 Auriane C. Ernault†Makiri Kawasaki†Benedetta FabriziPablo Montañés-AgudoShirley C. M. AmersfoorthRushd F. M. Al-ShamaRuben Coronel*‡Joris R. De Groot*‡
Auriane C. Ernault†Makiri Kawasaki†Benedetta FabriziPablo Montañés-AgudoShirley C. M. AmersfoorthRushd F. M. Al-ShamaRuben Coronel*‡Joris R. De Groot*‡- Department of Clinical and Experimental Cardiology, Heart Center, Amsterdam Cardiovascular Sciences, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
Background: Atrial fibrosis plays an important role in the development and persistence of atrial fibrillation by promoting reentry. Primary cilia have been identified as a regulator of fibroblasts (FB) activation and extracellular matrix (ECM) deposition. We hypothesized that selective reduction of primary cilia causes increased fibrosis and facilitates reentry.
Aim: The aim of this study was to disrupt the formation of primary cilia in FB and examine its consequences on ECM and conduction in a co-culture system of cardiomyocytes (CM) and FB.
Materials: Using short interfering RNA (siRNA), we removed primary cilia in neonatal rat ventricular FB by reducing the expression of Ift88 gene required for ciliary assembly. We co-cultured neonatal rat ventricular cardiomyocytes (CM) with FB previously transfected with Ift88 siRNA (siIft88) or negative control siRNA (siNC) for 48 h. We examined the consequences of ciliated fibroblasts reduction on conduction and tissue remodeling by performing electrical mapping, microelectrode, and gene expression measurements.
Results: Transfection of FB with siIft88 resulted in a significant 60% and 30% reduction of relative Ift88 expression in FB and CM-FB co-cultures, respectively, compared to siNC. Knockdown of Ift88 significantly increased the expression of ECM genes Fn1, Col1a1 and Ctgf by 38%, 30% and 18%, respectively, in comparison to transfection with siNC. Conduction velocity (CV) was significantly decreased in the siIft88 group in comparison to siNC [11.12 ± 4.27 cm/s (n = 10) vs. 17.00 ± 6.20 (n = 10) respectively, p < 0.05]. The fraction of sites with interelectrode activation block was larger in the siIft88 group than in the siNC group (6.59 × 10−2 ± 8.01 × 10−2 vs. 1.18 × 10−2 ± 3.72 × 10−2 respectively, p < 0.05). We documented spontaneous reentrant arrhythmias in two cultures in the siIft88 group and in none of the siNC group. Action potentials were not significantly different between siNC and siIft88 groups.
Conclusion: Disruption of cilia formation by siIft88 causes ECM remodeling and conduction abnormalities. Prevention of cilia loss could be a target for prevention of arrhythmias.
Introduction
Atrial fibrillation (AF) is the most common arrhythmia, with an estimated 46.3 million individuals suffering from this condition worldwide (Kornej et al., 2020). AF not only significantly reduces the quality of life, but also results in an increased morbidity and mortality (Benjamin et al., 1998). AF is a progressive disease which starts as a non-sustained arrhythmia originating from ectopic activity, and often progresses to a sustained state via structural remodeling of the atria (Krogh-Madsen et al., 2012; Heijman et al., 2021).
Atrial fibrosis, the concert of proliferation of fibroblasts and differentiation into myofibroblasts and the pathological accumulation of extracellular matrix (ECM), is the main component of structural remodeling in AF (Nattel et al., 2008). Fibrosis causes heterogeneous conduction delay, facilitating the occurrence and maintenance of AF. Cardiac (myo) fibroblasts are the main cell population responsible for ECM production and fibrosis formation in AF. TGF-β1 and angiotensin II are the main drivers for cardiac fibroblasts to form such pathological accumulation of ECM (Burstein and Nattel, 2008).
Primary cilium is a small organelle that extends from a cell’s surface, and functions as a chemo- and mechano-sensors. Our proteome and transcriptome analysis of atrial tissue of patients with or without AF (non-AF) shows that the gene-sets of ciliogenesis, including IFT88, are down-regulated in AF compared to non-AF patients (Kawasaki et al., 2021; van den Berg et al., 2021). The dysregulation of primary cilia has been implicated in the fibrosis formation in various tissues, such as liver and kidney (Teves et al., 2019; Collins and Wann, 2020). Furthermore, our simultaneous study has indicated that the proportion of atrial fibroblasts with primary cilia is decreased in the left atrial tissue of persistent AF patients compared to non-AF patients, and the disruption of primary cilia in human atrial fibroblasts enhances their capacity to produce ECM. Thus, disruption of primary cilia in FB could activate them and cause accumulation of ECM, resulting in the formation of fibrosis in AF. However, it remains unclear what the direct consequences of FB with disrupted primary cilia are on cardiac conduction and action potential properties in multicellular preparations.
The aim of this study was to selectively decrease primary cilia in FB and examine its consequences on conduction and action potential properties in a co-culture system of cardiomyocytes (CM) and FB isolated from neonatal rats.
Here, we report that the selective disruption of primary cilia in FB by RNAi targeting Ift88 induces ECM production, decreases conduction velocity, increases number of block lines and increases the risk or reentrant arrhythmias without changing the action potential (AP) characteristics of CM. Our data suggest that dysregulation of primary cilia causes fibrosis and hinders myocardial conduction, thereby facilitating the occurrence of re-entry and formation of the arrhythmogenic substrate.
Materials and methods
Neonatal rat ventricular myocytes and fibroblasts isolation
All animal experiments were approved by the local Animal Experiments Committee (Academic Medical Center, University of Amsterdam) and carried out in compliance with the Guide for the Care and Use of Laboratory Animals and in accordance with national and institutional guidelines. Neonatal rat ventricular CM and FB were isolated from 2-days-old Wistar rats (Janvier labs). Pups were anesthetized by isoflurane and hearts were excised after decapitation. Ventricles were cut in pieces and dissociated using trypsin (1 mg/ml; Sigma) and collagenase type 2 (1 mg/ml; Worthington). Cells were resuspended in TUNG culture medium (M199 medium, Gibco) supplemented with 10% heat inactivated fetal bovine serum (FB, Gibco), 1% HEPES (Gibco), 5,000 U/L penicillin-G (Sigma), 2 mg/L vitamin B12 (Sigma-Aldrich), 3.5 g/L glucose, 1% non-essential amino acids (Gibco), 1% L-glutamine (Gibco). The cell suspension was pre-plated for 2 h to physically separate the rapidly adhering FB from CM. CM in suspension were collected and plated on multi-electrode arrays (MEAs, Multi-Channel Systems MCS GmbH, Reutlingen, Germany) and on 24-well tissue culture plates coated with fibronectin (125 μg/ml BD Biosciences, Breda, Netherlands) at a density of 2.29 × 105 cells per cm2. CM and FB were cultured at 37°C and 5% CO2.
Transfection
In order to suppress the formation of primary cilia in FB, we downregulated the expression of the intraflagellar transport protein 88 gene (Ift88) required for ciliary assembly (Marshall, 2008). FB were transfected with 5 nM short interfering RNA (siRNA) for Ift88 (siIft88) (Silencer™ Select siIFT88, ThermoFisher Scientific, Cat#. 4390771, SiRNA ID. s157133) and with 5 nM negative control siRNA (siNC) (Silencer™ Negative Control, ThermoFisher Scientific, Cat#. AM4611) using Lipofectamine RNAiMAX reagent (Invitrogen, Cat#. 13778).
Co-culture of fibroblasts and cardiomyocytes
Twenty-four hours after transfection, cells were digested with trypsin, collected and counted for co-culture experiments. FB were resuspended in 1 ml of TUNG culture medium and added to the CM culture plates/MEAs at a ratio of 1:0.27 (CM:FB). After 48 h of co-culture, cells were collected for RNA isolation. Electrical mapping and microelectrode measurements were also performed.
qRT-PCR
Total RNA was extracted from co-cultured CM and FB with TRI reagent (Sigma-Aldrich) following manufacturer’s protocol. RNA was used to generate cDNA using Superscript II (Invitrogen). qRT-PCR was performed on a LightCycler 480 (Roche) with SYBR Green I Master (Roche). Results were analyzed using LinRegPCR software. Primer sequences are included in Table 1. Relative gene expression was calculated using reference gene Hprt. Gene expression in the siIFT88 experiments was normalized to siNC for each cell isolation, and expressed as fold change (FC).

TABLE 1. Primer sequences.
Protein analysis
Protein were isolated using TRI reagent (Sigma-Aldrich) following manufacturer’s protocol.
Protein concentration was measured by Pierce™ BCA Protein Assay Kit (Thermo Scientific) following manufacturer’s instructions.
Western blotting
Western blotting was performed following standard protocols. In short, proteins were separated by electrophoresis in 4%–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) and transferred to PVDF membranes with the TransBlot Turbo Transfer System (Bio-Rad). Membranes were blocked in TBST 5% milk Protifar (Nutricia) for 1 h at room temperature, then they were incubated with the primary antibodies overnight at 4°C, and finally with HRP-conjugated secondary antibodies for 1 h at room temperature. Western blots were developed with ECL prime western blotting detection agent (Amersham Biosciences) in ImageQuant LAS 4000 (GE Healthcare Life Sciences). Western blots were quantified using Fiji (ImageJ). Antibody references are listed in Table 2.

TABLE 2. Antibodies.
Simple western analysis
Proteins were separated by size and detected with a WES system (ProteinSimple, San Jose, CA, United States) using a 12–230 kDa separation module with the primary and secondary antibodies mentioned in Table 2. Protein signal analysis and quantification was performed with the Compass software v.4.0.0 (Protein Simple).
Immunofluorescence assays
Cells were fixed in 4% paraformaldehyde, washed and permeabilized (0.2% Triton X-100 in PBS), blocked with 4% goat serum and incubated overnight with the primary antibodies in a humidity chamber at 4°C. Coverslips were incubated for 1 h with secondary antibodies at room temperature, followed by DAPI (Molecular probes) staining. Images were acquired with a Leica DM6000B. The proportion of non-ciliated fibroblasts was quantified using ImageJ.
Electrical mapping
Electrical mapping was performed as previously described (Ernault et al., 2022). Briefly, 10 min before measurements, CM-FB culture media were replaced by a modified Tyrode’s solution (36.5°C) containing (mM): NaCl 140, KCl 5.4, CaCl2 1.8, MgCl2 1.0, glucose 5.5, HEPES 5.0; pH 7.4. Spontaneous electrical activity was recorded from CM-FB co-cultured on MEAs (60 electrodes terminals) (60EcoMEA-Glass-gr, Multi Channel Systems MCS GmbH). A reference electrode was place inside the MEA, not in contact with the cells. Unipolar electrograms were recorded simultaneously with a 256-channel amplifier (BioSemi, 24 bit dynamic range, 122.07 nV LSB, total noise 0.5 μV). Signals were recorded with a sampling frequency of 2048 Hz [bandwidth (−3 dB) DC–400 Hz].
Data analysis were performed using a custom-made data analysis program written in Matlab 2006b [The MathWorks Inc, Natick, MA, United States (Potse et al., 2002)]. At each local unipolar electrogram, activation time (AT) was determined as the interval from a reference time zero to the minimum derivative of the QRS-complex of the local unipolar electrogram, and used to construct activation maps. Conduction velocity (CV) was determined along lines perpendicular to isochronal lines by dividing the distance by the difference in local AT. Number of block lines was assessed by counting the number of 30 ms block lines separating adjacent electrodes within one spontaneously active monolayer. The number of 30 ms block line was normalized to the total number of recording electrodes.
Microelectrode measurements
Spontaneous APs were measured in CM-FB monolayers using glass pipette microelectrodes (Harvard apparatus GC100F- 10). These micro-electrodes were filled with 3M KCl, typical tip resistance was 15–25 MΩ. An AgCl-covered silver wire was used as a reference electrode. Resting membrane potential (RMP) was taken as the most negative membrane potential recorded at each recording position. Maximum AP upstroke velocity (Vmax) was measured and AP duration was determined at 40, 60, and 80% (APD40, APD60, APD80) of repolarization. All signal analyses were performed using a custom-made data analysis program written in Matlab 2006b [The MathWorks Inc, Natick, MA, United States (Potse et al., 2002)].
Statistics
Statistical analyses were performed with GraphPad Prism (GraphPad Software, San Diego, CA, United States). Data are given as mean ± SD unless indicated otherwise. Number of observations and repeated experiments are given in the figure legends. Data were tested for normality using a Shapiro-Wilk normality test. If normal distribution was verified, the 2-tailed Student’s t-test was performed. If the data were not normally distributed, statistical significance was assessed using the 2-tailed Mann-Whitney test. p < 0.05 was considered significant.
Results
Knockdown of Ift88 in fibroblasts is associated with extracellular matrix production
To evaluate the knockdown efficiency of siIft88 in FB, we measured Ift88 expression in FB only (mRNA and protein) and in CM-FB co-culture (mRNA). Transfection of FB with siIft88 resulted in a 60% significant reduction of (mRNA) Ift88 expression in FB after 48 h of culture [2.18 × 10−3 ± 1.89 × 10−4 (siIft88) vs. 5.50 × 10−3 ± 4.99 × 10−4 (siNC), p < 0 .001, n = 6] in comparison to the group transfected with siNC (Figure 1A). In addition, transfection of FB with siIft88 resulted in a 94% significant reduction of IFT88 protein expression in FB [0.06 ± 0.02 (siIft88) vs. 1.0 ± 0.13 (siNC), p < 0 .0001, n = 8] in comparison to siNC (Figures 1B,C; Supplementary Figure S1). Furthermore, transfection of FB with siIft88 resulted in a 30% significant reduction of relative Ift88 expression in the combined CM-FB co-culture, (FC = 0.07 ± 0.13, p < 0.0001, n = 12) in comparison to the group transfected with siNC (Figure 1B).
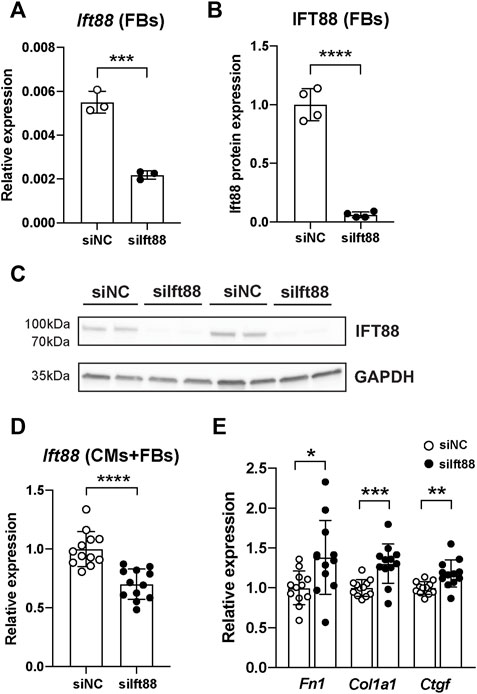
FIGURE 1. Reduction of Ift88 expression in FB leads to increased ECM genes expression in CM and FB co-culture. (A) Relative expression of (mRNA) Ift88 in FB previously transfected with siNC or siIft88. Data are mean ± SD. n = 6 from tree independent CM/FB isolation, Student’s t-test. (B,C) Relative expression of (protein) IFT88 in FB previously transfected with siNC or siIft88. Data are mean ± SD. n = 4 from two independent CM/FB isolation, Student’s t-test. Full unedited blots are provided in Supplementary Figure S1. (D) Relative expression of (mRNA) Ift88 in CM-FB co-cultures after FB transfection with siNC or siIft88. Data are mean ± SD. n = 12 from four independent CM/FB isolation, Student’s t-test. (E) Relative expression of ECM genes Fn1, Col1a1 and Ctfg after 48 h of co-culture of CM-FB. Data are mean ± SD. n = 12 from four independent CM/FB isolation, Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Next, we assessed whether Ift88 reduction in fibroblasts was associated with reduction of primary cilia. Knockdown of Ift88 in fibroblasts led to a significant increase in the proportion of non-ciliated fibroblasts in comparison to siNC (Supplementary Figure S2) [0.12 ± 0.03 (siIft88) vs. 0.07 ± 0.02 (siNC), p < 0.0001].
Knockdown of Ift88 significantly increased the relative expression of ECM genes Fn1, Col1a1 and Ctgf by 38%, 30% and 18%, respectively, in comparison to siNC (Fn1: FC = 1.38 ± 0.46, p < 0.05; Col1a1: FC = 1.30 ± 0.25, p < 0.001; Ctgf: FC = 1.18 ± 0.17, p < 0.01; n = 12) (Figure 1C).
Co-culture of cardiomyocytes and fibroblasts with reduced cilia show heterogeneous conduction slowing and reentry
To examine the effect of disrupted cilia formation in FB on conduction, we performed electrical mapping of CM co-cultured with FB pretreated with siIft88 or siNC. Activation maps generated from the siIft88 co-cultures showed isochrone crowding, revealing a slower conduction in siIft88 than in siNC co-cultures (Figures 2A,B).
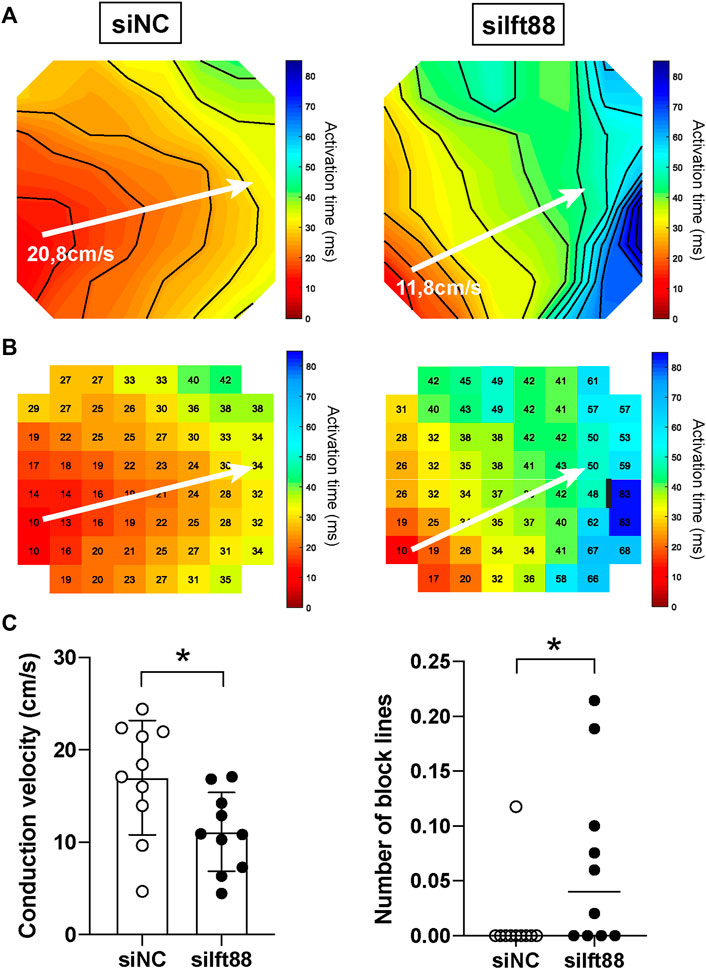
FIGURE 2. Reduced conduction velocity in CM monolayers co-cultured with decreased Ift88 FB. (A,B) Representative activation maps obtained after electrical mapping of spontaneously active CM co-cultured for 48 h with FB previously transfected with siNC or siIft88. Colors indicate activation times, according to the scale at right. Isochrones, 5 ms. White arrows indicate where conduction velocity was measured. Black line indicates 30 ms conduction block. Interelectrode distance of 700 μM. (C) Conduction velocity (CV) and number of block lines in spontaneously active CM monolayers co-cultured for 48 h with FB transfected with siNC or siIft88. Data are mean ± SD for CV and median for number of block lines, n = 10 monolayers from three independent CM/FB isolation. Student’s t-test for CV, Mann-Whitney test for number of block lines. Factor correction was carried out on CV to avoid inter-isolations differences.
On average, CV was 17.00 ± 6.20 cm/s in the siNC co-cultures, while it was 11.12 ± 4.27 cm/s in the siIft88 co-cultures (Figure 2C). CV was significantly lower in the siIft88 than in the siNC co-cultures (p < 0.05, n = 10).
Next, number of block lines was assessed by counting the number of 30 ms block lines in the activation maps, as shown in Figure 2B (right). Number of block lines was 1.18 × 10−2 ± 3.72 × 10−2 in the siNC co-cultures, while it was 6.59 × 10−2 ± 8.01 × 10−2 lines of block per electrode in the siIft88 co-cultures. Number of block lines was significantly higher in the siIft88 co-cultures than in the siNC (p < 0.05, n = 10).
Cardiomyocytes monolayers co-cultured with fibroblasts with reduced cilia are more prone to reentrant arrhythmias
Figure 3A shows example representative activation maps of a spontaneous rhythm in one MEA in the siIft88 group. Activation times (small numbers) in both panels indicate activation times relative to the same reference time. The white letters indicate electrodes from which selected unipolar electrograms are depicted in panel B. The activation maps show two cycles with reentrant activation patterns in one CM-FB co-culture from the siIft88 group. Activation is continuous from the last activated site in the left panel to the first activated site in the right panel. Electrograms selected along the reentrant circuit are shown in Figure 3B and show that the diastolic interval is spanned by local activation, as expected during reentry. Overall, we observed two spontaneous reentrant activation patterns in two different CM-FB co-cultures in the siIft88 group and none in the siNC group (Figure 3C).
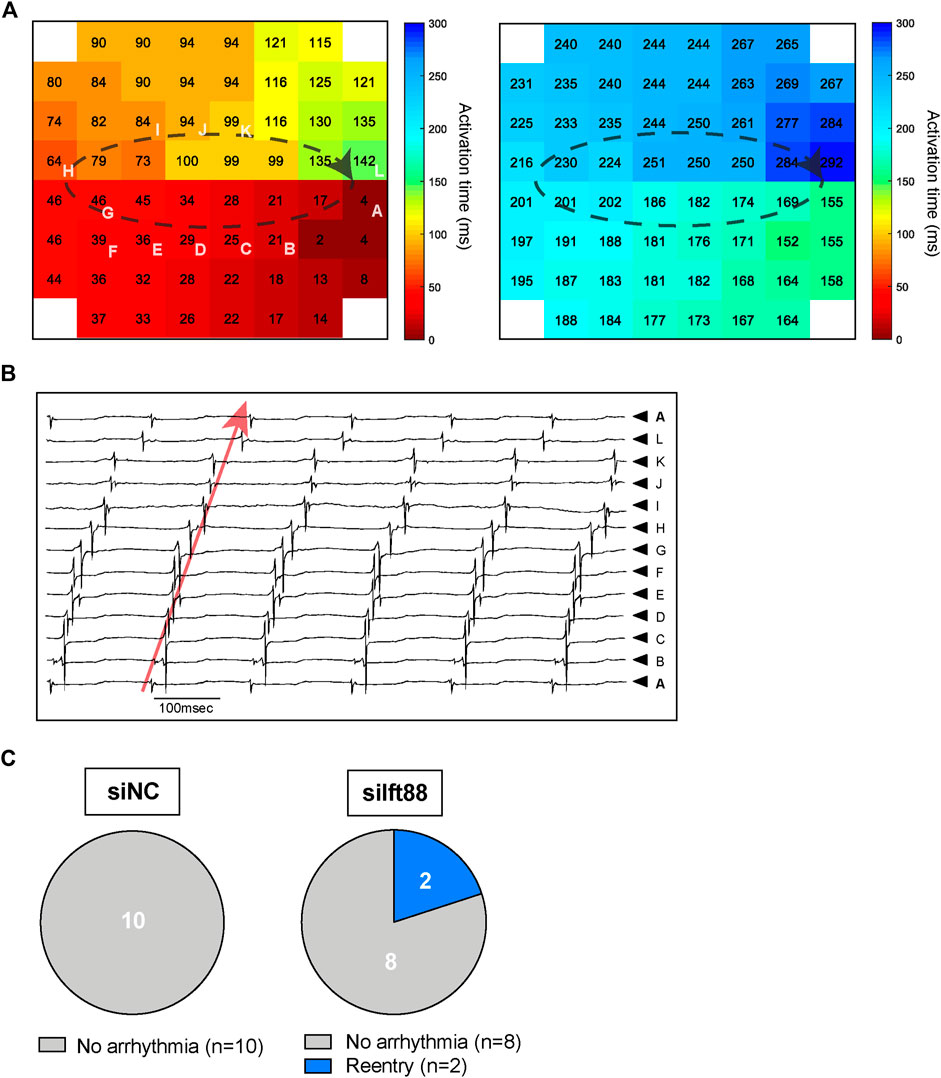
FIGURE 3. Reentry in CM co-cultured with siIft88 FB. (A) Activation maps of two following beats showing reentrant circuit and obtained from one CM monolayer co-cultured for 48 h with FB previously transfected with siIft88. Colors indicate activation times, according to the scale at right. Dashed black line indicated reentrant conduction pattern. Letters indicate electrodes from which selected unipolar electrograms are depicted in (B). (B) Electrograms selected along the reentry circuit. Red arrow shows that the diastolic interval is spanned within local activation (electrogram from electrode A is repeated on top and at the bottom) as expected during reentry. The letters next to the electrograms indicate electrodes from which selected unipolar electrograms are shown (A). (C) Proportion of CM-FB co-cultures showing reentrant arrhythmia or no arrhythmia in the siNC and siIft88 groups.
Knockdown of Ift88 in fibroblasts is not associated with changes of action potential characteristics nor reduced electrical coupling
Figure 4A shows representative examples of microelectrode AP measurements in CM-FB co-cultures from siNC and siIft88 groups. Overall, there were no significant differences in APD40 [92.03 ± 17.60 (siNC) vs. 82.24 ± 11.25 ms (siIft88)], APD60 [124.6 ± 20.21 (siNC) vs. 119.1 ± 14.83 ms (siIft88)], nor in APD80 [189.4 ± 20.54 (siNC) vs. 201.1 ± 39.50 ms (siIft88)] between siNC and siIft88 co-cultures (n ≥ 9) (Figure 4B).
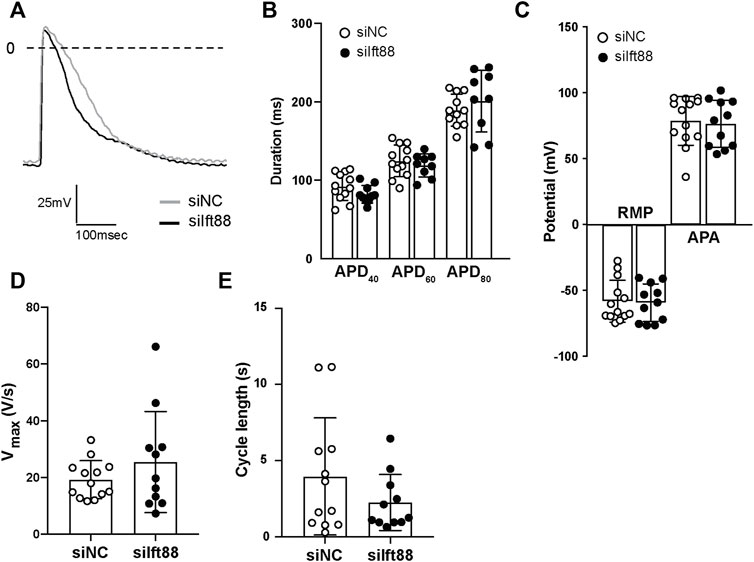
FIGURE 4. Co-culture between FB with reduced (mRNA) Ift88 expression and CM does not lead to changes of the action potential characteristics. (A) Representative spontaneous APs recorded in CM monolayers co-cultured for 48 h with FB previously transfected with siNC or siIft88. (B–E) Effect of co-culture on spontaneous APs duration at 40%, 60%, and 80% (APD40, APD60, and APD80) of repolarization (B), resting membrane potential (RMP), AP amplitude (APA) (C), maximal upstroke velocity (Vmax) (D) and cycle length (E). n ≥ 9 microelectrode measurements from two independent CM/FB isolation. Data are mean ± SD.
The observed reduction in conduction velocity (Figure 2) was not associated with significant depolarization of CM nor reduction of AP amplitude. Overall, the RMP was −57.96 ± 15.92 mV in the siNC co-cultures, while it was −59.23 ± 14.30 mV in the siIft88 co-cultures (n ≥ 9). The averaged AP amplitude was 78.94 ± 18.52 mV in the siNC co-cultures while it was 76.73 ± 17.83 mV in the siIft88 co-cultures (Figure 4C). Vmax and cycle length were also not significantly different between the two groups [19.24 ± 6.719 (siNC) vs. 25.43 ± 17.79 V/s (siIft88) and 3.96 ± 3.84 (siNC) vs. 2.25 ± 1.84 s (siIft88), respectively] (Figures 4D,E).
Connexin43 (main protein component of gap junctions in neonatal rat ventricular CM) expression and distribution were not different between siNC and siIft88 co-cultures (Supplementary Figures S3, S4). Finally, the expression of major cardiomyocytes ion channels (Kcnj11, Kcnq1, Kcnj2, Cacna1g, Scn1b, Scn5a) was not significantly different between siIft88 and siNC groups (Supplementary Figure S5).
Discussion
In this study, we show that the disruption of cilia formation in FB via targeting Ift88 with siRNA is associated with enhanced ECM gene expression and conduction delay in CM-FB co-cultures, without changes of AP characteristics.
The cardiac ECM is a dynamic and complex network which plays an important role in providing structural support to cardiac cells, and regulates gene expression as well as gap junction organization (Forte et al., 2012). Remodeling of the ECM under pathological conditions alters cardiac fibers organization, leading to heterogeneous conduction delay and enhanced anisotropy, increasing the risk of arrhythmias.
Dysregulation of primary cilia in cardiac diseases and fibrosis formation has been implicated in various studies. For instance, primary cilia are important for embryonic development (Komatsu and Mishina, 2013) and the patients affected by a ciliopathy, a genetic condition caused by dysfunction of primary cilia, often manifest congenital heart disease and multi-organ fibrosis (Seeger-Nukpezah and Golemis, 2012; Klena et al., 2017). Several age-dependent diseases, such as atherosclerosis was shown to be driven by loss of primary cilia in aortic endothelial cells (Dinsmore and Reiter, 2016), suggesting that primary cilia suppress pro-atherosclerotic signaling. Biological processes underlying atrial fibrosis formation, such as epithelial-mesenchymal transition, differentiation of fibroblasts into myofibroblasts, and ECM synthesis depend on primary cilia (Ten Dijke et al., 2012).
Co-culture of CM and FB with a reduction of primary cilia by knockdown of lft88 leads to increased ECM gene expression of Fn1, Col1a1 and Ctgf (Figure 1). This is in accordance with previous work showing that conditional knockout of Ift88 in mouse heart leads to increased production of ECM substrates collagen I and versican (Toomer et al., 2017). Consistently, silencing Ift88 in chondrocytes leads to increased expression of ECM components Mmp13, Adamts5, ColX, and Runx2 (Chang et al., 2012; Collins and Wann, 2020). These results suggest that primary cilia are dynamic cellular appendages which restrain and fine-tune FB activation and ECM production. Consistent with these previous reports, we previously observed that FB from patients with persistent AF express fewer primary cilia in association with more ECM production compared to FB from non-AF patients (data not shown).
Our results are in seemingly contradiction with the study by Villalobos et al. where the conditional knockout of polycistine1 (PC1 encoded by pkd1, pkd1-cKO) in activated cardiac fibroblasts reduces scar size in a myocardial infarction (MI) model. However, this reduction was not attributable to reduced fibrosis. PC1 together with polycistine2 (PC2, encoded by pkd2) form a transient receptor potential ion channel and mediate the mechano-sensation via primary cilia, but they are not involved in the formation of primary cilia (Surya M. Nauli et al. Nature Genetics, 2003). Therefore, from the paper by Villalobos et al. it remains unclear whether a loss of primary cilia in the activated cardiac fibroblasts increases or decreases cardiac fibrosis.
We observed differences in Ift88 transcript levels between FB cultures and CM-FB co-cultures (Figure 1). We speculate that these differences are caused by Ift88 expression in cardiomyocytes in which Ift88 was not knocked-down.
Co-culture of CM with FB transfected with siIft88 led to conduction abnormalities (slower CV, increased number of block lines) (Figure 2) and subsequent increased risk of reentrant arrhythmias (Figure 3), without changes in AP characteristics (Figure 4) and without reduction in Gja1 expression (Supplementary Figure S1). This demonstrates that the observed CV reduction and increased risk of arrhythmia is not subsequent to electrical remodeling of CM (i.e., reduction of intercellular electrical coupling between CM or depolarization of CM). Instead, the CV reduction and increased number of block lines observed in the siIft88 CM-FB co-cultures is likely due to the increased ECM production from activated fibroblasts following the dysregulation of primary cilia. Excessive ECM deposition separates CM and may lead to tortuous patterns of activation which results in conduction delay and blocks. These findings are in accordance with literature showing that conduction velocity is impaired when (myo)fibroblast concentration increases and fibrosis occurs (Spencer et al., 2017).
Study limitations
While knockdown of IFT88 is known to cause loss of primary cilia in fibroblasts, we cannot exclude that there are other, secondary, effects caused by this gene knockdown which could modify fibroblast behavior.
Extracellular matrix remodeling was in this study assessed by gene expression measurement, without quantifying the ECM architecture or composition. Further study should investigate and perform quantitative analysis on the ECM architecture and composition in the two groups.
Conclusion
Protecting the function of primary cilia and repairing the disrupted cilia is a potential therapy to prevent the onset of AF and to reverse the fibrosis formation in the advanced AF, respectively.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by the local Animal Experiments Committee (Academic Medical Center, University of Amsterdam).
Author contributions
AE, MK, RC, and JG designed the project. AE, MK, BF, PM-A, SA, and RA-S performed the experiments. AE, MK, PM-A, and BF acquired and analyzed the data. AE wrote the paper with input from all authors. All authors provided critical feedbacks on the manuscript and approved its submission.
Funding
This study was supported by Netherlands Organization for Health Research and Development (ZonMW/NWO, 106.146.310) awarded to JG.
Conflict of interest
JG received research grants through his institution from Abbott, Atricure, Boston Scientific, Bayer, Daiichi Sankyo, Johnson & Johnson, Medtronic Servier, and received speaker/consultancy fees from Atricure, Bayer, Daiichi Sankyo, Johnson & Johnson and Medtronic outside the submitted work. AHGD is a consultant for Atricure.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.1057200/full#supplementary-material
References
Benjamin E. J., Wolf P. A., D'agostino R. B., Silbershatz H., Kannel W. B., Levy D. (1998). Impact of atrial fibrillation on the risk of death: The framingham heart study. Circulation 98, 946–952. doi:10.1161/01.cir.98.10.946
Burstein B., Nattel S. (2008). Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 51, 802–809. doi:10.1016/j.jacc.2007.09.064
Chang C. F., Ramaswamy G., Serra R. (2012). Depletion of primary cilia in articular chondrocytes results in reduced Gli3 repressor to activator ratio, increased Hedgehog signaling, and symptoms of early osteoarthritis. Osteoarthr. Cartil. 20, 152–161. doi:10.1016/j.joca.2011.11.009
Collins I., Wann A. K. T. (2020). Regulation of the extracellular matrix by ciliary machinery. Cells 9, E278. doi:10.3390/cells9020278
Dinsmore C., Reiter J. F. (2016). Endothelial primary cilia inhibit atherosclerosis. EMBO Rep. 17, 156–166. doi:10.15252/embr.201541019
Ernault A. C., Verkerk A. O., Bayer J. D., Aras K., Montañés-Agudo P., Mohan R. A., et al. (2022). Secretome of atrial epicardial adipose tissue facilitates reentrant arrhythmias by myocardial remodeling. Heart rhythm. 19, 1461–1470. doi:10.1016/j.hrthm.2022.05.011
Forte G., Pagliari S., Ebara M., Uto K., Tam J. K., Romanazzo S., et al. (2012). Substrate stiffness modulates gene expression and phenotype in neonatal cardiomyocytes in vitro. Tissue Eng. Part A 18, 1837–1848. doi:10.1089/ten.TEA.2011.0707
Heijman J., Linz D., Schotten U. (2021). Dynamics of atrial fibrillation mechanisms and comorbidities. Annu. Rev. Physiol. 83, 83–106. doi:10.1146/annurev-physiol-031720-085307
Kawasaki M., Meulendijks E. R., Van Den Berg N. W. E., Nariswari F. A., Neefs J., Wesselink R., et al. (2021). Neutrophil degranulation interconnects over-represented biological processes in atrial fibrillation. Sci. Rep. 11, 2972. doi:10.1038/s41598-021-82533-5
Klena N. T., Gibbs B. C., Lo C. W. (2017). Cilia and ciliopathies in congenital heart disease. Cold Spring Harb. Perspect. Biol. 9, a028266. doi:10.1101/cshperspect.a028266
Komatsu Y., Mishina Y. (2013). Establishment of left-right asymmetry in vertebrate development: The node in mouse embryos. Cell. Mol. Life Sci. 70, 4659–4666. doi:10.1007/s00018-013-1399-9
Kornej J., Börschel C. S., Benjamin E. J., Schnabel R. B. (2020). Epidemiology of atrial fibrillation in the 21st century: Novel methods and new insights. Circ. Res. 127, 4–20. doi:10.1161/CIRCRESAHA.120.316340
Krogh-Madsen T., Abbott G. W., Christini D. J. (2012). Effects of electrical and structural remodeling on atrial fibrillation maintenance: A simulation study. PLoS Comput. Biol. 8, e1002390. doi:10.1371/journal.pcbi.1002390
Marshall W. F. (2008). Basal bodies platforms for building cilia. Curr. Top. Dev. Biol. 85, 1–22. doi:10.1016/S0070-2153(08)00801-6
Nauli S. M., Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. genet. 33 (2), 129–137. doi:10.1038/ng1076
Nattel S., Burstein B., Dobrev D. (2008). Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73. doi:10.1161/CIRCEP.107.754564
Potse M., Linnenbank A. C., Grimbergen C. A. (2002). Software design for analysis of multichannel intracardial and body surface electrocardiograms. Comput. Methods Programs Biomed. 69, 225–236. doi:10.1016/s0169-2607(02)00014-7
Seeger-Nukpezah T., Golemis E. A. (2012). The extracellular matrix and ciliary signaling. Curr. Opin. Cell Biol. 24, 652–661. doi:10.1016/j.ceb.2012.06.002
Spencer T. M., Blumenstein R. F., Pryse K. M., Lee S. L., Glaubke D. A., Carlson B. E., et al. (2017). Fibroblasts slow conduction velocity in a reconstituted tissue model of fibrotic cardiomyopathy. ACS Biomater. Sci. Eng. 3, 3022–3028. doi:10.1021/acsbiomaterials.6b00576
Ten Dijke P., Egorova A. D., Goumans M. J., Poelmann R. E., Hierck B. P. (2012). TGF-Β signaling in endothelial-to-mesenchymal transition: The role of shear stress and primary cilia. Sci. Signal. 5, pt2. doi:10.1126/scisignal.2002722
Teves M. E., Strauss J. F., Sapao P., Shi B., Varga J. (2019). The primary cilium: Emerging role as a key player in fibrosis. Curr. Rheumatol. Rep. 21, 29. doi:10.1007/s11926-019-0822-0
Toomer K. A., Fulmer D., Guo L., Drohan A., Peterson N., Swanson P., et al. (2017). A role for primary cilia in aortic valve development and disease. Dev. Dyn. 246, 625–634. doi:10.1002/dvdy.24524
Keywords: atrial fibrillation, fibrosis, fibroblasts, primary cilia, cardiac
Citation: Ernault AC, Kawasaki M, Fabrizi B, Montañés-Agudo P, Amersfoorth SCM, Al-Shama RFM, Coronel R and De Groot JR (2022) Knockdown of Ift88 in fibroblasts causes extracellular matrix remodeling and decreases conduction velocity in cardiomyocyte monolayers. Front. Physiol. 13:1057200. doi: 10.3389/fphys.2022.1057200
Received: 29 September 2022; Accepted: 07 November 2022;
Published: 17 November 2022.
Edited by:
Fu Siong Ng, Imperial College London, United KingdomReviewed by:
Robin Mark Shaw, The University of Utah, United StatesHenrique Girao, University of Coimbra, Portugal
Copyright © 2022 Ernault, Kawasaki, Fabrizi, Montañés-Agudo, Amersfoorth, Al-Shama, Coronel and De Groot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruben Coronel, rubencoronel@gmail.com; Joris R. De Groot, j.r.degroot@amsterdamumc.nl
†These authors have contributed equally to this work and share first authorship
‡These authors share last authorship