
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 04 October 2022
Sec. Cell Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.1036515
This article is part of the Research TopicOxidative Stress and Distinct Cell DeathView all 6 articles
 Li Long1†
Li Long1† Hongmei Guo1†
Hongmei Guo1† Xixi Chen1
Xixi Chen1 Yan Liu1,2Ruyi Wang1,3
Yan Liu1,2Ruyi Wang1,3 Xiaomei Zheng1,3Xiaobo Huang4*
Xiaomei Zheng1,3Xiaobo Huang4* Qiao Zhou1*
Qiao Zhou1* Yi Wang4*
Yi Wang4*Rheumatoid arthritis (RA) is a chronic, systemic disease of unknown etiology. The primary manifestation of RA is inflammatory synovitis, which eventually leads to deformity and functional loss. Ferroptosis is a non-apoptosis form of cell death that depends on intracellular iron accumulation. This leads to an increase in reactive oxygen species (ROS) induced-lipid peroxidation. The underlying mechanisms of ferroptosis are System Xc- and Glutathione metabolism, regulation of glutathione peroxidase 4 activity, and ROS generation. Recent studies have shown an association between the pathogenesis of RA and ferroptosis, suggesting the involvement of ferroptosis in the onset and progression of RA. In this review, we have focused on the mechanism of ferroptosis and its association with RA pathogenesis. Further, we discuss the status of therapeutics targeting ferroptosis in the treatment of patients with RA. Targeting ferroptosis could be a potential therapeutic approach for RA treatment.
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease. The primary clinical manifestation of RA is aggressive, symmetric polyarthritis. The pathological changes associated with RA are chronic inflammation of synovial joints, pannus formation, and gradual destruction of the articular cartilage and bone, ultimately leading to joint deformity (Kumar et al., 2016; Scherer et al., 2020). The incidence of RA is 0.5–1%, can occur at any age, and has a high disability rate (Cush, 2022; Hugon, 2022; Sun et al., 2022). Therefore, effective therapeutic strategies are crucial for a good prognosis for patients with RA. Non-steroidal anti-inflammatory drugs, glucocorticoids, and disease-modifying anti-rheumatic drugs (DMARDs), including conventional DMARDs, biological DMARDs, and targeted synthesis DMARDs, are used to treat RA patients. Despite the advancement in therapeutics, many patients do not achieve disease remission. “Ferroptosis” has recently been discovered and is iron-dependent cell death. Ferroptosis occurs due failure of the cells to remove excessive intracellular iron, reactive oxygen species (ROS), and lipid peroxides on time. Additionally, ferroptosis can also occur if the body’s antioxidant system is inhibited or inactivated, which leads to disruption in the redox balance of the cells, and the toxic lipid metabolites produced in the cells attack the biomolecules (Stockwell et al., 2017; Riegman et al., 2020; Jiang et al., 2021). Multiple studies have shown the involvement of ferroptosis in cancers (Zhao et al., 2022a; Gao et al., 2022; Lei et al., 2022), but there are relatively few reports on the association between ferroptosis and RA. In this study, we discuss the progress in ferroptosis and RA, thereby establishing an association between the incidence of RA and ferroptosis. Further, we shed light on the use of ferroptosis modulators as possible therapeutic strategies for the treatment of patients with RA.
The pathogenesis of RA is not completely understood. However, it is currently believed that RA occurs due to immune damage caused by a series of autoimmune reactions in genetically susceptible individuals under the influence of environmental factors. Risk factors for RA include smoking, silica exposure, disorders of periodontal and intestinal microbiota, etc. (Novella-Navarro et al., 2021). The immune cells and inflammatory mediators associated with RA mainly include T cells, B cells, macrophages, monocytes, interleukins (ILs), tumor necrosis factor-α (TNF-α), etc. (Figure 1). Fibroblast-like synoviocytes (FLS) are commonly found at the junction of RA pannus and cartilage. It has tumor-like effects and is considered a key factor in the onset and development of RA (Lefevre et al., 2009; Niu et al., 2019; Mueller et al., 2021; Jang et al., 2022).
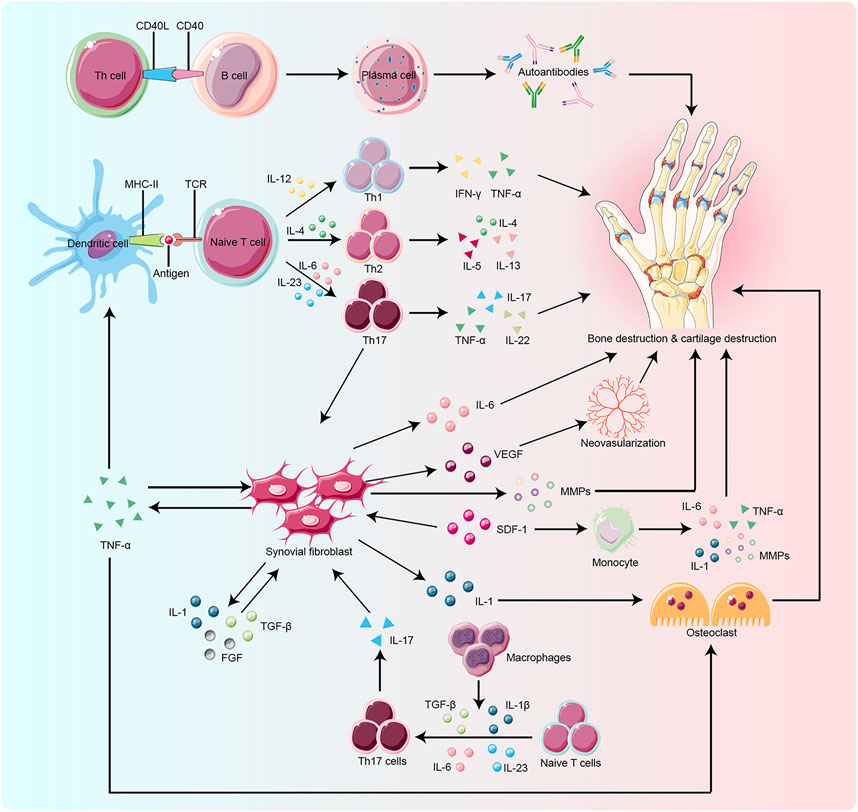
FIGURE 1. Key target and effector cells are involved in the pathogenesis of RA. Naïve T cell activation is induced by specific recognition of TCR and antigens presented by MHC-II molecules of antigen-presenting cells (APCs), such as dendritic cells, which are the most potent APCs. Cytokines produced by activated T cells and APCs influence T cell differentiation. IL-12, IL-4, and IL-23 play a key role in the differentiation of naïve T cells into Th1, Th2, and Th17 cells, respectively. Macrophages also promote Th17 differentiation by secreting cytokines such as TGF-β and IL-23. Upon activation and differentiation, more cytokines, including TNF-α, TGF-β, IL-1, SDF-1, and FGF, are secreted to promote the proliferation of RA-FLS. RA-FLS, in turn, secrete active substances like IL-6, MMPs, and TNF-α, to aggravate bone erosion and VEGF to promote neovascularization. The CD40/CD40L interactions activate B cells to produce autoantibodies which play a critical role in bone resorption. SDF-1 also activates monocytes to produce IL-1, IL-6, MMPs, and TNF-α, which lead to bone degradation. Osteoclasts are mainly activated by IL-1 and TNF-α, which ultimately promote bone destruction. TCR:T cell receptor, MHC-II:Major Histocompatibility Complex Class II, APCs: antigen-presenting cells,ILs:interleukins, Th:T helper cell, TGF-β:transforming growth factor-β, TNF-α:tumor necrosis factor-α, SDF-1:stromal cell-derived factor-1, FGF:fibroblast growth factor, RA-FLS:rheumatoid arthritis-fibroblast-like synoviocytes, MMPs:matrix metalloproteinases, VEGF:vascular endothelial growth factor, CD40:clusters of differentiation 40, CD40L:clusters of differentiation 40 ligand.
Rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) lead to the proliferation of synovial membrane, inflammatory reactions, extensive angiogenesis, the disintegration of cartilage matrix, and destruction of bones by producing abundant pro-inflammatory cytokines and chemokines (Bartok and Firestein, 2010; Aldridge et al., 2020; Bai et al., 2022; Ji et al., 2022); Figure 1). RA-FLS continuously produces many inflammatory factors like IL-6, IL-1β, TNF-α, vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMPs). ILs stimulate collagenase production by synovial cells and cartilage, thereby enhancing the inflammatory response in the joints. TNF-α activates macrophages to produce cytokines like IL-1, IL-6, and IL-8, which enhances inflammation and stimulates the differentiation of T cells, B cells, and NK cells. VEGF promotes the vascular opacification of the synovial membrane, which leads to joint destruction. MMPs degrade the synovial matrix. These inflammatory factors promote angiogenesis and proliferation of synovial membrane and activate the expression of stromal cells and osteoblasts, which are involved in inflammatory bone erosion (Davignon et al., 2018; Currie et al., 2019; Hattori et al., 2019; Nygaard and Firestein, 2020; Armaka et al., 2022; Micheroli et al., 2022).
Recent studies have shown that ferroptosis is another type of regulated cell death that depends on the accumulation of iron and highly lethal lipid peroxides. Morphologically, cells undergoing ferroptosis show a decrease in mitochondrial volume, increase in density of mitochondrial membrane, decrease or disappearance of mitochondrial cristae, and rupturing of the mitochondrial outer membrane (Stockwell et al., 2017; Chen et al., 2021). The biochemical features of ferroptotic cells include the accumulation of intracellular iron and ROS, activation of mitogen-activated protein kinases (MAPKs) signaling pathway, inhibition of the cystine/glutamate transporter system, and increase in nicotinamide adenine dinucleotide phosphate (NADPH) oxidation (Xie et al., 2016; Xia et al., 2021). Iron metabolism and lipid peroxidation are key factors leading to the ferroptosis of cells (Figure 2).
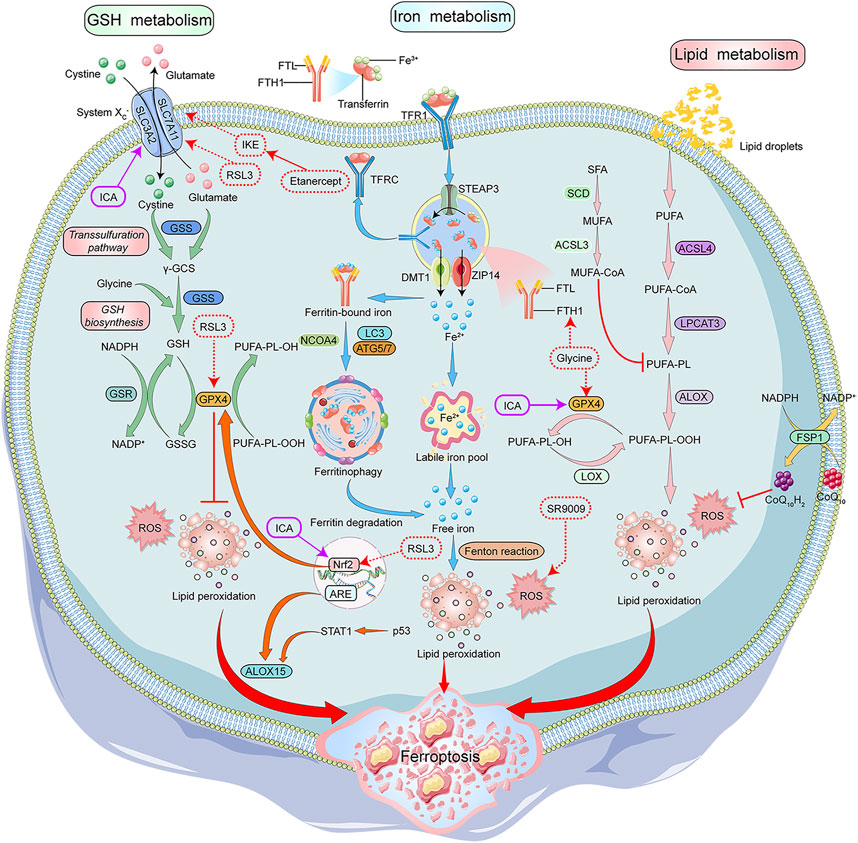
FIGURE 2. Mechanisms of Ferroptosis and its modulators in RA treatment. GSH, iron and lipid metabolism are the core components of ferroptosis. System Xc-/GPX4 axis is important for GSH synthesis. System Xc- transports cystine into the cells to generateγ-GCS along with glutamate, which is catalyzed by GSS. GSH is synthesized by γ-GCS and glycine. GSH acts as a GPX4 cofactor to convert PUFAs-OOH to PUFAs-OH. STEAP3 converted Fe3+ to Fe2+ in the endosomes, and the Influx of Fe2+ into the cytoplasm is mediated by DMT1 or ZIP14. In the cytoplasm, a small portion of Fe2+ remains in the LIP, and most of Fe2+ combines with ferritin for storage. NCOA4 aids in the autophagy of ferritin to provide Fe2+ by inducing ferritin degradation. The intracellular PUFAs-OOH reacts with Fe2+ to induce lipid peroxidation, eventually leading to ferroptosis. ALOX15 promotes lipid peroxidation, and CoQ10H2 inhibits lipid peroxidation. NADPH/FSP1/CoQ10 pathway is an antioxidant pathway parallel to GPX4. Glycine, SR9009, RSL3, and etanercept show positive effects on the treatment of RA patients by inducing ferroptosis, while G1dP3 plays a positive role in RA treatment by inhibiting ferroptosis. GSH:glutathione, System Xc-:glutamate reverse transporter, GPX4:glutathione peroxidase 4,γ-GCS:γ-glutamylcysteine synthetase, GSS:glutathione synthetase, PUFAs:Polyunsaturated fatty acids, STEAP3:Six-Transmembrane Epithelial Antigen of Prostate 3, DMT1:divalent metal ion transporter 1, ZIP14:ZRT- and IRT-like protein 14, LIP:labile iron pool, NCOA4:nuclear receptor coactivator 4, ALOX15:arachidonate lipoxygenase 15, CoQ10H2:Ubiquinol-10, FSP1:ferroptosis suppressor protein 1, CoQ10:coenzyme Q10, NAD(P)H:nicotinamide adenine dinucleotide phosphate, SR9009:REV-ERB agonist, RSL3:Ras-selective lethal small molecule 3, RA:rheumatoid arthritis, G1dP3:galectin-1 derived peptide 3.
Iron is indispensable for the body and involved in the transportation of cellular oxygen and the synthesis of adenosine triphosphate and deoxyribonucleic acid. It is also an important cofactor in the electron transport chain and MMPs in the mitochondria (Cai et al., 2019). A complex is formed between free Fe3+ in the blood and transferrin (TF), which transports Fe3+ into the cell, which forms endosomes by binding to transferrin receptors (TFR) on the cell membrane. Fe3+ is reduced to Fe2+ in the endosomes by the divalent metal ion transporter 1 (DMT1) or ZRT/IRT-like protein (ZIP) and then enters the cytoplasm. In the cells, most of Fe2+ is stored as cytoplasmic ferritin formed by ferritin light chain and ferritin heavy chain 1 (FTH1), and a small portion as labile iron pool (LIP). Autophagy of ferritin via nuclear receptor coactivator 4 (NCOA4) also contributes to active Fe2+ (Wang et al., 2021). Excess Fe2+ is oxidized to Fe3+ and pumped out of the cells (Bogdan et al., 2016). Some studies show that iron chelators like desferrioxamine can significantly inhibit cellular ferroptosis induced by Erastin, suggesting that iron homeostasis and lipid peroxidation are key factors associated with cellular ferroptosis (Dixon et al., 2012).
Lipid peroxidation is the loss of hydrogen atoms produced by lipids due to free radicals or lipid peroxidase, which induces oxidative degradation of lipids that ultimately damages the cells (Ayala et al., 2014). In ferroptosis, lipid peroxidation triggers oxidative degradation of two important biofilm components, polyunsaturated fatty acids (PUFAs) and phosphatidylethanolamine. PUFAs contain readily extractable diallyl hydrogen, which is prone to lipid peroxidation and is essential for the execution of ferroptosis. Acyl-coenzyme A synthetase long-chain family member 4 (ACSL4) is a key enzyme catalyzing the conversion of PUFAs into coenzyme A which regulates lipid composition. Studies have shown that lipid peroxidation, specifically in the cell membranes and plasma membranes, has PUFAs (Galluzzi et al., 2012; Yang et al., 2016; Xie et al., 2021). Multiple studies have shown that antioxidants inhibit Erastin-induced ferroptosis, which indicates lipid peroxidation could lead to ferroptosis of the cells (Dixon et al., 2012; Zhang et al., 2022).
Amino acid metabolism is an important component of cell metabolism and is closely associated with ferroptosis (Angeli et al., 2017). Glutathione (GSH) is a water-soluble tripeptide composed of amino acid residues like glutamate, cysteine, and glycine. Low GSH levels are an important antioxidant and free radical scavenger of the cells. The glutathione peroxidase 4 (GPX4) uses GSH as a cofactor to reduce lipid peroxide levels. GPX4 is the most important anti-lipid peroxidase in the cell and is a central regulator of ferroptosis (Miao et al., 2022). GSH depletion leads to GPX4 inactivation, which ultimately results in ferroptosis (Imai et al., 2017; Seibt et al., 2019).
GSH synthesis requires the exchange of extracellular cystine and intracellular glutamate through the cystine/glutamate reverse transporter (System Xc-). System Xc- consists of the solute carrier family 7 member 11 (SLC7A11), a 12-transmembrane protein transporter, and solute carrier family 3 member 2 (SLC3A2), a single transmembrane regulatory protein. Blocking the glutamate metabolic pathway or inhibiting System Xc- increases ROS-induced-lipid peroxidation, thereby inducing ferroptosis (Galluzzi et al., 2015). Glutaminase 2 (GLS2) converts glutamine to glutamate and is essential for ferroptosis (Gao et al., 2015). GLS2 up-regulation leads to p53-dependent ferroptosis, and p53 can limit ferroptosis by inhibiting the dipeptidyl peptidase 4 activity (Jennis et al., 2016). Studies have demonstrated that targeted glutamine decomposition therapy could effectively treat organ damage caused by ferroptosis (Li et al., 2017).
ROS are a group of molecules that contains partly reduced oxygen molecules/species like peroxides, superoxide, singlet oxygen, and free radicals, generated due the biomolecule destruction. Ferroptosis can be induced by ROS produced by iron-mediated Fenton reactions, NADPH acting on NADPH oxidases (NOXs), and depletion of GSH (Yang et al., 2014). At the structural level, lipids undergo extensive peroxidation forming thinner biofilms and increases the curvature. This further leads to oxidation reactions, ultimately destabilizing the membranes (Park and Chung, 2019).
The NADPH/ferroptosis-suppressor-protein 1 (FSP1)/coenzyme Q10 (CoQ10) pathway is another antioxidant system parallel to GPX4. The inhibition of this pathway can induce ferroptosis. In the plasma membrane, FSP1 acts as an oxidoreductase which reduces ubiquinone (CoQ) to dihydro ubiquinone (CoQH2). CoQH2 is an antioxidant that captures lipophilic free radicals and prevents the accumulation of lipid peroxides, thereby inhibiting ferroptosis (Doll et al., 2019). The GTP cyclohydrolase1 (GCH1)/tetrahydrobiopterin (BH4)/dihydrofolate reductase (DHFR) pathway is an antioxidant pathway independent of GPX4. Ferroptosis can be induced by blocking the GCH1/BH4/DHFR pathway. The initiation of the GCH1/BH4/DHFR pathway promotes CoQ synthesis and inhibits the accumulation of lipid peroxides, thereby preventing the ferroptosis of the cells (Kraft et al., 2020).
Previous studies have demonstrated an association between ferroptosis and RA development. Various studies have shown that several pathways or compounds are associated with ferroptosis in the pathogenesis of RA (Qiu et al., 2020; Ursini and Maiorino, 2020; Ferreira et al., 2021; Zhao et al., 2022b). Therefore, it is speculated that there could be more direct evidence that indicates the involvement of ferroptosis in the pathogenesis of RA in the future.
GPX4 belongs to the glutathione peroxidase family. GPX4 protects cells from damage caused by lipid peroxidation by degrading small peroxide molecules. Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that regulates the expression of various ferroptosis-related genes, including GPX4 and TFR1 (Chadha et al., 2020).
A study by Luo and Zhang (2021) shows that the lipopolysaccharide (LPS) stimulation reduces GPX4, SLC7A11, SLC3A2L, and NRF2 expression in human synovial cells HUM-CELL-0060, thereby confirming the association between ferroptosis and synovitis. Zhou et al. (2022) showed a significant increase in levels of ACSL4 and NOX4, whereas a significant decrease in GPX4 levels was observed in cartilage of RA patients. Similar results were observed in the adjuvant arthritis (AA) rat model, indicating an association between enhancement in ferroptosis of chondrocytes and RA progression. As opposed to other studies, Ling et al. (2022) reported a decrease in ACSL4 expression and an increase in FTH1, GPX4, and SLC7A11 expression, thereby indicating a reduction in ferroptosis in RA-synovium and FLS. The study also suggests the presence of fibroblastic subpopulations with different sensitivities to ferroptosis.
p53 is an important link between RA to ferroptosis. p53 regulates various metabolic pathways, such as amino acid, lipid, reactive oxygen, and iron metabolism, which are closely linked to ferroptosis (Liu and Gu, 2022). Additionally, p53 favors ferroptosis by downregulating SLC7A11 expression to increase ROS levels via spermidine/spermine N1-acetyl transferase (SAT1)-arachidonate15-lipoxygenase (ALOX15). Recent studies suggest that the p53/SLC7A11 signaling axis control ferroptosis in MH7A cells (Hu et al., 2022).
Previous studies have highlighted the correlation between iron metabolism and chronic anemia in RA patients (Stefanova et al., 2018; Zhao et al., 2022b; Chang et al., 2022). Recently, some studies have shown the involvement of iron metabolism in ferroptosis in RA pathogenies. An increase in lipid peroxidation and iron levels was observed in the synovial membrane and synovial fluid of RA patients with high disease activity compared to moderate disease activity. This confirms the accumulation of lipid peroxidation and iron overload in the synovial membrane and synovial fluid of patients with RA (Wu et al., 2022).
Ferritin is an important Fe2+ storage protein in ferroptosis. Recent studies have shown increased FTH1 expression in synovium and FLS of patients with RA (Ling et al., 2022). Follow-up studies have shown that Erastin (ferroptosis inducer) treatment reduces the expression of FTH1, thereby indicating the involvement of iron metabolism in the ferroptosis in RA-FLS.
Recent studies have demonstrated the involvement of ROS in multiple signal pathways, such as MAPK, Phosphatidylinositol-3-kinase-Akt, and Nuclear factor kappa B, which contribute to RA development (Shen et al., 2015; Phull et al., 2018). ROS inhibits the interaction between growth factors and chondrocytes, leading to the apoptosis of chondrocytes and cartilage damage. In addition, ROS destroys the extracellular and intracellular matrices, such as proteoglycans, which are essential for chondrocytes (Mateen et al., 2016). ROS and excessive lipid oxidation can also induce abnormal proliferation of RA-FLS (Xie et al., 2021).
Currently, ferroptosis modulators have been widely used for treating neoplastic diseases. RA-FLS have “tumor-like” characteristics, which are critical for the onset and development of RA. Furthermore, the role of ferroptosis modulators in the treatment of patients with RA has been investigated. We have summarized different ferroptosis modulators in RA treatment, based on their different mechanisms of action, as follows (Figure 2).
SLC7A11 is the main active subunit of System Xc- which regulates the dynamic balance of intracellular GSH. The inhibition of System Xc- activity reduces the influx of cystine into the cell. This further reduces the synthesis of intracellular GSH and the ability of GPX4 to scavenge peroxides, ultimately leading to ferroptosis of the cells (Song et al., 2018).
Food and Drug Administration has approved sulfasalazine, an anti-rheumatic drug, for the treatment of RA patients. In vitro studies have demonstrated that sulfasalazine inhibits System Xc- which significantly reduces lymphoma cell proliferation (Mao et al., 2021). However, no studies have shown how sulfasalazine directly affects ferroptosis in the treatment of RA patients.
A recent study has shown that RAS-selective lethal 3 (RSL3), a GPX4 inhibitor decreases the expression of GPX4, SLC7A11, and SLC3A2L in human synovial cells HUM-CELL-0060 and promotes cell death (Luo and Zhang, 2021). In collagen-induced arthritis (CIA) mice model, treatment with imidazole ketone erastin (IKE), a ferroptosis inducer, can reduce the number of fibroblasts in the synovial membrane (Wu et al., 2022). Further, treatment with the combination of low doses of IKE and etanercept (TNF antagonist) induced ferroptosis of fibroblasts and improved the symptoms in CIA mice. These results suggest that the combination of TNF inhibitors and ferroptosis inducers could be a potential strategy for the treatment of RA patients.
Glycine is a component of single-carbon metabolism. Glycine is also involved in the mechanism associated with the transfer of methyl groups. A study by Ling et al. has shown that glycine regulates the levels of S-adenosylmethionine (a direct methyl donor), which promotes the methylation of the GPX4 promoter, thereby enhancing the ferroptosis of RA-FLS (Ling et al., 2022).
Galectin-1 derived peptide 3 (G1dP3), a bioactive peptide derived from the galectin-1 structural domain, has potent anti-inflammatory effects on RA-FLS (Hu et al., 2020). A recent study suggests G1dP3 promotes ferroptosis in RA-FLS via the p53/SLC7A11 axis and has potential therapeutic effects (Hu et al., 2022).
Besides these ferroptosis inducers, a recent study has demonstrated that icariin inhibits ferroptosis by activating the Xc-/GPX4 axis, thereby protecting RA-FLS from LPS-induced cell death. This could be a new strategy for the treatment of RA patients (Luo and Zhang, 2021).
In vivo, the Fenton reaction is the main source of ROS. Sulfasalazine-induced ferroptosis is a classic example of a dual reaction. In addition to the direct inhibition of the System Xc-, Sulfasalazine induces ferroptosis by indirectly up-regulating the level of trivalent iron, thereby inducing the Fenton reaction (Chadha et al., 2020).
Auranofin is an early anti-rheumatic drug that is similar to sulfasalazine. In cancers, Auranofin exerts an anti-cancer effect by directly altering ferroptosis (Kato et al., 2020). Studies have shown that the high doses of auranofin (25 mg/kg) induce ferroptosis by inhibiting the thioredoxin reductase activity. At a low dose (5 mg/kg), auranofin triggers hepcidin, which reduces serum iron levels and transferrin saturation, thereby inhibiting ferroptosis in mice (Yang et al., 2020). Hence, it is necessary to choose an appropriate dose of auranofin to ensure cellular ferroptosis and reduce drug toxicity.
FTH1 is an essential Fe2+ storage protein in ferroptosis. Recent studies have demonstrated an increase in FTH1 levels in synovium and RA-FLS. On treatment with glycine, a decrease in FTH1 levels was observed in RA-FLS (Ling et al., 2022). These results suggest a reduction in FTH1 expression on glycine treatment, which subsequently participates in the Fenton reaction to increase ferroptosis during RA progression.
Effective binding of NRF2 to the antioxidant response element promotes the production of cytotoxic electrophiles and ROS (Dodson et al., 2019; Chadha et al., 2020). GPX4 inhibitor RSL3 reduces the production of GPX4 in synoviocytes and NRF2 levels, which ultimately reduces ROS production (Luo and Zhang, 2021).
Liu et al. (2019) show that the NK-1R antagonist, Aprepitant reduces TNF-α-induced expression of NOX-4 and ROS production in RA-FLS. Although the changes in ferroptosis-related markers were not evaluated in this study, it is likely that Aprepitant alters ferroptosis of RA-FLS. Further, the activation of nuclear receptor subfamily 1 group D member 1 (NR1D1) reduces ROS production and enhances the secretion of Nrf2-related enzymes (Liu et al., 2020). CIA mice treated with NR1D1 agonist SR9009 demonstrated a significant inhibition in synovial hyperplasia, cartilage, and bone destruction. This suggests that NR1D1 can potentially be used as therapeutics in the treatment of patients with RA. Various studies have indicated that ROS could be a new therapeutic target for RA treatment regarding ferroptosis (Xie et al., 2016; Xie et al., 2021; Chang et al., 2022; Lai et al., 2022).
Recently Zhou et al. (2022) showed a correlation between transient receptor potential melastatin 7 (TRPM7) levels and various core regulators of ferroptosis in chondrocytes of patients with RA and the AA rat model. Knockdown of TRPM7 expression or inhibiting its activity can protect chondrocytes from ferroptosis. These results suggest that TRPM7-mediated chondrocyte ferroptosis could be a promising target for RA treatment.
Recent studies suggest the involvement of FSP1 in ferroptosis, which is independent of the GSH-based GPX4 pathway. The FSP1-CoQ-dependent pathway depends on NADPH-induced enhancement of the antioxidant system. FSP1 reduces CoQ to block lipid peroxidation and may act as an important threshold for ferroptosis. Hence, FSP1 could be a potential therapeutic candidate for the treatment of patients with RA (Xie et al., 2021).
The pathogenesis of RA is still unclear. Some patients are insensitive to the currently available therapeutic modalities. There is a need for additional therapeutic targets in the treatment of patients with “refractory” RA. Recent studies have shown ferroptosis as a new type of cell death. Ferroptosis-related drugs have emerged as promising targeted drugs in cancer therapeutics. Studies have been conducted to investigate the therapeutic value of ferroptosis-targeting drugs in RA treatment, and preliminary results have been obtained. However, research on ferroptosis in RA is currently limited and needs to be further explored. For example, the primary focus of these studies has been on FLS and chondrocytes in RA. The role of immune cells has not been explored until now and requires further research. Moreover, there are discrepancies among the studies reporting the role and the levels of ferroptosis in RA. Further, there is a lack of direct evidence between ferroptosis and the most widely used anti-rheumatic drugs in the treatment of RA patients, which requires further investigation. Despite several studies indicating the therapeutic effects of ferroptosis inducers for treating RA patients, some studies have suggested that inhibition of ferroptosis may improve the symptoms of patients with RA. Such discrepancy in the results indicates the complex role of ferroptosis in RA, and additional studies are required to enhance our understanding. Furthermore, it is believed that drugs modulating the key molecules associated with ferroptosis will be available for RA treatment, thereby bringing hope to patients with RA.
YW, QZ, and XH contributed to the conception and design of the work. LL, HG, and XC drafted the manuscript. YL, RW, and XZ prepared the figure. YW, QZ, and LL substantively revised the manuscript. All authors read and approved the final manuscript.
This research was supported by the National Natural Science Foundation of China (81802504, 81872207), Sichuan Science and Technology Bureau (2019YFS0439, 2020JDJQ0067, 2020JDRC0118, 2021YJ0564, 2022YFH0005), the Science and Technology Innovation Project of Chengdu, China (No. 2021-YF05-00225-SN), a Sichuan Medical Association grant (No. Q19037), and a grant from Sichuan Provincial Health Commission (20PJ105).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aldridge J., Ekwall A. H., Mark L., Bergstrom B., Andersson K., Gjertsson I., et al. (2020). T helper cells in synovial fluid of patients with rheumatoid arthritis primarily have a Th1 and a CXCR3(+)Th2 phenotype. Arthritis Res. Ther. 22 (1), 245. doi:10.1186/s13075-020-02349-y
Angeli J. P. F., Shah R., Pratt D. A., Conrad M. (2017). Ferroptosis inhibition: Mechanisms and opportunities. Trends Pharmacol. Sci. 38 (5), 489–498. doi:10.1016/j.tips.2017.02.005
Armaka M., Konstantopoulos D., Tzaferis C., Lavigne M. D., Sakkou M., Liakos A., et al. (2022). Single-cell multimodal analysis identifies common regulatory programs in synovial fibroblasts of rheumatoid arthritis patients and modeled TNF-driven arthritis. Genome Med. 14 (1), 78. doi:10.1186/s13073-022-01081-3
Ayala A., Munoz M. F., Arguelles S. (2014). Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 360438. doi:10.1155/2014/360438
Bai L. K., Su Y. Z., Wang X. X., Bai B., Zhang C. Q., Zhang L. Y., et al. (2022). Synovial macrophages: Past life, current situation, and application in inflammatory arthritis. Front. Immunol. 13, 905356. doi:10.3389/fimmu.2022.905356
Bartok B., Firestein G. S. (2010). Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 233 (1), 233–255. doi:10.1111/j.0105-2896.2009.00859.x
Bogdan A. R., Miyazawa M., Hashimoto K., Tsuji Y. (2016). Regulators of iron homeostasis: New players in metabolism, cell death, and disease. Trends biochem. Sci. 41 (3), 274–286. doi:10.1016/j.tibs.2015.11.012
Cai Z., Min J., Wang H., Fang X., Wang F. (2019). Role of iron overload and ferroptosis in heart disease. Chin. Sci. Bull. 64 (28-29), 2974–2987. doi:10.1360/tb-2019-0242
Chadha S., Behl T., Kumar A., Khullar G., Arora S. (2020). Role of Nrf2 in rheumatoid arthritis. Curr. Res. Transl. Med. 68 (4), 171–181. doi:10.1016/j.retram.2020.05.002
Chang S., Tang M., Zhang B., Xiang D., Li F. (2022). Ferroptosis in inflammatory arthritis: A promising future. Front. Immunol. 13, 955069. doi:10.3389/fimmu.2022.955069
Chen K., Zhang S., Jiao J., Zhao S. (2021). Ferroptosis and its potential role in lung cancer: Updated evidence from pathogenesis to therapy. J. Inflamm. Res. 14, 7079–7090. doi:10.2147/JIR.S347955
Currie G. L., Angel-Scott H. N., Colvin L., Cramond F., Hair K., Khandoker L., et al. (2019). Animal models of chemotherapy-induced peripheral neuropathy: A machine-assisted systematic review and meta-analysis. PLoS Biol. 17 (5), e3000243. doi:10.1371/journal.pbio.3000243
Cush J. J. (2022). Rheumatoid arthritis: Early diagnosis and treatment. Rheum. Dis. Clin. North Am. 48 (2), 537–547. doi:10.1016/j.rdc.2022.02.010
Davignon J. L., Rauwel B., Degboe Y., Constantin A., Boyer J. F., Kruglov A., et al. (2018). Modulation of T-cell responses by anti-tumor necrosis factor treatments in rheumatoid arthritis: A review. Arthritis Res. Ther. 20 (1), 229. doi:10.1186/s13075-018-1725-6
Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dodson M., Castro-Portuguez R., Zhang D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107. doi:10.1016/j.redox.2019.101107
Doll S., Freitas F. P., Shah R., Aldrovandi M., da Silva M. C., Ingold I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575 (7784), 693–698. doi:10.1038/s41586-019-1707-0
Ferreira H. B., Melo T., Paiva A., Domingues M. D. R. (2021). Insights in the role of lipids, oxidative stress and inflammation in rheumatoid arthritis unveiled by new trends in lipidomic investigations. Antioxidants (Basel) 10 (1), E45. doi:10.3390/antiox10010045
Galluzzi L., Bravo-San Pedro J. M., Vitale I., Aaronson S. A., Abrams J. M., Adam D., et al. (2015). Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 22 (1), 58–73. doi:10.1038/cdd.2014.137
Galluzzi L., Vitale I., Abrams J. M., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., et al. (2012). Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 19 (1), 107–120. doi:10.1038/cdd.2011.96
Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59 (2), 298–308. doi:10.1016/j.molcel.2015.06.011
Gao W., Wang X., Zhou Y., Wang X., Yu Y. (2022). Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 7 (1), 196. doi:10.1038/s41392-022-01046-3
Hattori Y., Kida D., Kaneko A. (2019). Normal serum matrix metalloproteinase-3 levels can be used to predict clinical remission and normal physical function in patients with rheumatoid arthritis. Clin. Rheumatol. 38 (1), 181–187. doi:10.1007/s10067-017-3829-9
Hu J., Lu J., Zhang X., Wang C., Ren K., Chang Q., et al. (2020). Peptidomic analysis on synovial tissue reveals galectin-1 derived peptide as a potential bioactive molecule against rheumatoid arthritis. Cytokine 131, 155020. doi:10.1016/j.cyto.2020.155020
Hu J., Zhang R., Chang Q., Ji M., Zhang H., Geng R., et al. (2022). p53: A regulator of ferroptosis induced by galectin-1 derived peptide 3 in MH7A cells. Front. Genet. 13, 920273. doi:10.3389/fgene.2022.920273
Hugon J. (2022). Rheumatoid arthritis and cognitive decline. Jt. Bone Spine 89 (3), 105346. doi:10.1016/j.jbspin.2022.105346
Imai H., Matsuoka M., Kumagai T., Sakamoto T., Koumura T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi:10.1007/82_2016_508
Jang S., Kwon E-J., Lee J. J. (2022). Rheumatoid arthritis: Pathogenic roles of diverse immune cells. Int. J. Mol. Sci. 23 (2), 905. doi:10.3390/ijms23020905
Jennis M., Kung C. P., Basu S., Budina-Kolomets A., Leu J. I., Khaku S., et al. (2016). An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 30 (8), 918–930. doi:10.1101/gad.275891.115
Ji M., Ryu H. J., Baek H. M., Shin D. M., Hong J. H. (2022). Dynamic synovial fibroblasts are modulated by NBCn1 as a potential target in rheumatoid arthritis. Exp. Mol. Med. 54 (4), 503–517. doi:10.1038/s12276-022-00756-6
Jiang X., Stockwell B. R., Conrad M. (2021). Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22 (4), 266–282. doi:10.1038/s41580-020-00324-8
Kato I., Kasukabe T., Kumakura S. (2020). MeninMLL inhibitors induce ferroptosis and enhance the antiproliferative activity of auranofin in several types of cancer cells. Int. J. Oncol. 57 (4), 1057–1071. doi:10.3892/ijo.2020.5116
Kraft V. A. N., Bezjian C. T., Pfeiffer S., Ringelstetter L., Muller C., Zandkarimi F., et al. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6 (1), 41–53. doi:10.1021/acscentsci.9b01063
Kumar L. D., Karthik R., Gayathri N., Sivasudha T. (2016). Advancement in contemporary diagnostic and therapeutic approaches for rheumatoid arthritis. Biomed. Pharmacother. 79, 52–61. doi:10.1016/j.biopha.2016.02.001
Lai B., Wu C. H., Wu C. Y., Luo S. F., Lai J. H. (2022). Ferroptosis and autoimmune diseases. Front. Immunol. 13, 916664. doi:10.3389/fimmu.2022.916664
Lefevre S., Knedla A., Tennie C., Kampmann A., Wunrau C., Dinser R., et al. (2009). Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 15 (12), 1414–1420. doi:10.1038/nm.2050
Lei G., Zhuang L., Gan B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22 (7), 381–396. doi:10.1038/s41568-022-00459-0
Li Q., Han X., Lan X., Gao Y., Wan J., Durham F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2 (7), e90777. doi:10.1172/jci.insight.90777
Ling H., Li M., Yang C., Sun S., Zhang W., Zhao L., et al. (2022). Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis. Rheumatol. Oxf., keac069. doi:10.1093/rheumatology/keac069
Liu H., Zhu Y., Gao Y., Qi D., Zhao L., Zhao L., et al. (2020). NR1D1 modulates synovial inflammation and bone destruction in rheumatoid arthritis. Cell Death Dis. 11 (2), 129. doi:10.1038/s41419-020-2314-6
Liu X., Zhu Y., Zheng W., Qian T., Wang H., Hou X. (2019). Antagonism of NK-1R using aprepitant suppresses inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes. Artif. Cells Nanomed. Biotechnol. 47 (1), 1628–1634. doi:10.1080/21691401.2019.1573177
Liu Y., Gu W. (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29 (5), 895–910. doi:10.1038/s41418-022-00943-y
Luo H., Zhang R. (2021). Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the Xc-/GPX4 axis. Exp. Ther. Med. 21 (1), 72. doi:10.3892/etm.2020.9504
Mao C., Liu X., Zhang Y., Lei G., Yan Y., Lee H., et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593 (7860), 586–590. doi:10.1038/s41586-021-03539-7
Mateen S., Moin S., Zafar A., Khan A. Q. (2016). Redox signaling in rheumatoid arthritis and the preventive role of polyphenols. Clin. Chim. Acta. 463, 4–10. doi:10.1016/j.cca.2016.10.007
Miao Y., Chen Y., Xue F., Liu K., Zhu B., Gao J., et al. (2022). Contribution of ferroptosis and GPX4's dual functions to osteoarthritis progression. EBioMedicine 76, 103847. doi:10.1016/j.ebiom.2022.103847
Micheroli R., Elhai M., Edalat S., Frank-Bertoncelj M., Burki K., Ciurea A., et al. (2022). Role of synovial fibroblast subsets across synovial pathotypes in rheumatoid arthritis: A deconvolution analysis. RMD Open 8 (1), e001949. doi:10.1136/rmdopen-2021-001949
Mueller A. L., Payandeh Z., Mohammadkhani N., Mubarak S. M. H., Zakeri A., Alagheband Bahrami A., et al. (2021). Recent advances in understanding the pathogenesis of rheumatoid arthritis: New treatment strategies. Cells 10 (11), 3017. doi:10.3390/cells10113017
Niu H. Q., Zhao X. C., Zhao W. P., Li X. F. (2019). Metabolic changes of synovial fibroblasts and rheumatoid arthritis. Zhonghua Nei Ke Za Zhi 58 (1), 69–73. doi:10.3760/cma.j.issn.0578-1426.2019.01.013
Novella-Navarro M., Plasencia-Rodriguez C., Nuno L., Balsa A. (2021). Risk factors for developing rheumatoid arthritis in patients with undifferentiated arthritis and inflammatory arthralgia. Front. Med. 8, 668898. doi:10.3389/fmed.2021.668898
Nygaard G., Firestein G. S. (2020). Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 16 (6), 316–333. doi:10.1038/s41584-020-0413-5
Park E., Chung S. W. (2019). ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 10 (11), 822. doi:10.1038/s41419-019-2064-5
Phull A. R., Nasir B., Haq I. U., Kim S. J. (2018). Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem. Biol. Interact. 281, 121–136. doi:10.1016/j.cbi.2017.12.024
Qiu Y., Cao Y., Cao W., Jia Y., Lu N. (2020). The application of ferroptosis in diseases. Pharmacol. Res. 159, 104919. doi:10.1016/j.phrs.2020.104919
Riegman M., Sagie L., Galed C., Levin T., Steinberg N., Dixon S. J., et al. (2020). Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 22 (9), 1042–1048. doi:10.1038/s41556-020-0565-1
Scherer H. U., Haupl T., Burmester G. R. (2020). The etiology of rheumatoid arthritis. J. Autoimmun. 110, 102400. doi:10.1016/j.jaut.2019.102400
Seibt T. M., Proneth B., Conrad M. (2019). Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 133, 144–152. doi:10.1016/j.freeradbiomed.2018.09.014
Shen C., Cai G. Q., Peng J. P., Chen X. D. (2015). Autophagy protects chondrocytes from glucocorticoids-induced apoptosis via ROS/Akt/FOXO3 signaling. Osteoarthr. Cartil. 23 (12), 2279–2287. doi:10.1016/j.joca.2015.06.020
Song X., Zhu S., Chen P., Hou W., Wen Q., Liu J., et al. (2018). AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr. Biol. 28 (15), 2388–2399. doi:10.1016/j.cub.2018.05.094
Stefanova K. I., Delcheva G. T., Maneva A. I., Batalov A. Z., Geneva-Popova M. G., Karalilova R. V., et al. (2018). Pathobiochemical mechanisms relating iron homeostasis with parameters of inflammatory activity and autoimmune disorders in rheumatoid arthritis. Folia Med. Plovdiv. 60 (1), 124–132. doi:10.1515/folmed-2017-0068
Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171 (2), 273–285. doi:10.1016/j.cell.2017.09.021
Sun P., Su J., Wang X., Zhou M., Zhao Y., Gu H. (2022). Nucleic acids for potential treatment of rheumatoid arthritis. ACS Appl. Bio Mat. 5 (5), 1990–2008. doi:10.1021/acsabm.1c01205
Ursini F., Maiorino M. (2020). Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 152, 175–185. doi:10.1016/j.freeradbiomed.2020.02.027
Wang Y., Quan F., Cao Q., Lin Y., Yue C., Bi R., et al. (2021). Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231–243. doi:10.1016/j.jare.2020.07.007
Wu J., Feng Z., Chen L., Li Y., Bian H., Geng J., et al. (2022). TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat. Commun. 13 (1), 676. doi:10.1038/s41467-021-27948-4
Xia J., Si H., Yao W., Li C., Yang G., Tian Y., et al. (2021). Research progress on the mechanism of ferroptosis and its clinical application. Exp. Cell Res. 409 (2), 112932. doi:10.1016/j.yexcr.2021.112932
Xie Y., Hou W., Song X., Yu Y., Huang J., Sun X., et al. (2016). Ferroptosis: Process and function. Cell Death Differ. 23 (3), 369–379. doi:10.1038/cdd.2015.158
Xie Z., Hou H., Luo D., An R., Zhao Y., Qiu C. (2021). ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: A covert clue for potential therapy. Inflammation 44 (1), 35–47. doi:10.1007/s10753-020-01338-2
Yang L., Wang H., Yang X., Wu Q., An P., Jin X., et al. (2020). Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target. Ther. 5 (1), 138. doi:10.1038/s41392-020-00253-0
Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 113 (34), E4966–E4975. doi:10.1073/pnas.1603244113
Yang W. S., SriRamaratnam R., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156 (1-2), 317–331. doi:10.1016/j.cell.2013.12.010
Zhang H. L., Hu B. X., Li Z. L., Du T., Shan J. L., Ye Z. P., et al. (2022). PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 24 (1), 88–98. doi:10.1038/s41556-021-00818-3
Zhao L., Zhou X., Xie F., Zhang L., Yan H., Huang J., et al. (2022). Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 42 (2), 88–116. doi:10.1002/cac2.12250
Zhao T., Yang Q., Xi Y., Xie Z., Shen J., Li Z., et al. (2022). Ferroptosis in rheumatoid arthritis: A potential therapeutic strategy. Front. Immunol. 13, 779585. doi:10.3389/fimmu.2022.779585
Keywords: rheumatoid arthritis, pathogenesis, ferroptosis, reactive oxygen species, lipid peroxidation, glutathione (GSH) metabolism
Citation: Long L, Guo H, Chen X, Liu Y, Wang R, Zheng X, Huang X, Zhou Q and Wang Y (2022) Advancement in understanding the role of ferroptosis in rheumatoid arthritis. Front. Physiol. 13:1036515. doi: 10.3389/fphys.2022.1036515
Received: 04 September 2022; Accepted: 20 September 2022;
Published: 04 October 2022.
Edited by:
Quanlu Duan, Huazhong University of Science and Technology, ChinaReviewed by:
Wencong Song, Shenzhen Traditional Chinese Medicine Hospital, ChinaCopyright © 2022 Long, Guo, Chen, Liu, Wang, Zheng, Huang, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wang, d195aTIwMjJAMTYzLmNvbQ==; Xiaobo Huang, ZHJodWFuZ3hiQDE2My5jb20=; Qiao Zhou, MjI5MjI5MzRAcXEuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.