N. Engl. J. Med. 364, 928–938. doi:10.1056/NEJMoa1008816
Zhao Q., Zhang S., Zhao H., Zhang S., Dai Z., Qian Y., et al. (2018). Median Nerve Stimulation Prevents Atrial Electrical Remodelling and Inflammation in a Canine Model with Rapid Atrial Pacing. Europace 20, 712–718. doi:10.1093/europace/eux003
Zhao X.-H., Laschinger C., Arora P., Szászi K., Kapus A., Mcculloch C. A. (2007). Force Activates Smooth Muscle α-actin Promoter Activity through the Rho Signaling Pathway. J. Cel Sci 120, 1801–1809. doi:10.1242/jcs.001586
Zhou M., Wang H., Chen J., Zhao L. (2020). Epicardial Adipose Tissue and Atrial Fibrillation: Possible Mechanisms, Potential Therapies, and Future Directions. Pacing Clin. Electrophysiol. 43, 133–145. doi:10.1111/pace.13825
Keywords: atrial fibrillation, inflammation, fibrosis, cytokine, arrhythmogenic substrate
Citation: Ihara K and Sasano T (2022) Role of Inflammation in the Pathogenesis of Atrial Fibrillation. Front. Physiol. 13:862164. doi: 10.3389/fphys.2022.862164
Received: 25 January 2022; Accepted: 21 March 2022;
Published: 14 April 2022.
Edited by:
Sergey Popov, Tomsk National Research Medical Center (RAS), RussiaReviewed by:
Masahide Harada, Fujita Health University, JapanCopyright © 2022 Ihara and Sasano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetsuo Sasano, c2FzYW5vLmN2bUB0bWQuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.