 Declan Manning
Declan Manning Caroline Dart
Caroline Dart Richard L Evans
Richard L Evans- 1Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, United Kingdom
- 2Department of Physiology and Membrane Biology, School of Medicine, University of California, Davis, Davis, CA, United States
- 3Unilever Research and Development, Port Sunlight Laboratory, Bebington, Wirral, United Kingdom
The skin is a complex organ that acts as a protective layer against the external environment. It protects the internal tissues from harmful agents, dehydration, ultraviolet radiation and physical injury as well as conferring thermoregulatory control, sensation, immunological surveillance and various biochemical functions. The diverse cell types that make up the skin include 1) keratinocytes, which form the bulk of the protective outer layer; 2) melanocytes, which protect the body from ultraviolet radiation by secreting the pigment melanin; and 3) cells that form the secretory appendages: eccrine and apocrine sweat glands, and the sebaceous gland. Emerging evidence suggests that store-operated Ca2+ entry (SOCE), whereby depletion of intracellular Ca2+ stores triggers Ca2+ influx across the plasma membrane, is central to the normal physiology of these cells and thus skin function. Numerous skin pathologies including dermatitis, anhidrotic ectodermal dysplasia, hyperhidrosis, hair loss and cancer are now linked to dysfunction in SOCE proteins. Principal amongst these are the stromal interaction molecules (STIMs) that sense Ca2+ depletion and Orai channels that mediate Ca2+ influx. In this review, the roles of STIM, Orai and other store-operated channels are discussed in the context of keratinocyte differentiation, melanogenesis, and eccrine sweat secretion. We explore not only STIM1-Orai1 as drivers of SOCE, but also independent actions of STIM, and emerging signal cascades stemming from their activities. Roles are discussed for the elusive transient receptor potential canonical channel (TRPC) complex in keratinocytes, Orai channels in Ca2+-cyclic AMP signal crosstalk in melanocytes, and Orai isoforms in eccrine sweat gland secretion.
Introduction
Skin forms the largest organ system in the human body, and is comprised of a vast layer of dermal and epidermal tissues which protect the body from the surrounding environment. Ca2+ signals are utilised throughout the skin to control the functions of a range of cell types, including keratinocytes, melanocytes, eccrine sweat glands and cells from the immune and nervous systems. Each adopt a unique Ca2+ signalling toolkit to support their specific cellular functions. In recent decades, store-operated Ca2+ entry (SOCE), whereby depletion of intracellular Ca2+ stores triggers Ca2+ influx across the plasma membrane, has emerged as a critical process regulating differentiation, melanogenesis and sweat secretion. SOCE dysfunction in skin is linked to anhidrotic ectodermal dysplasia, hyperhidrosis, dermatitis, hair loss and cancer (Gwack et al., 2008; Stanisz et al., 2016; Lian et al., 2017; Evans et al., 2018; Yeh et al., 2020). To initiate SOCE, store-resident stromal interaction molecules (STIMs) sense Ca2+ depletion, cluster and activate plasma membrane store-operated Ca2+ (SOC) channels (Numaga-Tomita & Putney, 2013; Liou et al., 2005; Zhang et al., 2005). While the different cells of the skin and its appendages express several SOC isoforms, the predominant channels in this process are believed to be members of the Orai family, and principally Orai1 (Bovell et al., 2015, 2016; Lian et al., 2017; Yeh et al., 2020). Several transient receptor potential (TRP) proteins are also proposed to act as SOCs and TRP dysfunction is linked to numerous skin pathologies (Leuner et al., 2011; Caterina and Pang, 2016). This review will discuss the essential roles of STIM1 and Orai1 in keratinocyte, melanocyte and eccrine sweat gland function, as well as the emerging role for Orai1-driven TRP canonical channel (TRPC) activity in keratinocyte differentiation.
Keratinocytes: Differentiation
Keratinocytes form approximately 95% of all cells in the epidermis. Here, these cells differentiate to form a protective ‘cornified’ outer layer (Figure 1A). This process is tightly controlled by the transcriptional factor family activator protein-1 (AP1; Eckert et al., 1997). As cells differentiate, they largely lose their proliferative capacity, express keratinocyte-specific differentiation markers (such as involucrin), and form intercellular desmosome linkages which confer mechanical strength to the outer skin layer (Capone et al., 2000). An epidermal Ca2+ gradient, rising towards the surface layers, and maintained primarily via the sequestration of intracellular Ca2+ in cells of the stratum granulosum (Celli et al., 2016), drives the keratinocyte differentiation process (Tu et al., 2005). Cells detect elevated extracellular Ca2+ concentration ([Ca2+]o) by the Ca2+-sensing receptor (CaSR), a promiscuous G-protein coupled receptor that couples to several pathways (Figure 1B) including phospholipase C (PLC) activation and hence IP3 generation, store depletion and SOCE (Hofer and Brown, 2003; Tu et al., 2005).
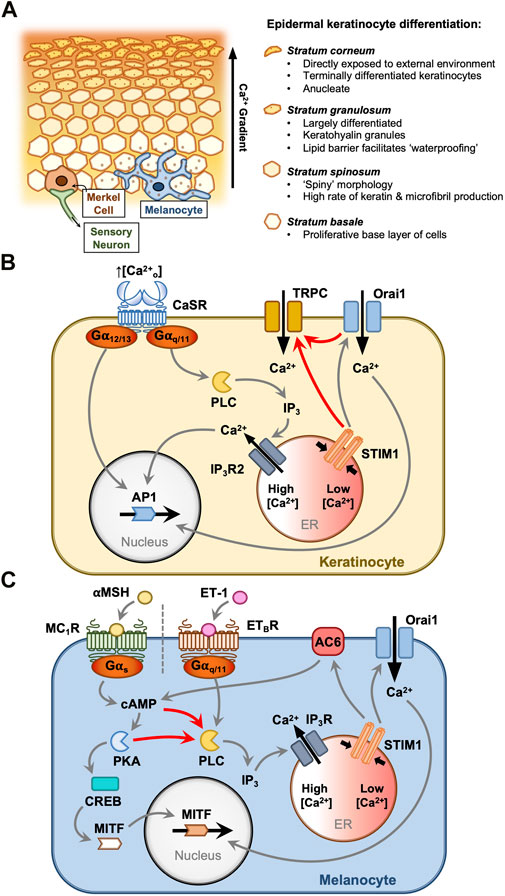
FIGURE 1. STIM1 and Orai1 drive keratinocyte and melanocyte functions in the epidermis. Keratinocytes and melanocytes are critical protective cells in the epidermal layer (A). Keratinocytes proliferate in the basal layer and differentiate in response to elevated extracellular [Ca2+]. This is driven by the Ca2+-sensing receptor (CaSR) (B), which activates Gα12/13 and Gαq/11 proteins. ER Ca2+ mobilisation triggers SOCE, and the resultant cytosolic Ca2+ signals contribute to AP1 activation and differentiation (B). Large currents carried by TRPC channels accompany the Orai channel Ca2+ influx, although the mechanism of TRPC channel activation in keratinocytes is not understood. Melanocytes protect the body from ultraviolet radiation by secreting melanin granules into the surrounding tissue. This is controlled by two key pathways (C), each triggered by paracrine hormones secreted from the surrounding cells (C). α-Melanocyte stimulating hormone (αMSH) and endothelin-1 (ET-1) stimulate melanocyte-inducing transcription factor (MITF)-driven melanogenesis via Gαs and Gαq/11 pathways, respectively. Both pathways prompt IP3-mediated ER Ca2+ store depletion and SOCE. Store depletion resulting from αMSH activity causes direct activation of adenylyl cyclase (AC6) by STIM1, producing a positive feedback loop where cyclic AMP production drives either protein kinase A (PKA) or EPAC activation to further activate phospholipase C (PLC), Ca2+ store depletion and SOCE.
In the HaCaT keratinocyte cell line, increasing the [Ca2+] in the culture media from 0.03 to 1.8 mM triggers a switch away from the proliferative phenotype that the cells adopt under low-confluence conditions in vitro. This ‘Ca2+ switch’ induces expression of keratin 1, which acts to slow cell growth (Tu et al., 2005). Knockdown of either STIM1 or Orai1 has been shown to ablate inward Ca2+ release-activated Ca2+ current (ICRAC) and reduce keratin one expression when cells are switched to 1.8 mM Cao2+. Additionally, HaCaT cells lacking STIM1 or Orai1 exhibit diminished proliferative responses to changes in Cao2+(Numaga-Tomita & Putney, 2013; Takei et al., 2016). SOCE via Orai1 has been shown to govern a range of functions in primary keratinocytes including proliferation, differentiation and migration (Vandenberghe et al., 2013) and Orai1 knockout mice consistently display a thin-skinned phenotype (Gwack et al., 2008). Ca2+ entry through STIM1-activated Orai1 channels thus seems vital for the routine functions of keratinocytes.
TRPC channels were first implicated in the keratinocyte SOC response in a study with primary human keratinocytes (Tu et al., 2005). Using siRNA knockdowns of TRPC1 and TRPC4, Tu and colleagues used Ca2+ imaging to demonstrate the involvement of these channels in keratinocyte SOCE. They also identified a physical interaction between TRPC1, IP3 receptors (IP3Rs) and PLCγ1 (Tu et al., 2005). TRPC1 and TRPC4 involvement was later confirmed in separate studies with primary human keratinocytes from gingiva (Cai et al., 2006; Fatherazi et al., 2007) and neonatal foreskin (Beck et al., 2008). Fatherazi et al. (2007) examined the biphasic SOC response with patch-clamp electrophysiology, highlighting an initial inward-rectifying current characteristic of ICRAC, followed by an ohmic current more characteristic of heteromeric TRPC channels. TRPC1, C5, C6 and C7 mRNA and protein were initially identified in gingival keratinocytes (Cai et al., 2005) compared with TRPC1, C4, C5 and C7 in HaCaT keratinocytes (Beck et al., 2006). Müller and colleagues demonstrated that specific activation of TRPC6 with hyperforin stimulated differentiation of HaCaT keratinocytes (Müller et al., 2008). Hence, there is uncertainty over the specific combinations of TRPC isoforms involved in keratinocyte SOC currents. Furthermore, the specific mechanism of action for this differentiation trigger event requires further clarification, with no link between Orai and TRPC channel activity identified in keratinocytes.
The consequences of impaired Ca2+ signalling in keratinocytes are also unclear. One group has demonstrated reduced TRPC1-7 expression and store-operated Ca2+ influx in primary keratinocytes harvested from psoriatic patients (Leuner et al., 2011). Celli et al. (2021) also demonstrated a reduction in store-operated Ca2+ influx in aged primary human keratinocytes, alongside reduced CaSR and PLCβ/γ expression, and impaired cell-to-cell adhesion. Recent studies in mice have also suggested that STIM1 may act as a primary thermo-sensor in keratinocytes via heat-induced regulation of Orai channels (Nwonkonko et al., 2019). However, it remains unclear whether STIM1 impacts thermosensation in human skin. Keratinocytes additionally play an essential role in superficial wound repair, and much evidence suggests that impaired SOCE processes would inhibit their ability to proliferate and repair damaged tissue (Tu et al., 2005; Darbellay et al., 2014; Numaga-Tomita & Putney, 2013). However, one in vivo study demonstrated that STIM1 knockout actually improves skin injury outcomes in mice (Putney et al., 2017). It is postulated that this is due to pro-inflammatory chemokine release from keratinocytes, which subsequently boosts protective neutrophil activity (Su and Richmond, 2015). Additional studies are required to resolve the contradictory evidence concerning SOCE processes in wound repair.
Melanocytes: Melanogenesis and proliferation
Melanocytes distribute melanin pigment among neighbouring keratinocytes to defend the immediate tissue from mutation and damage resulting from ultraviolet radiation (Figure 1A). The literature indicates that this skin pigmentation process is regulated by two different pathways (Figure 1C). One pathway facilitates ‘adaptive tanning’ in response to environmental ultraviolet radiation (UV) exposure (Stanisz et al., 2012; Nam & Lee, 2016; Kawakami & Fisher, 2017), and another pathway is linked more directly with baseline melanin production while also contributing to adaptive melanogenesis (Chakraborty et al., 1996; Scott et al., 2002; Cui et al., 2007). In this section, the emerging role of STIM and Orai will be discussed for each of these pathways.
Melanin production is induced by the activation of melanocyte-inducing transcription factor (also known as microphthalmia-associated transcription factor, MITF; Hodgkinson et al., 1993). MITF controls melanocyte differentiation, metabolism, proliferation and survival through its regulation of a range of genes. The most relevant in melanogenesis is its directed upregulation of tyrosinase (Tyr), which initiates the biosynthetic pathway for melanin (Kuzumaki et al., 1993). Both of the melanogenesis pathways described here converge on the upregulation of MITF in melanocytes, which is consistently found to drive skin pigmentation and melanocyte proliferation (reviewed by Kawakami & Fisher, 2017).
One distinct melanin production pathway is stimulated by α-melanocyte stimulating hormone (αMSH), and to a lesser extent, adrenocorticotropic hormone (ACTH), both of which are produced by neighbouring keratinocytes by cleaving proopiomelanocortin (POMC) (Chakraborty et al., 1996). αMSH activates melanocortin one receptor (MC1R) on nearby melanocytes to stimulate melanogenesis and proliferation through both Gαs signalling via cyclic AMP (cAMP) and Gβγ signalling via MAPK pathway activation (Cui et al., 2007). Point mutations in the POMC and MC1R genes can each produce a red-haired, fair-skinned phenotype characterised by low baseline melanin production (Valverde et al., 1995; Krude et al., 1998). Thus, αMSH-MC1R signalling is a factor in setting ‘baseline’ melanin production in the epidermis. Furthermore, UV radiation causes keratinocytes to increase production of αMSH, and elevated αMSH levels also stimulate melanocytes to upregulate MC1R expression (Chakraborty et al., 1996; Scott et al., 2002; Cui et al., 2007). As such, the αMSH-MC1R pathway appears capable of contributing to baseline melanin production and also stimulating an adaptive response to UV radiation (Cui et al., 2007).
cAMP has long been considered critical in the αMSH melanogenesis pathway for its role in activating PKA, which in turn activates nuclear cAMP response element-binding protein (CREB) which then activates MITF (D’Orazio et al., 2006) (Figure 1C). The role of cAMP signalling in melanocyte responses to αMSH is well characterised, however a role for Ca2+-dependent signalling proteins has only recently been proposed for this signalling network. Motiani et al. (2018) characterised SOCE in primary human melanocytes, B16 melanoma cells and a zebrafish melanogenesis model. Primary human melanocytes with greater melanin content were shown to undergo enhanced SOCE (Motiani et al., 2018). αMSH and the adenylyl cyclase (AC) activator, forskolin, each stimulated endoplasmic reticulum (ER) Ca2+ mobilisation, and Ca2+ release could be blocked by inhibitors of either AC or PLC, interlinking PLC activity and Ca2+ store depletion with the cAMP production caused by MC1R activation (Motiani et al., 2018). It is unclear whether cAMP indirectly activates PLC in these cells via phosphorylation events induced by PKA or via signalling through exchange protein directly activated by cAMP (EPAC). Orai1 knockdown in both primary melanocytes and B16 cell tumours inhibited αMSH-stimulated proliferation (Motiani et al., 2018). STIM1 knockdown also reduced melanocyte and melanoma growth, and furthermore knockdown of zebrafish STIM1a significantly reduced in vivo melanin production (Motiani et al., 2018). This result was mirrored in B16 melanoma cells where, surprisingly, STIM1 knockdown reduced melanin production but Orai1 knockdown had no effect (Motiani et al., 2018). Intriguingly, it was shown that this effect was exerted by direct activation of AC6 by STIM1, which interacts via the serine/proline-rich domain (SPD), generating cAMP (Motiani et al., 2018). Taken together, these data suggest a positive feedback loop whereby MC1R-generated cAMP causes ER Ca2+ store depletion and STIM1 oligomerisation, and independent of Orai1, STIM1 generates cAMP through AC6 activation to amplify the cAMP melanogenesis signal (Motiani et al., 2018).
When exposed to UV radiation, keratinocytes in the epidermis release several hormones to influence the growth and behaviour of neighbouring melanocytes. While one melanogenesis pathway is known to be activated by αMSH, locally-released endothelin-1 (ET-1) activates the melanocyte endothelin B receptor (ETBR) to trigger a distinct pathway in response to UV exposure (Kawakami & Fisher, 2017). ETBR stimulation activates PLC to generate diacylglycerol (DAG) and IP3-mediated Ca2+ store mobilisation, which concurrently stimulate the RAS/MAPK signalling pathway to drive MITF activation and melanogenesis (Imokawa et al., 1996; Sato-Jin et al., 2008) (Figure 1C). Stanisz et al. (2012) showed that Orai1 knockdown blunts in vitro primary melanocyte viability, and inhibition with 2-APB inhibits tyrosinase activity and UV- or ET-1-induced melanin production. This demonstrated that Orai1 activation and SOCE play a critical role in adaptive melanogenesis, specifically in response to ET-1 signalling. This was supported by Nam & Lee (2016), who showed that Orai channel inhibition with trans-anethole reduces UV-induced melanin production in B16 melanoma cells. Surprisingly, this study found that tyrosinase activity was preserved despite Orai1 inhibition, suggesting that SOCE might bypass MITF to upregulate melanin production in B16 cells (Nam & Lee, 2016). Thus, the precise role of Orai1 channels in ET-1-induced melanogenesis requires further clarification.
The role of STIM1 and Orai1 in melanocyte biology has been detailed by relatively few studies, which may be explained by the fact that melanocyte research is often linked to the goal of understanding melanoma pathology (reviewed by Stanisz et al., 2016 and Zhang et al., 2022). Studies have reported differing Orai1-3 and STIM1-2 expression profiles across model systems, and although STIM2 expression often exceeds that of STIM1, it appears to be STIM1 that activates Orai channels and AC6 in melanocytes (Stanisz et al., 2012, 2014; Motiani et al., 2018). Furthermore, the reported effects of STIM and Orai activity on melanocyte proliferation, migration, invasion and survival vary considerably between studies and according to the model system studied (Stanisz et al., 2012, 2014; Sun et al., 2014; Hooper et al., 2015).
The eccrine sweat gland: ‘Clear’ cell secretion
Human skin contains three secretory appendages: the eccrine and apocrine sweat glands, and the sebaceous gland. The former facilitates thermoregulation by secreting a clear salty fluid (sweat) directly onto the skin surface, whilst the latter two secrete more viscous lipid- and protein-containing secretions (apocrine sweat and sebum respectively) into the hair follicular space (Figure 2A). The processes involved in eccrine gland sweat production are well understood and have been reviewed extensively (Sato et al., 1989a, 1989b; Wilke et al., 2007; Bovell, 2018; Cui et al., 2018). In brief, in eccrine sweat gland secretory coil cells (notably the so-called ‘clear cells’, but also the ‘dark’ cells), sweating is initiated via a two-step process: an agonist-generated rise in [Ca2+]i (the ‘Ca2+ signal’), followed by the activation of Cl− transport across the epithelium from the plasma to the glandular lumen (‘Cl− transport’; Figure 2B). The principal agonist initiating thermoregulatory sweating is acetylcholine, with adrenaline and other catecholamines controlling stress-induced emotional sweating.
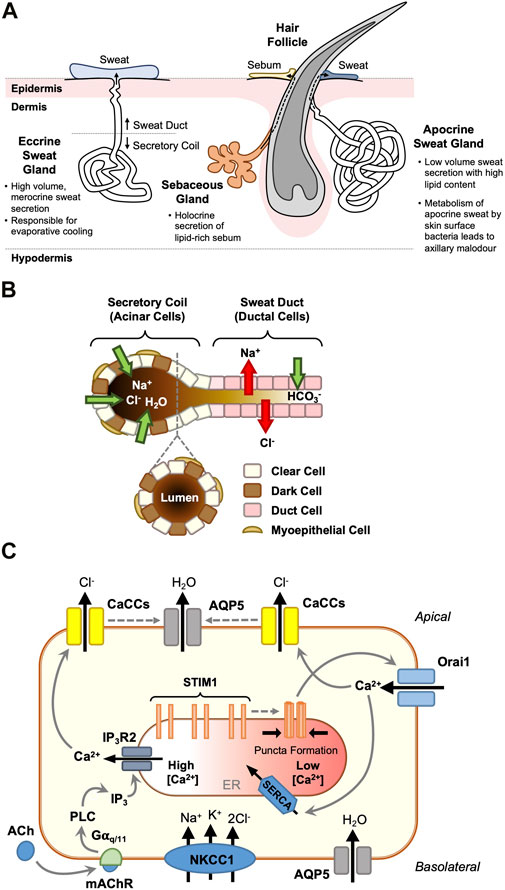
FIGURE 2. STIM1 and Orai1 are critical regulators of eccrine clear cell sweat secretion. Distinct from the apocrine sweat gland and the sebaceous gland which each associate with hair follicles (A), the eccrine sweat gland secretes dilute sweat directly onto the skin surface. It is responsible for salt and fluid secretion, through a two-phase merocrine secretory mechanism (B). Acinar cells in the secretory coil drive fluid secretion by actively transporting Cl−, with Na+ following across the tight junctions, and water osmotically. Myoepithelial cells provide structural support to the secretory cells, whilst upstream ductal cells modify the ionic composition of the hypertonic primary sweat, ready for release onto the skin surface. Clear cells are the key drivers of eccrine fluid secretion (C). In addition the transepithelial transport of Cl− ions (via the basolateral Na+-K+-2Cl- co-transporter, NKCC1, and Ca2+-activated Cl− channels, TMEM16A and Best2), these cells move water from the plasma to the gland lumen via basolateral and apical aquaporin 5 (AQP5) channels in response to stimulation from the sympathetic nerve termini (via mAChR activation). Gαq/11 proteins generate a biphasic cytosolic Ca2+ signal through sequential IP3-mediated ER Ca2+ release and store depletion resulting in STIM1-mediated Orai1 activation and SOCE. Elevated cytosolic [Ca2+] activates apical Ca2+-activated Cl− channels (CaCCs) and Cl− efflux, resulting in osmotically-driven water efflux into the sweat gland lumen (C).
The ion transport proteins involved in the Cl− transport process have been characterised at the molecular level, but it is only recently that insights into the protein components involved in generating the Ca2+ signal have been reported. Cl− influx is driven by the basolateral Na+-K+-2Cl- co-transporter NKCC1 (Zhang et al., 2014), and Cl− efflux via the Ca2+-activated Cl− channels (CaCCs) TMEM16A (Anoctamin-1) and bestrophin 2 (Ertongur-Fauth et al., 2014; Wilson and Metzler-Wilson, 2015; Concepcion et al., 2016) (see Figure 2C). The water channel aquaporin 5 (AQP5) is present in the apical membrane (Zhang et al., 2014) and knock-out studies in mice have suggested a central role for this channel in fluid secretion (Nejsum et al., 2002).
A key event in the generation of the ‘Ca2+ signal’ that triggers eccrine sweating is a biphasic increase in the level of [Ca2+]i comprising an initial agonist-induced release of Ca2+ from internal stores, followed by an influx of extracellular Ca2+. This SOCE process is widely documented in many exocrine cells (see Ambudkar, 2015; Concepcion & Feske, 2017). The first indication that SOCE in human eccrine gland cells may involve Ca2+-sensing STIM proteins and Ca2+-selective Orai channels was reported in the eccrine gland NCL-SG3 cell line (Bovell et al., 2015). Subsequent studies using freshly isolated human eccrine secretory coil cells showed that STIM1, Orai1 and Orai3 are expressed at both gene and protein levels (Bovell et al., 2016). Immunofluorescence demonstrated that STIM1 is located intracellularly (probably in the ER), and Orai one and three in the plasma membrane of secretory coil cells. Fluorescence imaging experiments showed that influx of extracellular Ca2+, triggered by the emptying of intracellular stores with thapsigargin, was perturbed using a variety of SOCE inhibitors (carboxyamidotriazole, diethylstilbesterol and BTP2; Bovell et al., 2016). Furthermore, thapsigargin-induced SOCE was reduced by ∼50% in an Orai1 knockdown, whilst Orai3 knockdown was ineffective. This suggested that with respect to SOCE, Orai3 can partially substitute for Orai1, and Orai1 can totally substitute for Orai3 in respective knockdowns (Bovell et al., 2016). A double Orai1-Orai3 knockdown nearly abolished SOCE. These data suggest that STIM1 and Orai1 are the principal drivers of SOCE and hence Cl− flux and sweat secretion in eccrine secretory coil cells, with Orai3 providing a supportive role which remains undefined. The suggestion that STIM1, and not STIM2, is the Ca2+ sensor critical for sweat secretion is supported by a mouse study, which shows that foot pad sweat secretion is significantly inhibited in a STIM1 knockout but is comparable to the wild type in a STIM2 knockout (Concepcion and Feske, 2017).
The significance of Orai channels in sweat gland function is further highlighted in human patients suffering loss-of-function mutations in their STIM1 and Orai1 genes, which results in anhidrosis (an inability to produce eccrine sweat) and hyperthermia at high ambient temperatures due to an intrinsic incapacity for evaporative cooling (Concepcion et al., 2016). Furthermore, NCL-SG3 cells lacking STIM1 and Orai1 expression are characterised by a loss of SOCE functionality and agonist-induced Cl− secretion via TMEM16A (Concepcion et al., 2016).
Finally, a study using freshly isolated eccrine secretory coil cells from patients suffering from hyperhidrosis (sweating that exceeds that required for normal body temperature regulation), showed that the [Ca2+]i response following stimulation with the muscarinic agonist carbachol was 2-fold that of cells isolated from non-hyperhidrotic individuals (Evans et al., 2018). Expression of STIM1 in hyperhidrotic cells was also double that of non-hyperhidrotics, while expression of Orai1, the muscarinic m3 receptor, IP3 isoforms 1 and 2, and AQP5 were not significantly changed. Over-expression of STIM1 at the protein level was confirmed by Western blotting and immunohistochemistry, whilst STIM2 was not over-expressed (Evans et al., 2018). Thapsigargin-induced SOCE was reduced in STIM1 and Orai1 knockdowns. These observations suggested that human hyperhidrotic eccrine gland secretory coil cells are characterised by an over-expression of STIM1 relative to non-hyperhidrotic individuals, resulting in enhanced SOCE, which presumably leads to higher sweat secretion.
Conclusion
Numerous skin cell types rely on Ca2+ entry channels to control their routine physiological functions. Here, the roles of SOCE channels have been summarised for a selection of key skin functions: keratinocyte differentiation, melanocyte proliferation and melanogenesis, and sweat secretion from ‘clear’ cells in the eccrine sweat gland. This selection is by no means exhaustive, with cells from the immune and nervous systems resident in the integumentary system playing vital roles governed by their own unique Ca2+ entry systems. STIM1 and Orai1 have been revealed as key drivers of all the skin functions discussed here, both through their well-characterised SOCE process, the independent actions of STIM1, and the uncharacterised signal cascades stemming from their activities. Clearly, much is yet to be determined about the role of non-Orai store-operated Ca2+ channels such as the elusive TRPC complex in keratinocytes, the role of Orai channels in Ca2+-cAMP signal crosstalk in melanocytes, and the interaction of different Orai isoforms in driving eccrine sweat gland fluid secretion.
Author contributions
DM and RE were responsible for defining the scope of the review. DM, CD, and RE were responsible for drafting of the article, revising and approving the final version. All figures were designed and created by DM.
Funding
This work was supported by a Biotechnology & Biological Sciences Research Council (BBSRC, UK)—Unilever Collaborative Training Partnership grant BB/R505432/1 (recipient: DM, award: CD).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ambudkar I. S. (2015). Calcium signalling in salivary gland physiology and dysfunction. J. Physiol. 594, 2813–2824. doi:10.1113/JP271143
Beck B., Lehen’kyi V., Roudbaraki M., Flourakis M., Charveron M., Bordat P., et al. (2008). TRPC channels determine human keratinocyte differentiation: New insight into basal cell carcinoma. Cell Calcium 43 (5), 492–505. doi:10.1016/j.ceca.2007.08.005
Beck B., Zholos A., Sydorenko V., Roudbaraki M., Lehen’kyi V., Bordat P., et al. (2006). TRPC7 is a receptor-operated DAG-activated channel in human keratinocytes. J. Invest. Dermatol. 126 (9), 1982–1993. doi:10.1038/sj.jid.5700352
Bovell D. L., Robertson J., Evans R. (2016). STIM and Orai proteins regulate Ca2+ entry into isolated human eccrine sweat gland secretory coil cells. Proc. Physiol. Soc. 37, PCA137.
Bovell D. L., Robertson J., Mohamed F. (2015). STIM and Orai proteins regulate calcium entry into human sweat gland cell line – NCL-SG3. Proc. Physiol. Soc. 34, PC050.
Bovell D. L. (2018). The evolution of eccrine sweat gland research towards developing a model for human sweat gland function. Exp. Dermatol. 27, 544–550. doi:10.1111/exd.13556
Cai S., Fatherazi S., Presland R. B., Belton C. M., Izutsu K. T. (2005). TRPC channel expression during calcium-induced differentiation of human gingival keratinocytes. J. Dermatol. Sci. 40 (1), 21–28. doi:10.1016/j.jdermsci.2005.06.005
Cai S., Fatherazi S., Presland R. B., Belton C. M., Roberts F. A., Goodwin P. C., et al. (2006). Evidence that TRPC1 contributes to calcium-induced differentiation of human keratinocytes. Pflugers Arch. 452 (1), 43–52. doi:10.1007/s00424-005-0001-1
Capone A., Visco V., Belleudi F., Marchese C., Cardinali G., Bellocci M., et al. (2000). Up-modulation of the expression of functional keratinocyte growth factor receptors induced by high cell density in the human keratinocyte HaCaT cell line. Cell Growth Differ 11 (11), 607–614.
Caterina M. J., Pang Z. (2016). TRP channels in skin biology and pathophysiology. Pharmaceuticals 9 (4), 77. doi:10.3390/ph9040077
Celli A., Crumrine D., Meyer J. M., Mauro T. (2016). Endoplasmic reticulum calcium regulates epidermal barrier response and desmosomal structure. J. Invest. Dermatol. 136 (9), 1840–1847. doi:10.1016/j.jid.2016.05.100
Celli A., Tu C., Lee E., Bikle D., Mauro T. (2021). Decreased calcium-sensing receptor expression controls calcium signaling and cell-to-cell adhesion defects in aged skin. J. Invest. Dermatol. 141 (11), 2577–2586. doi:10.1016/j.jid.2021.03.025
Chakraborty A. K., Funasaka Y., Slominski A., Ermak G., Hwang J., Pawelek J. M., et al. (1996). Production and release of proopiomelanocortin (POMC) derived peptides by human melanocytes and keratinocytes in culture: Regulation by ultraviolet B. Biochim. Biophys. Acta 1313 (2), 130–138. doi:10.1016/0167-4889(96)00063-8
Concepcion A. R., Feske S. (2017). Regulation of epithelial ion transport in exocrine glands by store-operated Ca2+ entry. Cell Calcium 63, 53–59. doi:10.1016/j.ceca.2016.12.004
Concepcion A. R., Vaeth M., Wagner L. E., Eckstein M., Hecht L., Yang J., et al. (2016). Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. J. Clin. Invest. 126, 4303–4318. doi:10.1172/JCI89056
Cui C-Y., Noh J. H., Michel M., Gorospe M., Schlessinger D. (2018). STIM1, but not STIM2, is the calcium sensor critical for sweat secretion. J. Invest. Dermatol. 138, 704–707. doi:10.1016/j.jid.2017.09.038
Cui R., Widlund H. R., Feige E., Lin J. Y., Wilensky D. L., Igras V. E., et al. (2007). Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 128 (5), 853–864. doi:10.1016/j.cell.2006.12.045
Darbellay B., Barnes L., Boehncke W. H., Saurat J. H., Kaya G. (2014). Reversal of murine epidermal atrophy by topical modulation of calcium signaling. J. Invest. Dermatol. 134 (6), 1599–1608. doi:10.1038/jid.2013.524
D’Orazio J. A., Nobuhisa T., Cui R., Arya M., Spry M., Wakamatsu K., et al. (2006). Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 443 (7109), 340–344. doi:10.1038/nature05098
Eckert R. L., Crish J. F., Banks E. B., Welter J. F. (1997). The epidermis: Genes on—genes off. J. Invest. Dermatol. 109 (4), 501–509. doi:10.1111/1523-1747.ep12336477
Ertongur-Fauth T., Hochheimer A., Buescher J. M., Rapprich S., Krohn M. (2014). A novel TMEM16A splice variant lacking the dimerization domain contributes to calcium-activated chloride secretion in human sweat gland epithelial cells. Exp. Dermatol. 23, 825–831. doi:10.1111/exd.12543
Evans R. L., Johnstone S. R., Elias M., Robertson J., Bovell D. L., Martin P. E. (2018). Enhanced Ca2+ signalling and over-expression of STIM1 are characteristic of human eccrine sweat gland secretory coil cells isolated from hyperhidrotic individuals. Proc. Physiol. Soc. 41, PCA104.
Fatherazi S., Presland R. B., Belton C. M., Goodwin P., Al-Qutub M., Trbic Z., et al. (2007). Evidence that TRPC4 supports the calcium selective ICRAC-like current in human gingival keratinocytes. Pflugers Arch. 453 (6), 879–889. doi:10.1007/s00424-006-0156-4
Gwack Y., Srikanth S., Oh-hora M., Hogan P. G., Lamperti E. D., Yamashita M., et al. (2008). Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol. Cell. Biol. 28 (17), 5209–5222. doi:10.1128/mcb.00360-08
Hodgkinson C. A., Moore K. J., Nakayama A., Steingrímsson E., Copeland N. G., Jenkins N. A., et al. (1993). Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 74 (2), 395–404. doi:10.1016/0092-8674(93)90429-T
Hofer A., Brown E. (2003). Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 4, 530–538. doi:10.1038/nrm1154
Hooper R., Zhang X., Webster M., Go C., Kedra J., Marchbank K., et al. (2015). Novel protein kinase C-mediated control of Orai1 function in invasive melanoma. Mol. Cell. Biol. 35 (16), 2790–2798. doi:10.1128/mcb.01500-14
Imokawa G., Yada Y., Kimura M. (1996). Signalling mechanisms of endothelin-induced mitogenesis and melanogenesis in human melanocytes. Biochem. J. 314 (1), 305–312. doi:10.1042/bj3140305
Kawakami A., Fisher D. E. (2017). The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Invest. 97 (6), 649–656. doi:10.1038/labinvest.2017.9
Krude H., Biebermann H., Luck W., Horn R., Brabant G., Gruters A. (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157. doi:10.1038/509
Kuzumaki T., Matsuda A., Wakamatsu K., Ito S., Ishikawa K. (1993). Eumelanin biosynthesis is regulated by coordinate expression of tyrosinase and tyrosinase-related protein-1 genes. Exp. Cell Res. 207, 33–40. doi:10.1006/excr.1993.1159
Leuner K., Kraus M., Woelfle U., Beschmann H., Harteneck C., Boehncke W-H., et al. (2011). Reduced TRPC channel expression in psoriatic keratinocytes is associated with impaired differentiation and enhanced proliferation. PLOS ONE 6 (2), e14716. doi:10.1371/journal.pone.0014716
Lian J., Cuk M., Kahlfuss S., Issekutz T., Unutmaz D., Feske S., et al. (2017). ORAI1 mutations abolishing store-operated Ca2+ entry cause anhidrotic ectodermal dysplasia with immunodeficiency. J. Allergy Clin. Immunol. 142 (4), 1297–1310. doi:10.1016/j.jaci.2017.10.031
Liou J., Kim M. L., Won D. H., Jones J. T., Myers J. W., Ferrell J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi:10.1016/j.cub.2005.05.055
Motiani R. K., Tanwar J., Raja D. A., Vashisht A., Khanna S., Sharma S., et al. (2018). STIM 1 activation of adenylyl cyclase 6 connects Ca2+ and cAMP signaling during melanogenesis. EMBO J. 37 (5), e97597. doi:10.15252/embj.201797597
Müller M., Essin K., Hill K., Beschmann H., Rubant S., Schempp C. M., et al. (2008). Specific TRPC6 channel activation, a novel approach to stimulate keratinocyte differentiation. J. Biol. Chem. 283 (49), 33942–33954. doi:10.1074/jbc.M801844200
Nam J. H., Lee D. U. (2016). Foeniculum vulgare extract and its constituent, trans-anethole, inhibit UV-induced melanogenesis via ORAI1 channel inhibition. J. Dermatol. Sci. 84 (3), 305–313. doi:10.1016/j.jdermsci.2016.09.017
Nejsum L. N., Kwon T-H., Jensen U. B., Fumagalli O., Frokiaer J., Krane C. M., et al. (2002). Functional requirement of aquaporin-5 in plasma membranes of sweat glands. Proc. Natl. Acad. Sci. U. S. A. 99, 511–516. doi:10.1073/pnas.012588099
Numaga-Tomita T., Putney J. W. (2013). Role of STIM1 and Orai1-mediated Ca2+ entry in Ca2+-induced epidermal keratinocyte differentiation. J. Cell Sci. 126, 605–612. doi:10.1242/jcs.115980
Nwonkonko R. M., Zhou Y., Gill D. L. (2019). STIM1 is a precise thermo-sensor in skin. Cell Res. 29, 259–260. doi:10.1038/s41422-019-0154-7
Putney J. W., Steinckwich-Besançon N., Numaga-Tomita T., Davis F. M., Desai P. N., D’Agostin D. M., et al. (2017). The functions of store-operated calcium channels. Biochim. Biophys. Acta. Mol. Cell Res. 1864 (6), 900–906. doi:10.1016/j.bbamcr.2016.11.028
Sato K., Kang W. H., Saga K., Sato K. T. (1989a). Biology of sweat glands and their disorders. I. Normal sweat gland function. J. Am. Acad. Dermatol. 20, 537–563. doi:10.1016/S0190-9622(89)70063-3
Sato K., Kang W. H., Saga K., Sato K. T. (1989b). Biology of sweat glands and their disorders. II. Disorders of sweat gland function. J. Am. Acad. Dermatol. 20, 713–726. doi:10.1016/S0190-9622(89)70081-5
Sato-Jin K., Nishimura E. K., Akasaka E., Huber W., Nakano H., Miller A., et al. (2008). Epistatic connections between microphthalmia‐associated transcription factor and endothelin signaling in Waardenburg syndrome and other pigmentary disorders. FASEB J. 22 (4), 1155–1168. doi:10.1096/fj.07-9080com
Scott M. C., Suzuki I., Abdel-Malek Z. A. (2002). Regulation of the human melanocortin 1 receptor expression in epidermal melanocytes by paracrine and endocrine factors and by ultraviolet radiation. Pigment. Cell Res. 15 (6), 433–439. doi:10.1034/j.1600-0749.2002.02051.x
Stanisz H., Saul S., Müller C. S. L., Kappl R., Niemeyer B. A., Vogt T., et al. (2014). Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment. Cell Melanoma Res. 27 (3), 442–453. doi:10.1111/pcmr.12222
Stanisz H., Stark A., Kilch T., Schwarz E. C., Müller C. S. L., Peinelt C., et al. (2012). ORAI1 Ca2+ channels control endothelin-1-induced mitogenesis and melanogenesis in primary human melanocytes. J. Invest. Dermatol. 132 (5), 1443–1451. doi:10.1038/jid.2011.478
Stanisz H., Vultur A., Herlyn M., Roesch A., Bogeski I. (2016). The role of Orai-STIM calcium channels in melanocytes and melanoma. J. Physiol. 594 (11), 2825–2835. doi:10.1113/JP271141
Su Y., Richmond A. (2015). Chemokine regulation of neutrophil infiltration of skin wounds. Adv. Wound Care 4 (11), 631–640. doi:10.1089/wound.2014.0559
Sun J., Lu F., He H., Shen J., Messina J., Mathew R., et al. (2014). STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 207 (4), 535–548. doi:10.1083/jcb.201407082
Takei K., Denda S., Nagayama M., Denda M. (2016). Role of STIM1-Orai1 system in intra-cellular calcium elevation induced by ATP in cultured human keratinocytes. Exp. Dermatol. 83 (2), 323–325. doi:10.1111/exd.12928
Tu C. L., Chang W., Bikle D. D. (2005). Phospholipase C gamma1 is required for activation of store-operated channels in human keratinocytes. J. Invest. Dermatol. 124 (1), 187–197. doi:10.1111/j.0022-202X.2004.23544.x
Valverde P., Healy E., Jackson I., Rees J. L., Thody A. J. (1995). Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet. 11, 328–330. doi:10.1038/ng1195-328
Vandenberghe M., Raphaël M., Lehen’kyi V., Gordienko D., Hastie R., Oddos T., et al. (2013). ORAI1 calcium channel orchestrates skin homeostasis. Proc. Nat. Acad. Sci. 110, E4839–E4848. doi:10.1073/pnas.1310394110
Wilke K., Matin A., Terstegen L., Biel S. S., Martin A. (2007). A short history of sweat gland biology. Int. J. Cosmet. Sci. 29, 169–179. doi:10.1111/j.1467-2494.2007.00387.x
Wilson T. E., Metzler-Wilson K. (2015). Sweating chloride bullets: Understanding the role of calcium in eccrine sweat glands and possible implications for hyperhidrosis. Exp. Dermatol. 24, 177–178. doi:10.1111/exd.12595
Yeh Y-H., Lin Y-P., Kramer H., Parekh A. B. (2020). Single-nucleotide polymorphisms in Orai1 associated with atopic dermatitis inhibit protein turnover, decrease calcium entry and disrupt calcium-dependent gene expression. Hum. Mol. Genet. 29 (11), 1808–1823. doi:10.1093/hmg/ddz223
Zhang H., Chen Z., Zhang A., Gupte A. A., Hamilton D. J. (2022). The role of calcium signaling in melanoma. Int. J. Mol. Sci. 23 (3), 1010–1017. doi:10.3390/ijms23031010
Zhang M., Zeng S., Zhang L., Li H., Chen L., Zhang X., et al. (2014). Localization of Na+-K+-ATPase α/β, Na+-K+-2Cl-cotransporter 1 and aquaporin-5 in human eccrine sweat glands. Acta Histochemica 116 (8), 1374–1381. doi:10.1016/j.acthis.2014.08.010
Keywords: skin, store-operated calcium entry, STIM, Orai channels, TRPC channels, keratinocytes, melanocytes, eccrine sweat gland
Citation: Manning D, Dart C and Evans RL (2022) Store-operated calcium channels in skin. Front. Physiol. 13:1033528. doi: 10.3389/fphys.2022.1033528
Received: 31 August 2022; Accepted: 12 September 2022;
Published: 05 October 2022.
Edited by:
Michael M. White, Drexel University College of Medicine, United StatesReviewed by:
Isaac Jardin, University of Extremadura, SpainCopyright © 2022 Manning, Dart and Evans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard L Evans, UmljaGFyZC5FdmFuc0B1bmlsZXZlci5jb20=