 Galina Yu Mironova
Galina Yu Mironova Nadia Haghbin
Nadia Haghbin Donald G. Welsh
Donald G. Welsh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 15 November 2022
Sec. Vascular Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.1058744
This article is part of the Research TopicMolecular Physiology of Smooth Muscle CellsView all 6 articles
Vascular smooth muscle contraction is intimately tied to membrane potential and the rise in intracellular Ca2+ enabled by the opening of L-type Ca2+ channels. While voltage is often viewed as the single critical factor gating these channels, research is starting to reveal a more intricate scenario whereby their function is markedly tuned. This emerging concept will be the focus of this three-part review, the first part articulating the mechanistic foundation of contractile development in vascular smooth muscle. Part two will extend this foundational knowledge, introducing readers to functional coupling and how neighboring L-type Ca2+ channels work cooperatively through signaling protein complexes, to facilitate their open probability. The final aspect of this review will discuss the impact of L-type Ca2+ channel trafficking, a process tied to cytoskeleton dynamics. Cumulatively, this brief manuscript provides new insight into how voltage, along with channel cooperativity and number, work in concert to tune Ca2+ responses and smooth muscle contraction.
Mechanical and chemical stimuli initiate vascular smooth muscle contraction through transduction pathways that enhance myosin light chain phosphorylation (Davis et al., 1999). This key biological event is set by the balance of two central enzymes, those being myosin light chain kinase and myosin light chain phosphatase (Takashima, 2009). Myosin light chain kinase is a serine/threonine-specific protein kinase responsible for phosphorylating Ser19 on the N-terminus of the regulatory light chain of the motor protein myosin-II (Figure 1). This enzyme’s activity is intimately tied to intracellular Ca2+ ([Ca2+]i) and its binding to low-affinity sites of kinase-bound Calmodulin, a messenger protein that interferes with the autoinhibitory domain (Herring et al., 2006; Schaub, 2007; Walsh, 2011). In contrast, myosin light chain phosphatase is a holoenzyme composed of three subunits: a 38 kDa catalytic subunit of type 1 protein phosphatase (PP1c), a 110–130 kDa regulatory subunit (MYPT1), and a small 20 kDa subunit of unknown function (Ding et al., 2006). MYPT1 is a key regulator of activity as phosphorylation at Thr853 or Thr696 inhibits PP1c, pushing the kinase-phosphatase balance towards enhanced myosin light chain phosphorylation and smooth muscle contraction. The control of MYTP1 phosphorylation is, in turn, set by signal transduction pathways tied to G-protein receptors, two of note being Gq/11 and G12/13. The RhoA/Rho-kinase pathway is particularly important, and when inhibited pharmacologically (e.g., Y27632 or H1152), MYPT1 phosphorylation, myosin light chain phosphatase activity and smooth muscle contraction are diminished (Fu et al., 1998; Anneloes Martinsen et al., 2012). Downstream signaling proteins such as PKC also limit myosin light chain phosphatase activity by phosphorylating CPI-17, a direct inhibitor of the catalytic unit PP1c (Deng et al., 2002).
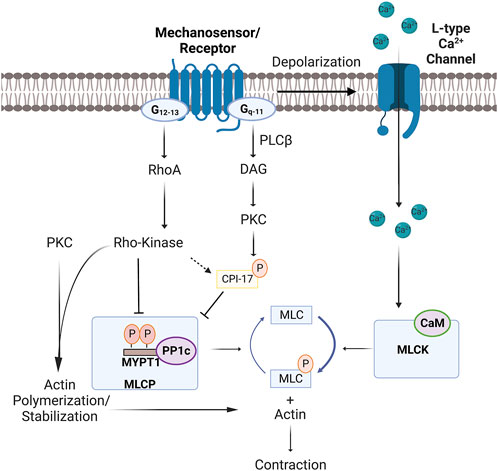
FIGURE 1. Mechanisms of cross-bridge cycling and MLC20 phosphorylation in vascular smooth muscle. Mechanosensor/receptor activation induces depolarization by activating transient receptor potential channels and inhibiting hyperpolarizing K+ currents. Depolarization opens L-type Ca2+ channels, with the resulting influx of Ca2+ enhancing MLCK and cross-bridge cycling through binding to CaM. Conversely, G-protein coupled receptors further enhance contraction by inhibiting MLCP (aka-Ca2+ sensitization) through two regulatory pathways. The first entails PKC activation by PLC-β and DAG, a series/threonine kinase that phosphorylates: 1) CPI-17 to inhibit PP1c; or 2) target proteins that set actin stabilization/polymerization. The second involves G12-13 activation of RhoA and Rho-kinase, the latter of which controls the phosphorylation state of: 1) MYPT1 (Threonine–696 and Threonine–853 (mouse numberings)); or 2) target proteins that set actin polymerization. Note, Ca2+ release from the sarcoplasmic reticulum, resulting from receptor-induced IP3 production, can also contribute to MLCK activation. Abbreviations: MLCK, Myosin light chain kinase; MLCP, Myosin light chain phosphatase, PLC, Phospholipase C; DAG, Diacylglycerol; CPI-17, Cytosolic phosphatase inhibitory protein of 17-kDa; IP3, Inositol trisphosphate; MYPT1, Myosin light chain phosphatase targeting subunit 1; MLC, Myosin regulatory light chain; CaM, Calmodulin; PKC, Protein Kinase C; PP1c, Type 1 protein phosphatase 1c.
Ca2+ sensitization is a colloquial term used in vascular biology to describe how force development can be tuned relative to the rise in [Ca2+]i, the latter set by membrane depolarization. While this term is traditionally tied to phosphatase modulation, its mechanistic underpinnings have expanded to regulatory processes linked to actin-myosin interaction and the structures responsible for force transmission. As to the former, consider proteins such as Caldesmon, which, when bound to actin, stabilize it and impairs myosin ATPase activity (Clark et al., 1986). G-protein-linked signaling proteins like PKC diminish Caldesmon’s inhibitory effects by loosening its physical binding to the thin filaments (Clark et al., 1986). Calponin is another example of an actin-myosin binding protein inhibiting ATPase activity through an interaction with and phosphorylation by RhoA/Rho-kinase (Kaneko et al., 2000). With respect to the latter, consider current experimental interest in cytoskeletal remodeling, an event strongly, although not exclusively, tied to the state of actin polymerization. This dynamic process is regulated by several transduction pathways, one of note to G-protein coupled receptors and downstream Rho/Rho-kinase signaling being LIM kinase regulation of Cofilin, a protein that guides actin depolymerization (Walsh et al., 2013).
The synopsis above briefly highlights how vascular smooth muscle, through multiple points of regulation, can tune contractile development to a voltage-dependent rise in [Ca2+]i. Decidedly absent from this discussion is whether L-type Ca2+ channels themselves can be functionally tuned. This idea was first raised by Fallon and colleagues who noted that these channel’s C-termini interact with one another (Fallon et al., 2009), facilitating a state whereby the opening of one channel fosters the opening of a companion. The result of said “functional coupling” would be enhanced Ca2+ influx and contraction at a defined voltage (Dixon et al., 2022). An alternative means of so called “tuning” would be to traffic additional L-type Ca2+ channels to the plasma membrane to enhance cluster formation and cooperative gating (Ghosh et al., 2018). Both aspects of regulatory control (i.e., Cooperative gating and Channel trafficking) will be discussed in the subsequent chapters by highlighting key literature and classic experiments.
Vascular L-type Ca2+ channels are comprised of a CaV1.2 α1 pore-forming subunit along with an auxiliary α2 (150 kDa), δ (17–25 kDa), β (50–78 kDa), and γ (32 kDa) subunit to ensure proper gating, regulation, and trafficking (Catterall et al., 2003; Feng et al., 2018). The CaV1.2 α1 subunit retains the transmembrane sequences that confer voltage-gating, and a C-terminus is notable for a diverse array of regulatory sites. L-type Ca2+ channels are the primary drivers of Ca2+ influx in vascular smooth muscle, and traditional physiology assumes their activity is nearly exclusively set by voltage, with each channel operating independently of one another. Observations collected over the past decade have begun to challenge this dogma by noting that subpopulations of closely situated L-type Ca2+ channels work cooperatively with one another to enhance their open probability. First described in 2005 by Navedo and others, L-type Ca2+ channels were rationalized to cluster on the plasma membrane in a configuration where the open probability of each individual channel was markedly higher (Navedo et al., 2005). The clustering of active channels creates regions on the plasma membrane of persistent Ca2+ influx, resulting from the generation of so-called Ca2+ sparklets (Figure 2A). Note, Ca2+ sparklets differ from Ca2+ sparks, events driven by ryanodine channels on the sarcoplasmic reticulum, as their duration is longer, their amplitude coupled to voltage, and their pharmacological profile distinct (Navedo et al., 2005). The latter is exemplified by the nifedipine block of Ca2+ sparklets, akin to that of L-type Ca2+ currents; in contrast, nifedipine has no effects on Ca2+ sparks. While data is limited, Amberg and others argued that ∼50% of SMC Ca2+ current is sparklets-related (Amberg et al., 2007), the remaining current being assigned to non-coupled independent channels.
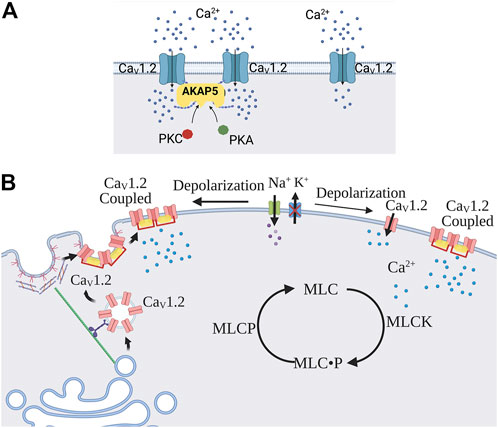
FIGURE 2. Cooperative gating and trafficking of L-type Ca2+ channels. (A) L-type Ca2+ channels can work as independent channels or in a cooperative gating manner due to proximate localization and the formation of connections at the CaV1.2 C-terminus. Cooperatively gated channels exhibit functional coupling properties, such as synchronous opening, higher Po, and more persistent current compared to individually gated channels. AKAP5 (mouse numbering), PKC, and PKA play a critical role in the promotion of functional coupling properties. (B) Depolarization enhances the trafficking of L-type Ca2+ channel subunits to the plasma membrane. This key biological process entails the movement of vesicles from the Golgi apparatus to caveolae strategically positioned on the plasma membrane. Abbreviations: AKAP5, A-kinase anchoring protein -5; PKA, Protein kinase A; PKC, Protein kinase C; MLCK, Myosin light chain kinase; MLCP, Myosin light chain phosphatase.
The regulatory elements driving Ca2+ sparklets activity have become a source of active experimental inquiry. Initial work argued that PKCα was an essential activator of Ca2+ sparklets, consistent with its impact on the whole cell L-type current (Wesselman et al., 2001; Korzick et al., 2004; Jarajapu et al., 2005; Amberg et al., 2007). Aligning with and building upon these findings, immunohistochemical and TIRF microscopy observations revealed close spatial membrane localization of Ca2+ sparklets, CaV1.2 clusters, and PKCα (Santana et al., 2008). The anchoring of PKCα in close proximity to CaV1.2 is achieved through A-kinase anchoring protein, AKAP5, also known as AKAP150 (mice) and AKAP79 (humans), being prominently expressed (Santana et al., 2009; Perino et al., 2012) and capable of binding to the C-terminus of the α1 subunit (Fallon et al., 2009; Dixon et al., 2015). PKCα release leads to displacement of Calmodulin from IQ domain which decreases Ca2+-induced inactivation of L-type Ca2+ channels (Faux and Scott, 1997; Santana et al., 2009). In light of AKAP5 bringing L-type Ca2+ channels in close apposition to PKCα (Coghlan et al., 1995; Oliveria et al., 2007), it follows that Ca2+ sparklets regulatory control is lost in AKAP150 deletion mice (Navedo et al., 2008). Subsequent studies have revealed that PKA and Calcineurin also bind AKAP5 in close apposition to L-type Ca2+ channels adding another regulatory layer to Ca2+ sparklets activity. PKA mobilization leads to phosphorylation of α1 subunit C-terminus at the Ser 1928, increasing the open probability of L-type Ca2+ channels (Nystoriak et al., 2017; Prada et al., 2019; Syed et al., 2019; Prada et al., 2020). In contrast, Calcineurin’s effects oppose PKCα, with its activation limiting Ca2+ sparklets activity (Navedo et al., 2006; Santana et al., 2009). This Yin-Yang relationship between PKA, Calcineurin, and PKCα can be functionally viewed as creating a flexible platform for Ca2+ sparklets regulation (Navedo et al., 2006; Santana et al., 2009).
Moving beyond cellular observations, the next logical question centers on the physiological and pathobiological role of Ca2+ sparklets. As to the former, experiments performed on AKAP−/− and PKCα−/− have observed that limited Ca2+ sparklets activity coincides with a marked reduction in myogenic tone, suggestive of the former driving the latter (Navedo et al., 2008; Navedo et al., 2010a). Secondly, it has been argued that clustered L-type Ca2+ channels are loosely coupled with ryanodine receptors and SERCA pump, thus impacting the Ca2+ load/release of the sarcoplasmic reticulum (Navedo et al., 2010b). This idea aligns with: 1) structural data showing close apposition of CaV1.2 clusters with sarcoplasmic reticulum release/uptake proteins; and 2) functional data noting that regions of CaV1.2-Ca2+ sparklet activity overlap with areas notable for the tranisent Ca2+ release from ryanodine receptors. Moreover, experimental data demonstrates that reducing Ca2+ sparklets activity notably slows Ca2+ refilling of the sarcoplasmic reticulum (Essin et al., 2007; Takeda et al., 2011).
Considering the preceding physiological observations, it follows that pathobiological processes impinging on Ca2+ sparklet activity will be destined to impact the contractile state of vascular smooth muscle. For example, the marked upregulation/AKAP binding of PKCα (hypertension) and PKA (hyperglycemia and diabetes type II) is notable for enhancing arterial tone (Navedo et al., 2008; Navedo et al., 2010a; Navedo et al., 2010b). The latter observation highlights the importance of localized PKA signaling as global activation will relax vascular smooth muscle through hyperpolarization (Navedo et al., 2010a; Morotti et al., 2017; Nystoriak et al., 2017; Prada et al., 2019). Genetic mutations to the CaV1.2 C-terminus, mimicking those observed with Timothy Syndrome, also impact functional coupling and vessel contractility (Navedo et al., 2010b; Napolitano et al., 2014; Priori et al., 2018; Han et al., 2019). So albeit in physiology or pathobiology, the dynamic balancing of PKCα, PKA, and Calcineurin activity is critical to the functional tuning of L-type Ca2+ channels as they respond to defined voltage stimuli. The recent review by Dixon and colleagues provides a more detailed examination of this phenomenon. (Dixon et al., 2022).
An alternative means to enhance the Ca2+ influx response to depolarization is to increase the number of L-type Ca2+ channels embedded in the plasma membrane. Protein trafficking is key, and work in expression systems provides foundational knowledge of how CaV1.2 subunits are chaperoned to and inserted into the plasma membrane (Figure 2B). Following synthesis in the sarcoplasmic reticulum, CaV1.2 subunits are packaged into vesicle structures which are then guided to the membrane along structural filaments, including actin fibers and microtubules (Simms et al., 2012). This movement is enabled by the key motor proteins, kinesin and dynein, and interestingly these vesicles can switch from actin to microtubules and visa versa, making the trafficking flexible and sensitive to changes in cytoskeletal reorganization (Ross et al., 2008; Smyth et al., 2010). Observing trafficking behavior in tsA-201 cells, Ghosh and others intriguingly noted that a resident pool of CaV1.2 containing vesicles displays a distinct pattern of movement and interaction with the plasma membrane (Ghosh et al., 2018). This included vesicular structures undergoing a dynamic fusion and fission on a second-time scale, with fusion processes displaying “kiss and stay” and “kiss and run” behavior. This study also noted that recently incorporated vesicles displayed Ca2+ sparklets activity, consistent with channel clustering, and this process was dependent on the cytoskeleton (Ghosh et al., 2018). While observations are limited in vascular smooth muscle cells, evidence points to caveolae being a site of convergence for L-type Ca2+ channels in resistance arteries (Martinsen et al., 2014; Suzuki et al., 2013). Little is known of the stimuli that foster L-type Ca2+ channel trafficking, but their targeted transport to caveolae suggests a potential regulatory role for mechanical forces like pressure. This perspective aligns with findings from acute hypertension models, where increased β and α2δ subunit expression (Herlitze et al., 2003; Klugbauer et al., 2003) is associated with enhanced surface expression of CaV1.2 subunits and vasoconstrictor drive (Bannister et al., 2012).
This mini-review summarizes current thinking on how the functional tuning of L-type Ca2+ channels could be tuned in vascular smooth muscle to impact Ca2+ influx and tissue contractility. Mechanisms of note include; 1) cooperative gating among neighboring L-type Ca2+ channels; and 2) stimulus-induced protein trafficking. Experimental research now defines how each mechanism is regulated by protein kinases, anchoring proteins, cytoskeletal structures, and initiating stimuli. Translating this knowledge into relevant biological settings is the next Frontier and one destined to intrigue the next generation of vascular biologists. Are, for example, the number and size of L-type Ca2+ channel clusters truly tuned in temporally concert with the changing physiological environment? Likewise, how do L-type Ca2+ channel clusters change in pathobiological settings like sepsis, where the proinflammatory environment progressively leads to circulatory collapse? This deeper understanding of L-type Ca2+ channels and its linkage to vascular tone is expected to reveal new conceptual avenues for therapeutic development.
GM: writing original draft, Figure 2, editing. NH: writing original draft, Figure 1. DW: editing, supervision.
This work is supported by an operating grant to the Canadian Institute of Health Research (FRN159667). DW is the Rorabeck Chair in Vascular Biology and Molecular Neuroscience. The figures for the article were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AKAP5, A-kinase anchoring protein-5; CPI-17, cytosolic phosphatase inhibitory protein of 17-kDa; MYPT1, Myosin light chain phosphatase targeting subunit 1; PKA, Protein kinase A; PKC, Protein Kinase C; PP1c, Type 1 protein phosphatase 1c.
Amberg G. C., Navedo M. F., Nieves-cintrón M., Molkentin J. D., Santana L. F. (2007). Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J. Physiol. 579 (1), 187–201. accessed September 21, 2022, from/pmc/articles/PMC2075382/, February 2. doi:10.1113/JPHYSIOL.2006.124420
Bannister J. P., Bulley S., Narayanan D., Thomas-Gatewood C., Luzny P., Pachuau J., et al. (1979). Transcriptional upregulation of α2δ-1 elevates arterial smooth muscle cell voltage-dependent Ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension 60 (4), 1006–1015. accessed September 25, 2022, from https://pubmed.ncbi.nlm.nih.gov/22949532/, October 2012. doi:10.1161/HYPERTENSIONAHA.112.199661
Catterall W. A., Striessnig J., Snutch T. P., Perez-Reyes E. (2003). International union of pharmacology. XL. Compendium of voltage-gated ion channels: Calcium channels. Pharmacol. Rev. 1, 579–581. doi:10.1124/pr.55.4.8
Clark T., Ngai P. K., Sutherland C., Gröschel-Stewart U., Walsh M. P., Groschel-Stewart U. (1986). Vascular smooth muscle Caldesmon. J. Biol. Chem. 261 (17), 8028–8035. accessed September 21, 2022, June 15. doi:10.1016/S0021-9258(19)57507-X
Coghlan V. M., Perrino B. A., Howard M., Langeberg L. K., Hicks J. B., Gallatin W. M., et al. (1995). Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Sci. (New York, N.Y.) 267 (5194), 108–111. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/7528941/. doi:10.1126/SCIENCE.7528941
Davis M. J., Hill M. A. (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79 (2), 387–423. accessed September 18, 2022. doi:10.1152/PHYSREV.1999.79.2.387
Deng J. T., Sutherland C., Brautigan D. L., Eto M., Walsh M. P. (2002). Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem. J. 367 (2), 517–524. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/12144526/, October 15. doi:10.1042/BJ20020522
Ding X., Murray P. A. (2006). Vascular smooth muscle. Found. Anesth. Basic Sci. Clin. Pract. 461, 461–469. accessed September 18, 2022, January 1. doi:10.1016/B978-0-323-03707-5.50044-9
Dixon R. E., Moreno C. M., Yuan C., Opitz-Araya X., Binder M. D., Navedo M. F., et al. (2015). Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. ELife 2015 (4). 05608. accessed July 4, 2022, February 25. doi:10.7554/ELIFE.05608
Dixon R. E., Navedo M. F., Binder M. D., Fernando Santana L. (2022). Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol. Rev. 102 (3), 1159–1210. accessed September 18, 2022, from https://pubmed.ncbi.nlm.nih.gov/34927454/, July 1. doi:10.1152/PHYSREV.00022.2021
Essin K., Welling A., Hofmann F., Luft F. C., Gollasch M., Moosmang S. (2007). Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J. Physiol. 584 (1), 205–219. accessed September 25, 2022, from https://pubmed.ncbi.nlm.nih.gov/17673505/, October 1. doi:10.1113/JPHYSIOL.2007.138982
Fallon J. L., Baker M. R., Xiong L., Loy R. E., Yang G., Dirksen R. T., et al. (2009). Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+·calmodulins. Proc. Natl. Acad. Sci. U. S. A. 106 (13), 5135–5140. accessed September 18, 2022, from https://pubmed.ncbi.nlm.nih.gov/19279214/, March. doi:10.1073/pnas.0807487106
Faux M. C., Scott J. D. (1997). Regulation of the AKAP79-protein kinase C interaction by Ca2+/calmodulin. J. Biol. Chem. 272 (27), 17038–17044. accessed September 21, 2022, from http://www.jbc.org/article/S0021925818393281/fulltext, July 4. doi:10.1074/jbc.272.27.17038
Feng T., Kalyaanamoorthy S., Barakat K. (2018). L-type calcium channels: Structure and functions. Ion Channels Health Sick. 77305. accessed June 22, 2022, from undefined/state.item.id, October 10. doi:10.5772/INTECHOPEN.77305
Fu X., Gong M. C., Jia T., Somlyo A. v., Somlyo A. P. (1998). The effects of the rho-kinase inhibitor Y-27632 on arachidonic acid-GTPgammaS-and phorbol ester-induced Ca2+-sensitization of smooth muscle. FEBS Lett. 440 (1–2), 183–187. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/9862451/, November 27. doi:10.1016/S0014-5793(98)01455-0
Ghosh D., Nieves-Cintrón M., Tajada S., Brust-Mascher I., Horne M. C., Hell J. W., et al. (2018). Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim. Biophys. Acta. Mol. Cell Res. 1865 (9), 1341–1355. accessed September 18, 2022, September 1. doi:10.1016/J.BBAMCR.2018.06.013
Han D., Xue X., Yan Y., Li G. (2019). Highlight article: Dysfunctional Cav1.2 channel in Timothy Syndrome, from cell to bedside. Exp. Biol. Med. 244 (12), 960–971. accessed September 21, 2022, from https://journals.sagepub.com/doi/full/10.1177/1535370219863149, September 1, 2019. doi:10.1177/1535370219863149
Herlitze S., Xie M., Han J., Hümmer A., Melnik-Martinez K. v., Moreno R. L., et al. (2003) Targeting mechanisms of high voltage-activated Ca2+ channels, J. Bioenerg. Biomembr., vol. 35, no. 6, pp. 621–637. accessed September 22, 2022, from https://pubmed.ncbi.nlm.nih.gov/15000523/, December 2003. doi:10.1023/b:jobb.0000008027.19384.c0
Herring B. P., El-Mounayri O., Gallagher P. J., Yin F., Zhou J. (2006). Regulation of myosin light chain kinase and telokin expression in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 291 (5), C817–C827. accessed September 18, 2022, from https://pubmed.ncbi.nlm.nih.gov/16774989/, 2006. doi:10.1152/ajpcell.00198.2006
Jarajapu Y. P. R., Knot H. J. (2005). Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am. J. Physiol. Heart Circ. Physiol. 289 (5), H1917–H1922. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/15980039/, November. doi:10.1152/AJPHEART.01012.2004
Kaneko T., Amano M., Maeda A., Goto H., Takahashi K., Ito M., et al. (2000). Identification of calponin as a novel substrate of rho-kinase. Biochem. Biophys. Res. Commun. 273 (1), 110–116. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/10873572/, June 24. doi:10.1006/BBRC.2000.2901
Klugbauer N., Marais E., Hofmann F. (2003). Calcium channel Alpha2delta subunits: Differential expression, function, and drug binding. J. Bioenerg. Biomembr. 35 (6), 639–647. accessed September 22, 2022, from https://pubmed.ncbi.nlm.nih.gov/15000524/, December. doi:10.1023/B:JOBB.0000008028.41056.58
Korzick D. H., Laughlin M. H., Bowles D. K., (2004) Alterations in PKC signaling underlie enhanced myogenic tone in exercise-trained porcine coronary resistance arteries, J. Appl. Physiol., vol. 96, no. 4, pp. 1425–1432. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/14672961/, April 2004. doi:10.1152/JAPPLPHYSIOL.01077.2003
Martinsen A., Dessy C., Morel N. (2014). Regulation of calcium channels in smooth muscle: New insights into the role of myosin light chain kinase. Channels 8 (5), 402–413. accessed September 21, 2022, from/pmc/articles/PMC4594426/, September 1. doi:10.4161/19336950.2014.950537
Martinsen A., Yerna X., Rath G., Gomez E. L., Dessy C., Morel N. (2012). Different effect of Rho kinase inhibition on calcium signaling in rat isolated large and small arteries. J. Vasc. Res. 49 (6), 522–533. accessed September 21, 2022, from https://www.karger.com/Article/FullText/341230, October. doi:10.1159/000341230
Morotti S., Nieves-Cintrón M., Nystoriak M. A., Navedo M. F., Grandi E. (2017). Predominant contribution of L-type Cav1.2 channel stimulation to impaired intracellular calcium and cerebral artery vasoconstriction in diabetic hyperglycemia. Channels (Austin, Tex.) 11 (4), 340–346. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/28631947/, July 4. doi:10.1080/19336950.2017.1293220
Napolitano C., Bloise R., Priori S. G. (2014). Timothy Syndrome. Cardiac Electrophysiol. Cell Bedside 1, 953–957. Sixth Edition. doi:10.1016/B978-1-4557-2856-5.00095-9
Navedo M. F., Amberg G. C. (2013). Local regulation of L-type Ca²⁺ channel sparklets in arterial smooth muscle. Microcirculation 20 (4), 290–298. accessed September 21, 2022, from/pmc/articles/PMC3577995/, May. doi:10.1111/MICC.12021
Navedo M. F., Amberg G. C., Nieves M., Molkentin J. D., Santana L. F., Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle, J. Gen. Physiol., vol. 127, no. 6, pp. 611–622. accessed July 4, 2022, from http://www.jgp.org/cgi/doi/10.1085/jgp.200609519, June 1, 2006. doi:10.1085/JGP.200609519
Navedo M. F., Amberg G. C., Votaw V. S., Santana L. F. (2005). Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. U. S. A. 102 (31), 11112–11117. accessed September 21, 2022, from https://www.pnas.org/doi/abs/10.1073/pnas.0500360102, August 2. doi:10.1073/PNAS.0500360102
Navedo M. F., Cheng E. P., Yuan C., Votaw S., Molkentin J. D., Scott J. D., et al. (2010a). Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy Syndrome. Circ. Res. 106 (4), 748–756. accessed September 21, 2022, from https://www.ahajournals.org/doi/abs/10.1161/circresaha.109.213363, March 5. doi:10.1161/CIRCRESAHA.109.213363
Navedo M. F., Nieves-Cintrón M., Amberg G. C., Yuan C., Votaw V. S., Lederer W. J., et al. (2008). AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II–induced hypertension. Circ. Res. 102 (2), e1–e11. accessed September 21, 2022, from https://www.ahajournals.org/doi/abs/10.1161/circresaha.107.167809, February 1, 2008. doi:10.1161/CIRCRESAHA.107.167809
Navedo M. F., Takeda Y., Nieves-Cintrón M., Molkentin J. D., Santana L. F. (2010b). Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 298 (2), C211–C220. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/19846755/, February. doi:10.1152/AJPCELL.00267.2009
Nystoriak M. A., Nieves-Cintrón M., Patriarchi T., Buonarati O. R., Prada M. P., Morotti S., et al. (2017). Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 10 (463), eaaf9647. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/28119464/, January 24. doi:10.1126/SCISIGNAL.AAF9647
Oliveria S. F., Dell’Acqua M. L., Sather W. A. (2007). AKAP79/150 anchoring of Calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55 (2), 261–275. accessed September 21, 2022, from/pmc/articles/PMC2698451/, July 7. doi:10.1016/J.NEURON.2007.06.032
Perino A., Ghigo A., Scott J. D., Hirsch E. (2012). Anchoring proteins as regulators of signaling pathways. Circ. Res. 111 (4), 482–492. accessed September 21, 2022, from https://www.ahajournals.org/doi/abs/10.1161/circresaha.111.262899, August 3. doi:10.1161/CIRCRESAHA.111.262899
Prada M. P., Syed A. U., Buonarati O. R., Reddy G. R., Nystoriak M. A., Ghosh D., et al. (2019). A gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. ELife 8, e42214. accessed September 21, 2022, March 1. doi:10.7554/ELIFE.42214
Prada M. P., Syed A. U., Reddy G. R., Martín-Aragón Baudel M., Flores-Tamez V. A., Sasse K. C., et al. (2020). AKAP5 complex facilitates purinergic modulation of vascular L-type Ca2+ channel CaV1.2. Nat. Commun. 11 (1), 5303. accessed September 21, 2022, from/pmc/articles/PMC7575592/, December 1. doi:10.1038/S41467-020-18947-Y
Priori S. G., Napolitano C. (2018). Timothy Syndrome, Cardiac Electrophysiol. Cell Bedside, 1, 910–916. Seventh Edition. doi:10.1016/B978-0-323-44733-1.00095-X
Ross J. L., Ali M. Y., Warshaw D. M. (2008). Cargo transport: Molecular motors navigate a complex cytoskeleton. Curr. Opin. Cell Biol. 20 (1), 41–47. accessed September 21, 2022, from/pmc/articles/PMC2688467/, February. doi:10.1016/J.CEB.2007.11.006
Santana L. F., Navedo M. F., Amberg G. C., Nieves-Cintrón M., Votaw V. S., Ufret-Vincenty C. A. (2008). Calcium sparklets in arterial smooth muscle. Clin. Exp. Pharmacol. Physiol. 35 (9), 1121–1126. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/18215181/, September. doi:10.1111/J.1440-1681.2007.04867.X
Santana L. F., Navedo M. F. (2009). Molecular and biophysical mechanisms of Ca2+ sparklets in smooth muscle. J. Mol. Cell. Cardiol. 47 (4), 436–444. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/19616004/, October. doi:10.1016/J.YJMCC.2009.07.008
Schaub M. C. (2007). Myosin light chain kinase. XPharm Compr. Pharmacol. Reference, 1–2. accessed September 18, 2022. doi:10.1016/B978-008055232-3.60529-X
Simms B. A., Zamponi G. W. (2012). Trafficking and stability of voltage-gated calcium channels. Cell. Mol. Life Sci. 69 (6), 843–856. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/21964928/, March. doi:10.1007/S00018-011-0843-Y
Smyth J. W., Shaw R. M. (2010). Forward trafficking of ion channels: What the clinician needs to know. Heart rhythm. 7 (8), 1135–1140. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/20621620/. doi:10.1016/J.HRTHM.2010.05.030
Suzuki Y., Yamamura H., Ohyac S., Imaizumi Y. (2013). Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+ (BKCa) and Cav1.2 Ca2+ channels and their clustering to regulate membrane excitability in vascular myocytes. J. Biol. Chem. 288 (51), 36750–36761. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/24202214/, December 20. doi:10.1074/JBC.M113.511485
Syed A. U., Reddy G. R., Ghosh D., Prada M. P., Nystoriak M. A., Morotti S., et al. (2019). Adenylyl cyclase 5-generated CAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J. Clin. Invest. 129 (8), 3140–3152. accessed September 21, 2022, from https://pubmed.ncbi.nlm.nih.gov/31162142/, August 1. doi:10.1172/JCI124705
Takashima S. (2009). Phosphorylation of myosin regulatory light chain by myosin light chain kinase, and muscle contraction. Circ. J. 73 (2), 208–213. accessed September 18, 2022, from https://pubmed.ncbi.nlm.nih.gov/19110504/. doi:10.1253/circj.cj-08-1041.CJ-08-1041
Takeda Y., Nystoriak M. A., Nieves-Cintrón M., Santana L. F., Navedo M. F. (2011). Relationship between Ca2+ sparklets and sarcoplasmic reticulum Ca2+ load and release in rat cerebral arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 301 (6), H2285–H2294. accessed September 25, 2022, from/pmc/articles/PMC3233819/, December. doi:10.1152/AJPHEART.00488.2011
Walsh M. P., Cole W. C. (2013). The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J. Cereb. Blood Flow. Metab. 33 (1), 1–12. accessed September 21, 2022, from/pmc/articles/PMC3597360/, January. doi:10.1038/JCBFM.2012.144
Walsh M. P. (2011). Vascular smooth muscle myosin light chain diphosphorylation: Mechanism, function, and pathological implications. IUBMB Life 63 (11), 987–1000. accessed September 18, 2022, from https://pubmed.ncbi.nlm.nih.gov/21990256/, November. doi:10.1002/IUB.527
Wesselman J. P., Spaan J. A. E., van der Meulen E. T., van Bavel E. (2001). Role of protein kinase C in myogenic calcium– contraction coupling of rat cannulated mesenteric small arteries. Clin. Exp. Pharmacol. Physiol. 28 (10), 848–855. accessed September 21, 2022, from https://onlinelibrary.wiley.com/doi/full/10.1046/j.1440-1681.2001.03534.x, October 30. doi:10.1046/J.1440-1681.2001.03534.X
Keywords: vascular smooth muscle, cooperative gating, functional coupling, L-type Ca2+ channels, Ca2+ sensitization, channel trafficking
Citation: Mironova GY, Haghbin N and Welsh DG (2022) Functional tuning of Vascular L-type Ca2+ channels. Front. Physiol. 13:1058744. doi: 10.3389/fphys.2022.1058744
Received: 30 September 2022; Accepted: 31 October 2022;
Published: 15 November 2022.
Edited by:
Ulrich Pohl, Ludwig Maximilian University of Munich, GermanyReviewed by:
Osama F. Harraz, University of Vermont, United StatesCopyright © 2022 Mironova, Haghbin and Welsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Galina Yu Mironova, Z21pcm9ub3ZAdXdvLmNh; Donald G. Welsh, ZHdlbHNoQHJvYmFydHMuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.