
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 29 August 2022
Sec. Red Blood Cell Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.796837
This article is part of the Research TopicImages from Red CellsView all 13 articles
 Sari Peretz1,2†
Sari Peretz1,2† Leonid Livshits1,3†
Leonid Livshits1,3† Etheresia Pretorius4
Etheresia Pretorius4 Asya Makhro3
Asya Makhro3 Anna Bogdanova3,5
Anna Bogdanova3,5 Max Gassmann3
Max Gassmann3 Ariel Koren1
Ariel Koren1 Carina Levin1,2*
Carina Levin1,2*Sickle cell disease (SCD) is caused by a point mutation in the beta-globin gene. SCD is characterized by chronic hemolytic anemia, vaso-occlusive events leading to tissue ischemia, and progressive organ failure. Chronic inflammatory state is part of the pathophysiology of SCD. Patients with SCD have extremely variable phenotypes, from mild disease to severe complications including early age death. The spleen is commonly injured in SCD. Early splenic dysfunction and progressive spleen atrophy are common. Splenomegaly and hypersplenism can also occur with the loss of the crucial splenic function. Acute, life-threatening spleen-related complications in SCD are well studied. The association of laboratory parameters with the spleen status including hyposplenism, asplenia, and splenomegaly/hypersplenism, and their implication in vaso-occlusive crisis and long-term complications in SCD remain to be determined. We evaluated the association between the spleen status with clinical and laboratory parameters in 31 SCD patients: Group a) Patients with asplenia/hyposplenism (N = 22) (including auto-splenectomy and splenectomized patients) vs. Group b) patients with splenomegaly and or hypersplenism (N = 9). Laboratory studies included: Complete Blood Count, reticulocyte count, iron metabolism parameters, C Reactive Protein (CRP), Hb variant distribution, and D-dimer. Metabolic and morphological red blood cell (RBC) studies included: density gradient (by Percoll), glucose consumption, lactate release, and K+ leakage, fetal RBC (F-Cells) and F-Reticulocytes, annexinV+, CD71+, oxidative stress measured by GSH presence in RBC and finally Howell Jolly Bodies count were all analyzed by Flow Cytometry. Scanning electron microscopy analysis of RBC was also performed. Patients with asplenia/hyposplenism showed significantly higher WBC, platelet, Hematocrit, hemoglobin S, CRP, D-dimer, Gamma Glutamyl Transferase (GGT), cholesterol, transferrin, annexin V+ RBCs, CD71+ RBCs, together with a markedly lower F Reticulocyte levels in comparison with splenomegaly/hypersplenism patients. In summary, important differences were also found between the groups in the studied RBCs parameters. Further studies are required to elucidate the effect of the spleen including hyper and hypo-splenia on laboratory parameters and in clinical manifestations, vascular pathology, and long-term complications of SCD. The benefits and risks of splenectomy compared to chronic transfusion need to be evaluated in clinical trials and the standard approach managing hypersplenism in SCD patients should be re-evaluated.
Sickle cell disease (SCD) is the most frequent worldwide hereditary hemoglobinopathy (Piel et al., 2013; Ware et al., 2017). The disorder is caused by the inheritance of abnormal beta-globin alleles carrying the sickle mutation on the HBB gene (HBB: c:20T>A- Glu6Val, βS). SCD results either by the homozygous state HbSS or by the combination of HbS with another abnormal HBB variants, mostly β+ or β0 thalassemia, both resulting in sickle red blood cells (RBC) (Bain 2008; Ware et al., 2017). Polymerization of deoxy-HbS generates abnormal dense and dehydrated RBC that play a central role in the acute and chronic manifestations of SCD. Intravascular sickling leads to impaired blood flow and vaso-occlusion with ischemic/reperfusion injury (Franceschi et al., 2011). Some organs, such as the spleen, are prone to be specifically vulnerable and damaged from HbS polymerization in SCD patients at early life (Platt 2000). Hyposplenism, asplenia, and progressive spleen atrophy (auto-splenectomy) are common. On the other hand, hypersplenism is characterized by an enlarged spleen (splenomegaly) which causes rapid and premature destruction of blood cells, resulting in thrombocytopenia, anemia, and or leukopenia, (Laws et al., 1979; Lv et al., 2016; Ladu et al., 2021). Splenomegaly and or hypersplenism can coexist with loss of function (Pearson 1969; O’Brien et al., 1976; Eichner 1979; Pearson et al., 1979; Serjeant et al., 1979; Zago et al., 1980; Brown et al., 1994).
In SCD, splenic dysfunction and hypersplenism are regularly observed during infancy; followed, in most cases, by spleen atrophy. In those cases, after repeated episodes of infarctions, the spleen is reduced to a fibrotic nodule (Diggs and Memphis 1935; Pearson et al., 1979). Another common and dangerous complication of SCD in young children is the sudden enlargement of the spleen defined as acute splenic sequestration crisis, associated with rapid decrease in hemoglobin levels of at least 2 g/dl, without signs of hemolysis (Lesher et al., 2009; Samuk et al., 2019).
Despite several postoperative complications, surgical splenectomy is recommended to treat the effects of hypersplenism in SCD subjects (Emond et al., 1984; Kyaw et al., 2006; Leone and Pizzigallo 2015; Iolascon et al., 2017; Luu et al., 2019; Tahir et al., 2020; Yacobovich et al., 2020).
One of the indicators for splenic dysfunction is the presence of Howell-Jolly bodies (HJB) inside RBCs. HJB are DNA inclusions that are usually formed at low frequency. Normally RBC containing HJB are removed from the peripheral blood by the spleen. In SCD patients, with functional asplenia, HJB are present in a higher amount than in healthy individuals. In addition to complete blood count (CBC), the presence of HJB is used as a splenic dysfunction marker (Harrod et al., 2007; El Hoss et al, 2019). Those results can provide a fast evaluation of spleen status.
Maintenance of normal spleen function is crucial in the prevention of morbidity and mortality in SCD patients. Since the spleen is involved in immune response and the clearance of senescent and aberrant blood cells, noninvasive, quantitative, and qualitative measurements of spleens’ function are required. In this study, we compared blood parameters and RBC properties of SCD patients and compared the results between SCD patients with asplenia, to patients with splenomegaly/hypersplenism. In addition, we implemented novel, non-routinely used methods for the evaluation of the functional status of the spleen in those patients.
The study included 31 SCD patients treated at the Pediatric Hematology Unit, Emek Medical Center, (EMC), Afula, Israel. To get a better understanding of the spleen influence in SCD, the studied group was divided into two subgroups: patients with asplenia/hyposplenism [group (a)] which included patients with non-visualization of the spleen on ultrasound or no palpable spleen or patients after surgical total splenectomy, In this sub group we included young patients with splenic abnormal function and patients that probably underwent auto-splenectomy, both conditions well known in patients with SCD and patients that underwent surgical splenectomy. The second group, [group (b)], included patients with splenomegaly and/or hypersplenism. This group included patients with an enlarged spleen, palpable by physical examination and/or detected by ultrasound, with or without cytopenias, persistent for more than 6 months of follow-up.
The asplenia/hyposplenism group (group a) included 22 patients: 12 females; mean age 25.1 ± 12.9 years; 12 HbSS genotype and 9 HbS/β-genotype [1- β+, 8- β0 and another one probably β0 based on High-performance liquid chromatography (HPLC) results], one HbS/D-genotype and seven patients after surgical total splenectomy. The hypersplenism group (group b) included 9 patients: 6 females; mean age 16.8 ± 10.3 years (4 HbSS and 5 HbS/β-genotype (4- β0 and one- β+), patient characteristics are presented in (Table 1). RBC membrane permeability and metabolic properties studied in 14 healthy individuals and, Percoll density gradients studied in 21 healthy individuals were used as control values for comparison with the results of the study groups.

TABLE 1. Patients’ characteristics. Demographic and clinical features of 31 SCD patients enrolled in this study and divided into two groups: Asplenia/hyposplenism and Hypersplenism. Events of VOC and hospitalizations presented here are per year prior to the study.
All subjects were under stable doses of hydroxyurea treatment (500–2,000 mg/daily, 22.8 ± 9.6 mg/kg with no significant differences between the cohorts). Any of the patients were receiving routine blood transfusions or sporadic blood transfusions for at least 3 months before the study’s tests. Informed consent was obtained from all patients or their parents and the study received the approval of the local Helsinki Committee (Registry Nos. EMC-0071-17 and EMC-0120-17).
Patients’ complete blood count (CBC) and blood biochemistry were analyzed on Advia2120 analyzer (SIEMENS, Germany) and AU5800 (Beckman Coulter, United States) and Clinical Chemistry Set-ups, respectively, at the EMC laboratory within 1 h after collection. High-performance liquid chromatography (HPLC) analysis of hemoglobin variant distribution was performed by Variant II analyzer (BIO-RAD, United States). The presented normal ranges are as accepted at the EMC, if not indicated differently.
A whole blood sample was fractionated on a Percoll-based gradient as described (Makhro et al., 2013) with some modifications. Briefly, whole blood sample was layered over a Percoll solution (90% commercial Percoll (17-0891-01, GE Healthcare) and 10% of the concentrated plasma-like medium (1.4 M NaCl, 40 mM KCl, 7.5 mM MgSO4, 100 mM glucose, 0.15 mM ZnCl2, 2 mM Glycine, 2 mM Sodium Glutamate, 2 mM Alanine, 1 mM Arginine, 6 mM Glutamine, 1% BSA, 200 mM HEPES imidazole, pH 7.4) supplemented with 2 mM CaCl2, and then centrifuged at 18,514 g for 60 min (minimal acceleration/deceleration) at 30°C (Eppendorf Centrifuge 5810R, F-34-6-38 Rotor supplemented with specific adapter for 15 ml Falcon tubes). Then, the distribution of blood components was captured by the 16 Mp camera (installed in the Samsung Galaxy S6 Model SM-G920F mobile phone (Samsung Electronics Co.) and further analyzed by the free Windows version of the ImageJ software (downloaded from https://imagej.nih.gov/ij/download.html). The total visualized fraction was divided to equal ten stations and the intensity of each subfraction was measured.
Isolated RBC samples were incubated in the plasma-like medium (140 mM NaCl, 4 mM KCl, 0.75 mM MgSO4, 10 mM glucose, 0.015 mM ZnCl2, 0.2 mM Glycine, 0.2 mM Sodium Glutamate, 0.2 mM Alanine, 0.1 mM Arginine, 0.6 mM Glutamine, 0.1% BSA, 20 mM HEPES imidazole, pH 7.4) supplemented with 2 mM CaCl2 as described previously (Makhro et al., 2017) for 4 h at 37°C in gentle shake (14,000 rpm). The medium’s concentrations of potassium, glucose, and lactate were measured prior (time 0 h s) and after incubation (4 h s) by GEM® Premier™ 4,000 blood gas analyzer. The 4 vs. 0 h s difference was normalized with total hemoglobin concentration measured simultaneously.
Morphological characteristics of RBC and reticulocytes, phosphatidylserine (PS) exposure on outer membrane leaflet representing early apoptotic RBCs was measured by annexin V+ RBCs, intracellular Ca2+ and oxidative stress measured by reduced glutathione (GSH) levels in RBC were investigated by FC as described elsewhere (Kuypers et al., 1996; Kämpf et al., 2019). Briefly, 2 µl of whole blood suspended in a 1 ml plasma-like medium was loaded with 1.5 µl of either:
- Intracellular Ca2+ dye, Fluo-4 AM [1 mM stock, (Thermo Fisher Scientific].
- GSH binding dye, monobromobimane (MBBR) (100 mM stock, Thermo Fisher Scientific).
- Anti-CD71-FITC (TfR) antibody (IQP-152F, IQ Products) for identification of reticulocyte fraction.
- eBioscience™ Annexin V Apoptosis Detection Kit eFluor 450 dye (Thermo Fisher Scientific) for PS externalization test.
The stained cells were incubated for 45 min in dark before FC measurements.
RBC sizes and morphology were examined in unstained samples.
In addition, the presence of DNA inclusions inside the RBCs, the Howell-Jolly bodies—HJB) (El Hoss et al., 2018) and fetal RBC (F-Cells), F Reticulocytes (representing early changes in HbF cells), were also examined using FC. HbF and F-Cells are used to monitor SCD patients’ response to Hydroxyurea treatment and to evaluate their chances to suffer from SCD-related complications (Lebman et al., 1982; Kono et al., 2009; Sato et al., 2010).
In brief, 20 μl of whole blood in an EDTA tube fixed in 1 ml of 25% glutaraldehyde solution (Sigma, United States) for 10 min. Immediately after fixation, 100 μl of the suspension was permeabilized in a new tube by resuspension in 400 μl of 0.1% TRITON X-100 (Sigma, United States) for 4 min. Following permeabilization, 20 μl of the suspension was taken in a new tube with 5 μl of 5 μM Hoechst 33342 (Sigma, United States), 5 μl 7-AAD (7-amino-actinomycin D) (Invitrogen, United States), 5 μl anti CD71 (transferrin receptor) APC conjugate (Invitrogen, United States), 5 μl intercellular anti-human fetal Hb (MHFH01) FITC conjugate (Life technologies, United States) and 70 μl PBS. After vortex and dark incubation for 15 min, 400 μl of PBS was added and gentle vortex before FC measurement.
The fluorescence intensity was measured on RBC gate using Navios EX flow cytometer (Beckman Coulter, United States). Double measurements (>50,000 total cells/measurement) were performed and averaged for each condition. All data were analyzed using Kaluza Analysis Software (Beckman Coulter, United States https://www.beckman.co.il/flow-cytometry/software/kaluza).
10 μl of whole blood was pipetted into a 10 mm round glass microscope cover slip and dried for 2 min at room temperature. The glass microscope cover slips were placed in a 24-well culture flask for further washing; PBS was added and removed after 15 min. Next, 4% formaldehyde was added to the sample for fixation for a minimum of 30 min., After three washes with PBS one drop of osmium tetra-oxide (OT) (Sigma, United States) was added directly onto the cover slip and then covered with dH2O for 15 min. Right after, the samples were washed three times with PBS for 3 min each wash. Next, dehydration was performed by a series of Ethanol-dH2O mixtures (30%, 50%, 70%, 90%, and finally 100%) for 3 min. The last wash with 100% ethanol was repeated twice. At this stage, the well plate was sealed with parafilm, and the samples were stored for 24–48 h. Next, the samples were covered with 99.9% hexamethyldisilazane (HMDS) (Sigma, United States) for 30 min. Then, the open plate was left overnight to evaporate HMDS. The prepared samples were coated with carbon and viewed with a Zeiss MERLIN field emission-SEM with the InLens detector using 1 kV (Carl Zeiss Microscopy, Munich, Germany).
Data for the entire study was analyzed using GraphPad 5 software. Differences between SCD patients with Asplenia/hyposplenism vs. Hypersplenism were tested using the Mann-Whitney test. For all analyses, a two-tailed test with p < 0.05 was accepted as statistically significant.
Comparison of the CBC and biochemistry parameters revealed significant differences between hypersplenic and asplenic/hyposplenic SCD subjects (Table 2).
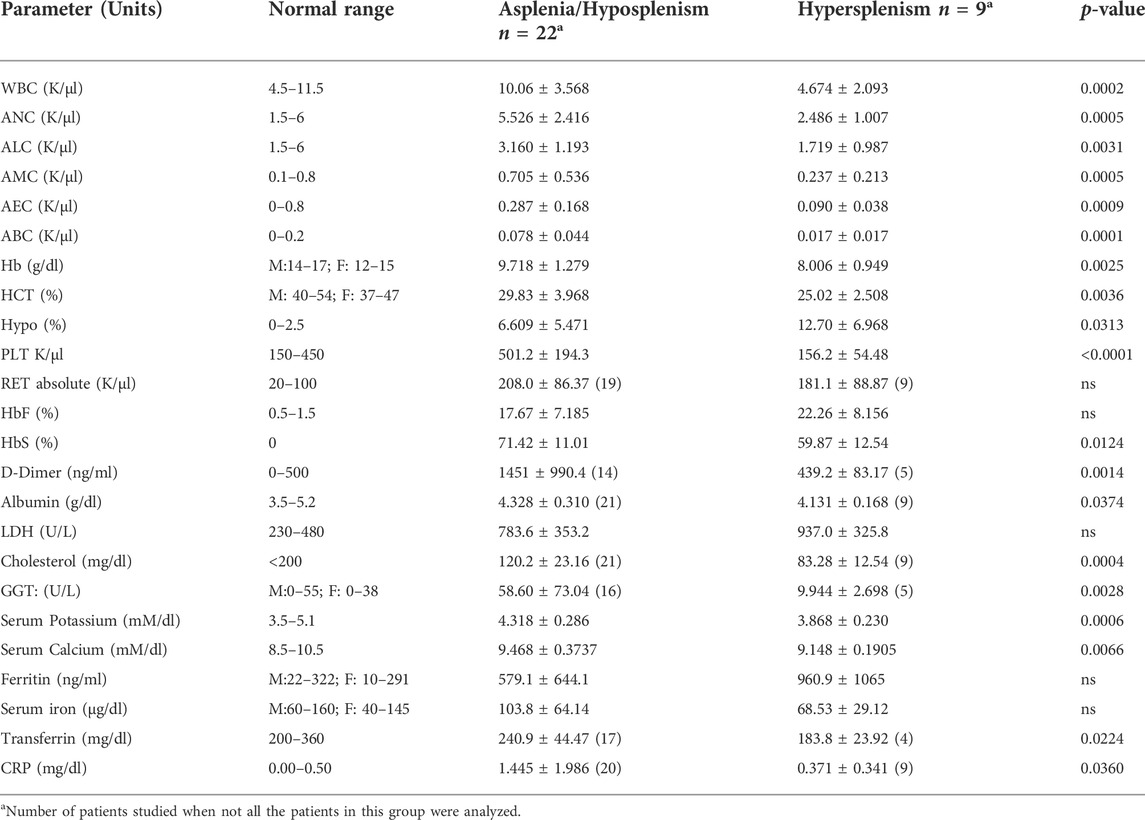
TABLE 2. CBC and biochemistry parameters in asplenic/hyposplenic compared to hypersplenic SCD subjects. Normal ranges are presented in accordance with the criteria accepted by the EMC Lab divisions. The data is presented as average ± Standard Deviation (SD); Significance was calculated using the two-tailed Mann-Whitney as p < 0.05. WBC, White blood count, ANC, Absolute Neutrophil count, ALC, Absolute lymphocyte count, AMC, Absolute monocyte count, AEC, Absolute eosinophil count, ABC, Absolute basophil count, Hb, Hemoglobin, HCT, Hematocrit, Hypo, Hypochromic Red Cells, PLT, Platelet, RET: Reticulocyte, HbF, Fetal hemoglobin, HbS, S hemoglobin, LDH, Lactic dehydrogenase, GGT, Gamma Glutamyl Transferase, CRP, C reactive protein. RBC, Red blood cells.
We found a significantly lower hematocrit and hemoglobin levels (approximately 2 g/dl reduction) in the hypersplenism vs. asplenia/hyposplenism group (p < 0.05) that was accompanied by an almost two-fold increase in the percentage of hypochromic RBC in the hypersplenism group (p = 0.03).
Lower white blood cell (WBC) and platelet counts were found in the hypersplenism group compared to the asplenia/hyposplenism group (p < 0.001), similarly to all the subsets of leukocytes (Table 2). The mean percent of HbF was higher in the hypersplenism group (an increase of 5%) but this difference was not significant. The mean percent of HbS was significantly lower in hypersplenism compared with the asplenia/hyposplenism group (p = 0.01).
The D-Dimer was significantly lower in the hypersplenism group but within the normal range, while in the asplenia/hyposplenism was increased (p < 0.05).
Serum Lactic dehydrogenase (LDH) levels were elevated in both groups, representing the chronic hemolytic state, with no differences between the groups (Table 2). Serum albumin, cholesterol, calcium, and potassium levels were within normal values in both groups but were significantly lower in the hypersplenism than in the asplenia/hyposplenism group (p = 0.05).
The CRP levels, representing the inflammation state, were within the normal range in the hypersplenism group, while in the asplenia/hyposplenism group were abnormally increased (p < 0.05).
No significant differences between the groups were found in ferritin and serum iron, however, the transferrin level was lower in the hypersplenism group (p < 0.05).
To specify the RBCs-related differences between the groups, blood samples were fractionated on Percoll density gradient as described. We found a significant reduction of dense/senescent RBC fractions (lower stations 9 and 10) in the hypersplenism compared with the asplenia/hyposplenism group. This demonstrates that the hypersplenism group contains more hydrated and less dense/dehydrated and sickled RBCs when compared to the asplenia/hyposplenism group (Figure 1).
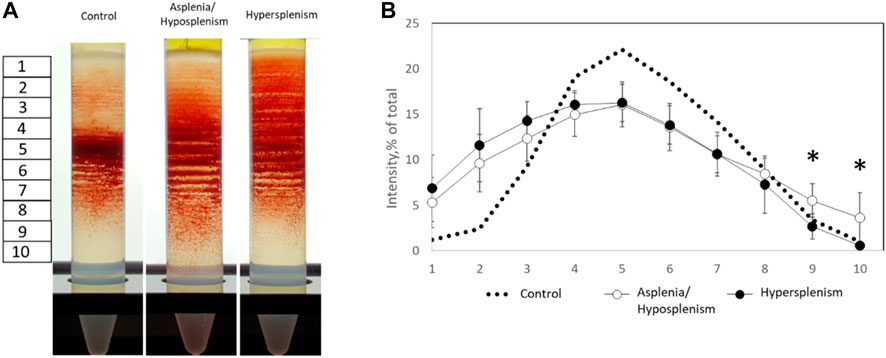
FIGURE 1. Percoll density gradients of control and SCD subjects for dense and hydrated RBC fractions. (A) Representative Percoll gradients for control, asplenic/hyposplenic, and hypersplenic subjects are demonstrated. (B) The RBC content in each station was estimated as a percentage from a sum of all sub-fraction intensity values. The correspondent examination of RBCs obtained from control subjects (n = 21; 10M/11F; 46.8 ± 14.2 years) was made. The data is presented in average ± SD; the significance is < 0.05.
Our next target was to study RBC membrane permeability and metabolic properties (Figure 2). We found no significant difference in the RBCs membrane permeability between the two groups measured by K+ leak and lactate released from RBCs. Also, the RBCs glucose consumption. was similar in the two groups, however, the values in the hypersplenism group were lower than in the asplenia/hyposplenism group and approximates to normal values of healthy controls.
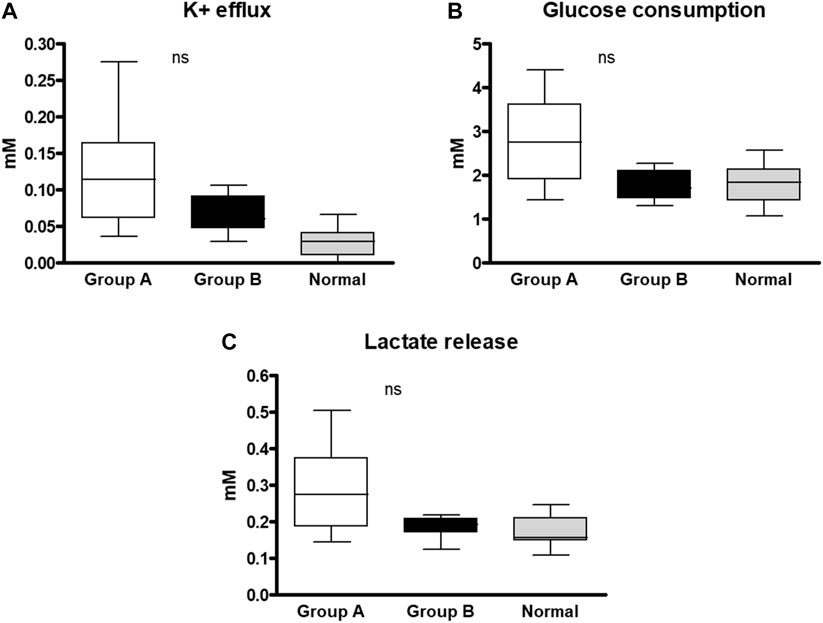
FIGURE 2. RBC membrane permeability and metabolic properties of SCD subjects and healthy control. Samples of each group were incubated for 4 h in a plasma-like medium and examined for: (A) K+ efflux, (B) Glucose consumption, and (C) Lactate release. The 4 vs. 0 h difference was normalized with total hemoglobin concentration. The correspondent examination of RBCs obtained from control subjects (n = 14; 8M/6F; 34.1 ± 16.1 years) was made. The data is presented in average ±SD; the significance is < 0.05. Group (A) patients with asplenia/hyposplenism, Group (B) patients with hypersplenism.
Using FC, we evaluated RBC and reticulocytes morphological characteristics, phosphatidylserine (PS) exposure on outer membrane leaflet, intracellular Ca2+, oxidative stress measured by reduced glutathione (GSH), reticulocytes identification, F- cells, and HJB levels. Representative scatter plots and gating strategy for RBC were shown for all fluorophores (Figure 3).
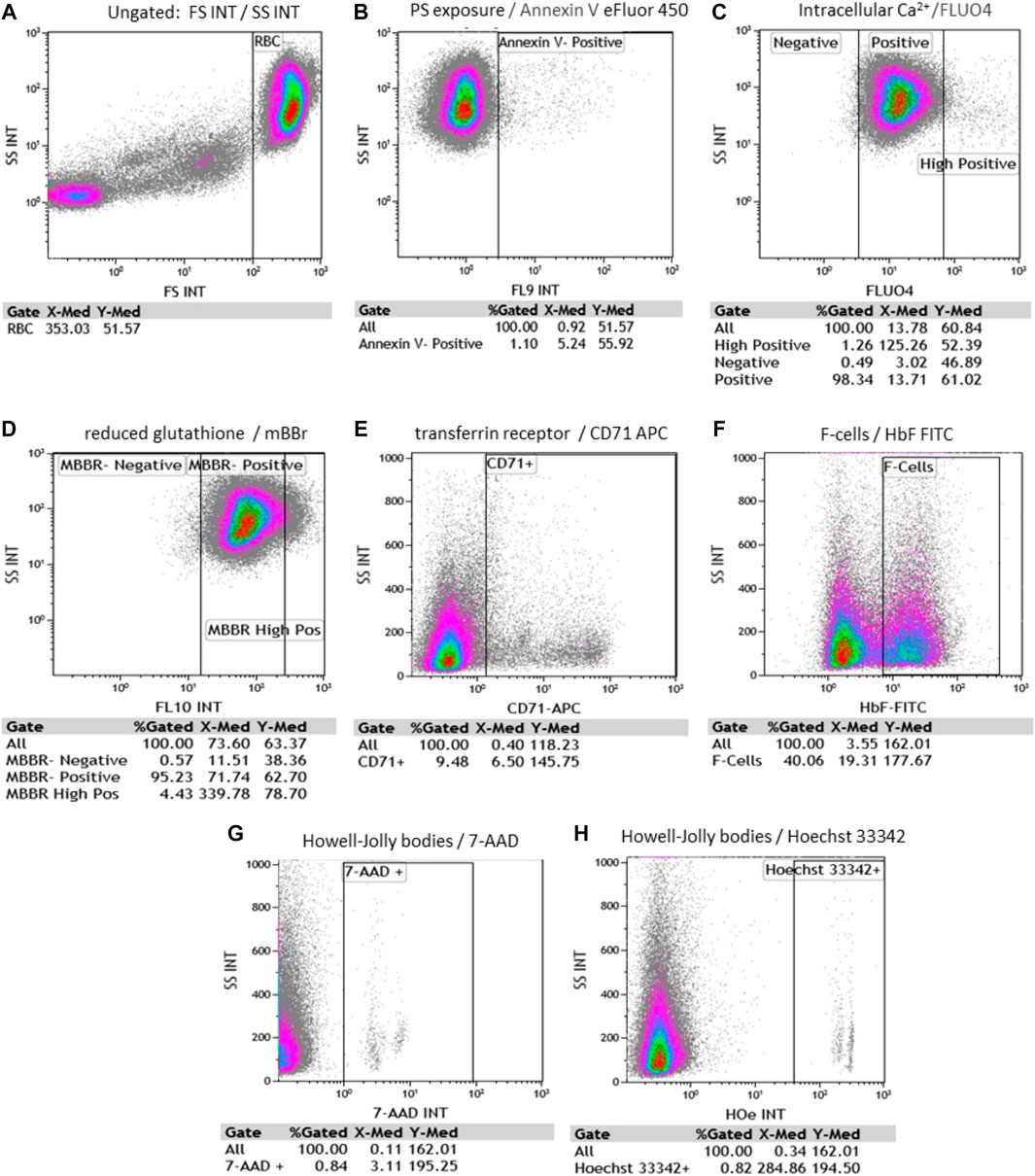
FIGURE 3. Representative flow cytometry scatter plots and gating strategy for RBC. Each evaluates % gated cells and median fluorescence intensity (MFI). (A) RBC side and forward scattering of unstained cells (SS and FS, accordingly); (B) Annexin V-positive RBC; (C) fluo-4 positive and fluo-4 high-positive RBC; (D) MBBR-positive and MBBR high-positive RBC; (E) CD71 positive RBC; (F) F- RBC (F-cells) positive fraction (G) 7-AAD (7-amino-actinomycin D) positive RBC and (H) Hoechst 33342 positive RBC. [(B–H) plots were all gated on RBC].
Side scatter (SSC) and forward scatter (FSC) median fluorescence intensity (MFI) of unstained RBC were 55.39 ± 5.6 and 399.6 ± 39.64; respectively in the hypersplenism group. and 64.29 ± 6.24 and 407.3 ± 45.89; respectively in the asplenia/hyposplenism group.
We found a lower percentage of Annexin V+ RBCs, in the hypersplenism group compared to the results in the asplenia/hyposplenism group (mean 0.34 ± 0.2% vs. 0.82 ± 0.5% respectively, p = 0.05). We also found lower, but not statistically significant, RBC intracellular Ca2+ levels as measured by fluo-4-high-positive RBCs, in the hypersplenism group compared to asplenia/hyposplenism (mean % 0.63 ± 0.3 vs. 1.63 ± 0.95; respectively p = ns. and MFI 96.56 ± 10.48 vs. 87.73 ± 44.88; respectively p = ns). Additionally, we observed decreased trend of MBBR-high-positive RBC in the hypersplenism group vs. the asplenia/hyposplenism group (mean % 0.97 ± 0.49 vs. 1.75 ± 0.93; respectively p = ns and MFI 356.66 ± 95.49 vs. 495.37 ± 107.81; respectively p = ns). Also, the total MBBR positive population MFI was lower in the hypersplenism group vs. the asplenia/hyposplenism group (mean MFI 68.23 ± 22.79 vs. 103.61 ± 28.04; respectively p = 0.04) (Figure 4).
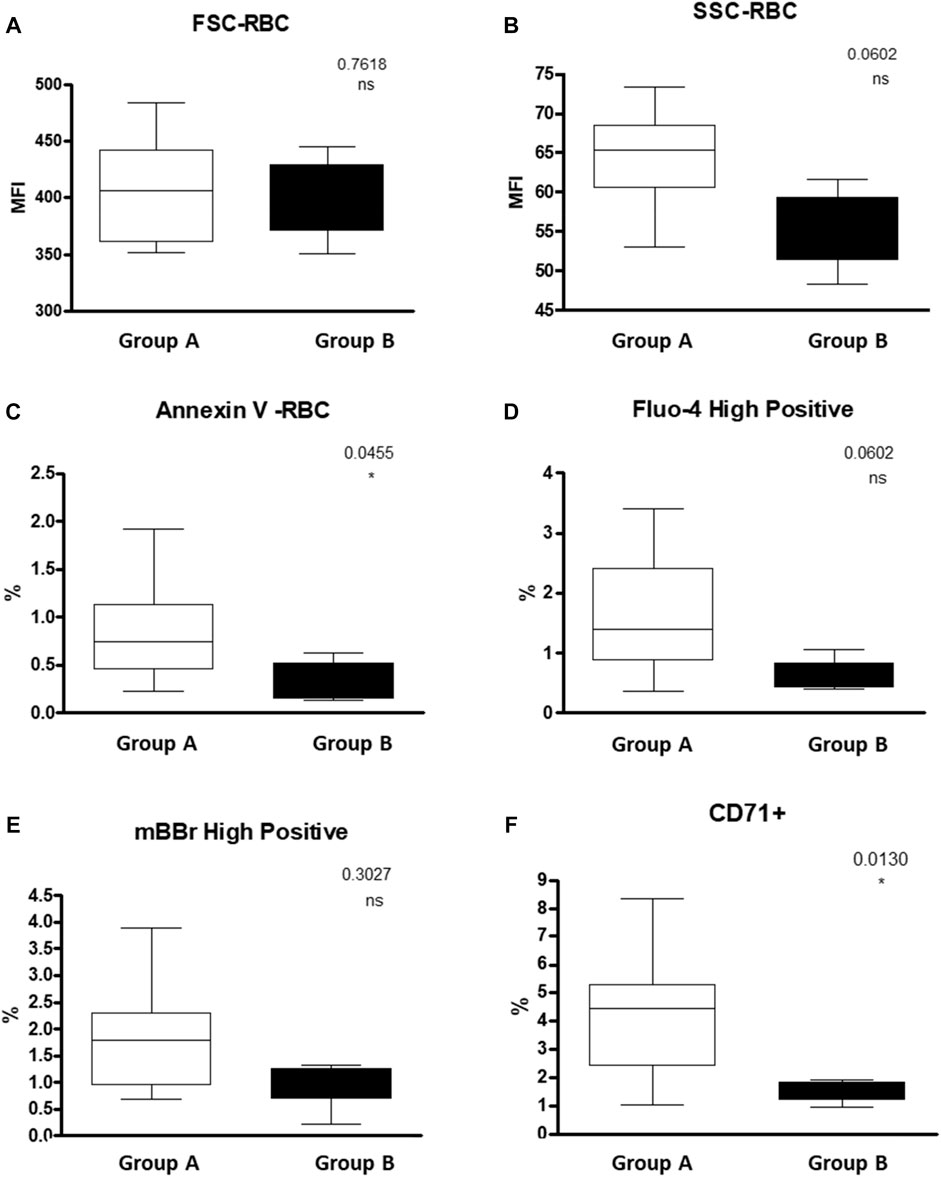
FIGURE 4. RBC properties of SCD patients Asplenic/hyposplenic compared to Hypersplenic using Flow cytometry: (A) + (B) FSC and SSC of RBC; (C) Annexin V positive RBC; (D) fluo-4 high-positive RBC; (E) MBBR high-positive RBC; (F) CD71 positive RBC. Group (A) patients with asplenia/hyposplenism, Group (B) patients with hypersplenism.
We found that the percent of immature RBCs CD71+, reticulocytes expressing the transferrin receptor on the cell surface was lower in the hypersplenism vs. asplenia/hyposplenism group (mean % 1.5 ± 0.4 vs. 4.2 ± 2.1; respectively, p = 0.013). These results correlate with a decreased trend in total reticulocyte count in the CBC in the hypersplenism group (Figure 4).
We found a higher, but not statistically significant, level of HbF measured by HPLC in the hypersplenism group compared to asplenia/hyposplenism (mean HbF % 22.26 ± 8.15 vs. 17.67 ± 7.18; respectively p = ns.) (Table 2), and percent of F-Cells measured by FC (Mean % 64.01 ± 19 vs. 52.77 ± 20; respectively p = ns). We show a positive correlation between these two parameters (Spearman, r = 0.8457) (Figure 5). Since reticulocytes circulate in the peripheral blood for 7 days, the calculation of F- reticulocyte can serve as a unique and important value for patient’s follow-up and treatment response. We found significantly higher F-reticulocyte counts in the hypersplenism compared to the asplenia/hyposplenism group (mean 33.2 ± 14.1 vs. 55.1 ± 12.6%; respectively p = 0.011) (Figure 5).
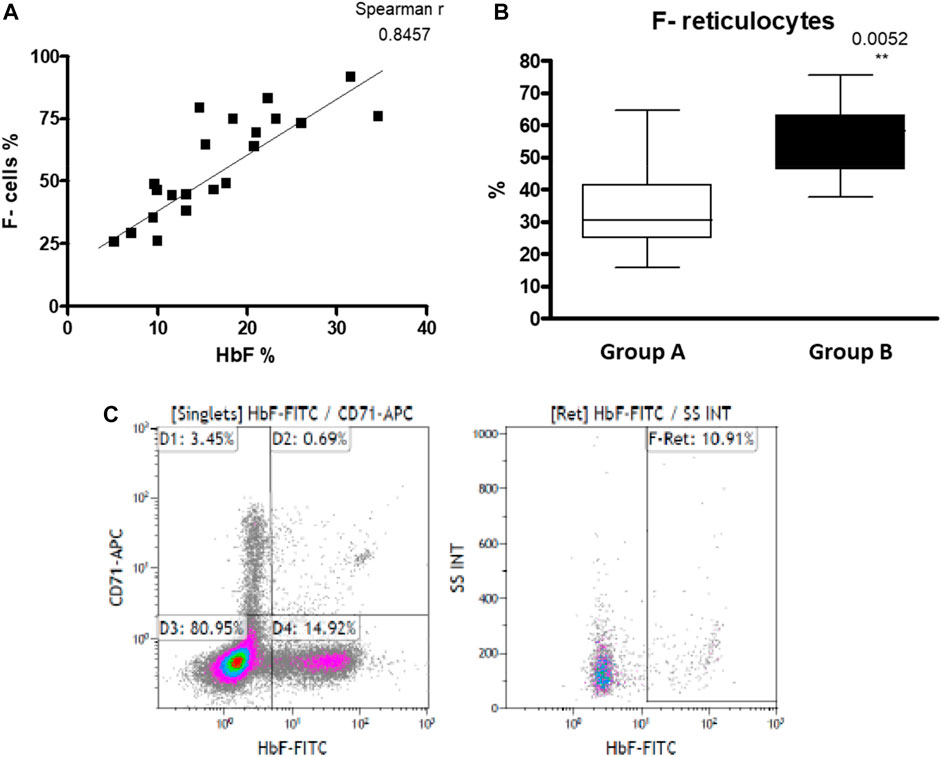
FIGURE 5. HbF in RBC of SCD patients: (A) HbF measured by HPLC and F- cells measured by FC positively correlates (n = 21). (B) F- reticulocytes count in asplenic/hyposplenic (n = 14) vs. hypersplenic (n = 7) SCD subjects. (C) representative FC gating strategy for F- Reticulocytes. Group (A) patients with asplenia/hyposplenism, Group (B) patients with hypersplenism.
Splenic dysfunction examined by the presence of HJB in the RBCs using FC revealed, as expected, a significantly lower count of HJB in the hypersplenism group compared to asplenia/hyposplenism patients in both nucleotides’ dyes; Hoechst 33342 and 7-AAD (0.27 ± 0.2 and 0.26 ± 0.2 vs. 0.43 ± 0.2 and 0.43 ± 0.2 respectively p = 0.01) (Figure 6).
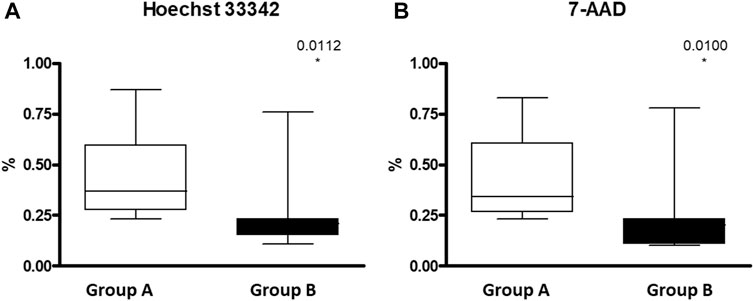
FIGURE 6. HJB inside RBC measured by FC in asplenic/hyposplenis vs. hypersplenic SCD subjects: (A) Hoechst 33342 for A and T nucleotides and (B) 7-AAD (7-amino-actinomycin D) for C and G nucleotides. Group (A) patients with asplenia/hyposplenism, (n = 13). Group (B) patients with hypersplenism, (n = 7).
Representative SEM images of RBCs from healthy and SCD subjects were shown including sickle shape RBC and RBCs with fibrin clot net in a patient with asplenia (Figure 7).
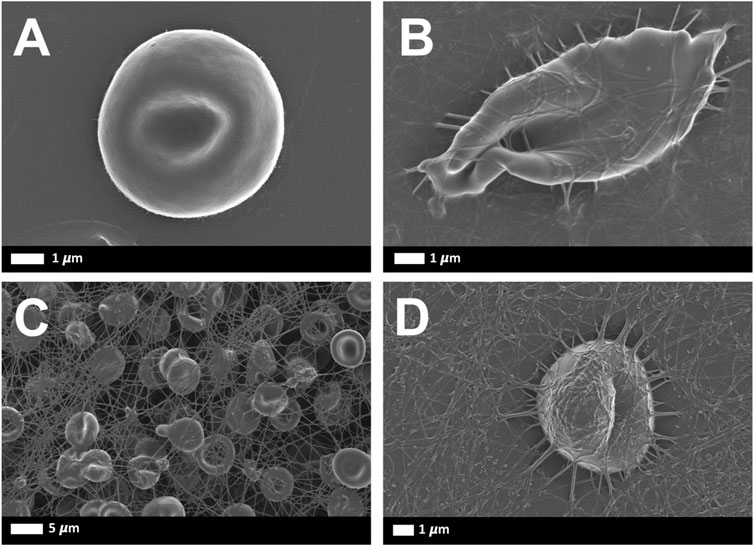
FIGURE 7. Representative scanning electron microscopy images of RBCs from healthy and SCD subjects: (A) Representative RBC from a healthy individual (B) Sickle RBC, (C) RBCs with fibrin net (D) single RBC with fibrin net that spontaneously formed.
The spleen has several functions including clearance of damaged blood cells and antibodies production (Lewis et al., 2019). On the one hand, SCD patients without a spleen suffer from the presence of damaged RBC which can increase the risk of vaso-occlusive crises and overwhelming severe bacterial infections (el Hoss and Brousse, 2019). On the other hand, the negative consequences of hypersplenism are associated with the increased clearance of blood cells and blood-borne antigens, leading to cytopenia and some degree of immunodeficiency caused by the leukopenia (Brousse et al., 2014). Excessive destruction of RBCs in SCD children with prolonged hypersplenism may lead to subsequent growth impairment and bone marrow hyperplasia. Also, transfusion efficiency is usually reduced in SCD patients with hypersplenism (Laws et al., 1979; Lv et al., 2016; Ladu et al., 2021). One of the targets of our study was to use simple tests to monitor spleen status in SCD patients. For example, the use of DNA dyes and the easy handling FC method can serve as a good alternative for HJB count, instead of a labore microscope examination.
The findings of the low Hb, HCT total WBC, WBC subtypes, and platelets in the hypersplenism group can be explained by spleen clearance of blood cells. On the other hand, the relative thrombocytosis in the asplenia/hyposplenism group is related to the lack of platelet clearance by the absent or nonfunctional spleen. Our results related to the blood cells counts confirm most of the previously reported data (al-Salem et al., 1996; Fasola and Adekanmi 2019; el Hoss and Brousse, 2019).
Lower percent of HbS in hypersplenism group compared with the asplenia/hyposplenism group, is probably related to the more effective clearance of sickled RBC by the spleen (Brousse et al., 2014)., It is well known that the HbS content and the percentage of HbF are the main modulators of clinical severity in SCD, and even small changes in sickle RBCs hemoglobin composition may reduce or delay polymerization with great significance on clinical manifestations (Hebert et al., 2020). The slightly higher mean percent of HbF and the F-Cells in the hypersplenism group suggest less Hb polymerization.
Chronic inflammatory state that is part of the pathophysiology of the SCD (Adekile 2013), we observed increased acute-phase reactant proteins in patients with non-functional spleen, the asplenia/hyposplenism group. Among those findings we can mention the higher D Dimer the lower transferrin that are common in anemia of chronic diseases (Costa et al., 2008), and the higher and abnormally elevated CRP and ferritin levels all observed in the asplenia/hyposplenism group.
Also, the high abnormal D-Dimer that we found in the asplenia/hyposplenism group probably indicated a prothrombotic state and not only a marker of inflammation. It is known that in a healthy individual, the main clotting plasma protein, fibrinogen, is in a soluble state. In those subjects’ fibrin network formation can be induced if thrombin is added to whole blood (Palta et al., 2014). Fibrin deposits that form without the addition of thrombin are only noted during pathological clotting (Lipinski et al., 2012). However, in the presence of circulating inflammatory molecules, in inflammatory conditions, the plasma proteins may be induced to form spontaneous fibrin networks, without the presence of thrombin. Since SCD is a chronic inflammatory state, fibrin production can be spontaneously seen (Figures 7C,D). Hypercoagulation and clotting pathologies are well-known accompaniments to inflammatory conditions (Pretorius et al., 2017; Olumuyiwa-Akeredolu et al., 2019; Randeria et al., 2019).
In the current study, we observed normal electrolyte values in our SCD patients, however, the serum potassium and calcium were lower in the hypersplenism group compared to the asplenia/hyposplenism, group probably reflecting reduced intravascular hemolysis. The increase in serum potassium has been described in sickle cell patients, with higher levels during an acute crisis (Pandey et al., 2012; Mansoor et al., 2021). In addition to an increased release caused by the chronic hemolysis, serum K+ abnormalities may be related to the status of hydration, the degree of acidosis during acute events, abnormalities in renal function, and alterations in RBCs ion transporting systems. Some of these RBCs systems such as the Gardos channel, K-Cl cotransporter, and the Na/K pump are directly involved in the maintenance of RBC hydration; their increased activity contributes to RBC dehydration in SCD subjects (Vitoux et al., 1989; Gibson et al., 1998; Izumo et al., 1987; Luthra and Sears 1982; Hannemann et al., 2015).
Percoll density gradient was used as an indication of RBCs dehydration-rehydration state (Hänggi et al., 2014). The reduction of dense/senescent RBC fractions observed in patients with hypersplenism compared with the asplenia/hyposplenism group demonstrates that the hypersplenism group contains more hydrated and less sickled RBCs; which can be considered a beneficial effect related to increased clearance of abnormal RBCs by the spleen.
Nicotinamide adenine dinucleotide (NAD) is a ubiquitous oxidation-reduction (redox) cofactor in RBCs. The synthesis of NAD and its reduced form, NADH, play major roles in maintaining redox balance (Zerez et al., 1988). Sickle RBCs have a lower redox ratio [(NADH):(NAD+ + NADH)] than normal RBCs. Increased reduced glutathione (GSH) availability can decrease oxidative stress status and potentially reduce sickled RBC (Niihara et al., 2018). MBBR interaction with GSH forms a highly fluorescent derivative detected by FC. Here we showed decreased MBBR-high-positive RBC both percentage and MFI in the hypersplenism group compared with the asplenia/hyposplenism group. These results were contrary to what we expected and to the literature. One of our study limitations is the low sample size in both groups. We believe that increasing the sample size can reveal more genuinely the MBBR results and then facilitate explaining those facts. In addition, since reticulocytes contain increased amounts of reduced glutathione than mature RBC (Roth et al., 1979) and we found a 3-fold increase of reticulocytes count in the asplenia/hyposplenism group compared to hypersplenism, we hypothesis that the clearance of reticulocyte by the spleen of hypersplenic patients may have led to decreased MBBR-high-positive RBC which also included reticulocytes.
Regarding the results of the flow cytometry analysis, as previously done (Warang et al., 2011; More et al., 2020), we used SSC and FSC values to assess the RBCs size.
Here we observed an interesting tendency for the RBC hypochromia and a decrease in RBC side scatter value, in patients with hypersplenism compared with the asplenia/hyposplenism group. This suggests on the one hand a reduced number of sickled and hyperchromic RBCs. And on the other hand, less senescent RBCs, normally removed by the spleen, appear to be selectively cleared by the spleen in patients with hypersplenism (Girasole et al., 2012).
This is in line with the significantly lower number of early apoptotic RBCs (annexin V+) in this patient group. Additionally, we show that these morphological changes appear in view of the decrease in the dense RBCs at lower fractions of Percoll density gradient separation as well as with a decrease in the number of RBCs high-positive intracellular Ca2+ concentrations. These findings confirm the previously reported data of higher intracellular Ca2+ content in dense RBCs than in light RBC fractions (Romero et al. 1997). Furthermore, such an increase of RBCs with elevated Ca2+ may be directly connected with the correspondent increase in PS-exposing erythrocytes as shown by Wesseling (Wesseling et al., 2016).
Normally, PS is confined to the cytoplasmic RBC membrane leaflet, PS external expression represents a signal for clearance and early apoptosis of RBCs, known to be increased in SC RBCs.(Hannemann et al., 2018; Thiagarajan et al., 2021; Anke). In accordance with the known evidence that PS externalization is realized by the scramblase activated by an increase of intracellular Ca2+ content (Wesseling et al., 2016), we demonstrate a lower percent of RBCs with abnormal elevated intracellular Ca2+ together with a significant reduction in annexin V + RBCs in hypersplenic subjects.
The fluctuation in intracellular Ca2+ and the increased externalization of PS in RBCs may also contribute to hypercoagulability and the pathological clotting that was noted in the SEM analysis (Figure 7) (Hood et al., 2018; Swieringa et al., 2018; Toorn et al., 2019).
Another parameter used to monitor the severity and drug treatment response in SCD patients is the HbF level analyzed by HPLC, or the F-Cells measured by FC. Hydroxyurea (HU) treatment for SCD patients leads to an increase in HbF and the number of F-Cells RBC concomitant with the improvement of clinical symptoms (Hebert et al., 2020). Hydroxyurea is a safe and effective treatment for SCD, however a proportion of SCA patients are not responding to HU treatment (Pule et al., 2015), in any case, it is important to evaluate patient’s response to this treatment, titrate the dosage and prevent unnecessary overdose. It is known that the clinical response to HU treatment can be evident a few days after treatment initiation and on the opposite sudden stop of HU can provoke an acute pain crisis long before the HbF level drops (Pule et al., 2015).
Measurement of F Reticulocytes using FC, after a few days of HU treatment, will provide a fast understanding of each patient’s response to HU treatment instead of the routinely used HbF measurement analyzed by HPLC, which shows HbF changes usually after 3–4 months (Steinberg et al., 2014). In our study, we found that F Reticulocytes count was significantly higher in hypersplenic subjects compared to asplenia/hyposplenism. We found that the percent of immature RBCs CD71+, reticulocytes expressing the transferrin receptor on the cell surface was lower in the hypersplenism vs. asplenia/hyposplenism group. These results correlate with a decreased trend in total reticulocyte count in the CBC suggesting an accelerated maturation of reticulocytes in hypersplenic subjects and less hemolysis (Lebman et al., 1982; Kono et al., 2009). The impact of the number of circulating reticulocytes as well as its influence on sickling and oxidative stress mechanism in SCD patients need further research.
In patients with functional spleen and hypersplenism the enhanced clearance of sickled RBC (especially due to its pathological morphological and rheological properties), together with the specific slow and open microcirculation in the spleen provides the best platform to favor deoxygenation, promoting RBC sickling and subsequent, accelerating RBC destruction (Brousse et al., 2014). Thus, despite the improvement in RBC rheology and metabolism, these features may be counteracted with aggravation of the anemia and anemia-related complications.
One of the study limitations is the relatively small number of patients studied and more than that this fact did not allow comparison inside each subgroup. In addition, the results of our study should be compared with other clinical parameters like age and clinical severity. The patients with hypersplenism are younger than the patients with asplenia/hyposplenism but they are in the second decade of age, beyond the age when usually asplenia develops in SCD patients. We also suggest, that in the future, a study that will use the power of electron microscopy at single cell level, to make a robust comparative RBCs morphological evaluation of asplenia/hyposplenism vs hypersplenism.
In conclusion, in addition to the known laboratory manifestations of hypersplenism such as severe pancytopenia, our results show some significant “beneficial” laboratory findings including lower HbS, CRP, and D-dimmer values and lower morphological and metabolic pathological properties of the RBCs. The results of this study may provide other tools to define more precisely the spleen function in SCD patients. Given our results, it is extremely important to make any efforts to preserve the spleen in patients with SCD and preserve the immunological and hematological functions including the eradication of abnormal and senescent blood cells. New treatments should be focused on the prevention of functional asplenia and hypersplenism and avoid performing surgical splenectomy.
The benefits and risks of total splenectomy compared to chronic transfusion need to be evaluated in clinical trials and the standard approach to managing hypersplenism in SCD patients should be re-evaluated.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the EMC Emek Medical Center Local Helsinky Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
CL, AK, and MG contributed to the conception and design of the study. AK and CL treated and recruited the patients, and obtained the samples for the study, SP, LL, and EP performed the experiments; SP and LL organized the database and performed the statistical analysis. SP and LL wrote the first draft of the manuscript. AM, AB, and EP wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This project was partially funded by the Fondation Botnar as well as by the Baugarten Stiftung, Susanne & René Braginsky Stiftung and Ernst Göhner Stiftung and the Israeli Ministry for Development of the Negev and the Galilee. The authors thank the staff of Emek Medical Center’s Hematology Laboratory Division for their technical help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adekile A D. (2013)., What’s new in the pathophysiology of sickle cell disease? Med. Princ. Pract. Health Sci. Centre 22. 311–312. doi:10.1159/0003502834
al-Salem A. H., Qaisaruddin S., Nasserallah Z., al Dabbous I., al Jam’a A. (1996). Splenectomy in patients with sickle-cell disease. Am. J. Surg. 172 (3), 254–258. doi:10.1016/S0002-9610(96)00158-4
Bain B J. (2008). Haemoglobinopathy diagnosis. Hemoglobin and the genetic of hemoglobin synthesis. Chapter 1 2nd Edition. John Wiley & Sons. Blackwell Publishing Ltd. 2nd ed. John Wiley & Sons. Blackwell publishing Ltd.
Brousse V, Buffet P, Rees D (2014). The spleen and sickle cell disease: The sick(led) spleen. Br. J. Haematol. 166 (2), 165–176. doi:10.1111/bjh.12950
Brown A. K., Sleeper L. A., Miller S. T., Pegelow C. H., Gill F. M., Waclawiw M. A. (1994). Reference values and hematologic changes from birth to 5 Years in patients with sickle cell disease. Cooperative study of sickle cell disease. Arch. Pediatr. Adolesc. Med. 148 (8), 796–804. doi:10.1001/archpedi.1994.02170080026005
Costa E., Rocha S., Rocha-Pereira P., Reis F., Teixeira F., Miranda V., et al. (2008). Cross-talk between inflammation, coagulation/fibrinolysis and vascular access in hemodialysis patients. J. Vasc. Access 9 (4), 248–253. doi:10.1177/112972980800900405
Diggs L. W., Memphis Tenn (1935). Siderofibrosis of the spleen in sickle cell anemia. J. Am. Med. Assoc. 104, 538–541. doi:10.1001/jama.1935.02760070020005
Eichner E. R. (1979). Splenic function: Normal, too much and too little. Am. J. Med. 66 (2), 311–320. doi:10.1016/0002-9343(79)90554-0
El Hoss S., Cochet S., Marin M., Lapouméroulie C., Dussiot M., et al. (2019). Insights into determinants of spleen injury in sickle cell anemia. Blood Adv. 3 (15), 2328–2336. doi:10.1182/bloodadvances.2019000106
El Hoss S., Dussiot M., Renaud O., Brousse V., El Nemer W. (2018). A novel non-invasive method to measure splenic filtration function in humans. Haematologica 103 (10), e436–39. doi:10.3324/haematol.2018.188920
Emond A. M., Morais P., Venugopal S., Carpenter R. G., Serjeant G. R. (1984). Role of splenectomy in homozygous sickle cell disease in childhood. Lancet (London, Engl. 1 (8368), 88–91. doi:10.1016/s0140-6736(84)90014-x
Fasola F. A., Adekanmi A. J. (2019). Haematological profile And blood transfusion pattern of patients with sickle cell anaemia vary with spleen size. Ann. Ib. Postgrad. Med. 17 (1), 30
Franceschi Lucia de, Domenica Cappellini Maria, Olivieri Oliviero (2011). Thrombosis and sickle cell disease. Semin. Thromb. Hemost. 37 (3), 226–236. doi:10.1055/s-0031-1273087
Gibson J. S., Speake P. F., Ellory J. C. (1998). Differential oxygen sensitivity of the K+-Cl- cotransporter in normal and sickle human red blood cells. J. Physiol. 511, 225–234. doi:10.1111/j.1469-7793.1998.225bi.x
Girasole M., Dinarelli S., Boumis G. (2012). Structural, morphological and nanomechanical characterisation of intermediate states in the ageing of erythrocytes. J. Mol. Recognit. 25 (5), 285–291. doi:10.1002/jmr.2170
Hänggi P., Makhro A., Gassmann M., Schmugge M., Goede J. S., Speer O., et al. (2014). Red blood cells of sickle cell disease patients exhibit abnormally high abundance of N-methyl D-aspartate receptors mediating excessive calcium uptake. Br. J. Haematol. 167 (2), 252–264. doi:10.1111/bjh.13028
Hannemann A., Rees D. C., Tewari S., Gibson J. S. (2015). Cation homeostasis in red cells from patients with sickle cell disease heterologous for HbS and HbC (HbSC genotype). EBioMedicine 2 (11), 1669–1676. doi:10.1016/j.ebiom.2015.09.026
Hannemann A, Rees D C., Brewin J N., Noe A, Low B, Gibson J S. (2018). Oxidative stress and phosphatidylserine exposure in red cells from patients with sickle cell anaemia. Br. J. Haematol. 182 (4), 567–578. doi:10.1111/bjh.15441
Harrod V L., Howard T A., Zimmerman S A., Dertinger S D., RussellWare E. (2007). Quantitative analysis of howell-jolly bodies in children with sickle cell disease. Exp. Hematol. 35 (2), 179–183. doi:10.1016/j.exphem.2006.09.013
Hebert N, Rakotoson M G, Bodivit G, Audureau E, Bencheikh L, Kiger L, et al. (2020). Individual red blood cell fetal hemoglobin quantification allows to determine protective thresholds in sickle cell disease. Am. J. Hematol. 95 (11), 1235–1245. doi:10.1002/ajh.25937
Hood J E., Yesudasan S, Averett R (2018). Glucose concentration affects fibrin clot structure and morphology as evidenced by fluorescence imaging and molecular simulations. Clin. Appl. Thromb. Hemost. 24, 104S-116S–116S. doi:10.1177/1076029618792304
el Hoss S., Brousse V. (2019). Considering the spleen in sickle cell disease. Expert Rev. Hematol. 12 (7), 563–573. doi:10.1080/17474086.2019.1627192
Iolascon A, Andolfo I, Barcellini W, Corcione F, Garçon L, de Franceschi L, et al. (2017). Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica 102 (8), 1304–1313. doi:10.3324/haematol.2016.161166
Izumo H., Lear S., Williams M., Rosa R., Epstein F. H. (1987). Sodium-potassium pump, ion fluxes, and cellular dehydration in sickle cell anemia. J. Clin. Invest. 79 (6), 1621–1628. doi:10.1172/JCI112998
Kämpf S, Seiler E, Bujok J, Hofmann-Lehmann R, Riond B, Makhro A, et al. (2019). Aging markers in equine red blood cells. Front. Physiol. 10, 893. doi:10.3389/fphys.2019.00893
Kono M, Kondo T, Takagi Y, Wada A, Fujimoto K (2009). Morphological definition of CD71 positive reticulocytes by various staining techniques and electron microscopy compared to reticulocytes detected by an automated Hematology analyzer. Clin. Chim. Acta. 404 (2), 105–110. doi:10.1016/j.cca.2009.03.017
Kuypers F. A., Lewis R. A., Hua M., Schott M. A., Discher D., Ernst J. D., et al. (1996). Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood 87 (3), 1179–1187. doi:10.1182/blood.v87.3.1179.bloodjournal8731179
Kyaw M H., Holmes E M., Toolis F, Wayne B, Chalmers J., Jones Ian G., et al. (2006). Evaluation of severe infection and survival after splenectomy. Am. J. Med. 119 (3), 276e1–e7. doi:10.1016/j.amjmed.2005.07.044
Ladu A I., O Aiyenigba A, Adekunle A, Bates I (2021). The spectrum of splenic complications in patients with sickle cell disease in africa: A systematic review. Br. J. Haematol. 193 (1), 26–42. doi:10.1111/bjh.17179
Laws H. L., Burlingame M. W., Carpenter J. T., Prchal J. T., Conrad M. E. (1979). Splenectomy for hematologic disease. Surg. Gynecol. Obstet. 149 (4), 509
Lebman D., Trucco M., Bottero L., Lange B., Pessano S., Rovera G. (1982). A monoclonal antibody that detects expression of transferrin receptor in human erythroid precursor cells. Blood 59 (3), 671–678. doi:10.1182/blood.v59.3.671.bloodjournal593671
Leone G, Pizzigallo E (2015). Bacterial infections following splenectomy for malignant and nonmalignant hematologic diseases. Mediterr. J. Hematol. Infect. Dis. 7 (1), e2015057–e21. doi:10.4084/MJHID.2015.057
Lesher A P., Ram K, Glenn J B., Jackson S M., Andre H (2009). Outcome of splenectomy in children younger than 4 Years with sickle cell disease. J. Pediatr. Surg. 44 (6), 1134–1138. doi:10.1016/j.jpedsurg.2009.02.016
Lewis S M., Williams A, Eisenbarth S C. (2019). Structure and function of the immune system in the spleen. Sci. Immunol. 4 (33), eaau6085. doi:10.1126/sciimmunol.aau6085
Lipinski B, Pretorius E, Oberholzer H M., J van der Spuy W (2012). Interaction of fibrin with red blood cells: The role of iron. Ultrastruct. Pathol. 36 (2), 79–84. doi:10.3109/01913123.2011.627491
Luthra M. G., Sears D. A. (1982). Increased Ca++, Mg++, and Na+ + K+ ATPase activities in erythrocytes of sickle cell anemia. Blood 60 (6), 1332–1336. doi:10.1182/blood.v60.6.1332.bloodjournal6061332
Luu S, Spelman D, Woolley I J. (2019). Post-splenectomy sepsis: Preventative strategies, challenges, and solutions. Infect. Drug Resist. 12, 2839–2851. doi:10.2147/IDR.S179902
Lv Y, Lau W Y, Li Y, Deng J, Han X, Gong X, et al. (2016). Hypersplenism: History and current status. Exp. Ther. Med. 12 (4), 2377–2382. doi:10.3892/etm.2016.3683
Makhro A, Kaestner L, Anna B (2017)., NMDA receptor activity in circulating red blood cells: Methods of detection. Methods Mol. Biol. 1677, 265–282. doi:10.1007/978-1-4939-7321-7_15
Makhro A, Pascal H, Goede J S., Wang J, Brüggemann A, Gassmann M., et al. (2013). N-Methyl-D-Aspartate receptors in human erythroid precursor cells and in circulating red blood cells contribute to the intracellular calcium regulation. Am. J. Physiol. Cell Physiol. 305 (11), c1123–38. doi:10.1152/ajpcell.00031.2013
Mansoor F, Bai P, Kaur N, Sultan S, Sharma S, Dilip A, et al. (2021). Evaluation of serum electrolyte levels in patients with anemia. Cureus 13 (10), e18417. doi:10.7759/cureus.18417
More T A, Dalal B, Devendra R, Warang P, Shankarkumar A, Kedar P (2020). Applications of imaging flow cytometry in the diagnostic assessment of red cell membrane disorders. Cytom. B Clin. Cytom. 98 (3), 238–249. doi:10.1002/cyto.b.21857
Niihara Y, ScottMiller T., Kanter J, Lanzkron S, Smith W R., Hsu L L., et al. (2018). A phase 3 trial of L-glutamine in sickle cell disease. N. Engl. J. Med. 379 (3), 226–235. doi:10.1056/NEJMoa1715971
O’Brien R T., Sue McIntosh G T. A, Pearson H A., Pearson H. A. (1976). Prospective study of sickle cell anemia in infancy. J. Pediatr. 89 (2), 205–210. doi:10.1016/S0022-3476(76)80449-0
Olumuyiwa-Akeredolu O. O., Page M. J., Soma P., Pretorius E. (2019). Platelets: Emerging facilitators of cellular crosstalk in rheumatoid arthritis. Nat. Rev. Rheumatol. 15 (4), 237–248. doi:10.1038/s41584-019-0187-9
Palta S, Saroa R, Anshu P (2014). Overview of the coagulation system. Indian J. Anaesth. 58 (5), 515–523. doi:10.4103/0019-5049.144643
Pandey S., Sharma A., Dahia S., Shah V., Sharma V., Mishra R. M., et al. (2012). Biochemical indicator of sickle cell disease: Preliminary report from India. Indian J. Clin. biochem. 27 (2), 191–195. doi:10.1007/s12291-011-0162-y
Pearson H. A., McIntosh S., Ritchey A. K., Lobel J. S., Rooks Y., Johnston D. (1979). Developmental aspects of splenic function in sickle cell diseases. Blood 53 (3), 358–365. doi:10.1182/blood.v53.3.358.358
Pearson H. A., Spencer R. P., Cornelius E. A. (1969). Functional asplenia in sickle-cell anemia. N. Engl. J. Med. 281 (17), 923–926. doi:10.1056/NEJM196910232811703
Piel F B., Hay S I., Gupta S, Weatherall D J., Williams T N. (2013). Global burden of sickle cell anaemia in children under five, 2010-2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 10 (7), e1001484. doi:10.1371/journal.pmed.1001484
Platt O. S. (2000). The acute chest syndrome of sickle cell disease. N. Engl. J. Med. 342 (25), 1904–1907. doi:10.1056/NEJM200006223422510
Pretorius E, Swanepoel A C., DeVilliers S, Bester J (2017). Blood clot parameters: Thromboelastography and scanning electron microscopy in research and clinical practice. Thromb. Res. 154, 59–63. doi:10.1016/j.thromres.2017.04.005
Pule G D., Mowla S, Novitzky N, Wiysonge C S., Ambroise W (2015). A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev. Hematol. 8 (5), 669–679. doi:10.1586/17474086.2015.1078235
Randeria Shehan N., Thomson Greig J. A., Timothy Roberts Theo A. Nell, Pretorius Etheresia, Pretorius E. (2019). Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc. Diabetol. 18 (1), 72. doi:10.1186/s12933-019-0870-9
Romero P. J., Romero E. A., Winkler M. D. (1997). Ionic calcium content of light dense human red cells separated by Percoll density gradients. Biochim. Biophys. Acta 1323 (1), 23–28. doi:10.1016/s0005-2736(96)00141-1
Roth E. F., Grossman H. A. H. T, Nagel R. L. (1979). Reticulocytes contain increased amounts of reduced glutathione except when produced by acetylphenylhydrazine. Biochem. Med. 21 (3), 333–341. doi:10.1016/0006-2944(79)90087-5
Samuk I, Seguier-Lipszyc E, Baazov A, Tamary H, Nahum E, Steinberg R, et al. (2019). Emergency or urgent splenectomy in children for non-traumatic reasons. Eur. J. Pediatr. 178 (9), 1363–1367. doi:10.1007/s00431-019-03424-6
Sato S., Kozuma Y., Hasegawa Y., Kojima H., Chiba S., Ninomiya H. (2010). Enhanced expression of CD71, transferrin receptor, on immature reticulocytes in patients with paroxysmal nocturnal hemoglobinuria. Int. J. Lab. Hematol. 32, e137–43. doi:10.1111/j.1751-553X.2009.01148.x
Serjeant G. R., Sommereux A. M., Stevenson M., Mason K., Serjeant B. E. (1979). Comparison of sickle cell-beta0 thalassaemia with homozygous sickle cell disease. Br. J. Haematol. 41 (1), 83–93. doi:10.1111/j.1365-2141.1979.tb03684.x
Steinberg M H., Chui D H. K., GeorgeDover J., Paola S, Alsultan A (2014). Fetal hemoglobin in sickle cell anemia: A glass half full? Blood 123 (4), 481–485. doi:10.1182/blood-2013-09-528067
Swieringa F, Spronk Hi M. H., Heemskerk J W. M., van der Meijden Paola E. J. (2018). Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2 (3), 450–460. doi:10.1002/rth2.12107
Tahir F, Ahmed J, Malik F (2020). Post-splenectomy sepsis: A review of the literature. Cureus 12 (2), e6898. doi:10.7759/cureus.6898
Thiagarajan P, Parker C J., JosefPrchal T. (2021). How do red blood cells die? Front. Physiol. 12, 655393. doi:10.3389/fphys.2021.655393
Toorn F A. v d, de Mutsert R, WillemLijfering M., FritsRosendaal R., Vlieg A v H (2019). Glucose metabolism affects coagulation factors: The NEO study. J. Thromb. Haemost. 17 (11), 1886–1897. doi:10.1111/jth.14573
Vitoux D, Olivieri O, Garayt R P., Cragoe E J., Galacteros F, Beuzard Y (1989). Inhibition of K+ efflux and dehydration of sickle cells by [(dihydroindenyl)oxy]alkanoic acid: An inhibitor of the K+ Cl- cotransport system. Proc. Natl. Acad. Sci. U. S. A. 86, 4273–4276. doi:10.1073/pnas.86.11.4273
Warang P, Gupta M, Kedar P, Ghosh K, Colah R (2011). Flow cytometric osmotic fragility--an effective screening approach for red cell membranopathies. Cytom. B Clin. Cytom. 80 (3), 186–190. doi:10.1002/cyto.b.20583
Ware Rl E., de Montalembert M, Tshilolo L, Abboud M R. (2017). Sickle cell disease. Lancet (London, Engl. 390 (10091), 311–323. doi:10.1016/S0140-6736(17)30193-9
Wesseling M C., Wagner-Britz L, Huppert H, Benjamin H, Hertz L, Nguyen D B, et al. (2016). Phosphatidylserine exposure in human red blood cells depending on cell age. Cell. Physiol. biochem. 38 (4), 1376–1390. doi:10.1159/000443081
Yacobovich J, Barzilai-Birenboim S, Steinberg-Shemer Oa, Stark P, Pazgal I, Tamary H (2020). Splenectomy in childhood for non-malignant haematologic disorders - long-term follow-up shows minimal adverse effects. Br. J. Haematol. 190 (6), 909–915. doi:10.1111/bjh.16657
Zago M. A., Costa F. F., Freitas T. C., Bottura C. (1980). Clinical, hematological and genetic features of sickle-cell anemia and sickle cell-beta thalassemia in a Brazilian population. Clin. Genet. 18 (1), 58–64. doi:10.1111/j.1399-0004.1980.tb01366.x
Keywords: hypersplenism, asplenia, fetal hemoglobin, reticulocytes, sickle cell disease, hyposplenism
Citation: Peretz S, Livshits L, Pretorius E, Makhro A, Bogdanova A, Gassmann M, Koren A and Levin C (2022) The protective effect of the spleen in sickle cell patients. A comparative study between patients with asplenia/hyposplenism and hypersplenism. Front. Physiol. 13:796837. doi: 10.3389/fphys.2022.796837
Received: 17 October 2021; Accepted: 02 August 2022;
Published: 29 August 2022.
Edited by:
Eitan Fibach, Hadassah Medical Center, IsraelReviewed by:
Alessandro Matte’, University of Verona, ItalyCopyright © 2022 Peretz, Livshits, Pretorius, Makhro, Bogdanova, Gassmann, Koren and Levin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carina Levin, bGV2aW5fY0BjbGFsaXQub3JnLmls,TGV2aW5jYXJpbmFAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.