
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 09 March 2022
Sec. Cardiac Electrophysiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.850117
This article is part of the Research TopicInherited Cardiac ArrhythmiasView all 3 articles
 Yukun Li1,2
Yukun Li1,2 Xiaodong Peng1,2
Xiaodong Peng1,2 Rong Lin3Xuesi Wang1,2Xinmeng Liu1,2
Rong Lin3Xuesi Wang1,2Xinmeng Liu1,2 Rong Bai4
Rong Bai4 Changsheng Ma1,2
Changsheng Ma1,2 Ribo Tang1,2*Yanfei Ruan1,2*
Ribo Tang1,2*Yanfei Ruan1,2* Nian Liu1,2*
Nian Liu1,2*Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a severe yet rare inherited arrhythmia disorder. The cornerstone of CPVT medical therapy is the use of β-blockers; 30% of patients with CPVT do not respond well to optimal β-blocker treatment. Studies have shown that flecainide effectively prevents life-threatening arrhythmias in CPVT. Flecainide is a class IC antiarrhythmic drug blocking cardiac sodium channels. RyR2 inhibition is proposed as the principal mechanism of antiarrhythmic action of flecainide in CPVT, while it is highly debated. In this article, we review the current progress of this issue.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare inherited arrhythmia syndrome characterized by bidirectional or polymorphic ventricular tachycardia (VT) provoked by emotional stress and/or physical activity. Clinical phenotypes include catecholamine-associated syncope and a characteristic pattern of bidirectional VT in the absence of structural heart disease (Leenhardt et al., 1995; Liu et al., 2008; Priori et al., 2013). The primary treatment strategy for CPVT is the use of β-blockers due to the catecholamine-dependent onset of VT, while insufficient protection from cardiac events has been reported despite optimal β-blockers therapy (Padfield et al., 2016; Yang et al., 2016). Flecainide, a classic antiarrhythmic agent, has been gaining the interest of clinicians in the treatment of CPVT. Accumulating clinical evidence shows that flecainide, alone and combined with β-blocker therapy, effectively prevents VT in patients with CPVT and has been recommended in the international guidelines (Liu et al., 2012; Steinfurt et al., 2015; Baltogiannis et al., 2019). In the initial study, the antiarrhythmic mechanism of flecainide in CPVT was the suppression of abnormal calcium release from the sarcoplasmic reticulum (SR) by targeting the cardiac ryanodine receptor (RyR2; Watanabe et al., 2009; Hilliard et al., 2010; Hwang et al., 2011). However, not all studies support this hypothesis (Liu et al., 2011; Sikkel et al., 2013; Bannister et al., 2015, 2016). In the last decade, the therapeutic mechanisms of flecainide in CPVT have become a major topic of debate in this field. In this review, we summarize and discuss the current progress in this field.
CPVT has been mainly related to mutations in genes encoding the cardiac ryanodine receptor (RyR2) and cardiac calsequestrin (CASQ2), which can be identified in 60–70% of CPVT patients (Wleklinski et al., 2020). RyR2 and CASQ2 are responsible for calcium homeostasis in cardiomyocytes.
The delicate balance of Ca2+ fluxes between the intracellular compartment and the extracellular space in cardiac myocytes is crucial for normal excitation-contraction (EC) coupling (Wier and Balke, 1999; Bers, 2002). During the plateau phase of the action potential, a small amount of Ca2+ enters the cytosol of cardiac myocytes via voltage-dependent L-type Ca2+ channels, resulting in a large amount of Ca2+ release into the cytosol via the RyR2 channel, which is called Ca2+-induced Ca2+ release (CICR). The cytosolic Ca2+ sharply from 150 nM to 1 μM activates the contractile apparatus. Then, the elevated cytosolic Ca2+ promptly resumed to 150 nM during the diastolic phase to ensure regular relaxation properties of the myocytes. The majority of Ca2+ is reuptake into the SR by Ca2+ ATPase isoform 2a (SERCA2a). The remaining Ca2+ is extruded into the extracellular fluid via a forward-mode Na+/Ca2+ exchanger (NCX).
The mutations of RyR2 and CASQ2 disrupt normal Ca2+ handling in the SR, enhancing the open probability of RyR2 and leading to spontaneous Ca2+ release events during the diastolic period. Under adrenergic stress, Ca2+ overload in the SR can facilitate abnormal Ca2+ leakage during relaxation. Elevated intracellular Ca2+ levels during the diastolic period would activate the forward mode of NCX, which extrudes Ca2+ in exchange for Na+ with a stoichiometry of 1:3, generating a net inward current. The transient inward current (Iti) produces delayed afterdepolarizations (DADs) and causes triggered activity once it reaches the threshold of the Na+ channel. Taken together, the molecular pathophysiology of arrhythmia occurrence in CPVT involves two critical steps: (1) spontaneous Ca2+ release from SR during the diastolic period, which could be exaggerated by adrenergic stimulation and (2) triggered activity activated by Iti, which is induced by spontaneous Ca2+ release.
The insufficient protection of β-blockers in CPVT has been reported (Padfield et al., 2016; Yang et al., 2016). Almost 30% of patients with CPVT still experience cardiac arrhythmias despite optimal β-blocker therapy. Therefore, it is important to explore alternative treatment options for CPVT. Knollman and his collaborators first reported that flecainide monotherapy or flecainide combined with β-blockers exhibited striking efficacy in preventing ventricular arrhythmias in two CPVT patients who did not respond to the combination therapy with β-blockers and verapamil (Watanabe et al., 2009). Subsequently, in a retrospective cohort study, the efficacy of flecainide was assessed in 33 patients with CPVT who were unprotected by conventional therapy (van der Werf et al., 2011). Ventricular arrhythmias were effectively controlled by flecainide in 22 patients (76%) and were completely suppressed in 14 patients (63%). In a randomized clinical trial, 14 patients with CPVT using maximally tolerated β-blockers demonstrated that ventricular arrhythmias during exercise were significantly reduced by flecainide, with complete suppression observed in 11 of 13 patients, and serious adverse events did not differ between the flecainide and placebo arms (Kannankeril et al., 2017). Table 1 lists the clinical efficacy and safety of flecainide treatment for CPVT in the literature (van der Werf et al., 2011; Khoury et al., 2013; Miyake et al., 2013; Watanabe et al., 2013; Roses-Noguer et al., 2014; Roston et al., 2015; Padfield et al., 2016; Kannankeril et al., 2017; Wangüemert Pérez et al., 2018). Overall, flecainide effectively prevented ventricular arrhythmias in patients with CPVT without apparent adverse events. Consequently, flecainide has been recommended for CPVT patients with ventricular arrhythmias who already have optimized β-blocker treatment.
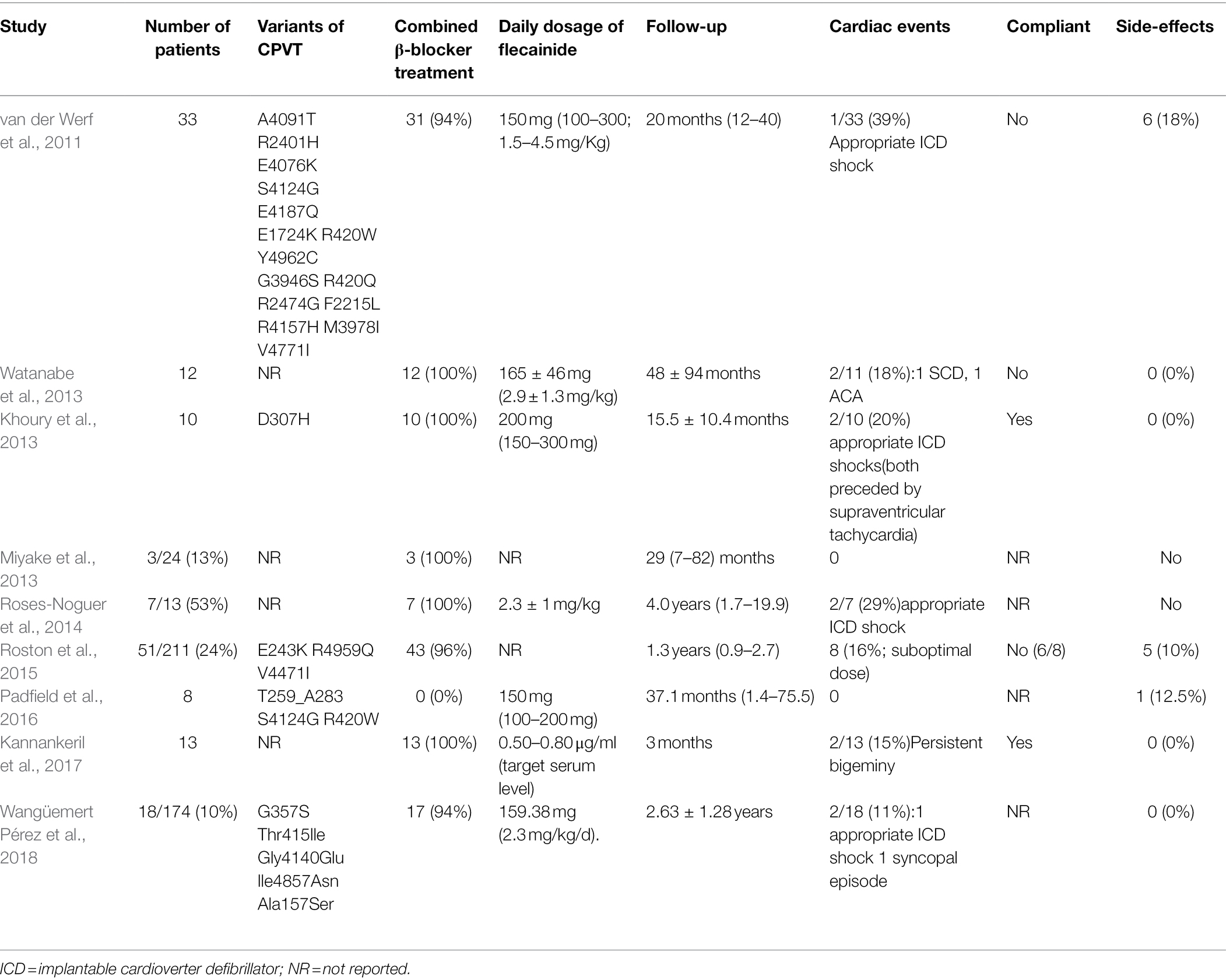
Table 1. Clinical efficacy and safety of flecainide treatment in CPVT.
The abnormal Ca2+ leak events from RyR2 are the essence of molecular arrhythmogenic mechanism in CPVT. Theoretically, direct RyR2 blockers are promising mechanism-based therapies for CPVT. Tetracaine, a sodium channel blocker, is a RyR2 blocker that effectively inhibits Ca2+ leak from SR. Thus, Knollmann and his collaborators screened the RyR2 inhibiting effects of clinically available sodium channel blockers in a lipid bilayer study and found that flecainide reduced the duration of RyR2 channel openings, but not its closed channel duration. They then tested the effects of flecainide on CASQ2 knockout mice. Intraperitoneal administration of flecainide completely suppressed exercise-induced VT in vivo, and incubation with flecainide significantly ameliorated the spontaneous Ca2+ release from SR induced by isoproterenol in isolated myocytes. In contrast, without RyR2 blocking action in the lipid bilayer, lidocaine did not show therapeutic effects in vivo and in vitro. Therefore, they propose that the underlying antiarrhythmic mechanism of flecainide in CPVT attributes to its RyR2 blockade but not its intrinsic sodium channel inhibiting action.
Subsequently, the Knollman group performed a series of experiments to reinforce the concept. Galimberti and Knollmann (2011) reported that flecainide suppressed the spontaneous Ca2+ wave with IC50 12.8 μM in permeabilized ventricular myocytes. The blocking action of flecainide is use-dependent, suggesting that RyR2 activity determines the potency and efficacy of flecainide. Given flecainide’s blocking features of the sodium channel and RyR2 channel, it is challenging to dissect the antiarrhythmic mechanisms of flecainide in CPVT. Kryshtal et al. (2021) synthesized a flecainide analogues, named N-methyl flecainide, which has the sodium channel blocking action but without the RyR2 inhibiting effect. They reported that flecainide, but not N-methyl flecainide, significantly reduced arrhythmias in CPVT transgenic mice and decreased spontaneous calcium-release events in intact and membrane-permeabilized myocytes. Therefore, they concluded that RyR2 channel inhibition, but not sodium channel blockade, is likely the principal mechanism of the antiarrhythmic action of flecainide in CPVT.
Flecainide is a hydrophilic sodium channel blocker with a pKa of 9.3. At pH 7.4, only 1% of flecainide is neutral and is available for diffusion across the membrane of myocytes (Liu et al., 2012). The intrinsic feature of flecainide makes it difficult to quickly achieve sufficient concentration to block RyR2, which is located in the intracellular space. Thus, the RyR2 blocking action of flecainide cannot fully explain the rapid amelioration of spontaneous Ca2+ release after the acute administration of flecainide in isolated cardiac myocytes.
Liu et al. (2011) tested the effects of flecainide in a CPVT RyR2-R4496C+/− mouse model. Flecainide significantly reduced ventricular arrhythmias induced by adrenaline and caffeine in vivo. In isolated intact RyR2R4496C+/− myocytes, flecainide did not affect Ca2+ transient amplitude, decay, or SR Ca2+ content. In permeabilized RyR2R4496C+/− myocytes, flecainide did not alter the frequency of spontaneous Ca2+ sparks. In contrast, when the dosage of flecainide reached 6 μM, the upstroke of action potential was blunted significantly at the pace of 5 Hz (Figure 1). Flecainide effectively prevented isoproterenol-induced triggered activity but had little effect on spontaneous Ca2+ transients (SCaTs) elicited by isoproterenol (Figure 2). The threshold for action potential induction increased significantly after acute administration of flecainide. Based on the above data, Liu et al. suggested that the antiarrhythmic mechanism of flecainide was mediated by its Na+ channel blockade.
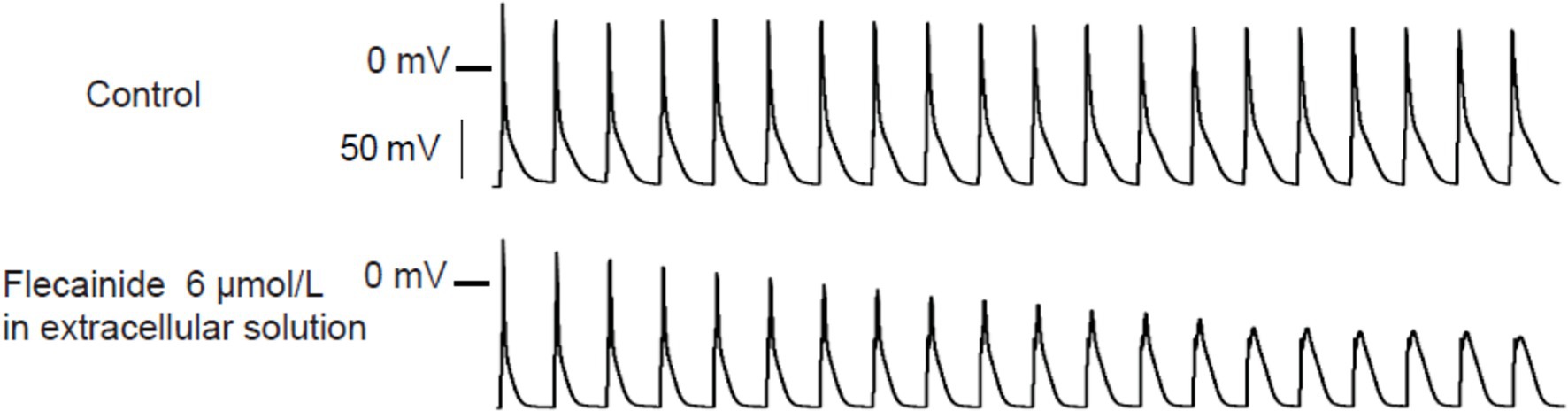
Figure 1. Representative action potential recordings of RyR2R4496C+/ myocytes at 5 Hz pacing. Flecainide (6 μM) almost abolished the upstroke of the action potential. Reproduced with permission by Lippincott Williams and Wilkins from Liu et al. (2011).
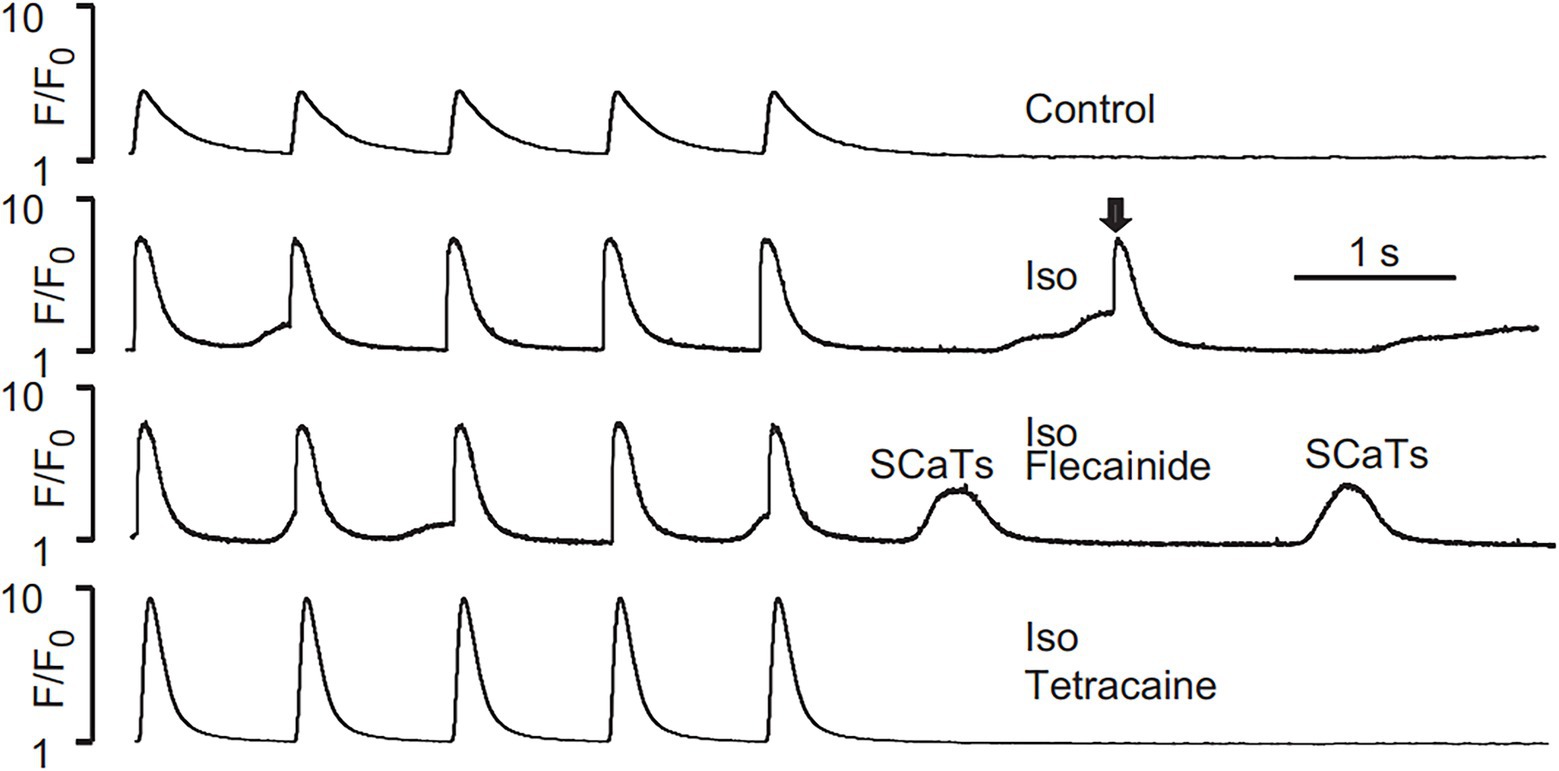
Figure 2. SCaTs elicited in an RyR2R4496C+/− myocyte in the presence of isoproterenol (Iso) at 1 Hz pacing. The arrow shows a triggered SCaT, presumably from a triggered electric beat induced by SCaTs. Flecainide (incubation for 30 min) did not prevent isoproterenol-induced SCaTs in an RyR2R4496C+/− myocyte. Reproduced with permission by Lippincott Williams and Wilkins from Liu et al. (2011).
Sikkel et al. (2013) performed a study to explore the effects of flecainide on Ca2+ handling in isolated rat ventricular myocytes. They found that sodium channel blockers (flecainide, tetrodotoxin, propafenone, and lidocaine) could reduce spontaneous Ca2+ release events under their experimental conditions. After inactivation of the sodium channel using the voltage-clamp approach, flecainide could not reduce Ca2+ waves. Therefore, they proposed that Na+ channel blockade by flecainide could reduce Na+ influx into cardiac myocytes, resulting in the enhancement of Ca2+ efflux through NCX and decrease of Ca2+ in the vicinity of the RyR2 channels, ultimately reducing the frequency of spontaneous Ca2+ release events. In the HEK293 cell line expressing hRyR2, which is devoided of Na+ channels in the cellular membrane, Bannister et al. reported that flecainide did not affect spontaneous Ca2+ release events (Bannister et al., 2015, 2016). Thus, these studies suggest that flecainide’s antiarrhthymic action in CPVT relies on its Na+ channel blockade but not RyR2 inhibition.
Neuronal sodium channels are expressed in the cellular membranes of cardiac myocytes. Radwański et al. (2015a) demonstrated that 100 nM TTX, which blocks neuronal sodium channels but not NaV1.5, significantly reduced and desynchronized spontaneous Ca2+ release events in isolated myocytes. Next, they demonstrated that the NaV1.6 blocker riluzole ameliorated spontaneous Ca2+ release events in vitro and reduced arrhythmias in vivo in a CPVT mouse model. Cardiac Na+ and Ca2+ cycling interplay in the nanodomains beneath the membrane. They speculated that targeting neuronal sodium channels may be a promising therapeutic strategy for Ca2+ dysregulation-associated heart diseases such as CPVT and heart failure, which would not compromise electrical excitability, which is proarrhythmic (Radwański et al., 2015b).
There is a hot debate on the antiarrhythmic mechanisms of flecainide in CPVT. The critical issue is whether flecainide is a RyR2 blocker, as the major mechanism responsible for the efficacy of flecainide in CPVT has been observed in clinical practice. There are multiple targets of flecainide in cardiac myocytes, including sodium channel, potassium channel, and RyR2 channel et al. (Table 2). It is challenging to dissect the therapeutic mechanisms of flecainide in intact cardiac myocytes because of its influence on Na+, Ca2+, and K+ homeostasis at the cellular level simultaneously.
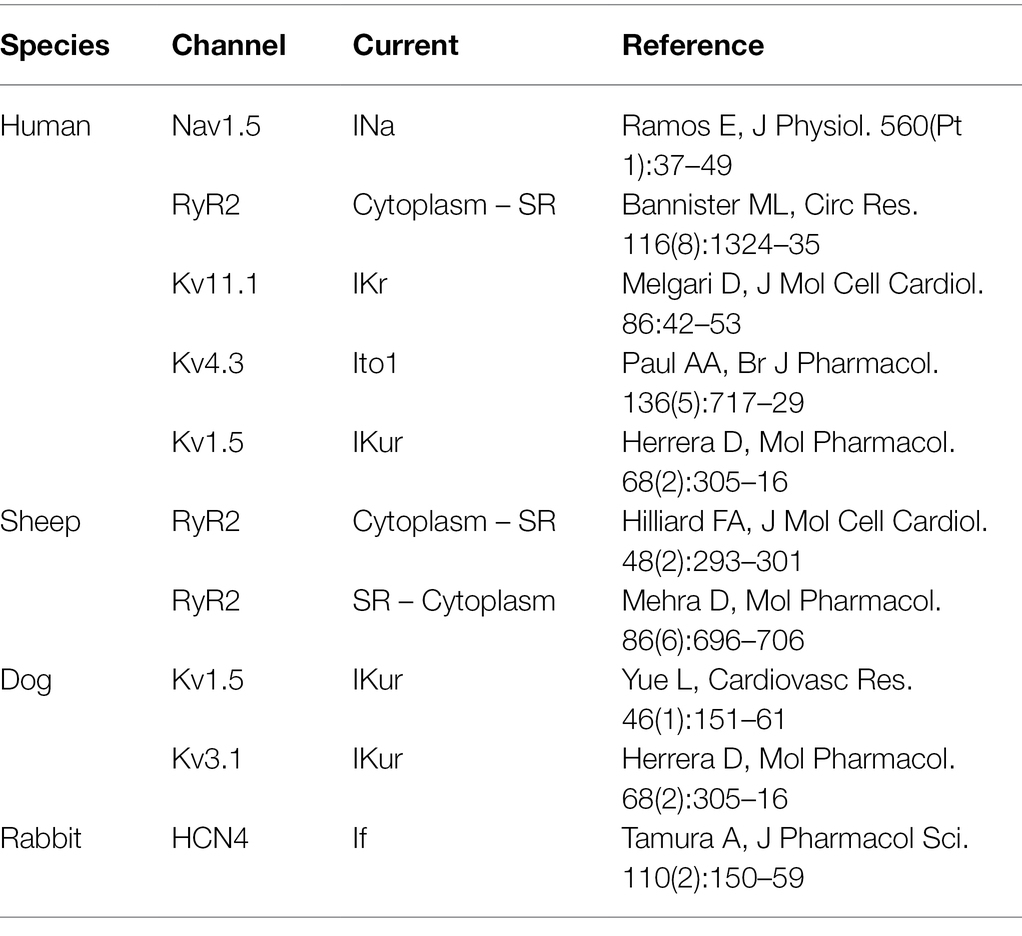
Table 2. Blocking effects of flecainide on ion channels.
Single RyR2 channel experiments in artificial lipid bilayers appear to resolve this issue directly. The initial study by Knollmann et al. tested the inhibitory potency of flecainide on the current flow in the cytoplasm-to-lumen direction in sheep RyR2 channels and found that it inhibited the duration of channel openings and did not affect closed channel duration (Watanabe et al., 2009). The reduction in open-channel probability was concentration-dependent, with an IC50 of 55 ± 8.1 μM. Later, this group presented a detailed analysis of the kinetics of RyR2 inhibition by flecainide. Flecainide inhibited RyR2 by two distinct modes: a fast block consisting of brief substrate and closed events with a mean duration of ∼1 ms, and a slow block consisting of closed events with a mean duration of ∼1 s (Mehra et al., 2014). These two modes are independent mechanisms for RyR2 inhibition.
Under physiological conditions, the current flow of cardiac RyR2 is directed from the lumen to the cytoplasm. Bannister et al. (2015) tested the effects of flecainide on the luminal-to-cytosolic flux of cations through human RyR2 in a lipid bilayer study and reported that flecainide, even at supraphysiological concentrations, did not inhibit the open probability of RyR2. Since the ion fluxes across the SR membrane are bidirectional, the authors also explored whether flecainide modulates cytoplasm-to-SR luminal “countercurrent.” They found that 50 μM flecainide had a negligible effect on the mechanisms responsible for the SR charge-compensating counter current (Bannister et al., 2015, 2016). More recently, the study by Salvage et al. showed that low cytoplasmic concentrations (0.5–10 μM) of flecainide activated isolated mouse RyR2 channels. In contrast, high cytoplasmic concentrations (50–100 μM) of flecainide showed an inhibitory action (Salvage et al., 2021).
RyR2 is a macro-molecular complex, and numerous accessory proteins modulate RyR2’s function, such as FKBP12, FKBP12.6, calmodulin, and S100A1 (Ritterhoff et al., 2014, 2015). Flecainide may affect RyR2’s function directly or indirectly by binding to accessory proteins. The procedure for purifying RyR2 for single-channel recording may disrupt the interaction between accessory proteins and RyR2. This might be an interpretation of the controversial results from different laboratories.
Permeabilized myocytes, devoid of the influence of the cellular membrane, allow direct exploration of the effects of flecainide on Ca2+ handling in SR. Hilliard et al. (2010) demonstrated that 25 μM flecainide significantly reduced the spontaneous Ca2+ wave frequency in permeabilized ventricular myocytes. Savio-Galimberti and Knollmann (2015) showed that flecainide suppressed spontaneous Ca2+ wave with IC50 15.6 ± 3.4 μM in permeabilized CASQ2−/− myocytes. On the contrary, in permeabilized RyR2R4496C myocytes, Liu et al. (2011) reported that 6 μM flecainide did not affect the frequency of spontaneous Ca2+ sparks; Bannister et al. (2015) failed to confirm the reduction of spontaneous Ca2+ wave frequency after administration of 25 μM flecainide in permeabilized rat cardiac myocytes. However, it is difficult to reconcile these conflicting results. Permeabilized myocytes are indispensable for disrupting the intracellular structure and loss of cytosolic proteins, which depend on the degree of membrane permeabilization induced by the concentration of saponin or β-escin and the duration of exposure to the agent. This may differ among laboratories, leading to controversial results (Smith and MacQuaide, 2015).
Flecainide has a narrow therapeutic window between the effective dose and the dose that can produce adverse toxic effects. The target range for flecainide concentration is 0.5–2.4 μM in the clinical practice (Melgari et al., 2015; Rabêlo Evangelista et al., 2021; Yang et al., 2021). Despite conflicting results in single-channel recordings, the therapeutic concentration of flecainide cannot block RyR2 effectively. Based on the IC50 of flecainide for inhibiting RyR2, a high flecainide concentration (25 μM) was used in intact or permeabilized myocytes to elucidate antiarrhythmic mechanisms. Watanabe et al. (2009) reported that flecainide concentration in cardiac tissue is 33 ± 0.8 μM 1 h after injection in mice, suggesting that flecainide can accumulate in cardiac tissue. Thus, it seems reasonable to use very high concentrations of flecainide in the experiments. In a study by Liu et al. (2011), 6 μM flecainide completely abolished the upstroke of action potentials in mouse ventricular myocytes, consistent with the adverse toxic effects of high-dose flecainide administration. In this scenario, it is unlikely that the blockade of RyR2 by high concentrations of flecainide (25 μM) is the primary mechanism underlying its dramatic efficacy in CPVT (Liu et al., 2011). Moreover, despite the high concentration of flecainide in cardiac tissue, it cannot be arbitrarily inferred that flecainide in the cytoplasm of cardiac myocytes reaches a sufficient concentration to block RyR2. It is almost impossible for high concentrations of flecainide in cardiac tissue to exclusively affect RyR2, but not cardiac sodium channels.
Flecainide is a multiple potassium channel blocker. Class III antiarrhythmic drugs are effective in the reentrant arrhythmias but not trigger arrhythmias. They are not effective in CPVT in the clinical setting. Potassium channel blockers prolong the action potential duration, leading to Ca2+ overload, and are detrimental to CPVT (Němec et al., 2010). In this scenario, potassium channel block is unlikely responsible for the flecainide efficacy in CPVT.
Most of the studies in CPVT are from small animal models, and mouse has a small heart size and fast heart rate. We need to be aware of the limitations to generalize the results of the small animal research to humans (Joukar, 2021).
Bidirectional VT is a typical arrhythmic phenotype observed in patients with CPVT (van der Werf et al., 2012). It has been proposed that bidirectional VT in CPVT, digitalis toxicity, and Andersen-Tawil Syndrome (ATS) share a similar underlying electrophysiological mechanism, which is associated with alternating ectopic foci originating from the distal His-Purkinje system in the left and/or right ventricle, induced by Ca2+ overload in Purkinje cells (Smith et al., 2006; Tristani-Firouzi and Etheridge, 2010).
Digitalis intoxication is manifested by Na+-K+ pump inhibition, resulting in intracellular Ca2+ overload, which causes triggered arrhythmias, such as bidirectional VT. The sodium channel blockers lidocaine and phenytoin have been recommended for the effective treatment of dysrhythmias associated with digitalis intoxication (French et al., 1984; Antman and Smith, 1985). In isolated myocytes, sodium channel blockers can ameliorate intracellular Ca2+ overload induced by digitalis and reduce spontaneous Ca2+ release events. Since there is no RyR2 blocking action of lidocaine and phenytoin, the sole sodium channel blockade is responsible for the antiarrhythmic effects of lidocaine and phenytoin in digitalis intoxication.
ATS, which is mainly caused by KCNJ2 mutations, phenocopies CPVT and may manifest the typical adrenergically mediated bidirectional VT (Zhang et al., 2005). The underlying arrhythmogenic mechanism is triggered arrhythmias induced by Ca2+ dysregulation in cardiac myocytes. A series of cases have shown that flecainide effectively prevents arrhythmias in patients with ATS (Pellizzón et al., 2008; Kuroda et al., 2017). Given that ATS presents bidirectional VT, which is usually observed in digitalis toxicity, and phenytoin is used to treat arrhythmia in digitalis toxicity, Maneesh et al. tested phenytoin in three ATS patients who did not respond to conventional therapy (β-blockers, flecainide, and verapamil). They reported that phenytoin completely suppressed ventricular arrhythmias in two patients and significantly reduced ventricular arrhythmias burden in one patient (Rai et al., 2019). Thus, sodium channel inhibition is likely the principal mechanism of flecainide action in ATS.
Another interesting issue is the diverse responses to sodium channel blockers in CPVT. Use and frequency dependence are common properties of class I antiarrhythmic agents. Flecainide, but not lidocaine, preferentially blocks sodium channel in the open state (Kojima et al., 1989; Ramos and O’Leary, 2004). The electrophysiological mechanism of CPVT is that DAD reaches the threshold of Na+ channel and results in triggered activity, which is also frequency-dependent. Therefore, flecainide is more effective than other sodium channel blockers during fast heart rates, such as conditions in which patients with CPVT develop cardiac arrhythmias. In CPVT, the efficacy of other sodium channel blockers with the strong use dependence block, such as propafenone and pilsicainide, needs further investigation.
Recently, loss of function (LOF) of RyR2 mutations has been identified as a new clinical entity, termed cardiac ryanodine receptor calcium-release syndrome (CRDS), which is characterized by ventricular fibrillation and sudden death. However, it does not manifest ventricular tachyarrhythmias during stress testing (Roston et al., 2021). CRDS is a mirror image of CPVT due to the opposite of the RyR2 function. It is logical to infer that flecainide might exacerbate the CRDS phenotype if it can inhibit RyR2. To date, flecainide has proven to be a promising therapeutic agent for CRDS (Tester et al., 2020; Ormerod et al., 2021). The programmed electrical stimulation protocol with a pattern of long-burst, long-pause, and short-coupled (LBLPS) can induce ventricular arrhythmias in transgenic mice with RyR2 LOF mutations. Sun et al. (2021) demonstrated that treatment with flecainide abolished LBLPS-induced ventricular arrhythmias in model mice. In the induced pluripotent stem cell cardiomyocytes carrying homozygous RYR2 duplication, which presented LOF, Tester et al. (2020) reported that flecainide significantly reduced arrhythmic activity caused by isopropanol. Ormerod et al. (2021) tested flecainide in nine CRDS patients and found that the administration of flecainide substantially reduced arrhythmia inducibility in one subject and abolished arrhythmia in all others. Sun et al. (2021) proposed that the therapeutic mechanisms of flecainide in CRDS are attributable to its multiple blocking of membrane channels.
Flecainide has a significant impact on the clinical management of patients with CPVT. Efforts have been made to explore the underlying mechanisms of flecainide therapy for CPVT. There is a hot debate regarding the effects of flecainide on RyR2. Understanding the mechanisms of flecainide in CPVT will improve our knowledge of Ca2+ dysregulation in cardiac myocytes and help develop a more specific therapeutic strategy for CPVT.
YR and NL defined the theme of review. YL wrote the manuscript. XP, RL, XW, and XL took part in preparing the manuscript. YR, NL, RT, CM, and RB prepared and reviewed the manuscript before publication. All authors confirmed that they have read and approved the manuscript and they have met the criteria for authorship.
This work was supported by the National Science Foundation of China (grant nos. 81770318, 82170318, and 81870244) and Beijing Municipal Natural Science Foundation (grant no. 7192051).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Antman, E. M., and Smith, T. W. (1985). Digitalis toxicity. Annu. Rev. Med. 36, 357–367. doi: 10.1146/annurev.me.36.020185.002041
Baltogiannis, G. G., Lysitsas, D. N., di Giovanni, G., Ciconte, G., Sieira, J., Conte, G., et al. (2019). CPVT: arrhythmogenesis, therapeutic management, and future perspectives. A brief review of the literature. Front. Cardiovasc. Med. 6:92. doi: 10.3389/fcvm.2019.00092
Bannister, M., Alvarez-Laviada, A., Thomas, N., Mason, S., Coleman, S., du Plessis, C., et al. (2016). Effect of flecainide derivatives on sarcoplasmic reticulum calcium release suggests a lack of direct action on the cardiac ryanodine receptor. Brit. J. Pharmacol. 173, 2446–2459. doi: 10.1111/bph.13521
Bannister, M. L., Thomas, N. L., Sikkel, M. B., Mukherjee, S., Maxwell, C., MacLeod, K. T., et al. (2015). The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ. Res. 116, 1324–1335. doi: 10.1161/CIRCRESAHA.116.305347
Bers, D. M. (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
French, J. H., Thomas, R. G., Siskind, A. P., Brodsky, M., and Iseri, L. T. (1984). Magnesium therapy in massive digoxin intoxication. Ann. Emerg. Med. 13, 562–566. doi: 10.1016/S0196-0644(84)80534-X
Galimberti, E. S., and Knollmann, B. C. (2011). Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin. J. Mol. Cell. Cardiol. 51, 760–768. doi: 10.1016/j.yjmcc.2011.07.002
Hilliard, F. A., Steele, D. S., Laver, D., Yang, Z., Le Marchand, S. J., Chopra, N., et al. (2010). Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J. Mol. Cell. Cardiol. 48, 293–301. doi: 10.1016/j.yjmcc.2009.10.005
Hwang, H. S., Hasdemir, C., Laver, D., Mehra, D., Turhan, K., Faggioni, M., et al. (2011). Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 4, 128–135. doi: 10.1161/CIRCEP.110.959916
Joukar, S. (2021). A comparative review on heart ion channels, action potentials and electrocardiogram in rodents and human: extrapolation of experimental insights to clinic. Lab. Anim. Res. 37:25. doi: 10.1186/s42826-021-00102-3
Kannankeril, P. J., Moore, J. P., Cerrone, M., Priori, S. G., Kertesz, N. J., Ro, P. S., et al. (2017). Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol. 2, 759–766. doi: 10.1001/jamacardio.2017.1320
Khoury, A., Marai, I., Suleiman, M., Blich, M., Lorber, A., Gepstein, L., et al. (2013). Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 10, 1671–1675. doi: 10.1016/j.hrthm.2013.08.011
Kojima, M., Hamamoto, T., and Ban, T. (1989). Sodium channel-blocking properties of flecainide, a class IC antiarrhythmic drug, in Guinea-pig papillary muscles. An open channel blocker or an inactivated channel blocker. Naunyn Schmiedeberg's Arch. Pharmacol. 339, 441–447. doi: 10.1007/BF00736059
Kryshtal, D. O., Blackwell, D. J., Egly, C. L., Smith, A. N., Batiste, S. M., Johnston, J. N., et al. (2021). RYR2 channel inhibition is the principal mechanism of Flecainide action in CPVT. Circ. Res. 128, 321–331. doi: 10.1161/CIRCRESAHA.120.316819
Kuroda, Y., Yuasa, S., Watanabe, Y., Ito, S., Egashira, T., Seki, T., et al. (2017). Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem. Biophys. Rep. 9, 245–256. doi: 10.1016/j.bbrep.2017.01.002
Leenhardt, A., Lucet, V., Denjoy, I., Grau, F., Ngoc, D. D., and Coumel, P. (1995). Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91, 1512–1519. doi: 10.1161/01.cir.91.5.1512
Liu, N., Denegri, M., Ruan, Y., Avelino-Cruz, J. E., Perissi, A., Negri, S., et al. (2011). Short communication: Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ. Res. 109, 291–295. doi: 10.1161/CIRCRESAHA.111.247338
Liu, N., Napolitano, C., Venetucci, L. A., and Priori, S. G. (2012). Flecainide and antiarrhythmic effects in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Trends Cardiovasc. Med. 22, 35–39. doi: 10.1016/j.tcm.2012.06.008
Liu, N., Ruan, Y., and Priori, S. G. (2008). Catecholaminergic polymorphic ventricular tachycardia. Prog. Cardiovasc. Dis. 51, 23–30. doi: 10.1016/j.pcad.2007.10.005
Mehra, D., Imtiaz, M. S., van Helden, D. F., Knollmann, B. C., and Laver, D. R. (2014). Multiple modes of ryanodine receptor 2 inhibition by flecainide. Mol. Pharmacol. 86, 696–706. doi: 10.1124/mol.114.094623
Melgari, D., Zhang, Y., El Harchi, A., Dempsey, C. E., and Hancox, J. C. (2015). Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J. Mol. Cell. Cardiol. 86, 42–53. doi: 10.1016/j.yjmcc.2015.06.021
Miyake, C. Y., Webster, G., Czosek, R. J., Kantoch, M. J., Dubin, A. M., Avasarala, K., et al. (2013). Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: success depends on substrate. Circ. Arrhythm. Electrophysiol. 6, 579–587. doi: 10.1161/CIRCEP.113.000170
Němec, J., Kim, J. J., Gabris, B., and Salama, G. (2010). Calcium oscillations and T-wave lability precede ventricular arrhythmias in acquired long QT type 2. Heart Rhythm. 7, 1686–1694. doi: 10.1016/j.hrthm.2010.06.032
Ormerod, J. O. M., Ormondroyd, E., Li, Y., Taylor, J., Wei, J., Guo, W., et al. (2021). Provocation testing and therapeutic response in a newly described channelopathy: RyR2 calcium release deficiency syndrome. Circ. Genom. Precis. Med. 15:e003589. doi: 10.1161/CIRCGEN.121.003589
Padfield, G. J., AlAhmari, L., Lieve, K. V. V., AlAhmari, T., Roston, T. M., Wilde, A. A., et al. (2016). Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of β-blockade. Heart Rhythm. 13, 609–613. doi: 10.1016/j.hrthm.2015.09.027
Pellizzón, O. A., Kalaizich, L., Ptácek, L. J., Tristani-Firouzi, M., and Gonzalez, M. D. (2008). Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J. Cardiovasc. Electrophysiol. 19, 95–97. doi: 10.1111/j.1540-8167.2007.00910.x
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963. doi: 10.1016/j.hrthm.2013.05.014
Rabêlo Evangelista, A. B., Monteiro, F. R., Nearing, B. D., Belardinelli, L., and Verrier, R. L. (2021). Flecainide-induced QRS complex widening correlates with negative inotropy. Heart Rhythm. 18, 1416–1422. doi: 10.1016/j.hrthm.2021.04.007
Radwański, P. B., Brunello, L., Veeraraghavan, R., Ho, H.-T., Lou, Q., Makara, M. A., et al. (2015a). Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc. Res. 106, 143–152. doi: 10.1093/cvr/cvu262
Radwański, P. B., Brunello, L., Veeraraghavan, R., Ho, H.-T., Lou, Q., Makara, M. A., et al. (2015b). Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc. Res. 106, 143–152. doi: 10.1093/cvr/cvu262
Rai, M. K., Pai, R., Prabhu, M. A., Pasha, S. W., Kedambadi, R. C., Kamath, P., et al. (2019). Short-term response to phenytoin sodium in Andersen-Tawil syndrome-1 with a cardiac-dominant phenotype. Pacing Clin. Electrophysiol. 42, 201–207. doi: 10.1111/pace.13569
Ramos, E., and O’Leary, M. E. (2004). State-dependent trapping of flecainide in the cardiac sodium channel. J. Physiol. 560, 37–49. doi: 10.1113/jphysiol.2004.065003
Ritterhoff, J., Völkers, M., Seitz, A., Spaich, K., Gao, E., Peppel, K., et al. (2015). S100A1 DNA-based inotropic therapy protects against proarrhythmogenic ryanodine receptor 2 dysfunction. Mol. Ther. 23, 1320–1330. doi: 10.1038/mt.2015.93
Ritterhoff, J., Völkers, M., Seitz, A., Spaich, K., Peppel, K., Pleger, S., et al. (2014). Abstract 37: The positive inotropic S100a1 prevents arrhythmogenic sarcoplasmic reticulum Ca2+ leak and ventricular arrhythmias. Circ. Res. 115, A37–A37. doi: 10.1161/res.115.suppl_1.37
Roses-Noguer, F., Jarman, J. W. E., Clague, J. R., and Till, J. (2014). Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 11, 58–66. doi: 10.1016/j.hrthm.2013.10.027
Roston, T. M., Vinocur, J. M., Maginot, K. R., Mohammed, S., Salerno, J. C., Etheridge, S. P., et al. (2015). Catecholaminergic polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ. Arrhythm. Electrophysiol. 8, 633–642. doi: 10.1161/CIRCEP.114.002217
Roston, T. M., Wei, J., Guo, W., Li, Y., Zhong, X., Wang, R., et al. (2021). Clinical and functional characterization of ryanodine receptor 2 variants implicated in calcium-release deficiency syndrome. JAMA Cardiol. 7, 84–92. doi: 10.1001/jamacardio.2021.4458
Salvage, S. C., Gallant, E. M., Fraser, J. A., Huang, C. L.-H., and Dulhunty, A. F. (2021). Flecainide paradoxically activates cardiac ryanodine receptor channels under low activity conditions: a potential pro-arrhythmic action. Cells 10:2101. doi: 10.3390/cells10082101
Savio-Galimberti, E., and Knollmann, B. C. (2015). Channel activity of cardiac ryanodine receptors (RyR2) determines potency and efficacy of flecainide and R-propafenone against arrhythmogenic calcium waves in ventricular cardiomyocytes. PLoS One 10:e0131179. doi: 10.1371/journal.pone.0131179
Sikkel, M. B., Collins, T. P., Rowlands, C., Shah, M., O’Gara, P., Williams, A. J., et al. (2013). Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc. Res. 98, 286–296. doi: 10.1093/cvr/cvt012
Smith, A. H., Fish, F. A., and Kannankeril, P. J. (2006). Andersen-Tawil syndrome. Indian Pacing Electrophysiol. J. 6, 32–43.
Smith, G. L., and MacQuaide, N. (2015). The direct actions of flecainide on the human cardiac ryanodine receptor: keeping open the debate on the mechanism of action of local anesthetics in CPVT. Circ. Res. 116, 1284–1286. doi: 10.1161/CIRCRESAHA.115.306298
Steinfurt, J., Dechant, M.-J., Böckelmann, D., Zumhagen, S., Stiller, B., Schulze-Bahr, E., et al. (2015). High-dose flecainide with low-dose β-blocker therapy in catecholaminergic polymorphic ventricular tachycardia: a case report and review of the literature. J. Cardiol. Cases 11, 10–13. doi: 10.1016/j.jccase.2014.08.009
Sun, B., Yao, J., Ni, M., Wei, J., Zhong, X., Guo, W., et al. (2021). Cardiac ryanodine receptor calcium release deficiency syndrome. Sci. Transl. Med. 13:eaba7287. doi: 10.1126/scitranslmed.aba7287
Tester, D. J., Kim, C. J., Hamrick, S. K., Ye, D., O’Hare, B. J., Bombei, H. M., et al. (2020). Molecular characterization of the calcium release channel deficiency syndrome. JCI Insight 5:e135952. doi: 10.1172/jci.insight.135952
Tristani-Firouzi, M., and Etheridge, S. P. (2010). Kir 2.1 channelopathies: the Andersen–Tawil syndrome. Pflugers Arch. - Eur. J. Physiol. 460, 289–294. doi: 10.1007/s00424-010-0820-6
van der Werf, C., Kannankeril, P. J., Sacher, F., Krahn, A. D., Viskin, S., Leenhardt, A., et al. (2011). Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 57, 2244–2254. doi: 10.1016/j.jacc.2011.01.026
van der Werf, C., Zwinderman, A. H., and Wilde, A. A. M. (2012). Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace 14, 175–183. doi: 10.1093/europace/eur277
Wangüemert Pérez, F., Hernández Afonso, J. S., Groba Marco, M. D. V., Caballero Dorta, E., Álvarez Acosta, L., Campuzano Larrea, O., et al. (2018). Flecainide reduces ventricular arrhythmias in patients with genotype RyR2-positive catecholaminergic polymorphic ventricular tachycardia. Rev. Esp. Cardiol. 71, 185–191. doi: 10.1016/j.rec.2017.04.032
Watanabe, H., Chopra, N., Laver, D., Hwang, H., Davies, S., Roach, D., et al. (2009). Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 15, 380–383. doi: 10.1038/nm.1942
Watanabe, H., van der Werf, C., Roses-Noguer, F., Adler, A., Sumitomo, N., Veltmann, C., et al. (2013). Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 10, 542–547. doi: 10.1016/j.hrthm.2012.12.035
Wier, W. G., and Balke, C. W. (1999). Ca2+ release mechanisms, Ca2+ Sparks, and local control of excitation-contraction coupling in Normal heart muscle. Circ. Res. 85, 770–776. doi: 10.1161/01.RES.85.9.770
Wleklinski, M. J., Kannankeril, P. J., and Knollmann, B. C. (2020). Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 598, 2817–2834. doi: 10.1113/JP276757
Yang, P.-C., Giles, W. R., Belardinelli, L., and Clancy, C. E. (2021). Mechanisms of flecainide induced negative inotropy: an in silico study. J. Mol. Cell Cardiol. 158, 26–37. doi: 10.1016/j.yjmcc.2021.05.007
Yang, P.-C., Moreno, J. D., Miyake, C. Y., Vaughn-Behrens, S. B., Jeng, M.-T., Grandi, E., et al. (2016). In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 594, 567–593. doi: 10.1113/JP271282
Keywords: flecainide, antiarrhythmic mechanism, sodium channel, ryanodine receptor, catecholaminergic polymorphic ventricular tachycardia
Citation: Li Y, Peng X, Lin R, Wang X, Liu X, Bai R, Ma C, Tang R, Ruan Y and Liu N (2022) The Antiarrhythmic Mechanisms of Flecainide in Catecholaminergic Polymorphic Ventricular Tachycardia. Front. Physiol. 13:850117. doi: 10.3389/fphys.2022.850117
Edited by:
Yanmin Zhang, Xi’an Jiaotong University, ChinaReviewed by:
Hiroki Takanari, Tokushima University, JapanCopyright © 2022 Li, Peng, Lin, Wang, Liu, Bai, Ma, Tang, Ruan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nian Liu, bGl1bmlhbjE5NzNAaG90bWFpbC5jb20=; Yanfei Ruan, cnVhbnlhbmZlaUBob3RtYWlsLmNvbQ==; Ribo Tang, dGFuZ3JpYm9AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.