 Klaus Schicker
Klaus Schicker Clemens V. Farr
Clemens V. Farr Danila Boytsov
Danila Boytsov Michael Freissmuth
Michael Freissmuth Walter Sandtner
Walter Sandtner
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 03 February 2022
Sec. Membrane Physiology and Membrane Biophysics
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.817886
This article is part of the Research TopicCombining Computational and Experimental Approaches to Characterize Ion Channels and TransportersView all 9 articles
The diversity in solute carriers arose from evolutionary pressure. Here, we surmised that the adaptive search for optimizing the rate of substrate translocation was also shaped by the ambient extracellular and intracellular concentrations of substrate and co-substrate(s). We explored possible solutions by employing kinetic models, which were based on analytical expressions of the substrate uptake rate, that is, as a function of the microscopic rate constants used to parameterize the transport cycle. We obtained the defining terms for five reaction schemes with identical transport stoichiometry (i.e., Na+: substrate = 2:1). We then utilized an optimization algorithm to find the set of numeric values for the microscopic rate constants, which provided the largest value for the substrate uptake rate: The same optimized rate was achieved by different sets of numerical values for the microscopic rate constants. An in-depth analysis of these sets provided the following insights: (i) In the presence of a low extracellular substrate concentration, a transporter can only cycle at a high rate, if it has low values for both, the Michaelis–Menten constant (KM) for substrate and the maximal substrate uptake rate (Vmax). (ii) The opposite is true for a transporter operating at high extracellular substrate concentrations. (iii) Random order of substrate and co-substrate binding is superior to sequential order, if a transporter is to maintain a high rate of substrate uptake in the presence of accumulating intracellular substrate. Our kinetic models provide a framework to understand how and why the transport cycles of closely related transporters differ.
Cellular membranes are diffusion barriers for polar solutes. Uptake of these solutes into a cell or a subcellular compartment is, therefore, contingent on solute carriers (SLC). For this reason, SLCs are vital for many physiological functions. The latter include cellular uptake of nutrients and extrusion of toxic compounds from the interior of a cell (Hediger et al., 2004; Omote et al., 2006; Sano et al., 2020). In addition, SLCs are involved in higher order functions, such as neurotransmission (e.g., reuptake of neurotransmitters subsequent to their vesicular release; Rudnick and Sandtner, 2019; Bhat et al., 2021). Many of the SLCs can harvest the energy contained in the transmembrane ion gradients to drive uphill transport of their substrate against an opposing substrate gradient (Mitchell, 1979). These are termed concentrative or secondary active transporters, which either work as symporters or antiporters (Jennigs, 2018). Another class of SLCs only facilitates passive diffusion of a polar solute by providing an aqueous pathway, via which the solute can enter or leave the cell. The latter are termed facilitating or equilibrative transporters. Both, the concentrative and the equilibrative SLC operate by the alternate access mechanism (Jardetzky, 1966) which entails the following sequence of events: Extracellular substrate first binds to the transporter in its outward-facing conformation. On substrate binding the transporter rearranges to adopt the inward-facing conformation. From there the substrate is released into the cytosol. Subsequent to this, the carrier rearranges again to return to the substrate-free outward-facing conformation. From this point on, this series of reactions can repeat all over. Substrate uptake by a solute carrier is, therefore, a process, which encompasses several partial reactions. These include conformational change and binding/unbinding reactions of substrate and (co)-substrates to and from the transporter. These partial reactions form a closed loop, which is also referred to as the transport cycle.
We have recently described an approach to kinetic modeling of a solute carrier, which allows for deriving analytical expressions for its functional descriptors (Schicker et al., 2021). These include the KM and the Vmax for substrate uptake, the rate of basal substrate release from the interior of the cell, etc. The corresponding terms express these descriptors as a function of the microscopic rate constants used to parameterize the kinetic model. In the present study, we derived the defining terms for the substrate uptake rate of a sodium symporter for five different reactions scheme, which all adhere to the same transporter stoichiometry (Na+:substrate = 2:1). The rationale for obtaining these analytical terms was as follows: The substrate uptake rate is the only functional descriptor of a transporter, for which compelling arguments can be made that it has been optimized (i.e., maximized) by evolution. These are: the magnitude of solute flux through SLCs into a cell or a cell organelle is determined by the number of transporter units expressed on the cell or organelle surface and the substrate uptake rate (i.e., turnover rate) of the individual transporters. Accordingly, to maintain a substrate flux, which is commensurate with the physiological needs, the cell can either increase the number of transporters or the rate of substrate turnover. The former is associated with two problems: (i) Protein synthesis is energetically costly (Millward and Garlick, 1976; Waterlow et al., 1978; Siems et al., 1984) and (ii) additional transporters occupy space in the membrane. Membranes cannot be infinitely crowded by transmembrane proteins (Bar-Even et al., 2011). Having to have fewer transporters, thus, increases the energy efficiency of a cell/organism. This is expected to improve fitness at conditions in which nutrients are scarce. Accordingly, the substrate uptake rate of a solute carrier fulfills all criteria of a trait subject to evolutionary selection.
We emulated the evolutionary pressure on the substrate uptake rate by relying on an optimization algorithm. This searched for the set of microscopic rate constants, which returned the largest value for the substrate uptake rate at given intra- and extracellular concentrations of Na+ and substrate. The resulting sets of values provided by the optimization algorithm were subjected to an in-depth analysis. This analysis showed how a solute carrier must adjust its operation to cycle at a high rate at the various conditions/challenges, which it may encounter.
Time-dependent changes in state occupancies of the model in Figure 1A were evaluated by numerical integration of the resulting system of differential equations using the Systems Biology Toolbox (Schmidt and Jirstrand, 2006) and MATLAB 2018a (MathWorks, Natick, MA, United States).
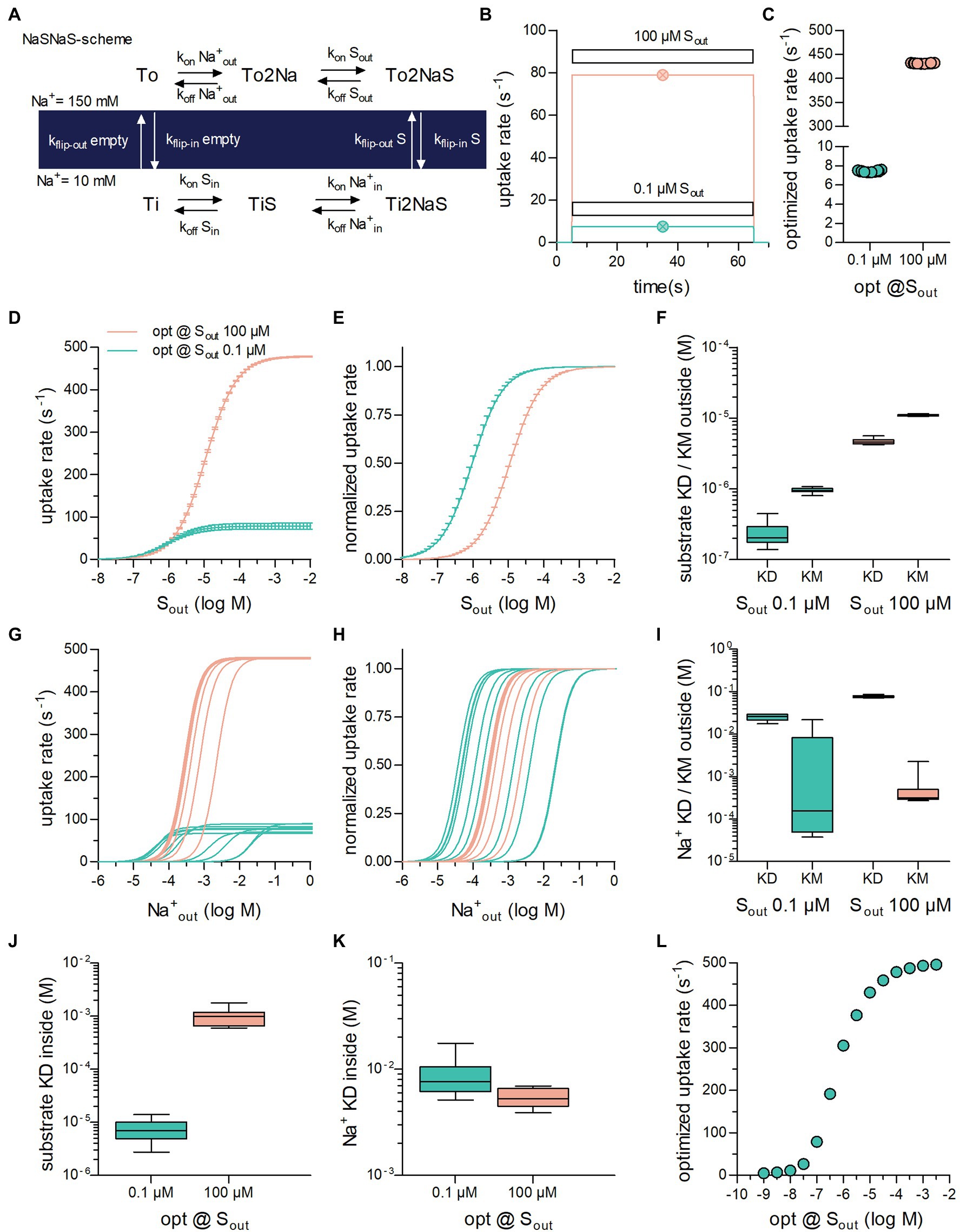
Figure 1. Optimization of secondary active transporters operating in a sequential binding mode for translocation of substrate at a low and high concentration. (A) Reaction scheme of a Na+ symporter in the sequential binding mode (“first in, first out”) referred to as NaSNaS: The apo-outward-facing transporter (To) first binds two Na+ ions (To2Na). On substrate binding, (To2NaS) the transporter rearranges to adopt the inward-facing conformation (Ti2NaS). Subsequent to the release of its cargo into the cytosol (TiNa2S → TiS → Ti) the substrate-free inward-facing transporter (Ti) undergoes a conformational change upon which the transporter returns to To. (B) Simulated substrate uptake rate of the symporter operating according to the scheme outlined in (A) after application of.1 μM (green trace) and 100 μM (magenta trace) of substrate (Sout). For this simulation the microscopic rate constants (i.e., konNa+out and kflip-inS) were parameterized with the values for the optimized transporter T1 shown in Table 1. The symbols in green and in magenta show the substrate uptake rates computed for T1 with the analytical expression (see supplement) for.1 μM and 100 μM Sout, respectively. The uptake rates obtained with the two different approaches were identical. (C) Substrate uptake rates of transporters optimized for.1 μM Sout (green open circles) and for 100 μM Sout (magenta open circles). The data points in the graph show the substrate uptake rates obtained from 10 optimization runs. The corresponding sets of numeric values for the microscopic rate constants are listed in Table 1. (D) The curves represent the substrate uptake rate as a function of Sout of transporters optimized for.1 μM Sout (in green) and 100 μM Sout (in magenta). The data are means from the 10 independently optimized transporters (T1–T10 and T11–T20), error bars indicate S.D. (E) The data in panel (D) were plotted as normalized values (Vmax = 1). (F) Plotted are the KD values for binding of the substrate to the outward-facing conformation of the transporter and the corresponding KM values of transporters optimized for.1 μM and 100 μM Sout, respectively. The coefficients of variation for the KDs were:.43 and.094 and for the KMs:.094 and.03 at.1 μM and 100 μM Sout, respectively. (G) Substrate uptake rate as a function of of transporters optimized for.1 μM Sout (in green) and for 100 μM Sout (in magenta). The concentration dependence for differed considerably between transporters optimized for the same Sout. (H) The same data as in (G) but normalized (I) Plotted are the KD values for Na+ binding to the outward-facing conformation of the transporter and the corresponding KM values of transporters optimized for.1 μM Sout and for 100 μM Sout. The variation in the KD values was much less than the variation in the KM values. The coefficients of variation for the KDs were:.17 and.06 and for the KMs: 1.81 and 1.12 at.1 μM and 100 μM Sout, respectively. (J) Shown are the KD values of substrate binding to the inward-facing conformation of transporters optimized for.1 μM and 100 μM Sout, respectively (coefficients of variation:.47 and.36). (K) Plotted are the KD values of Na+ binding to the inward-facing conformation of transporters optimized for.1 μM Sout and for 100 μM Sout (coefficients of variation:.43 and.2). (L) Plotted are the optimized substrate uptake rates as a function of the Sout, for which they were optimized. Each data point is the means ± SD of the substrate uptake rate obtained from 10 optimization runs. The optimized rate rose upon increase of Sout. It leveled out at 500 s−1 because of the constraints imposed in the optimization.
Explicit expressions for the substrate uptake rate were derived as described previously (Burtscher et al., 2019; Schicker et al., 2021). Numerical sets of values for the microscopic rate constants maximizing these expressions were generated by a simulated annealing algorithm (Metropolis et al., 1953; Tsallis and Stariolo, 1996). In brief, in an initial step, a set of values for the microscopic rate constants was randomly drawn from independent normal distributions, centered at chosen start values with SDs of the same size. The substrate uptake rate for the drawn set was then calculated and compared with the substrate uptake rate calculated from the original set (i.e., the start values). The probability of accepting the new set was: prob. = exp.[−(−current Value + best Value)/T(iter)], with T being a temperature parameter, which was chosen to decrease exponentially with the number of iterations. If accepted, the new set was used for the next iteration, if not, the old set was retained. This procedure was repeated for 5,000 iterations. We safeguarded against trapping in a local maximum by reinitiating the algorithm with the maximum T value increased by 10% using the best parameter set found in the first round of iterations. If this yielded a better overall value for the substrate uptake rate, the next run was reinitiated with the original T. Otherwise T was increased by additional 10%. This procedure was repeated until reheating was unsuccessful in obtaining a better set of values for 10 times. On completion, the algorithm reported the best parameter set (i.e., the set of values which gave the largest substrate uptake rate).
In the optimization of the sequential binding order schemes, we kept the detailed balance constraint, by allowing the algorithm to vary all microscopic rate constants except one. This rate constant was then calculated from the other rate constants such that detailed balance was maintained. In the case of the random order binding scheme, it was necessary to calculate three rate constants because of the larger number of loops. Each set of microscopic rate constants was also evaluated for adherence to the other imposed constraints (e.g., diffusion limit for the association rates of substrate and co-substrate). Only if a set of values complied with the imposed constraints it was passed on to the annealing algorithm.
Figure 1A shows the reaction scheme of a hypothetical symporter: In each cycle, the transporter translocates one substrate molecule through the membrane together with two Na+ ions. We selected this stoichiometry, because it is frequently observed: For instance, sodium-dependent glucose (SGLT1/SLC5A1 and SGLT2/SLC5A2; Wright et al., 2011) and phosphate transporters (PiT-1/SLC20A1 and PiT-2/SLC20A2; Forster et al., 2013) operate with this stoichiometry. For the sake of simplicity, we assumed binding of the two sodium ions to occur in a single reaction. In Figure 1B, we used this model to predict the rate of substrate uptake through the transporter by assuming that two different concentrations of extracellular substrate (Sout) were applied, that is,.1 μM (magenta line in Figure 1B) and 100 μM (blue line in Figure 1B). As seen, on exposure of the cell to the substrate, the substrate uptake rate rose. The rise was large on application of 100 μM Sout and small on application of.1 μM Sout. In the simulation, the substrate was removed after 60 s upon which the substrate uptake rate dropped to zero.
The data in Figure 1B were obtained by numerically solving the system of differential equations underlying the kinetic model. An alternative approach to compute the substrate uptake rate relies on deriving its defining analytical term. For the sake of space, we show the term in the supplement. Figure 1B also displays the substrate uptake rates obtained by this second approach in the presence of.1 μM (open circle in magenta) and 100 μM Sout (open circle in blue). It is evident that the substrate uptake rates predicted by the two methods were identical.
The extracellular concentration of a substrate (Sout) is a given quantity, that is, it is typically not subject to control by a single cell. Accordingly, SLC, which are tasked with transporting a substrate into the interior of a cell, must adjust their operation to the substrate concentration they encounter. The substrate concentration surrounding a cell can therefore, be assumed to exert evolutionary pressure. To emulate optimization of the substrate uptake rate by evolution, we maximized this rate, utilizing its defining function. For this purpose, we employed an optimization algorithm, which can approximate global minima/maxima of a function (i.e., simulated annealing—for details see the method section). The optimization algorithm can find the set of numeric values for the microscopic rate constants, which returns the largest value for the substrate uptake rate at given intra- and extracellular concentrations of Na+ and substrate.
We note that microscopic rate constants are a priori not mathematically constrained: They can assume values between zero and infinity. If they are permitted to vary across the entire mathematically possible range, the optimized substrate uptake rate will also adopt values between zero and infinity. It is a futile and meaningless exercise to maximize a function, for which it is known that no maximum exists. Fortunately, however, there are limits to the values of the microscopic rate constants. For instance, the association rates of co-substrates and substrate cannot be larger than the diffusion limit, which therefore imposes an upper limit on these rates. Likewise, the dissociation rates of substrate and co-substrate must also have an upper limit, because raising the dissociation rate constant results in affinity loss, which, when substantial, prevents the co-substrate and the substrate from interacting with the transporter in their physiological concentration ranges. It is also clear that a conformational change cannot occur with infinite velocity. We selected 1,000 s−1 as the upper limit for conformational transition rates. The choice of this value was based on information obtained from the literature (Zhang et al., 2007; Schicker et al., 2011; Hasenhuetl et al., 2018; Erdem et al., 2019). Another necessary constraint was to ensure that every set of optimized values complied with the rule of microscopic reversibility: The product of the rates in the forward direction in a loop must equal the product of the rates in the opposite direction. The combined constraints reshape the parameter space of the function, such that it harbors critical points, which do not exist in the unconstrained parameter space.
We performed ten optimization runs in which we assumed that Sout was.1 μM and 100 μM (Figure 1C). In all runs, we set the extra- and intracellular Na+ concentration to 150 mM and 10 mM and the intracellular substrate concentration (Sin) to zero. The data points show the substrate uptake rates, to which the optimization algorithm converged when Sout was set to.1 μM (left column) and 100 μM (right column). The rates were low at.1 μM Sout (7.44 s−1 ± .08 s−1) and high at 100 μM Sout (431.40 s−1 ± .66 s−1). We emphasize that each point in Figure 1C represents a unique set of numeric values for the microscopic rate constants. In Table 1, we show the ten sets, which we obtained from the optimization runs where Sout was set to.1 μM and 100 μM. Although the values of the microscopic rate constants differed between sets, they all gave essentially the same substrate uptake rate when optimized for the same substrate concentration. We therefore conclude that the optimized rate can be realized by different sets of numeric values for the microscopic rate constants. Because of the large differences in these values, each set can be viewed to define a transporter with an individual phenotype. For this reason, we will from here on treat the term “transporter” and “a set of optimized values” as a synonymous description.
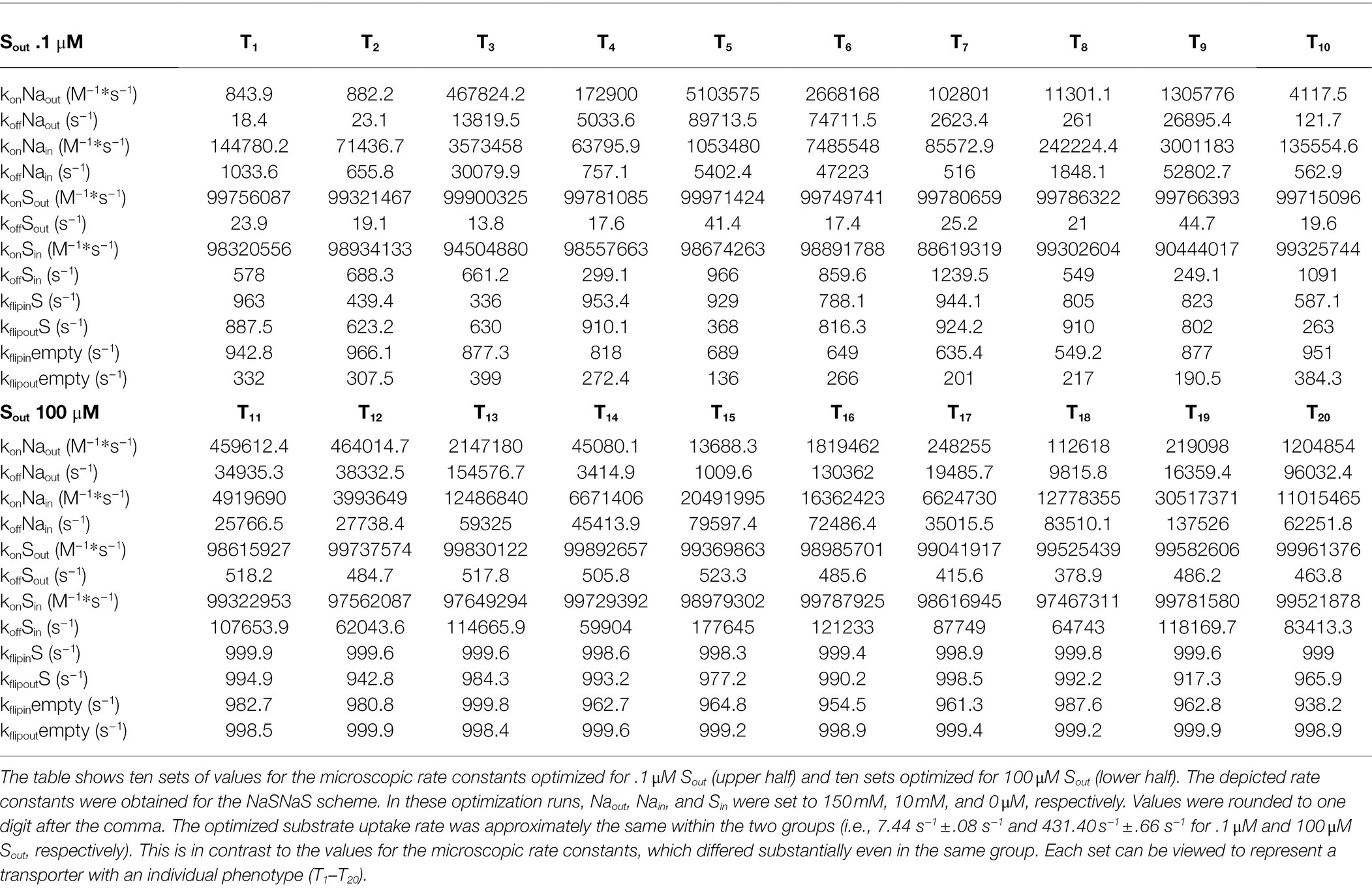
Table 1. Microscopic rate constants of optimized transporters.
The observations summarized in Table 1 warranted further scrutiny. They suggest that not all reactions, which a transporter undergoes, require the same extent of fine-tuning to support a high substrate uptake rate. We analyzed the transporters in Table 1 to understand, which reactions in the transport cycle do and do not require precise adjustment. Accordingly, we examined for each individual transporter, the values for a collection of descriptors of transporter function: (i) These included descriptors, which can be computed with the kinetic model but also obtained experimentally (i.e., Vmax and the KM for substrate/co-substrate) and (ii) descriptors, which can only be extracted from the kinetic model (i.e., KDs for substrate and co-substrate to the outward- and inward-facing conformation of the transporter). The rationale was as follows: If the values of a descriptor are all similar for transporters optimized for the same substrate concentration, we can conclude that fine-tuning of the reactions, which affect this descriptor, is essential for the realization of the optimized rate. Conversely, this is not the case, if the values are vastly different.
In Figure 1D, we plotted the substrate uptake rate as a function of Sout for the transporters in Table 1: The Vmax values were low and high for the group of transporters, which were optimized for.1 μM and 100 μM Sout, respectively. Within the two groups of transporters, the Vmax values varied, but the magnitude of this variation was small: Vmax of transporter optimized for.1 μM Sout and 100 μM Sout, were 78.40 ± 6.61 s−1 and 478.38 ± 1.58 s−1, respectively. In Figure 1E, the data were normalized to maximum velocity, because differences in the apparent affinity of the optimized transporters for the substrate can be more readily appreciated in this representation. It is evident that the transporters, which were optimized for.1 μM Sout displayed a higher apparent affinity for the substrate than those optimized for 100 μM Sout. The KM values within groups fell into narrow ranges, that is,.95 ± .08 μM and 11.1 ± .31 μM for transporters optimized for.1 μM and 100 μM Sout, respectively. Our analysis therefore indicates that transporters, must have low KM and low Vmax values to support a high substrate uptake rate when Sout is low, but a high KM and a high Vmax value when Sout is high.
We computed the KD values for substrate binding to the outward-facing conformation of the individual transporters (Figure 1F): It is evident that the KD values for the substrate were low and high for transporters, which were optimized for.1 μM and 100 μM Sout, respectively (.1 μM Sout:.24 μM ± .03 μM; 100 μM Sout: 4.72 μM ± .42 μM). It also evident that the KD values were smaller and that they covered a larger range than the corresponding KM values (Figure 1F).
Inspection of Table 1 shows that the rate constants, which govern Na+ binding to the outward-facing state, are subject to large variations in transporters 1–10; in contrast, these rate constants differed substantially less in transporters 11–20, which were optimized to cope with 100 μM Sout. We illustrated the resulting difference in apparent affinity for Na+ by plotting the absolute (Figure 1G) and normalized substrate uptake rate (Figure 1H) of the optimized transporters as function of the Na+ concentration: for those transporters, which were optimized for.1 μM Sout the apparent affinity for Na+ varied over five orders of magnitude (blue curves in Figures 1G,H). In contrast, for transporters optimized for 100 μM Sout, the apparent affinity for Na+ fell into a narrow range (magenta curve in Figures 1G,H). Thus, if the extracellular substrate concentration is low, the Na+ binding reaction does not need to be fine-tuned to obtain optimal rates. However, at a higher substrate concentration, the Na+ binding reaction is subject to stringent constraints. This observation can be rationalized by taking into account that, in the optimization, Na+ was assumed to be present at a high concentration (i.e., 150 mM). At this concentration Na+ binding is unlikely to become rate limiting for substrate transport if Sout is low. At a higher substrate concentration, the apparent association rate of the substrate (kapp) is expected to increase. In this scenario, Na+ binding becomes rate limiting, if it occurs at too low a rate.
We compared the KD values for Na+ binding to the outward-facing conformation of the transporter t the corresponding KM values (Figure 1I): Within each group, the variation in the KD values was small, that is, KD = 25.10 ± 3.91 mM and 79.21 ± 3.30 mM for transporters optimized for Sout.1 μM and Sout 100 μM, respectively. This contrasted with the large variation in the corresponding KM values for Na+ seen for transporters optimized for.1 μM Sout. This discrepancy can be explained as follows: The KD values are determined by the ratio of the dissociation and association rates for Na+ but not by the absolute values of these rates. Conversely, given that the KD values and the KM values for Na+ were found to differ, it is safe to conclude that KM values are highly dependent on the absolute values of these rates. These observations therefore imply that the affinity for Na+—rather than the velocity of Na+ binding to the transporter is subject to precise adjustment for supporting optimal uptake rates at low substrate concentrations.
Figures 1J,K summarize the KD values for binding of substrate and of Na+ to the inward-facing conformation of the transporter, respectively. The coefficients of variation in KD values of the inward-facing state were larger by a factor of 2 to 4 than the corresponding KD values of the outward-facing state: KDSin was 7.46 μM ± 3.4 μM and 701 μM ± 155 μM for transporters optimized for.1 μM Sout and for 100 μM Sout, respectively. Likewise, the was 8.81 mM ± 3.63 mM and 6.46 mM ± .70 mM for transporters optimized for.1 μM and 100 μM Sout, respectively. The high KDSin at 100 μM Sout was dictated by the low substrate affinity for the outward-facing state of transporters, which had been optimized for this condition, and the requirement to maintain microscopic reversibility. However, the larger variation in KDSin and indicates that the reactions, which define the substrate and co-substrate affinities for the inward-facing conformation, do not require as stringent an adjustment as those, which define the corresponding affinities to the outward-facing conformation. Finally, we surveyed transporter optimization over a large range of extracellular substrate concentration (1 nM to 10 mM; Figure 1L): The resulting optimized substrate uptake rate of the transporters increased as a function of Sout but leveled off at an uptake rate of about 500 s−1. This upper limit reflect the constraint imposed by the boundary conditions of the optimization (i.e., a diffusion-limited kon and an upper limit of 1,000 s−1 for the rate of conformational transitions, see above). It was 249.4, 499.4, and 997.8 s−1 when the upper limit of the rate of conformational transitions was set to 500, 1,000, and 2,000 s−1, respectively.
The reaction scheme in Figure 1A describes a hypothetical symporter, which binds co-substrate and substrate in a sequential order: The two Na+ ions are the first to bind when the transporter adopts the outward-facing conformation and the first to dissociate upon conversion of the transporter to the inward-facing conformation. In the subsequent description, we will refer to this reaction scheme as the NaSNaS scheme. This notation lists from left to right the order of the binding/unbinding events starting at the outward-facing apo-state (To) in clockwise direction. In Figures 2A–C, we show the reaction schemes for the three (sequential) alternatives. According to our notation, we refer to these as NaSSNa, SNaNaS and SNaSNa schemes. In addition, we also examined the reaction scheme of a transporter, in which the two Na+ ions and the substrate are allowed to bind in random order (Figure 2D).
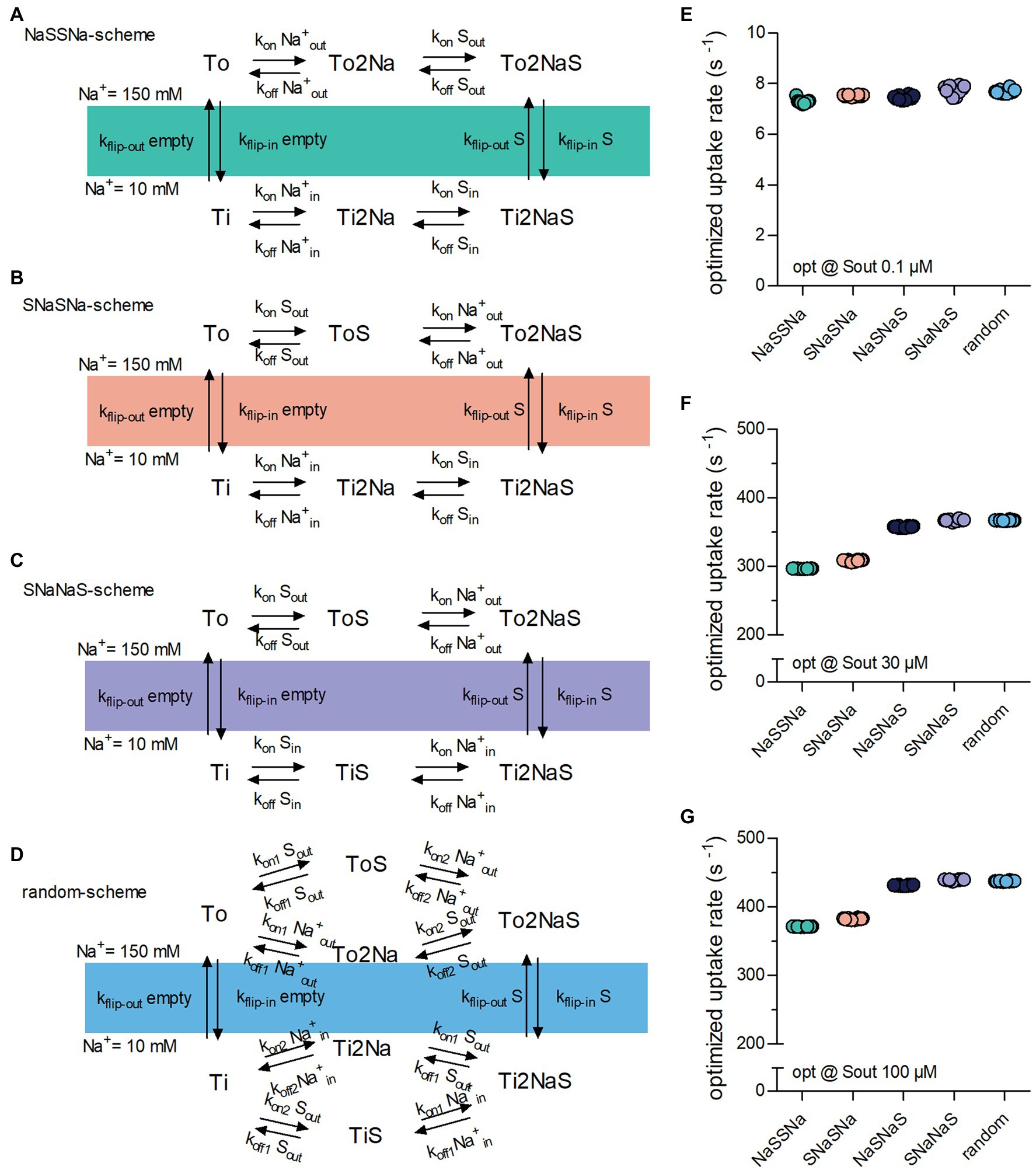
Figure 2. Binding order of co-substrate and substrate affects the optimized substrate uptake rate. (A) NaSSNa scheme (sequential). Na+ is the first to bind when the transporter adopts the outward-facing conformation and the last to dissociate from the inward-facing conformation. (B) SNaSNa scheme (sequential). Substrate is the first to bind to the outward-facing conformation and the first to dissociate from the inward-facing conformation. (C) SNaNaS scheme (sequential). Substrate is the first to bind to the outward-facing conformation and the last to dissociate from the inward-facing conformation (D). Random binding order scheme. Na+ and substrate bind in random order. (E) Plotted is the optimized substrate uptake rate for all schemes of transporter optimized for.1 μM Sout. At this low substrate concentration the optimized rate is approximately the same for all schemes. (F) Shown is the optimized substrate uptake rate for the different schemes optimized for 30 μM Sout. The height of the optimized rate differed between schemes (G) the same as in (F) but of transporter optimized for 100 μM Sout. The rank order of the optimized rates among schemes was the same as in (F).
We explored the impact of these five reaction schemes (Figures 1A, 2A–D) on the optimized substrate uptake rates by raising Sout from.1 (Figure 2E) to 30 (Figure 2F) and 100 μM (Figure 2G) and by conducting 10 optimization runs for each reaction scheme. As evident from Figure 2E, the substrate uptake rate was roughly the same for all schemes, when Sout was low (i.e.,.1 μM). However, when optimized for a higher Sout, the schemes differed in the magnitude of the optimized substrate uptake rates, which they were able to support. The rank order was as follows: NaSSNa < SNaSNa < SNaSNa < SNaNaS = random. The rank order was the same for transporters optimized for 30 μM and 100 μM Sout, (cf. Figures 2F,G). Thus, random order of substrate and co-substrate binding and SNaNaS are best suited to support a large substrate uptake rate.
Due to the way SLC operate, the intracellular concentration of the substrate and the substrate uptake rate are inversely correlated. This can be explained as follows: The substrate must be released into the cytosol to complete a full cycle. As the intracellular substrate concentration (Sin) increases progressively during uptake, rebinding of the substrate to the inward-facing conformation occurs at a more frequent rate. This hampers progression through the transport cycle and thus reduces the substrate uptake rate.
Here we propose that—similar to the extracellular substrate concentration (Sout)—Sin can also exert an evolutionary pressure on the operation of a solute carrier. Depending on the physiological context, transporters may encounter intracellular concentrations of their cognate substrate, which range from low to high levels. This can be illustrated by two examples in the SLC6 family: The cytosolic concentrations of the monoamines dopamine, norepinephrine, and dopamine are expected to be low. This is because of the presence of vesicular monoamine transporters (vMAT1/SLC18A1 & vMAT2/SLC18A2), which shuffle cytosolic monoamines into vesicles (Yaffe et al., 2018). Accordingly, under physiological conditions, monoamine transporters for dopamine (DAT/SLC6A3), norepinephrine (NET/SLC6A2) and (SERT/SLC6A4) are unlikely to encounter high intracellular concentrations of their substrate. Conversely, the creatine transporter-1 (SLC6A8) must maintain substrate influx in the presence of millimolar intracellular creatine (Snow and Murphy, 2001). It is clear that a solute carrier, which must support a high substrate uptake rate against high Sin, must adjust its operation differently than a transporter, which does not need to overcome the hurdle imposed by frequent rebinding of the substrate to the inward-facing conformation.
We first optimized the substrate uptake rate of a transporter operating according to the NaSNaS scheme (illustrated in Figure 1A) by performing 20 optimization runs each, where Sout was 30 μM and Sin was set at 0,.1, 1, and 5 mM. It is evident from Figure 3A that the optimized substrate uptake rate decreased by raising Sin. This is in line with the inverse correlation of Sin and the substrate uptake rate discussed above. Figure 3B illustrates the range of KD values for substrate binding to the inward-facing conformation of the transporters, which had been optimized to cope with different concentrations of Sin: KD values of substrate for the inward-facing conformation increased as Sin was raised. This was to be expected, because lowering the intracellular affinity for substrate reduces the extent by which Sin can rebind, and it thus allows for a higher substrate uptake rate in the presence of Sin.
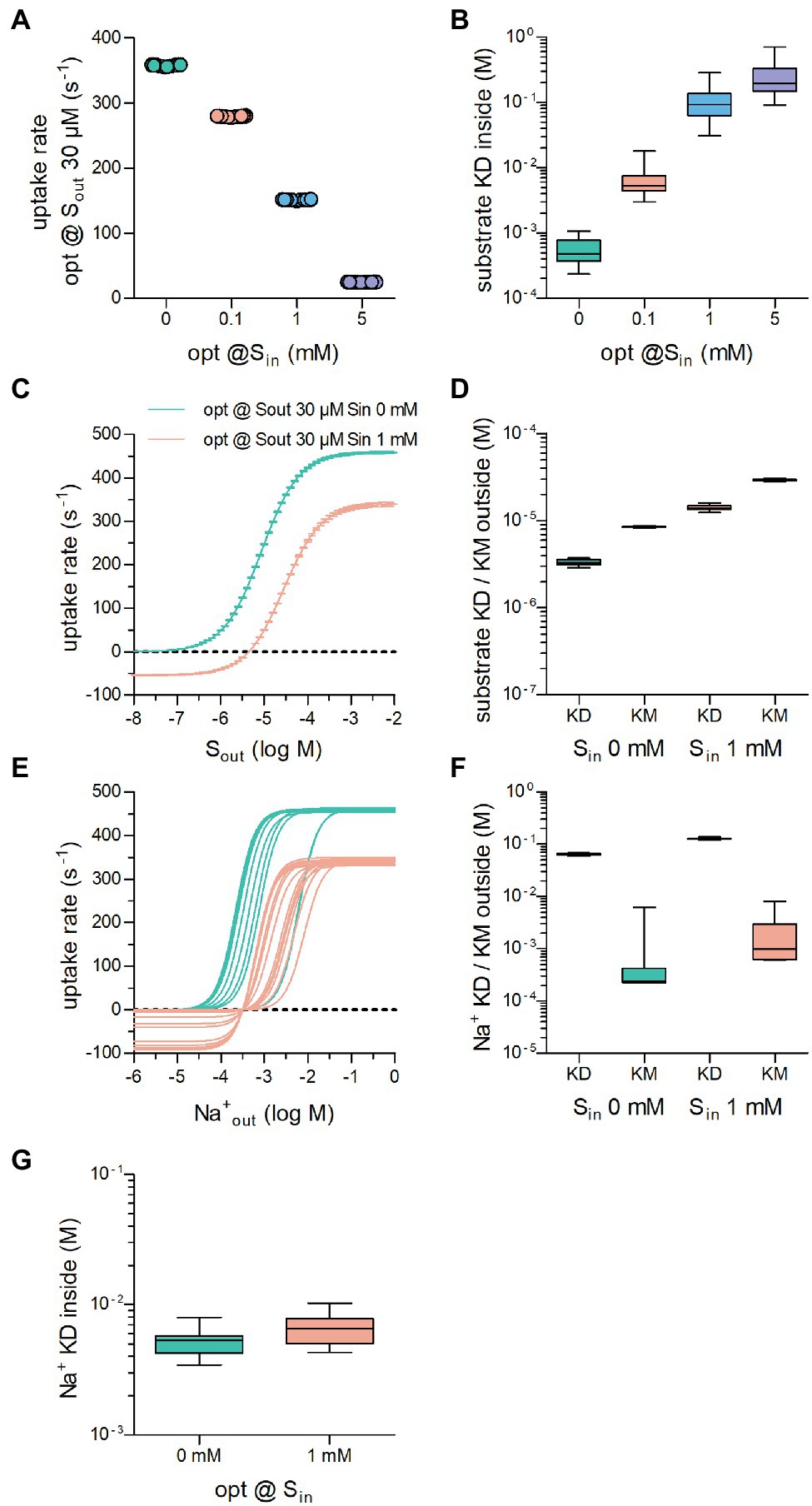
Figure 3. The optimized substrate uptake rate depends on the intracellular substrate concentration (Sin). (A) Plotted are the optimized substrate uptake rates for transporters operating according to the NaSNaS scheme. Sout was set to 30 μM in all optimization runs. The data show the substrate uptake rate of transporters optimized for 0 mM,.1 mM, 1 mM, and 5 mM Sin, respectively. The rate decreased with rising Sin. Twenty optimization runs were carried out for each condition. (B) Shown is the KD of substrate binding to the inward-facing conformation of transporters in (A). The KD decreased as Sin increased. (C) The curves show the substrate uptake rate as a function of Sout of transporters optimized for 0 mM (green) and 1 mM Sin (magenta). In the presence of Sin, the substrate uptake rate assumed negative values when Sout was low. In this range of Sout, the transporters cycled in the reverse direction. In the presence of 1 mM Sin, Vmax was reduced. (D) Plotted are the KD values for substrate binding to the outward-facing conformation of the transporter and the corresponding KM values of transporters optimized for 0 mM Sin and for 1 mM Sin. At high Sin both the KM and the KD values rose. The coefficients of variation for the KDs were.080 and.070 and for the KMs.020 and.026 at.1 μM and 100 μM Sout, respectively. (E) Shown is the concentration dependence of the substrate uptake rate for of transporters optimized for 0 mM Sin and 1 mM Sin. The dependence on the Na+ concentration was highly variable between transporters optimized for the same Sin. (F) Plotted are the KD values for Na+ binding to the outward-facing conformation of the transporter and the corresponding KM values of transporters optimized for 0 mM Sin and for 1 mM Sin. The variation in the KM values was larger than the variation in the KD values. The coefficients of variation for the KDs were.041 and.033 and for the KMs 2.03 and.97 at.1 μM and 100 μM Sout, respectively. At high Sin both the KM and the KD values for Na+ increased. (G) KDs for Na+ for the inward facing conformation of the transporters optimized for 0 mM and 1 mM Sin, respectively.
We then compared the uptake rate of transporters optimized for 30 μM Sout and 0 mM or 1 mM Sin over a large range of extracellular substrate concentration. As can be seen from Figure 3C, it was inevitable that transporters optimized in the presence of 1 mM Sin had negative uptake rates at low extracellular substrate concentrations, that is, the transporters cycled in the backward rather than the forward mode and hence mediated substrate efflux from the cell. It is also clear that the presence of 1 mM Sin reduced the maximum achievable uptake rate Vmax in the forward transport mode and shifted the KM. Because KM and KD differ (cf. Figure 1F), we examined the range of KD values for substrate binding to the outward-facing conformation of transporters optimized for 0 and 1 mM Sin; these are illustrated together with the corresponding KM values in Figure 3D: Both the KD values and the KM values for substrate increased, if the transporter had to cope with a high intracellular substrate concentration. The variation in these parameters was low (coefficient of variations = .070 and.025 for KD and KM, respectively, of transporters optimized in the presence of 30 μM Sout and 1 mM Sin). We conclude that the decrease in the apparent (KM) and the true affinity (KD) for the substrate, is required to allow for rapid cycling of the transporters in the presence of high Sin.
Finally, we examined how the selective pressure exerted by high intracellular substrate affected the affinity of the transporters to the co-substrate ion. As can be seen from Figure 3E, many different solutions emerged: On average, transporters optimized to cope with 1 mM Sin (magenta lines in Figure 3E) required higher extracellular Na+ concentrations to support substrate uptake than those optimized in the absence of intracellular substrate (green lines in Figure 3E). However, in both groups, the optimized transporters displayed highly variable responses to . This is reflected in the large range of the KM values for Na+ (Figure 3F). In contrast, the KD of Na+ for binding the outward-facing conformation did not vary to any substantial extent. This observation is consistent with our conclusion from Figure 1I, namely, that the constraint is imposed by the true affinity for Na+ rather than by its association rate (see above). We also computed KDs for Na+ binding to the inward-facing conformation of the transporters optimized for 0 mM and 1 mM Sin: There was an overlap in the range of KD values for (Figure 3G). In addition, their variation was larger than that of KD values for (cf. Figures 3F,G). Hence, we conclude that Na+ binding to the inward-facing conformation need not be stringently adjusted to allow for high substrate uptake rates.
We next addressed the question, if the binding order of substrate and co-substrate determined the cycle rate of a transporter challenged with high concentrations of intracellular substrate. Optimization runs were carried out with Sout = 30 μM and Sin = 1 mM for all five schemes (cf. Figures 1A, 2A). It is evident from Figure 4A that these schemes differed in the magnitude of the optimized substrate uptake rates, which they were able to support. The rank order was NaSSNa < NaSNaS < SNaSNa < SNaNaS = random. This rank order differed from that observed in the absence of Sin (cf. Figure 2F). At constant Sout = 30 μM, we also varied the internal substrate concentration by lowering to Sin 100 μM and raising it to the point, where net uptake rat was zero (Figure 4B). For all schemes we found an optimized substrate uptake rate of zero when Sin was 6.75 mM. This was to be expected because at the chosen concentrations of Na+ (i.e., 150 mM and 10 mM ) the concentrative power (Sin/Sout) of the transporter is 225. This numerical value is identical for all schemes, because they are governed by the same transport stoichiometry. Accordingly, the transporters cannot further cycle productively in a forward direction, when Sin becomes 225 times larger than Sout (30 μM * 225 = 6.75 mM). For all schemes, we extracted the KD of substrate binding to the inward-facing conformation of the transporters optimized at varying Sin (Figure 4C). In all instances, this KD increased with increasing Sin. However, the magnitude of the drop in affinity depended on the reaction scheme.
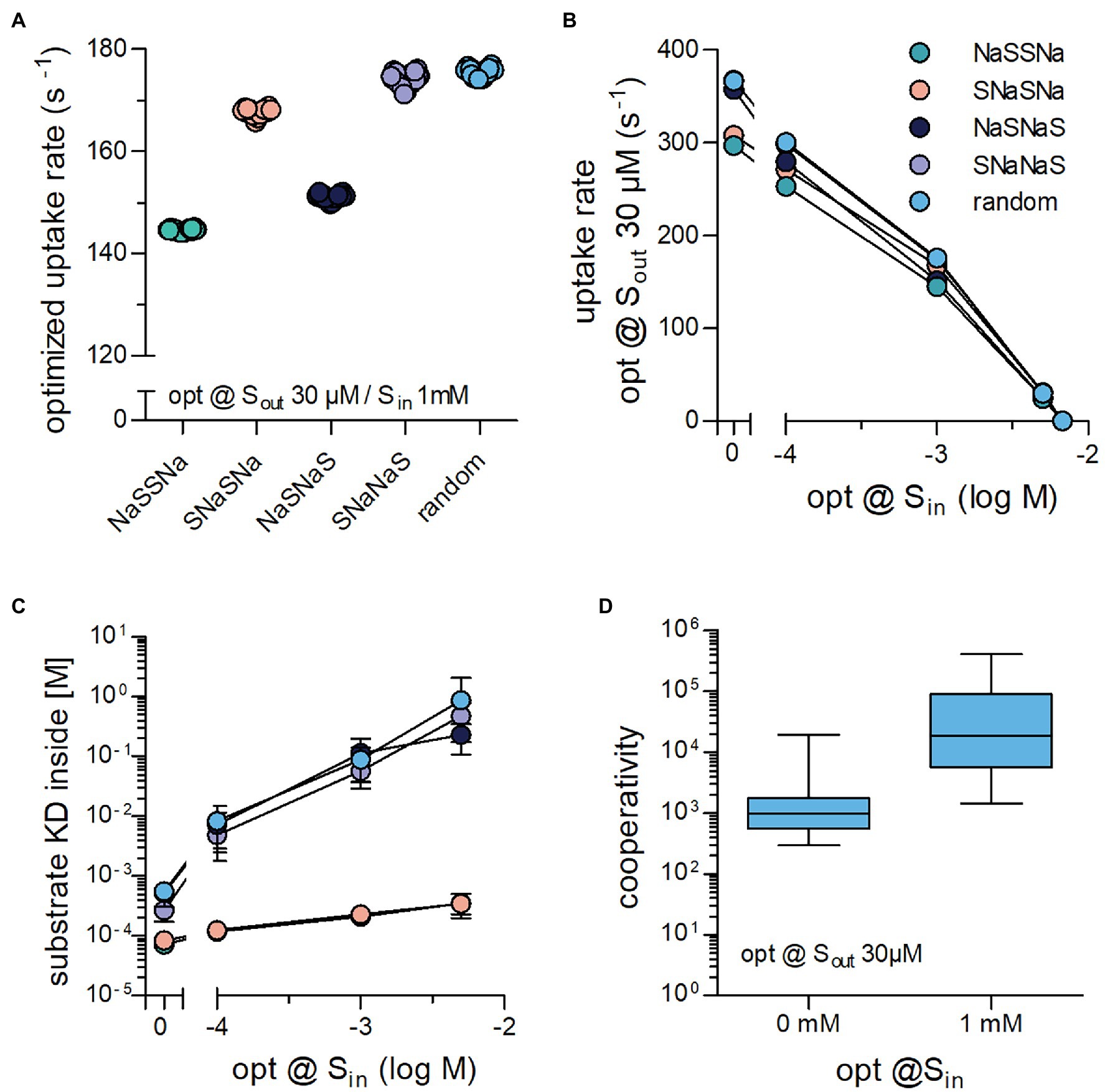
Figure 4. Binding order of co-substrate and substrate affects the optimized substrate uptake rate, when Sin is high. (A) Shown are the substrate uptake rates for transporters, which operate according to the reaction schemes illustrated in Figures 1A, 2A–D and which were optimized for 30 μM Sout and 1 mM Sin. The optimized substrate uptake rate differed between reaction schemes. The data are the rates obtained from 20 optimization runs. (B) Plotted are the substrate uptake rates for all reaction schemes optimized for 0 mM,.1 mM, 1 mM, 5 mM, and 6.75 mM Sin, respectively. The data are means ± SD of the substrate uptake rates obtained from 20 optimization runs. At 6.75 mM Sin, the substrate uptake rate was zero for all reaction schemes. This is due to the fact that the concentrative power is defined by the transport stoichiometry (Na+: substrate = 2:1) and the Na+ gradient rather than the reaction scheme: With a gradient of 150 mM external to 10 mM internal Na+, the concentrative power of the transporters is 225, which yields 6.75 mM Si at 30 μM Sout (C) Shown are KD values for the substrate binding to the inward-facing conformation for all reaction schemes of transporters optimized to 0 mM,.1 mM, 1 mM, and 5 mM Sin, respectively. While the absolute values of this KD differed between schemes, they all rose with increasing Sin. (D) The random binding order scheme, allows for Na+ and substrate to bind in a cooperative manner: The affinity for substrate is high when Na+ is bound and low in its absence. Cooperativity was defined as the ratio of the KD of substrate binding to the inward-facing conformation in the absence and presence of Na+. Transporters optimized for 1 mM Sin displayed larger cooperativity values than those optimized for 0 mM Sin (p < .0001; Wilcoxon signed rank test).
For several secondary active transporters, binding of substrate and co-substrate was shown to occur in a cooperative manner: The apparent substrate affinity for the transporter depended on the concentration of the co-substrate (Meinild and Forster, 2012; Perez et al., 2014; Hasenhuetl et al., 2018; Erdem et al., 2019). It was low and high when the concentration of the co-substrate was low and high, respectively. Thus, the substrate can bind with higher affinity to transporters, when they are bound to the co-substrate (e.g., Na+). In this way, the concentration of the co-substrate determines the abundance of high and low affinity states for the substrate. Under physiological conditions, the co-substrate concentration is lower on the intracellular than on the extracellular side. Accordingly, cooperative binding is predicted to promote the forward cycling mode by reducing the substrate affinity to the inward-facing conformation. In fact, the drop in intracellular affinity resulting from cooperative binding is a requirement for maintaining a large substrate uptake rate at high Sin (Erdem et al., 2019). Notably, cooperative binding is contingent on a random binding order for substrate and co-substrate. For this reason cooperative binding can only be assessed in the random binding order scheme. Based on this consideration, a rise in Sin is predicted to increase the extent of cooperativity. We verified this prediction in optimization runs and extracted the cooperativity for transporters optimized at 30 μM Sout and 0 mM Sin or 1 mM Sin by calculating the ratio KDSin in the absence of bound Na+/KDSin in the presence of bound Na+. It is evident from Figure 4D that there is a large range of optimized solutions, but on average cooperativity was more pronounced at higher Sin.
Evolution also optimized SLCs for substrate specificity. Selective transporters presumably arose from unselective ancestors. As a starting point, we posited that an unselective SLC must display low affinity for the various substrates: It is difficult to envisage a substrate binding site, which can provide strong bonding interactions to accommodate many distinct molecular scaffolds. We also assumed that, in the evolutionary trajectory from an unselective to a specific transporter, an increase in substrate specificity ought to translate in higher uptake rates for the substrate. Accordingly, in the optimization, we modeled an unselective solute carrier as a transporter, which had a low (true) affinity (i.e., a high KD) for substrate by implementing a constraint, which prevented the substrate KD from dropping below a user-defined arbitrary value (e.g., 10 mM). With this constraint in place, the optimization algorithm only returned sets of values for the microscopic rate constants, which defined transporters with a high KD for substrate. In the subsequent description, we refer to such sets as unselective transporters. In contrast, sets generated in optimization runs, in which the KD for substrate was not constrained, are referred to as selective transporters. For the optimizations summarized in Figure 5, we employed the random binding order scheme, we assumed zero-trans conditions and the presence of 1 μM Sout. Figure 5A shows the result for 20 selective and unselective transporters (KD ≥ 10 mM): It is evident that the optimized substrate uptake rat of the unselective transporters (magenta symbols, Figure 5A) was lower by about three orders of magnitude than that of the selective SLCs (green symbols, Figure 5A). In Figure 5B, we examined the Michaelis–Menten kinetics of the substrate uptake rate of these optimized transporters. Figure 5C shows the same data normalized to Vmax to illustrate the distribution of KM. The Vmax values of the unselective transporters were lower than those of the selective SLCs but they varied over about orders of magnitude (Figure 5D). Similarly, the KM values of the unselective transporters, which were consistently higher than those of the selective SLCs, were again highly variable. In Figure 5E, we show optimized rates as a function of the concentration of substrate, for which the rates were optimized. Displayed in this plot are the data for selective transporters (unconstrained substrate KD) and unselective transporters (constrained at KD > .1 mM, 1 mM, and 10 mM). It is evident that at the various [Sout] tested the selective transporters had larger substrate uptake rates than the unselective ones.
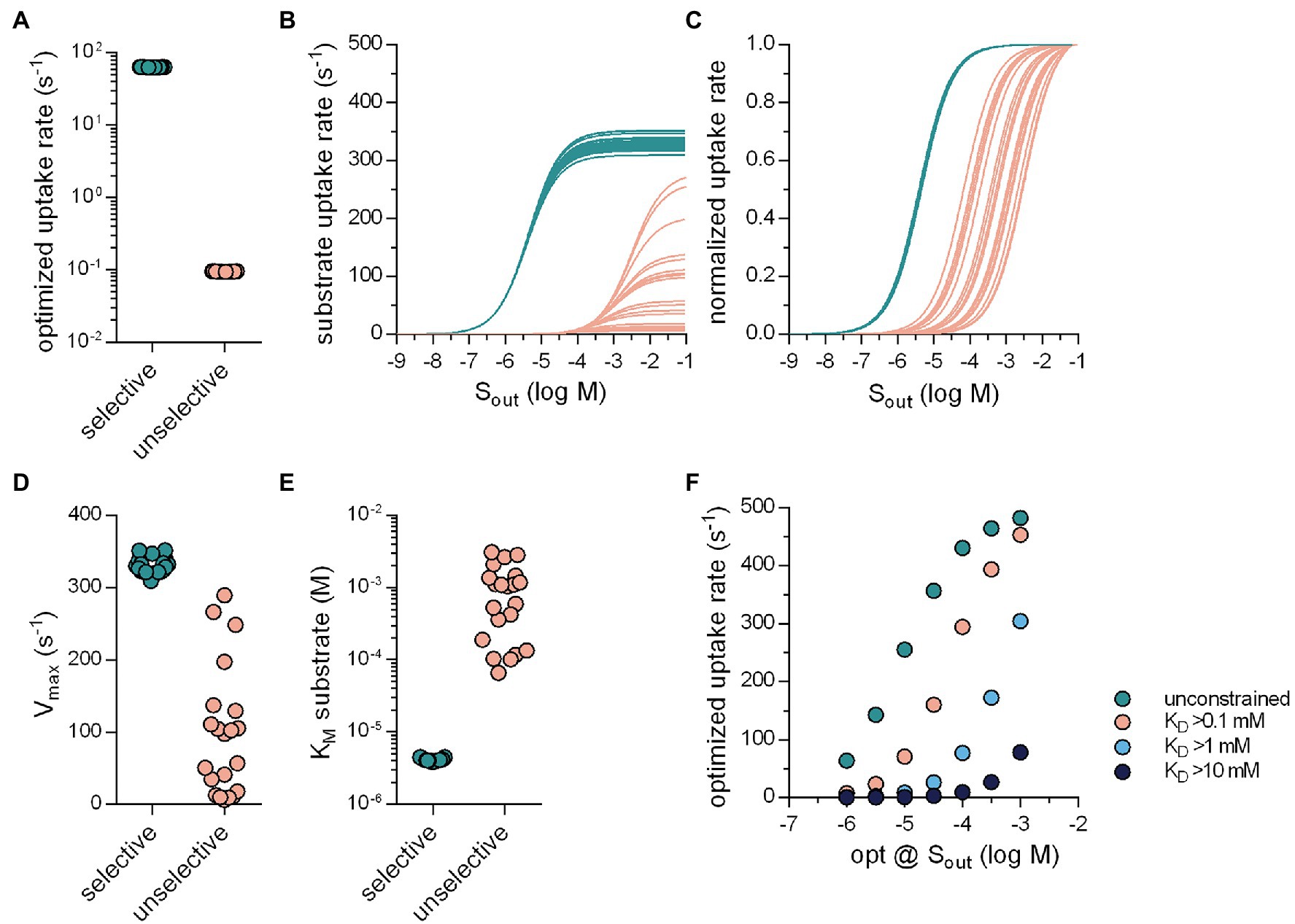
Figure 5. Selective transporters can support larger substrate uptake rates. (A) Optimized substrate uptake rates for selective (green circles; 63.79 ± .19 s−1; n = 20) and unselective transporters (pink circles;.095 ± .0006 s−1; n = 20). The transporters were optimized for 1 μM Sout. The substrate KD of the unselective transporters was not allowed to drop below 10 mM. For the optimizations we used the random binding order scheme. (B) Substrate uptake rate as a function of Sout for the selective (green lines) and the unselective transporters (pink lines). (C) The same data as in (B) but normalized (D) Vmax values of selective and unselective transporters. The coefficient of variation of the Vmax values was.034 and.91 for selective and unselective transporters, respectively (E) KM values of selective and unselective transporters. The coefficient of variation of the KM values was.042 and.90 for the selective and unselective transporters, respectively. (F) The substrate uptake rate as a function of the concentrations of Sout for which these rates were optimized. The green circles are the optimized rates of the selective transporters. The circles in pink, light blue, and dark blue are the optimized rates of unselective transporters, for which the substrate KD was not allowed to drop below.1 mM, 1 mM, and 10 mM, respectively (n = 10 for each Sout). The optimized rates of the unselective transporters were lower than that of the selective ones.
These results confirm that SLCs can raise their transport capacity by becoming more specific for their cognate substrates. We, therefore, consider it plausible that specific SLCs arose from ancestors, which were unselective and that this transformation was driven by the need to support high substrate uptake rates. Conversely, there are transporters, which are under evolutionary pressure to remain unselective, because they support the disposition of xenobiotics. This is exemplified by members of the SLC22 family, which recognize diverse substrates to mediate disposition of drugs and xenobiotics: Both organic cation (OCT1-3/SLC22A1-3) and anion transporters (OAT1-3/SLC22A6-8) translocate most of their substrates with KM values in the high μM range (VanWert et al., 2010; Motohashi and Inui, 2013). This is despite the fact that, in most instances, they are faced with substrate concentrations in the low micromolar range. However, we find this in good agreement with our results, which showed that the unselective transporters that we optimized for 1 μM Sout, displayed KMs in the submillimolar range (see Figure 5E). It is worth noting that the SLC22 family also encompasses members, which have a narrow substrate specificity; these have KM value in the low micromolar range.
Solute carriers have a long evolutionary history: More than 50% of SLC subfamilies, which are present in the human genome, are also found in prokaryotes (Hoglund et al., 2011). Eukaryotic transporters have longer N- and C-termini than bacterial transporters. This presumably reflects evolutionary adaptation to the increase in complexity: The N- and C-termini harbor site for posttranslational modifications (e.g., phosphorylation by protein kinases) and docking sites for the protein machinery required for trafficking between cellular compartments (Chiba et al., 2014). The evolutionary history also suggests that individual SLC subfamilies expanded and contracted during phylogenesis (Caveney et al., 2006; Denecke et al., 2020). Expansion was not only driven by the adaptation to new substrates but also by the requirements to optimize concentrative power and uptake rate: Phosphate transporters of the SLC34 subfamily differ in their concentrative power and in their electrogenicity (Forster et al., 2013). Similarly, the three closely related monoamine transporters provide different solution to the trade-off between harvesting the membrane potential and maintaining constant uptake at variable voltage (Bhat et al., 2021). Here, we explored how the concentration of substrate, which a solute carrier encounters on both, the extra- and intracellular side can exert evolutionary pressure on the operating mode of a transporter. Our approach relied on analytical expressions for descriptors of transporter function (i.e., KM and Vmax of substrate transport) as a function of the microscopic rate constants, which parameterize the kinetic models of SLC. Arguably, the outcome of evolutionary adaptation must maximize substrate uptake rate at the prevailing conditions. Accordingly, our optimization algorithm searched for the microscopic rate constants, which yielded the largest possible value for the substrate uptake rate. The pertinent insights can be summarized as follows: (i) low extracellular substrate concentrations select for transporters, which have low KM and Vmax. Only this combination allows for a high rate in the transport cycle, but there is a surprisingly broad range of microscopic rate constants, which support this solution. (ii) In contrast, a transporter operating at high extracellular substrate concentrations has a substantially more restricted parameter space and maintains a high uptake rate only if it has a high KM and a high Vmax for substrate. (iii) Random order of substrate and co-substrate binding is superior to all possible sequential orders, if a transporter is to maintain a high rate of substrate uptake in the presence of accumulating intracellular substrate, because it allows for cooperative binding.
Solute carriers have long been known to fall into two categories, that is, high-affinity–low-capacity transporters and low affinity–high capacity transporters. It is important to note, however, that there is not any relation between the KM value and the Vmax value, which a priori dictates that these two parameters must move in the same direction. We examined the relation between turnover rates and KM in the SLC6 family, because turnover rates have been determined with high precision by electrophysiological recordings (Bicho and Grewer, 2005; Erdem et al., 2019; Bhat et al., 2021; Shi et al., 2021) and the individual steps of the transport cycle have been analyzed in detail. In addition, the KM values for cognate substrate span more than two orders of magnitude. It is evident from Figure 6A that there is a good correlation (r2 = .915) between turnover rate and KM. Similarly, the monoamine transporter of the earthworm Lumbricus terrestris can translocate several substrates albeit with substantial differences in Vmax and KM: The KM for norepinephrine is 20-fold higher than for tyramine (Caveney et al., 2006). Again, there is a remarkable correlation (r2 = .981) between uptake velocity and KM (Figure 6B). It is, therefore, safe to conclude that the existing dichotomy—in the real world and in our data sets—is a consequence of the optimization of the substrate uptake rates.
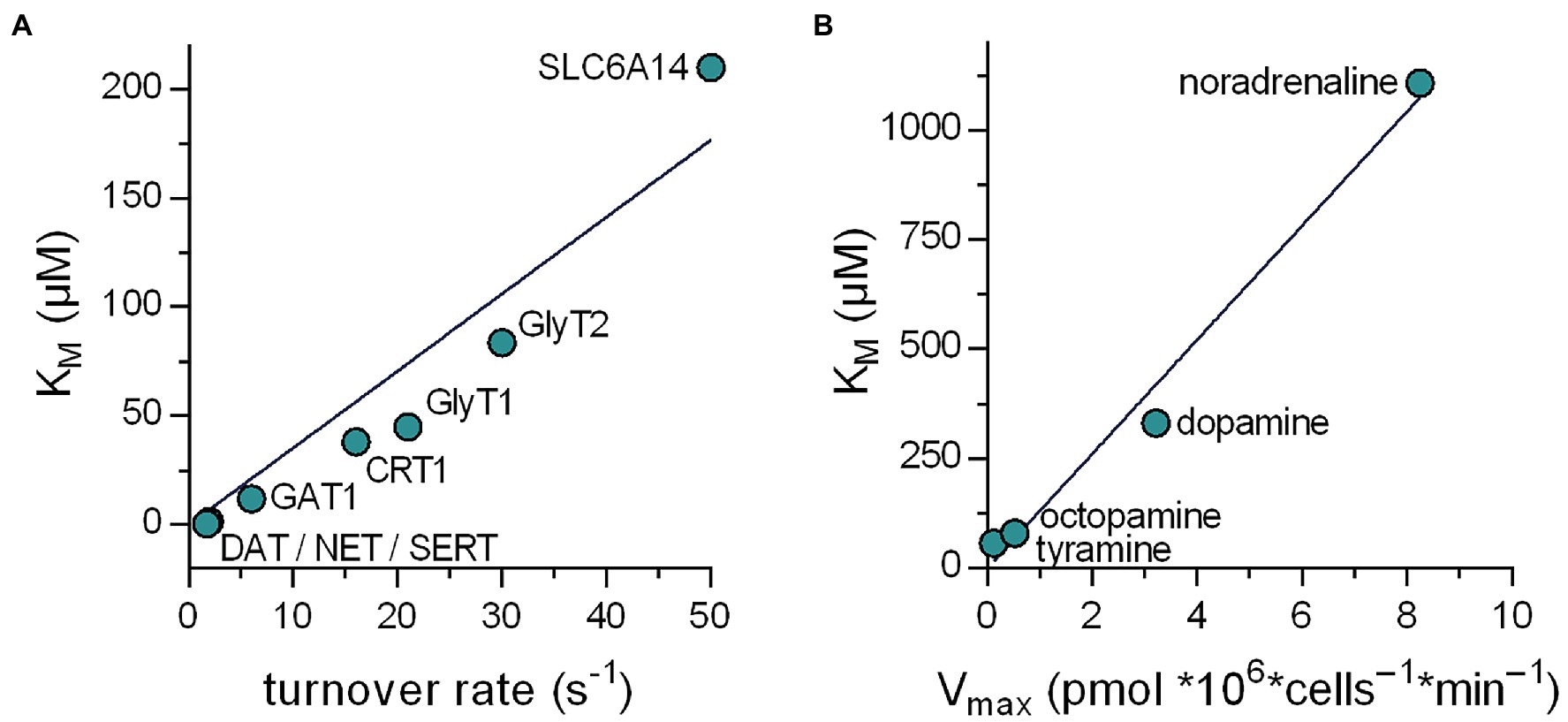
Figure 6. Relation between the KM and the substrate turnover rate of transporters in the SLC6 family. (A) Plotted are data obtained from literature of eight members of the SLC6 family. The line in the graph is a linear fit to the data points (r2 = .915). It is evident that in the real world a positive correlation exists between the KM and the turnover rate. (B) Relation between KM and Vmax for the various substrates of the Lumbricus terrestris monoamine transporter. The data were taken from Caveney et al. (2006). The line in the graph is a linear fit to the data points (r2 = .981).
Similarly, our optimization algorithm required boundary conditions to identify realistic maxima. Table 2 provides a compilation of substrate turnover rates reported for a collection of SLC. The list includes carriers, which cycle at a rate of 3 s−1 as well as such that cycle at a rate of about 700 s−1. These rates are reasonably close to those, which the optimization algorithm returned, that is, about 500 s−1 and 4 s−1 for Sout 100 μM and.1 μM, respectively. This confirms that the constraints, which we imposed in the optimization, were realistic. Importantly, our analysis establishes a relation between the turnover rate of the transporter and the substrate concentration for which it was optimized. Thus, the substrate turnover rate allows for inferring the concentration range, in which a candidate solute carrier operates under physiological conditions.
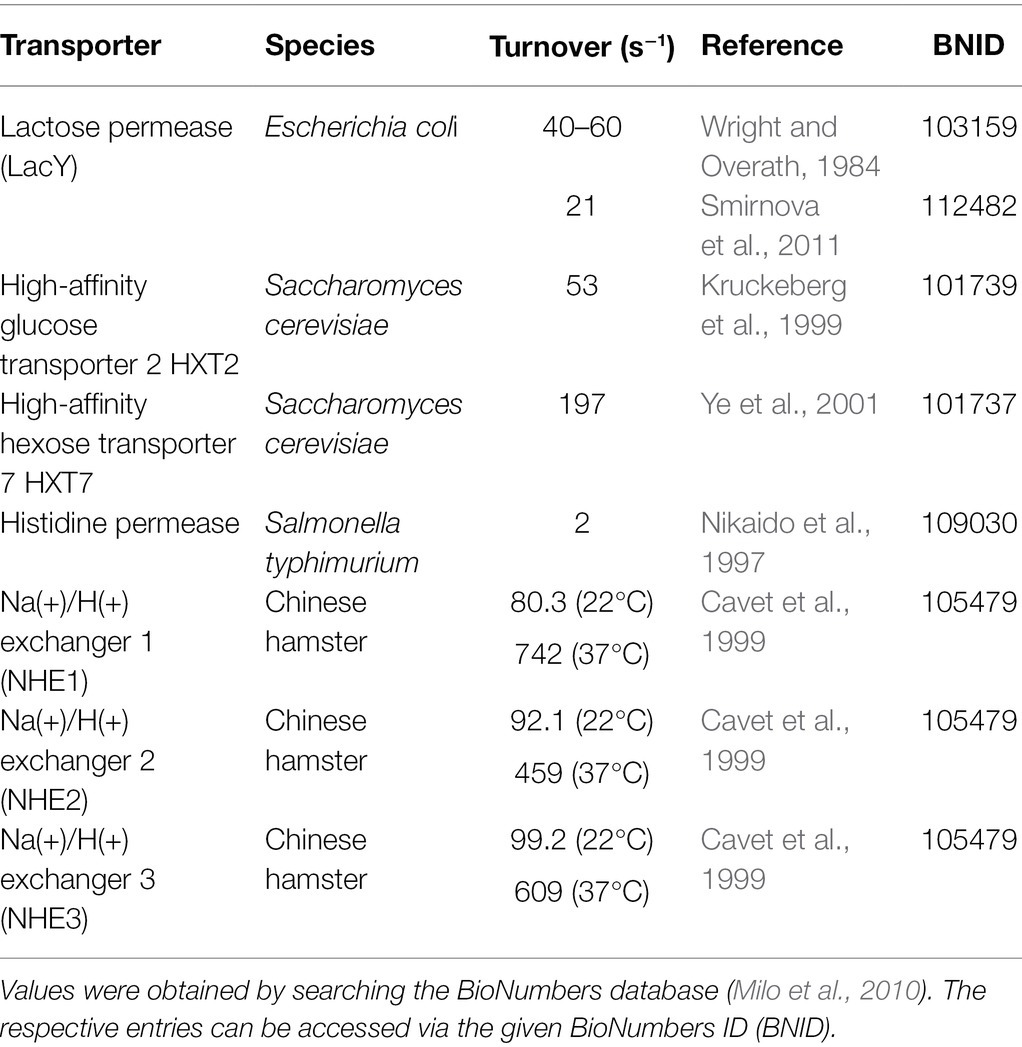
Table 2. Substrate turnover rates of transporters from various species.
We subjected transporters to a selection by optimization at low substrate concentration but at high concentrations of Na+ co-substrate Nevertheless, some of the solutions produced hypothetical transporters, which were optimized to operate at very low Na+ concentrations (cf. Figures 1G,H). While these are obviously of little benefit to a multicellular organism with homeostatic control of extracellular ion composition, these transporters are optimally adapted to support nutrient uptake of a unicellular organism invading ecological niches with low ambient salt concentrations. Bacterial transport is thought to rely mainly on the proton motive force. However, there are several examples of bacterial Na+-dependent symporters (Wilson and Ding, 2001). In addition, our analysis showed that KM values for substrate and co-substrate differed substantially from the KD values for their binding to the outward-facing conformation (cf. Figures 1F,I). This highlights the fact that experimentally determined KM values for substrate and co-substrate are not necessarily adequate measures of true affinity (i.e., koff/kon). Transporters are forced to adjust ratios of binding and unbinding reactions, which keep the optimum KD values in a narrow range at both, low and high substrate concentrations. In contrast, the initiation of the transport cycle is limited by the apparent on rates of substrate and the co-substrate ion(s). It is worth noting that all solutions resulted in transporters, which bound substrate at a rate close to the diffusion-imposed limit (cf. Table 1). As a consequence, this allows for the emergence of high-affinity/low-capacity SLC, which translocate substrate effectively at very low co-substrate concentrations, because large variations in kon for Na+ are tolerated.
Substrate accumulation on the intracellular side also exerts a selective pressure. It is low, if the transporter operates in a relay with another transporter, which sequesters the substrate, or with an enzyme, which modifies the substrate: The monoamine transporters for dopamine (DAT/SLC6A3), norepinephrine (NET/SLC6A2), and (SERT/SLC6A4) need not cope with rising intracellular level of their cognate substrate because of the action of vesicular monoamine transporters (vMAT1/SLC18A1 & vMAT2/SLC18A2) which shuffle cytosolic monoamines into vesicles (Hou and Matherly, 2014; Sitte and Freissmuth, 2015). Similarly, the reduced folate carrier (RFC/SLC19A1) and the proton-coupled folate transporter (PCFT/SLC46A4) are unlikely to face inhibition by accumulation of intracellular folate, because it is converted to polyglutamylated folate by folylpolyglutamate synthase (Raz et al., 2016). In fact, it has been argued that, in several instances, transporters and metabolizing enzymes are spatially organized to promote sustained influx of substrate: Direct or indirect tethering of enzymes to SLCs creates membrane transport metabolons, which effectively lower [Sin] to preclude inhibition on the intracellular side (Moraes and Reithmeier, 2012). In contrast, the creatine transporter-1 (CrT-1/SLC6A8) must maintain the forward transporter mode, although intracellular concentrations are in the range of 5–7 mM (Wyss and Kaddurah-Daouk, 2000). Our analysis allows for understanding how the adaptation to high intracellular substrate is achieved. It is obvious that the transporter can only progress in the forward cycle mode, if the inward-facing conformation of the transporter has a low affinity for the substrate, because a low affinity precludes rebinding of the substrate. However, to afford a lower substrate affinity to the inward-facing conformation, the KM for substrate must increase and, as a corollary, the true affinities of the substrate and the Na+ ions for the outward-facing must decrease. In this context, it is worth mentioning that the three monoamine transporter (NET/SLC6A2, DAT/SLC6A3, and SERT/SLC6A4) display high KM for substrate and Na+, that is, in the low micromolar and millimolar range, respectively, (Bulling et al., 2012; Li et al., 2017; Bhat et al., 2021). In contrast, the glycine transporter-1 (GlyT1/SLC6A9) and CrT1/SLC6A8 feature about 3–20-fold lower apparent affinities for their cognate substrate and co-substrate Na+ (Boehm et al., 2003; Erdem et al., 2019). This is consistent with the fact that these transporters support influx of their cognate substrates in the presence of millimolar concentrations of Sin, while the monoamine transporters do not. Thus, the solutions, which were explored by our optimization algorithm, reflect parameter space visited during the evolutionary adaptation of transporters.
Our study has several limitations: (i) We restricted our analysis to sodium symporters with a 2 Na+ and 1 substrate stoichiometry. It is evident, however, that the approach can be extended to any other stoichiometry of symport. Our approach is also applicable to antiporters although incorporating the kon and koff of the counter-transported solute substantially increases the parameter space and thus the computational effort. (ii) We did not examine the impact of allosteric regulation, which is a major computational challenge. It is clear, however, that evolution also selected for transporters for allostery: DAT/SLC6A3, for instance, harbors an allosteric Zn2+ binding site, while its next relatives NET/SLC6A2 and SERT/SLC6A4 do not (Li et al., 2017). (iii) While modeling the evolutionary trajectory to substrate specificity, we assumed that an ancestral unselective transporter gave rise to a specific transporter. This need not be the case. In fact, two rounds of genome duplication events are thought to have occurred after the split of vertebrates from cephalochordates (Wolfe, 2001; Simakov et al., 2020). Duplicated transporters have two possible evolutionary fates: They can be subject to inactivating mutations and lost. Alternatively, they can accumulate mutations, which initially relax their substrate specificity and eventually evolve by further mutations into transporters with novel substrate specificity. We also did not explore the structural basis for evolution in substrate affinity. It is worth noting that solute carries can achieve the substrate translocation step by three different mechanisms, that is, based on a rocker switch, a rocking bundle and a sliding elevator (Diallinas, 2021). In transporters, which operate via a sliding elevator mechanism, the substrate binding site must reside in the elevator, which moves along the scaffold domain in a direction perpendicular to the membrane plane and thus translocates the substrate. Surprisingly, in the uric acid-xanthine permease UapA of Aspergillus nidulans, which operates via a sliding elevator mechanism, selectivity can be relaxed by several mutations, which are in the scaffold domain and thus outside of the binding site proper (Diallinas, 2021). Two non-mutually exclusive interpretations have been provided for the effect of the mutations: They may eliminate a selectivity filter, which restricts access to the binding site (Vlanti et al., 2006), and/or they may relax a brake and thus facilitate triggering of the elevator movement (Diallinas, 2021). In transporters, which operate by a rocker switch or a rocking bundle mechanism, the binding site is more deeply buried in the hydrophobic core of the protein than in those operating as sliding elevators. Accordingly, it is readily conceivable that restrictions can be imposed on access to the binding site. In fact, the selective binding to the closely related DAT/SLC6A3 and SERT/SLC6A4 is determined by the association rate constant rather than the dissociation rate constant (Hasenhuetl et al., 2015); access to the binding site is limited—at least in part—by the extracellular loops (Esendir et al., 2021). Exchanging the N-terminus of DAT/SLC6A3 with that of SERT/SLC6A4 reduces the KM of the resulting chimaera for dopamine (Sweeney et al., 2017). Taken together, these observations suggest that changes in transporter selectivity and uptake rates can be brought about by mutations of many residues, which are not necessarily confined to the substrate binding site. During evolution the sequential impact of mutations is likely to change microscopic rate constants along trajectories as diverse as explored by our computational optimization. A long-term challenge is to delineate such an evolutionary path through the landscape of possible microscopic rate constants by sequential mutagenesis. This should be gratifying, because it is expected shed light on the evolutionary history underlying transporter diversity.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
KS, MF, and WS conceptualized and wrote the manuscript. KS scripted the optimization algorithm. CF, DB, KS, and WS analyzed the data. All authors contributed to the article and approved the submitted version.
This work was funded by a grant from the Austrian Science Fund/FWF (P31813 to WS) and the Vienna Science and Technology Fund/WWTF (LSC17-026 to MF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.817886/full#supplementary-material
Bar-Even, A., Noor, E., Savir, Y., Liebermeister, W., Davidi, D., Tawfik, D. S., et al. (2011). The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410. doi: 10.1021/bi2002289
Bhat, S., El-Kasaby, A., Freissmuth, M., and Sucic, S. (2021). Functional and biochemical consequences of disease variants in neurotransmitter transporters: a special emphasis on folding and trafficking deficits. Pharmacol. Ther. 222:107785. doi: 10.1016/j.pharmthera.2020.107785
Bicho, A., and Grewer, C. (2005). Rapid substrate-induced charge movements of the GABA transporter GAT1. Biophys. J. 89, 211–231. doi: 10.1529/biophysj.105.061002
Boehm, E., Chan, S., Monfared, M., Wallimann, T., Clarke, K., and Neubauer, S. (2003). Creatine transporter activity and content in the rat heart supplemented by and depleted of creatine. Am. J. Physiol. Endocrinol. Metab. 284, E399–E406. doi: 10.1152/ajpendo.00259.2002
Bulling, S., Schicker, K., Zhang, Y.-W., Steinkellner, T., Stockner, T., Gruber, C., et al. (2012). The mechanistic basis for non-competitive ibogaine inhibition of serotonin and dopamine transporters. J. Biol. Chem. 87, 18524–18534. doi: 10.1074/jbc.M112.343681
Burtscher, V., Schicker, K., Freissmuth, M., and Sandtner, W. (2019). Kinetic models of secondary active transporters. Int. J. Mol. Sci. 20:5365. doi: 10.3390/ijms20215365
Caveney, S., Cladman, W., Verellen, L., and Donly, C. (2006). Ancestry of neuronal monoamine transporters in the Metazoa. J. Exp. Biol. 209, 4858–4868. doi: 10.1242/jeb.02607
Cavet, M. E., Akhter, S., de Medina, F. S., Donowitz, M., and Tse, C. M. (1999). Na(+)/H(+) exchangers (NHE1-3) have similar turnover numbers but different percentages on the cell surface. Am. J. Physiol. 277, C1111–21. doi: 10.1152/ajpcell.1999.277.6.C1111
Chiba, P., Freissmuth, M., and Stockner, T. (2014). Defining the blanks – pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol. Res. 83, 63–73. doi: 10.1016/j.phrs.2013.11.009
Denecke, S. M., Driva, O., Luong, H. N. B., Ioannidis, P., Linka, M., Nauen, R., et al. (2020). The identification and evolutionary trends of the solute carrier superfamily in arthropods. Genome Biol. Evol. 12, 1429–1439. doi: 10.1093/gbe/evaa153
Diallinas, G. (2021). Transporter specificity: a tale of loosened elevator-sliding. Trends Biochem. Sci. 46, 708–717. doi: 10.1016/j.tibs.2021.03.007
Erdem, F. A., Ilic, M., Koppensteiner, P., Gołacki, J., Lubec, G., Freissmuth, M., et al. (2019). A comparison of the transport kinetics of glycine transporter 1 and glycine transporter 2. J. Gen. Physiol. 151, 1035–1050. doi: 10.1085/jgp.201912318
Esendir, E., Burtscher, V., Coleman, J. A., Zhu, R., Gouaux, E., Freissmuth, M., et al. (2021). Extracellular loops of the serotonin transporter act as a selectivity filter for drug binding. J. Biol. Chem. 297:100863. doi: 10.1016/j.jbc.2021.100863
Forster, I. C., Hernando, N., Biber, J., and Murer, H. (2013). Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 34, 386–395. doi: 10.1016/j.mam.2012.07.007
Hasenhuetl, P. S., Bhat, S., Mayer, F. P., Sitte, H. H., Freissmuth, M., and Sandtner, W. (2018). A kinetic account for amphetamine-induced monoamine release. J. Gen. Physiol. 150, 431–451. doi: 10.1085/jgp.201711915
Hasenhuetl, P. S., Schicker, K., Koenig, X., Li, Y., Sarker, S., Stockner, T., et al. (2015). Ligand selectivity among the dopamine and serotonin transporters specified by the forward binding reaction. Mol. Pharmacol. 88, 12–18. doi: 10.1124/mol.115.099036
Hediger, M. A., Romero, M. F., Peng, J.-B., Rolfs, A., Takanaga, H., and Bruford, E. A. (2004). The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch. Eur. J. Physiol. 447, 465–468. doi: 10.1007/s00424-003-1192-y
Hoglund, P. J., Nordstrom, K. J. V., Schioth, H. B., and Fredriksson, R. (2011). The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 28, 1531–1541. doi: 10.1093/molbev/msq350
Hou, Z., and Matherly, L. H. (2014). Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. Curr. Top. Membr. 73, 175–204. doi: 10.1016/B978-0-12-800223-0.00004-9
Jardetzky, O. (1966). Simple allosteric model for membrane pumps. Nature 211, 969–970. doi: 10.1038/211969a0
Jennigs, M. L. (2018). Carriers, exchangers, and cotransporters in the first 100 years of the journal of general physiology. J. Gen. Physiol. 150, 1063–1080. doi: 10.1085/jgp.201812078
Kruckeberg, A. L., Ye, L., Berden, J. A., and van Dam, K. (1999). Functional expression, quantification and cellular localization of the Hxt2 hexose transporter of Saccharomyces cerevisiae tagged with the green fluorescent protein. Biochem. J. 339, 299–307.
Li, Y., Mayer, F. P., Hasenhuetl, P. S., Burtscher, V., Schicker, K., Sitte, H. H., et al. (2017). Occupancy of the zinc-binding site by transition metals decreases the substrate affinity of the human dopamine transporter by an allosteric mechanism. J. Biol. Chem. 292, 4235–4243. doi: 10.1074/jbc.M116.760140
Meinild, A.-K., and Forster, I. C. (2012). Using lithium to probe sequential cation interactions with GAT1. Am. J. Physiol. Cell Physiol. 302, C1661–C1675. doi: 10.1152/ajpcell.00446.2011
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H., and Teller, E. (1953). Equation of state calculations by fast computing machines. J. Chem. Phys. 21, 1087–1092. doi: 10.1063/1.1699114
Millward, D. J., and Garlick, P. J. (1976). The energy cost of growth. Proc. Nutr. Soc. 35, 339–349. doi: 10.1079/PNS19760054
Milo, R., Jorgensen, P., Moran, U., Weber, G., and Springer, M. (2010). BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–3. doi: 10.1093/nar/gkp889
Mitchell, P. (1979). Compartmentation and communication in living systems. Ligand conduction: a general catalytic principle in chemical, osmotic and chemiosmotic reaction systems. Eur. J. Biochem. 95, 1–20. doi: 10.1111/j.1432-1033.1979.tb12934.x
Moraes, T. F., and Reithmeier, R. A. (2012). Membrane transport metabolons. Biochim. Biophys. Acta 1818, 2687–2706. doi: 10.1016/j.bbamem.2012.06.007
Motohashi, H., and Inui, K. (2013). Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 15, 581–588. doi: 10.1208/s12248-013-9465-7
Nikaido, K., Liu, P. Q., and Ames, G. F. (1997). Purification and characterization of HisP, the ATP-binding subunit of a traffic ATPase (ABC transporter), the histidine permease of Salmonella typhimurium. Solubility, dimerization, and ATPase activity. J. Biol. Chem. 272, 27745–52. doi: 10.1074/jbc.272.44.27745
Omote, H., Hiasa, M., Matsumoto, T., Otsuka, M., and Moriyama, Y. (2006). The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 27, 587–593. doi: 10.1016/j.tips.2006.09.001
Perez, C., Faust, B., Mehdipour, A. R., Francesconi, K. A., Forrest, L. R., and Ziegler, C. (2014). Substrate-bound outward-open state of the betaine transporter BetP provides insights into Na+ coupling. Nat. Commun. 5:4231. doi: 10.1038/ncomms5231
Raz, S., Stark, M., and Assaraf, Y. G. (2016). Folylpoly-γ-glutamate synthetase: a key determinant of folate homeostasisand antifolate resistance in cancer. Drug Resist. Updat. 28, 43–64. doi: 10.1016/j.drup.2016.06.004
Rudnick, G., and Sandtner, W. (2019). Serotonin transport in the 21st century. J. Gen. Physiol. 151, 1248–1264. doi: 10.1085/jgp.201812066
Sano, R., Shinozaki, Y., and Ohta, T. (2020). Sodium–glucose cotransporters: functional properties and pharmaceutical potential. J. Diabetes Investig. 11, 770–782. doi: 10.1111/jdi.13255
Schicker, K., Bhat, S., Farr, C., Burtscher, V., Horner, A., Freissmuth, M., et al. (2021). Descriptors of secondary active transporter function and how they relate to partial reactions in the transport cycle. Membranes 11:178. doi: 10.3390/membranes11030178
Schicker, K., Uzelac, Z., Gesmonde, J., Bulling, S., Stockner, T., Freissmuth, M., et al. (2011). A unifying concept of serotonin transporter associated currents. J. Biol. Chem. 287, 438–445. doi: 10.1074/jbc.M111.304261
Schmidt, H., and Jirstrand, M. (2006). Systems biology toolbox for MATLAB: a computational platform for research in systems biology. Bioinformatics 22, 514–515. doi: 10.1093/bioinformatics/bti799
Shi, Y., Wang, J., Ndaru, E., and Grewer, C. (2021). Pre-steady-state kinetic analysis of amino acid transporter SLC6A14 reveals rapid turnover rate and substrate translocation. Front. Physiol. 12:777050. doi: 10.3389/fphys.2021.777050
Siems, W., Dubiel, W., Dumdey, R., Müller, M., and Rapoport, S. M. (1984). Accounting for the ATP-consuming processes in rabbit reticulocytes. Eur. J. Biochem. 139, 101–107. doi: 10.1111/j.1432-1033.1984.tb07982.x
Simakov, O., Marlétaz, F., Yue, J. X., O'Connell, B., Jenkins, J., Brandt, A., et al. (2020). Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830. doi: 10.1038/s41559-020-1156-z
Sitte, H. H., and Freissmuth, M. (2015). Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 36, 41–50. doi: 10.1016/j.tips.2014.11.006
Smirnova, I., Kasho, V., Sugihara, J., and Kaback, H. R. (2011). Opening the periplasmic cavity in lactose permease is the limiting step for sugar binding. Natl. Acad. Sci. U. S. A. 108, 15147–51. doi: 10.1073/pnas.1112157108
Snow, R. J., and Murphy, R. M. (2001). Creatine and the creatine transporter: a review. Mol. Cell. Biochem. 224, 169–181. doi: 10.1023/A:1011908606819
Sweeney, C. G., Tremblay, B. P., Stockner, T., Sitte, H. H., and Melikian, H. E. (2017). Dopamine transporter amino and carboxyl termini synergistically contribute to substrate and inhibitor affinities. J. Biol. Chem. 292, 1302–1309. doi: 10.1074/jbc.M116.762872
Tsallis, C., and Stariolo, D. A. (1996). Generalized simulated annealing. Phys. Stat. Mech. Appl. 233, 395–406. doi: 10.1016/S0378-4371(96)00271-3
VanWert, A. L., Gionfriddo, M. R., and Sweet, D. H. (2010). Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 31, 1–71. doi: 10.1002/bdd.693
Vlanti, A., Amillis, S., Koukaki, M., and Diallinas, G. (2006). A novel-type substrate-selectivity filter and ER-exit determinantsin the UapA purine transporter. J. Mol. Biol. 357, 808–819. doi: 10.1016/j.jmb.2005.12.070
Waterlow, J. C., Golden, M. H., and Garlick, P. J. (1978). Protein turnover in man measured with 15N: comparison of end products and dose regimes. Am. J. Phys. 235, E165–E174. doi: 10.1152/ajpendo.1978.235.2.E165
Wilson, T. H., and Ding, P. Z. (2001). Sodium-substrate cotransport in bacteria. Biochim. Biophys. Acta 1505, 121–130. doi: 10.1016/s0005-2728(00)00282-6
Wolfe, K. H. (2001). Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2, 333–341. doi: 10.1038/35072009
Wright, E. M., Loo, D. D. F. L., and Hirayama, B. A. (2011). Biology of human sodium glucose transporters. Physiol. Rev. 91, 733–794. doi: 10.1152/physrev.00055.2009
Wright, J. K., and Overath, P. (1984). Purification of the lactose:H+ carrier of Escherichia coli and characterization of galactoside binding and transport. Eur. J. Biochem. 138, 497–508. doi: 10.1111/j.1432-1033.1984.tb07944.x
Wyss, M., and Kaddurah-Daouk, R. (2000). Creatine and creatinine metabolism. Physiol. Rev. 80, 1107–1213. doi: 10.1152/physrev.2000.80.3.1107
Yaffe, D., Forrest, L. R., and Schuldiner, S. (2018). The ins and outs of vesicular monoamine transporters. J. Gen. Physiol. 150, 671–682. doi: 10.1085/jgp.201711980
Ye, L., Berden, J. A., van Dam, K., and Kruckeberg, A. L. (2001). Expression and activity of the Hxt7 high-affinity hexose transporter of Saccharomyces cerevisiae. Yeast 18, 1257–67. doi: 10.1002/yea.771
Keywords: solute carriers, kinetic model, optimization, evolution, secondary active transporters, substrate uptake
Citation: Schicker K, Farr CV, Boytsov D, Freissmuth M and Sandtner W (2022) Optimizing the Substrate Uptake Rate of Solute Carriers. Front. Physiol. 13:817886. doi: 10.3389/fphys.2022.817886
Edited by:
Christof Grewer, Binghamton University, United StatesReviewed by:
Reinhart Reithmeier, University of Toronto, CanadaCopyright © 2022 Schicker, Farr, Boytsov, Freissmuth and Sandtner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter Sandtner, d2FsdGVyLnNhbmR0bmVyQG1lZHVuaXdpZW4uYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.