 Zineb Bouredji
Zineb Bouredji Anteneh Argaw1
Anteneh Argaw1 Jérôme Frenette
Jérôme Frenette
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 23 November 2022
Sec. Striated Muscle Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.1032450
Skeletal muscle makes up almost half the body weight of heathy individuals and is involved in several vital functions, including breathing, thermogenesis, metabolism, and locomotion. Skeletal muscle exhibits enormous plasticity with its capacity to adapt to stimuli such as changes in mechanical loading, nutritional interventions, or environmental factors (oxidative stress, inflammation, and endocrine changes). Satellite cells and timely recruited inflammatory cells are key actors in muscle homeostasis, injury, and repair processes. Conversely, uncontrolled recruitment of inflammatory cells or chronic inflammatory processes leads to muscle atrophy, fibrosis and, ultimately, impairment of muscle function. Muscle atrophy and loss of function are reported to occur either in physiological situations such as aging, cast immobilization, and prolonged bed rest, as well as in many pathological situations, including cancers, muscular dystrophies, and several other chronic illnesses. In this review, we highlight recent discoveries with respect to the molecular mechanisms leading to muscle atrophy caused by modified mechanical loading, aging, and diseases. We also summarize current perspectives suggesting that the inflammatory process in muscle homeostasis and repair is a double-edged sword. Lastly, we review recent therapeutic approaches for treating muscle wasting disorders, with a focus on the RANK/RANKL/OPG pathway and its involvement in muscle inflammation, protection and regeneration processes.
Skeletal muscle is an essential organ of the human body. It is characterized by its great plasticity and capacity to adapt to numerous hormonal, nervous, and mechanical stimuli (Vittorio and Marcella, 2004). Skeletal muscle derives from a population of progenitor cells that originate in the somites of the paraxial mesoderm (Buckingham et al., 2003). Skeletal muscle plasticity is based on a balance between muscle hypertrophy, which results from an increase in organelles, cytoplasm, proteins and, consequently, an increase in the cross-section of muscle fibers, and muscle atrophy, which is associated with a decrease in cellular constituents and an increase in protein breakdown. Muscle breakdown involves several mechanisms, including the ubiquitin-proteasome system (UPS), which is regulated by E3 ubiquitin ligases, the autophagy-lysosome system (ALS), calpains, and caspases (Bonaldo and Sandri, 2013) (Yin et al., 2021). Many factors and situations cause protein degradation and/or a reduction in protein synthesis leading to muscle atrophy and wasting. Primary skeletal muscle atrophy is linked to genetic muscle disorders while secondary skeletal muscle atrophy is a consequence of physiological situations such as age-related sarcopenia, bed rest, and unloading, and pathological situations such as cancers, AIDS, osteoporosis, and heart failure (HF) (Yin et al., 2021). Muscle atrophy is also accelerated by the uncontrolled immune response observed in chronic diseases such as muscular dystrophies, including Duchenne muscular dystrophy (DMD), where the immune system promotes muscle cell death and fibrosis as the disease progresses (Tidball et al., 2018). However, the involvement of timely recruited inflammatory cells is a prerequisite for the activation and differentiation of satellite cells during the regeneration process (Tidball et al., 2018) (Dufresne et al., 2016). In this review, we highlight recent discoveries on the molecular mechanisms leading to muscle atrophy caused by modified mechanical loading, aging, and diseases. We also summarize current perspectives suggesting that the inflammatory process in muscle homeostasis and repair is a double-edged sword. Lastly, recent therapeutic approaches for treating muscle wasting disorders are reviewed, including pathways with potential impacts on skeletal muscle and bones.
Prolonged bedrest and spaceflight-related unloading induce impaired muscle function, mainly due to the breakdown of myofibrillar proteins and the impairment of protein synthesis. In spaceflight or ground-based models of microgravity, antigravity muscles such as postural muscles atrophy due to a loss of myofibrillar proteins (Qaisar et al., 2020). Postural muscles, which are predominantly composed of slow-twitch type I fibers, are more affected than fast-twitch type II fibers following a period of unloading (Baldwin et al., 2013). Mechanistically, it has been suggested that selective atrophy is in part related to pre-transcriptional and transcriptional changes involving the downregulation of pre-mRNA and mRNA of myosin heavy chain (MHC) type I, actin, transcriptional enhancer factor-1 (TEF-1), and myogenin, which are mainly involved in protein synthesis (Baldwin et al., 2013). In addition to type I fiber atrophy, a switch in fiber phenotype from slow to fast is observed following hindlimb suspension (HS) and microgravity conditions. Epigenetic modifications of histones or non-coding RNAs would play a role in the changes in muscle phenotype (Pandorf et al., 2009) (Haddad et al., 2003). However, in humans, muscles with predominantly fast-twitch fibers appear to be as affected by microgravity as type I fiber muscles (Fitts et al., 2001). For example, the vastus lateralis muscle is predominantly composed of fast-twitch type II fibers, which are significantly more affected than slow-twitch type I fibers when humans are exposed to spaceflight for 11 days (Edgerton et al., 1995), suggesting that the preponderance of certain types of muscle fibers in a muscle may be more important than the individual fiber type in its responsiveness to unloading stimuli (Baldwin et al., 2013). In addition to muscle mass, a 20-day bedrest of human volunteers induced a significant decrease in quadriceps muscle thickness and cross-sectional area (CSA) and an increase in the expression of two ubiquitin ligase genes, Cbl-b and atrogin-1, which are involved in muscle proteolysis (Ogawa et al., 2006), suggesting that the mechanisms of muscle atrophy may differ depending on muscle location and function, fiber type, or the condition leading to the atrophy. In general, slow-twitch fibers are mostly affected by denervation, microgravity, and immobilization due to mechanical discharge while fast-twitch fibers are more affected by aging or disease conditions. As the oxidative metabolism of slow twitch fibers is mainly regulated by the transcription factor peroxisome proliferator-activated receptor-g coactivator-1 (PGC1a), the downstream calcineurin/nuclear factor of activated T cells (NFAT) may play a role in the protection of slow-twitch fibers from muscle atrophy in some pathological conditions (Wang and Pessin, 2013). Nonetheless, an imbalance between protein degradation and protein synthesis pathways remains at the origin of muscle atrophy.
The insulin/IGF-1/Akt-mTOR pathway is the main pathway regulating muscle growth (Figure 1). When insulin and IGF-1 bind to their respective receptors, they initiate a phosphorylation cascade that leads to the activation of Akt, also known as protein kinase B. Akt enhances protein synthesis via the activation of the mammalian target of rapamycin (mTOR) pathway and the inhibition of glycogen synthase kinase β (GSK3β). Additionally, Akt blocks forkhead box Os (FoxOs), a family of transcription factors, preventing protein degradation (Sartori et al., 2021) (Schiaffino and Mammucari, 2011). It has been reported that the IGF-1/PI3K/Akt signaling pathway reduces denervation-, unloading-, and joint immobilization-induced muscle atrophy, and that the injection or overexpression of IGF-1 counteracts denervation- and age-related muscle atrophy (Timmer et al., 2018). On the other hand, several cellular systems are responsible for ensuring the turnover of muscle proteins and are finely regulated by complex regulatory mechanisms (Figure 1). UPS remains the primary system for protein breakdown in all tissues (Rock et al., 1994). The labeling of proteins by a ubiquitin molecule leads to their recognition and degradation by the 26S proteasome. Ubiquitination is ensured by three enzymes (E-1, E-2, and E-3). E3 is the key enzyme as it recognizes the substrate and catalyzes the insertion of ubiquitin (Lecker et al., 2006). The cellular level of E3 ligases determines protein degradation. Two E3 ligases, atrogin-1 and MuRF-1, are specific constituents of muscle, and their expression increases considerably in catabolic states (Lecker et al., 2006). Accordingly, FoxOs are key players in the regulation of muscle atrophy. FoxO 1, 3, and 4 regulate the induction of atrophy-related genes by controlling the ALS and UPS (atrogin-1 and MuRF1) (Brocca et al., 2017) (Bonaldo and Sandri, 2013). Akt phosphorylates FoxOs, inducing their inhibition and exportation from the nucleus thereby preventing muscle atrophy (Calnan and Brunet, 2008). The expression of FoxO-dependent atrophy-related genes varies in different muscles under different conditions. FoxO-1, -3, or -4 deletion in mice significantly prevents the loss of muscle mass following unloading (Brocca et al., 2017) but only partially spares skeletal muscle following denervation (Milan et al., 2015), highlighting the divergence in the mechanisms of the atrophy program in each catabolic condition. Calpains are also involved in inactivity-induced muscle atrophy since their inhibition attenuates muscle atrophy in rodents (Talbert et al., 2013) (Hyatt and Powers, 2020). Transgenic overexpression of calpastatin, the endogenous calpain inhibitor, protects skeletal muscle against atrophy suggesting their involvement in muscle wasting (Salazar et al., 2010) (Tidball and Spencer, 2002). Disruption in cytosolic Ca2+ homeostasis plays a role in initiating calpain activation following physical inactivity (Ingalls et al., 2001), and reactive oxygen species (ROS) generation exacerbates the rise in cytosolic Ca2+ leading to further calpain activation (Hyatt and Powers, 2020). More recently, Hayden and colleagues provided evidence that calpains play an important function in inactivity-induced mitochondrial dysfunction and oxidative stress in skeletal muscle fibers (Hyatt et al., 2021). They showed that the overexpression of calpastatin inhibits calpain activation thereby reducing mechanical ventilation-induced oxidative stress and mitochondrial dysfunction, fiber atrophy and contractile dysfunction (Hyatt et al., 2021).
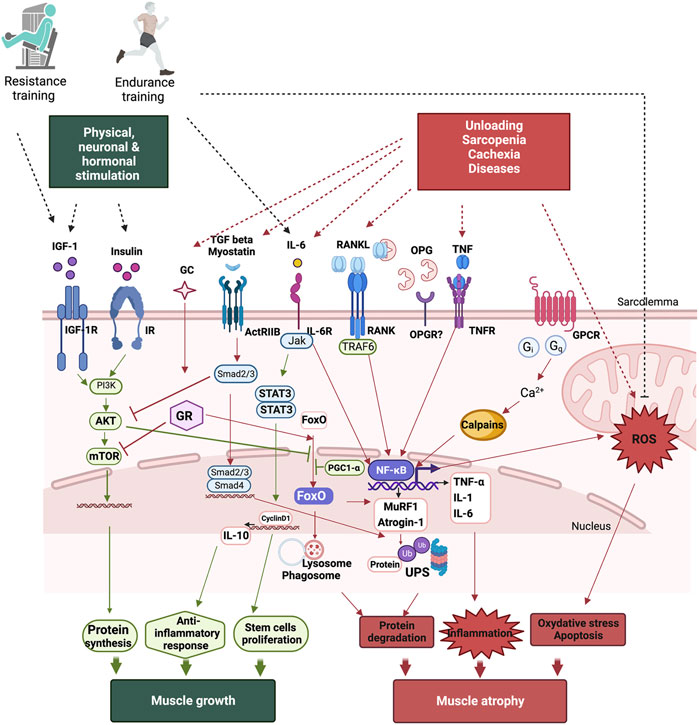
FIGURE 1. Signaling pathways leading to protein turnover in skeletal muscles. Muscle growth occurs following physical, neuronal, and hormonal stimulations. While a gain of muscle mass is mainly associated with resistance training, endurance training also improves muscle function by improving mitochondrial function, reducing oxidative stress, and modulating inflammation. Following unloading, age-related muscle wasting, sarcopenia, or diseases such as cachexia induces muscle atrophy. Muscle atrophy occurs as result of an increase in protein degradation, inflammation, oxidative stress, and apoptosis. The main pathway involved in protein synthesis is the PI3K/Akt/mTOR pathway, which is activated both by growth hormone (GH) and insulin-like growth factor IGF-1. Protein breakdown involves the activation of the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALS), which are mainly regulated by FoxOs and NF-κB, leading to muscle atrophy. NF-κB also inceases pro-inflammatory cytokine production and ROS, accentuating muscle loss. TGF beta, myostatin, RANKL, TNF-α, and IL-6 induce NF-κB activation. However, IL-6 can act as both a pro- and an anti-inflammatory cytokine as it induces anti-inflammatory cytokine IL-10 production and is mainly involved in stem cell proliferation leading to muscle growth.
In addition to the dysregulation of protein turnover, muscle stem cell proliferation and differentiation also decrease, which may contribute to atrophy under unloading conditions. Muscle atrophy leads to a significant decrease in the number of stem cells, with alterations in their capacity to proliferate and differentiate, accentuating muscle wasting in a mouse model of HS (Mitchell and Pavlath, 2004). Furthermore, the Notch signaling pathway, which tightly regulates stem cell proliferation and quiescence but not differentiated myofibers, is also involved in the alteration of regeneration and muscle atrophy in aging and muscular diseases (Gioftsidi et al., 2022). A recent study has shown that the inhibition of Notch signaling attenuates atrophy and fibrosis and reduces fibroblast levels in denervated gastrocnemius muscles (Feng et al., 2019). The mechanisms of muscle atrophy are thus complex and involve a panoply of signaling pathways. The specificities of the molecular mechanisms are still being investigated.
Sarcopenia is characterized by a progressive age-related loss in muscle mass and strength limiting motor function and a decrease in quality of life. Muscle atrophy and muscle cell death are the primary cause of the loss in muscle mass due to many molecular and cellular changes, including changes in mitochondria function, oxidative stress, hormonal signaling, and inflammatory cytokine secretion (Roubenoff, 2000). At the cellular level, mitochondrial function deteriorates in sarcopenia. Mitochondrial dysfunction involves various processes, including the production of ROS, mitochondrial biogenesis and turnover, Ca2+ dynamics, energy sensing, and apoptosis (Gonzalez-Freire et al., 2015). Recently, Grevendonk et al. (2021) examined mitochondrial function in young and elderly patients and determined whether physical activity interferes with mitochondrial dysfunction. The elderly patients exhibited a lower mitochondrial capacity compared to young adults with a similar mitochondrial content and who had performed comparable physical exercise. The muscle strength, aerobic capacity, exercise efficiency, insulin sensitivity, and gait stability were lower in elderly adults with higher body fat. However, an increase in physical activity partially reversed these observations (Grevendonk et al., 2021). In addition to mitochondrial dysfunctions, many studies have reported that age-related decreases in nicotinamide adenine dinucleotide (NAD+) accentuate muscle wasting (Yaku et al., 2018) (Castro-Portuguez and Sutphin, 2020). NAD+ is an important co-enzyme that regulates the energy balance, oxidative stress and protein posttranslational modifications. Aging disrupts both NAD + synthesis and degradation. Camacho-Pereira and colleagues shows that the decrease in NAD+ in aging is related to an increased level of CD38; the main enzyme involved in NAD + degradation (Camacho-Pereira et al., 2016). In addition, they showed that CD38 is also involved in mitochondrial dysfunction through a mechanism involving SIRT3; a regulator of mitochondrial homeostasis. In murine, the deletion of Nampt, an essential enzyme in the NAD + salvage pathway, induces an 80% decrease in intramuscular NAD + content which is accompanied by muscle degeneration and progressive loss of muscle strength and function. In contrast, the lifelong overexpression of Nampt preserved muscle NAD + levels and exercise capacity in aged mice, highlighting the important role played by NAD+ in muscle homeostasis (Frederick et al., 2016). In other tissues, PARP-mediated NAD+ depletion and loss of SIRT1 activity have been associated with an increase in oxidative nuclear damage and oxidative stress in both rodents and human (Massudi et al., 2012). Autophagy is another cellular mechanism that is modified with aging and that results in muscle atrophy (Wohlgemuth et al., 2010). Autophagy ensures that cellular components are delivered into lysosomes to be degraded and recycled, ensuring cellular homeostasis (Barbosa et al., 2019). However, autophagic activity decreases with age, increasing the production of ROS and oxidative stress causing sarcopenia (Barbosa et al., 2019) and reducing the ability of satellite cells to participate in muscle regeneration during aging (see review Chen et al., 2022). Activating transcription factor 4 (ATF4) also participates in age-related skeletal muscle atrophy (Ebert et al., 2015). ATF4 interacts with both C/EBPβ and the ATF–C/EBP composite site to activate Gadd45a, a gene involved in stress-related growth arrest and muscle atrophy (Ebert et al., 2020). In terms of muscle phenotype, type II muscle fibers are mostly atrophied in aging, also causing a marked conversion from the fast-to the slow-twitch phenotype (Dao et al., 2020) (Nilwik et al., 2013) (Ciciliot et al., 2013). The decline in satellite cell content may in part explain age-related type II muscle fiber atrophy (Verdijk et al., 2014). Thus, age-related muscle atrophy is very complex and multiple pathways converge toward muscle atrophy along with a reduced muscle regeneration capacity.
The age-related impairment of the motor unit (MU) is also responsible for the loss of muscle mass and function due to motoneuron loss, dysfunction in the process of compensatory reinnervation, and alterations at the neuromuscular junctions (NMJ). Larsson and colleagues (2019) recently presented a detailed review demonstrating the age-related changes in the MU in animals and humans (Larsson et al., 2019). It has been reported that the number of MUs is reduced by more than 40% after the age of 71, causing progressive muscle fiber denervation (Piasecki et al., 2016). A reinnervation mechanism is often set up at an advanced age favoring an inhomogeneity in the distribution of MUs (Larsson et al., 2019). However, this compensatory system is often insufficient to counteract the loss of motoneurons leading to the death of muscle fibers. In addition, the neuronal conduction and the quality of transmission are impaired in aging motor neurons (Larsson et al., 2019). Structural and molecular changes such as the reduction in the stability of transmission, the presence of fragmentation and the alteration of primary signaling molecules like the nicotinic acetylcholine receptor and agrin have been reported in aging NMJ (Rudolf et al., 2014) (Hepple and Rice, 2016). Skeletal muscle capillarization is also reduced during aging and the distance between satellite cells and capillaries increases potentially impacting the capacity for muscle regeneration in humans (Joanisse et al., 2017) (Larsson et al., 2019). Muscle capillarization is accompanied by a decrease in mitochondrial enzyme function, highlighting the limitations in oxidative capacity (Coggan et al., 1992). Thus, aging-related morphological changes occurring at the MU and capillarization may contribute directly or indirectly to molecular mechanisms responsible for muscle atrophy.
Changes in anabolic hormone secretion also play an important and easily observed role in sarcopenia. Growth hormone (GH), testosterone, and insulin-like growth factor (IGF-1) secretion decline gradually with age (Bian et al., 2020). GH binds directly to its receptor, activating the JAK2/STAT pathway and myocyte proliferation (Skorupska, 2018) (Velloso, 2008) and maintains muscle mass by promoting IGF-1 secretion by the liver, a key anabolic hormone (Chikani and Ho, 2014) (Widdowson and Gibney, 2008). In addition to the decrease in anabolic hormone secretion, anabolic resistance slowly develops as the anabolic response to stimulation is attenuated with aging, reducing the sensitivity to IGF-1 stimuli and the Akt-mTOR cascade thus limiting the capacity of muscles to grow (Dao et al., 2020) (Funai et al., 2006). Moreover, IGF-1 modulates mitochondrial functions through the activation of the Akt pathway and the inhibition of FoxO proteins (Poudel et al., 2020). The local expression of IGF-1 protects against the age-related decline in muscle force and mass. Transgenic mice revealed that the local expression of IGF acts by increasing the ALS and the expression of PGC1-α, which modulates mitochondrial function and ROS detoxification in aged mice (Ascenzi et al., 2019). In addition, the levels of myostatin, a member of the TGF-β superfamily and a negative regulator of skeletal muscle mass, increase in aging men and women (Yarasheski et al., 2002) and may potentially be an important regulator of muscle wasting. Several alterations that occur with aging disturb the protein turnover process and lead to protein breakdown and muscle atrophy. All these changes in the hormonal profile mainly influence muscle growth and lead to sarcopenia during aging.
Sarcopenia is characterized by chronic low-level inflammation that develops with advanced age, which is also called inflammaging. This inflammation may contribute to muscle loss associated with aging. Pro-inflammatory cytokines, including members of the TNF family, as well as downstream transcription factor NF-κB, are significant regulators of muscle atrophy (Thoma and Lightfoot, 2018). The NF-κB signaling pathway increases the expression of several proteins of the UPS involved in the degradation of specific muscle contractile proteins and can also interfere with myogenic differentiation (Li et al., 2008) (Thoma and Lightfoot, 2018) (Cai et al., 2004). Furthermore, Pasco et al. (2020) recently summarized the mechanisms of muscle atrophy involving the weak inducer of TNF-like apoptosis (TWEAK) and its associated receptor, fibroblast growth factor 14 (Fn14). The TWEAK/Fn14 signaling pathway maintains chronic inflammation and induces the secretion of profibrotic cytokines (Zhang et al., 2021). Fn14 levels increase in aged mice while the genetic deletion of Fn14 increases specific muscle protein levels and reduces age-related fiber atrophy, most likely by decreasing the DNA-binding activity of NF-κB (Tajrishi et al., 2014). Additionally, TNF-α enhances ROS generation in muscle cells, causing muscle wasting, while antioxidant treatments inhibit TNF-α-induced atrophy (Xu et al., 2018). NF-κB and chronic inflammatory pathways thus play significant roles in skeletal muscle atrophy under physiological and pathological conditions. Acute and well-controlled inflammation remains essential for muscle repair, regeneration, and growth. Its dual function is discussed in the following section.
The presence of satellite cells and inflammatory cells plays a major role in the skeletal muscle regenerative process (Tidball, 2017). The participation of sentinel inflammatory cells such as mast cells are important for the initiation of the inflammatory process following an injury (Perandini et al., 2018) (Bentzinger et al., 2013) (Gaudenzio et al., 2016). Mast cells secrete inflammatory mediators and lead to a well-orchestrated sequential activation and recruitment of neutrophils that play an important role in fighting pathogens (Kawanishi et al., 2016). However, in cases of severe injuries, the release of ROS and the secretion of proteolytic enzymes resulting from prolonged activation of neutrophils may cause collateral damage (Duchesne et al., 2017) (Dumont et al., 2008). The pro-inflammatory environment is crucial for the arrival of monocytes that differentiate into pro-inflammatory M1 macrophages in injured skeletal muscle. M1 macrophages secrete pro-inflammatory cytokines. Including TNF-α and IL-1β, and play a key role by ensuring the phagocytosis of cell debris and apoptotic neutrophils, the activation and proliferation of satellite cells, and the recruitment of other immune actors (Locati et al., 2013) (Mosser and Edwards, 2008) (Otis et al., 2014). Pro-inflammatory M1 macrophages switch to the anti-inflammatory M2 macrophage phenotype, which promotes the differentiation of satellite cells and the formation of mature myofibers. M2 macrophages initiate muscle repair by secreting anti-inflammatory cytokines (IL-4, IL-10) and growth factors (TGF-β, IGF-1, and HGF) (Arnold et al., 2007) (Chazaud et al., 2003) (Deng et al., 2012) (Oishi and Manabe, 2018). They are important for promoting muscle regeneration and repair following acute muscle injuries (Chazaud et al., 2003) (Arnold et al., 2007) (Chazaud, 2020) (Bouredji et al., 2021). In addition to cytokines, lipid mediators generated via cyclooxygenase-2 (COX-2) also play a prominent role by orchestrating the inflammatory process and contributing to the macrophage switch and inflammation resolution (Duchesne et al., 2017). The depletion of immune cells or COX-2 inhibition impairs muscle regeneration and induces fibrosis during acute inflammatory processes, highlighting the predominant role of inflammation in muscle healing. This puts into question the relevance of using anti-inflammatory treatments for acute muscle injuries (Toumi et al., 2006) (Summan et al., 2006) (Duchesne et al., 2017) (Bondesen et al., 2004).
Discoveries over the past 20 years have significantly improved our understanding of myokines, i.e., interleukins, growth factors, and the numerous peptides secreted by muscles (Pedersen, 2013). Interleukin 6 (IL-6) is one of the most thoroughly studied myokines. Considered, in part, as a pro-inflammatory cytokine, human muscle contractions release a significant amount of IL-6, depending on the duration and intensity of the physical activity (Pedersen, 2013). IL-6 secreted during exercise has anabolic properties and participates in the regulation of glucose and lipid metabolism (Ellingsgaard et al., 2019). IL-6 is an essential regulator of satellite cell-mediated muscle growth while the deletion of the IL-6 gene attenuates muscle hypertrophy and reduces the proliferation of satellite cells via STAT3 as well as the expression of its target, cyclin D1 (Serrano et al., 2008). Chowdhury et al. (2020) recently showed that the high levels of IL-6 found in circulation during physical exercise mainly originates from muscle. Furthermore, muscle-released IL-6 increases RANKL secretion by osteoblasts, promoting osteoclast differentiation and osteocalcin release. In turn, circulating osteocalcin potentiates IL-6 released by muscle cells, increasing nutrient uptake and exercise capacity in rodents (Chowdhury et al., 2020). IL-6 is also an anti-inflammatory cytokine and promotes the expression of another anti-inflammatory myokine, IL-10, which inhibits the production of proinflammatory cytokines, including IL-1 and TNF-α, during regular exercise (Petersen and Pedersen, 2005), favoring the switch of macrophages to anti-inflammatory M2 macrophages during muscle repair and regeneration (Deng et al., 2012). However, IL-6 is also associated with numerous skeletal muscle alterations such as atrophy and muscle wasting. The IL-6/JAK/STAT3 signaling pathway is strongly activated during denervated skeletal muscle atrophy. IL-6 enhances C2C12 myotube atrophy while pharmacological blocking of IL-6 reduces the expression of atrophic and autophagy-related genes (Huang et al., 2020) (Madaro et al., 2018). Another study has recently shown that IL-6 is upregulated in the skeletal muscles of limb-immobilized mice and that a systemic IL-6 deficiency protects against immobilization-induced muscle atrophy (Hirata et al., 2022). IL-15 is also abundantly expressed and secreted by skeletal muscle in response to exercise (Huang et al., 2015) (Yang H et al., 2013) and protects muscle against sepsis-induced muscle wasting (Kim et al., 2012). In vitro, IL-15 overexpression induces myotube hypertrophy similar to the overexpression of IGF-1 (Quinn et al., 2002) while the co-incubation of IL-15 and TNF-α reduces the detrimental effects of TNF-α on myotube development (O’Leary et al., 2017), highlighting the potential of IL-15 to prevent muscle wasting. However, the hypertrophic effects of IL-15 seem to be effective in pathological situations, and IL-15 is not an anabolic factor in homeostasis and steady state conditions (Nadeau and Aguer, 2019) where anabolic effects on the muscle mass of mice overexpressing IL-15 are not observed (Pistilli and Quinn, 2013). IL-15 is also involved in bone development, and mice lacking IL-15 receptor alpha (IL-15 RA) exhibit a decrease in bone mineralization (Loro et al., 2017). Myostatin, another myokine and a member of the TGF-β superfamily, negatively regulates muscle growth. The myostatin/TGF-β/activin pathway via activin receptor type II (ActRIIB) induces the activation of SMAD2/3 causing the inhibition of Akt, protein synthesis, and cell differentiation (Morissette et al., 2009) (Trendelenburg et al., 2009). In addition, myostatin deletion increases both muscle mass and bone density (DiGirolamo et al., 2015). More recently, ActRIIB signaling has been shown to directly and negatively regulate bone mass in osteoblasts, suggesting that myostatin neutralization would benefit bones (Goh et al., 2017). Although the receptor activator of nuclear factor kappa B (RANK), its ligand (RANKL), and osteoprotegerin (OPG), a decoy soluble receptor of RANKL, constitute the primary pathway regulating bone homeostasis, we have shown over the last 10 years that this triad plays significant roles in skeletal muscle disease and repair. RANKL, through the activation of the NF-κB pathway, is involved in the activation of inflammatory-atrophic pathways in skeletal muscle (Hamoudi et al., 2019). Muscle cells in culture also produce and secrete OPG (Dufresne et al., 2015), and its deletion in mice causes muscle atrophy (Hamoudi et al., 2020). Pharmaceutical treatments with recombinant OPG markedly improve muscle integrity and function in dystrophic mice (Dufresne et al., 2015) (Dufresne et al., 2018) and in a mouse model of acute muscle injury (Bouredji et al., 2021). Our recent review discusses the involvement of the RANK/RANKL/OPG triad in skeletal and cardiac muscles in greater detail (Marcadet et al., 2022).
While sarcopenia is characterized by a loss of muscle mass and strength with aging, cachexia is a condition characterized by a continuous decrease in skeletal muscle mass, involuntary weight loss, and the deterioration of an individual’s nutritional status, and is mainly observed in chronic diseases such as cancer (Anker et al., 2019) (Dolly et al., 2020). Perturbations of cellular and inflammatory processes, including the activation of inflammation and proteolytic mechanisms, UPS and the ALS are at the origin of cancer-associated cachexia (Baracos et al., 2018). The central mechanism behind cancer-associated cachexia is inflammation (Cortiula et al., 2022) involving the overproduction of inflammatory cytokines, especially IL-6, TNF-α, and IL-1β. The simultaneous secretion of IL-6 and TNF-α induces muscle weakness as well as the activation of pro-inflammatory pathways, including the NF-kB pathway (Webster et al., 2020). Cachexia is associated with high levels of circulating cytokines, and preclinical studies have shown that there is an increase in UPS and a decrease in protein synthesis due to the downregulation of Akt and FoxO3 phosphorylation (Webster et al., 2020). A recent clinical study involving gastric cancer patients with cachexia has shown that the patients exhibit a significant decrease in the CSA of skeletal muscle associated with a significant increase in UPS and ALS (Zhang et al., 2020). The current hypothesis regarding cancer-associated cachexia suggests that mitochondrial dysfunction combined with lipid droplet accumulation and oxidative stress increases proteolysis and the loss of muscle mass through UPS and ALS (Dolly et al., 2020). The RANKL/RANK interaction has recently been associated with muscle wasting and cachexia in a mouse model of non-metastatic cancer. Fabrizio et al. (2022) have shown that elevated levels of circulating RANKL are sufficient to cause skeletal muscle atrophy and bone resorption whereas anti-RANKL treatments improve muscle mass and function in cancer-associated cachexia (Pin et al., 2022). The NF-kB pathway is also involved in impairing satellite cell differentiation in cancer patients and animal models by inducing PAX7 dysregulation, impairing muscle regeneration, and promoting muscle wasting (He et al., 2013). Moreover, therapeutic approaches, including chemotherapy or radiotherapy, can potentially induce muscle atrophy and dysfunction. Chemotherapeutic compounds induce muscle wasting in mice through the activation of the NF-kB pathway (Damrauer et al., 2018) (Coletti, 2018). Some 218 proteins are downregulated in C26-tumor bearing mice and chemotherapy-treated mice. A pathway analysis has shown showed that these two conditions both lead to the dysregulation of mitochondrial functions, the Krebs cycle, and fatty acid metabolism as well as Ca2+ dysfunctions and that both are associated with muscle loss (Barreto et al., 2016). Furthermore, cancer cells and the administration of cytotoxic chemotherapeutics induce a rapid inflammatory response, leading to the dysregulation of the neuroendocrine system, more precisely the hypothalamic-GH-IGF-1 axis. The inhibition of this axis contributes to skeletal muscle atrophy and cachexia (Martín et al., 2021). In addition, a rapid inflammatory response induced by cytotoxic chemotherapy treatment leads to endogenous glucocorticoid (GC) secretion, which increases in the UPS-, lysosome- and autophagy-genes, resulting in muscle loss (Braun et al., 2014). Conversely, the muscle-specific deletion of the GC receptor blocks treatment-induced muscle atrophy, highlighting the involvement of GCs in cachexia (Braun et al., 2014) (Braun et al., 2013). However, the mechanisms of chemotherapy-induced cachectic myopathy may be specific to each chemical agent and has recently been reviewed by Campelj et al. (2021)(Campelj et al., 2021). Overall, cancer-related cachexia is a consequence of many disorders caused by cancerous cells and the side effects of therapeutic approaches, including the reaction of the body against the tumor, the inflammatory reaction, the impairment of metabolism, and the loss in appetite.
Muscle wasting often occurs concomitant with chronic diseases. Patients suffering from chronic HF present marked muscle atrophy, with a reduction in exercise capacity and muscle strength (Fülster et al., 2013). Similarly, 47% of young patients with HF experience muscle wasting (Hajahmadi et al., 2017). Metabolic and molecular changes, in particular protein breakdown and an increase in the production of ROS, may explain the muscle dysfunction/exercise intolerance following HF (Springer et al., 2017) (Nambu et al., 2021) (Wood et al., 2021). MuRF-1 is increased in the skeletal muscle of HF patients and physical activity attenuates its expression (Gielen et al., 2012). The inhibition of MuRF1 has a protective effect on muscle atrophy in mice with HF (Adams et al., 2019). In addition, the expression of IGF-1 in skeletal muscle is reduced (Hambrecht et al., 2002) and, consistent with these findings, the phospho-Akt/Akt levels and phospho-mTOR/mTOR ratios are diminished in patients with HF compared with healthy controls of a similar age, muscle mass, and physical activity levels. Further investigations are clearly needed to better understand the molecular mechanisms regulating HF-induced muscle wasting and exercise intolerance.
A common characteristic of genetic muscle diseases, including congenital muscular dystrophy and mitochondrial and genetic myopathies, is a major impairment of muscle integrity and the loss of Ca2+ homeostasis, which in turn leads to a loss of muscle function (Yin et al., 2021). In myotonic dystrophy type 1 (DM1), a genetic disease that causes myotonia, muscle atrophy, and insulin resistance, insulin receptor type 1 is expressed at lower levels in skeletal muscle, which leads to a decrease in mTOR signaling and an increase in UPS protein expression, giving rise to muscle atrophy (Renna et al., 2019). Congenital muscular dystrophy caused by genetic changes in the LAMA2 gene is mainly characterized by severe muscle wasting. Preclinical studies have shown that the increase in protein degradation is the result of an increase in UPS protein expression and a reduction in Akt phosphorylation and protein synthesis (Carmignac et al., 2011) (Gawlik et al., 2019). DMD is one of the most common forms of muscular dystrophy. Although the alterations in muscle integrity and function lead to progressive muscle atrophy in DMD, certain muscles may grow as a result of an accumulation of fat and fibrosis and an increase in myofiber size called compensatory hypertrophy (Kornegay et al., 2012). In the mdx mouse model of DMD, an initial phase of muscle hypertrophy is followed by atrophy associated with a decline in regenerative potential and changes in the activation of the mTOR signaling pathway (Mouisel et al., 2010). In both animals and humans, dystrophic muscles are not uniformly affected, with some being atrophied others hypertrophied, adding a greater challenge for therapeutic healthcare (Kornegay et al., 2012). Additionally, membrane fragility due to the absence of dystrophin leads to chronic inflammation, which aggravates the pathology (Petrof et al., 1993) (Wehling et al., 2001). Patients affected by DMD exhibit an increased expression of inflammatory cells (Abdel-Salam et al., 2009) while chronic inflammation alters the macrophage switch. Macrophages express high levels of transforming growth factor β1 (TGF-β1), which disrupts muscle regeneration and increases connective tissue deposition (Lemos et al., 2015) (Tidball, 2017). Neutrophils and mastocytes also play a harmful role in dystrophic muscles, and their depletion reduces necrosis (Hodgetts et al., 2006) (Radley and Grounds, 2006), whereas regulatory T cells modulate the progression of muscular dystrophy by suppressing the pro-inflammation environment in dystrophic muscles (Villalta et al., 2014). A recent review summarizes the harmful role of inflammatory cells in muscular dystrophies and describes an immune response involving neutrophils, macrophages, helper T-lymphocytes, and cytotoxic T-lymphocytes (Tidball et al., 2018).
The standard of care for patients with muscular dystrophies remains the chronic use of GCs. Although GCs are the only therapeutic agents with proven benefits for quality of life, mobility, and life expectancy, they are associated with several adverse effects, including potential muscle atrophy (Goemans and Buyse, 2014). GCs induce selective fast-twitch glycolytic fiber atrophy typified by a reduction in CSA and protein content. GCs modulate IGF-1 and myostatin signaling and induce the increase in protein breakdown through the UPS and lysosomal systems involving Murf-1, Atrogin-1, and FoxO (Schakman et al., 2013). Furthermore, GCs interfere with protein synthesis by inhibiting the mTOR/S6 kinase 1 pathway (Schakman et al., 2013) (Braun and Marks, 2015). Other mechanisms that contribute to GC-induced muscle atrophy involve the downregulation of the transcription factors myoD and myogenin, and the increase in insulin resistance and Ca2+ levels through activated store-operated Ca2+channels (SOCs) (Hasselgren et al., 2010). A recent study has further highlighted the adverse effects of GCs on bone integrity (Box et al., 2022). GC-related bone demineralization is mainly caused by a reduction in bone formation combined with an increase in bone resorption (Box et al., 2022). GCs inhibit osteoblastogenesis and the activity of osteoblasts and osteocytes (Weinstein et al., 1998). They also inhibit OPG secretion by transrepressing the OPG gene, which increases the RANKL/OPG ratio, and by promoting osteoclast proliferation and bone resorption (Kondo et al., 2008) (Jia et al., 2006). Prolonged and chronic use of GCs in DMD is thus potentially associated with muscle atrophy and bone demineralization.
Osteoporosis is characterized by a reduction in bone mass associated with microarchitectural deterioration of bone tissue due to an imbalance between bone resorption and bone formation, which occur simultaneously with sarcopenia (Reginster et al., 2016). The occurrence of osteoporosis was, for a long time, considered a consequence of sarcopenia associated with an age-related reduction in mobility. However, major advances point to bidirectional molecular communication between bone and muscle that share common signaling pathways, suggesting a concomitant wasting of bone and muscle (Reginster et al., 2016). Terracciano et al. (2013) have shown that patients suffering from osteoporosis present a preferential and diffuse type II fiber atrophy that is proportional to the degree of bone loss and possibly linked to a reduction in Akt expression (Terracciano et al., 2013). Our laboratory has investigated bone-muscle bi-directional communication, with a focus on RANK/RANKL/OPG, the main pathway regulating bone homeostasis. The dysregulation of this pathway is directly associated with the onset of osteoporosis (Ono et al., 2020). Preclinical studies in mice have shown that RANKL/RANK interactions are involved in muscle wasting, atrophy, and dysfunction (Box et al., 2022) (Hamoudi et al., 2020) (Dufresne et al., 2018) (Bonnet et al., 2019). Our work highlights the involvement of the RANK/RANKL/OPG pathway in mitigating DMD. Moreover, OPG knockout mice exhibit selective atrophy of fast-twitch-type IIb myofibers, the most powerful muscle fibers, which may occur through the activation of the NF-kB pathway (Hamoudi et al., 2020). In vitro, RANKL stimulation of myotubes in culture increases the expression of NF-kB, atrogin-1, and MuRF-1 (Hamoudi et al., 2020). Furthermore, mice overexpressing RANKL exhibit decreased muscle mass, force, and glucose uptake associated with an upregulation of inflammatory genes (Bonnet et al., 2019). In a mouse model of sarcopenia, RANKL neutralization increases muscle volume and force and normalizes insulin signaling and inflammatory genes in skeletal muscle (Bonnet et al., 2019). Our recent review summarizes the current knowledge on the involvement of RANK/RANKL/OPG signaling in cardiac, skeletal, and smooth muscles in health and disease (Marcadet et al., 2022). The molecular mechanisms concertedly regulating muscle wasting and bone diseases are now being intensively investigated and important findings will soon emerge.
Exercise is the most effective treatment for counteracting muscle wasting and atrophy (Glass and Roubenoff, 2010). Resistance training induces muscle hypertrophy in part by increasing the expression of IGF-1 (Glass and Roubenoff, 2010). The overexpression of IGF-1 or its addition to cultures of myotubes increases myofiber size. IGF-1 induces an increase in muscle mass by promoting protein synthesis through PI3K-Akt-mTOR signaling and inhibiting FoxO transcription factors involved in myofibril degradation. The expression of hypertrophic factor PGC1α increases with exercise training, reducing the FoxO3-dependent loss in muscle mass (Sandri et al., 2006). While resistance exercise induces skeletal muscle hypertrophy by promoting growth signaling pathways, endurance training is also beneficial for improving the capillary and mitochondrial networks and promoting the switch in muscle fiber phenotype towards more oxidative fiber isoforms thereby improving muscle physiology in HF (Nijholt et al., 2022). More recently, swimming has been shown to attenuate tumor growth and cancer-related muscle atrophy by downregulating the expression of proinflammatory proteins including NF-κB, p-NF-κB, TNF-α, IL-1β, and IL-6 (Li et al., 2021). The anti-inflammatory effects of exercise are increasingly being investigated and are of major interest for the treatment of chronic inflammation and muscle atrophy (Suzuki, 2019) (He and Ye, 2020). However, resistance training cannot be prescribed for all patients, in particular to those who are bed-ridden with acute disease or frail elderly sarcopenic individuals. In addition, exercise must be performed on a regular, long-term basis to maintain its effectiveness. Additionally, the transcutaneous neuromuscular electrical stimulation (NMES), a biophysical agent, may be beneficial to slow down muscle atrophy in deconditioning patients. For example, NMES was found to have benefits in terms of improved muscle mass and function and health status for patients with limited physical capacity (Gerovasili et al., 2009). In bed-ridden patients suffering from a severe COPD, NMES associated with an active limb mobilization significantly improve muscle strength and life quality (Zanotti et al., 2003). The same results were obtained in a subsequent study (Vivodtzev et al., 2006) confirming the benefice of NMES when combined with rehabilitation program. In patient with advanced chronic heart failure, the use of a low-frequency NMES helps to increase exercise capacity and to counteract skeletal muscle deterioration (Nuhr et al., 2004). Furthermore, the NMES of lower extremities preserves the muscle mass in critically ill patients (Gerovasili et al., 2009). Although NMES seems to improve muscle function particularly in patients with limited physical capacity, the methodological quality is generally poor and the heterogeneity in the study design render it difficult to draw solid conclusion about the efficacy of NMES.
Nutritional therapies that have emerged lately show that amino acid (AA) supplementation is beneficial for muscle health. A recent study has shown that taurine, a non-essential AA, has a therapeutic effect in the treatment of age-related sarcopenia. Taurine improves skeletal muscle regeneration, reduces chronic inflammation, and decreases high levels of oxidative stress thereby counteracting the effects of aging (Barbiera et al., 2022). Another recent study has shown that supplementation with essential AA reduces the loss in muscle volume in immobilized older adults recovering from total knee arthroplasty (Dreyer et al., 2018). The branched-chain AA leucine and its metabolite, β-hydroxy-β- methylbutyrate, activate mTORC1, increasing protein synthesis (Kamei et al., 2020). β-alanine levels are reduced in the skeletal muscles of aged mice, while β-alanine supplementation increases physical performance and improves endurance exercise-induced executive function in middle-aged individuals (Kamei et al., 2020). In elderly subjects with sarcopenia, an 18-month supplementation with a mixture of AAs significantly increases lean body mass associated with a significant decrease in circulating TNF levels and a marked increase in IGF-1 levels (Solerte et al., 2008). Preclinical evidence has shown that, in addition to AA, various nutrients including fatty acids (omega-3 and omega-6) and vitamins (C, D, E) are involved in modulating muscle protein synthesis and degradation and mitochondrial metabolism and energy production, thus improving muscle mass, strength, and function (Lee et al., 2022). Currently, the standard treatment approaches for sarcopenia are resistance exercise, protein supplementation, and vitamin D administration (Morley, 2016).
As inflammation is an important biological process involved in muscle wasting, especially in cancer-associated cachexia, several preclinical studies on rodent cancer models have reported that inhibiting IL-6 and its signaling pathways may have beneficial effect on muscle wasting (Belizário et al., 2016). The earliest studies over 20 years ago have shown that blocking the IL-6 receptor prevents muscle atrophy in a tumor-bearing mouse model via the modulation of the lysosomal and ATP-ubiquitin-dependent proteolytic pathways (Fujita et al., 1996). The study conducted in mice bearing human tumors shows that tumor cell-secreted IL-6 directly contributes to body weight loss while the administration of a human-mouse chimeric monoclonal antibody reverses muscle wasting, highlighting the potential role for IL-6 as pro-cachectic agent and the potential of its neutralization for treating cachexia (Zaki et al., 2004). In the same vein, targeting an IL-6 downstream signaling pathway has been shown to have a beneficial effect on protecting muscle against cachexia (Belizário et al., 2016). Bonetto et al. (2012) have shown that the inhibition of the STAT3/JAK pathway, downstream from IL-6 signaling, reduces cancer-induced cachexia and may be an interesting therapeutic target (Bonetto et al., 2012). Furthermore, in a model of denervation-induced skeletal muscle atrophy, blocking IL-6 with tocilizumab or the pharmacological/genetic inhibition of the JAK/STAT3 pathway in skeletal muscle both suppress muscle atrophy and inhibit mitophagy, leading to a decrease in the expression of atrophic genes (MuRF1 and MAFbx) and autophagy-related genes (PINK1, BNIP3, Beclin 1, ATG7, and LC3B) (Huang et al., 2020). Targeting IL-6 or its downstream mediators is thus a promising therapeutic approach for preventing muscle atrophy.
Targeting the NF-κB pathway and the UPS system are also of interest in preventing muscle atrophy as their dysregulation is associated with different pathologies that cause muscle atrophy. Preclinical studies using MG132, a specific reversible proteasome inhibitor, have shown that MG132 blocks the degradation of ubiquitin-conjugated IκB, thereby inhibiting NF-κB activation and reducing cachexia (Inoue et al., 2009) (Zhang et al., 2013). The RANKL/RANK interaction activates NF-κB and we have previously shown, in dystrophic mice, that an anti-RANKL treatment improves muscle function and integrity and increases the proportion of anti-inflammatory and non-cytotoxic M2 macrophages (Hamoudi et al., 2019). In postmenopausal women, neutralizing RANKL using a monoclonal antibody (denosumab) treatment for more than 3 years improves appendicular lean mass and handgrip strength compared with an untreated group (Bonnet et al., 2019). In a mouse model of non-metastatic ovarian cancer cachexia, where elevated RANKL levels cause muscle atrophy, the administration of anti-RANKL has been shown to reduce muscle mass loss and dysfunction (Pin et al., 2022). Interestingly, full-length OPG, which contains four RANKL-binding domains, two TRAIL-binding domains, and one heparin-binding domain has been shown to be effective in preserving muscle function and integrity in dystrophic mice and is even superior to anti-RANKL, suggesting that full length OPG also acts via a mechanism that is independent of RANKL (Dufresne et al., 2018). In dystrophic mice, OPG improves muscle function, attenuates muscle inflammation, and improves muscle integrity by modulating Ca2+ homeostasis (Dufresne et al., 2015) (Dufresne et al., 2018). Alternative approaches targeting NF-κB/TNF-α or UPS could thus reduce muscle atrophy (Fang et al., 2021).
Megestrol acetate (MA) is currently the only drug used to fight both cachexia by stimulating appetite and certain cancers by acting as an antineoplastic agent (Wen et al., 2012). The combination of MA and thalidomide, which is known to have immunosuppressive effects, is superior to MA alone and improves the condition of patients with cachexia by reducing TNF-α levels (Wen et al., 2012). Lastly, targeting oxidative stress in the elderly appears be another potential therapeutic strategy. Allopurinol is in the xanthine oxidase inhibitor family of medications and reduces oxidative stress. A retrospective study in elderly subjects taking allopurinol as a medication has shown that they exhibit a greater degree of improvement in muscle function during rehabilitation, highlighting the potential beneficial effect of allopurinol on skeletal muscle (Beveridge et al., 2013). As inflammatory pathways are directly involved in inducing protein breakdown, targeting these pathways is a major focus of developing new therapeutic approaches to fight muscle atrophy.
Therapies targeting endogenous regulators of muscle atrophy such as TGF-β-like molecules, including the myostatin/TGF-β/activin pathway, have been tested preclinically given their interaction with activin receptor complex (ActRIIB), which induces the activation of SMAD2/3, which in turn causes the inhibition of Akt and results in a decrease in protein synthesis and cell differentiation (Morissette et al., 2009) (Trendelenburg et al., 2009). Different strategies have been developed to target endogenous regulators of muscle atrophy, including the neutralization of myostatin using myostatin antibody, the memetic endogenous neutralization of myostatin by follistatin, or the blockage of ActRIIB (Smith and Lin, 2013). However, clinical results have been very disappointing as they are not very efficacious and have significant adverse effects (Sartori et al., 2021) (Golan et al., 2018). Nonetheless, a novel strategy for inhibiting myostatin using a monoclonal antibody that selectively binds to the autoinhibited precursor form of myostatin and prevents its maturation has shown promising results in animals, with a reduction in muscle atrophy and an improvement in muscle strength and function (Pirruccello-Straub et al., 2018). In cachexia-associated muscle atrophy, the pharmacological blockade of myostatin/activin has been found to be the most effective therapy (Estelle et al., 2014). The use of β-blockers may be a potential strategy for treating cachexia given that the pathology is associated with weight loss and that β-blockers have effects on survival and weight gain by inhibiting catecholamine-induced lipolysis and decreasing insulin sensitivity (Kung et al., 2010). The β-blocker carvedilol has been shown to attenuate and partially reverse cachexia in patients with chronic HF (Clark et al., 2017). For GC-induced skeletal muscle atrophy, an emerging approach targeting urotensin-II (U-II) has shown interesting results as a novel therapeutic strategy. As GC increases the expression of U-II, antagonizing urotensin signaling improves muscle atrophy by increasing PI3K/Akt/mTOR levels and inhibiting UPS (Yin et al., 2022). Lastly, the use of anabolic hormones as therapeutic agents to treat muscle atrophy has attracted interest but often the effects have been associated with significant adverse effects (Morley, 2016). For example, the administration of high doses of testosterone has been shown to improve muscle mass and function but has several potentially limiting side effects (Morley, 2016). The same applies to ghrelin as its adverse effects need to be taken into consideration given that it increases IGF-1 levels, which may induce diabetes or insulin resistance (Yin et al., 2021). IGF-1 has shown very interesting effects on various neuromuscular diseases in preclinical studies. However, clinical trials have failed to reproduce these results (Song et al., 2013). Given the lack of effective treatments, major research efforts are ongoing to develop new therapeutic strategies that target muscle atrophy. The combination of exercise, nutraceutical, and pharmacological approaches that target multiple pathways may be required to effectively reduce muscle loss in aging and disease conditions.
Muscle wasting and atrophy occur under different physiological and pathological conditions and involve a variety of cellular and molecular changes that may differ depending on the situation and the muscle fiber phenotype. Muscle atrophy in microgravity and the HS model primarily affects postural and slow-twitch muscle fibers. The mechanisms involved are complex and are not yet fully understood. Age-related sarcopenia is a morbidity that causes many complications and that is often associated with other co-morbidities such as osteoporosis, diabetes, and HF. Each of these morbidities causes muscle atrophy by modulating protein synthesis and degradation pathways and hormonal signaling, and by impairing mitochondrial and lysosomal functions. Pathological situations such as cancer cachexia involve an impairment of the protein degradation pathway, the modulation of inflammation, and apoptosis, whereas, in muscular dystrophies, the rise in cytosolic Ca2+ levels, muscle damage, and chronic inflammation are mainly responsible for muscle atrophy and wasting. While we have reviewed the mechanisms involved in the atrophy of skeletal muscle, we have also referenced the double-edged sword of inflammation in muscle repair. Although much is known about muscle wasting, it remains a focus of much ongoing research, and while many therapeutic approaches have been investigated in numerous preclinical studies, the clinical therapeutic armamentarium for treating muscle disorders, especially muscle wasting, has to be expanded, hence the need for more investigations.
ZB wrote the manuscript with support from AA and JF. ZB conceived the figure under the supervision of AA.
This work was supported by a grant to JF from the Natural Sciences and Engineering Research Council of Canada (NSERC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Salam E., Abdel-Meguid I., Korraa S. S. (2009). Markers of degeneration and regeneration in Duchenne muscular dystrophy. Acta Myol. 28 (3), 94–100.
Adams V., Bowen T. S., Werner S., Barthel P., Amberger C., Konzer A., et al. (2019). Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 10 (5), 1102–1115. doi:10.1002/jcsm.12448
Anker M. S., Holcomb R., Muscaritoli M., von Haehling S., Haverkamp W., Jatoi A., et al. (2019). Orphan disease status of cancer cachexia in the USA and in the European union: a systematic review. J. Cachexia Sarcopenia Muscle 10 (1), 22–34. doi:10.1002/jcsm.12402
Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., et al. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204 (5), 1057–1069. doi:10.1084/jem.20070075
Ascenzi F., Barberi L., Dobrowolny G., Villa Nova Bacurau A., Nicoletti C., Rizzuto E., et al. (2019). Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 18 (3), e12954. doi:10.1111/acel.12954
Baldwin K. M., Haddad F., Pandorf C. E., Roy R. R., Edgerton V. R. (2013). Alterations in muscle mass and contractile phenotype in response to unloading models: role of transcriptional/pretranslational mechanisms. Front. Physiol. 4, 284. doi:10.3389/fphys.2013.00284
Baracos V. E., Martin L., Korc M., Guttridge D. C., Fearon K. C. H. (2018). Cancer-associated cachexia. Nat. Rev. Dis. Prim. 4 (1), 17105. doi:10.1038/nrdp.2017.105
Barbiera A., Sorrentino S., Fard D., Lepore E., Sica G., Dobrowolny G., et al. (2022). Taurine administration counteracts aging-associated impingement of skeletal muscle regeneration by reducing inflammation and oxidative stress. Antioxidants 11 (5), 1016. doi:10.3390/antiox11051016
Barbosa M. C., Grosso R. A., Fader C. M. (2019). Hallmarks of aging: An autophagic perspective. Front. Endocrinol. 9, 790. doi:10.3389/fendo.2018.00790
Barreto R., Mandili G., Witzmann F. A., Novelli F., Zimmers T. A., Bonetto A. (2016). Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front. Physiol. 7, 472. doi:10.3389/fphys.2016.00472
Belizário J. E., Fontes-Oliveira C. C., Borges J. P., Kashiabara J. A., Vannier E. (2016). Skeletal muscle wasting and renewal: a pivotal role of myokine IL-6. SpringerPlus 5 (1), 619. doi:10.1186/s40064-016-2197-2
Bentzinger C. F., Wang Y. X., Dumont N. A., Rudnicki M. A. (2013). Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 14 (12), 1062–1072. doi:10.1038/embor.2013.182
Beveridge L. A., Ramage L., McMurdo M. E. T., George J., Witham M. D. (2013). Allopurinol use is associated with greater functional gains in older rehabilitation patients. Age Ageing 42 (3), 400–404. doi:10.1093/ageing/aft046
Bian A., Ma Y., Zhou X., Guo Y., Wang W., Zhang Y., et al. (2020). Association between sarcopenia and levels of growth hormone and insulin-like growth factor-1 in the elderly. BMC Musculoskelet. Disord. 21 (1), 214. doi:10.1186/s12891-020-03236-y
Bonaldo P., Sandri M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6 (1), 25–39. doi:10.1242/dmm.010389
Bondesen B. A., Mills S. T., Kegley K. M., Pavlath G. K. (2004). The COX-2 pathway is essential during early stages of skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 287 (2), C475–C483. doi:10.1152/ajpcell.00088.2004
Bonetto A., Aydogdu T., Jin X., Zhang Z., Zhan R., Puzis L., et al. (2012). JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303 (3), E410–E421. doi:10.1152/ajpendo.00039.2012
Bonnet N., Bourgoin L., Biver E., Douni E., Ferrari S. (2019). RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J. Clin. Invest. 129 (8), 3214–3223. doi:10.1172/JCI125915
Bouredji Z., Hamoudi D., Marcadet L., Argaw A., Frenette J. (2021). Testing the efficacy of a human full-length OPG-Fc analog in a severe model of cardiotoxin-induced skeletal muscle injury and repair. Mol. Ther. Methods Clin. Dev. 21, 559–573. doi:10.1016/j.omtm.2021.03.022
Box C. D., Cronin O., Hauser B. (2022). The impact of high dose glucocorticoids on bone health and fracture risk in systemic vasculitides. Front. Endocrinol. 13, 806361. doi:10.3389/fendo.2022.806361
Braun T. P., Grossberg A. J., Krasnow S. M., Levasseur P. R., Szumowski M., Zhu X. X., et al. (2013). Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. Fed. Am. Soc. Exp. Biol. 27 (9), 3572–3582. doi:10.1096/fj.13-230375
Braun T. P., Marks D. L. (2015). The regulation of muscle mass by endogenous glucocorticoids. Front. Physiol. 6, 12. doi:10.3389/fphys.2015.00012
Braun T. P., Szumowski M., Levasseur P. R., Grossberg A. J., Zhu X., Agarwal A., et al. (2014). Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PloS one 9 (9), e106489. doi:10.1371/journal.pone.0106489
Brocca L., Toniolo L., Reggiani C., Bottinelli R., Sandri M., Pellegrino M. A. (2017). FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 595 (4), 1143–1158. doi:10.1113/JP273097
Buckingham M., Bajard L., Chang T., Daubas P., Hadchouel J., Meilhac S., et al. (2003). The formation of skeletal muscle: from somite to limb. J. Anat. 202 (1), 59–68. doi:10.1046/j.1469-7580.2003.00139.x
Cai D., Frantz J. D., Tawa N. E., Melendez P. A., Oh B. C., Lidov H. G. W., et al. (2004). IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119 (2), 285–298. doi:10.1016/j.cell.2004.09.027
Camacho-Pereira J., Tarrago M. G., Chini C. C. S., Nin V., Escande C., Warner G. M., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23 (6), 1127–1139. doi:10.1016/j.cmet.2016.05.006
Campelj D. G., Goodman C. A., Rybalka E. (2021). Chemotherapy-induced myopathy: The dark side of the cachexia sphere. Cancers 13 (14), 3615. doi:10.3390/cancers13143615
Carmignac V., Quéré R., Durbeej M. (2011). Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum. Mol. Genet. 20 (3), 541–552. doi:10.1093/hmg/ddq499
Castro-Portuguez R., Sutphin G. L. (2020). Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 132, 110841. doi:10.1016/j.exger.2020.110841
Chazaud B. (2020). Inflammation and skeletal muscle regeneration: Leave it to the macrophages!. Trends Immunol. 41 (6), 481–492. doi:10.1016/j.it.2020.04.006
Chazaud B., Sonnet C., Lafuste P., Bassez G., Rimaniol A. C., Poron F., et al. (2003). Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 163 (5), 1133–1143. doi:10.1083/jcb.200212046
Chen M., Wang Y., Deng S., Lian Z., Yu K. (2022). Skeletal muscle oxidative stress and inflammation in aging: Focus on antioxidant and anti-inflammatory therapy. Front. Cell Dev. Biol. 10, 964130. doi:10.3389/fcell.2022.964130
Chikani V., Ho K. K. Y. (2014). Action of GH on skeletal muscle function: molecular and metabolic mechanisms. J. Mol. Endocrinol. 52 (1), R107–R123. doi:10.1530/JME-13-0208
Chowdhury S., Schulz L., Palmisano B., Singh P., Berger J. M., Yadav V. K., et al. (2020). Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J. Clin. Invest. 130 (6), 2888–2902. doi:10.1172/JCI133572
Ciciliot S., Rossi A. C., Dyar K. A., Blaauw B., Schiaffino S. (2013)., 45. Netherlands, 2191–2199. doi:10.1016/j.biocel.2013.05.016Muscle type and fiber type specificity in muscle wastingInt. J. Biochem. Cell Biol.10
Coggan A. R., Spina R. J., King D. S., Rogers M. A., BrownM. , Nemeth P. M., et al. (1992). Histochemical and enzymatic comparison of the gastrocnemius muscle of young and elderly men and women. J. Gerontol. 47 (3), B71–B76. doi:10.1093/geronj/47.3.B71
Coletti D. (2018). Chemotherapy-induced muscle wasting: an update. Eur. J. Transl. Myol. 28 (2), 7587. doi:10.4081/ejtm.2018.7587
Cortiula F., Hendriks L. E. L., van de Worp W. R. P. H., Schols A. M. W. J., Vaes R. D. W., Langen R. C. J., et al. (2022). Physical exercise at the crossroad between muscle wasting and the immune system: implications for lung cancer cachexia. J. Cachexia Sarcopenia Muscle 13 (1), 55–67. doi:10.1002/jcsm.12900
Damrauer J. S., Stadler M. E., Acharyya S., Baldwin A. S., Couch M. E., Guttridge D. C. (2018). Chemotherapy-induced muscle wasting: association with NF-κB and cancer cachexia. Eur. J. Transl. Myol. 28 (2), 7590. doi:10.4081/ejtm.2018.7590
Dao T., Green A. E., Kim Y. A., Bae S. J., Ha K. T., Gariani K., et al. (2020). Sarcopenia and muscle aging: A brief overview. Endocrinol. Metab. 35 (4), 716–732. doi:10.3803/EnM.2020.405
Deng B., Wehling-Henricks M., Villalta S. A., Wang Y., Tidball J. G. (2012). IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 189, 3669–3680. doi:10.4049/jimmunol.1103180
DiGirolamo D. J., Singhal V., Chang X., Lee S. J., Germain-Lee E. L. (2015). Administration of soluble activin receptor 2B increases bone and muscle mass in a mouse model of osteogenesis imperfecta. Bone Res. 3 (1), 14042. doi:10.1038/boneres.2014.42
Dolly A., Dumas J.-F., Servais S. (2020). Cancer cachexia and skeletal muscle atrophy in clinical studies: what do we really know? J. Cachexia Sarcopenia Muscle 11 (6), 1413–1428. doi:10.1002/jcsm.12633
Dreyer H. C., Owen E. C., Strycker L. A., Smolkowski K., Muyskens J. B., Kirkpatrick T. K., et al. (2018). Essential amino acid supplementation mitigates muscle atrophy after total knee arthroplasty: A randomized, double-blind, placebo-controlled trial. JB JS Open Access 3 (2), e0006. doi:10.2106/JBJS.OA.18.00006
Duchesne E., Dufresne S. S., Dumont N. A. (2017). Impact of inflammation and anti-inflammatory modalities on skeletal muscle healing: From fundamental research to the clinic. Phys. Ther. 97 (8), 807–817. doi:10.1093/ptj/pzx056
Dufresne S. S., Boulanger-Piette A., Bosse S., Argaw A., Hamoudi D., Marcadet L., et al. (2018). Genetic deletion of muscle RANK or selective inhibition of RANKL is not as effective as full-length OPG-fc in mitigating muscular dystrophy. Acta Neuropathol. Commun. 6 (1), 31. doi:10.1186/s40478-018-0533-1
Dufresne S. S., Dumont N. A., Bouchard P., Lavergne E., Penninger J. M., Frenette J. (2015). Osteoprotegerin protects against muscular dystrophy. Am. J. Pathol. 185 (4), 920–926. doi:10.1016/j.ajpath.2015.01.006
Dufresne S. S., Frenette J., Dumont N. A. (2016). Inflammation and muscle regeneration, a double-edged sword. Med. Sci. 32 (6–7), 591–597. doi:10.1051/medsci/20163206022
Dumont N., Bouchard P., Frenette J. (2008). Neutrophil-induced skeletal muscle damage: a calculated and controlled response following hindlimb unloading and reloading.’. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295 (6), R1831–R1838. doi:10.1152/ajpregu.90318.2008
Ebert S. M., Bullard S. A., Basisty N., Marcotte G. R., Skopec Z. P., Dierdorff J. M., et al. (2020). Activating transcription factor 4 (ATF4) promotes skeletal muscle atrophy by forming a heterodimer with the transcriptional regulator C/EBPβ. J. Biol. Chem. 295 (9), 2787–2803. doi:10.1074/jbc.RA119.012095
Ebert S. M., Dyle M. C., Bullard S. A., Dierdorff J. M., Murry D. J., Fox D. K., et al. (2015). Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. J. Biol. Chem. 290 (42), 25497–25511. doi:10.1074/jbc.M115.681445
Edgerton V. R., Zhou M. Y., Ohira Y., Klitgaard H., Jiang B., Bell G., et al. (1995). Human fiber size and enzymatic properties after 5 and 11 days of spaceflight. J. Appl. Physiol. (Bethesda, Md, 1985) 78 (5), 1733–1739. doi:10.1152/jappl.1995.78.5.1733
Ellingsgaard H., Hojman P., Pedersen B. K. (2019). Exercise and health — emerging roles of IL-6. Curr. Opin. Physiology 10, 49–54. doi:10.1016/j.cophys.2019.03.009
Estelle L.-T., Giulia C. M., KellyAnn S., Jerome N.-F., Steffen H., David J. G., et al. (2014). An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy’, molecular and cellular biology. Mol. Cell Biol. 34 (4), 606–618. doi:10.1128/MCB.01307-13
Fang W.-Y., Tseng Y. T., Lee T. Y., Fu Y. C., Chang W. H., Lo W. W., et al. (2021). Triptolide prevents LPS-induced skeletal muscle atrophy via inhibiting NF-κB/TNF-α and regulating protein synthesis/degradation pathway. Br. J. Pharmacol. 178 (15), 2998–3016. doi:10.1111/bph.15472
Feng F., Shan L., Deng J. X., Luo L. L., Huang Q. S. (2019). Role of the Notch signaling pathway in fibrosis of denervated skeletal muscle. Curr. Med. Sci. 39 (3), 419–425. doi:10.1007/s11596-019-2053-z
Fitts R. H., Riley D. R., Widrick J. J. (2001). Functional and structural adaptations of skeletal muscle to microgravity. J. Exp. Biol. 204 (18), 3201–3208. doi:10.1242/jeb.204.18.3201
Frederick D. W., Loro E., Liu L., Davila A., Chellappa K., Silverman I. M., et al. (2016). Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 24 (2), 269–282. doi:10.1016/j.cmet.2016.07.005
Fujita J., Yano M., Saito H., Monden M., Katsume A., Ohsugi Y., et al. (1996). Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int. J. Cancer 68 (5), 637–643. doi:10.1002/(SICI)1097-0215(19961127)68:5<637::AID-IJC14>3.0.CO;2-Z
Fülster S., Tacke M., Sandek A., Ebner N., Tschope C., Doehner W., et al. (2013). Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur. Heart J. 34 (7), 512–519. doi:10.1093/eurheartj/ehs381
Funai K., Parkington J. D., Carambula S., Fielding R. A. (2006). Age-associated decrease in contraction-induced activation of downstream targets of Akt/mTor signaling in skeletal muscle.Regulatory, integrative and comparative physiology. Am. J. Physiol. Regul. Integr. Comp. Physiol.United States 290 (4), R1080–R1086. doi:10.1152/ajpregu.00277.2005
Gaudenzio N., Sibilano R., Marichal T., Starkl P., Reber L. L., Cenac N., et al. (2016). Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Invest. 126 (10), 3981–3998. doi:10.1172/JCI85538
Gawlik K. I., Korner Z., Oliveira B. M., Durbeej M. (2019). Early skeletal muscle pathology and disease progress in the dy3K/dy3K mouse model of congenital muscular dystrophy with laminin α2 chain-deficiency. Sci. Rep. 9 (1), 14324. doi:10.1038/s41598-019-50550-0
Gerovasili V., Stefanidis K., Vitzilaios K., Karatzanos E., Politis P., Koroneos A., et al. (2009). Electrical muscle stimulation preserves the muscle mass of critically ill patients: a randomized study. Crit. Care 13 (5), R161. doi:10.1186/cc8123
Gielen S., Sandri M., Kozarez I., Kratzsch J., Teupser D., Thiery J., et al. (2012). Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized leipzig exercise intervention in chronic heart failure and aging catabolism study. Circulation 125 (22), 2716–2727. doi:10.1161/CIRCULATIONAHA.111.047381
Gioftsidi S., Relaix F., Mourikis P. (2022). The Notch signaling network in muscle stem cells during development, homeostasis, and disease. Skelet. Muscle 12 (1), 9. doi:10.1186/s13395-022-00293-w
Glass D., Roubenoff R. (2010). Recent advances in the biology and therapy of muscle wasting. Ann. N. Y. Acad. Sci. 1211 (1), 25–36. doi:10.1111/j.1749-6632.2010.05809.x
Goemans N., Buyse G. (2014). Current treatment and management of dystrophinopathies. Curr. Treat. Options Neurol. 16 (5), 287. doi:10.1007/s11940-014-0287-4
Goh B. C., Singhal V., Herrera A. J., Tomlinson R. E., Kim S., Faugere M. C., et al. (2017). Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J. Biol. Chem. 292 (33), 13809–13822. doi:10.1074/jbc.M117.782128
Golan T., Geva R., Richards D., Madhusudan S., Lin B. K., Wang H. T., et al. (2018). LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J. Cachexia Sarcopenia Muscle 9 (5), 871–879. doi:10.1002/jcsm.12331
Gonzalez-Freire M., de Cabo R., Bernier M., Sollott S. J., Fabbri E., Navas P., et al. (2015). Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 70 (11), 1334–1342. doi:10.1093/gerona/glv070
Grevendonk L., Connell N. J., McCrum C., Fealy C. E., Bilet L., Bruls Y. M. H., et al. (2021). Impact of aging and exercise on skeletal muscle mitochondrial capacity, energy metabolism, and physical function. Nat. Commun. 12 (1), 4773. doi:10.1038/s41467-021-24956-2
Haddad F., Bodell P. W., Qin A. X., Giger J. M., Baldwin K. M. (2003). Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J. Biol. Chem. 278 (39), 37132–37138. doi:10.1074/jbc.M305911200
Hajahmadi M., Shemshadi S., Khalilipur E., Amin A., Taghavi S., Maleki M., et al. (2017). Muscle wasting in young patients with dilated cardiomyopathy. J. Cachexia Sarcopenia Muscle 8 (4), 542–548. doi:10.1002/jcsm.12193
Hambrecht R., Schulze P. C., Gielen S., Linke A., Mobius-Winkler S., Yu J., et al. (2002). Reduction of insulin-like growth factor-I expression in the skeletal muscle of noncachectic patients with chronic heart failure. J. Am. Coll. Cardiol. 39 (7), 1175–1181. doi:10.1016/s0735-1097(02)01736-9
Hamoudi D., Bouredji Z., Marcadet L., Yagita H., Landry L. B., Argaw A., et al. (2020). Muscle weakness and selective muscle atrophy in osteoprotegerin-deficient mice. Hum. Mol. Genet. 29 (3), 483–494. doi:10.1093/hmg/ddz312
Hamoudi D., Marcadet L., Piette Boulanger A., Yagita H., Bouredji Z., Argaw A., et al. (2019). An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum. Mol. Genet. 28 (18), 3101–3112. doi:10.1093/hmg/ddz124
Hasselgren P.-O., Alamdari N., Aversa Z., Gonnella P., Smith I. J., Tizio S. (2010). Corticosteroids and muscle wasting: role of transcription factors, nuclear cofactors, and hyperacetylation. Curr. Opin. Clin. Nutr. Metab. Care 13 (4), 423–428. doi:10.1097/MCO.0b013e32833a5107
He N., Ye H. (2020). Exercise and muscle atrophy BT - physical exercise for human health. Editor J. Xiao (Singapore: Springer Singapore), 255–267. doi:10.1007/978-981-15-1792-1_17
He W. A., Berardi E., Cardillo V. M., Acharyya S., Aulino P., Thomas-Ahner J., et al. (2013). NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123 (11), 4821–4835. doi:10.1172/JCI68523
Hepple R. T., Rice C. L. (2016). Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 594 (8), 1965–1978. doi:10.1113/JP270561
Hirata Y., Nomura K., Kato D., Tachibana Y., Niikura T., Uchiyama K., et al. (2022). A Piezo1/KLF15/IL-6 axis mediates immobilization-induced muscle atrophy. J. Clin. Invest. 132 (10), 1–13. doi:10.1172/JCI154611
Hodgetts S., Radley H., Davies M., Grounds M. D. (2006). Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul. Disord. 16 (9–10), 591–602. doi:10.1016/j.nmd.2006.06.011
Huang P.-L., Hou M. S., Wang S. W., Chang C. L., Liou Y. H., Liao N. S. (2015). Skeletal muscle interleukin 15 promotes CD8+ T-cell function and autoimmune myositis. Skelet. Muscle 5 (1), 33. doi:10.1186/s13395-015-0058-2
Huang Z., Zhong L., Zhu J., Xu H., Ma W., Zhang L., et al. (2020). Inhibition of IL-6/JAK/STAT3 pathway rescues denervation-induced skeletal muscle atrophy. Ann. Transl. Med. 8 (24), 1681. doi:10.21037/atm-20-7269
Hyatt H. W., Ozdemir M., Yoshihara T., Nguyen B. L., Deminice R., Powers S. K. (2021). Calpains play an essential role in mechanical ventilation-induced diaphragmatic weakness and mitochondrial dysfunction. Redox Biol. 38, 101802. doi:10.1016/j.redox.2020.101802
Hyatt H. W., Powers S. K. (2020). The role of calpains in skeletal muscle remodeling with exercise and inactivity-induced atrophy. Int. J. Sports Med. 41 (14), 994–1008. doi:10.1055/a-1199-7662
Ingalls C. P., Wenke J. C., Armstrong R. B. (2001). Time course changes in [Ca2+]i, force, and protein content in hindlimb-suspended mouse soleus muscles. Aviat. Space Environ. Med. 72 (5), 471–476.
Inoue S., Nakase H., MatsuuraM. , Mikami S., Ueno S., UzaN. , et al. (2009). The effect of proteasome inhibitor MG132 on experimental inflammatory bowel disease. Clin. Exp. Immunol. 156 (1), 172–182. doi:10.1111/j.1365-2249.2008.03872.x
Jia D., O'Brien C. A., Stewart S. A., Manolagas S. C., Weinstein R. S. (2006). Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology 147 (12), 5592–5599. doi:10.1210/en.2006-0459
Joanisse S., Nederveen J. P., Snijders T., McKay B. R., Parise G. (2017). Skeletal muscle regeneration, repair and remodelling in aging: The importance of muscle stem cells and vascularization. Gerontology 63 (1), 91–100. doi:10.1159/000450922
Kamei Y., Hatazawa Y., Uchitomi R., Yoshimura R., Miura S. (2020). Regulation of skeletal muscle function by amino acids. Nutrients 12, E261. doi:10.3390/nu12010261
Kawanishi N., Mizokami T., Niihara H., Yada K., Suzuki K. (2016). Neutrophil depletion attenuates muscle injury after exhaustive exercise. Med. Sci. Sports Exerc. 48 (10), 1917–1924. doi:10.1249/MSS.0000000000000980
Kim H. C., Cho H.-Y., Hah Y.-S. (2012). Role of IL-15 in sepsis-induced skeletal muscle atrophy and proteolysis. Tuberc. Respir. Dis. 73 (6), 312–319. doi:10.4046/trd.2012.73.6.312
Kondo T., Kitazawa R., Yamaguchi A., Kitazawa S. (2008). Dexamethasone promotes osteoclastogenesis by inhibiting osteoprotegerin through multiple levels. J. Cell. Biochem. 103 (1), 335–345. doi:10.1002/jcb.21414
Kornegay J. N., Childers M. K., Bogan D. J., Bogan J. R., Nghiem P., Wang J., et al. (2012). The paradox of muscle hypertrophy in muscular dystrophy. Phys. Med. Rehabil. Clin. N. Am. 23 (1), 149–172. doi:10.1016/j.pmr.2011.11.014
Kung T., Springer J., Doehner W., Anker S. D., von Haehling S. (2010). Novel treatment approaches to cachexia and sarcopenia: highlights from the 5th cachexia conference. Expert Opin. Investig. Drugs 19 (4), 579–585. doi:10.1517/13543781003724690
Larsson L., Degens H., Li M., Salviati L., Lee Y. I., Thompson W., et al. (2019). Sarcopenia: Aging-Related loss of muscle mass and function. Physiol. Rev. 99 (1), 427–511. doi:10.1152/physrev.00061.2017
Lecker S. H., Goldberg A. L., Mitch W. E. (2006). Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17 (7), 1807–1819. doi:10.1681/ASN.2006010083
Lee M.-K., Jeong H. H., Kim M. J., Ryu H., Baek J., Lee B. (2022). Nutrients against glucocorticoid-induced muscle atrophy. Foods 11, 687. doi:10.3390/foods11050687
Lemos D. R., Babaeijandaghi F., Low M., Chang C. K., Lee S. T., Fiore D., et al. (2015). Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 21 (7), 786–794. doi:10.1038/nm.3869
Li H., Malhotra S., Kumar A. (2008). Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 86, 1113–1126. doi:10.1007/s00109-008-0373-8
Li J., Xie Q., Liu L., Cheng Y., Han Y., Chen X., et al. (2021). Swimming attenuates muscle wasting and mediates multiple signaling pathways and metabolites in CT-26 bearing mice. Front. Mol. Biosci. 8, 812681. doi:10.3389/fmolb.2021.812681
Locati M., Mantovani A., Sica A. (2013). Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 120, 163–184. doi:10.1016/B978-0-12-417028-5.00006-5
Loro E., Ramaswamy G., Chandra A., Tseng W. J., Mishra M. K., Shore E. M., et al. (2017). IL15RA is required for osteoblast function and bone mineralization. Bone 103, 20–30. doi:10.1016/j.bone.2017.06.003
Madaro L., Passafaro M., Sala D., Etxaniz U., Lugarini F., Proietti D., et al. (2018). Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 20 (8), 917–927. doi:10.1038/s41556-018-0151-y
Marcadet L., Bouredji Z., Argaw A., Frenette J. (2022). The roles of RANK/RANKL/OPG in cardiac, skeletal, and smooth muscles in health and disease. Front. Cell Dev. Biol. 10, 903657. doi:10.3389/fcell.2022.903657
Martín A. I., Priego T., Moreno-Ruperez A., Gonzalez-Hedstrom D., Granado M., Lopez-Calderon A. (2021). IGF-1 and IGFBP-3 in inflammatory cachexia. Int. J. Mol. Sci. 22, 9469. doi:10.3390/ijms22179469
Massudi H., Grant R., Braidy N., Guest J., Farnsworth B., Guillemin G. J. (2012). Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7 (7), e42357. doi:10.1371/journal.pone.0042357
Milan G., Romanello V., Pescatore F., Armani A., Paik J. H., Frasson L., et al. (2015). Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6 (1), 6670. doi:10.1038/ncomms7670
Mitchell P. O., Pavlath G. K. (2004). Skeletal muscle atrophy leads to loss and dysfunction of muscle precursor cells. Am. J. Physiol. Cell Physiol. 287 (6), C1753–C1762. doi:10.1152/ajpcell.00292.2004
Morissette M. R., Cook S. A., Buranasombati C., Rosenberg M. A., Rosenzweig A. (2009). Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am. J. Physiol. Cell Physiol. 297 (5), C1124–C1132. doi:10.1152/ajpcell.00043.2009
Morley J. E. (2016). Pharmacologic options for the treatment of sarcopenia. Calcif. Tissue Int. 98 (4), 319–333. doi:10.1007/s00223-015-0022-5
Mosser D. M., Edwards J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8 (12), 958–969. doi:10.1038/nri2448
Mouisel E., Ferry A., Hourde C., Vignaud A., Butler-Browne G. (2010). Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18–24-month-old) dystrophic mdx mice. Muscle & Nerve 41 (6), 809–818. doi:10.1002/mus.21624
Nadeau L., Aguer C. (2019). Reply to "discussion of 'interleukin-15 as a myokine: mechanistic insight into its effect on skeletal muscle metabolism' - interleukin-15 and interleukin-15Rα-dependent/-independent functions in human skeletal muscle are largely unknown". NRC Res. Press 44 (3), 338–339. doi:10.1139/apnm-2018-0823
Nijholt K. T., Sánchez-Aguilera P. I., Vorrips S. N., de Boer R. A., Westenbrink B. D. (2022). Exercise: a molecular tool to boost muscle growth and mitochondrial performance in heart failure? Eur. J. Heart Fail. 24 (2), 287–298. doi:10.1002/ejhf.2407
Nilwik R., Snijders T., Leenders M., Groen B. B. L., van Kranenburg J., Verdijk L. B., et al. (2013). The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp. Gerontol. 48 (5), 492–498. doi:10.1016/j.exger.2013.02.012
Nuhr M. J., Pette D., Berger R., Quittan M., Crevenna R., Huelsman M., et al. (2004). Beneficial effects of chronic low-frequency stimulation of thigh muscles in patients with advanced chronic heart failure. Eur. Heart J. 25 (2), 136–143. doi:10.1016/j.ehj.2003.09.027
Ogawa T., Furochi H., Mameoka M., Hirasaka K., Onishi Y., Suzue N., et al. (2006). Ubiquitin ligase gene expression in healthy volunteers with 20-day bedrest. Muscle Nerve 34 (4), 463–469. doi:10.1002/mus.20611
Oishi Y., Manabe I. (2018). Macrophages in inflammation, repair and regeneration. Int. Immunol. 30 (11), 511–528. doi:10.1093/intimm/dxy054
O’Leary M. F., Wallace G. R., Bennett A. J., Tsintzas K., Jones S. W. (2017). IL-15 promotes human myogenesis and mitigates the detrimental effects of TNFα on myotube development. Sci. Rep. 7 (1), 12997. doi:10.1038/s41598-017-13479-w
Ono T., Hayashi M., Sasaki F., Nakashima T. (2020). RANKL biology: bone metabolism, the immune system, and beyond. Inflamm. Regen. 40 (1), 2–16. doi:10.1186/s41232-019-0111-3
Otis J. S., Niccoli S., Hawdon N., Sarvas J. L., Frye M. A., Chicco A. J., et al. (2014). Pro-inflammatory mediation of myoblast proliferation. PloS one 9 (3), e92363. doi:10.1371/journal.pone.0092363
Pandorf C. E., Haddad F., Wright C., Bodell P. W., Baldwin K. M. (2009). Differential epigenetic modifications of histones at the myosin heavy chain genes in fast and slow skeletal muscle fibers and in response to muscle unloading. Am. J. Physiol. Cell Physiol. 297 (1), C6–C16. doi:10.1152/ajpcell.00075.2009
Pedersen B. K. (2013). Muscle as a secretory organ. Compr. Physiol. 3, 1337–1362. (Major Reference Works). doi:10.1002/cphy.c120033
Perandini L. A., Chimin P., da Siva D., Olsen N. (2018). Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: can physical exercise restore the satellite cell niche? FEBS J. 285 (11), 1973–1984. doi:10.1111/febs.14417
Petersen A. M. W., Pedersen B. K. (2005). The anti-inflammatory effect of exercise. J. Appl. Physiology 98 (4), 1154–1162. doi:10.1152/japplphysiol.00164.2004
Petrof B. J., Shrager J. B., Stedman H. H., Kelly A. M., Sweeney H. L. (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A. 90 (8), 3710–3714. doi:10.1073/pnas.90.8.3710
Piasecki M., Ireland A., Jones D. A., McPhee J. S. (2016). Age-dependent motor unit remodelling in human limb muscles. Biogerontology 17 (3), 485–496. doi:10.1007/s10522-015-9627-3
Pin F., Jones A. J., Huot J. R., Narasimhan A., Zimmers T. A., Bonewald L. F., et al. (2022). RANKL blockade reduces cachexia and bone loss induced by non-metastatic ovarian cancer in mice. J. Bone Min. Res. 37 (3), 381–396. doi:10.1002/jbmr.4480
Pirruccello-Straub M., Jackson J., WawerSik S., Webster M. T., SaLta L., Long K., et al. (2018). Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci. Rep. 8 (1), 2292. doi:10.1038/s41598-018-20524-9
Pistilli E. E., Quinn L. S. (2013). From anabolic to oxidative: reconsidering the roles of IL-15 and IL-15rα in skeletal muscle. Exerc. Sport Sci. Rev. 41 (2), 100–106. doi:10.1097/JES.0b013e318275d230
Qaisar R., Karim A., Elmoselhi A. B. (2020). Muscle unloading: A comparison between spaceflight and ground-based models. Acta Physiol. 228 (3), e13431. doi:10.1111/apha.13431
Quinn L. S., Anderson B. G., Drivdahl R. H., Alvarez B., Argiles J. M. (2002). Overexpression of interleukin-15 induces skeletal muscle hypertrophy in vitro: implications for treatment of muscle wasting disorders. Exp. Cell Res. 280 (1), 55–63. doi:10.1006/excr.2002.5624
Radley H. G., Grounds M. D. (2006). Cromolyn administration (to block mast cell degranulation) reduces necrosis of dystrophic muscle in mdx mice. Neurobiol. Dis. 23 (2), 387–397. doi:10.1016/j.nbd.2006.03.016
Reginster J.-Y., Beaudart C., Buckinx F., Bruyere O. (2016). Osteoporosis and sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 19 (1), 31–36. doi:10.1097/MCO.0000000000000230
Renna L. V., Bose F., Brigonzi E., Fossati B., Meola G., Cardani R. (2019). Aberrant insulin receptor expression is associated with insulin resistance and skeletal muscle atrophy in myotonic dystrophies. PLoS One 14 (3), e0214254. doi:10.1371/journal.pone.0214254
Rock K. L., Gramm C., Rothstein L., ClarK K., Stein R., Dick L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78 (5), 761–771. doi:10.1016/s0092-8674(94)90462-6
Roubenoff R. (2000). Sarcopenia and its implications for the elderly. Eur. J. Clin. Nutr. 54, S40–S47. doi:10.1038/sj.ejcn.1601024
Rudolf R., Khan M. M., Labeit S., Deschenes M. R. (2014). Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 6, 99. doi:10.3389/fnagi.2014.00099
Salazar J. J., Michele D. E., Brooks S. V. (2010). Inhibition of calpain prevents muscle weakness and disruption of sarcomere structure during hindlimb suspension. J. Appl. Physiol. (Bethesda, Md, 1985) 108 (1), 120–127. doi:10.1152/japplphysiol.01080.2009
Sandri M., Lin J., Handschin C., Yang W., Arany Z. P., Lecker S. H., et al. (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U. S. A. 103 (44), 16260–16265. doi:10.1073/pnas.0607795103
Sartori R., Romanello V., Sandri M. (2021). Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat. Commun. 12 (1), 330. doi:10.1038/s41467-020-20123-1
Schakman O., KaliSta S., Barbe C., LoumAye A., Thissen J. P. (2013)., 45. Netherlands, 2163–2172. doi:10.1016/j.biocel.2013.05.036Glucocorticoid-induced skeletal muscle atrophyInt. J. Biochem. Cell Biol.10
Schiaffino S., Mammucari C. (2011). Regulation of skeletal muscle growth by the IGF1-akt/PKB pathway: insights from genetic models. Skelet. Muscle 1 (1), 4. doi:10.1186/2044-5040-1-4
Serrano A. L., Baeza-Raja B., Perdiguero E., Jardi M., Munoz-Canoves P. (2008). Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7 (1), 33–44. doi:10.1016/j.cmet.2007.11.011
Skorupska E. (2018). Muscle atrophy measurement as assessment method for low back pain patients. Adv. Exp. Med. Biol. 1088, 437–461. doi:10.1007/978-981-13-1435-3_20
Smith R. C., Lin B. K. (2013). Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 7 (4), 352–360. doi:10.1097/SPC.0000000000000013
Solerte S. B., Gazzaruso C., Bonacasa R., Rondanelli M., Zamboni M., Basso C., et al. (2008). Nutritional supplements with oral amino acid mixtures increases whole-body lean mass and insulin sensitivity in elderly subjects with sarcopenia. Am. J. Cardiol. 101 (11A), 69E–77E. doi:10.1016/j.amjcard.2008.03.004
Song Y.-H., Song J. L., Delafontaine P., Godard M. P. (2013). The therapeutic potential of IGF-I in skeletal muscle repair. Trends Endocrinol. Metab. 24 (6), 310–319. doi:10.1016/j.tem.2013.03.004
Springer J., Springer J.-I., Anker S. D. (2017). Muscle wasting and sarcopenia in heart failure and beyond: update 2017. Esc. Heart Fail. 4 (4), 492–498. doi:10.1002/ehf2.12237
Summan M., Warren G. L., Mercer R. R., Chapman R., Hulderman T., Van Rooijen N., et al. (2006). Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290 (6), R1488–R1495. doi:10.1152/ajpregu.00465.2005
Suzuki K. (2019). Chronic inflammation as an immunological abnormality and effectiveness of exercise. Biomolecules 9, E223. doi:10.3390/biom9060223
Tajrishi M. M., Sato S., Shin J., Zheng T. S., Burkly L. C., Kumar A. (2014). The TWEAK–Fn14 dyad is involved in age-associated pathological changes in skeletal muscle. Biochem. Biophys. Res. Commun. 446 (4), 1219–1224. doi:10.1016/j.bbrc.2014.03.084
Talbert E. E., Min K., Powers S. K., Kwon O. S., Smuder A. J. (2013). Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J. Appl. physiology (Bethesda, Md 1985) 114 (10), 1482–1489. doi:10.1152/japplphysiol.00925.2012
Terracciano C., CeliM. , Lecce D., Baldi J., RastElli E., Lena E., et al. (2013). Differential features of muscle fiber atrophy in osteoporosis and osteoarthritis. Osteoporos. Int. 24 (3), 1095–1100. doi:10.1007/s00198-012-1990-1
Thoma A., Lightfoot A. P. (2018). NF-kB and inflammatory cytokine signalling: Role in skeletal muscle atrophy. Adv. Exp. Med. Biol. 1088, 267–279. doi:10.1007/978-981-13-1435-3_12
Tidball J. G. (2017). Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 17 (3), 165–178. doi:10.1038/nri.2016.150
Tidball J. G., Spencer M. J. (2002). Expression of a calpastatin transgene slows muscle wasting and obviates changes in myosin isoform expression during murine muscle disuse. J. Physiol. 545 (3), 819–828. doi:10.1113/jphysiol.2002.024935
Tidball J. G., Welc S. S., Wehling-Henricks M. (2018). Immunobiology of inherited muscular dystrophies. Compr. Physiol., 1313–1356. doi:10.1002/cphy.c170052
Timmer L. T., Hoogaars W. M. H., Jaspers R. T. (2018). The role of IGF-1 signaling in skeletal muscle atrophy BT - muscle atrophy. Editor J. Xiao (Singapore: Springer Singapore), 109–137. doi:10.1007/978-981-13-1435-3_6
Toumi H., F’guyer S., Best T. M. (2006). The role of neutrophils in injury and repair following muscle stretch. J. Anat. 208 (4), 459–470. doi:10.1111/j.1469-7580.2006.00543.x
Trendelenburg A. U., Meyer A., Rohner D., Boyle J., Hatakeyama S., Glass D. J. (2009). Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 296 (6), C1258–C1270. doi:10.1152/ajpcell.00105.2009
Velloso C. P. (2008). Regulation of muscle mass by growth hormone and IGF-I. Br. J. Pharmacol. 154 (3), 557–568. doi:10.1038/bjp.2008.153
Villalta S. A., Rosenthal W., Martinez L., Kaur A., Sparwasser T., Tidball J. G., et al. (2014). Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 6 (258), 258ra142. doi:10.1126/scitranslmed.3009925
Vittorio S., Marcella F. (2004). Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy’, science’s STKE. Am. Assoc. Adv. Sci. 2004 (244), re11. doi:10.1126/stke.2442004re11
Vivodtzev I., Pepin J. L., Vottero G., Mayer V., Porsin B., Levy P., et al. (2006). Improvement in quadriceps strength and dyspnea in daily tasks after 1 month of electrical stimulation in severely deconditioned and malnourished COPD. Chest 129 (6), 1540–1548. doi:10.1378/chest.129.6.1540
Wang Y., Pessin J. E. (2013). Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 16 (3), 243–250. doi:10.1097/MCO.0b013e328360272d
Webster J. M., Kempen L. J. A. P., Hardy R. S., Langen R. C. J. (2020). Inflammation and skeletal muscle wasting during cachexia. Front. Physiol. 11, 597675. doi:10.3389/fphys.2020.597675
Wehling M., Spencer M. J., Tidball J. G. (2001). A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 155 (1), 123–131. doi:10.1083/jcb.200105110
Weinstein R. S., Jilka R. L., Parfitt A. M., Manolagas S. C. (1998). Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Invest. 102 (2), 274–282. doi:10.1172/JCI2799
Wen H.-S., Li X., Cao Y. Z., Zhang C. C., Yang F., Shi Y. M., et al. (2012). Clinical studies on the treatment of cancer cachexia with megestrol acetate plus thalidomide. Chemotherapy 58 (6), 461–467. doi:10.1159/000346446
Widdowson W. M., Gibney J. (2008). The effect of growth hormone replacement on exercise capacity in patients with GH deficiency: A metaanalysis. J. Clin. Endocrinol. Metab. 93 (11), 4413–4417. doi:10.1210/jc.2008-1239
Wohlgemuth S. E., Seo A. Y., Marzetti E., Lees H. A., Leeuwenburgh C. (2010). Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp. Gerontol. 45 (2), 138–148. doi:10.1016/j.exger.2009.11.002
Wood N., Straw S., Witte K. K., Roberts L. D., Bowen D. S., Scalabrin M. (2021). Skeletal muscle atrophy in heart failure with diabetes: from molecular mechanisms to clinical evidence. Esc. Heart Fail. 8 (1), 3–15. doi:10.1002/ehf2.13121
Xu T., Yang X., Wu C., Qiu J., Fang Q., Wang L., et al. (2018). Pyrroloquinoline quinone attenuates cachexia-induced muscle atrophy via suppression of reactive oxygen species. J. Thorac. Dis. 10 (5), 2752–2759. doi:10.21037/jtd.2018.04.112
Yaku K., Okabe K., Nakagawa T. (2018). NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 47, 1–17. doi:10.1016/j.arr.2018.05.006
Yarasheski K. E., BhaSin S., Sinha-HikIm I., Pak-Loduca J., Gonzalez-Cadavid N. F. (2002). Serum myostatin-immunoreactive protein is increased in 60-92 year old women and men with muscle wasting. J. Nutr. Health Aging 6 (5), 343–348.
Yin L., Jia W., Wang N., Liang M., Shang J. (2022). Antagonizing urotensin receptor is a novel therapeutic strategy for glucocorticoid-induced skeletal muscle atrophy. Clin. Transl. Med. 12, e879. doi:10.1002/ctm2.879
Yin L., Li N., Jia W., Wang N., Liang M., Yang X., et al. (2021). Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 172, 105807. doi:10.1016/j.phrs.2021.105807
Zaki M. H., Nemeth J. A., Trikha M. (2004). CNTO 328, a monoclonal antibody to IL-6, inhibits human tumor-induced cachexia in nude mice. Int. J. Cancer 111 (4), 592–595. doi:10.1002/ijc.20270
Zanotti E., Felicetti G., Maini M., Fracchia C. (2003). Peripheral muscle strength training in bed-bound patients with COPD receiving mechanical ventilation: effect of electrical stimulation. Chest 124 (1), 292–296. doi:10.1378/chest.124.1.292
Zhang L., Tang H., Kou Y., Li R., Zheng Y., Wang Q., et al. (2013). MG132-mediated inhibition of the ubiquitin–proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 139 (7), 1105–1115. doi:10.1007/s00432-013-1412-6
Zhang Y., Wang J., Wang X., Gao T., Tian H., Zhou D., et al. (2020). The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am. J. Clin. Nutr. 111 (3), 570–579. doi:10.1093/ajcn/nqz347
Keywords: muscle atrophy, sarcopenia, cachexia, inflammation, muscle-bone crosstalk
Citation: Bouredji Z, Argaw A and Frenette J (2022) The inflammatory response, a mixed blessing for muscle homeostasis and plasticity. Front. Physiol. 13:1032450. doi: 10.3389/fphys.2022.1032450
Received: 30 August 2022; Accepted: 10 November 2022;
Published: 23 November 2022.
Edited by:
Hualin Sun, Nantong University, ChinaReviewed by:
Adam P. Lightfoot, Liverpool John Moores University, United KingdomCopyright © 2022 Bouredji, Argaw and Frenette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jérôme Frenette, amVyb21lLmZyZW5ldHRlQGNyY2h1ZGVxdWViZWMudWxhdmFsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.