 Sarah M. Nwia
Sarah M. Nwia Xiao Chun Li
Xiao Chun Li Ana Paula de Oliveira Leite
Ana Paula de Oliveira Leite Rumana Hassan1,2
Rumana Hassan1,2 Jia Long Zhuo
Jia Long Zhuo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 19 April 2022
Sec. Membrane Physiology and Membrane Biophysics
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.861659
This article is part of the Research TopicThe Forever Young Na+/H+ Exchanger Family: New Insights in its Structure, Function and RegulationView all 9 articles
The sodium (Na+)/hydrogen (H+) exchanger 3 (NHE3) is one of the most important Na+/H+ antiporters in the small intestines of the gastrointestinal tract and the proximal tubules of the kidney. The roles of NHE3 in the regulation of intracellular pH and acid–base balance have been well established in cellular physiology using in vitro techniques. Localized primarily on the apical membranes in small intestines and proximal tubules, the key action of NHE3 is to facilitate the entry of luminal Na+ and the extrusion of intracellular H+ from intestinal and proximal tubule tubular epithelial cells. NHE3 is, directly and indirectly, responsible for absorbing the majority of ingested Na+ from small and large intestines and reabsorbing >50% of filtered Na+ in the proximal tubules of the kidney. However, the roles of NHE3 in the regulation of proximal tubular Na+ transport in the integrative physiological settings and its contributions to the basal blood pressure regulation and angiotensin II (Ang II)-induced hypertension have not been well studied previously due to the lack of suitable animal models. Recently, novel genetically modified mouse models with whole-body, kidney-specific, or proximal tubule-specific deletion of NHE3 have been generated by us and others to determine the critical roles and underlying mechanisms of NHE3 in maintaining basal body salt and fluid balance, blood pressure homeostasis, and the development of Ang II-induced hypertension at the whole-body, kidney, or proximal tubule levels. The objective of this invited article is to review, update, and discuss recent findings on the critical roles of intestinal and proximal tubule NHE3 in maintaining basal blood pressure homeostasis and their potential therapeutic implications in the development of angiotensin II (Ang II)-dependent hypertension.
High blood pressure or hypertension has recently become a national epidemic of proportion in the public health. According to American Heart Association and American College of Cardiology’s recent estimates, 46% of American adults suffer from hypertension, which is defined as a systolic and diastolic blood pressure ≥130/80 mmHg (Whelton and Carey, 2017; Whelton et al., 2018). Important risk factors that increase an individual’s chances of developing hypertension include genetic predisposition, unhealthy diet, lack of physical activities or excess salt intake (Intersalt, 1988; Crowley et al., 2005; Coffman, 2011; Whelton and Carey, 2017; Whelton et al., 2018; Carey, 2019; Mattson, 2019; Harrison et al., 2021; Zhuo et al., 2021). A variety of treatments are available to treat hypertension, but nonpharmacological approaches are usually recommended first. Along with recommendations to lose weight, patients are also counseled to reduce sodium intake, increase potassium intake, and reduce alcohol consumption (Svetkey et al., 1999; Sacks et al., 2001; Siervo et al., 2015; Whelton et al., 2018). The average American sodium intake for those 1 year and older totals 3,440 mg/day, almost 1,500 mg/day higher than the recommended limit of 2,300 mg/day (Svetkey et al., 1999; Sacks et al., 2001; Siervo et al., 2015; DeSalvo, 2016). The effect of high dietary sodium intake on blood pressure has been well-studied and the causative relationship between increased dietary salt and increased blood pressure has been well-established by salt-sensitive hypertension animal models (Rapp, 1982; Intersalt, 1988; Joe and Shapiro, 2012; DeSalvo, 2016; Zhuo and Li, 2018; Mattson, 2019; Filippini et al., 2021; Harrison et al., 2021). Dietary sodium is absorbed in the gastrointestinal tract primarily by the Na+/H+ exchanger 3 (NHE3) and when the blood is filtered by the kidneys, NHE3 in the proximal tubule is chiefly responsible for >50% of sodium reabsorption, highlighting its important role in the regulation of physiological sodium and fluid balance, blood pressure homeostasis, and the pathophysiology of hypertension (Schultheis et al., 1998a,b; Wang et al., 1999; Zhuo and Li, 2013; Li et al., 2015a,b).
If non-pharmacological approaches fail to control blood pressure, there are several classes of pharmacological agents available to treat hypertension. These antihypertensive therapeutics include diuretics targeting distal nephron Na+ transport, angiotensin-converting enzyme (ACE) inhibitors to inhibit the formation of a vasoactive peptide angiotensin II (Ang II), angiotensin II type 1 receptor blockers (ARBs), beta-blockers, and calcium channel blockers (Calhoun et al., 2008a; Whelton et al., 2018; Carey, 2019). For most hypertensive patients, treatment with a diuretic, ACE inhibitor, or ARB alone, or with a number of different classes of antihypertensive drugs will be able to control hypertension (Calhoun et al., 2008a,b; Muntner et al., 2017; Whelton et al., 2018; Carey, 2019). If a patient still has uncontrolled blood pressure while taking 3 different classes of medications or they require 4 or more different medications to control their blood pressure, they are considered to have resistant hypertension, or apparent treatment resistant hypertension (Calhoun et al., 2008b; Carey, 2013, 2016, 2019; Whelton et al., 2018). About 13% of United States adults have been diagnosed with resistant hypertension (Calhoun et al., 2008a,b; Carey, 2013, 2019; Whelton et al., 2018). This data indicates that the mechanisms of hypertension have yet to be completely understood and patients with resistant hypertension may benefit greatly from new knowledges and the development of novel hypertension treatments.
Against this background, we and others have recently hypothesized that the Na+/H+ exchanger 3 (NHE3) in small intestines and the proximal tubule of the kidney plays a key role in maintaining physiological blood pressure homeostasis and the development of Ang II-induced hypertension, and may serve a potential new therapeutic target in treating hypertension. To test our hypothesis, we and other laboratories have used novel mutant mouse models with whole body- (Schultheis et al., 1998a,b; Lorenz et al., 1999; Wang et al., 1999; Wang et al., 2001; Li et al., 2015b), kidney- (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015a, 2019b; Fenton et al., 2017; Zhuo et al., 2021), or proximal tubule-specific deletion of NHE3 to study the important roles and underlying mechanisms of NHE3 in maintaining basal blood pressure homeostasis and the development of Ang II-induced hypertension (Li et al., 2018, 2019b; Zhuo et al., 2021). The objective of this invited article is to review, update, and discuss the localization, physiological function, and the roles of intestinal and proximal tubule NHE3 in maintaining basal blood pressure homeostasis and the development of Ang II-dependent hypertension.
The sodium (Na+) and proton (H+) antiport cross the cell membranes plays a fundamental role in maintaining normal cellular volume and acid–base homeostasis (Yun et al., 1995; Donowitz et al., 2013; Pedersen and Counillon, 2019). The presence of a direct coupling between Na+ and H+ flux across the brush border membranes of rat small intestines and the kidney was demonstrated in 1970s (Murer et al., 1976). The genes encoding a Na+/H+ exchanger was subsequently cloned in humans (Sardet et al., 1989; Takaichi et al., 1992), rabbits (Tse et al., 1992, 1993; Collins et al., 1993), and rats (Orlowski et al., 1992; Wang et al., 1993). The Na+/H+ exchanger family is now known to have nine distinct isoforms (NHE1-NHE9), based on the order of their molecular cloning, each serving a different role. These NHE isoforms range in size from 645 to 898 amino acids long, inserting into the plasma membrane with the N-terminal facing the extracellular space and the C terminal within the cytoplasmic domain (Sardet et al., 1989; Orlowski et al., 1992; Takaichi et al., 1992; Tse et al., 1992, 1993; Collins et al., 1993). The N-terminal is responsible for the exchange of solutes, while the cytosolic C-terminal mediates the hormonal regulation of the exchangers (Wakabayashi et al., 1992; Counillon et al., 1994; Yun et al., 1995; Pedersen and Counillon, 2019). Each isoform varies in its distinct tissue distribution, subcellular location, and level of expression in different tissues under different regulations (Figure 1; Counillon et al., 1994; Yun et al., 1995; Pedersen and Counillon, 2019).
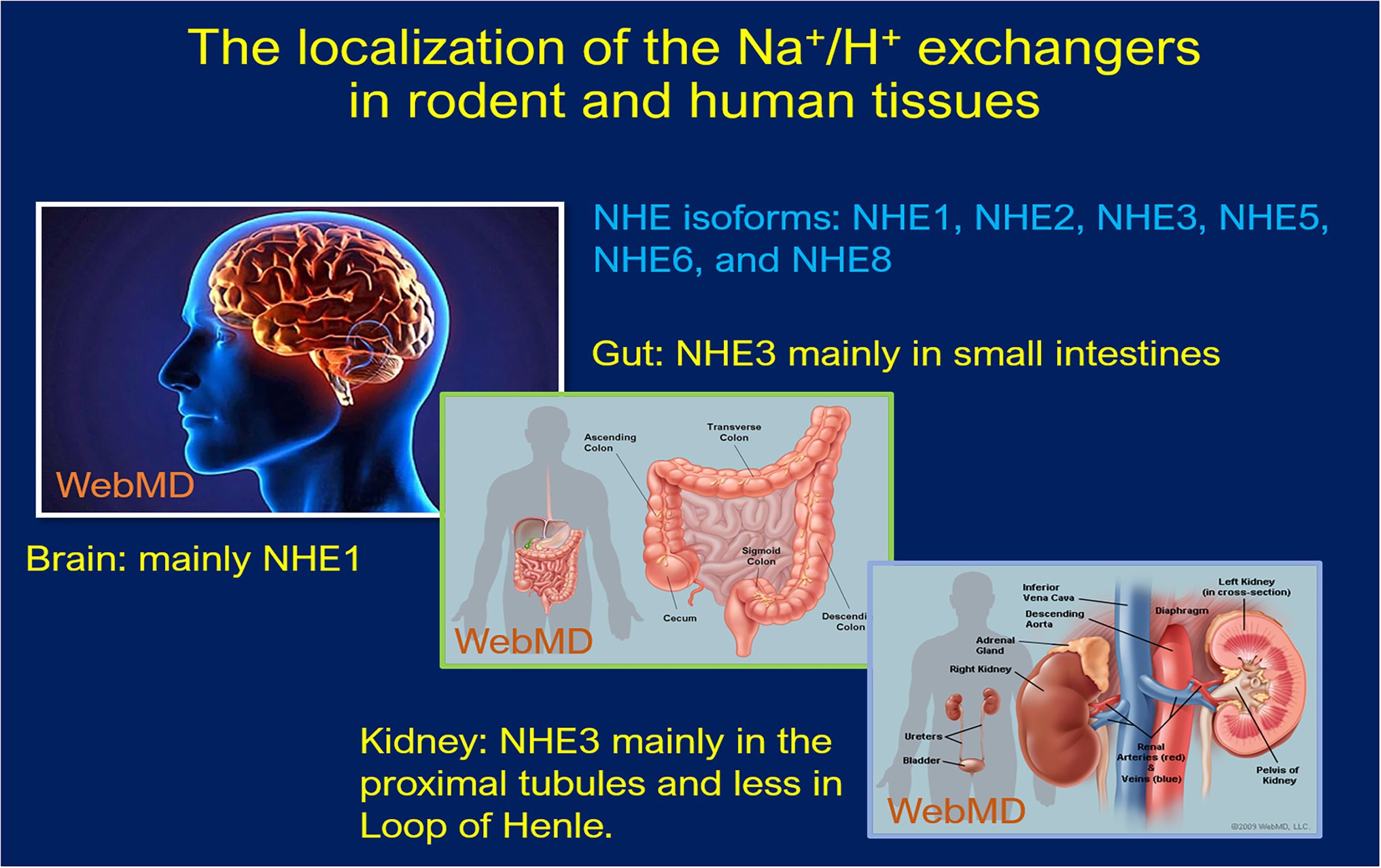
Figure 1. Schematic localization of the Na+/H+ exchanger expression in human and rodent tissues. NHE1 and NHE2 are expressed in almost every tissue type, including intestinal, kidney, testicular, gastric parietal, and skeletal muscle cells etc. NHE4 expression occurs in the GI tract, whereas NHE5 is most prominently expressed in the brain on glioma cells. NHE6–9 are specific in their intracellular localization found in endosomes, while NHE7 is localized to the trans-Golgi network along with early and recycling endosomes. NHE8 is also an intracellular NHE protein, especially in late endosomes and facilitates alkalinization of the organelle.
NHE1 is expressed in almost every tissue type, responsible for maintaining appropriate intracellular volume and cytosolic pH (Biemesderfer et al., 1992; Yun et al., 1995; Evans et al., 1999; Pedersen and Counillon, 2019). NHE2 has been localized to intestinal, kidney, testicular, gastric parietal, and skeletal muscle cells, particularly on the plasma membranes (Bookstein et al., 1997; Chambrey et al., 1998; Schultheis et al., 1998b; Malakooti et al., 1999; Bachmann et al., 2004). NHE4 can be found along the GI tract, as well as the basolateral membranes of the thick ascending limb and distal convoluted tubule of the nephron, where it plays an important role in maintaining cellular pH homeostasis through aldosterone signaling (Chambrey et al., 2001; Bourgeois et al., 2010; Arena et al., 2012; Pedersen and Counillon, 2019). NHE5 is most prominently expressed in the brain on glioma cells and plays a role in the regulation of growth factor and integrin signaling (Attaphitaya et al., 1999; Kurata et al., 2019). NHE6-9 are specific in their intracellular localization. NHE6 and 9 are found in endosomes, while NHE7 has been localized to the trans-Golgi network along with early and recycling endosomes (Xinhan et al., 2011; Pedersen and Counillon, 2019; Kucharava et al., 2020). NHE8 is also an intracellular NHE protein but is a component of late endosomes and facilitates alkalinization of the organelle (Goyal et al., 2005; Lawrence et al., 2010; Pedersen and Counillon, 2019).
The roles of NHE1, NHE2, and NHE8 along the GI tract or the kidney remain poorly understood, but it is most likely that their respective contributions to overall salt and fluid, acid and base balance, Na+ absorption in the gastrointestinal tract, and Na+ reabsorption in the kidney may be very limited (Yun et al., 1995; Zachos et al., 2005; Bourgeois et al., 2010; Pedersen and Counillon, 2019). Indeed, the role of NHE1 was investigated through a whole-body genetic deletion model (Nhe1−/−), but the results yielded CNS pathologies instead such as ataxia and seizures and no evidence of abnormal Na+ reabsorption or blood pressure phenotypes were noted (Cox et al., 1997). NHE2 has also been investigated as a potential contributor to sodium and water reabsorption along the GI tract; however, a Nhe2−/− knockout model demonstrated no differences in GI tract reabsorption when compared to the wild-type (WT), with similar results emerging from a pharmacological inhibition study (Schultheis et al., 1998b; Ledoussal et al., 2001a,b; Gawenis et al., 2002; Bachmann et al., 2004; Bailey et al., 2004; Guan et al., 2006; Engevik et al., 2013). Like Nhe1−/− and Nhe2−/− models, Nhe8−/− mouse models also demonstrated no significant difference in serum Na+ levels and exhibited a lack of diarrhea phenotype indicating the limited role of NHE8 in intestinal Na+ absorption and fluid balance (Xu et al., 2012; Xu et al., 2013). Thus, there is clear lack of evidence supporting any important role of these NHE isoforms in mediating or regulating sodium absorption and maintaining body salt and fluid balance or blood pressure homeostasis. Because there is no direct evidence in the literature for these above-mentioned NHEs to play a significant role in regulating intestinal and kidney Na+ transport and maintaining basal body salt and fluid balance and blood pressure homeostasis, this article will only focus on NHE3 in the gastrointestinal tract and the kidney.
NHE3 is an antiporter that is expressed mainly on the apical membrane but can be sequestered intracellularly within endosomes depending on body salt and fluid, acid and base balance, blood pressure homeostasis, or hemodynamic factors (Wu et al., 1996; Chow, 1999; Donowitz et al., 2013; Pedersen and Counillon, 2019; Zhuo et al., 2021). NHE3 has been localized primarily in abundance to the small intestines of the gastrointestinal tract and the proximal tubules and the thick ascending limbs of the kidney (Biemesderfer et al., 1993; Kulaksiz et al., 2001; He and Yun, 2010; Shawki et al., 2016; Pedersen and Counillon, 2019). Large intestines also express NHE3 but with a much lower level than small intestines (Bookstein et al., 1997; Gawenis et al., 2002; Bradford et al., 2009; He and Yun, 2010; Pedersen and Counillon, 2019). Indeed, the levels of NHE3 expression in small and large intestines are corresponding to their respective contributions to absorbing ingested sodium loads. In the kidney, NHE3 is expressed primarily along the apical membranes of the proximal tubules, especially in early S1 and S2 segments, and its expression gradually decreases throughout the thick ascending limb of the loop of Henle (Biemesderfer et al., 1993; Wu et al., 1996; Cox et al., 1997; Chow, 1999; Brooks et al., 2001; Wade et al., 2001; Bacic et al., 2003; Zhuo et al., 2021).
The expression of NHE3 in the gastrointestinal tract and the kidney may be regulated by diverse molecular mechanisms and hormonal factors (Wu et al., 1996; D’Souza et al., 1998; Hu et al., 2001; Leong et al., 2002; Fuster et al., 2007; Li et al., 2009a, 2012; Kemp et al., 2014, 2016; Castelo-Branco et al., 2017). A number of mechanisms have been shown to regulate the level of NHE3 expression, but the majority of these regulators can be divided into four groups—phosphorylation, trafficking, protein–protein interaction, and transcriptional regulation (Zhao et al., 1999, 2004; He and Yun, 2010; Pedersen and Counillon, 2019). NHE3 can be phosphorylated by a variety of protein kinases including protein kinase A, which downregulates signal transduction pathways, and casein kinase 2, which upregulates NHE3 trafficking to the plasma membranes (Kurashima et al., 1997; Zhao et al., 1999; Kocinsky et al., 2005; Sarker et al., 2008). NHE3 trafficking describes the ability of NHE3 to be recycled between intracellular compartments and the plasma membrane via a clathrin-mediated pathway (D'Souza et al., 1998). However, there is accumulating evidence that under physiological conditions, NHE3 is equally distributed between the body (active) and base (less active) of the microvilli in brush border membranes (Biemesderfer et al., 2001). In response to the changes in renal perfusion pressure, hormonal, or pharmacological factors, NHE3 may be translocated between the body and base of the microvilli (Yip et al., 1998; Yang et al., 2007; Riquier-Brison et al., 2010). Indeed, acute hypertension or acute ACE inhibition induced NHE3 translocation from the body to the base of microvilli in the rat kidney (Yip et al., 1998; Yang et al., 2007). The vasoactive peptide hormone angiotensin II (Ang II) appears to regulate membrane NHE3 abundance in a biphasic manner (Leong et al., 2002; Yang et al., 2007; Li et al., 2009b, 2012; Riquier-Brison et al., 2010; Li and Zhuo, 2011). A physiological concentration of Ang II increases apical membrane NHE3 protein abundance, while a high pressor concentration of Ang II increases NHE3 translocation from the membranes and reduces the number of transporters on the membranes (Leong et al., 2002; He et al., 2010; Riquier-Brison et al., 2010; Li and Zhuo, 2011).
Another mechanism by which the expression of NHE3 on the plasma membrane can be regulated is via protein–protein interactions. One example of this in the proximal tubules is the Na+/H+ exchanger regulatory factor (NHERF1), which is activated by dopamine and results in the inactivation of the NHE3 transporter through the formation of a multiprotein signaling complex (Wade et al., 2001; Weinman et al., 2001; He et al., 2010; Giral et al., 2012). Finally, the regulation of NHE3 expression may involve transcriptional regulation, which is an insidious process when compared to the other mechanisms (He and Yun, 2010; Pedersen and Counillon, 2019). In addition to their effect on exocytosis, insulin, dexamethasone and glucocorticoids activate (Bobulescu et al., 2005; Fuster et al., 2007; Wang et al., 2007); whereas dopamine, cAMP, and cGMP inhibit the exocytosis or transcription levels of NHE3 (Yun et al., 1997; Zizak et al., 1999; Hu et al., 2001; Bacic et al., 2003; Cha et al., 2005). Acute treatment with parathyroid hormone (PTH) appeared to inhibit NHE3 translocation from the base of microvilli (Yang et al., 2004), whereas long-term treatment with PTH decreased NHE3 mRNA levels due to a PKA-dependent inhibitory effect on the NHE3 promoter (Bezerra et al., 2008). Taken together, the expression of NHE3 on the plasma membrane is regulated by a complex system, allowing it to adapt to body salt and fluid balance, vasoactive hormones, and molecular and cellular stressors to efficiently regulate cellular acid and base, maintain body salt and fluid balance and blood pressure homeostasis.
In contrast to other NHE isoforms, NHE3 is the major isoform in the NHE family and plays the most significant role in regulating the absorption of sodium within the gastrointestinal tract (Schultheis et al., 1998a; Gawenis et al., 2002; Woo et al., 2003; Bradford et al., 2009; Li et al., 2015a,b; Shawki et al., 2016; Zhuo et al., 2021). This is strongly supported by integrative studies with generation of global or whole-body NHE3 deletion mouse models (Nhe3−/−) to investigate its role in the intestinal absorption of sodium. Schultheis et al. (1998a) were the first to generate and reported the gastrointestinal phenotypes of the Nhe3−/− mouse model. Although these Nhe3−/− mice were able to survive, they developed severe diarrhea and Na+ wasting phenotypes, with a large increase in 24-h fecal Na+ excretion when compared to the WT or proximal tubule-specific Nhe3−/− mice, PT- Nhe3−/− (Figure 2; Schultheis et al., 1998a; Li et al., 2015b, 2018b). Additionally, there was a major upregulation in epithelial Na+ channel (ENaC) and H+/K+ ATPase activity, but with near-normal blood gas, pH, and electrolyte balance, indicating that a compensatory mechanism might be at work (Schultheis et al., 1998a). These results indicated that NHE3 is the primary transporter responsible for the absorption of dietary sodium in the intestines and therefore contributes considerably to the maintenance of sodium and fluid balance. Despite its key role in the absorption of dietary sodium, however, deletion of NHE3 along the gastrointestinal tract had no significant impacts on the natriuresis response in Nhe3−/− mice (Li et al., 2015a,b). This is because the kidney acts to compensate for the loss of NHE3 in the proximal tubules and the loop of Henle by upregulating the expression of the renin–angiotensin–aldosterone system and other Na+ cotransporters, such as sodium and glucose cotransporter 2 (SGLT2), Na+/K+-ATPase, sodium bicarbonate cotransporter, and aquaporin 1 (AQP1; Li et al., 2015a,b). Nevertheless, basal systolic, diastolic, and mean arterial blood pressure are consistently about 15 to 20 mmHg lower in adult male and female global Nhe3−/− mice than WT mice (Figure 3), therefore affirming an important role of intestinal and kidney NHE3 in maintaining physiological blood pressure homeostasis (Schultheis et al., 1998a; Li et al., 2015b, 2018b).
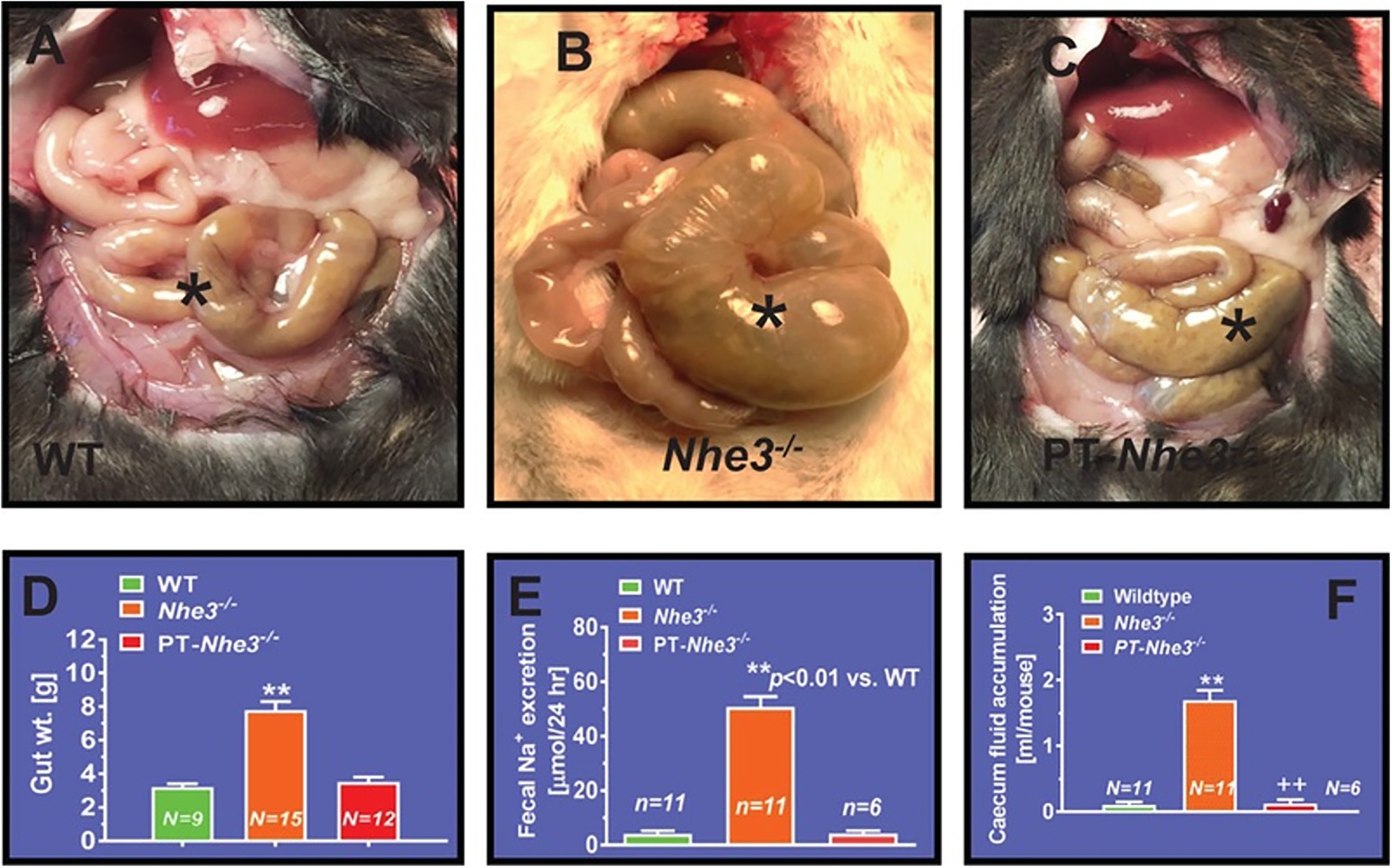
Figure 2. Proximal tubule-specific deletion of NHE3 does not significantly alter the intestinal structural and absorptive phenotypes in PT-Nhe3−/− mice. (A) A representative normal caecum segment between small and large intestines in a WT mouse (*). (B) A representative cecum segment between small and large intestines in a global Nhe3−/− mouse, showing the extremely enlarged cecum segment accumulated with a large volume of fluid content inside (*). (C) A representative cecum segment between small and large intestines in a PT-Nhe3−/− mouse, showing the lack of enlarged cecum segment and no accumulation of a large volume of fluid content in the cecum segment (*). (D) The overall gut weight more than doubled in global Nhe3−/− mice than WT and PT-Nhe3−/− mice (p < 0.01). (E) Twenty-four hours fecal Na+ excretion was ~10-time higher in global Nhe3−/− mice than WT and PT-Nhe3−/− mice (p < 0.01). (F) Accumulation of fluid content in the cecum segment was ~10 times higher in global Nhe3−/− mice than WT and PT-Nhe3−/− mice (p < 0.01). There were no differences in the overall gut weight and 24 h fecal Na+ excretion between WT and PT-Nhe3−/− mice. Reproduced from Li et al., 2018 with permission from the copyright holder.
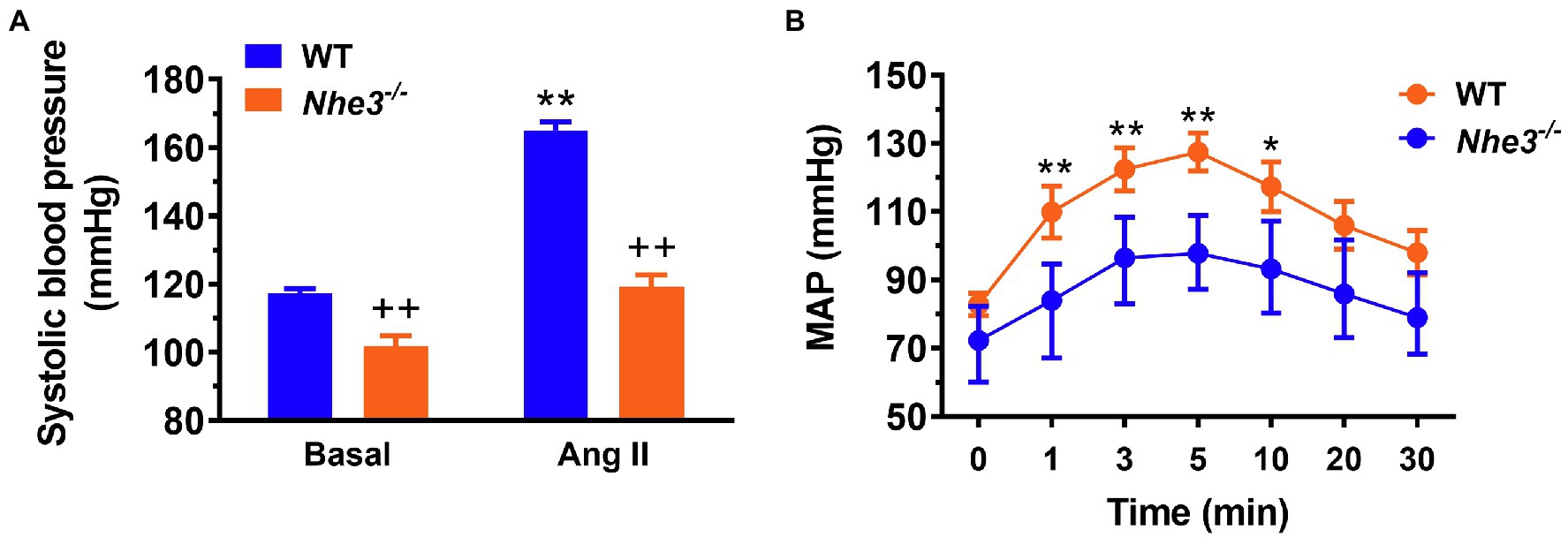
Figure 3. Angiotensin II-induced hypertension is attenuated in global Nhe3−/− mice with transgenic rescue of the NHE3 gene in small intestines using the small intestine-specific IFABP promoter, tgNhe3−/− (A,B). Note that basal blood pressure remains significantly lower in tgNhe3−/− mice than WT mice (A). *p < 0.05, and **p < 0.01 vs. their basal level; ++p < 0.01 vs. WT mice. Reproduced from Li et al. (2019a) with permission from the copyright holder.
To further differentiate the roles of intestinal and kidney NHE3 in the regulation of Na+ absorption and reabsorption and blood pressure, Woo et al. (2003) and Li et al. (2015a) have generated and studied a different Nhe3−/− mouse model, tgNhe3−/−, with transgenic rescue of the NHE3 gene selectively in small intestines using the small intestine-specific IFABP promoter (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015a). In this mouse model, overall salt wasting phenotypes including severe diarrhea, marked increased fecal Na+ excretion, and lowered basal blood pressure were moderately but significantly improved in tgNhe3−/− mice (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015a), but all these abnormal phenotypes persisted in tgNhe3−/− mice (Li et al., 2015a; Zhuo et al., 2021). Indeed, diarrhea persisted, and basal blood pressure remained significantly lower in tgNhe3−/− mice (Figure 4; Li et al., 2015a; Zhuo et al., 2021). Recently, Xue et al. (2020, 2022) have generated a new inducible intestinal epithelial cell-specific NHE3 knockout mouse model, which showed similar intestinal abnormal absorptive phenotypes, mimicking congenital sodium diarrhea with enhanced phosphate. Thus, the development of moderate to severe diarrhea due to intestinal Na+ absorptive defect is a major phenotype in global and intestine-specific Nhe3−/− mice (Schultheis et al., 1998a; Li et al., 2015a,b; Xue et al., 2020, 2022).
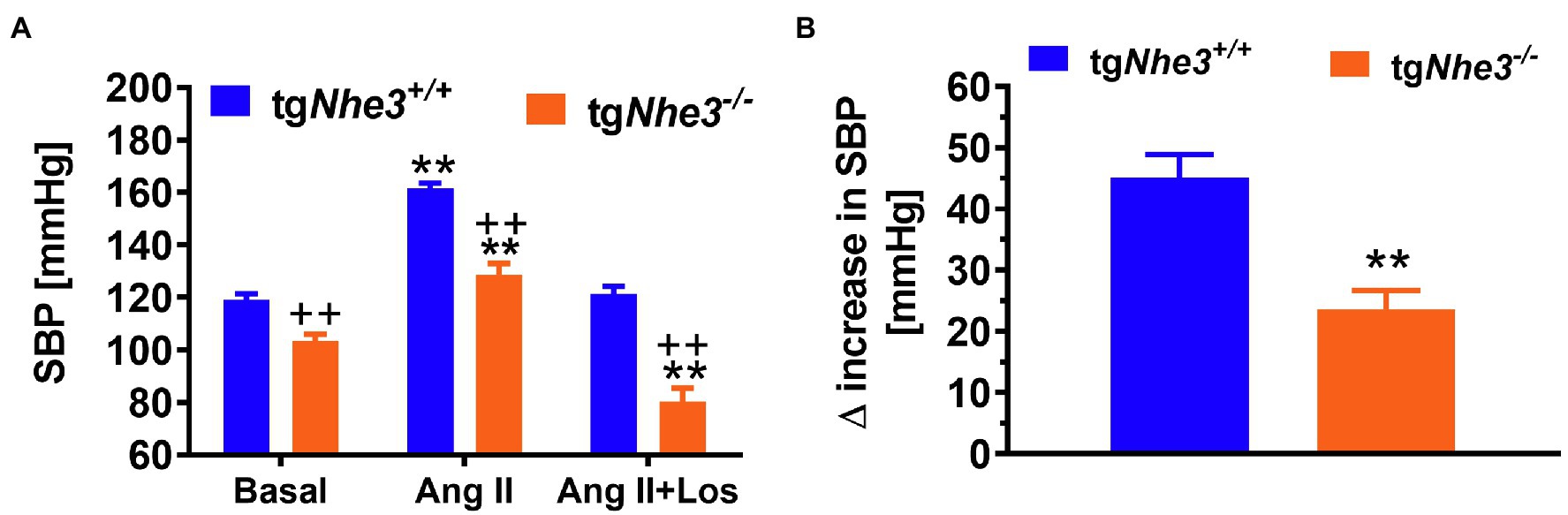
Figure 4. Angiotensin II-induced hypertension is attenuated in global Nhe3−/− mice with transgenic rescue of the NHE3 gene in small intestines using the small intestine-specific IFABP promoter, tgNhe3−/− (A,B). Note that basal blood pressure remains significantly lower in tgNhe3−/− mice than WT mice (A). **p < 0.01 vs. their basal level; ++p < 0.01 vs. WT mice. Reproduced from Li et al. (2019a) with permission from the copyright holder.
Another interesting and consistent finding in studying global Nhe3−/− and tgNhe3−/− mice is that these mice develop striking structural abnormalities in small intestines (Schultheis et al., 1998a,b; Li et al., 2015a,b). The structural abnormalities show that all intestinal segments were significantly enlarged in Nhe3−/− mice, with extremely enlarged cecum segment containing a large amount of accumulated fluid (Schultheis et al., 1998a; Li et al., 2015a,b). These abnormalities were also found in Nhe3−/− mice with transgenic rescue of the NHE3 gene selectively in small intestines (Li et al., 2015a). Since mice with deletion of NHE3 selectively in the proximal tubules of the kidney do not develop these intestinal structural abnormalities, and severe diarrhea and Na+ wasting phenotypes (see below; Li et al., 2018, 2019a), our studies suggest that NHE3 in the gastrointestinal tract may likely play an important role in the development of the gut in addition to its major role in mediating Na+ absorption. However, a recently developed, inducible intestine-specific Nhe3−/− mouse model does not show any intestinal structural abnormalities as Nhe3−/− or tgNhe3−/− mice do (Xue et al., 2020, 2022). The reasons underlying these differences between global Nhe3−/−, transgenic tgNhe3−/−, and intestine-specific Nhe3−/− mice remain unknown. One likely explanation may be that the inducible intestine-specific Nhe3−/− model is a short-term deletion model in adult mice lasting only for a few weeks, which may not be long enough to reveal any structural development problems. Further studies are necessary to uncover the molecular mechanisms underlying the development of structural abnormalities in small intestines in Nhe3−/− and tgNhe3−/− mice.
It is well recognized that among all Na+ transporters and cotransporters, NHE3 is the most critical Na+ transporter responsible for Na+ reabsorption within the kidney (Lorenz et al., 1999; Wang et al., 1999; Zhuo and Li, 2013; Li et al., 2018, 2019a; Zhuo et al., 2021). Specifically, the proximal tubules are responsible for reabsorbing 65%–70% of glomerular filtered Na+ and water loads, whereas NHE3 in the proximal tubules is directly and indirectly responsible for >50% of glomerular filtered Na+ reabsorption (Schultheis et al., 1998a; Lorenz et al., 1999; Wang et al., 1999; Li et al., 2015a,b; Zhuo et al., 2021). To further investigate the physiological function of NHE3 in the kidney, pan-renal tubule-specific and proximal tubule-specific Nhe3−/− mouse models have been generated and studied, respectively, during recent years. A whole-kidney tubule epithelia-specific Nhe3−/− mouse model was generated by cross breeding NHE3-floxed mice with Pax8-Cre mice (Fenton et al., 2015, 2017). This scientific approach is based on the premise that Pax8 is only expressed along the renal tubules, thus in theory, NHE3 is expected to be deleted from all nephron segments in the kidney, including proximal tubule segments (S1–S3) and the ascending limb of the loop of Henle in this Pax8-Cre/NHE3-floxed mouse model. These mice were found to have fully compensated plasma ion concentrations, osmolality, and pH when compared to the WT counterpart. These findings of normal blood biochemical phenotypes in Pax8-Cre/NHE3-floxed mice have effectively alleviated a major concern that genetic deletion or therapeutically targeting of NHE3 in the kidney may cause body acid-base, and salt and fluid imbalance. Alternatively, these results strongly suggest that these mice are able to adequately compensate for the loss of NHE3 function in the kidney to maintain normal body salt and fluid, acid and base balance (Fenton et al., 2015, 2017). However, despite these similar blood biochemistries, these mice increased fluid intake and urine flow rate, with decreased urine osmolality and increased urine pH (Fenton et al., 2015, 2017). Intra-arterial blood pressure was found to be significantly lower in Pax8-Cre/NHE3-floxed mice likely due to increased natriuretic responses. Furthermore, these mice demonstrated an increased sensitivity to dietary NaCl, along with a 20% decrease in GFR (Fenton et al., 2017). Taken together, this study using Pax8-Cre/NHE3-floxed mice provides important evidence for an important role of NHE3 along the entire nephron segments contributing to body salt and fluid balance and blood pressure homeostasis.
However, Pax8-Cre/NHE3-floxed mice is at best a whole kidney tubular epithelia-specific Nhe3−/− mouse model, the contributions of NHE3 in the proximal tubules and the loop of Henle are difficult to be separated in this mouse model. In previous micropuncture studies, Vallon et al. (2000); Gekle et al. (2004) showed that luminal perfusion of the rat proximal tubule and loop of Henle with a NHE3 inhibitor S3226 inhibited proximal tubular reabsorption by ~30% without altering fluid and Na+ reabsorption in the Loop of Henle. This study is largely consistent with the micropuncture findings that proximal tubular fluid or bicarbonate reabsorption was decreased by about 50% in global Nhe3−/− mice (Lorenz et al., 1999; Wang et al., 1999). Since the proximal tubule is responsible for 65%–70% of Na+ reabsorption in the kidney, whereas NHE3 in the proximal tubule is directly and indirectly responsible for over 50% of Na+ reabsorption in the kidney, our group recently generated and studied a new Nhe3−/− mouse model with proximal-tubule specific deletion of NHE3 using the SGLT2-Cre/NHE3-floxed approach (PT-Nhe3−/−; Li et al., 2018, 2019b; Zhuo et al., 2021). This new mutant mouse model allowed us to directly test the hypothesis that NHE3 in the proximal tubule is primarily responsible for maintaining body salt and fluid balance and blood pressure homeostasis in part by modulating the pressure-natriuresis response (Li et al., 2018, 2019b; Zhuo et al., 2021). In contrast to the whole-body Nhe3−/− (Schultheis et al., 1998a; Li et al., 2015a,b) or the tgNhe3−/− mouse model (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015a), this proximal tubule-specific Nhe3−/− mouse model did not show the abnormalities in both structures and Na+ absorption in the gastrointestinal tract (Li et al., 2018, 2019b, 2021). No diarrhea phenotype was developed in this proximal tubule-specific Nhe3−/− mouse model since no difference was found in 24 h fecal Na+ excretion between WT and PT-Nhe3−/− mice (Figure 2). Additionally, there were no differences in plasma pH, plasma ions or bicarbonate, and hematocrit between WT and PT-Nhe3−/− mice, suggesting that loss of NHE3 function in the proximal tubules does not cause abnormal body acid and base, as well as body electrolyte and fluid balance (Li et al., 2018, 2019b, 2021). However, 24-h urine (diuresis) and urinary Na+ (natriuresis) and K+ excretion were significantly increased under basal physiological conditions. These kidney phenotypes were associated significantly decreased basal systolic, diastolic, and mean arterial blood pressure (−12 to −15 mmHg) in both male and female PT-Nhe3−/− mice when measured via telemetry (Figure 5; Li et al., 2018, 2019b, 2021). When compared to the wild type, the pressure-natriuresis response was significantly augmented in PT-Nhe3−/− mice in response to similar increases in renal perfusion pressure. These findings strongly support the hypothesis that NHE3 in the proximal tubule of the kidney is responsible for a significant bulk of Na+ and water reabsorption, and that the presence or loss of NHE3 function in the proximal tubules alone is sufficient to alter basal blood pressure homeostasis.
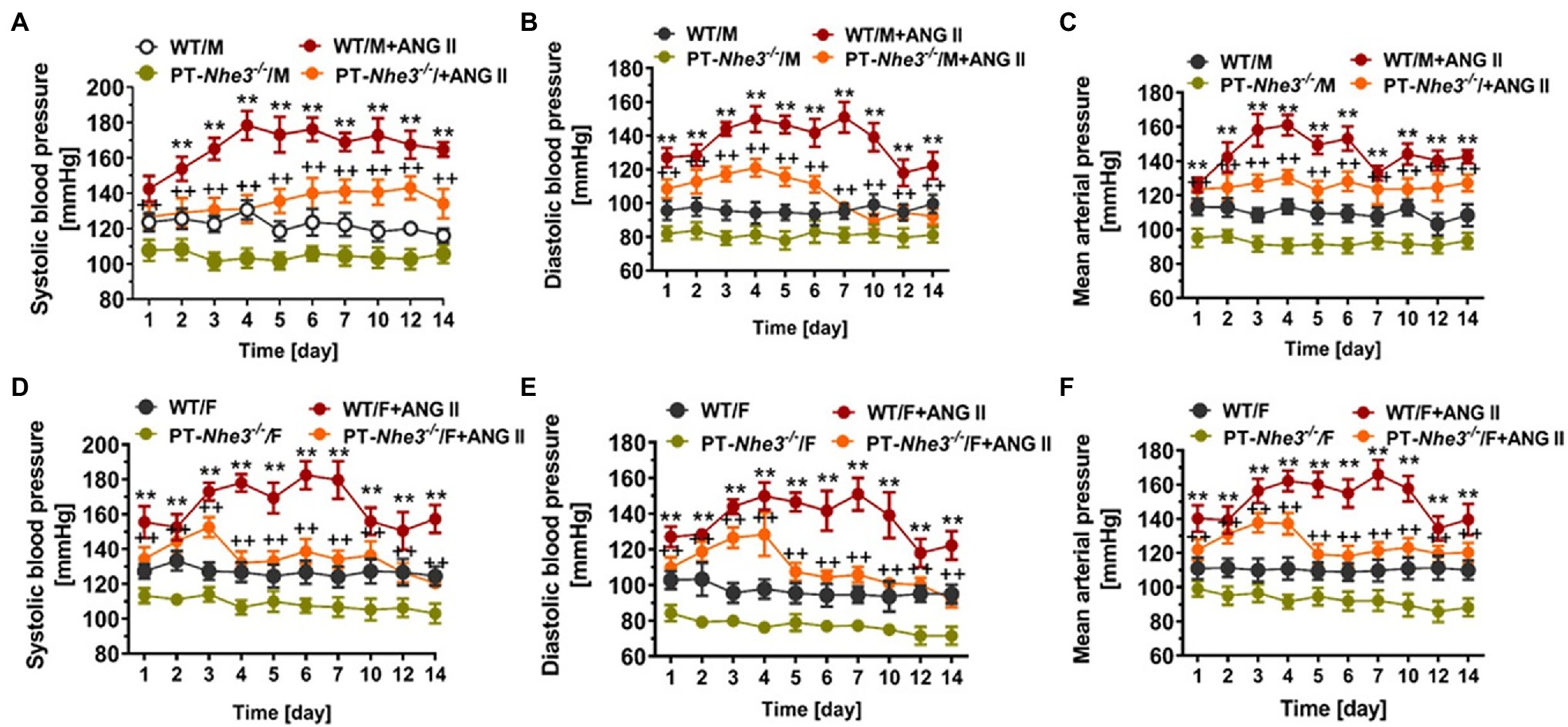
Figure 5. Systolic, diastolic and mean arterial blood pressure responses to a high pressor dose of ANG II infusion, 1.5 mg/kg/day, i.p., in conscious adult male (A-C) and female (D-F) WT and PT-Nhe3−/− mice, as measured continuously for 14 days using the direct implanted telemetry technique. Note that basal blood pressure levels were about 12–15 mmHg lower in PT-Nhe3−/− than WT mice, and that Ang II-induced hypertension was attenuated in male (A-C) and female (D-F) PT-Nhe3−/− mice. **p < 0.01 vs. WT time-control group; ++p < 0.01 vs. PT-Nhe3−/− time-control group, respectively. N = 5–12 per group. Reproduced from Li et al. (2019b) with permission.
The renin–angiotensin–aldosterone system (RAAS) has been well established as an important vasoactive and humoral system in the regulation of blood pressure, with its major components widely expressed in different tissues (De Gasparo et al., 2000; Kobori et al., 2007; Navar et al., 2011; Forrester et al., 2018). However, the kidney RAAS plays a dominant role (Harris and Navar, 1985; Crowley et al., 2005, 2006; Li et al., 2009b, 2011, 2018, 2021; Gurley et al., 2011; Yang and Xu, 2017; Kemp et al., 2022). The juxtaglomerular (JG) cells in the renal cortex express prorenin, which is cleaved into renin when the JG cells are activated in response to stimuli like decreased blood pressure, sympathetic activation, or decreased sodium delivery to the macula densa (Kobori et al., 2007; Chappell, 2012; Zhuo et al., 2013; Sequeira-Lopez and Gomez, 2021). Renin is released into the kidney as well as in the circulation to cleave the liver-derived substrate angiotensinogen (AGT) to produce angiotensin I (Ang I), which is the precursor for the active peptide Ang II (Kobori et al., 2007; Forrester et al., 2018). Ang I must then be converted into the biologically active Ang II by the angiotensin-converting enzyme (ACE; Kobori et al., 2007; Chappell, 2012; Zhuo et al., 2013; Sequeira-Lopez and Gomez, 2021). While other Ang peptides may possess different biological activities, Ang II is the key active peptide in the regulation of blood pressure via acting upon AT1 and AT2 receptors, which have counter-regulatory properties (Harris and Navar, 1985; Crowley et al., 2005, 2006; Li et al., 2009b, 2011, 2018, 2021; Gurley et al., 2011; Carey, 2017; Kemp et al., 2022).
In the proximal tubules of the kidney, Ang II activates G protein-coupled AT1 (AT1a) receptors on apical and basolateral membranes, which mediates Gq/11/phospholipase C/IP3/protein kinase C signaling pathways and increases the expression of NHE3 and Na+ reabsorption in the proximal tubules (De Gasparo et al., 2000; Kobori et al., 2007; Li et al., 2009b; Li and Zhuo, 2011; Navar et al., 2011; Forrester et al., 2018; Li et al., 2018). Ang II may also indirectly activate the Gi/cAMP/protein kinase A signaling pathways to inhibit NHE3 activity in proximal tubule cells, as forskolin significantly increased cAMP production in OK cells and Ang II attenuated these receptors (Thekkumkara et al., 1998). Systemically, activation of AT1 receptors by Ang II leads to vasoconstriction, increased salt and water retention, sympathetic stimulation, and increased aldosterone synthesis (De Gasparo et al., 2000; Kobori et al., 2007; Li et al., 2009a; Navar et al., 2011; Forrester et al., 2018). Conversely, activation of AT2 receptors by Ang II appears to provide counteract and protective effects against AT1 receptor-mediated effects by inducing vasodilation, diuresis and natriuresis (Siragy et al., 1999; Siragy and Carey, 1999; Kemp et al., 2014, 2016; Carey, 2017). Nevertheless, it is the AT1 receptor-mediated effects by Ang II play a predominant role in maintaining basal blood pressure homeostasis and the development of Ang II-dependent hypertension.
It is well-recognized that Ang II-induced or Ang II-dependent hypertension is caused by complex genetic, neural, and hormonal mechanisms involving AT1 receptor-mediated systemic, neural, adrenal, cardiovascular, gastrointestinal, and renal responses (Harris and Young, 1977; Harris and Navar, 1985; Navar et al., 1987; Crowley et al., 2005; Kobori et al., 2007; Forrester et al., 2018; Rucker et al., 2018; Li et al., 2019a). In both small intestines and the proximal tubules of the kidney, NHE3 is a recognized downstream target of Ang II-induced AT1 receptor activation, which increases NHE3 expression or activity and promotes Na+ absorption from small intestines and Na+ reabsorption from the proximal tubules (Levens et al., 1980, 1981; Saccomani et al., 1990 Jin et al., 1998; Li et al., 2009b, 2015a,b, 2019a; Zhuo et al., 2021). Increased Na+ absorption from the gut and reabsorption from the kidney without proportional increases in fecal and urinary Na+ excretion leads to body salt and fluid retention, one important mechanism underlying the development and progression of Ang II-dependent hypertension (Li et al., 2015a,b, 2019a). In a previous study, Gurley et al. (2011) showed that genetic deletion of AT1a receptors in the renal proximal tubules protected against Ang II-induced hypertension in part by decreasing NHE3 expression in the kidney. However, whether and to what extent NHE3 in the gastrointestinal tract and the kidney directly contribute to the development of Ang II-induced hypertension have not been investigated previously.
Against this background, our group has tested this hypothesis by comparing systolic and mean arterial blood pressure responses to Ang II infusion in WT, whole-body Nhe3−/−, tgNhe3−/− and PT-Nhe3−/− mice, respectively (Li et al., 2015a,b, 2018, 2019b, 2021). We tested two methods of Ang II infusion—an acute systemic infusion under anesthesia, and a chronic osmotic minipump infusion in conscious animals. Under both conditions, WT mice were found to have significantly increased systolic and mean arterial pressure, as expected. By contrast, whole-body Nhe3−/−, tgNhe3−/− and PT-Nhe3−/− mice demonstrated significantly attenuated responses to acute or chronic Ang II infusions, with the decreases reaching about 50% of WT responses (Figures 35; Li et al., 2015a,b, 2018, 2019b, 2021). Our results provide strong evidence for the 1st time an important role of NHE3 in the gastrointestinal tract and the proximal tubules of the kidney not only in maintaining basal blood pressure homeostasis but also in the development of Ang II-induced hypertension.
The Na+/H+ exchanger 3 in the proximal tubules of the kidney has long been implicated in the development of hypertension in spontaneously hypertension in rats (SHRs; Thomas et al., 1988, 1990; Kelly et al., 1997; Aldred et al., 2000; LaPointe et al., 2002; Kemp et al., 2014, 2016, 2022). Whether NHE3 in the proximal tubules plays any role in hypertension in SHRs remains incompletely understood (Yip et al., 1998; Magyar et al., 2000; Panico et al., 2009; Crajoinas et al., 2014). Early in vivo micropuncture studies suggested that the development of hypertension in young SHRs up to 8 weeks of age involves sodium retention with significantly increased proximal tubule reabsorption (Jv; Thomas et al., 1988, 1990). Increased NHE3 expression and activity has been reported in the proximal tubules of young prehypertensive and adult hypertensive SHRs (Morduchowicz et al., 1989; Dagher and Sauterey, 1992; Hayashi et al., 1997; Kelly et al., 1997; Aldred et al., 2000; LaPointe et al., 2002; Crajoinas et al., 2010). These studies suggest that increased NHE3 expression and activity in the proximal tubules may contribute to the development of hypertension in SHRs by promoting proximal tubule Na+ reabsorption and inducing salt retention.
However, there is evidence that the expression and activity of NHE3 in the proximal tubules are decreased rather than increased in the development of hypertension in SHRs, and therefore NHE3 unlikely plays an important role in the development of spontaneous hypertension (Yip et al., 1998; Magyar et al., 2000; Panico et al., 2009; Crajoinas et al., 2014). In a micropuncture study, Panico et al. (2009) compared proximal tubule fluid reabsorption (Jv) between adult Wistar–Kyoto rats (WKY) and SHRs and found that Jv was in fact decreased by >50% in the proximal tubules of SHRs. The same study further showed that microperfusion of the proximal tubules with a highly potent NHE3 inhibitor, S-1611, decreased Jv in WKY rats but not in SHRs, suggesting that NHE3 stimulates proximal tubule Na+ reabsorption in WKY rather than in SHRs (Panico et al., 2009). In another studies, Crajoinas et al. (2010, 2014) microperfused proximal tubules to determine NHE3-mediated bicarbonate reabsorption between young prehypertensive SHR (5-wk-old) and adult SHR (14-wk-old), and age-matched WKY rats. NHE3 transport activity was found to increase significantly in the proximal tubules of pre-hypertensive SHRs, but not in adult SHRs, when compared with age matched WKY rats (Crajoinas et al., 2010). The lower NHE3 activity in adult SHRs may probably be due to higher phosphorylation of NHE3 at serine 552 (Crajoinas et al., 2010, 2014). More importantly, however, lower NHE3 activity and reduced proximal tubule Na+ reabsopriton in adult SHRs may be due to the redistribution of NHE3 proteins from the brush border (or apical membranes) to the base of the microvilli of proximal tubules (Zhang et al., 1996; Yip et al., 1998; Magyar et al., 2000). Using confocal immunofluorescent microscopic imaging, Yip et al. (1998) demonstrated that NHE3 was localized in the brush border of the microvilli in the proximal tubules of young prehypertensive SHRs, whereas in adult SHRs with established hypertension, it was found at the base of the microvilli of the proximal tubules. Alternatively, Magyar et al. (2000) used the subcellular fractionation of the renal cortex for subcellular localization of NHE3 proteins in the proximal tubules of young and adult SHRs, and compared with that of age matched Sprague–Dawley (SD) rats in response to the development of acute or chronic hypertension. In adult SD rats with acute hypertension, cortical apical NHE3 was found to redistribute from fractions 4–6 to fractions 5–7, even to fractions 8–10, likely corresponding to the base of the microvilli (Magyar et al., 2000). In adult SHRs with further acute increases in blood pressure, the redistribution peak of renal cortical NHE3 was similar to that of SD rats with acute hypertension. Similar subcellular redistribution of NHE3 was found in adult SHRs with chronic hypertension (Magyar et al., 2000). These studies suggest that NHE3 proteins in the proximal tubules redistribute constantly between the brush border and the base of the microvilli in response to the changes in blood pressure. However, how the increased or decreased NHE3 expression and activity correlates with the subcellular redistribution of NHE3 proteins in the proximal tubules before and after the development of hypertension in SHRs remains incompletely understood.
In addition to well-recognized AT1 receptor-mediated effects on NHE3 expression and activity, a defect in AT2 receptor-mediated inhibition of NHE3 expression and activity has been reported in the proximal tubules of SHRs, which may contribute to the development of hypertension in SHRs (Kemp et al., 2014, 2016, 2022; Zhuo and Li, 2019). Although Ang II also bind the AT2 receptors, the predominant agonist for this receptor may be the downstream metabolite of Ang II, i.e., Ang III (Padia et al., 2009; Kemp et al., 2012). After being cleaved from Ang II by the enzyme aminopeptidase A (APA), Ang III primarily binds and activates AT2 receptors to induce the Ang III/AT2/NHE3 signaling (Kemp et al., 2012; Zhuo and Li, 2019). This hypothesis is further supported by a recent study that intrarenal infusion of an AT2 receptor agonist C21 compound induced the subcellular translocation of membrane NHE3 in the proximal tubules of WKY but not SHRs (Kemp et al., 2020). This NHE3 internalization or translocation was associated with significantly increased natriuretic and vasoprotective responses in WKY, suggesting that the Ang III/AT2/NHE3 signaling pathway may play a counteract regulatory role in the regulation of proximal tubule Na+ reabsorption. The angiotensin-converting enzyme 2 (ACE2)/Ang-(1–7)/Mas receptor axis may also regulate NHE3 expression and activity in the kidney of SHRs (Castelo-Branco et al., 2017). Ang (1–7) is derived from Ang I or Ang II by the action of ACE2 and binds to the Mas receptor (Chappell, 2012; Karnik et al., 2017). Ang (1–7) elicits an antagonistic and protective response to Ang II primarily by promoting NO production and inducing vasodilation. Administration of high doses of Ang 1–7 has been shown to inhibit NHE3 transporter activity and decrease kidney Na+ reabsorption in SHRs (Castelo-Branco et al., 2017). Nevertheless, the roles of these alternative pathways in the regulation of NHE3 expression and activity and subsequent Na+ reabsorption from the proximal tubules, blood pressure control, and the development of Ang II-dependent hypertension may be much smaller than those of the predominant Ang II/AT1 (AT1a)/NHE3 signaling pathways.
Sex differences occur in every genetic, genomic, biological, physiological, and diseased responses, as expected. Indeed, it is well-established that the prevalence of hypertension varies between the sexes and increases with age (Silva-Antonialli et al., 2004; Choi et al., 2017; Ramirez and Sullivan, 2018). Males generally have a higher prevalence than premenopausal females, but this changes after menopause, with prevalence increasing in postmenopausal females (Choi et al., 2017; Ramirez and Sullivan, 2018). In vivo studies using SHRs have demonstrated sex differences in the RAS showing that female rats showed lower AT1 receptor expression and higher AT2 receptor expression in the kidney and vasculatures when compared to male SHRs (Silva-Antonialli et al., 2004). Hilliard et al. (2011, 2012) have likewise shown gender differences in AT2 receptor-mediated pressure-natriuresis and renal autoregulation. Additionally, Ang 1–7 was found to be increased in female SHRs despite no difference being found in Ang II levels between the sexes (Sullivan et al., 2010). We have recently studied sex differences in the hypertensive response to Ang II infusions, finding no differences in hypertensive responses between the male and female WT mice, but noting a significant decrease in hypertensive response to Ang II infusion between male and female PT-Nhe3−/− mice (Li et al., 2019b). In female PT-Nhe3−/− mice, the hypertensive response to chronic Ang II infusion was attenuated after 3 days, while in males the same response persisted for 7 days (Li et al., 2019b). Veiras et al. (2017) determined that in female rat proximal tubules, phosphorylation of NHE3 was markedly increased, but the expression of other sodium transporters further down the nephron segments was increased, resulting in a similar natriuresis response due to compensation. The mechanisms behind some but not all sex differences remain unclear, but female hormones, along with other unknown modifiers, may play a protective role in the development of hypertension (Seely et al., 1999; Mirabito et al., 2014; Hu et al., 2021).
As reviewed and discussed above, the evidence presented in this review has highlighted the importance of NHE3 in the gastrointestinal tract, mainly small intestines, and the kidney, primarily the proximal tubules, in maintaining physiological Na+ and fluid balance, basal blood pressure homeostasis, the pressure-natriuresis response, and its role in the development of Ang II-induced hypertension. By studying the roles and the mechanisms by which NHE3 regulates Na+ absorption in the gut and Na+ reabsorption in the proximal tubules, which contributes not only to maintain normal blood pressure but also to the development of Ang II-induced hypertension, a translational relevance may be discovered to pharmacologically target NHE3 in hypertension. With currently available antihypertensive drugs, appropriately 50% of hypertensive patients have their blood pressure adequately controlled. However, some patients fail to control their hypertension even treated simultaneously with three to four different antihypertensive drugs and develop so-called resistant hypertension or apparent treatment resistant hypertension (aTRH; Calhoun et al., 2008; Carey, 2013, 2019; Whelton et al., 2017; Muntner et al., 2018). The prevalence of resistant hypertension or aTRH remains as high as 13%–30% in American adult population, implying that other mechanisms are at play.
Against this background, we have hypothesized that therapeutically targeting NHE3 may present us a new additional pathway to treat human hypertension. As proof-of-concept studies, gastrointestinal NHE3 inhibitors have been developed to treat several disease targets including hypertension, constipation, and hyperphosphatemia in elder patients (Linz et al., 2012, 2016, 2020; Li et al., 2019b; Kovesdy et al., 2021). One NHE3 blocker, SAR218034 (SAR), has been shown to increase fecal sodium excretion and decrease systolic blood pressure in lean old SHRs (Linz et al., 2012). Another NHE3 inhibitor derived from SAR218034, tenapanor, was tested in both rats and healthy human volunteers. This human clinical trial demonstrated an increase in stool Na+, indicating that the drug was working to block the absorption of Na+ from the gut effectively (Spencer et al., 2014). Both drugs, which are nonabsorbable after oral ingestion and only target NHE3 in intestinal apical membranes, exhibited minimally changed plasma concentrations, indicating minimal systemic effects and allowing other NHE proteins to function as they would physiologically (Linz et al., 2012; Spencer et al., 2014). Combinations of tenapanor and enalapril, an ACE inhibitor, yielded even more impressive results in nephrectomized rats, decreasing systolic blood pressure by more than 30 mmHg when co-administered (Spencer et al., 2014). These results suggest that nonabsorbable, intestine-targeting NHE3 inhibitors may be promising for the treatment of resistant hypertension in elder patients with constipation when combined with other already existing antihypertensive drugs by selectively inhibiting NHE3 and Na+ absorption from the gastrointestinal tract. However, tenapanor is reportedly contraindicated in young patients due to salt wasting and decreases in blood pressure (Spencer et al., 2014).
A different class of NHE3 inhibitor, AVE-0657, has been developed by Sanofi-Aventis to treat sleep apnea (Wang et al., 2014), and related clinical trials have unfortunately abandoned. Unlike nonabsorbable SAR218034 and tenapanor (Linz et al., 2012; Spencer et al., 2014), AVE-0657 is absorbable from the gut after oral administration, enters the circulation, and is filtered by the glomerulus and expected to inhibit apical membrane NHE3 in the kidney, primarily the proximal tubules and less the thick ascending limb of the loop of Henle. We recently tested this hypothesis that AVE-0657 attenuates Ang II-induced hypertension in mice primarily by inhibiting NHE3 and Na+ reabsorption in the proximal tubules (Figure 6; Li et al., 2019b; Zhuo et al., 2021). Our studies demonstrated that AVE-0657 administration did not increase fecal Na+ excretion from the gastrointestinal tract suggesting that AVE-0657 does not inhibit NHE3 in small intestines as SAR218034 and tenapanor do. More importantly, AVE-0657 induced natriuresis and significantly attenuated the hypertensive response in Ang II-infused, 2% high salt-fed C57BL/6 J mice (Figure 6; Li et al., 2019b; Zhuo et al., 2021). When the ARB losartan was added to the AVE-0657 treatment, Ang II-infused, high salt-fed-indued hypertension was normalized to the control level. Interesting, the results of this study used AVE-0657 to treat Ang II-infused, high salt-fed-indued hypertensive mice are similar to or consistent with PT-Nhe3−/− mice with or without Ang II-infused, high salt-fed-indued hypertension (Li et al., 2018, 2019b). Taken together, these results are very promising, and we propose to further study and confirm this proof-of-concept hypothesis in different animal models of hypertension, especially in aTRH humans, by therapeutically targeting NHE3 in the proximal tubules of the kidney with or without other classes of antihypertensive drugs (Li et al., 2018, 2019b; Zhuo et al., 2021).
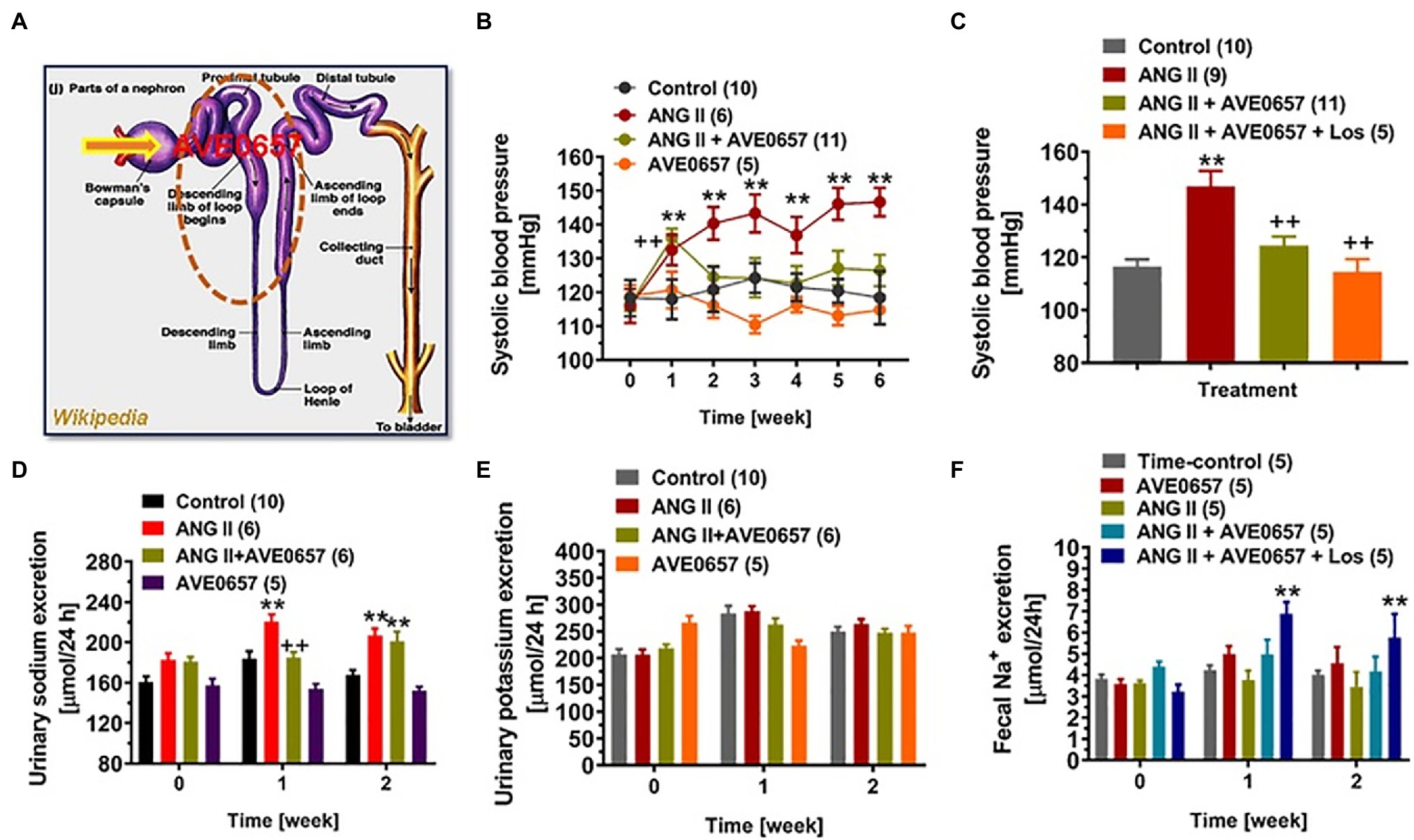
Figure 6. An orally absorbable NHE3 inhibitor AVE0657 (20 mg/kg/day, p.o., Sanofi-Aventis) significantly attenuated ANG II-induced hypertension in male C57BL/6 J mice infused with a slow pressor dose of ANG II at 0.5 mg/kg/day, i.p., for 2 weeks. Upon oral ingestion, AVE0657 is absorbable from the gut into the circulation, which is filtered by the glomerulus into the proximal tubules to inhibit NHE3 in the apical membranes (A). (B) AVE0657 attenuated the development of Ang II-induced hypertension. (C) Concurrent treatments with AVE0657 and Ang II receptor blocker losartan completely blocked the development of Ang II-induced hypertension. (D) The effect of AVE0657 on natriuretic response. (E) AVE0657 had no effect on urinary potassium excretion. (F) AVE0657 had no effect on fecal sodium excretion. **p < 0.01 vs. time-control group; ++p < 0.01 vs. ANG II group. Reproduced from Li et al. (2019b) with permission from the copyright holder.
JZ and XL: conceptualization, review, and editing. SN: writing draft preparations. AL and RH: participants in experiments. SN, JZ, and XL: finalization. All authors contributed to the article and approved the submitted version.
This work was supported in part by grants from National Institute of Diabetes and Digestive and Kidney Diseases (2R01DK102429-03A1, 2R01DK067299-10A1, and 1R01DK102429-01) and National Heart, Lung, and Blood Institute (1R56HL130988-01) to JZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All animal experiments were carried out, and data collected in the laboratory of JZ at the University of Mississippi Medical Center in Jackson, Mississippi, and Tulane University School of Medicine, in New Orleans, Louisiana, respectively. We thank Gary Shull of the University of Cincinnati College of Medicine for providing breeding pairs of global Nhe3−/− and tgNhe3−/− mice, Manoocher Soleimani of the University of Cincinnati School of Medicine for providing breeding pairs of NHE3-floxed mice, and Isabelle Rubera and Michell Tauc from the Laboratoire de Physiomédecine Moléculaire, LP2M, UMR-CNRS 7370, Université Côte d’Azur, Nice Cedex 2, France (I.R., M.T.) for providing breeding pairs of iL1-sglt2-Cre mouse strain, and Sanofi-Aventis for providing the absorbable NHE3 inhibitor AVE-0657 for our studies. The contributions of our unnamed past and present technicians and postdoctoral fellows from their excellent technical assistance are greatly appreciated.
Aldred, K. L., Harris, P. J., and Eitle, E. (2000). Increased proximal tubule NHE-3 and H+-ATPase activities in spontaneously hypertensive rats. J. Hypertens. 18, 623–628. doi: 10.1097/00004872-200018050-00016
Arena, E. A., Longo, W. E., Roberts, K. E., Geibel, P., Nateqi, J., Brandstetter, M., et al. (2012). Functional role of NHE4 as a pH regulator in rat and human colonic crypts. Am. J. Physiol. Cell Physiol. 302, C412–C418. doi: 10.1152/ajpcell.00163.2011
Attaphitaya, S., Park, K., and Melvin, J. E. (1999). Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J. Biol. Chem. 274, 4383–4388. doi: 10.1074/jbc.274.7.4383
Bachmann, O., Riederer, B., Rossmann, H., Groos, S., Schultheis, P. J., Shull, G. E., et al. (2004). The Na+/H+ exchanger isoform 2 is the predominant NHE isoform in murine colonic crypts and its lack causes NHE3 upregulation. Am. J. Physiol. 287, G125–G133. doi: 10.1152/ajpgi.00332.2003
Bacic, D., Kaissling, B., McLeroy, P., Zou, L., Baum, M., and Moe, O. W. (2003). Dopamine acutely decreases apical membrane Na/H exchanger NHE3 protein in mouse renal proximal tubule. Kidney Int. 64, 2133–2141. doi: 10.1046/j.1523-1755.2003.00308.x
Bailey, M. A., Giebisch, G., Abbiati, T., Aronson, P. S., Gawenis, L. R., Shull, G. E., et al. (2004). NHE2-mediated bicarbonate reabsorption in the distal tubule of NHE3 null mice. J. Physiol. 561, 765–775. doi: 10.1113/jphysiol.2004.074716
Bezerra, C. N., Girardi, A. C., Carraro-Lacroix, L. R., and Rebouças, N. A. (2008). Mechanisms underlying the long-term regulation of NHE3 by parathyroid hormone. Am. J. Physiol. Ren. Physiol. 294, F1232–F1237. doi: 10.1152/ajprenal.00025.2007
Biemesderfer, D., DeGray, B., and Aronson, P. S. (2001). Active (9.6 s) and inactive (21 s) oligomers of NHE3 in microdomains of the renal brush border. J. Biol. Chem. 276, 10161–10167. doi: 10.1074/jbc.M008098200
Biemesderfer, D., Pizzonia, J., Abu-Alfa, A., Exner, M., Reilly, R., Igarashi, P., et al. (1993). NHE3: a Na+/H+ exchanger isoform of renal brush border. Am. J. Physiol. 265, F736–F742.
Biemesderfer, D., Reilly, R. F., Exner, M., Igarashi, P., and Aronson, P. S. (1992). Immunocytochemical characterization of Na+/H+ exchanger isoform NHE-1 in rabbit kidney. Am. J. Physiol. 263, F833–F840.
Bobulescu, I. A., Dwarakanath, V., Zou, L., Zhang, J., Baum, M., and Moe, O. W. (2005). Glucocorticoids acutely increase cell surface Na+/H+ exchanger-3 (NHE3) by activation of NHE3 exocytosis. Am. J. Physiol. Ren. Physiol. 289, F685–F691. doi: 10.1152/ajprenal.00447.2004
Bookstein, C., Xie, Y., Rabenau, K., Musch, M. W., McSwine, R. L., Rao, M. C., et al. (1997). Tissue distribution of Na+/H+ exchanger isoforms NHE2 and NHE4 in rat intestine and kidney. Am. J. Physiol. 273, C1496–C1505.
Bourgeois, S., Meer, L. V., Wootla, B., Bloch-Faure, M., Chambrey, R., Shull, G. E., et al. (2010). NHE4 is critical for the renal handling of ammonia in rodents. J. Clin. Invest. 120, 1895–1904. doi: 10.1172/JCI36581
Bradford, E. M., Sartor, M. A., Gawenis, L. R., Clarke, L. L., and Shull, G. E. (2009). Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G886–G898. doi: 10.1152/ajpgi.90520.2008
Brooks, H. L., Sorensen, A. M., Terris, J., Schultheis, P. J., Lorenz, J. N., Shull, G. E., et al. (2001). Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J. Physiol. 530, 359–366. doi: 10.1111/j.1469-7793.2001.0359k.x
Calhoun, D. A., Jones, D., Textor, S., Goff, D. C., Murphy, T. P., Toto, R. D., et al. (2008a). Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for high blood pressure research. Hypertension 51, 1403–1419. doi: 10.1161/HYPERTENSIONAHA.108.189141
Calhoun, D. A., Jones, D., Textor, S., Goff, D. C., Murphy, T. P., Toto, R. D., et al. (2008b). Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for high blood pressure research. Circulation 117, e510–e526. doi: 10.1161/CIRCULATIONAHA.108.189141
Carey, R. M. (2013). Resistant hypertension. Hypertension 61, 746–750. doi: 10.1161/HYPERTENSIONAHA.111.00601
Carey, R. M. (2016). Resistant hypertension: mineralocorticoid receptor ntagonist or renal denervation? Hypertension 67, 278–280. doi: 10.1161/HYPERTENSIONAHA.115.06616
Carey, R. M. (2017). AT2 receptors: potential therapeutic targets for hypertension. Am. J. Hypertens. 30, 339–347. doi: 10.1093/ajh/hpw121
Carey, R. M. (2019). Resistant hypertension. Hypertension 73, 299–300. doi: 10.1161/HYPERTENSIONAHA.118.12270
Castelo-Branco, R. C., Leite-Dellova, D. C. A., Fernandes, F. B., Malnic, G., and de Mello-Aires, M. (2017). The effects of angiotensin-(1-7) on the exchanger NHE3 and on [Ca(2+)]i in the proximal tubules of spontaneously hypertensive rats. Am. J. Physiol. Ren. Physiol. 313, F450–F460. doi: 10.1152/ajprenal.00557.2016
Cha, B., Kim, J. H., Hut, H., Hogema, B. M., Nadarja, J., Zizak, M., et al. (2005). cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem. 280, 16642–16650. doi: 10.1074/jbc.M500505200
Chambrey, R., St John, P. L., Eladari, D., Quentin, F., Warnock, D. G., Abrahamson, D. R., et al. (2001). Localization and functional characterization of Na+/H+ exchanger isoform NHE4 in rat thick ascending limbs. Am. J. Physiol. Ren. Physiol. 281, F707–F717. doi: 10.1152/ajprenal.2001.281.4.F707
Chambrey, R., Warnock, D. G., Podevin, R. A., Bruneval, P., Mandet, C., Belair, M. F., et al. (1998). Immunolocalization of the Na+/H+ exchanger isoform NHE2 in rat kidney. Am. J. Physiol. 275, F379–F386. doi: 10.1152/ajprenal.1998.275.3.F379
Chappell, M. C. (2012). Nonclassical renin-angiotensin system and renal function. Compr. Physiol. 2, 2733–2752. doi: 10.1002/cphy.c120002
Choi, H. M., Kim, H. C., and Kang, D. R. (2017). Sex differences in hypertension prevalence and control: analysis of the 2010–2014 Korea National Health and Nutrition Examination Survey. PLoS One 12:e0178334. doi: 10.1371/journal.pone.0178334
Chow, C. W. (1999). Regulation and intracellular localization of the epithelial isoforms of the Na+/H+ exchangers NHE2 and NHE3. Clin. Invest. Med. 22, 195–206.
Coffman, T. M. (2011). Under pressure: the search for the essential mechanisms of hypertension. Nat. Med. 17, 1402–1409. doi: 10.1038/nm.2541
Collins, J. F., Honda, T., Knobel, S., Bulus, N. M., Conary, J., DuBois, R., et al. (1993). Molecular cloning, sequencing, tissue distribution, and functional expression of a Na+/H+ exchanger (NHE-2). Proc. Natl. Acad. Sci. U. S. A. 90, 3938–3942. doi: 10.1073/pnas.90.9.3938
Counillon, L., Pouyssegur, J., and Reithmeier, R. A. (1994). The Na+/H+ exchanger NHE-1 possesses N- and O-linked glycosylation restricted to the first N-terminal extracellular domain. Biochemistry 33, 10463–10469. doi: 10.1021/bi00200a030
Cox, G. A., Lutz, C. M., Yang, C. L., Biemesderfer, D., Bronson, R. T., Fu, A., et al. (1997). Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91, 139–148. doi: 10.1016/S0092-8674(01)80016-7
Crajoinas, R. O., Lessa, L. M., Carraro-Lacroix, L. R., Davel, A. P., Pacheco, B. P., Rossoni, L. V., et al. (2010). Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am. J. Physiol. Ren. Physiol. 299, F872–F881. doi: 10.1152/ajprenal.00654.2009
Crajoinas, R. O., Pessoa, T. D., Rodrigues, M. V., Malnic, G., and Girardi, A. C. (2014). Changes in the activity and expression of protein phosphatase-1 accompany the differential regulation of NHE3 before and after the onset of hypertension in spontaneously hypertensive rats. Acta Physiol. 211, 395–408. doi: 10.1111/apha.12288
Crowley, S. D., Gurley, S. B., Herrera, M. J., Ruiz, P., Griffiths, R., Kumar, A. P., et al. (2006). Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. U. S. A. 103, 17985–17990.
Crowley, S. D., Gurley, S. B., Oliverio, M. I., Pazmino, A. K., Griffiths, R., Flannery, P. J., et al. (2005). Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J. Clin. Invest. 115, 1092–1099. doi: 10.1172/JCI23378
Dagher, G., and Sauterey, C. (1992). H+ pump and Na+/H+ exchange in isolated single proximal tubules of spontaneously hypertensive rats. J. Hypertens. 10, 969–978.
de Gasparo, M., Catt, K. J., Inagami, T., Wright, J. W., and Unger, T. (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472.
DeSalvo, K. B. (2016). Public health 3.0: applying the 2015-2020 dietary guidelines for Americans. Public Health Rep. 131, 518–521. doi: 10.1177/0033354916662207
Donowitz, M., Ming Tse, C., and Fuster, D. (2013). SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Asp. Med. 34, 236–251. doi: 10.1016/j.mam.2012.05.001
D'Souza, S., Garcia-Cabado, A., Yu, F., Teter, K., Lukacs, G., Skorecki, K., et al. (1998). The epithelial sodium-hydrogen antiporter Na+/H+ exchanger 3 accumulates and is functional in recycling endosomes. J. Biol. Chem. 273, 2035–2043. doi: 10.1074/jbc.273.4.2035
Engevik, M. A., Hickerson, A., Shull, G. E., and Worrell, R. T. (2013). Acidic conditions in the NHE2−/− mouse intestine result in an altered mucosa-associated bacterial population with changes in mucus oligosaccharides. Cell. Physiol. Biochem. 32, 111–128. doi: 10.1159/000356632
Evans, R. L., Bell, S. M., Schultheis, P. J., Shull, G. E., and Melvin, J. E. (1999). Targeted disruption of the Nhe1 gene prevents muscarinic agonist-induced up-regulation of Na+/H+ exchange in mouse parotid acinar cells. J. Biol. Chem. 274, 29025–29030. doi: 10.1074/jbc.274.41.29025
Fenton, R. A., Poulsen, S. B., de la Mora Chavez, S., Soleimani, M., Busslinger, M., Dominguez Rieg, J. A., et al. (2015). Caffeine-induced diuresis and natriuresis is independent of renal tubular NHE3. Am. J. Physiol. Ren. Physiol. 308, F1409–F1420. doi: 10.1152/ajprenal.00129.2015
Fenton, R. A., Poulsen, S. B., de la Mora Chavez, S., Soleimani, M., Dominguez Rieg, J. A., and Rieg, T. (2017). Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 92, 397–414. doi: 10.1016/j.kint.2017.02.001
Filippini, T., Malavolti, M., Whelton, P. K., Naska, A., Orsini, N., and Vinceti, M. (2021). Blood pressure effects of sodium reduction: dose-response meta-analysis of experimental studies. Circulation 143, 1542–1567. doi: 10.1161/CIRCULATIONAHA.120.050371
Forrester, S. J., Booz, G. W., Sigmund, C. D., Coffman, T. M., Kawai, T., Rizzo, V., et al. (2018). Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98, 1627–1738. doi: 10.1152/physrev.00038.2017
Fuster, D. G., Bobulescu, I. A., Zhang, J., Wade, J., and Moe, O. W. (2007). Characterization of the regulation of renal Na+/H+ exchanger NHE3 by insulin. Am. J. Physiol. Ren. Physiol. 292, F577–F585. doi: 10.1152/ajprenal.00240.2006
Gawenis, L. R., Stien, X., Shull, G. E., Schultheis, P. J., Woo, A. L., Walker, N. M., et al. (2002). Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G776–G784. doi: 10.1152/ajpgi.00297.2001
Gekle, M., Volker, K., Mildenberger, S., Freudinge, R. R., Shull, G. E., and Wiemann, M. (2004). NHE3 Na+/H+ exchanger supports proximal tubular protein reabsorption in vivo. Am. J. Physiol. Ren. Physiol. 287, F469–F473. doi: 10.1152/ajprenal.00059.2004
Giral, H., Cranston, D., Lanzano, L., Caldas, Y., Sutherland, E., Rachelson, J., et al. (2012). NHE3 regulatory factor 1 (NHERF1) modulates intestinal sodium-dependent phosphate transporter (NaPi-2b) expression in apical microvilli. J. Biol. Chem. 287, 35047–35056. doi: 10.1074/jbc.M112.392415
Goyal, S., Mentone, S., and Aronson, P. S. (2005). Immunolocalization of NHE8 in rat kidney. Am. J. Physiol. Ren. Physiol. 288, F530–F538. doi: 10.1152/ajprenal.00229.2004
Guan, Y., Dong, J., Tackett, L., Meyer, J. W., Shull, G. E., and Montrose, M. H. (2006). NHE2 is the main apical NHE in mouse colonic crypts but an alternative Na+-dependent acid extrusion mechanism is upregulated in NHE2-null mice. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G689–G699. doi: 10.1152/ajpgi.00342.2005
Gurley, S. B., Riquier-Brison, A. D. M., Schnermann, J., Sparks, M. A., Allen, A. M., Haase, V. H., et al. (2011). AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 13, 469–475. doi: 10.1016/j.cmet.2011.03.001
Harris, P. J., and Navar, L. G. (1985). Tubular transport responses to angiotensin. Am. J. Physiol. 248, F621–F630.
Harris, P. J., and Young, J. A. (1977). Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Arch. 367, 295–297. doi: 10.1007/BF00581370
Harrison, D. G., Coffman, T. M., and Wilcox, C. S. (2021). Pathophysiology of hypertension: the mosaic theory and beyond. Circ. Res. 128, 847–863. doi: 10.1161/CIRCRESAHA.121.318082
Hayashi, M., Yoshida, T., Monkawa, T., Yamaji, Y., Sato, S., and Saruta, T. (1997). Na+/H+-exchanger 3 activity and its gene in the spontaneously hypertensive rat kidney. J. Hypertens. 15, 43–48.
He, P., Klein, J., and Yun, C. C. (2010). Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 285, 27869–27878. doi: 10.1074/jbc.M110.133066
He, P., and Yun, C. C. (2010). Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J. Biomed. Biotechnol. 2010:238080. doi: 10.1155/2010/238080
Hilliard, L. M., Jones, E. S., Steckelings, U. M., Unger, T., Widdop, R. E., and Denton, K. M. (2012). Sex-specific influence of angiotensin type 2 receptor stimulation on renal function: a novel therapeutic target for hypertension. Hypertension 59, 409–414. doi: 10.1161/HYPERTENSIONAHA.111.184986
Hilliard, L. M., Nematbakhsh, M., Kett, M. M., Teichman, E., Sampson, A. K., Widdop, R. E., et al. (2011). Gender differences in pressure-natriuresis and renal autoregulation: role of the angiotensin type 2 receptor. Hypertension 57, 275–282. doi: 10.1161/HYPERTENSIONAHA.110.166827
Hu, M. C., Fan, L., Crowder, L. A., Karim-Jimenez, Z., Murer, H., and Moe, O. W. (2001). Dopamine acutely stimulates Na+/H+ exchanger (NHE3) endocytosis via clathrin-coated vesicles: dependence on protein kinase A-mediated NHE3 phosphorylation. J. Biol. Chem. 276, 26906–26915. doi: 10.1074/jbc.M011338200
Hu, R., McDonough, A. A., and Layton, A. T. (2021). Sex differences in solute and water handling in the human kidney: modeling and functional implications. IScience 24:102667. doi: 10.1016/j.isci.2021.102667
Intersalt (1988). Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ 297, 319–328. doi: 10.1136/bmj.297.6644.319
Jin, X. H., Wang, Z. Q., Siragy, H. M., Guerrant, R. L., and Carey, R. M. (1998). Regulation of jejunal sodium and water absorption by angiotensin subtype receptors. Am. J. Physiol. 275, R515–R523. doi: 10.1152/ajpregu.1998.275.2.R515
Joe, B., and Shapiro, J. I. (2012). Molecular mechanisms of experimental salt-sensitive hypertension. J. Am. Heart Assoc. 1:e002121.
Karnik, S. S., Singh, K. D., Tirupula, K., and Unal, H. (2017). Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR review. Br. J. Pharm. 174, 737–753. doi: 10.1111/bph.13742
Kelly, M. P., Quinn, P. A., Davies, J. E., and Ng, L. L. (1997). Activity and expression of Na+/H+ exchanger isoforms 1 and 3 in kidney proximal tubules of hypertensive rats. Circ. Res. 80, 853–860. doi: 10.1161/01.RES.80.6.853
Kemp, B. A., Bell, J. F., Rottkamp, D. M., Howell, N. L., Shao, W., Navar, L. G., et al. (2012). Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension 60, 387–395. doi: 10.1161/HYPERTENSIONAHA.112.191403
Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., Brautigan, D. L., and Carey, R. M. (2022). Renal AT2 receptors mediate natriuresis via protein phosphatase PP2A. Circ. Res. 130, 96–111. doi: 10.1161/CIRCRESAHA.121.319519
Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., and Carey, R. M. (2020). Identification of a primary renal AT2 receptor defect in spontaneously hypertensive rats. Circ. Res. 28, 644–659. doi: 10.1161/CIRCRESAHA.119.316193
Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., Padia, S. H., and Carey, R. M. (2014). AT2 receptor activation induces natriuresis and lowers blood pressure. Circ. Res. 115, 388–399. doi: 10.1161/CIRCRESAHA.115.304110
Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., Padia, S. H., and Carey, R. M. (2016). AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II-dependent hypertension. Circ. Res. 119, 532–543. doi: 10.1161/CIRCRESAHA.116.308384
Kobori, H., Nangaku, M., Navar, L. G., and Nishiyama, A. (2007). The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 59, 251–287. doi: 10.1124/pr.59.3.3
Kocinsky, H. S., Girardi, A. G., Biemesderfer, D., Nguyen, T., Mentone, S., and Orlowski, J. (2005). Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am. J. Physiol. Ren. Physiol. 289, F249–F258. doi: 10.1152/ajprenal.00082.2004
Kovesdy, C. P., Adebiyi, A., Rosenbaum, D., Jacobs, J. W., and Quarles, L. D. (2021). Novel treatments from inhibition of the intestinal sodium–hydrogen exchanger 3. Int. J. Nephrol. Renov. Dis. 14, 411–420. doi: 10.2147/IJNRD.S334024
Kucharava, K., Brand, Y., Albano, G., Sekulic-Jablanovic, M., Glutz, A., Xian, X., et al. (2020). Sodium-hydrogen exchanger 6 (NHE6) deficiency leads to hearing loss, via reduced endosomal signalling through the BDNF/Trk pathway. Sci. Rep. 10:3609. doi: 10.1038/s41598-020-60262-5
Kulaksiz, H., Bektas, H., and Cetin, Y. (2001). Expression and cell-specific and membrane-specific localization of NHE-3 in the human and Guinea pig upper gastrointestinal tract. Cell Tissue Res. 303, 337–343. doi: 10.1007/s004410000329
Kurashima, K., Yu, F. H., Cabado, A. G., Szabo, E. Z., Grinstein, S., and Orlowski, J. (1997). Identification of sites required for down-regulation of Na+/H+ exchanger NHE3 activity by cAMP-dependent protein kinase. Phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 272, 28672–28679. doi: 10.1074/jbc.272.45.28672
Kurata, T., Rajendran, V., Fan, S., Ohta, T., Numata, M., and Fushida, S. (2019). NHE5 regulates growth factor signaling, integrin trafficking, and degradation in glioma cells. Clin. Exp. Metastasis 36, 527–538. doi: 10.1007/s10585-019-10001-6
LaPointe, M. S., Sodhi, C., Sahai, A., and Batlle, D. (2002). Na+/H+ exchange activity and NHE-3 expression in renal tubules from the spontaneously hypertensive rat. Kidney Int. 62, 157–165. doi: 10.1046/j.1523-1755.2002.00406.x
Lawrence, S. P., Bright, N. A., Luzio, J. P., and Bowers, K. (2010). The sodium/proton exchanger NHE8 regulates late endosomal morphology and function. Mol. Biol. Cell 21, 3497–3615. doi: 10.1091/mbc.E09-12-1053
Ledoussal, C., Lorenz, J. N., Nieman, M. L., Soleimani, M., Schultheis, P. J., and Shull, G. E. (2001a). Renal salt wasting in mice lacking NHE3 Na+/H+ exchanger but not in mice lacking NHE2. Am. J. Physiol. Ren. Physiol. 281, F718–F727. doi: 10.1152/ajprenal.2001.281.4.F718
Ledoussal, C., Woo, A. L., Miller, M. L., and Shull, G. E. (2001b). Loss of the NHE2 Na+/H+ exchanger has no apparent effect on diarrheal state of NHE3-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1385–G1396. doi: 10.1152/ajpgi.2001.281.6.G1385
Leong, P. K., Yang, L. E., Holstein-Rathlou, N. H., and McDonough, A. A. (2002). Angiotensin II clamp prevents the second step in renal apical NHE3 internalization during acute hypertension. Am. J. Physiol. Ren. Physiol. 283, F1142–F1150. doi: 10.1152/ajprenal.00178.2002
Levens, N. R., Peach, M. J., Carey, R. M., Poat, J. A., and Munday, K. A. (1980). Stimulation of intestinal sodium and water transport in vivo by angiotensin II and analogs. Endocrinology 107, 1946–1953. doi: 10.1210/endo-107-6-1946
Levens, N. R., Peach, M. J., Carey, R. M., Poat, J. A., and Munday, K. A. (1981). Response of rat jejunum to angiotensin II: role of norepinephrine and prostaglandins. Am. J. Physiol. 240, G17–G24. doi: 10.1152/ajpgi.1981.240.1.G17
Li, X. C., Hopfer, U., and Zhuo, J. L. (2009a). AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway. Am. J. Physiol. Ren. Physiol. 297, F1342–F1352.
Li, X. C., Hopfer, U., and Zhuo, J. L. (2012). Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 303, F1617–F1628. doi: 10.1152/ajprenal.00219.2012
Li, X. C., Leite, A. P. O., Zheng, X., Zhao, C., Chen, X., Zhang, L., et al. (2021). Proximal tubule-specific deletion of angiotensin II type 1a receptors in the kidney attenuates circulating and intratubular angiotensin II-induced hypertension in PT-Agtr1a−/− mice. Hypertension 77, 1285–1298. doi: 10.1161/HYPERTENSIONAHA.120.16336
Li, X. C., Shao, Y., and Zhuo, J. L. (2009b). AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int. 76, 169–177. doi: 10.1038/ki.2009.134
Li, X. C., Shull, G. E., Miguel-Qin, E., Chen, F., and Zhuo, J. L. (2015a). Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol. Rep. 3:e12605. doi: 10.14814/phy2.12605
Li, X. C., Shull, G. E., Miguel-Qin, E., and Zhuo, J. L. (2015b). Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol. Genomics 47, 479–487. doi: 10.1152/physiolgenomics.00056.2015
Li, X. C., Soleimani, M., Zhu, D., Rubera, I., Tauc, M., Zheng, X., et al. (2018). Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension 72, 1328–1336. doi: 10.1161/HYPERTENSIONAHA.118.10884
Li, H., Weatherford, E. T., Davis, D. R., Keen, H. L., Grobe, J. L., Daugherty, A., et al. (2011). Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1067–R1077. doi: 10.1152/ajpregu.00124.2011
Li, X. C., Zheng, X., Chen, X., Zhao, C., Zhu, D., Zhang, J., et al. (2019a). Genetic and genomic evidence for an important role of the Na+/H+ exchanger 3 in blood pressure regulation and angiotensin II-induced hypertension. Physiol. Genomics 51, 97–108. doi: 10.1152/physiolgenomics.00122.2018
Li, X. C., Zhu, D., Chen, X., Zheng, X., Zhao, C., Zhang, J., et al. (2019b). Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 74, 526–535. doi: 10.1161/HYPERTENSIONAHA.119.13094
Li, X. C., Zhu, D., Zheng, X., Zhang, J., and Zhuo, J. L. (2018b). Intratubular and intracellular renin-angiotensin system in the kidney: a unifying perspective in blood pressure control. Clin. Sci. (Lond). 132, 1383–1401. doi: 10.1042/CS20180121
Li, X. C., and Zhuo, J. L. (2011). Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int. 80, 620–632. doi: 10.1038/ki.2011.161
Linz, B., Hohl, M., Reil, J. C., Bohm, M., and Linz, D. (2016). Inhibition of NHE3-mediated sodium absorption in the gut reduced cardiac end-organ damage without deteriorating renal function in obese spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 67, 225–231. doi: 10.1097/FJC.0000000000000336
Linz, B., Saljic, A., Hohl, M., Gawalko, M., Jespersen, T., Sanders, P., et al. (2020). Inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut: a new antihypertensive concept. Int. J. Cardio. Heart Vasc. 29:100591. doi: 10.1016/j.ijcha.2020.100591
Linz, D., Wirth, K., Linz, W., Heuer, H. O., Frick, W., Hofmeister, A., et al. (2012). Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension 60, 1560–1567. doi: 10.1161/HYPERTENSIONAHA.112.201590
Lorenz, J. N., Schultheis, P. J., Traynor, T., Shull, G. E., and Schnermann, J. (1999). Micropuncture analysis of single-nephron function in NHE3-deficient mice. Am. J. Physiol. 277, F447–F453. doi: 10.1152/ajprenal.1999.277.3.F447
Magyar, C. E., Zhang, Y., Holstein-Rathlou, N. H., and McDonough, A. A. (2000). Proximal tubule Na transporter responses are the same during acute and chronic hypertension. Am. J. Physiol. Ren. Physiol. 279, F358–F369. doi: 10.1152/ajprenal.2000.279.2.F358
Malakooti, J., Dahdal, R. Y., Schmidt, L., Layden, T. J., Dudeja, P. K., and Ramaswamy, K. (1999). Molecular cloning, tissue distribution, and functional expression of the human Na+/H+ exchanger NHE2. Am. J. Physiol. 277, G383–G390. doi: 10.1152/ajpgi.1999.277.2.G383
Mattson, D. L. (2019). Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat. Rev. Nephrol. 15, 290–300. doi: 10.1038/s41581-019-0121-z
Mirabito, K. M., Hilliard, L. M., Kett, M. M., Brown, R. D., Booth, S. C., Widdop, R. E., et al. (2014). Sex- and age-related differences in the chronic pressure-natriuresis relationship: role of the angiotensin type 2 receptor. Am. J. Physiol. Ren. Physiol. 307, F901–F907. doi: 10.1152/ajprenal.00288.2014
Morduchowicz, G. A., Sheikh-Hamad, D., Jo, O. D., Nord, E. P., Lee, D. B., and Yanagawa, N. (1989). Increased Na+/H+ antiport activity in the renal brush border membrane of SHR. Kidney Int. 36, 576–581. doi: 10.1038/ki.1989.233
Muntner, P., and Whelton, P. K. (2017). Using predicted cardiovascular disease risk in conjunction with blood pressure to guide antihypertensive medication treatment. J. Am. Coll. Cardiol. 69, 2446–2456. doi: 10.1016/j.jacc.2017.02.066
Muntner, P., Whelton, P. K., Woodward, M., and Carey, R. M. (2018). A comparison of the 2017 American College of Cardiology/American Heart Association blood pressure guideline and the 2017 American Diabetes Association diabetes and hypertension position statement for U.S. adults with diabetes. Diabetes Care 41, 2322–2329. doi: 10.2337/dc18-1307
Murer, H., Hopfer, U., and Kinne, R. (1976). Sodium/proton antiport in brush-border-membrane vesicles isolated from rat small intestine and kidney. Biochem. J. 154:942389, 597–604.
Navar, L. G., Carmines, P. K., Huang, W. C., and Mitchell, K. D. (1987). The tubular effects of angiotensin II. Kidney Int. Suppl. 20, S81–S88.
Navar, L. G., Prieto, M. C., Satou, R., and Kobori, H. (2011). Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr. Opin. Pharmacol. 11, 180–186. doi: 10.1016/j.coph.2011.01.009.
Noonan, W. T., Woo, A. L., Nieman, M. L., Prasad, V., Schultheis, P. J., Shull, G. E., et al. (2005). Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R685–R691. doi: 10.1152/ajpregu.00209.2004
Orlowski, J., Kandasamy, R. A., and Shull, G. E. (1992). Molecular cloning of putative members of the Na/H exchanger gene family. cDNA cloning, deduced amino acid sequence, and mRNA tissue expression of the rat Na/H exchanger NHE-1 and two structurally related proteins. J. Biol. Chem. 267, 9331–9339. doi: 10.1016/S0021-9258(19)50428-8
Padia, S. H., Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., and Carey, R. M. (2009). Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 53, 338–343. doi: 10.1161/HYPERTENSIONAHA.108.124198
Panico, C., Luo, Z., Damiano, S., Artigiano, F., Gill, P., and Welch, W. J. (2009). Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension 54, 1291–1297. doi: 10.1161/HYPERTENSIONAHA.109.134783
Pedersen, S. F., and Counillon, L. (2019). The SLC9A-C mammalian Na+/H+ exchanger family: molecules, mechanisms, and physiology. Physiol. Rev. 99, 2015–2113. doi: 10.1152/physrev.00028.2018
Ramirez, L. A., and Sullivan, J. C. (2018). Sex differences in hypertension: where we have been and where we are going. Am. J. Hypertens. 31, 1247–1254. doi: 10.1093/ajh/hpy148
Rapp, J. P. (1982). Dahl salt-susceptible and salt-resistant rats: a review. Hypertension 4, 753–763. doi: 10.1161/01.HYP.4.6.753
Riquier-Brison, A. D., Leong, P. K., Pihakaski-Maunsbach, K., and McDonough, A. A. (2010). Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am. J. Physiol. Ren. Physiol. 298, F177–F186. doi: 10.1152/ajprenal.00464.2009
Rucker, A. J., Rudemiller, N. P., and Crowley, S. D. (2018). Salt, hypertension, and immunity. Annu. Rev. Physiol. 80, 283–307. doi: 10.1146/annurev-physiol-021317-121134
Saccomani, G., Mitchell, K. D., and Navar, L. G. (1990). Angiotensin II stimulation of Na+/H+ exchange in proximal tubule cells. Am. J. Physiol. 258, F1188–F1195.
Sacks, F. M., Svetkey, L. P., Vollmer, W. M., Appel, L. J., Bray, G. A., Harsha, D., et al. (2001). Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. DASH-sodium collaborative research group. N. Engl. J. Med. 344, 3–10. doi: 10.1056/NEJM200101043440101
Sardet, C., Franchi, A., and Pouysségur, J. (1989). Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell 56, 271–280. doi: 10.1016/0092-8674(89)90901-X
Sarker, R., Gronborg, M., Cha, B., Mohan, S., Chen, Y., Pandey, A., et al. (2008). Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol. Biol. Cell 19, 3615–4018. doi: 10.1091/mbc.e08-01-0019
Schultheis, P. J., Clarke, L. L., Meneton, P., Harline, M., Boivin, G. P., Stemmermann, G., et al. (1998a). Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J. Clin. Invest. 101, 1243–1253. doi: 10.1172/JCI1249
Schultheis, P. J., Clarke, L. L., Meneton, P., Miller, M. L., Soleimani, M., Gawenis, L. R., et al. (1998b). Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 19, 282–285.
Seely, E. W., Walsh, B. W., Gerhard, M. D., and Williams, G. H. (1999). Estradiol with or without progesterone and ambulatory blood pressure in postmenopausal women. Hypertension 33, 1190–1194. doi: 10.1161/01.HYP.33.5.1190
Sequeira-Lopez, M. L. S., and Gomez, R. A. (2021). Renin cells, the kidney, and hypertension. Circ. Res. 128, 887–907. doi: 10.1161/CIRCRESAHA.121.318064
Shawki, A., Engevik, M. A., Kim, R. S., Knight, P. B., Baik, R. A., Anthony, S. R., et al. (2016). Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G423–G430. doi: 10.1152/ajpgi.00167.2016
Siervo, M., Lara, J., Chowdhury, S., Ashor, A., Oggioni, C., and Mathers, J. C. (2015). Effects of the dietary approach to stop hypertension (DASH) diet on cardiovascular risk factors: a systematic review and meta-analysis. Br. J. Nutr. 113, 1–15. doi: 10.1017/S0007114514003341
Silva-Antonialli, M. M., Rita, C. A., Tostes, L. F., Fior-Chadi, D. R., Akamine, E. H., Carvalho, M. H. C., et al. (2004). A Lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc. Res. 62, 587–593. doi: 10.1016/j.cardiores.2004.01.020
Siragy, H. M., and Carey, R. M. (1999). Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension 33, 1237–1242.
Siragy, H. M., Inagami, T., Ichiki, T., and Carey, R. M. (1999). Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc. Natl. Acad. Sci. U. S. A. 96, 6506–6510.
Spencer, A. G., Labonte, E. D., Rosenbaum, D. P., Plato, C. F., Carreras, C. W., Leadbetter, M. R., et al. (2014). Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci. Transl. Med. 6, 227ra36. doi: 10.1126/scitranslmed.3007790
Sullivan, J. C., Bhatia, K., Yamamoto, T., and Elmarakby, E. A. (2010). Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 56, 658–666. doi: 10.1161/HYPERTENSIONAHA.110.153668
Svetkey, L. P., Sacks, F. M., Obarzanek, E., Vollmer, W. M., Appel, L. J., Lin, P. H., et al. (1999). The DASH diet, sodium intake and blood pressure trial (DASH-sodium): rationale and design. DASH-sodium collaborative research group. J. Am. Diet. Assoc. 99(8 Suppl), S96–S104. doi: 10.1016/s0002-8223(99)00423-x
Takaichi, K., Wang, D., Balkovetz, D. F., and Warnock, D. G. (1992). Cloning, sequencing, and expression of Na+/H+ antiporter cDNAs from human tissues. Am. J. Physiol. 262, C1069–C1076. doi: 10.1152/ajpcell.1992.262.4.C1069
Thekkumkara, T. J., Cookson, R., and Linas, S. L. (1998). Angiotensin (AT1A) receptor-mediated increases in transcellular sodium transport in proximal tubule cells. Am. J. Physiol. 274, F897–F905. doi: 10.1152/ajprenal.1998.274.5.F897
Thomas, D., Harris, P. J., and Morgan, T. O. (1988). Age-related changes in angiotensin II-stimulated proximal tubule fluid reabsorption in the spontaneously hypertensive rat. J. Hypertens. Suppl. 6, S449–S451. doi: 10.1097/00004872-198812040-00141
Thomas, D., Harris, P. J., and Morgan, T. O. (1990). Altered responsiveness of proximal tubule fluid reabsorption of peritubular angiotensin II in spontaneously hypertensive rats. J. Hypertens. 8, 407–410. doi: 10.1097/00004872-199005000-00002
Tse, C. M., Brant, S. R., Walker, M. S., Pouyssegur, J., and Donowitz, M. (1992). Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney-specific Na+/H+ exchanger isoform (NHE-3). J. Biol. Chem. 267, 9340–9346. doi: 10.1016/S0021-9258(19)50429-X
Tse, C. M., Levine, S. A., Yun, C. H., Montrose, M. H., Little, P. J., Pouyssegur, J., et al. (1993). Cloning and expression of a rabbit cDNA encoding a serum-activated ethylisopropylamiloride-resistant epithelial Na+/H+ exchanger isoform (NHE-2). J. Biol. Chem. 268, 11917–11924. doi: 10.1016/S0021-9258(19)50287-3
Vallon, V., Schwark, J. R., Richter, K., and Hropot, M. (2000). Role of Na+/H+ exchanger NHE3 in nephron function: micropuncture studies with S3226, an inhibitor of NHE3. Am. J. Physiol. Ren. Physiol. 278, F375–F379. doi: 10.1152/ajprenal.2000.278.3.F375
Veiras, L. C., Girardi, A. C. C., Curry, J., Pei, L., Ralph, D. L., Tran, A., et al. (2017). Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J. Am. Soc. Nephrol. 28, 3504–3517. doi: 10.1681/ASN.2017030295
Wade, J. B., Welling, P. A., Donowitz, M., Shenolikar, S., and Weinman, E. J. (2001). Differential renal distribution of NHERF isoforms and their colocalization with NHE3, ezrin, and ROMK. Am. J. Physiol. Cell Physiol. 280, C192–C198. doi: 10.1152/ajpcell.2001.280.1.C192
Wakabayashi, S., Fafournoux, P., Sardet, C., and Pouysségur, J. (1992). The Na+/H+ antiporter cytoplasmic domain mediates growth factor signals and controls "H+-sensing". Proc. Natl. Acad. Sci. U. S. A. 89, 2424–2428. doi: 10.1073/pnas.89.6.2424
Wang, Z., Orlowski, J., and Shull, G. E. (1993). Primary structure and functional expression of a novel gastrointestinal isoform of the rat Na/H exchanger. J. Biol. Chem. 268, 11925–11928. doi: 10.1016/S0021-9258(19)50288-5
Wang, T., Yang, C. L., Abbiati, T., Schultheis, P. J., Shull, G. E., Giebisch, G., et al. (1999). Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. Am. J. Physiol. 277, F298–F302. doi: 10.1152/ajprenal.1999.277.2.F298
Wang, T., Yang, C. L., Abbiati, T., Shull, G. E., Giebisch, G., and Aronson, P. S. (2001). Essential role of NHE3 in facilitating formate-dependent NaCl absorption in the proximal tubule. Am. J. Physiol. Ren. Physiol. 281, F288–F292.
Wang, D., Zhang, H., Lang, F., and Yun, C. C. (2007). Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am. J. Physiol. Cell Physiol. 292, C396–C404. doi: 10.1152/ajpcell.00345.2006
Wang, Q., Zhou, R., Zhang, C., Dong, H., Ma, J., and Wang, G. (2014). Inhibition of central Na+/H+ exchanger type 3 can alleviate sleep apnea in Sprague-Dawley rats. Chin. Med. J. 127, 48–53.
Weinman, E. J., Steplock, D., and Shenolikar, S. (2001). Acute regulation of NHE3 by protein kinase A requires a multiprotein signal complex. Kidney Int. 60, 450–454. doi: 10.1046/j.1523-1755.2001.060002450.x
Whelton, P. K., Chen, J., and Krousel-Wood, M. (2017). Lessons learned from Systolic Blood Pressure Intervention Trial. Curr. Opin. Cardiol. 32, 407–412. doi: 10.1097/HCO.0000000000000396
Whelton, P. K., and Carey, R. M. (2017). The 2017 clinical practice guideline for high blood pressure. JAMA 318, 2073–2074. doi: 10.1001/jama.2017.18209
Whelton, P. K., Carey, R. M., Aronow, W. S., Casey, D. E. Jr., Collins, K. J., Dennison Himmelfarb, C., et al. (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation 138, e426–e483.
Woo, A. L., Noonan, W. T., Schultheis, P. J., Neumann, J. C., Manning, P. A., Lorenz, J. N., et al. (2003). Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. Am. J. Physiol. Ren. Physiol. 284, F1190–F1198. doi: 10.1152/ajprenal.00418.2002
Wu, M. S., Biemesderfer, D., Giebisch, G., and Aronson, P. S. (1996). Role of NHE3 in mediating renal brush border Na+/H+ exchange. Adaptation to metabolic acidosis. J. Biol. Chem. 271, 32749–32752. doi: 10.1074/jbc.271.51.32749
Xinhan, X., Matsushita, M., Numaza, M., Taguchi, A., Mitsui, K., and Kanazawa, H. (2011). Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin. Am. J. Physiol. Cell Physiol. 301, C1431–C1444. doi: 10.1152/ajpcell.00154.2011
Xu, H., Li, J., Chen, H., Wang, C., and Ghishan, F. K. (2013). NHE8 plays important roles in gastric mucosal protection. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G257–G261. doi: 10.1152/ajpgi.00433.2012
Xu, H., Zhang, B., Li, J., Wang, C., Chen, H., and Ghishan, F. K. (2012). Impaired mucin synthesis and bicarbonate secretion in the colon of NHE8 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G335–G343. doi: 10.1152/ajpgi.00146.2012
Xue, J., Thomas, L., Murali, S. K., Levi, M., Fenton, R. A., Dominguez Rieg, J. A., et al. (2022). Enhanced phosphate absorption in intestinal epithelial cell-specific NHE3 knockout mice. Acta Physiol. 234:e13756. doi: 10.1111/apha.13756
Xue, J., Thomas, L., Tahmasbi, M., Valdez, A., Dominguez Rieg, J. A., Fenton, R. A., et al. (2020). An inducible intestinal epithelial cell-specific NHE3 knockout mouse model mimicking congenital sodium diarrhea. Clin. Sci. 134, 941–953. doi: 10.1042/CS20200065
Yang, L. E., Leong, P. K., and McDonough, A. A. (2007). Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am. J. Physiol. Ren. Physiol. 293, F1197–F1208. doi: 10.1152/ajprenal.00040.2007
Yang, L. E., Maunsbach, A. B., Leong, P. K., and McDonough, A. A. (2004). Differential traffic of proximal tubule Na+ transporters during hypertension or PTH: NHE3 to base of microvilli vs. NaPi2 to endosomes. Am. J. Physiol. Ren. Physiol. 287, F896–F906. doi: 10.1152/ajprenal.00160.2004
Yang, T., and Xu, C. (2017). Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J. Am. Soc. Nephrol. 28, 1040–1049. doi: 10.1681/ASN.2016070734
Yip, K. P., Tse, C. M., McDonough, A. A., and Marsh, D. J. (1998). Redistribution of Na+/H+ exchanger isoform NHE3 in proximal tubules induced by acute and chronic hypertension. Am. J. Physiol. 275, F565–F575.
Yun, C. H., Oh, S., Zizak, M., Steplock, D., Tsao, S., Tse, C. M., et al. (1997). cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc. Natl. Acad. Sci. U. S. A. 94, 3010–3015.
Yun, C. H., Tse, C. M., Nath, S. K., Levine, S. A., Brant, S. R., and Donowitz, M. (1995). Mammalian Na+/H+ exchanger gene family: structure and function studies. Am. J. Physiol. 269, G1–G11. doi: 10.1152/ajpgi.1995.269.1.G1
Zachos, N. C., Tse, M., and Donowitz, M. (2005). Molecular physiology of intestinal Na+/H+ exchange. Annu. Rev. Physiol. 67, 411–443. doi: 10.1146/annurev.physiol.67.031103.153004
Zhang, Y., Mircheff, A. K., Hensley, C. B., Magyar, C. E., Warnock, D. G., Chambrey, R., et al. (1996). Rapid redistribution and inhibition of renal sodium transporters during acute pressure natriuresis. Am. J. Physiol. 270, F1004–F1014. doi: 10.1152/ajprenal.1996.270.6.F1004
Zhao, H., Shiue, H., Palkon, S., Wang, Y., Cullinan, P., Burkhardt, J. K., et al. (2004). Ezrin regulates NHE3 translocation and activation after Na+−glucose cotransport. Proc. Natl. Acad. Sci. U. S. A. 101, 9485–9490. doi: 10.1073/pnas.0308400101
Zhao, H., Wiederkehr, M. R., Fan, L., Collazo, R. L., Crowder, L. A., and Moe, O. W. (1999). Acute inhibition of Na+/H+ exchanger NHE-3 by cAMP. Role of protein kinase A and NHE-3 phosphoserines 552 and 605. J. Biol. Chem. 274, 3978–3987. doi: 10.1074/jbc.274.7.3978.
Zhuo, J. L., and De Li, X. C. (2018). Novo DNA (de)methylation in the kidney. Hypertension 72, 1084–1086. doi: 10.1161/HYPERTENSIONAHA.118.11755
Zhuo, J. L., Ferrao, F. M., Zheng, Y., and Li, X. C. (2013). New frontiers in the intrarenal renin-angiotensin system: a critical review of classical and new paradigms. Front. Endocrinol. 4:166. doi: 10.3389/fendo.2013.00166
Zhuo, J. L., and Li, X. C. (2013). Proximal nephron. Compr. Physiol. 3, 1079–1123. doi: 10.1002/cphy.c110061
Zhuo, J. L., and Li, X. C. (2019). Angiotensin III/AT2 receptor/NHE3 signaling pathway in the proximal tubules of the kidney: a novel natriuretic and antihypertensive mechanism in hypertension. J. Am. Heart Assoc. 8:e012644. doi: 10.1161/JAHA.119.012644
Zhuo, J. L., Soleimani, M., and Li, X. C. (2021). New insights into the critical importance of intratubular Na+/H+ exchanger 3 and its potential therapeutic implications in hypertension. Curr. Hypertens. Rep. 23:34. doi: 10.1007/s11906-021-01152-7
Zizak, M., Lamprecht, G., Steplock, D., Tariq, N., Shenolikar, S., Donowitz, M., et al. (1999). cAMP-induced phosphorylation and inhibition of Na+/H+ exchanger 3 (NHE3) are dependent on the presence but not the phosphorylation of NHE regulatory factor. J. Biol. Chem. 274, 24753–24758. doi: 10.1074/jbc.274.35.24753
Keywords: angiotensin II, Na+/H+ exchanger 3, hypertension, kidney, proximal tubule
Citation: Nwia SM, Li XC, Leite APO, Hassan R and Zhuo JL (2022) The Na+/H+ Exchanger 3 in the Intestines and the Proximal Tubule of the Kidney: Localization, Physiological Function, and Key Roles in Angiotensin II-Induced Hypertension. Front. Physiol. 13:861659. doi: 10.3389/fphys.2022.861659
Edited by:
Francesca Di Sole, Des Moines University, United StatesReviewed by:
Adriana Castello Costa Girardi, University of São Paulo, BrazilCopyright © 2022 Nwia, Li, Leite, Hassan and Zhuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Long Zhuo, anpodW9AdHVsYW5lLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.