 Peng Yan
Peng Yan Ben Ke
Ben Ke Xiangdong Fang
Xiangdong Fang- Department of Nephrology, The Second Affiliated Hospital of Nanchang University, Nanchang, China
Renal ion channel transport and electrolyte disturbances play an important role in the process of functional impairment and fibrosis in the kidney. It is well known that there are limited effective drugs for the treatment of renal fibrosis, and since a large number of ion channels are involved in the renal fibrosis process, understanding the mechanisms of ion channel transport and the complex network of signaling cascades between them is essential to identify potential therapeutic approaches to slow down renal fibrosis. This review summarizes the current work of ion channels in renal fibrosis. We pay close attention to the effect of cystic fibrosis transmembrane conductance regulator (CFTR), transmembrane Member 16A (TMEM16A) and other Cl− channel mediated signaling pathways and ion concentrations on fibrosis, as well as the various complex mechanisms for the action of Ca2+ handling channels including Ca2+-release-activated Ca2+ channel (CRAC), purinergic receptor, and transient receptor potential (TRP) channels. Furthermore, we also focus on the contribution of Na+ transport such as epithelial sodium channel (ENaC), Na+, K+-ATPase, Na+-H+ exchangers, and K+ channels like Ca2+-activated K+ channels, voltage-dependent K+ channel, ATP-sensitive K+ channels on renal fibrosis. Proposed potential therapeutic approaches through further dissection of these mechanisms may provide new therapeutic opportunities to reduce the burden of chronic kidney disease.
1 Introduction
Renal fibrosis is a process described by excessive proliferation of fibroblasts and deposition of extracellular matrix (ECM), which collectively lead to a broad maladaptive repair of affected renal tissue (Duffield, 2014). Under pathological conditions, the fibrotic process is triggered and coordinated by cross-talk between multiple cell types (Gewin et al., 2017). In particular, myofibroblasts, which are a key population for interstitial collagen matrix deposition, typically have the properties of contraction, proliferation, enhanced secretion, and expression of α smooth muscle actin (αSMA), a cytoskeletal protein of highly contractile microfilaments. In addition to primarily resident mesenchymal cells (fibroblasts and pericytes), other sources of myofibroblasts include perivascular fibroblasts, circulating fibroblasts, and epithelial-mesenchymal transition (EMT) transdifferentiated renal tubular epithelial cells (Mack and Yanagita, 2015). Interstitial inflammatory cell infiltration is also one of the characteristic histological features of renal fibrosis, which is observed in almost all types of renal disease and fibrosis (Huen and Cantley, 2017). Activated lymphocytes and macrophages as well as damaged epithelial cells secrete a variety of pro-inflammatory and pro-fibrotic factors, among which transforming growth factor-β (TGF-β) is recognized to be particularly prominent in renal fibrosis [for more factors references (Meng et al., 2016; Gewin et al., 2017)]. These cytokines promote the initiation of the fibrotic response by favoring fibroblast activation, inflammatory cell recruitment and endothelial cell loss, and achieve crosstalk between these cells. New evidence suggests that endothelial cell damage may be involved in endothelial-to-mesenchymal transition as well as in complex secretory synthesis (Lipphardt et al., 2017; Yang et al., 2019a). In addition, the kidney is a highly vascularized organ, and dysfunction of the interstitial capillary network mediating hypoxia and oxidative stress production is one of the important mechanisms driving renal fibrosis. Furthermore, when repeated epithelial injury occurs, apoptosis of renal tubular cells can develop, often resulting in tubular atrophy at a later stage. Interstitial fibrosis and tubular atrophy are common endpoints in almost all forms of kidney injury. Although a plethora of literature with in-depth and extensive mechanistic research, unfortunately there is no effective treatment for renal fibrosis, and the only proven method to slow the decline of renal function remains blockade of the renin-angiotensin system with angiotensin converting enzyme inhibitors (ACEI), angiotensin receptor blockers, or renin inhibitors (Francois and Chatziantoniou, 2018). Late development to end-stage renal disease (ESRD) still requires replacement therapy.
Ion channels are a class of pore-forming proteins found in all living cells that provide energetically favorable passage for ions to diffuse rapidly and passively according to their electrochemical potential (Roux, 2017). By mediating the influx or efflux of essential ions transported across the cell membrane, they can modulate the cytoplasmic or extracellular ion concentration, membrane potential and cell volume, which are fundamental to the survival and functional state of all cells. Regulation of changes in ion fluxes and channel activity in response to changing environmental requirements and stimuli is required for processes including proliferation, apoptosis, invasion, secretion and migration, and other cellular behavioral processes. Ions play an important role in different signaling pathways, since many extracellular molecules target ions for related functions, and in the last decade or so, the field of ion channels has developed rapidly. Indeed, the term “channelopathy” has been extended to describe a growing range of diseases associated with ion channel dysfunction. Dysregulated ion channels are involved in pathological conditions including hypertension, hyperglycemia, obesity, electrolyte disturbances, and cellular mechanical changes, all of which accelerate renal fibrosis phenotypes. Although the pathological factors and mechanisms of renal fibrosis remain incompletely understood, increasing evidence suggests that gene expression and signaling pathway activation associated with renal fibrosis are closely linked to the regulation of voltage- and non-voltage-gated ion channels. In this review, we summarize most recent data regarding the involvement of four major classes of Cl−, Ca2+, Na+, and K+ ion channels in the regulation of renal fibrosis. Continued efforts to explore their interactions and mechanisms, and to assess as candidate pharmacological targets of delay progression of renal failure will help develop more effective treatments.
2 Chloride channels
Cl− channels are a group of functionally and structurally diverse anion-selective channels that have recently gained a considerable amount of interest. In the past, it was always thought that Cl− was in electrochemical equilibrium on the membrane due to less research and the high resting Cl− permeability, but it is now clear that in most cells, Cl− is actively transported and out of electrochemical equilibrium (Duran et al., 2010), for example, some Cl− channels or transporters like Cl−-HCO3− exchangers, Na+-Cl− cotransporters pump Cl− into the cell, thus able to work and signal (Schmick and Bastiaens, 2014). Cl− channels exhibit regulation of various physiological functions, including fluid secretion, cell volume, intracellular pH, and are involved in processes such as proliferation, trans-epithelial transport, cell cycle and electrical excitability (Stauber et al., 2012). Different Cl− channels have been described in several categories based on structural and biological properties and gating characteristics: cystic fibrosis transmembrane conductance regulator (CFTR); Ca2+-activated Cl− channels; voltage-activated Cl− channels; volume-regulated anion channels (VRAC), ligand-gated channels, and other chloride channels (Suzuki et al., 2006). Here we focus on the most recent data regarding the involvement of these Cl− channels in the behavior of renal fibrosis.
2.1 Cystic fibrosis transmembrane conductance regulator(CFTR)
CFTR, a member of the ATP-binding cassette transporter protein superfamily, is widely expressed in apical epithelial membranes including the kidney, lung, liver, and reproductive tract (Csanady et al., 2019). CFTR is a cAMP-dependent anion channel consisting of a dimer with six transmembrane structures per subunit that regulate fluid transport and electrolyte balance. Dysfunction of CFTR leads to abnormal anion secretion, causing a series of epithelial dysfunctions and the development of chronic inflammatory. Cystic fibrosis (CF) is the result of a CFTR mutation, which is well described in terms of lung symptoms (Gibson et al., 2003). Some studies have shown that CFTR has a tumor suppressive effect in various types of cancer (Maisonneuve et al., 2013; Liu et al., 2020). In addition, CFTR is abundantly expressed on the apical surface of renal tubules, and CF patients had more prominent proteinuria, which may be caused by tubular dysfunction and interstitial injury (Jouret and Devuyst, 2009), thus suggesting that CFTR is closely associated with renal fibrotic disease.
Functional CFTR deficiency result in epithelial cells transforming into a more proliferative, less differentiated state that is more sensitive to EMT stimuli such as TGF-β1. Mesenchymal markers such as N- cadherin, vimentin, collagen I, and fibronectin are significantly upregulated in the native human CF airways compared to non CF airways (Quaresma et al., 2020), and increase CFTR activity using the potent CFTR modulator (HECM) drug restores the mutated CFTR-induced epithelial phenotype and confers direct protection against EMT (Narayanan et al., 2020). In the kidney, the mouse δF508 CFTR mutation exacerbates the fibrotic phenotype induced by unilateral ureteral obstruction (UUO), which is a well-established animal model of renal fibrosis, and in vitro, inhibition of CFTR activity using the inhibitors inh-172 or GlyH101 is sufficient to trigger the EMT process in renal cells (Zhang et al., 2017). In addition, it has been recognized that CFTR deficiency may lead to disruption of the cytoskeleton and reduced formation of cellular tight junctions in renal tubular epithelial cells by reducing direct Zonula Occludens-1 (ZO-1) interactions (Ruan et al., 2014; Castellani et al., 2017), which is one of the characteristics of EMT. Yes-associated protein 1 (YAP1) is an important mammalian transcriptional effector regulating the Hippo pathway and has been shown to play a key role in organ development, fibrosis, and wound healing (Dey et al., 2020). YAP1 was found to be aberrantly active in the presence of mutant CFTR, and is an important mediator of CFTR-related fibrosis/EMT processes (Quaresma et al., 2022). However, further elaboration is needed regarding its more precise mechanism.
CFTR dysfunction activates canonical Wnt/β-catenin signaling to mediate tubular epithelial-mesenchymal fibroblast transition (Zhang et al., 2017). Wnt/β-catenin is an evolutionary conserved signaling pathway that regulates cell fate, homeostasis and regeneration, and is activated following kidney injury in various animal models and human kidney diseases. β-catenin overexpression induces fibrotic features, including epithelial cell dedifferentiation and EMT (Clevers and Nusse, 2012). Interestingly, CFTR expression was downregulated in the UUO mouse model and human fibrotic kidney, and the UUO-induced deltaF508 fibrosis mouse model has significantly higher β-catenin protein activity and renal fibrosis aggregation, which can be rescued by overexpression of CFTR, implying that CFTR regulation appears to be a potential therapeutic target for anti-fibrosis via Wnt/β-catenin signaling (Zhang et al., 2017). This result was further demonstrated by Liu and his colleagues in a rat model of diabetic nephropathy (Liu et al., 2021). Mechanistically, CFTR can interact with Dishevelled2 (Dvl2), a vital component of Wnt signaling, to inhibit β-catenin activation via the PDZ structural domain, as Dvl2 has PDZ domain and CFTR has a PDZ binding domain (Simons et al., 2009; Zhang et al., 2017). However, the nature of how this interaction inhibits β-catenin activity has not been fully elucidated. Moreover, it may also be related to the pH and charge changes of the Wnt pathway. Loss of CFTR function leads to elevated intracellular PH due to impaired Cl− and HCO3− secretion and dysregulation of the H+ pump (membrane potential changes induced by Cl− flux contribute to the acidification of H+-ATPase) (Massey et al., 2021). It has been shown that intracellular alkalinization enhances the interaction of DvL with the Wnt receptor Frizzled proteins (FZD) by addressing DvL to negatively charged phospholipids on the plasma membrane through its positively charged DEP structural domain, thereby inhibiting β-catenin phosphate degradation and promoting Wnt signaling (Simons et al., 2009) (see Figure 1).
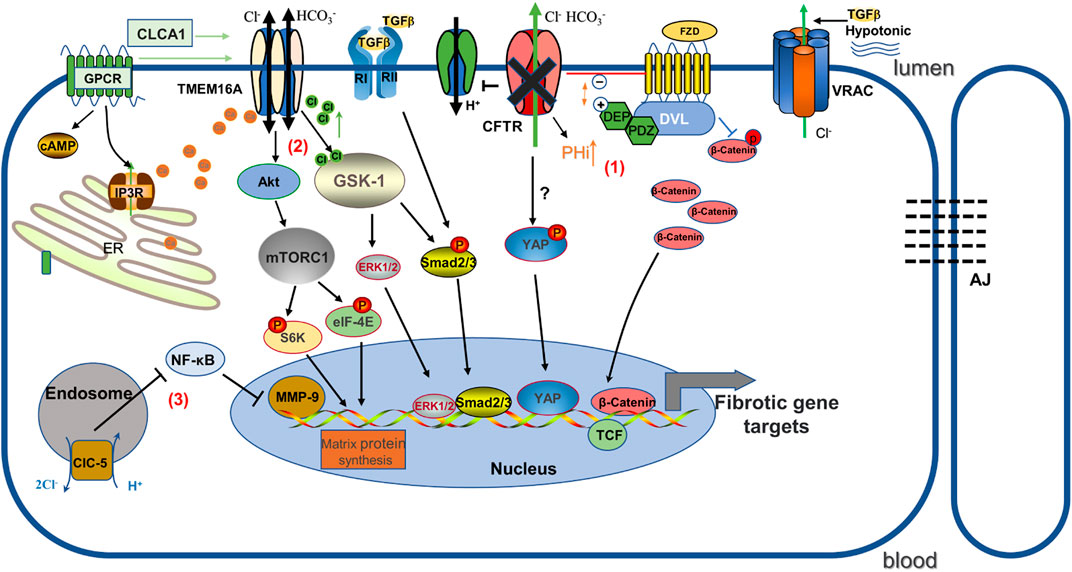
FIGURE 1. Schematic diagram illustrating the mechanisms by which several chloride channels affect gene expression in renal fibrosis. (1) CFTR deficiency causes increased intracellular pH. It promotes the binding of positively charged DEP and negatively charged phospholipids at the plasma membrane, and as a result, increased association of FZD and Dvl inhibits the phosphorylated degradation of β-catenin and promotes Wnt/β-catenin signaling. In addition, YAP may play a role in promoting renal fibrosis as one of the mechanisms. (2) TMEM16A stimulated by chloride channel accessory 1 (CLCA1) activates mTORC1-mediated matrix protein synthesis, and GSK-1 induces pro-fibrotic expression in response to TMEM16A-induced activation by increased Cl-concentration. (3)ClC-5 overexpression inhibits NF-κB/MMP-9. FZD: Frizzled; DVL: Dishevelled; AJ: Adherens junctions; ClC-5: voltage-gated chloride channel-5.
Notably, CFTR positively regulates β-catenin protein activity in the hematopoietic system and embryonic stem cells (Liu et al., 2017; Sun et al., 2018), implying that CFTR seems to respond differently to β-catenin signaling in cellular context, possibly by acting on different components of the Wnt/β-catenin pathway.
With more clinical focus regarding CFTR in autosomal dominant polycystic kidney disease (ADPKD), the importance of CFTR in fibrosis is increasingly recognized. Increased cAMP levels stimulate apical CFTR choreography, which contributes to epithelial cell proliferation and Cl−-dependent fluid secretion, while ECM gradually accumulates in the kidney with cystogenesis and the production of pro-fibrotic factors (Ramasubbu et al., 1998; Norman, 2011). Moreover, the cyst wall contains an extensive and abnormal capillary network and the absence of peritubular capillaries is a feature of tubulointerstitial fibrosis in chronic kidney disease (CKD). While deeply investigating the role of CFTR in kidney disease, independently of ADPKD, applying this target to chronic kidney disease or fibrotic kidney disease would be quite an attractive therapeutic treatment in terms of prospect.
2.2 Transmembrane member 16A
Transmembrane member (TMEM) 16A (also known as Anoctamin-1,ANO1), a Ca2+-activated Cl− channel, belongs to the 10-member of the TMEM16 family and is known to transport chlorine and bicarbonate (Pedemonte and Galietta, 2014). Structurally TMEM16A is a revealed homodimer membrane protein with each subunit containing a ten-transmembrane helix structure (TM1-10), in which an ion-selective pore and two Ca2+-sensitive binding sites were identified. When Ca2+ directly binds to a site TM6-8 located near the cytoplasmic end of the membrane pore, the TM6 conformation is triggered to change, causing the pore to dilate and thus leaving the channel in an open state (Dang et al., 2017; Paulino et al., 2017). TMEM16A is widely expressed in various cells and mediates a variety of fundamental physiological functions, such as epithelial regulation-secretion, smooth muscle contraction, cell volume regulation, cell proliferation and sensory transduction (Yang et al., 2008; Hartzell et al., 2009; Oh and Jung, 2016). TMEM16A activity may be involved in the regulation of epithelial CFTR-dependent Cl− transport. Recently, information about the relationship between TMEM16A and renal fibrosis was reported.
TMEM16A contributes to renal fibrosis through increased intracellular Cl− concentration and TGF-β1-dependent pathways (Li et al., 2022a). Previous studies have indicated that TMEM16A expression is strongly expressed in the kidneys of patients with IgA nephropathy, renal cyst models and mice with high-fat diet/streptozotocin-induced diabetic nephropathy (Buchholz et al., 2014; Lian et al., 2017; Li et al., 2022a), suggesting an important role for TMEM16A in kidney disease. Similarly, Li et al. found that TMEM16A expression was markedly increased in fibrotic kidneys from UUO and high-fat diet mouse models, and that inhibition of TMEM16A activity in vivo with specific inhibitors or knockdown by shRNA effectively attenuated UUO-induced renal fibrosis and macrophage infiltration (Li et al., 2022a). Also, in cultured human proximal renal tubular epithelial (HK2) cells, inhibition of TMEM16A effectively reduced TGF-β1-induced EMT and restored E-cadherin abundance. Mechanistically, high levels of TMEM16A respond to TGF-β1-induced increases in [Cl]i, and mediate the pro-fibrotic effects of TGF-β1 in a Cl− sensitive serum- and glucocorticoid-inducible protein kinase 1 (SGK1) dependent manner through the intracellular effectors Smad2/3 and extracellular signal-regulated kinases 1 and 2 (ERK1/2) pathways (Li et al., 2022a) (Figure 1). However, we recognize that TMEM16A is an outward rectifier current that promotes Cl− secretion. The mechanism regarding TMEM16A mediating the elevation of [Cl]i in renal fibrosis has not yet been fully elucidated, and may be due to 1) differences in Cl− concentration on renal tubular epithelial cells that predispose to inward flow, 2) indirect regulation of other Cl− reabsorption pathways, 3) pathological conditions may shift Cl− outflow to influx when the membrane potential reaches the Cl− reverse potential (Deba and Bessac, 2015). Notably, nephron-specific TMEM16A knockout mice cause reduced glomeruli numbers and subsequently albuminuria and tubular injury (Faria et al., 2014; Schenk et al., 2018), which may be related to TMEM16A regulation of albumin uptake and endosomal acidification functions (Faria et al., 2014). Therefore, TMEM16A contributes to the regulation of renal function, but also plays an important role in response to environmental stimuli such as obstruction and high fat in renal injury.
Recently, an unexpected association between TMEM16A and senescence-associated secretory phenotype (SASP)-associated renal fibrosis was found. Aging kidney injury is an important driver of interstitial fibrosis, in part because senescent cells secrete important pro-inflammatory and pro-fibrotic mediators in the senescence-associated secretory phenotype, which directly affects the surrounding microenvironment, thereby triggering the persistent fibrotic process (Docherty et al., 2019; Docherty et al., 2020). Cellular senescence can therefore be considered as a pathological feature of renal fibrosis. It was found that chloride channel accessory 1 (CLCA1) expression was increased in aged mice and its overexpression enhanced TMEM16A activity in a paracrine manner (Sala-Rabanal et al., 2015), subsequently activating mTORC1 in the process. Activation of mTORC1 signaling is not only involved in renal fibroblast activation and collagen synthesis, but also increases the expression of TGF-β1, which mediates the development of fibrosis, and finally mTORC1 regulates oxidative stress injury and stem cell failure to accelerate cell and tissue aging (Zoncu et al., 2011; Jiang et al., 2013; Fantus et al., 2016). Inhibition of TMEM16A reduced mTORC1 activation and matrix protein synthesis in CLCA1 overexpressing cells, while also inhibiting SASP (Lee et al., 2021). Based on the above findings, TMEM16A is considered a potential target to prevent senescence-associated renal injury. However, the mechanism of how the process of cell surface anion secretion should trigger SASP remains unclear.
In diabetic nephropathy, TMEM16A activation also promoted podocyte apoptosis in diabetic mice by activating the P38/c-jun N-terminal kinase (JNK) signaling pathway (Lian et al., 2017). In conclusion, the important role of TMEM16A in promoting kidney injury through the regulation of multiple signaling pathways suggests that is TMEM16A may be a potential new molecular target for preventing the progression of renal fibrosis and chronic kidney disease.
2.3 Voltage-gated Cl− channels
The voltage-gated chloride channel (ClC) family is a class of dimers consisting of nine isomers in mammals further divided into three groups: 1) ClC-1, ClC-2, ClC -Ka, and ClC -Kb; 2) ClC-3 to ClC-5; and 3) ClC-6 and ClC-7 (Jentsch and Pusch, 2018). It is found that ClC-3 to ClC-7 are mainly present in the endolysosomal membrane where they actually function as 2 Cl−/H+ exchangers, whereas the other types are plasma membrane Cl− channels (Picollo and Pusch, 2005; Scheel et al., 2005; Jentsch and Pusch, 2018). By exchanging chlorine for hydrogen, ClCs provide a Cl− shunt conductance in the lysosomes to neutralize the positive charge and promotes acidification (Gunther et al., 2003; Hara-Chikuma et al., 2005). Recently, the role of ClC in renal fibrosis has been gradually recognized.
As a members of the ClC superfamily, ClC-5 is an intracellular Cl− channel encoded by the chloride voltage-gated channel 5 (CLCN5) gene, which is involved in cell proliferation, apoptosis, cellular electrical activity and volume homeostasis in addition to controlling acidification (Devuyst and Luciani, 2015). Like all other eukaryotic ClCs, ClC-5 is homodimeric with 18 transmembrane domains per subunit containing an independent ion-permeable pore where it is labeled by three anion-binding sites (Dutzler, 2004). ClC-5 is highly expressed in different renal tubular segments as well as in podocytes, and is upregulated in glomeruli of proteinuric nephropathy patients, suggesting that ClC-5 may play a role in the formation of proteinuric nephropathy (Ceol et al., 2012; Solanki et al., 2018). Previous studies indicated that aging ClC-5 knockout mice significantly increased renal tubular atrophy, interstitial fibrosis, renal calcinosis and had elevated TGF-β1 compared to wild-type mice, and that high citrate food feeding protected renal function and delayed the progression of renal disease (Cebotaru et al., 2005). In contrast, Yang’s group showed that mice with ClC-5 upregulation using a specialized adeno-associated virus vector largely protected against the development of renal fibrosis and inflammatory lesions after unilateral ureteral obstruction (UUO), and restored E-cadherin synthesis and reduced vimentin, α-SMA, and collagen fibril expression in the renal cortex (Yang et al., 2019b). Moreover, in renal tubular epithelial cells, ClC-5 overexpression prevented the epithelial-to-mesenchymal transition induced by TGF-β1 and matrix metalloproteinase-9 (MMP-9) expression (Yang et al., 2019b). Matrix metalloproteinases (MMP) are a large class of proteins that require metal ions as active forms of cofactors and have different roles in different pathological conditions (Zitka et al., 2010). MMP-9 has been shown to be critically involved in the pro-fibrotic microenvironment in the obstructed kidney by promoting growth factor release and communication between the epithelial and interstitial compartments (Tan et al., 2010; Wang et al., 2019a). ClC-5 expression inhibits immune cell infiltration and inflammatory cytokine release and ameliorates renal fibrosis by inhibiting NF-κB-mediated activation of MMP-9 pathway signaling (Yang et al., 2019b) (see Figure 1).
Interestingly, MMP9 appears to have a strong positive correlation with ClC-3, which regulates the extracellular environment and promotes the migration and invasion of cancer cells through multiple pathways of upregulation of MMP9 expression (Guan et al., 2016; Wang et al., 2017; Guan et al., 2019). ClC-3 and ClC-5 are very similar in basic biophysical properties, both of them are expressed intracellularly in almost all cell types and have been shown by several studies to be actually Cl−/H+ reverse transporter proteins, but ClC-3 is more widely distributed compared to ClC-5 (Steinmeyer et al., 1995). In human keratinocytes and human fetal lung fibroblasts, ClC-3 overexpression showed significantly more α-smooth muscle actin (α-SMA) expression as well as increased myofibroblast activation (Yin et al., 2008), suggesting a role for ClC-3 in fibroblast transformation. It is well known that renal oxidative stress production is the key to the development of renal fibrosis. Although ClC-3 is mainly expressed in endosomes and lysosomes, extracellular production of superoxide flux can mediate intracellular signaling through plasma membrane ClC-3 channels, further activating the production of mitochondrial reactive oxygen species (ROS) (Hawkins et al., 2007; Jha et al., 2016). However, the exact role and mechanism of ClC-3-mediated renal oxidative stress and fibrosis remains unclear. Because ClC-3 or ClC-5 are expressed primarily on organelle membranes, it is difficult to record their currents, and thus many of their proposed roles in a variety of biological processes may need to be reevaluated, although it is an interesting and relatively unexplored target in renal fibrosis.
It is worth mentioning that other ClC members, such as ClC-2 channels promote the migration transition of myofibroblasts and ECM synthesis (collagen I and fibronectin) via PI3K/Akt signaling (Sun et al., 2016), and that deletion of ClC2 alters the integrity of adherens junctions, leading to the release of membrane-bound β-catenin and activation of Wnt target genes (Jin et al., 2018a).
2.4 Volume-regulated anion channel (VRAC)
VRAC is an important anion channel that regulates cell volume in response to swelling stress, and was initially identified primarily as a way for cells to release Cl− ions or specific organic osmolytes, such as halides or glutamate, followed by osmotic water, during regulated volume reduction (RVD) (Pedersen et al., 2015; Jentsch, 2016). Under these conditions, the Cl− current through the VRAC, named IClswell, displays a slight outward rectification and is independent of voltage activation and time (Schlichter et al., 2011). Recent studies have identified the leucine-rich-repeat-containing 8A (LRRC8A), a hexamer consisting of four transmembrane helices per subunit, was an essential contributor to VRAC current triggering. To show functional gating selectivity and volume sensitivity, LRRC8A was usually found to act in conjunction with at least one other LRRC8 member (LRRC8B-E) (Voss et al., 2014; Gaitan-Penas et al., 2016). A multitude of studies have proposed that LRRC8A is involved in a variety of pathophysiological processes, including cell apoptosis, proliferation, migration, metabolism and secretion, all of which involve changes in local cell volume (Chen et al., 2019a). VRAC facilitate cell survival under hypotonic conditions as detailed above. Increased cell volume has been reported to lead to increased membrane tension, activation of mechanosensitive and Ca2+ permeable channels, leading to cell damage and death, and increased deposition of matrix proteins (Warntges et al., 2001). Also, increased proximal tubular hyperosmotic stress responds to mechanical stress and osmotic pressure, rearranging focal adhesions in tubular epithelial cells to induce EMT, which in turn leads to renal fibrosis (Miyano et al., 2021). Regardless of hypo- or hyperosmotic stress conditions, cell volume recovery is critical for homeostasis and renal function of the organism. However, the mechanism between increased cell volume and VRAC activation is not yet fully understood, which may be partly through downstream signaling or sensing changes in intracellular ionic strength (Voets et al., 1999; Syeda et al., 2016). Also, the actin cytoskeleton may be involved in this process. In RVD, the actin cytoskeleton (filaments) depolymerizes during cell swelling, which also stimulates microtubule expression and is involved in VRAC stimulation (Burow et al., 2015). The functional integrity of actin filaments and microtubules is a prerequisite in maintaining the effective RVD responses. Additionally, disruption of the cytoskeleton of glomerular and tubular cells, especially of the actin and microtubule network, is strongly associated with pro-fibrotic effects (Parrish, 2016). Therefore, we hypothesized that VRAC may have an important role in mediating cytoskeletal protein-driven renal fibrosis through regulation of cell volume.
In addition, in the absence of osmotic challenge, other stimuli such as hypoxia, serum, intracellular Ca2+, ATP, phospholipids and other intracellular signals have been reported to stimulate VRAC (Patel et al., 1998; Catacuzzeno et al., 2011; Kunzelmann, 2015). Friard et al. (2019) found that TGF-β1 also activated VRAC/LRRC8A channels to trigger chloride currents, but at a relatively slow rate and with weak current amplitude. Using a biochemical or pharmacological approach, they further showed that inhibition of VRAC/LRRC8A attenuated TGF-β1-induced expression of the EMT phenotype and associated markers in human proximal tubular epithelial cells, and that this mechanism may be related to its ability to permeabilize glutathione (GSH) and thus counteract increased intracellular ROS levels (Friard et al., 2019). Studies on the association of volume-sensitive Cl− -channels in renal tubular epithelial cells may provide interesting models for better understanding the process of renal epithelial mesenchymal transition under stress. Furthermore, establishing a new approach based on VRAC/LRRC8A for the treatment of renal fibrosis has great potential, which requires more research.
2.5 Voltage-dependent anion channels
The voltage-dependent anion channel (VDAC) is a multifunctional channel that controls cellular energy, metabolism, oxidative stress and apoptosis (Shoshan-Barmatz et al., 2010). They are a class of bidirectional transport porins located in the mitochondrial outer membrane different from classical ion channels. Three types of VDAC (VDAC1-3) have been identified in higher eukaryotes, of which VDAC1 is highly expressed in most cell types. Nowak et al. (2020) recently found that VDAC1 expression was reduced in mitochondria of an ischemia/reperfusion (I/R) kidney injury mouse model compared to WT mice, and that VDAC1 deficiency resulted in reduced mitochondrial respiration and ATP levels with increased mitochondrial fission in non-injured kidneys, supporting the role of VDAC1 in maintaining mitochondrial dynamics and energy metabolism. Importantly, the overall absence of VDAC1 impedes the morphological regeneration of proximal tubules and the recovery of renal function, increases collagen deposition in the post-ischemic kidney and exacerbates interstitial renal fibrosis (Nowak et al., 2020). In contrast, Li et al. (2022b) found that knockdown of VDAC1 significantly attenuated ischemic injury-induced apoptosis and mitochondrial damage in renal tubular cells. This discrepancy may be related to complex mechanisms, differences in disease and cell types. Interestingly, Tf-D-LP4, a peptide targeting VDAC1, was found to be effective in treating nonalcoholic fatty liver disease (NAFLD), reducing inflammation, liver fibrosis, and normalizing liver enzymes (Pittala et al., 2019), and VBIT-4, an inhibitor of VDAC1 oligomerization, attenuates fibrosis caused by increased cardiac aldosterone (Klapper-Goldstein et al., 2020). Thus, VDAC1 appears to be a promising treatment for fibrotic disease. Although the role of VDAC1 in different renal diseases has been established, valuable evidence to support the claim that VDAC perturbation causes or exacerbates renal fibrosis remains to be determined.
2.6 Chloride intracellular channels
Chloride intracellular channel 4 (CLIC4) belongs to a highly conserved and most extensively studied member of the CLIC family of proteins (Shukla and Yuspa, 2010). It is present in the inner cell membrane and abundant in the cytoplasm as the soluble form. CLIC4 is involved in various cellular functions, including regulation of cell proliferation, apoptosis, pH homeostasis, cell cycle, angiogenesis and differentiation (Shukla et al., 2009; Shukla and Yuspa, 2010). In the kidney, CLIC4 null mice support vacuolar acidification and are associated with abnormal dilatation of proximal tubules (Ulmasov et al., 2009). Using biochemical and morphological analysis, CLIC4 was reported to be overexpressed in the proximal tubular region of rats with hypertensive nephropathy compared to normal rats (Hatziioanou et al., 2018), inhibition of CLIC4 largely reduced TGF-β1-induced transdifferentiation of fibroblasts from myofibroblasts and α-SMA as well as ECM component expression (Shukla et al., 2014). Indeed, CLIC4 directly enhances TGF-β signaling by binding to Smad2/3 and preventing their dephosphorylation (Shukla et al., 2009). Furthermore, CLIC4 is able to regulate the matrix degradation activity of MMP-14 (Hsu et al., 2019), which has been shown to be a relevant mediator of vascular senescent renal fibrosis (Vasko et al., 2014). We are looking forward to new research in this area as it is an interesting and relatively unexplored target in renal fibrosis.
2.7 Claudin proteins-associated channels
The paracellular channel through the tight junctions are an important pathway for transepithelial Cl− transport in the kidney, which provides approximately 70% of Cl− reabsorption (Sansom et al., 1984; Schild et al., 1988). The claudin proteins (CLDNs), forming the channels that connect two extracellular compartments by passing perpendicular to the membrane plane, are the main components of tight junction generation (Hou et al., 2013). Paracellular permeability depend on the complement of CLDNs in each nephron segment, where they are expressed in a nephron-specific manner. The localization of CLDNs in the nephron varies among mammals, with several CLDNs normally expressed in the renal tubule (2, 10a, 17), podocytes (5, 6) and collecting ducts (3, 4, 7 and 8) (Hou et al., 2013). Claudins are reported to be altered during cellular EMT and are involved in the feedback regulation of epithelial cell phenotypic transformation (Quaresma et al., 2020). Dan et al, (2019) showed that CLDN-2 expression was initially increased and then decreased in a mouse obstructive nephropathy model, and silencing of CLDN-2 enhanced Ras homolog family member A (RHOA)-mediated activation of myocardin-related transcription factor (MRTF), a major regulator of EMT, to promote epithelial reprogramming in renal fibrosis. In addition, CLDN5-specific deletion in podocytes significantly exacerbated interstitial renal fibrosis after UUO by blocking WNT inhibitory factor-1 (WIF1) secretion (Sun et al., 2022). Therefore, it is possible that deletion of CLDNs contributes to the pathogenesis of renal fibrosis. See Table 1 for a full listing of ion channels in renal fibrosis.
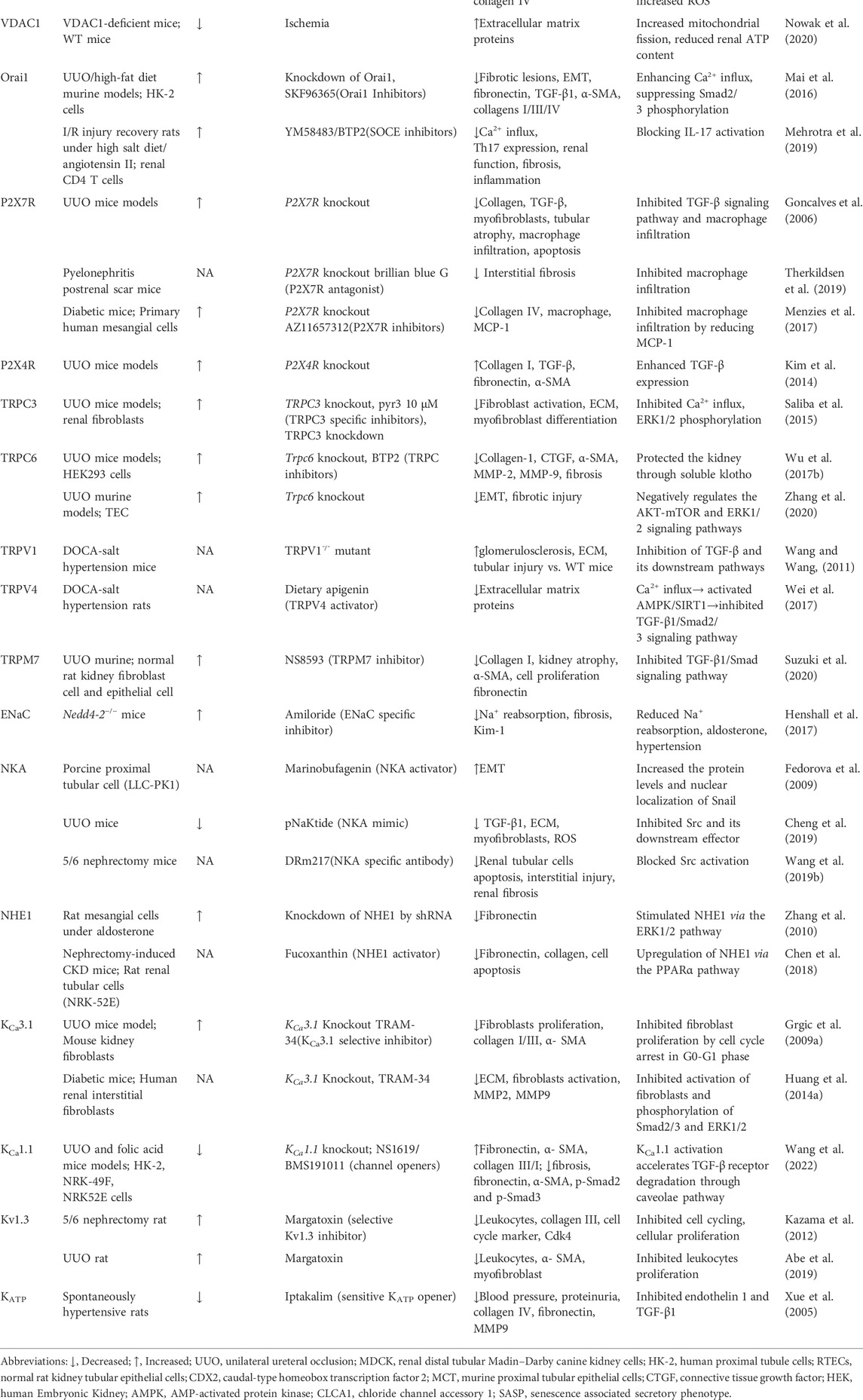
TABLE 1. The expression and function of various ion channels for renal fibrosis.
3 Ca2+ channels
Ca2+ signaling is a key determinant of homeostasis and cellular function. As a ubiquitous and essential second messenger, the cytosolic Ca2+ is involved in the regulation of various cellular functions, such as cell activation, proliferation, development, differentiation, survival, homeostasis and effector functions (Berridge, 2016). In the kidney, Ca2+ is also involved in the endocrine regulation of renal blood flow, glomerular filtration, and tubular handling of water and electrolytes. Dysregulation of Ca2+ signaling is often thought to play an important role in the development of renal diseases such as polycystic kidney, acute kidney injury, glomerular disease, and diabetic nephropathy (Yang and Yang, 2013; Ning et al., 2021). In addition, critical regulation of intracellular Ca2+ levels during driving EMT is necessary for the translation of extracellular signals into gene expression effects and execution of cellular behavior. In this scenario, Ca2+-permeable ion channels that regulate Ca2+ signaling can have a dramatic impact on cellular phenotypes. This review will discuss the role of Ca2+-release-activated Ca2+ channels (CRACs), purinergic P2 receptors, and transient receptor potential (TRP) channels in the process of renal fibrosis.
3.1 Ca2+ release-activated Ca2+ channels
The influx of extracellular Ca2+ or the release of intracellular Ca2+ stores (about 90% stored in the endoplasmic reticulum (ER) and mitochondria) is the main source of increased cytoplasmic Ca2+ levels (Prakriya and Lewis, 2015). As a major pathway mediating critical Ca2+ entry in several non-excitable and excitable cells, store-operated Ca2+ entry (SOCE), triggered by Ca2+-dependent depletion in the ER through the store-operated Ca2+ (SOC) channel (mechanistically we also call Ca2+ release-activated Ca2+ channel (CRAC), has been explored in detail. In this process, two highly conserved molecular proteins play a decisive role: one is stromal interaction molecule (STIM) responsible for sensing ER Ca2+, including STIM1 and STIM2, and another is Orai, the store-operated channel protein, divided into three subtypes, Orai1 to Orai3, which are all important components of SOCE. Specifically, when the inositol 1,4,5-trisphosphate receptor (IP3) receptor is activated, the intracellular space in the ER is depleted by Ca2+ release, which is sensed by STIM1 and subsequently relocated by binding to the microtubule plus-end binding protein EB1 (shifted to the Orai1 protein in the plasma membrane), activating CRAC and thus facilitating Ca2+ influx (Prakriya and Lewis, 2015; Chen et al., 2019b). SOCE plays an important role in regulating cell migration, proliferation, apoptosis, gene regulation, and secretion (Kim et al., 2011) (see Figure 2).
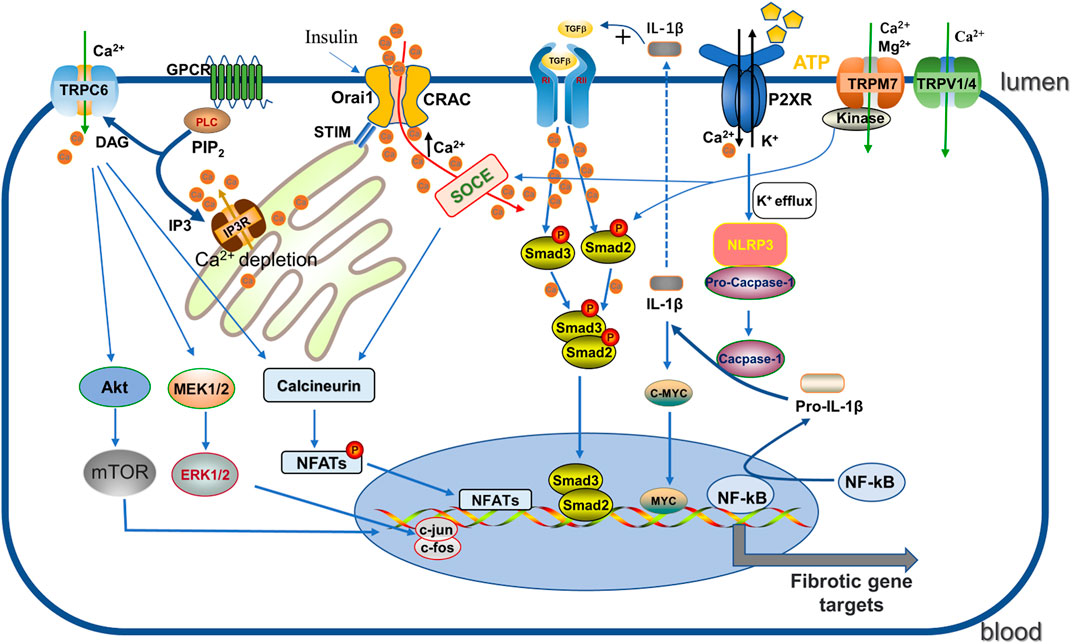
FIGURE 2. Regulation of CRAC, P2XR, TRP channels. (1) GPCR activates PLC, then further decompose the phosphorylated PIP2 into DAG and IP3. IP3 acts on IP3R on the ER, triggering Ca2+ release leading to depletion, which is sensed by STIM and then binds and activates the ORAI pore-forming channel CRAC, facilitating Ca2+ entry. Enhanced Ca2+ promotes the phosphorylation of Smad2/3 to enhance the TGF-β1 signaling pathway, and binds to calcineurin and promotes NFAT activity. (2) Activated P2X7R induces NLRP3 inflammasome activation by K+ efflux and catalyzes pro-IL -1β cleavage to IL-1β, which not only promotes the TGF signaling pathway but also enhances c-myc transcriptional enhancement of fibrosis. (3) TRPC6 promotes the fibrotic process by enhancing AKT-mTOR and ERK1/2 pathways as well as the calcineurin/NFAT pathway. DAG: diacylglycerol; GPCR: G protein coupled receptor; IP3: 1, 4, 5-triphosphoinositol; IP3R: inositol 1,4,5-triphate receptor; PIP2: phosphorylated phosphatidylinositol 4, 5-diphosphate; PLC: phospholipase C; NFATs: nuclear factor of activated T cells; IL-1β: Interleukin-1β; NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing-3.
STIM/Orai-dependent SOCE is closely associated with the progression of renal fibrosis. It was shown that Orai1 expression was upregulated in the kidneys of UUO and high-fat diet (HDF)-induced renal fibrosis mouse models and in renal tubular epithelial cells from kidney biopsies of patients with fibrotic nephropathy, such as focal proliferative sclerosis and tubulointerstitial nephritis (Mai et al., 2016). Investigations in HK2 cells revealed that TGF-β1-driven EMT as well as fibronectin and α-SMA expression were significantly reduced by Orai1 silencing that was attributed to the prevention of abnormal Ca2+ influx and inhibition of Smad2/3 phosphorylation (Mai et al., 2016; Ma et al., 2018). The renoprotective effect of Orai1 deficiency was similarly confirmed in vitro experiments in UUO mice and high-fat fed ApoE−/− mice (Mai et al., 2016). Studies on podocytes from diabetic nephropathy mice suggest that specific Orai1 deletion prevents insulin-stimulated SOCE, slit-diaphragm disruption and proteinuria, possibly due to chronic stimulation of Orai1 activation or aberrant Ca2+ signaling, which in turn activates Ca2+-regulated phosphatases leading to remodeling of the actin cytoskeleton (Kim et al., 2021). Furthermore, using siRNA to down-regulate STIM1 expression in cultured podocytes from diabetic nephropathy rats serum could reverse the decrease in autophagy and inhibit EMT by restoring Ca2+ homeostasis (Jin et al., 2018b; Jin et al., 2019). Spontaneously hypertensive rats with truncated STIM1 exhibit albuminuria, glomerular and interstitial fibrosis (Dhande et al., 2020). Notably, in cultured human mesangial cells, activation of glucagon-like peptide-1 receptor (GLP-1R) or thapsigargin on SOCE inhibited high glucose- and TGF-β1-stimulated matrix protein synthesis (Wu et al., 2015; Huang et al., 2019). In contrast, knockdown of Orai1 using targeted nanoparticle siRNA delivery significantly increased fibronectin and type IV collagen expression in mesangial cells as well as mesangial expansion by a mechanism running through inhibition of pre-fibrotic Smad1 and Smad3 phosphate activation (Wu et al., 2017a; Chaudhari et al., 2017). This suggests that SOCE appears to have complex functions in mesangial cells, with different functions in response to high glucose or high fat stimuli in different cell types and technical approaches. Collectively, these results support that STIM/Orai-dependent SOCE may be an important therapeutic approach to study the progression of renal fibrosis.
Strong T helper 17 (Th17) cells inflammatory response exacerbates ischemia-reperfusion-induced acute kidney injury (AKI) and mediates inflammatory cell infiltration promoting renal fibrosis (Peng et al., 2015; Mehrotra et al., 2017). In studies of Th17 differentiation in AKI rats, Mehrotra et al. (2019) revealed the important role of orai1-dependent SOCE involved in Th17 differentiation and inflammatory responses during AKI and AKI to CKD transformation. This was manifested as Orai1 mutation leading to Th17 cell damage, and almost no IL-17 was expressed in Oria1−/− cells, whereas stimulated IL-17 expression was inhibited by SOCE antagonist. In addition, Orai1 was consistently expressed in CD4 T cells after recovery from AKI and the use of the SOCE pathway inhibitor (YM58483/BPT2) significantly attenuated ischemia-reperfusion kidney injury, which was associated with reduced Th17 cells. Similarly, YM58483/BPT2 attenuated the progression of inflammation, proteinuria and interstitial fibrosis induced by exposure to angiotensin II or high salt diet I/R recovery in mice, which is thought to be reactivated by Th17 cells (Mehrotra et al., 2019). Thus, continued Orai1 expression may underlie the susceptible activation of Th17 cells, suggesting that Orai-mediated Ca2+ mechanisms may be an attractive therapeutic target against CKD progression or immune-mediated inflammatory renal fibrosis.
3.2 Rurinergic P2 receptors
Adenosine 5′-triphosphate (ATP) is not only an important intracellular energy carrier, but its extracellular release also has a role as a signaling molecule (Linden et al., 2019), for example, as a damage-associated molecular pattern (DAMP) signal involved in the inflammatory response to kidney tissue injury (Arulkumaran et al., 2013). ATP signaling acts through purinergic P2 receptors, including metabolic P2Y and ionotropic P2X, to participate in intracellular Ca2+ regulation, fluid secretion, glomerular filtration rate and epithelial transport (Ralevic and Burnstock, 1998; Vallon et al., 2020). P2Y receptors have eight subtypes (P2Y1, 2, 4, 6 and 11–14) and act as a class of G protein-coupled receptors that initiate second messenger cascade signaling by binding ATP, thereby increasing intracellular Ca2+ release (Geyti et al., 2008), and the P2X receptor family has seven subtypes (P2X1∼7), which are widely distributed in various cell types and form permeable Ca2+ non-selective cation channels. In renal tubular epithelial cells, stimuli triggered by a range of factors including cellular mechanical stretch (e.g., increased sensory blood flow and tubular fluid flow), pathogen invasion, cell injury, or agonist binding can induce the release of ATP into the extracellular space, activating the P2X receptor and allowing the influx of Ca2+, Na+, and other cations, thereby triggering biological effects (Monaghan et al., 2021). P2X7R and P2X4R are key subtypes in the pathophysiology of the kidney, while they share some commonalities including structural commonalities and physical and functional interactions but may even have opposite properties (Craigie et al., 2013).
P2X7R was initially identified in macrophages and monocytes and plays an important role in the regulation of pro-inflammation and pro-apoptosis (Arulkumaran et al., 2011). Persistent interstitial inflammation drives worsening kidney injury, fibrosis and functional impairment. P2X7R is involved in the rapid processing and release of cytokines IL-1β and IL-18 and promotes apoptosis and necrosis, which is associated with the activation of the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing-3 (NLRP3) inflammasome (Baron et al., 2015). Recently, it has been observed that P2X7R serves as a possible target with potential pro-fibrotic function in several types of fibrotic diseases (Gentile et al., 2015). The distribution of transient expression of P2X7R was identified in fibrotic kidneys of UUO-induced mouse model (Goncalves et al., 2006), whereas P2X7R is usually expressed at low levels in normal kidneys (Turner et al., 2003). Using genetic tools, P2X7 knockout mice showed significantly reduced renal interstitial macrophage infiltration and myofibroblast numbers as well as TGF-β1 expression compared to wild-type mice in a UUO model (Goncalves et al., 2006). Consistent with this finding, Therkildsen et al. studied sporadic renal cortical fibrosis in mice exposed to E. coli producing the virulence factor alpha-hemolysin (HlyA), and the functional significance of P2X7R was demonstrated by using the most potent antagonist, brillian blue G, which significantly reduced fibrosis and macrophage infiltration in mice with renal scarring caused by pyelonephritis (Therkildsen et al., 2019). These results suggest that deletion of P2X7R effectively inhibit renal fibrosis after obstruction and infection, and macrophage infiltration seems to be an important part of the process.
In diabetic patients, extensive renal expression of P2X7R is associated with severe mesangial expansion and fibrosis (Menzies et al., 2017). P2X7R activation is involved in TGF-β secretion and ECM production in mesangial cells (Solini et al., 2005), in turn P2X7R deficiency prevents glomerular macrophage accumulation and collagen IV deposition (Menzies et al., 2017). Long-term high-fat feeding of mice resulted in renal inflammation and oxidative stress as well as alterations in renal structure, which were associated with NLRP3 inflammasome activation (Stienstra et al., 2011). It was found that P2X7R activation, NLRP3 inflammasome formation, caspase-1 induction, pro-IL-1β, pro-IL-18 activation all increased after “metabolic” renal injury and were all inhibited by P2X7R silencing (Solini et al., 2013). IL-1β activates autophagic flux to promote transcription factor MYC accumulation, and up-regulation of MYC target genes are essential for driving progressive renal tubulointerstitial fibrosis (Lemos et al., 2018). Therefore, blocking the axis of P2X7R-NLRP3 inflammasome may be an important target to protect kidney function from fibrosis progression. This mechanism has also been validated in the UUO model (Nam et al., 2019) (see Figure 2). Interstitial fibroblasts in early kidney injury contribute to kidney repair. After acute kidney injury, ATP released from necrotic tubular cells rapidly acts on adjacent interstitial fibroblasts by binding to P2X7R, inducing cell death (Ponnusamy et al., 2011). P2X7 receptor blockade not only reduces interstitial fibroblast loss but, interestingly, also promotes renal recovery by reducing the infiltration of innate and adaptive effector cells, increasing the infiltration of regulatory T cells (Tregs) during recovery, and delaying renal fibrosis and scar formation (Ponnusamy et al., 2011; Koo et al., 2017). Thus, these reports confirm that P2X7R plays a key role in the development and progression of renal fibrosis, providing a critical target and direction for the treatment of renal fibrosis.
In contrast to P2X7R, P2X4R has a nephroprotective effect in the UUO mouse model (Kim et al., 2014). Vallon et al. (2020) speculated that the reason may be related to the fact that P2X4R controls T cell migration and induces its toxic function subsequently promoting fibroblast apoptosis. However, the role of P2X4R in fibrosis progression needs to be carefully understood, as ATP-P2X4 signaling can likewise induce activation of the NLRP3 inflammasome, exacerbating the progression of renal tubulointerstitial inflammation and fibrosis (Chen et al., 2013; Han et al., 2020).
3.3 TRP channels
Transient receptor potential (TRP) channels are a large family of Ca2+ permeable channels that are widely expressed in a variety of cells types. Since the original TRP genes appeared in Drosophila, at least 27 TRP genes have been identified in mammals (Wu et al., 2010). As an important pathway for Ca2+ to flow into the cytoplasm, TRP channels regulate Ca2+ to depolarize cell membranes, change enzyme activities, and achieve signal cascade response (Zheng, 2013). Based on differences in their amino acid sequences and topologies, TRP channels can be divided into seven subfamilies (canonical TRPC, vanilloid TRPV, melastatin TRPM, polycystin TRPP, ankyrin TRPA, mucolipin TRPML, and NompC-like TRP) (Nelson et al., 2011). Although TRP channels belong to the non-voltage gated Ca2+ superfamily, they share similarities in structural features, including six transmembrane segments (S1-S6) as well as intracellular NH2 and COOH terminations and a pore lining (composed of S5 and S6), which subunits complement each other to form a tetramer (Hardie, 2011). As an integral membrane protein of cells, TRP channels integrate multiple stimuli including temperature, smell, pH, pressure, mechanical stimuli, chemical reagents, herbs as well as poisons and respond to intracellular Ca2+ signaling, participating in a variety of pathophysiological processes (Vriens et al., 2008; Moran et al., 2011). Several studies have linked different fibrotic disease types, including renal fibrosis, to TRP channels (Inoue et al., 2019; Okada et al., 2021). Here, we will describe a few typical examples of the role of TRP channels in renal fibrosis, due to the large size of the TRP channel family and limited space.
TRPC3/6/7 is a constitutively active receptor-operated channel that can be regulated by various signaling molecules such as phosphatidylinositol 4,5-bisphosphate (PIP2), diacylglycerol (DAG), ATP, calmodulin and reactive oxygen species (Lemonnier et al., 2008). These TRPC proteins can be activated by stretching under mechanical stress and amplify downstream cellular signals coupled by the integrated stimulus via calcium permeation and membrane depolarization (Scott et al., 2006). The potential role of TRPC3 and TRPC6 in renal fibrosis has been studied in an animal model of UUO induction, and both of them are upregulated in the obstructed kidney (Saliba et al., 2015; Wu et al., 2017b). TRPC3 channels, accompanied by direct Ca2+ influx, have been shown to promote renal fibroblast proliferation, myofibroblast differentiation, and extracellular matrix remodeling upon the DAG and the DAG generating angiotensin II (Ang II), which was notably reduced by the TRPC3 channels inhibitor pyr3 or siRNA knockdown (Saliba et al., 2015). The mechanism underlying increased interstitial fibroblasts proliferation and inflammation may involve Ca2+ entry via TRPC3, which activates and phosphorylates extracellular signal-regulated kinase (ERK1/2), a major regulator of the cell cycle (Saliba et al., 2015). TRPC3−/− mice exhibit diminished renal injury, inflammation, and protection against UUO-induced renal fibrosis (Saliba et al., 2015). The contribution of TRPC3 to the fibroblast fibrogenic response may also be related to its physical interaction with NADPH oxidase 2 (NOX2), a membrane-bound reactive oxygen species (ROS)-producing enzyme (Numaga-Tomita et al., 2017).
TRPC6, as described above, is considered to be another key TRPC channel subtype in the progression of renal fibrosis. The functional significance of TRPC6 channels in pathogenesis of renal diseases, including focal segmental glomerulosclerosis (FSGS), diabetic nephropathy, immune-related nephropathy and chronic kidney disease was extensively studied [more details see the review (Hall et al., 2019)]. TRPC6 expression is significantly increased in UUO induced fibrotic mice as compared to normal renal tissue (Wu et al., 2017b), and also activated under ROS produced by NADPH oxidase (NOX) as TRPC3 (Kim et al., 2013; Ilatovskaya et al., 2018). Directly tested for its involvement in the activation of EMT by AKT-mTOR and ERK1/2 pathways in UUO mouse kidneys, BTP2 (a nonselective TRPC6 inhibitor) or TRPC6 knockdown was found to blunt this effect (Kong et al., 2019; Zhang et al., 2020) (Figure 2). Blockade of TRPC6 with another selective inhibitor BI-749327 observed similar protection in obstructed kidneys (Lin et al., 2019). Indeed, TRPC6-dependent elevation of cellular solute Ca2+ activates calcineurin, which stimulates the nuclear factor of activated T cells (NFAT) transcriptional pathway to induce fibroblast differentiation (Davis et al., 2012; Lin et al., 2019; Gu et al., 2020). Studies have demonstrated that TRPC6 and NFAT form a mutual positive feedback loop that aggravates the renal fibrosis (Nijenhuis et al., 2011). TRPC6 channel also affects components of the innate immune response. It has been reported that TRPC6 channels regulate CXC chemokine receptor 2 (CXCR2)-related chemotaxis by mediating Ca2+ influx (Lindemann et al., 2013), promoting renal tubular cell senescence and renal fibrosis by inducing mitochondrial dysfunction (Meng et al., 2022). Interestingly, Klotho, a single-channel type 1 transmembrane protein with anti-renal fibrotic effects, had no effect on obstruction-induced fibrosis in TRPC6 knockout mice, suggesting that the renal protective effect of Klotho in UUO is partly mediated through inhibition of TRPC6 (Satoh et al., 2012; Wu et al., 2017b). In diabetic nephropathy, excessive activation of TRPC6 channel activity plays an important role in podocyte apoptotic injury (Staruschenko et al., 2019), and Wnt/β-catenin signaling pathway may be active in this process (Li et al., 2013). Notably, TRPC6 knockdown had no effect on tubulointerstitial inflammation and fibrosis in autoimmune glomerulonephritis and aging rats, although it significantly reduced glomerular sclerosis (Kim et al., 2019; Kim and Dryer, 2021), indicating that targeting TRPC6 may have disease specificity in the treatment of glomerular diseases. Overall, the understanding of TRPC3 and TRPC6 in renal fibrosis pathogenesis has expanded in recent years and may facilitate the development of emerging therapeutic strategies.
The TRPV1 channel subtype is a polymodal cation channel involved in cellular environmental crosstalk and has emerged as an important player in the regulation of related diseases such as inflammation, cancer and immune diseases through the integration of physical or chemical stimuli (Bujak et al., 2019). TRPV1 is abundantly expressed in renal tissues, especially in the renal pelvis, and is involved in the regulation of renal hemodynamics and excretory function (Feng et al., 2008; Li and Wang, 2008). Capsaicin activation of TRPV1 greatly alleviated renal fibrosis in UUO and hyperadenine-fed mouse models by reducing myofibroblast activation and preventing phenotypic alterations in renal tubular epithelial cells, and this mechanism is associated with inhibition of TGF-β1-Smad2/3 signaling (Liu et al., 2022). In a mouse model of deoxycorticosterone acetate (DOCA) -salt induced hypertension, TRPV1 knockdown exaggerated renal injury including renal cortical tubulointerstitial injury, fibrosis, and macrophage infiltration, accompanied by increased ECM protein and activation of TGF-β signaling pathway (Wang et al., 2008; Wang and Wang, 2011). In addition, the nephroprotective effect of TRPV1 activation has also been demonstrated in models of diabetic nephropathy and ischemic renal injury (Chen et al., 2014; Wei et al., 2020). Interestingly, TRPV1 knockout mice exhibit a younger metabolism as well as a longer lifespan, predicting that TRPV1 is associated with metabolic disorders, obesity, and aging (Riera et al., 2014).
The vanilloid TRPV4 channel appears to act differently in different models of fibrosis. Increasing evidence suggests that key vanilloid TRPV4 channel subtypes regulate myofibroblast differentiation in cardiac fibrosis, pulmonary fibrosis by integrating mechanical and soluble signals from ECM stiffness and TGF-β1 (Adapala et al., 2013; Rahaman et al., 2014). However, in hypertensive kidney, Wei et al. found that TRPV4-dependent rise of cytosolic Ca2+ activated by apigenin triggered AMP-activated protein kinase (AMPK)/sirtuin 1 (SIRT1) pathway and further inhibited TGF-β1/Smad2/3 signaling pathway and extracellular protein expression in renal mesangial and tubular epithelial cells. TRPV4 knockdown abolished this beneficial effect of hypertension-induced renal fibrosis (Wei et al., 2017). The underlying mechanism between TRPV4 and renal fibrosis still needs further elaboration.
The melastatin TRPM7 channel subtype is a bifunctional protein comprised of a cation channel segment linked to an α-type protein kinase domain (Takezawa et al., 2004). TRPM7 promotes SOCE enhancement through its kinase function and increases Ca2+-dependent pro-inflammatory and pro-proliferative cytokine effects (Schilling et al., 2014; Faouzi et al., 2017). TRPM7 is significantly upregulated in UUO kidneys compared to normal mice (Suzuki et al., 2020), and its upregulation reportedly contributes to fibrosis (Xu et al., 2015a). Suzuki et al. (2020) showed that inhibition of TRPM7 using NS8593, a small conductance K+ channel inhibitor, directly blocked TRPM7 currents, reduced UUO kidney injury, and attenuated renal fibrosis and atrophy. Also, the TGF-β1/Smad signaling pathway was inhibited in this process. Indeed, Smad2 is a substrate of TRPM7 kinase, and inhibition of TRPM7 reduces the expression of TGF-β1/Smad signaling pathway, which contributes to the reduction of renal interstitial and tubular epithelial cell proliferation and ECM production. TRPM7 channel activity is critical for intracellular Mg2+ homeostasis (Ryazanova et al., 2010), and alterations in Mg2+ metabolism may be associated with TRPM7 downregulation (Sontia et al., 2008). Aldosterone mediates blood pressure-independent renal fibrosis and inflammation via Mg2+-sensitive pathways (Sontia et al., 2008), suggesting that TRPM7 is involved in the pathogenesis of aldosterone-associated renal fibrosis. However, additional fundamental research is needed to identify specific mechanisms of TRPM7 and Mg2+ in renal fibrosis.
4 Na+ transport
Na+ is an important extracellular cation that maintains cellular excitability and is involved in the regulation of water-electrolyte homeostasis, acid-base balance, vascular, neurological, and secretory functions (Greger, 2000). Renal Na+ reabsorption is a precisely regulated and controlled process by multiple physiological mechanisms, and has recently received widespread attention. In particular, mechanistic studies of sodium-glucose co-transport have been used as a promising approach for treatment in diabetic nephropathy, and in addition, Na+ transport is an important pathological mechanism in salt-sensitive hypertension and cystic kidney fibrosis. Na+ channels are usually altered under pathological regulation, for example, TGF-β1-induced EMT is accompanied by an increase in intracellular Na+ and water content, suggesting that Na+ channels may be relevant in mediating the EMT process (Lamouille and Derynck, 2007; Rajasekaran et al., 2010). Here, we focus on the role of the epithelial sodium channel (ENaC), Na+, K+-ATPase (NKA), and Na+-H+ exchangers (NHE) in renal fibrosis.
4.1 ENaC
ENaC is a non-voltage gated amiloride-sensitive ion channel and is involved in maintenance of Na+ homeostasis and fluid balance by controlling Na+ transport in the extracellular fluid of the lumen to epithelial cell (Rotin and Staub, 2021). ENaC is typically composed of structurally similar α, β and γ subunits and formed a heterotrimer expressed in several epithelia, including those of the renal collecting duct, urinary bladder, distal colon, and lung (Hanukoglu and Hanukoglu, 2016). In addition to the regulation of aldosterone sensitivity, extracellular Na+, proteases, lipids, angiotensin and other factors also regulate ENaC activity through complex mechanisms (Rotin and Staub, 2021). Chronic ENaC dysregulation has been shown to be the perfect culprit for salt-sensitive hypertension in humans, given its power to accelerate Na+-induced damage and water movement through the plasma membrane (Mutchler et al., 2021). However. the functional significance of ENaC channels in renal fibrosis independent of hypertension has been less studied.
In pulmonary and cardiac fibrotic diseases, increased ENaC activity has been shown to be an important mechanistic participant (Jia et al., 2018; Duerr et al., 2020). Increased Na+ flux uptake in response to ENaC activation promotes skin fibroblast activation and collagen marker synthesis through the PI3K/Akt signaling pathway, which can be reduced by ENaC blockade (Xu et al., 2015b). However, the deletion of ENaC subunit reportedly causes decreased myogenic self-regulation, leading to renal inflammation and injury with elevated TGF-β1 and collagen III (Drummond et al., 2011), and in turn increased TGF-β1 can reduce ENaC functional activity in epithelial cells (Chang et al., 2008), implying that ENaC decreases in a positive feedback manner in response to renal injury. Li et al. (2007) previously showed that the expression of α, β and γ-ENaC is significantly decreased in the kidney of ureteral obstruction rats when compared to normal tissues, and these changes may lead to the dysfunction of water and Na+ metabolism partly through upregulation of cyclooxygenase-2 (COX-2) signaling (Norregaard et al., 2005). Using the same UUO rat model in another study, Yang et al. demonstrated that breviscapine (a flavonoid derived from the herb) prevented the downregulation of γ-ENaC in the obstructed kidney induced by UUO, and significantly reduced the response to renal tubular interstitial fibrosis (Mei et al., 2016). To date, the exact mechanisms of ENaC regulation and release in renal fibrosis remain uncertain, and there are numerous controversies, particularly regarding ENaC expression in different fibrotic disease states and at different stages of fibrosis development.
ENaC on the plasma membrane of the distal nephron, as mentioned above, is normally regulated by various hormones such as aldosterone. As an important regulator of salt reabsorption by mineralocorticoids, serum- and glucocorticoid-inducible protein kinase1(SGK1) triggers a cascade reaction that phosphorylate NEDD4-2, a Nedd4 family ubiquitin protein ligase, to interacts with ENaC via the carboxy-terminal Pro-Tyr motif on the channel subunit. NEDD4-2 deletion prevents this interaction and serves as a ligand for increased ENaC activity (Harvey et al., 2001). For example, in ADPKD, increased apical ENaC expression and enhanced Na+ reabsorption due to mislocalization of NEDD4-2 are important pathogenic mechanisms (Kaimori et al., 2017). Henshall et al. (2017) showed that renal-tubule-specific NEDD4-2-deficient mice, accompanied by increased Na+ reabsorption, resulted in significant fibrotic kidney injury, inflammation and apoptosis of renal tubular epithelial cells as compared to normal mice, and using amiloride, a specific inhibitor of ENaC, inhibited ENaC activity and reduced the extent of kidney lesions. In addition, NEDD4-2-deficient mice are sensitive to dietary Na+ due to dysregulation of ENaC (Manning et al., 2020), which further drives the progression of renal interstitial injury and fibrosis through activation of Wnt/β-catenin and TGF-β signaling under a high-Na+ diet, and interestingly low Na+ diets can rescue this effect, suggesting an important role of ENaC-regulated Na+ homeostasis in renal fibrosis (Manning et al., 2021) (Figure 3). High levels of Na+ also affect vascular tissue remodeling and fibrosis by impairing phenotypic changes in endothelial cells. Studies have shown that amiloride inhibition of ENaC improves endothelial function, reduces cortical stiffness, and significantly reduces arterial fibrosis and sclerosis in mice (Martinez-Lemus et al., 2017). Moreover, ENaCα subunit deficiency in endothelial cells protects the kidney from ischemic injury by promoting eNOS activation, increasing dependent NO production, and renal perfusion (Tarjus et al., 2019). However, the mechanism by which this elevated Na+ reabsorption by ENaC in renal tubules and endothelial cells causes kidney injury and fibrosis remain to be fully understood.
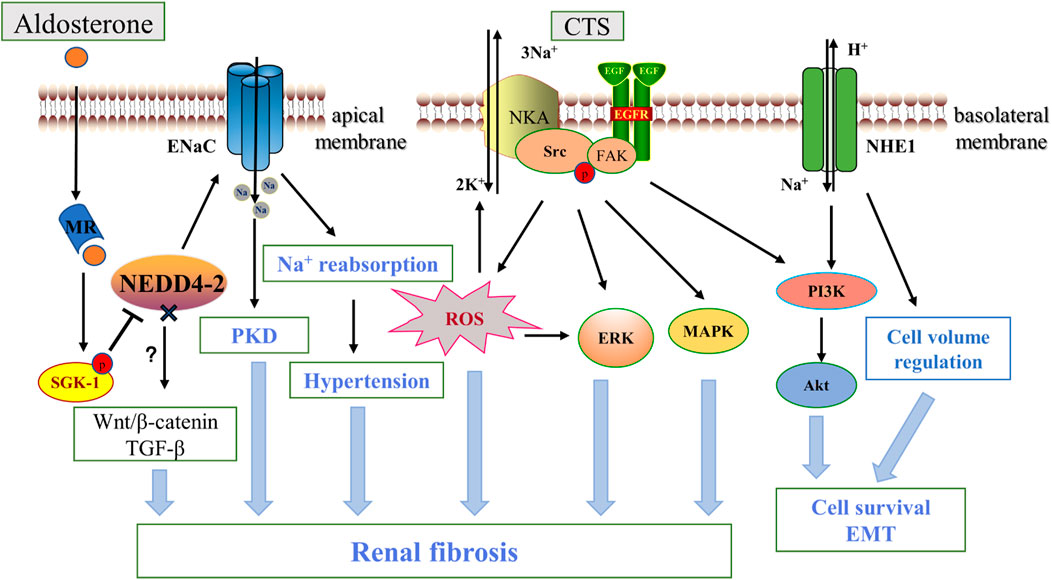
FIGURE 3. ENaC, NKA, NHE1 and renal fibrosis. Aldosterone binds to the intracellular salt corticosteroid receptor (MR), induces the expression of Na+ transport regulators such as SGK1, and phosphorylation inhibits NEDD4-2, which ubiquitinates and degrades ENaC. Increased ENaC activity contributes to PKD disease progression and affects blood pressure through Na+ reabsorption, and NEDD4-2 deficiency also promotes fibrosis via the Wnt/β-catenin and TGF-β pathways. Na+, K+-ATPase acts as a scaffolding protein that interacts with CTS to activate Src and then trans-activates EGFR involving a cascade reaction leading to reactive oxygen species generation, which leads to downstream activation of ERK, AKT, MAPK in addition to further activation of NKA. NHE1 promotes renal tubule survival through PI3K/Akt and regulation of cell volume. In addition, ENaC is located in the apical membrane, while NKA and NHE -1 are mainly located in the basolateral membrane. CTS, cardiotonic steroids; PKD, polycystic kidney disease; EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; FAK, focal adhesion kinase; ERK, extracellular-signal-regulated kinase; MAPK, mitogen-activated protein kinase; PI3K, Phosphatidylinositol 3-kinase.
Furthermore, ENaC dysfunction can promote renal fibrosis by facilitating the development of polycystic kidney disease (PKD), and the mechanism may be related to the elimination of antisecretory absorption and purinergic signaling regulation (Pavlov et al., 2015; Sudarikova et al., 2021). Although inhibition of ENaC such as amiloride is not a routine treatment for hypertension because of its lower efficacy compared to other diuretics, the refracted regulatory mechanism of ENaC in fibrosis cannot be ignored and may become a new tool in the treatment of renal fibrosis.
4.2 Na+, K+-ATPase
Na+, K+-ATPase (NKA) is a cell membrane P-type active cationic hetero-oligomer composed of three subunits, the catalytic α-subunit, the glycosylated β-subunit and the γ-subunit (Feraille and Dizin, 2016). NKA is located primarily in the basolateral membrane and provides the driving force for Na+ reabsorption in the apical renal epithelium. ATP-dependent transport of one pump enables three Na+ outputs in exchange for two K+ entering the cell (Dyla et al., 2020). In addition to acting as an ion transporter protein to maintain cellular ion homeostasis, NKA is also involved in other important cellular processes such as signaling through protein interactions and cell adhesion.
Long-term and systematic studies emphasized the importance of NKA in the pathology of renal fibrosis. Firstly, NKA activity and expression were significantly reduced in renal fibrous tissue from patients with diabetic nephropathy and in the kidneys of acute UUO-induced animal models (Li et al., 2003; Rajasekaran et al., 2010). The cardiotonic steroid (CTS) marinobufagenin (MBG) stimulates NKA signaling has many adverse pathological effects on kidney disease, such as upregulating the expression of the EMT-related transcription factor Snail (Fedorova et al., 2009). Elkareh et al. (2007) also found that MBG stimulates fibroblast collagen synthesis and lead to fibrosis in renal and cardiovascular tissues, which appears to be associated with amplification of ROS production in feed-forward mechanisms via activation of Src-EGFR (Yan et al., 2013). Passive immunization against MBG improves renal function and attenuates renal fibrosis in a model of kidney disease (Haller et al., 2014). In addition, Cheng et al. (2019) found that targeting NKA-mediated signaling with pNaKtide (a peptide inhibiting NKA) markedly attenuated UUO-induced renal fibrosis. They further found that inhibition of Src activation and its downstream ERK1/2, p38 mitogen-activated protein kinase (MAPK) and AKT signaling pathways were the main mechanisms of the antifibrotic effects of pNaKtide. Telocinobufagin is a novel cardiotonic steroid with similar mechanistic effects in the kidney (Kennedy et al., 2018). Further study demonstrated that target-specific NKA was also effective. In a 5/6 nephrectomized rat model, using the NKA antagonist DRm217 alleviated glomerular atrophy and inhibited tubulointerstitial injury and fibrosis (Wang et al., 2019b). Therefore, targeting NKA inhibition is an important target for renal fibrosis (see Figure 3). CD40 receptor activation in the renal tubular epithelium is known to contribute to kidney injury and fibrosis (Haller et al., 2017). Recent studies have shown that knockdown of the NKA α1 heterodimer or loss of the functional NKA/Src cascade complex leads to a decrease in CD40, whereas rescue of the α1 heterodimer restores CD40 expression in renal epithelial cells (Xie et al., 2018), suggesting that NKA is a novel signaling mechanism for CD40 in the pathogenesis of renal injury and fibrosis.
Notably, in renal epithelial cells, RNAi-mediated specific knockdown of NKA-β1 induced loss of fibroblast phenotype, whereas ectopic expression of NKA-β1 reduced TGF-β1-mediated EMT, suggesting that NKA plays an important role in maintaining and shaping a well-differentiated phenotype of epithelial cells (Rajasekaran et al., 2010). Indeed, this may be related to the involvement of NKA-β1 in the assembly of tight junctions and the generation of epithelial cell polarity by including intracellular ion gradients, synergistic effects of E-cadherin, and regulation of MAPK, RhoA GTPase, and stress fibers (Rajasekaran and Rajasekaran, 2003).
4.3 NHE
Na+-H+ exchangers (NHEs) are membrane proteins widely present in mammals and are directly or indirectly involved in maintaining intracellular pH, cell volume regulation, cell proliferation migration and apoptosis (Orlowski and Grinstein, 1997). Nine well-characterized isoforms have been identified, of which NHE1-NHE5 are mainly located on the plasma membranes of different cell types and NHE6-NHE9 are restricted to intracellular organelles (Zhao et al., 2016). All NHE members appear to exist in the form of dimers, although the transfer function is in the form of monomers. NHE1 was the first to be identified, and in the kidney it is commonly expressed in the proximal tubule along the basolateral membrane of polarized epithelial cells (Coupaye-Gerard et al., 1996).
Apoptosis of renal tubular epithelial cells usually leads to tubular atrophy and renal fibrosis associated with CKD progression (Grgic et al., 2012; Schelling, 2016). It is well known that an important feature of apoptosis is a decrease in cell volume. Well-characterized ion channels and transporter proteins such as Cl−/HCO3−-exchanger and the Na+-K+-2Cl− cotransporters (NKCC) are largely not expressed in the proximal tubule, making NHE1 particularly important as a regulatory volume increase (RVI)-mediated defense of renal tubular epithelial cells against apoptosis (Okada et al., 2001). Reduced expression of NHE1 in proximal tubules was noted in ureteral obstruction models (Manucha et al., 2007). A recent study found that fucoxanthin (extracted from brown seaweed) increased NHE1 expression, reduced tubular apoptosis and interstitial fibrosis, and improved renal function in CKD mice. This process involves the peroxisome activated receptor alpha (PPARα) pathway (Chen et al., 2018). The pro-survival effect of NHE1 has been demonstrated in various animal models of kidney disease (Wu et al., 2003; Khan et al., 2006; Manucha et al., 2007). In addition, Hydrogen ion extrusion-induced cytoplasmic alkalinization (Lagadic-Gossmann et al., 2004) and activation of the PI3K/Akt pathway induced by NHE1 (Wu et al., 2004) are equally important pro-survival mechanisms. As late NHE1 activity decreases, sustained apoptotic stimulation overcomes the pre-NHE1 effect, allowing the cells to move toward apoptosis and fibrosis.
In addition to facilitating Na+-H+ exchange and acting as a pro-survival factor, NHE1 is able to act as a molecular scaffolding platform to direct the formation of signaling complexes (Karydis et al., 2009). Indeed, NHEs are common targets of various inflammatory, oxidative stress stimuli and the increase in cell volume itself induces multiple changes in cell function and gene expression through the activation of osmotic signaling pathways (Lang et al., 1998). It was reported that NHE1 expression was stimulated after aldosterone treatment and that NHE1 was able to mediates aldosterone-associated fibronectin accumulation in rat mesangial cells through the ERK1/2 pathway, this direct effect could be ameliorated by shRNA-NHE1 (Zhang et al., 2010). Another study stresses the critical role of NHE-1 in an adriamycin (ADR)-induced glomerulosclerosis rat model, where NHE-1 mRNA expression was significantly enhanced, and high sodium diet accelerated interstitial fibrosis and further increased NHE1 expression, this effect could be prevented by amiloride (although not a specific NHE1 inhibitor) (Okuda et al., 1994). In addition, long-term lithium exposure is associated with chronic interstitial fibrosis, and the use of amiloride to partially reduce NHE1 activity can attenuate lithium-induced interstitial fibrosis, but the exact mechanism is not clear (Kalita-De Croft et al., 2018).
Interestingly, activated NHE1 is required for early cardiac hypertrophy and hepatic stellate cell proliferation in mice, yet its effect on renal fibroblasts has been poorly studied (Benedetti et al., 2001; Mraiche et al., 2011). Thus, we believe that NHE is a promising but understudied new direction for renal fibrosis research, since NHE1 can act as a predictor of fibrosis and targeting NHE1 does not inhibit basal exchange activity, which is the basis of ion homeostasis, but the mechanism needs to be better understood by more studies.
5 K+ channels
K+ channels are the most common and diverse superfamily of ion channels that are widely distributed in a variety of cell types and selectively allow the movement of K+ ions across the cell. K+ channels play an important role in the physiological and pathophysiological processes of cells, are involved in regulating cell proliferation, apoptosis, inflammation, immunity, and epithelial transport across a broad spectrum of the kidney (Welling, 2016). Importantly, K+ channels regulate resting membrane potential and therefore play a key role in cellular excitability. As an important contributor to setting membrane potential, K+ channels regulate cell cycle progression (membrane potential is not constant in the cell cycle, e.g., plasma membrane hyperpolarization occurs between G1 and S phases, while depolarization is necessary for cells to move from G2 to M) and help ensure the driving force of Ca2+ entry to influence cell cycle progression (Urrego et al., 2014). Dysregulation of the renal cell cycle is closely associated with renal fibrosis (Wu et al., 2021), especially renal tubular epithelial cells (TEC), and therefore we can speculate that K+ channels play an important role in renal fibrosis. Furthermore, as previously mentioned, the promotion of K+ efflux through K+ channels promotes the activation of NLRP3 inflammasome, which release inflammatory factors and thereby promote fibrogenesis (Solini et al., 2013; Di et al., 2018). There are generally four functional classes of K+ channels defined based on their structure, biophysical properties, and physiology: Ca2+-activated K+ channels (called KCa), voltage-gated K+ channels (called Kv), and others (inwardly rectifying K+ channels (Kir)and tandem pore domain K+ channels (K2P)channels) (Gonzalez et al., 2012).
5.1 Ca2+-activated K+ channels
As the name implies, KCa channels, a subgroup of K+ channels, are activated by intracellular micromolar Ca2+ and display different single-channel conductance to K+ ions, serving as an important link between cellular Ca2+ and electrical signaling (Rothberg, 2012). KCa channels are widely distributed in almost all cell types where they regulate a variety of cellular functions, including vascular tone, blood pressure, transmitter delivery, cell volume, membrane potential, and proliferation (Stocker, 2004). Based on their single channel conductance, the KCa channels were initially divided into three main types: large conductance Ca2+ -activated K+ channels (BK or KCa1.1), small (SK or KCa2.1–2.3), and intermediate conductance (IK or KCa3.1) (Wei et al., 2005).
Differences regarding the mechanisms of Ca2+ activation have revealed the division of KCa channels into two well-defined classes, as KCa1.1 is usually activated by the synergistic effect of voltage and cytoplasmic Ca2+ increase, while KCa2.1–2.3 and KCa3.1 channels, which share the same Ca2+/calmodulin (CaM)-mediated gating mechanism, are only gated by cytoplasmic Ca2+ increase (Fanger et al., 1999; Kaczmarek et al., 2017). To obtain the properties required for targeted activation, KCa channels are usually positioned close to physiological Ca2+ release sites, such as cell surface, endoplasmic reticulum storage release sites, and ultimately controlled by their switches. When a Ca2+ channel opens, the increased Ca2+ concentration activates all nearby KCa channels. The opening of KCa channels such as KCa3.1 regulates the rapid K+ efflux, leading to a high degree of hyperpolarization of the plasma membrane (a negative shift of the membrane potential towards the K+ equilibrium potential), which in turn increases the electrochemical driving force of Ca2+. Importantly this has been shown to occur during renal fibrosis, with increased KCa3.1 expression reportedly recorded from fibrotic kidneys of patients with diabetic nephropathy compared to normal (Huang et al., 2013), as well as KCa1.1 in fibrotic kidneys of mice (Wang et al., 2022). Here in this review we will discuss the intermediate conductance KCa3.1 channels and the large conductance BK channels that are most relevant to the particular topic.
5.1.1 The KCa3.1 channel
The KCa3.1 channel is a tetrameric trans-membrane protein encoded by the gene KCNN4. Each subunit consists of six transmembrane structural domain with a pore between the fifth and sixth domain. The channel is insensitive to voltage due to the lack of a voltage-sensing structural domain, and is gated solely by internal Ca2+ ranging from 100 to 300 nM (Mcmanus, 1991; Ishii et al., 1997). Unlike KCa1.1 channels (binding directly to the channel), Ca2+ is normally bound to CaM, which is constitutively associated with the intracellular channel through the C-terminus, leading to a conformational change and subsequent opening of the KCa3.1 channel (Fanger et al., 1999). Functionally, KCa3.1 is involved in the regulation of membrane potential, Ca2+ signaling and cell volume in almost all cells. It was found that KCa3.1 channels play an important role in the RVD response in different cell types (Khanna et al., 1999; Wang et al., 2003), deletion of KCNN4 inhibits the ability of T lymphocytes and erythrocytes to regulate osmotic changes in mice (Begenisich et al., 2004), and as mentioned before, activation of KCa3.1 channels and the resulting K+ efflux would cause an exaggerated Ca2+ influx by hyperpolarization/repolarization of the plasma membrane and loss of electric potential against Ca2+ influx (see Figure 4A). KCa3.1-mediated Ca2+ influx is pathologically associated with various diseases including inflammation, atherosclerosis, autoimmune diseases, and cancer, and has been shown to play an important role in promoting mitogenic and proliferative features of tissues (Chou et al., 2008; Feske et al., 2015).
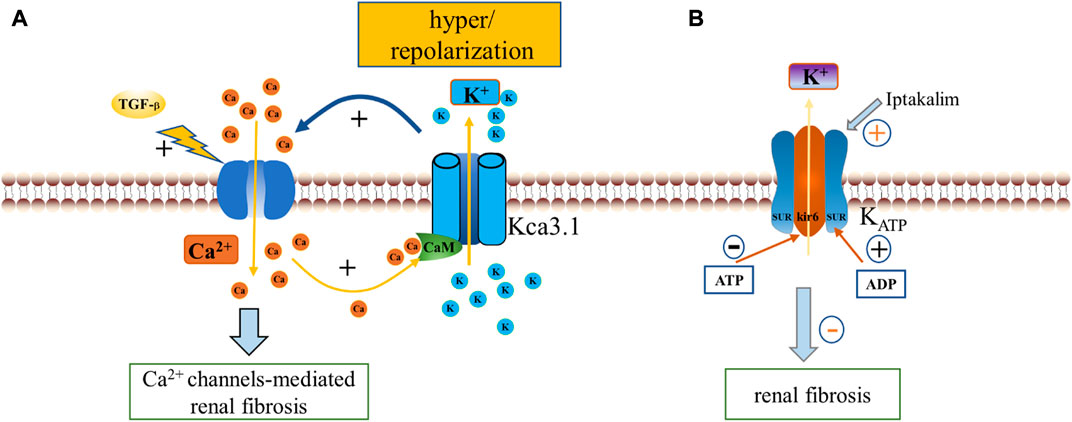
FIGURE 4. Diagram illustrating KCa3.1 channels activity regulating Ca2+ influx-dependent fibrosis pathway and regulation of KATP channels opening. (A). KCa3.1 channels form a positive feedback loop with Ca2+ channels. TGF -β exposure promotes Ca2+ inward flow, and Ca2+ binding to the KCa3.1 channel CAM allosterically opens the channel, creating a membrane hyperpolarization or repolarization that further promotes Ca2+ inward flow, thereby triggering Ca2+ channels-mediated renal fibrosis. (B). ATP binds to Kir6 subunits to inhibit the channel, while KATP opens in response to ADP dissociation at the SUR subunit. The iptakalim (a novel KATP channel opener) can open KATP channels to inhibit renal fibrosis. CAM, calmodulin; KCa3.1, Ca2+-activated K+ channel 3.1; KATP, ATP-sensitive K+ channels.
Recent studies have examined the role of KCa3.1 in mediating renal fibrosis. Grgic and colleagues showed that in mouse renal fibroblasts, mitogenic stimulation with the mitogen basic fibroblast growth factor (bFGF) upregulated KCa3.1 expression, and the KCa3.1-specific inhibitor TRAM-34 suppressed renal fibroblast proliferation in a dose-dependent manner through cell cycle arrest in the G0/G1 phase, which normally requires membrane hyperpolarization and increased Ca2+ inward flow (Grgic et al., 2009a). They also indicated that KCa3.1 was upregulated in fibrotic kidneys of mice induced by UUO, and again KCa3.1-deficient mice or mice treated with TRAM-34 markedly reduced the number of myofibroblasts and delayed renal fibrosis in mice after ureteral obstruction when compared to control mice (Grgic et al., 2009a). Huang et al. (2014a) further demonstrated that blockade of KCa3.1 attenuated renal fibrosis in a diabetic mouse model, and the downregulation of collagen synthesis, α-smooth muscle actin as well as the reduction in fibroblast activation supported this result. Importantly, in human renal interstitial fibroblasts, TRAM34 inhibited cell activation induced by TGF-β1 and also reduced the expression of fibrosis-related genes matrix metalloproteinase-2 (MMP2) and MMP9 (Huang et al., 2014a), suggesting a prominent role of targeting KCa3.1 in reversing renal fibroblast activation.
KCa3.1 plays an important role in TGF-β1 signaling and may be mediated through Smad3, P38, or ERK1/2 phosphorylation pathways (Huang et al., 2014b). Huang et al. (2014b) demonstrated that TGF-β1-induced current through KCa3.1 channels was inhibited by TRAM34 using patch clamp technique in human proximal tubule cells. Actually, TGF-β enhanced KCa3.1 activity, which in turn contributed to the activation of mitogen-activated protein kinase signaling and increased expression of monocyte chemoattractant protein-1 (MCP-1), which is critical to the pathogenesis of renal fibrosis (Huang et al., 2014b). Furthermore, both in human proximal tubule cells and in mouse kidney, high glucose-induced elevation of cytokine CCL20 expression and NF-κB binding activity were significantly associated with KCa3.1 (Huang et al., 2014c), suggesting that cytokine-induced renal injury may be mediated through modulation of K-channel activity. At the immune level, KCa3.1-mediated intracellular Ca2+ influx and membrane potential are required for T cell, macrophage and mast cell migration and inflammatory chemokine and cytokine production (Cruse et al., 2006; Toyama et al., 2008), and inhibition of KCa3.1 using TRAM34 reduces renal fibrosis in diabetic mice by inhibiting the generation of pro-inflammatory cytokines and macrophage infiltration (Huang et al., 2014c). KCa3.1 is also associated with TGF-β1-induced premature senescence and mesangial cell proliferation (Fu et al., 2014).
Taken together, these results suggest that KCa3.1 may represent a highly promising approach for the future treatment of renal tubulointerstitial fibrosis, and that inhibition of KCa3.1 may have more benefits than inhibition of myofibroblasts, possibly through direct effects on a variety of cells including fibroblasts, but also through indirect effects on inflammatory and immune cells.
5.1.2 BK channels
BK channels, also called as KCa1.1, maxiK, slo1, are activated in response to elevated intracellular Ca2+ and membrane depolarization. Structurally BK channels are homotetramers in which the four α-subunits of the pore formation are encoded by the gene KCNMA1 (Lee and Cui, 2010). Each α subunit consists of seven transmembrane regions (S0-S6), with these different structural domains responsible for various functions. The S0 region at the N-terminus on the outer side of the membrane is usually associated with regulatory BKβ subunits, and the S1-S4 regions contain voltage sensors (with several positively charged residues) that make these channels sensitive to voltage. Four structural domains (S7- S10) carrying hydrophobic fragments extend from the intracellular C-terminus. the S7 and S8 domains form the regulator of conductance of K (RCK1) region, while the S9 and S10 regions allow Ca2+ binding in the so-called “Ca2+ bowl” (Cui et al., 2009; Ge et al., 2014). Activated K+ fluxes are variably modulated by intracellular voltage or ligand sensors through pore structures in response to various stimuli, thereby linking cellular signaling and membrane excitability.
A recent study indicates that BK channels play a key role in the prevention of renal fibrosis. Wang et al. (2022) reported that BK protein expression was reduced in renal tissue of UUO- or folate-induced fibrotic mice when compared to control mice, and that BK-deficient mice were more susceptible to renal fibrosis. Pharmacological activation of BK channels effectively prevented the development of renal fibrosis and protected renal function by inhibiting TGF-β/Smad signaling. Their further study of the mechanism revealed that BK channels could accelerate the degradation of TGF-β receptors through caveolae pathway, thereby achieving inhibition of the TGF-β signaling pathway (Wang et al., 2022). Inconsistent with these studies, BK channels appear to mediate glomerular pathological processes in high glucose cultured rat mesangial cells and induce the expression of collagen IV, fibronectin, TGF-β1 and Smad2/3 (Wu et al., 2020). This may be related to BK cell specificity as well as complex mechanisms. Studies on how BK channels alter epithelial cell responses to TGF-β via ion flux or downstream signaling mediators and which are the key cell types remain poorly understood.
5.2 Kv1.3
Kv1.3 is a member of the large and common voltage-dependent K+ channel (Kv) superfamily that is widely expressed in all organisms and is known to consist of several subfamilies (Kv1-4), of which Kv1.1-1.7 has been extensively studied. Kv1.3 was originally identified in T lymphocytes, and consists of four identical α subunits forming a homotetramer, each consisting of six transmembrane structural domains (S1-S6)and a P-loop. Among them, S4 segment is responsible for channel membrane voltage sensing, while S5 and S6 form a central pore structural domain, which has a highly sensitive selectivity for K+ ions (Perez-Verdaguer et al., 2016). Kv 1.3 is involved in a wide range of physiological and pathological processes, including Ca2+ signaling, cell volume regulation, cytokine secretion, energy homeostasis, cell proliferation and migration, and plays an important role in chronic inflammation, cancer progression, autoimmune diseases and other processes (Wulff et al., 2009; Serrano-Albarras et al., 2018; Wang et al., 2019c).
Kv1.3 is a key regulatory protein in the immune response and is highly expressed in activated effector T cells. Selective blockade of Kv1.3 channels modulates Ca2+ signaling influx patterns and inhibits T cell transcription and proliferation, thereby exerting immunosuppressive actions (Orban et al., 2013; Perez-Garcia et al., 2018). Kv1.3-mediated renal fibrosis is closely associated with inflammatory immune responses and fibrogenic cytokines. Previous studies have shown that Kv1.3 in T lymphocytes is involved in the pathogenesis of renal diseases, including acute glomerulonephritis, lupus nephritis and chronic kidney disease, and that its expression would contribute to disease progression (Grgic et al., 2009b; Hyodo et al., 2010; Kazama et al., 2012; Khodoun et al., 2020). In the advanced stages of congestive heart failure (CHF), upregulation of Kv1.3 channels activates the proliferation of regulatory T cells (Tregs), which promotes cardiac fibrosis by secreting the fibrotic cytokine TGF-β (Shao et al., 2018). Kazama et al. (2012), Kazama (2015) found diffuse interstitial fibrosis with leukocyte infiltration in the kidney of 5/6 nephrectomy rats, and Kv1.3 channel expression was upregulated in proliferating leukocytes in fibrotic kidneys. Importantly, inhibition of Kv1.3 channels using the selective blocker margatoxin significantly inhibited renal lymphocyte proliferation and ameliorated the progression of renal fibrosis. Their team further demonstrated the effectiveness of targeting lymphocyte Kv1.3 channels in the treatment of renal fibrosis using the UUO mouse model (Abe et al., 2019). Furthermore, stimulation with TGF-β significantly induced an increase in Kv1.3 density and outward K+ current amplitude, and the Kv1.3 channel inhibitor regulated the expression of the TGF-β in mouse microglia (Schilling et al., 2000), hypothesizing that Kv1.3 channel inhibitors have a similar mechanism to alleviate renal fibrosis by regulating the expression of the cytokine TGF-β.
In conclusion, it is necessary to conduct a more thorough study on the protective effect of Kv1.3 on renal fibrosis through the regulation of immune cell function to provide attractive ideas for the prevention and treatment of renal diseases.
5.3 ATP-sensitive K+ channels
The typical ATP-sensitive K+ (KATP) channels are ubiquitous hetero-octameric complexs consisted of at least two proteins: the pore-forming subunit belonging to the Kir6.0 family (Kir6.1 or Kir6.2) and the sulfonyl binding regulatory subunit (SUR1 or SUR2) (Rodrigo and Standen, 2005; Tinker et al., 2018). Structurally, the tetramer composed by the kir6 family subunits forms a K+-selective pore-forming channel that is surrounded by four SUR protein subunits. Energy switch located at the cytoplasmic end of the channel lumen, where ATP binds to the Kir6 subunit to provide energy for channel closure, while SUR has an ADP binding site and ADP dissociation leads to a sustained “activation state” (Nichols et al., 2013). In other words, KATP channels open in response to a decrease in cellular ATP (see Figure 4B). However, cells ordinarily contain millimolar ATP, while channels open in response to micromolar concentrations, which makes channel opening rare. In this way, as endogenous metabolic sensors, KATP channels have been suggested to play a key role in matching membrane electrical excitability to the energy metabolic state, as well as in processes including maintenance of glucose homeostasis, energy, systemic blood pressure, and vascular tone regulation (Seino and Miki, 2003; Flagg et al., 2010). The evidence on the role of KATP in renal fibrosis is limited and remains of particular interest. Xue et al. (2005) reported that KATP channel subunit SUR2, Kir6.1 mRNA expression was upregulated in renal tissue of hypertensive rats. Iptakalim, a novel KATP channel opener, regulated Kir6.1 overexpression (no effect on SUR2) and inhibited the accumulation of extracellular matrix components [fibronectin, collagen IV, MMP-9 and tissue inhibitor of MMP 1 (TIMP-1)] in the kidney of hypertensive rats, protecting renal function and preventing fibrosis progression. They also found that this effect was accompanied by a decrease in the expression of TGF-β1, endothelin 1 in the kidney during hypertensio (Xue et al., 2005). Despite the limited data, we believe that KATP is a promising new direction for research in renal fibrosis and look forward to new research findings in this area.
6 Conclusion
Undoubtedly, years of research established that studies regarding physiology, pathology and mechanism of ion channels in renal fibrosis have gradually become a hotspot in our description, and the feasibility of ion channels in the treatment and intervention of diseases has been confirmed by an increasing number of trials. In addition to their function as channels, the interactions between ion channels and proteins indicate their involvement in different processes such as proliferation, apoptosis, atrophy, cell cycle, cell acidity, cytoskeletal structure, immune and cell volume regulation, all of which are related to renal fibrosis. Although efforts to characterize how ion channels perform these important functions have somewhat provided us with mechanistic insight, there are still numerous unanswered questions and even controversy. It is important to emphasize that chloride channels are a promising and novel direction in renal fibrosis research, but the potential relationship between them and intracellular acidification, how they connect as channel regulators and integrate cellular signaling and function still needs to be catalogued. The availability of new pharmacology based on the genetic properties, structural differences and gating mechanisms of different chloride channels may allow for more successful therapies in the future. Although STIM and Orai have emerged as core elements of highly evolutionarily conserved Ca2+ channels, there are still large gaps in signaling pathways, isoform differences as well as cell and tissue specificity. There is an urgent need to develop specific drugs targeting the exciting P2X7/TRPC channel, however, research on the differential regulatory mechanisms of different subtypes and drug development are still worth exploring. The role of Na channels and transporters in the renal fibrosis pathway should not be ignored. We are looking forward to the pharmacological and genetic targeting studies of ENaC protein on renal fibrosis. An equally exciting pathways in renal fibrosis is potassium handling; in the future, hopefully more K+ channel agonists or inhibitors will be used in this area. It must be emphasized that the crosstalk between various ion channels is complex and the coordinated functioning of the entire channel network determines ion homeostasis in the kidney, which is reflected in the complex pathological features of most renal diseases. Understanding the rules of channel interactions remains an important challenge for successful intervention in various forms of renal fibrosis. In addition, the abundance of scientific research tools is essential for the accurate measurement of channel regulation within some cells. At last, since ion channels are widely expressed in most cells and tissues, drugs designed for channel-related diseases should focus more on tissue and subtype specificity to avoid safety issues, and precise intracellular delivery of novel modulators based on nanoplatforms might be a promising option. Overall, continually refining our understanding of the ion channel transport mechanisms that underlie renal fibrosis will undoubtedly expand therapeutic possibilities, and more research is needed to decipher the complex chain of events in renal fibrosis.
Author contributions
PY conceived the review and drafted the manuscript. BK and XF provided funding and revised the paper. All authors approved the final version of the manuscript submitted.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82160143, 81860133), and the Natural Science Foundation of Jiangxi Province (General Program 20192BAB205022).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe N., Toyama H., Saito K., Ejima Y., Yamauchi M., Mushiake H., et al. (2019). Delayed rectifier K(+)-channel is a novel therapeutic target for interstitial renal fibrosis in rats with unilateral ureteral obstruction. Biomed. Res. Int. 2019, 7567638. doi:10.1155/2019/7567638
Adapala R. K., Thoppil R. J., Luther D. J., Paruchuri S., Meszaros J. G., Chilian W. M., et al. (2013). TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 54, 45–52. doi:10.1016/j.yjmcc.2012.10.016
Arulkumaran N., Turner C. M., Sixma M. L., Singer M., Unwin R., Tam F. W. (2013). Purinergic signaling in inflammatory renal disease. Front. Physiol. 4, 194. doi:10.3389/fphys.2013.00194
Arulkumaran N., Unwin R. J., Tam F. W. (2011). A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 20 (7), 897–915. doi:10.1517/13543784.2011.578068
Baron L., Gombault A., Fanny M., Villeret B., Savigny F., Guillou N., et al. (2015). The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 6 (2), e1629. doi:10.1038/cddis.2014.576
Begenisich T., Nakamoto T., Ovitt C. E., Nehrke K., Brugnara C., Alper S. L., et al. (2004). Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J. Biol. Chem. 279 (46), 47681–47687. doi:10.1074/jbc.M409627200
Benedetti A., Di Sario A., Casini A., Ridolfi F., Bendia E., Pigini P., et al. (2001). Inhibition of the NA(+)/H(+) exchanger reduces rat hepatic stellate cell activity and liver fibrosis: An in vitro and in vivo study. Gastroenterology 120 (2), 545–556. doi:10.1053/gast.2001.21203
Berridge M. J. (2016). The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96 (4), 1261–1296. doi:10.1152/physrev.00006.2016
Buchholz B., Faria D., Schley G., Schreiber R., Eckardt K. U., Kunzelmann K. (2014). Anoctamin 1 induces calcium-activated chloride secretion and proliferation of renal cyst-forming epithelial cells. Kidney Int. 85 (5), 1058–1067. doi:10.1038/ki.2013.418
Bujak J. K., Kosmala D., Szopa I. M., Majchrzak K., Bednarczyk P. (2019). Inflammation, cancer and immunity-implication of TRPV1 channel. Front. Oncol. 9, 1087. doi:10.3389/fonc.2019.01087
Burow P., Klapperstuck M., Markwardt F. (2015). Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflugers Arch. 467 (6), 1215–1226. doi:10.1007/s00424-014-1561-8
Castellani S., Favia M., Guerra L., Carbone A., Abbattiscianni A. C., Di Gioia S., et al. (2017). Emerging relationship between CFTR, actin and tight junction organization in cystic fibrosis airway epithelium. Histol. Histopathol. 32 (5), 445–459. doi:10.14670/HH-11-842
Catacuzzeno L., Aiello F., Fioretti B., Sforna L., Castigli E., Ruggieri P., et al. (2011). Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J. Cell. Physiol. l226 (7), 1926–1933. doi:10.1002/jcp.22523
Cebotaru V., Kaul S., Devuyst O., Cai H., Racusen L., Guggino W. B., et al. (2005). High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent's disease. Kidney Int. 68 (2), 642–652. doi:10.1111/j.1523-1755.2005.00442.x
Ceol M., Tiralongo E., Baelde H. J., Vianello D., Betto G., Marangelli A., et al. (2012). Involvement of the tubular ClC-type exchanger ClC-5 in glomeruli of human proteinuric nephropathies. PLoS One 7 (9), e45605. doi:10.1371/journal.pone.0045605
Chang C. T., Hung C. C., Chen Y. C., Yen T. H., Wu M. S., Yang C. W., et al. (2008). Transforming growth factor-beta1 decreases epithelial sodium channel functionality in renal collecting duct cells via a Smad4-dependent pathway. Nephrol. Dial. Transpl. 23 (4), 1126–1134. doi:10.1093/ndt/gfm786
Chaudhari S., Li W., Wang Y., Jiang H., Ma Y., Davis M. E., et al. (2017). Store-operated calcium entry suppressed the TGF-β1/Smad3 signaling pathway in glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 313 (3), F729–F39. doi:10.1152/ajprenal.00483.2016
Chen K., Zhang J., Zhang W., Zhang J., Yang J., Li K., et al. (2013). ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 45 (5), 932–943. doi:10.1016/j.biocel.2013.02.009
Chen L., Konig B., Liu T., Pervaiz S., Razzaque Y. S., Stauber T. (2019). More than just a pressure relief valve: Physiological roles of volume-regulated LRRC8 anion channels. Biol. Chem. 400 (11), 1481–1496. doi:10.1515/hsz-2019-0189
Chen L., Marko L., Kassmann M., Zhu Y., Wu K., Gollasch M. (2014). Role of TRPV1 channels in ischemia/reperfusion-induced acute kidney injury. PLoS One 9 (10), e109842. doi:10.1371/journal.pone.0109842
Chen Y. C., Cheng C. Y., Liu C. T., Sue Y. M., Chen T. H., Hsu Y. H., et al. (2018). Alleviative effect of fucoxanthin-containing extract from Brown seaweed Laminaria japonica on renal tubular cell apoptosis through upregulating Na(+)/H(+) exchanger NHE1 in chronic kidney disease mice. J. Ethnopharmacol. 224, 391–399. doi:10.1016/j.jep.2018.06.023
Chen Y. F., Chen L. H., Shen M. R. (2019). The distinct role of STIM1 and STIM2 in the regulation of store-operated Ca(2+) entry and cellular function. J. Cell. Physiol. 234 (6), 8727–8739. doi:10.1002/jcp.27532
Cheng X., Song Y., Wang Y. (2019). pNaKtide ameliorates renal interstitial fibrosis through inhibition of sodium-potassium adenosine triphosphatase-mediated signaling pathways in unilateral ureteral obstruction mice. Nephrol. Dial. Transpl. 34 (2), 242–252. doi:10.1093/ndt/gfy107
Chou C. C., Lunn C. A., Murgolo N. J. (2008). KCa3.1: Target and marker for cancer, autoimmune disorder and vascular inflammation? Expert Rev. Mol. diagn. 8 (2), 179–187. doi:10.1586/14737159.8.2.179
Clevers H., Nusse R. (2012). Wnt/β-catenin signaling and disease. Cell 149 (6), 1192–1205. doi:10.1016/j.cell.2012.05.012
Coupaye-Gerard B., Bookstein C., Duncan P., Chen X. Y., Smith P. R., Musch M., et al. (1996). Biosynthesis and cell surface delivery of the NHE1 isoform of Na+/H+ exchanger in A6 cells. Am. J. Physiol. 271 (1), C1639–C1645. doi:10.1152/ajpcell.1996.271.5.C1639
Craigie E., Birch R. E., Unwin R. J., Wildman S. S. (2013). The relationship between P2X4 and P2X7: A physiologically important interaction? Front. Physiol. 4, 216. doi:10.3389/fphys.2013.00216
Cruse G., Duffy S. M., Brightling C. E., Bradding P. (2006). Functional KCa3.1 K+ channels are required for human lung mast cell migration. Thorax 61 (10), 880–885. doi:10.1136/thx.2006.060319
Csanady L., Vergani P., Gadsby D. C. (2019). Structure, gating, and regulation of the cftr anion channel. Physiol. Rev. 99 (1), 707–738. doi:10.1152/physrev.00007.2018
Cui J., Yang H., Lee U. S. (2009). Molecular mechanisms of BK channel activation. Cell. Mol. Life Sci. 66 (5), 852–875. doi:10.1007/s00018-008-8609-x
Dan Q., Shi Y., Rabani R., Venugopal S., Xiao J., Anwer S., et al. (2019). Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells. J. Biol. Chem. 294 (42), 15446–15465. doi:10.1074/jbc.RA118.006484
Dang S., Feng S., Tien J., Peters C. J., Bulkley D., Lolicato M., et al. (2017). Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 552 (7685), 426–429. doi:10.1038/nature25024
Davis J., Burr A. R., Davis G. F., Birnbaumer L., Molkentin J. D. (2012). A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 23 (4), 705–715. doi:10.1016/j.devcel.2012.08.017
Deba F., Bessac B. F. (2015). Anoctamin-1 Cl(-) channels in nociception: Activation by an N-aroylaminothiazole and capsaicin and inhibition by t16a[inh]-A01. Mol. pain 11, 55. doi:10.1186/s12990-015-0061-y
Devuyst O., Luciani A. (2015). Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J. Physiol. 593 (18), 4151–4164. doi:10.1113/JP270087
Dey A., Varelas X., Guan K. L. (2020). Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 19 (7), 480–494. doi:10.1038/s41573-020-0070-z
Dhande I. S., Zhu Y., Kneedler S. C., Joshi A. S., Hicks M. J., Wenderfer S. E., et al. (2020). Stim1 polymorphism disrupts immune signaling and creates renal injury in hypertension. J. Am. Heart Assoc. 9 (5), e014142. doi:10.1161/JAHA.119.014142
Di A., Xiong S., Ye Z., Malireddi R. K. S., Kometani S., Zhong M., et al. (2018). The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49 (1), 56–65. doi:10.1016/j.immuni.2018.04.032
Docherty M. H., Baird D. P., Hughes J., Ferenbach D. A. (2020). Cellular senescence and senotherapies in the kidney: Current evidence and future directions. Front. Pharmacol. 11, 755. doi:10.3389/fphar.2020.00755
Docherty M. H., O'sullivan E. D., Bonventre J. V., Ferenbach D. A. (2019). Cellular senescence in the kidney. J. Am. Soc. Nephrol. 30 (5), 726–736. doi:10.1681/ASN.2018121251
Drummond H. A., Grifoni S. C., Abu-Zaid A., Gousset M., Chiposi R., Barnard J. M., et al. (2011). Renal inflammation and elevated blood pressure in a mouse model of reduced {beta}-ENaC. Am. J. Physiol. Ren. Physiol. 301 (2), F443–F449. doi:10.1152/ajprenal.00694.2010
Duerr J., Leitz D. H. W., Szczygiel M., Dvornikov D., Fraumann S. G., Kreutz C., et al. (2020). Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 11 (1), 2012. doi:10.1038/s41467-020-15743-6
Duffield J. S. (2014). Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest. 124 (6), 2299–2306. doi:10.1172/JCI72267
Duran C., Thompson C. H., Xiao Q., Hartzell H. C. (2010). Chloride channels: Often enigmatic, rarely predictable. Annu. Rev. Physiol. 72, 95–121. doi:10.1146/annurev-physiol-021909-135811
Dutzler R. (2004). Structural basis for ion conduction and gating in ClC chloride channels. FEBS Lett. 564 (3), 229–233. doi:10.1016/S0014-5793(04)00210-8
Dyla M., Kjaergaard M., Poulsen H., Nissen P. (2020). Structure and mechanism of P-type ATPase ion pumps. Annu. Rev. Biochem. 89, 583–603. doi:10.1146/annurev-biochem-010611-112801
Elkareh J., Kennedy D. J., Yashaswi B., Vetteth S., Shidyak A., Kim E. G., et al. (2007). Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 49 (1), 215–224. doi:10.1161/01.HYP.0000252409.36927.05
Fanger C. M., Ghanshani S., Logsdon N. J., Rauer H., Kalman K., Zhou J., et al. (1999). Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem. 274 (9), 5746–5754. doi:10.1074/jbc.274.9.5746
Fantus D., Rogers N. M., Grahammer F., Huber T. B., Thomson A. W. (2016). Roles of mTOR complexes in the kidney: Implications for renal disease and transplantation. Nat. Rev. Nephrol. 12 (10), 587–609. doi:10.1038/nrneph.2016.108
Faouzi M., Kilch T., Horgen F. D., Fleig A., Penner R. (2017). The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 595 (10), 3165–3180. doi:10.1113/JP274006
Faria D., Rock J. R., Romao A. M., Schweda F., Bandulik S., Witzgall R., et al. (2014). The calcium-activated chloride channel Anoctamin 1 contributes to the regulation of renal function. Kidney Int. 85 (6), 1369–1381. doi:10.1038/ki.2013.535
Fedorova L. V., Raju V., El-Okdi N., Shidyak A., Kennedy D. J., Vetteth S., et al. (2009). The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: Implication of epithelial-to-mesenchymal transition. Am. J. Physiol. Ren. Physiol. 296 (4), F922–F934. doi:10.1152/ajprenal.90605.2008
Feng N. H., Lee H. H., Shiang J. C., Ma M. C. (2008). Transient receptor potential vanilloid type 1 channels act as mechanoreceptors and cause substance P release and sensory activation in rat kidneys. Am. J. Physiol. Ren. Physiol. 294 (2), F316–F325. doi:10.1152/ajprenal.00308.2007
Feraille E., Dizin E. (2016). Coordinated control of ENaC and Na+, K+-ATPase in renal collecting duct. J. Am. Soc. Nephrol. 27 (9), 2554–2563. doi:10.1681/ASN.2016020124
Feske S., Wulff H., Skolnik E. Y. (2015). Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353. doi:10.1146/annurev-immunol-032414-112212
Flagg T. P., Enkvetchakul D., Koster J. C., Nichols C. G. (2010). Muscle KATP channels: Recent insights to energy sensing and myoprotection. Physiol. Rev. 90 (3), 799–829. doi:10.1152/physrev.00027.2009
Francois H., Chatziantoniou C. (2018). Renal fibrosis: Recent translational aspects. Matrix Biol. 68-69, 318–332. doi:10.1016/j.matbio.2017.12.013
Friard J., Corinus A., Cougnon M., Tauc M., Pisani D. F., Duranton C., et al. (2019). LRRC8/VRAC channels exhibit a noncanonical permeability to glutathione, which modulates epithelial-to-mesenchymal transition (EMT). Cell Death Dis. 10 (12), 925. doi:10.1038/s41419-019-2167-z
Fu R. G., Zhang T., Wang L., Du Y., Jia L. N., Hou J. J., et al. (2014). Inhibition of the K+ channel K(Ca)3.1 reduces TGF-β1-induced premature senescence, myofibroblast phenotype transition and proliferation of mesangial cells. PLoS One 9 (1), e87410. doi:10.1371/journal.pone.0087410
Gaitan-Penas H., Gradogna A., Laparra-Cuervo L., Solsona C., Fernandez-Duenas V., Barrallo-Gimeno A., et al. (2016). Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys. J. 111 (7), 1429–1443. doi:10.1016/j.bpj.2016.08.030
Ge L., Hoa N. T., Wilson Z., Arismendi-Morillo G., Kong X. T., Tajhya R. B., et al. (2014). Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int. Immunopharmacol. 22 (2), 427–443. doi:10.1016/j.intimp.2014.06.040
Gentile D., Natale M., Lazzerini P. E., Capecchi P. L., Laghi-Pasini F. (2015). The role of P2X7 receptors in tissue fibrosis: A brief review. Purinergic Signal. 11 (4), 435–440. doi:10.1007/s11302-015-9466-3
Gewin L., Zent R., Pozzi A. (2017). Progression of chronic kidney disease: Too much cellular talk causes damage. Kidney Int. 91 (3), 552–560. doi:10.1016/j.kint.2016.08.025
Geyti C. S., Odgaard E., Overgaard M. T., Jensen M. E., Leipziger J., Praetorius H. A. (2008). Slow spontaneous [Ca2+] i oscillations reflect nucleotide release from renal epithelia. Pflugers Arch. 455 (6), 1105–1117. doi:10.1007/s00424-007-0366-4
Gibson R. L., Burns J. L., Ramsey B. W. (2003). Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168 (8), 918–951. doi:10.1164/rccm.200304-505SO
Goncalves R. G., Gabrich L., Rosario A., Takiya C. M., Ferreira M. L., Chiarini L. B., et al. (2006). The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int. 70 (9), 1599–1606. doi:10.1038/sj.ki.5001804
Gonzalez C., Baez-Nieto D., Valencia I., Oyarzun I., Rojas P., Naranjo D., et al. (2012). K(+) channels: Function-structural overview. Compr. Physiol. 2 (3), 2087–2149. doi:10.1002/cphy.c110047
Greger R. (2000). Physiology of renal sodium transport. Am. J. Med. Sci. 319 (1), 51–62. doi:10.1097/00000441-200001000-00005
Grgic I., Campanholle G., Bijol V., Wang C., Sabbisetti V. S., Ichimura T., et al. (2012). Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 82 (2), 172–183. doi:10.1038/ki.2012.20
Grgic I., Kiss E., Kaistha B. P., Busch C., Kloss M., Sautter J., et al. (2009). Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc. Natl. Acad. Sci. U. S. A. 106 (34), 14518–14523. doi:10.1073/pnas.0903458106
Grgic I., Wulff H., Eichler I., Flothmann C., Kohler R., Hoyer J. (2009). Blockade of T-lymphocyte KCa3.1 and Kv1.3 channels as novel immunosuppression strategy to prevent kidney allograft rejection. Transpl. Proc. 41 (6), 2601–2606. doi:10.1016/j.transproceed.2009.06.025
Gu L. F., Ge H. T., Zhao L., Wang Y. J., Zhang F., Tang H. T., et al. (2020). Huangkui capsule ameliorates renal fibrosis in a unilateral ureteral obstruction mouse model through TRPC6 dependent signaling pathways. Front. Pharmacol. 11, 996. doi:10.3389/fphar.2020.00996
Guan Y., Luan Y., Xie Y., Zhou H., Li W., Zhang X., et al. (2019). Chloride channel-3 is required for efficient tumour cell migration and invasion in human cervical squamous cell carcinoma. Gynecol. Oncol. 153 (3), 661–669. doi:10.1016/j.ygyno.2019.03.006
Guan Y. T., Huang Y. Q., Wu J. B., Deng Z. Q., Wang Y., Lai Z. Y., et al. (2016). Overexpression of chloride channel-3 is associated with the increased migration and invasion ability of ectopic endometrial cells from patients with endometriosis. Hum. Reprod. 31 (5), 986–998. doi:10.1093/humrep/dew034
Gunther W., Piwon N., Jentsch T. J. (2003). The ClC-5 chloride channel knock-out mouse - an animal model for Dent's disease. Pflugers Arch. 445 (4), 456–462. doi:10.1007/s00424-002-0950-6
Hall G., Wang L., Spurney R. F. (2019). TRPC channels in proteinuric kidney diseases. Cells 9 (1), E44. doi:10.3390/cells9010044
Haller S. T., Drummond C. A., Yan Y., Liu J., Tian J., Malhotra D., et al. (2014). Passive immunization against marinobufagenin attenuates renal fibrosis and improves renal function in experimental renal disease. Am. J. Hypertens. 27 (4), 603–609. doi:10.1093/ajh/hpt169
Haller S. T., Kumarasamy S., Folt D. A., Wuescher L. M., Stepkowski S., Karamchandani M., et al. (2017). Targeted disruption of Cd40 in a genetically hypertensive rat model attenuates renal fibrosis and proteinuria, independent of blood pressure. Kidney Int. 91 (2), 365–374. doi:10.1016/j.kint.2016.08.015
Han S. J., Lovaszi M., Kim M., D'agati V., Hasko G., Lee H. T. (2020). P2X4 receptor exacerbates ischemic AKI and induces renal proximal tubular NLRP3 inflammasome signaling. FASEB J. 34 (4), 5465–5482. doi:10.1096/fj.201903287R
Hanukoglu I., Hanukoglu A. (2016). Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579 (2), 95–132. doi:10.1016/j.gene.2015.12.061
Hara-Chikuma M., Yang B., Sonawane N. D., Sasaki S., Uchida S., Verkman A. S. (2005). ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J. Biol. Chem. 280 (2), 1241–1247. doi:10.1074/jbc.M407030200
Hardie R. C. (2011). A brief history of trp: Commentary and personal perspective. Pflugers Arch. 461 (5), 493–498. doi:10.1007/s00424-011-0922-9
Hartzell H. C., Yu K., Xiao Q., Chien L. T., Qu Z. (2009). Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J. Physiol. 587 (10), 2127–2139. doi:10.1113/jphysiol.2008.163709
Harvey K. F., Dinudom A., Cook D. I., Kumar S. (2001). The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J. Biol. Chem. 276 (11), 8597–8601. doi:10.1074/jbc.C000906200
Hatziioanou D., Barkas G., Critselis E., Zoidakis J., Gakiopoulou H., Androutsou M. E., et al. (2018). Chloride intracellular channel 4 overexpression in the proximal tubules of kidneys from the spontaneously hypertensive rat: Insight from proteomic analysis. Nephron 138 (1), 60–70. doi:10.1159/000479169
Hawkins B. J., Madesh M., Kirkpatrick C. J., Fisher A. B. (2007). Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol. Biol. Cell 18 (6), 2002–2012. doi:10.1091/mbc.e06-09-0830
Henshall T. L., Manning J. A., Alfassy O. S., Goel P., Boase N. A., Kawabe H., et al. (2017). Deletion of Nedd4-2 results in progressive kidney disease in mice. Cell Death Differ. 24 (12), 2150–2160. doi:10.1038/cdd.2017.137
Hou J., Rajagopal M., Yu A. S. (2013). Claudins and the kidney. Annu. Rev. Physiol. 75, 479–501. doi:10.1146/annurev-physiol-030212-183705
Hsu K. S., Otsu W., Li Y., Wang H. C., Chen S., Tsang S. H., et al. (2019). CLIC4 regulates late endosomal trafficking and matrix degradation activity of MMP14 at focal adhesions in RPE cells. Sci. Rep. 9 (1), 12247. doi:10.1038/s41598-019-48438-0
Huang C., Day M. L., Poronnik P., Pollock C. A., Chen X. M. (2014). Inhibition of KCa3.1 suppresses TGF-β1 induced MCP-1 expression in human proximal tubular cells through Smad3, p38 and ERK1/2 signaling pathways. Int. J. Biochem. Cell Biol. 47, 1–10. doi:10.1016/j.biocel.2013.11.017
Huang C., Pollock C. A., Chen X. M. (2014). High glucose induces CCL20 in proximal tubular cells via activation of the KCa3.1 channel. PLoS One 9 (4), e95173. doi:10.1371/journal.pone.0095173
Huang C., Shen S., Ma Q., Chen J., Gill A., Pollock C. A., et al. (2013). Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-β1/Smad pathway in diabetic mice. Diabetes 62 (8), 2923–2934. doi:10.2337/db13-0135
Huang C., Shen S., Ma Q., Gill A., Pollock C. A., Chen X. M. (2014). KCa3.1 mediates activation of fibroblasts in diabetic renal interstitial fibrosis. Nephrol. Dial. Transpl. 29 (2), 313–324. doi:10.1093/ndt/gft431
Huang L., Ma R., Lin T., Chaudhari S., Shotorbani P. Y., Yang L., et al. (2019). Glucagon-like peptide-1 receptor pathway inhibits extracellular matrix production by mesangial cells through store-operated Ca(2+) channel. Exp. Biol. Med. 244 (14), 1193–1201. doi:10.1177/1535370219876531
Huen S. C., Cantley L. G. (2017). Macrophages in renal injury and repair. Annu. Rev. Physiol. 79, 449–469. doi:10.1146/annurev-physiol-022516-034219
Hyodo T., Oda T., Kikuchi Y., Higashi K., Kushiyama T., Yamamoto K., et al. (2010). Voltage-gated potassium channel Kv1.3 blocker as a potential treatment for rat anti-glomerular basement membrane glomerulonephritis. Am. J. Physiol. Ren. Physiol. 299 (6), F1258–F1269. doi:10.1152/ajprenal.00374.2010
Ilatovskaya D. V., Blass G., Palygin O., Levchenko V., Pavlov T. S., Grzybowski M. N., et al. (2018). A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J. Am. Soc. Nephrol. 29 (7), 1917–1927. doi:10.1681/ASN.2018030280
Inoue R., Kurahara L. H., Hiraishi K. (2019). TRP channels in cardiac and intestinal fibrosis. Semin. Cell Dev. Biol. 94, 40–49. doi:10.1016/j.semcdb.2018.11.002
Ishii T. M., Silvia C., Hirschberg B., Bond C. T., Adelman J. P., Maylie J. (1997). A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U. S. A. 94 (21), 11651–11656. doi:10.1073/pnas.94.21.11651
Jentsch T. J., Pusch M. (2018). CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev. 98 (3), 1493–1590. doi:10.1152/physrev.00047.2017
Jentsch T. J. (2016). VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 17 (5), 293–307. doi:10.1038/nrm.2016.29
Jha J. C., Banal C., Chow B. S., Cooper M. E., Jandeleit-Dahm K. (2016). Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 25 (12), 657–684. doi:10.1089/ars.2016.6664
Jia G., Habibi J., Aroor A. R., Hill M. A., Demarco V. G., Lee L. E., et al. (2018). Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism. 78, 69–79. doi:10.1016/j.metabol.2017.08.008
Jiang L., Xu L., Mao J., Li J., Fang L., Zhou Y., et al. (2013). Rheb/mTORC1 signaling promotes kidney fibroblast activation and fibrosis. J. Am. Soc. Nephrol. 24 (7), 1114–1126. doi:10.1681/ASN.2012050476
Jin J., Wu D., Zhao L., Zou W., Shen W., Tu Q., et al. (2018). Effect of autophagy and stromal interaction molecule 1 on podocyte epithelial-mesenchymal transition in diabetic nephropathy. Int. J. Clin. Exp. Pathol. 11 (5), 2450–2459.
Jin J., Ye M., Hu K., Gong J., He Q. (2019). STIM promotes the epithelial-mesenchymal transition of podocytes through regulation of FcγRII activity in diabetic nephropathy. Histol. Histopathol. 34 (6), 671–682. doi:10.14670/HH-18-068
Jin Y., Ibrahim D., Magness S. T., Blikslager A. T. (2018). Knockout of ClC-2 reveals critical functions of adherens junctions in colonic homeostasis and tumorigenicity. Am. J. Physiol. Gastrointest. Liver Physiol. 315 (6), G966–G79. doi:10.1152/ajpgi.00087.2018
Jouret F., Devuyst O. (2009). CFTR and defective endocytosis: New insights in the renal phenotype of cystic fibrosis. Pflugers Arch. 457 (6), 1227–1236. doi:10.1007/s00424-008-0594-2
Kaczmarek L. K., Aldrich R. W., Chandy K. G., Grissmer S., Wei A. D., Wulff H. (2017). International union of basic and clinical pharmacology. C. Nomenclature and properties of calcium-activated and sodium-activated potassium channels. Pharmacol. Rev. 69 (1), 1–11. doi:10.1124/pr.116.012864
Kaimori J. Y., Lin C. C., Outeda P., Garcia-Gonzalez M. A., Menezes L. F., Hartung E. A., et al. (2017). NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology. Sci. Rep. 7 (1), 7733. doi:10.1038/s41598-017-08284-4
Kalita-De Croft P., Bedford J. J., Leader J. P., Walker R. J. (2018). Amiloride modifies the progression of lithium-induced renal interstitial fibrosis. Nephrol. Carlt. 23 (1), 20–30. doi:10.1111/nep.12929
Karydis A., Jimenez-Vidal M., Denker S. P., Barber D. L. (2009). Mislocalized scaffolding by the Na-H exchanger NHE1 dominantly inhibits fibronectin production and TGF-beta activation. Mol. Biol. Cell 20 (8), 2327–2336. doi:10.1091/mbc.E08-08-0842
Kazama I., Maruyama Y., Endo Y., Toyama H., Ejima Y., Matsubara M., et al. (2012). Overexpression of delayed rectifier K(+) channels promotes in situ proliferation of leukocytes in rat kidneys with advanced chronic renal failure. Int. J. Nephrol. 2012, 581581. doi:10.1155/2012/581581
Kazama I. (2015). Roles of lymphocyte kv1.3-channels in the pathogenesis of renal diseases and novel therapeutic implications of targeting the channels. Mediat. Inflamm. 2015, 436572. doi:10.1155/2015/436572
Kennedy D. J., Khalaf F. K., Sheehy B., Weber M. E., Agatisa-Boyle B., Conic J., et al. (2018). Telocinobufagin, a novel cardiotonic steroid, promotes renal fibrosis via Na⁺/K⁺-ATPase profibrotic signaling pathways. Int. J. Mol. Sci. 9 (9), E2566. doi:10.3390/ijms19092566
Khan S., Wu K. L., Sedor J. R., Abu Jawdeh B. G., Schelling J. R. (2006). The NHE1 Na+/H+ exchanger regulates cell survival by activating and targeting ezrin to specific plasma membrane domains. Cell. Mol. Biol. 52 (8), 115–121.
Khanna R., Chang M. C., Joiner W. J., Kaczmarek L. K., Schlichter L. C. (1999). hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J. Biol. Chem. 274 (21), 14838–14849. doi:10.1074/jbc.274.21.14838
Khodoun M., Chimote A. A., Ilyas F. Z., Duncan H. J., Moncrieffe H., Kant K. S., et al. (2020). Targeted knockdown of Kv1.3 channels in T lymphocytes corrects the disease manifestations associated with systemic lupus erythematosus. Sci. Adv. 6 (47), eabd1471. doi:10.1126/sciadv.abd1471
Kim E. Y., Anderson M., Wilson C., Hagmann H., Benzing T., Dryer S. E. (2013). NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: Essential role of podocin in formation of this complex. Am. J. Physiol. Cell Physiol. 305 (9), C960–C971. doi:10.1152/ajpcell.00191.2013
Kim E. Y., Dryer S. E. (2021). Effects of TRPC6 inactivation on glomerulosclerosis and renal fibrosis in aging rats. Cells 10 (4), 856. doi:10.3390/cells10040856
Kim E. Y., Shotorbani P. Y., Dryer S. E. (2019). TRPC6 inactivation does not affect loss of renal function in nephrotoxic serum glomerulonephritis in rats, but reduces severity of glomerular lesions. Biochem. Biophys. Rep. 17, 139–150. doi:10.1016/j.bbrep.2018.12.006
Kim J. H., Hwang K. H., Dang B. T. N., Eom M., Kong I. D., Gwack Y., et al. (2021). Insulin-activated store-operated Ca(2+) entry via Orai1 induces podocyte actin remodeling and causes proteinuria. Nat. Commun. 12 (1), 6537. doi:10.1038/s41467-021-26900-w
Kim K. D., Srikanth S., Yee M. K., Mock D. C., Lawson G. W., Gwack Y. (2011). ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J. Immunol. 187 (7), 3620–3630. doi:10.4049/jimmunol.1100847
Kim M. J., Turner C. M., Hewitt R., Smith J., Bhangal G., Pusey C. D., et al. (2014). Exaggerated renal fibrosis in P2X4 receptor-deficient mice following unilateral ureteric obstruction. Nephrol. Dial. Transpl. 29 (7), 1350–1361. doi:10.1093/ndt/gfu019
Klapper-Goldstein H., Verma A., Elyagon S., Gillis R., Murninkas M., Pittala S., et al. (2020). An implantable system for long-term assessment of atrial fibrillation substrate in unanesthetized rats exposed to underlying pathological conditions. Sci. Rep. 10 (1), 553. doi:10.1038/s41598-020-57528-3
Kong W., Haschler T. N., Nurnberg B., Kramer S., Gollasch M., Marko L. (2019). Renal fibrosis, immune cell infiltration and changes of TRPC channel expression after unilateral ureteral obstruction in Trpc6-/- mice. Cell. Physiol. biochem. 52 (6), 1484–1502. doi:10.33594/000000103
Koo T. Y., Lee J. G., Yan J. J., Jang J. Y., Ju K. D., Han M., et al. (2017). The P2X7 receptor antagonist, oxidized adenosine triphosphate, ameliorates renal ischemia-reperfusion injury by expansion of regulatory T cells. Kidney Int. 92 (2), 415–431. doi:10.1016/j.kint.2017.01.031
Kunzelmann K. (2015). TMEM16, LRRC8A, bestrophin: Chloride channels controlled by Ca(2+) and cell volume. Trends biochem. Sci. 40 (9), 535–543. doi:10.1016/j.tibs.2015.07.005
Lagadic-Gossmann D., Huc L., Lecureur V. (2004). Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 11 (9), 953–961. doi:10.1038/sj.cdd.4401466
Lamouille S., Derynck R. (2007). Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178 (3), 437–451. doi:10.1083/jcb.200611146
Lang F., Busch G. L., Ritter M., Volkl H., Waldegger S., Gulbins E., et al. (1998). Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 78 (1), 247–306. doi:10.1152/physrev.1998.78.1.247
Lee H. J., Donati A., Feliers D., Sun Y., Ding Y., Madesh M., et al. (2021). Chloride channel accessory 1 integrates chloride channel activity and mTORC1 in aging-related kidney injury. Aging Cell 20 (7), e13407. doi:10.1111/acel.13407
Lee U. S., Cui J. (2010). BK channel activation: Structural and functional insights. Trends Neurosci. 33 (9), 415–423. doi:10.1016/j.tins.2010.06.004
Lemonnier L., Trebak M., Putney J. W. (2008). Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4, 5-bisphosphate. Cell calcium 43 (5), 506–514. doi:10.1016/j.ceca.2007.09.001
Lemos D. R., Mcmurdo M., Karaca G., Wilflingseder J., Leaf I. A., Gupta N., et al. (2018). Interleukin-1β activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 29 (6), 1690–1705. doi:10.1681/ASN.2017121283
Li C., Wang W., Kwon T. H., Knepper M. A., Nielsen S., Frokiaer J. (2003). Altered expression of major renal Na transporters in rats with unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 284 (1), F155–F166. doi:10.1152/ajprenal.00272.2002
Li C., Wang W., Norregaard R., Knepper M. A., Nielsen S., Frokiaer J. (2007). Altered expression of epithelial sodium channel in rats with bilateral or unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 293 (1), F333–F341. doi:10.1152/ajprenal.00372.2006
Li J., Wang D. H. (2008). Increased GFR and renal excretory function by activation of TRPV1 in the isolated perfused kidney. Pharmacol. Res. 57 (3), 239–246. doi:10.1016/j.phrs.2008.01.011
Li X. L., Liu J., Chen X. S., Cheng L. M., Liu W. L., Chen X. F., et al. (2022). Blockade of TMEM16A protects against renal fibrosis by reducing intracellular Cl(-) concentration. Br. J. Pharmacol. 179 (12), 3043–3060. doi:10.1111/bph.15786
Li X., Pan J., Li H., Li G., Liu B., Tang X., et al. (2022). DsbA-L interacts with VDAC1 in mitochondrion-mediated tubular cell apoptosis and contributes to the progression of acute kidney disease. EBioMedicine 76, 103859. doi:10.1016/j.ebiom.2022.103859
Li Z., Xu J., Xu P., Liu S., Yang Z. (2013). Wnt/β-catenin signalling pathway mediates high glucose induced cell injury through activation of TRPC6 in podocytes. Cell Prolif. 46 (1), 76–85. doi:10.1111/cpr.12010
Lian H., Cheng Y., Wu X. (2017). TMEM16A exacerbates renal injury by activating P38/JNK signaling pathway to promote podocyte apoptosis in diabetic nephropathy mice. Biochem. Biophys. Res. Commun. 487 (2), 201–208. doi:10.1016/j.bbrc.2017.04.021
Lin B. L., Matera D., Doerner J. F., Zheng N., Del Camino D., Mishra S., et al. (2019). In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. U. S. A. 116 (20), 10156–10161. doi:10.1073/pnas.1815354116
Lindemann O., Umlauf D., Frank S., Schimmelpfennig S., Bertrand J., Pap T., et al. (2013). TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J. Immunol. 190 (11), 5496–5505. doi:10.4049/jimmunol.1201502
Linden J., Koch-Nolte F., Dahl G. (2019). Purine release, metabolism, and signaling in the inflammatory response. Annu. Rev. Immunol. 37, 325–347. doi:10.1146/annurev-immunol-051116-052406
Lipphardt M., Song J. W., Matsumoto K., Dadafarin S., Dihazi H., Muller G., et al. (2017). The third path of tubulointerstitial fibrosis: Aberrant endothelial secretome. Kidney Int. 92 (3), 558–568. doi:10.1016/j.kint.2017.02.033
Liu C., Song C., Li J., Sun Q. (2020). CFTR functions as a tumor suppressor and is regulated by DNA methylation in colorectal cancer. Cancer Manag. Res. 12, 4261–4270. doi:10.2147/CMAR.S248539
Liu H., Yan R., Liang L., Zhang H., Xiang J., Liu L., et al. (2021). The role of CDX2 in renal tubular lesions during diabetic kidney disease. Aging 13 (5), 6782–6803. doi:10.18632/aging.202537
Liu Z., Guo J., Wang Y., Weng Z., Huang B., Yu M. K., et al. (2017). CFTR-beta-catenin interaction regulates mouse embryonic stem cell differentiation and embryonic development. Cell Death Differ. 24 (1), 98–110. doi:10.1038/cdd.2016.118
Liu Z., Wang W., Li X., Tang S., Meng D., Xia W., et al. (2022). Capsaicin ameliorates renal fibrosis by inhibiting TGF-β1-Smad2/3 signaling. Phytomedicine. 100, 154067. doi:10.1016/j.phymed.2022.154067
Ma J., Zhang L., Hao J., Li N., Tang J., Hao L. (2018). Up-regulation of microRNA-93 inhibits TGF-β1-induced EMT and renal fibrogenesis by down-regulation of Orai1. J. Pharmacol. Sci. 136 (4), 218–227. doi:10.1016/j.jphs.2017.12.010
Mack M., Yanagita M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87 (2), 297–307. doi:10.1038/ki.2014.287
Mai X., Shang J., Liang S., Yu B., Yuan J., Lin Y., et al. (2016). Blockade of Orai1 store-operated calcium entry protects against renal fibrosis. J. Am. Soc. Nephrol. 27 (10), 3063–3078. doi:10.1681/ASN.2015080889
Maisonneuve P., Marshall B. C., Knapp E. A., Lowenfels A. B. (2013). Cancer risk in cystic fibrosis: A 20-year nationwide study from the United States. J. Natl. Cancer Inst. 105 (2), 122–129. doi:10.1093/jnci/djs481
Manning J. A., Shah S. S., Henshall T. L., Nikolic A., Finnie J., Kumar S. (2020). Dietary sodium modulates nephropathy in Nedd4-2-deficient mice. Cell Death Differ. 27 (6), 1832–1843. doi:10.1038/s41418-019-0468-5
Manning J. A., Shah S. S., Nikolic A., Henshall T. L., Khew-Goodall Y., Kumar S. (2021). The ubiquitin ligase NEDD4-2/NEDD4L regulates both sodium homeostasis and fibrotic signaling to prevent end-stage renal disease. Cell Death Dis. 12 (4), 398. doi:10.1038/s41419-021-03688-7
Manucha W., Carrizo L., Ruete C., Valles P. G. (2007). Apoptosis induction is associated with decreased NHE1 expression in neonatal unilateral ureteric obstruction. BJU Int. 100 (1), 191–198. doi:10.1111/j.1464-410X.2007.06840.x
Martinez-Lemus L. A., Aroor A. R., Ramirez-Perez F. I., Jia G., Habibi J., Demarco V. G., et al. (2017). Amiloride improves endothelial function and reduces vascular stiffness in female mice fed a western diet. Front. Physiol. 8, 456. doi:10.3389/fphys.2017.00456
Massey M. K., Reiterman M. J., Mourad J., Luckie D. B. (2021). Is CFTR an exchanger?: Regulation of HCO3 (-)Transport and extracellular pH by CFTR. Biochem. Biophys. Rep. 25, 100863. doi:10.1016/j.bbrep.2020.100863
Mcmanus O. B. (1991). Calcium-activated potassium channels: Regulation by calcium. J. Bioenerg. Biomembr. 23 (4), 537–560. doi:10.1007/BF00785810
Mehrotra P., Collett J. A., Mckinney S. D., Stevens J., Ivancic C. M., Basile D. P. (2017). IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: Compensatory role of natural killer cells in athymic rats. Am. J. Physiol. Ren. Physiol. 312 (3), F385–F97. doi:10.1152/ajprenal.00462.2016
Mehrotra P., Sturek M., Neyra J. A., Basile D. P. (2019). Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J. Clin. Invest. 129 (11), 4951–4961. doi:10.1172/JCI126108
Mei Y., Yangyang Z., Shuai L., Hao J., Yirong Y., Yong C., et al. (2016). Breviscapine prevents downregulation of renal water and sodium transport proteins in response to unilateral ureteral obstruction. Iran. J. Basic Med. Sci. 19 (5), 573–578.
Meng P., Huang J., Ling X., Zhou S., Wei J., Zhu M., et al. (2022). CXC chemokine receptor 2 accelerates tubular cell senescence and renal fibrosis via beta-catenin-induced mitochondrial dysfunction. Front. Cell Dev. Biol. 10, 862675. doi:10.3389/fcell.2022.862675
Meng X. M., Nikolic-Paterson D. J., Lan H. Y. (2016). TGF-Β: The master regulator of fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338. doi:10.1038/nrneph.2016.48
Menzies R. I., Booth J. W. R., Mullins J. J., Bailey M. A., Tam F. W. K., Norman J. T., et al. (2017). Hyperglycemia-induced renal P2X7 receptor activation enhances diabetes-related injury. EBioMedicine 19, 73–83. doi:10.1016/j.ebiom.2017.04.011
Miyano T., Suzuki A., Sakamoto N. (2021). Hyperosmotic stress induces epithelial-mesenchymal transition through rearrangements of focal adhesions in tubular epithelial cells. PLoS One 16 (12), e0261345. doi:10.1371/journal.pone.0261345
Monaghan M. T., Bailey M. A., Unwin R. J. (2021). Purinergic signalling in the kidney: In physiology and disease. Biochem. Pharmacol. 187, 114389. doi:10.1016/j.bcp.2020.114389
Moran M. M., Mcalexander M. A., Biro T., Szallasi A. (2011). Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 10 (8), 601–620. doi:10.1038/nrd3456
Mraiche F., Oka T., Gan X. T., Karmazyn M., Fliegel L. (2011). Activated NHE1 is required to induce early cardiac hypertrophy in mice. Basic Res. Cardiol. 106 (4), 603–616. doi:10.1007/s00395-011-0161-4
Mutchler S. M., Kirabo A., Kleyman T. R. (2021). Epithelial sodium channel and salt-sensitive hypertension. Hypertension 77 (3), 759–767. doi:10.1161/HYPERTENSIONAHA.120.14481
Nam S. A., Kim W. Y., Kim J. W., Park S. H., Kim H. L., Lee M. S., et al. (2019). Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-beta and NLRP3 inflammasome signaling pathway. Cell Death Dis. 10 (2), 78. doi:10.1038/s41419-019-1356-0
Narayanan V., Schappell L. E., Mayer C. R., Duke A. A., Armiger T. J., Arsenovic P. T., et al. (2020). Osmotic gradients in epithelial acini increase mechanical tension across E-cadherin, drive morphogenesis, and maintain homeostasis. Curr. Biol. 30 (4), 624–633. doi:10.1016/j.cub.2019.12.025
Nelson P. L., Beck A., Cheng H. (2011). Transient receptor proteins illuminated: Current views on TRPs and disease. Vet. J. 187 (2), 153–164. doi:10.1016/j.tvjl.2010.01.020
Nichols C. G., Singh G. K., Grange D. K. (2013). KATP channels and cardiovascular disease: Suddenly a syndrome. Circ. Res. 112 (7), 1059–1072. doi:10.1161/CIRCRESAHA.112.300514
Nijenhuis T., Sloan A. J., Hoenderop J. G., Flesche J., Van Goor H., Kistler A. D., et al. (2011). Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am. J. Pathol. 179 (4), 1719–1732. doi:10.1016/j.ajpath.2011.06.033
Ning B., Guo C., Kong A., Li K., Xie Y., Shi H., et al. (2021). Calcium signaling mediates cell death and crosstalk with autophagy in kidney disease. Cells 10 (11), 3204. doi:10.3390/cells10113204
Norman J. (2011). Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim. Biophys. Acta 1812 (10), 1327–1336. doi:10.1016/j.bbadis.2011.06.012
Norregaard R., Jensen B. L., Li C., Wang W., Knepper M. A., Nielsen S., et al. (2005). COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 289 (2), F322–F333. doi:10.1152/ajprenal.00061.2005
Nowak G., Megyesi J., Craigen W. J. (2020). Deletion of VDAC1 hinders recovery of mitochondrial and renal functions after acute kidney injury. Biomolecules 10 (4), E585. doi:10.3390/biom10040585
Numaga-Tomita T., Oda S., Shimauchi T., Nishimura A., Mangmool S., Nishida M. (2017). TRPC3 channels in cardiac fibrosis. Front. Cardiovasc. Med. 4, 56. doi:10.3389/fcvm.2017.00056
Oh U., Jung J. (2016). Cellular functions of TMEM16/anoctamin. Pflugers Arch. 468 (3), 443–453. doi:10.1007/s00424-016-1790-0
Okada Y., Maeno E., Shimizu T., Dezaki K., Wang J., Morishima S. (2001). Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol. 532 (1), 3–16. doi:10.1111/j.1469-7793.2001.0003g.x
Okada Y., Sumioka T., Reinach P. S., Miyajima M., Saika S. (2021). Roles of epithelial and mesenchymal TRP channels in mediating inflammatory fibrosis. Front. Immunol. 12, 731674. doi:10.3389/fimmu.2021.731674
Okuda S., Tamaki K., Ando T., Nagashima A., Nakayama M., Fukuda K., et al. (1994). Increased expression of Na+/H+ exchanger in the injured renal tissues of focal glomerulosclerosis in rats. Kidney Int. 46 (6), 1635–1643. doi:10.1038/ki.1994.463
Orban C., Biro E., Grozdics E., Bajnok A., Toldi G. (2013). Modulation of T lymphocyte calcium influx patterns via the inhibition of kv1.3 and ikca1 potassium channels in autoimmune disorders. Front. Immunol. 4, 234. doi:10.3389/fimmu.2013.00234
Orlowski J., Grinstein S. (1997). Na+/H+ exchangers of mammalian cells. J. Biol. Chem. 272 (36), 22373–22376. doi:10.1074/jbc.272.36.22373
Parrish A. R. (2016). The cytoskeleton as a novel target for treatment of renal fibrosis. Pharmacol. Ther. 166, 1–8. doi:10.1016/j.pharmthera.2016.06.006
Patel A. J., Lauritzen I., Lazdunski M., Honore E. (1998). Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J. Neurosci. 18 (9), 3117–3123. doi:10.1523/jneurosci.18-09-03117.1998
Paulino C., Kalienkova V., Lam A. K. M., Neldner Y., Dutzler R. (2017). Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 552 (7685), 421–425. doi:10.1038/nature24652
Pavlov T. S., Levchenko V., Ilatovskaya D. V., Palygin O., Staruschenko A. (2015). Impaired epithelial Na+ channel activity contributes to cystogenesis and development of autosomal recessive polycystic kidney disease in PCK rats. Pediatr. Res. 77 (1-1), 64–69. doi:10.1038/pr.2014.145
Pedemonte N., Galietta L. J. (2014). Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94 (2), 419–459. doi:10.1152/physrev.00039.2011
Pedersen S. F., Klausen T. K., Nilius B. (2015). The identification of a volume-regulated anion channel: An amazing odyssey. Acta Physiol. 213 (4), 868–881. doi:10.1111/apha.12450
Peng X., Xiao Z., Zhang J., Li Y., Dong Y., Du J. (2015). IL-17A produced by both γδ T and Th17 cells promotes renal fibrosis via RANTES-mediated leukocyte infiltration after renal obstruction. J. Pathol. 235 (1), 79–89. doi:10.1002/path.4430
Perez-Garcia M. T., Cidad P., Lopez-Lopez J. R. (2018). The secret life of ion channels: Kv1.3 potassium channels and proliferation. Am. J. Physiol. Cell Physiol. 314 (1), C27–C42. doi:10.1152/ajpcell.00136.2017
Perez-Verdaguer M., Capera J., Serrano-Novillo C., Estadella I., Sastre D., Felipe A. (2016). The voltage-gated potassium channel Kv1.3 is a promising multitherapeutic target against human pathologies. Expert Opin. Ther. Targets 20 (5), 577–591. doi:10.1517/14728222.2016.1112792
Picollo A., Pusch M. (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436 (7049), 420–423. doi:10.1038/nature03720
Pittala S., Krelin Y., Kuperman Y., Shoshan-Barmatz V. (2019). A mitochondrial VDAC1-based peptide greatly suppresses steatosis and NASH-associated pathologies in a mouse model. Mol. Ther. 27 (10), 1848–1862. doi:10.1016/j.ymthe.2019.06.017
Ponnusamy M., Ma L., Gong R., Pang M., Chin Y. E., Zhuang S. (2011). P2X7 receptors mediate deleterious renal epithelial-fibroblast cross talk. Am. J. Physiol. Ren. Physiol. 300 (1), F62–F70. doi:10.1152/ajprenal.00473.2010
Prakriya M., Lewis R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95 (4), 1383–1436. doi:10.1152/physrev.00020.2014
Quaresma M. C., Botelho H. M., Pankonien I., Rodrigues C. S., Pinto M. C., Costa P. R., et al. (2022). Exploring YAP1-centered networks linking dysfunctional CFTR to epithelial-mesenchymal transition. Life Sci. Alliance 5 (9), e202101326. doi:10.26508/lsa.202101326
Quaresma M. C., Pankonien I., Clarke L. A., Sousa L. S., Silva I. A. L., Railean V., et al. (2020). Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 11 (10), 920. doi:10.1038/s41419-020-03119-z
Rahaman S. O., Grove L. M., Paruchuri S., Southern B. D., Abraham S., Niese K. A., et al. (2014). TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Invest. 124 (12), 5225–5238. doi:10.1172/JCI75331
Rajasekaran A. K., Rajasekaran S. A. (2003). Role of Na-K-ATPase in the assembly of tight junctions. Am. J. Physiol. Ren. Physiol. 285 (3), F388–F396. doi:10.1152/ajprenal.00439.2002
Rajasekaran S. A., Huynh T. P., Wolle D. G., Espineda C. E., Inge L. J., Skay A., et al. (2010). Na, K-ATPase subunits as markers for epithelial-mesenchymal transition in cancer and fibrosis. Mol. Cancer Ther. 9 (6), 1515–1524. doi:10.1158/1535-7163.MCT-09-0832
Ralevic V., Burnstock G. (1998). Receptors for purines and pyrimidines. Pharmacol. Rev. 50 (3), 413–492.
Ramasubbu K., Gretz N., Bachmann S. (1998). Increased epithelial cell proliferation and abnormal extracellular matrix in rat polycystic kidney disease. J. Am. Soc. Nephrol. 9 (6), 937–945. doi:10.1681/ASN.V96937
Riera C. E., Huising M. O., Follett P., Leblanc M., Halloran J., Van Andel R., et al. (2014). TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 157 (5), 1023–1036. doi:10.1016/j.cell.2014.03.051
Rodrigo G. C., Standen N. B. (2005). ATP-sensitive potassium channels. Curr. Pharm. Des. 11 (15), 1915–1940. doi:10.2174/1381612054021015
Rothberg B. S. (2012). The BK channel: A vital link between cellular calcium and electrical signaling. Protein Cell 3 (12), 883–892. doi:10.1007/s13238-012-2076-8
Rotin D., Staub O. (2021). Function and regulation of the epithelial Na(+) channel ENaC. Compr. Physiol. 11 (3), 2017–2045. doi:10.1002/cphy.c200012
Roux B. (2017). Ion channels and ion selectivity. Essays Biochem. 61 (2), 201–209. doi:10.1042/EBC20160074
Ruan Y. C., Wang Y., Da Silva N., Kim B., Diao R. Y., Hill E., et al. (2014). CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 127 (20), 4396–4408. doi:10.1242/jcs.148098
Ryazanova L. V., Rondon L. J., Zierler S., Hu Z., Galli J., Yamaguchi T. P., et al. (2010). TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat. Commun. 1, 109. doi:10.1038/ncomms1108
Sala-Rabanal M., Yurtsever Z., Nichols C. G., Brett T. J. (2015). Secreted CLCA1 modulates TMEM16A to activate Ca(2+)-dependent chloride currents in human cells. eLife 4. doi:10.7554/eLife.05875
Saliba Y., Karam R., Smayra V., Aftimos G., Abramowitz J., Birnbaumer L., et al. (2015). Evidence of a role for fibroblast transient receptor potential canonical 3 Ca2+ channel in renal fibrosis. J. Am. Soc. Nephrol. 26 (8), 1855–1876. doi:10.1681/ASN.2014010065
Sansom S. C., Weinman E. J., O'neil R. G. (1984). Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am. J. Physiol. 247 (2), F291–F302. doi:10.1152/ajprenal.1984.247.2.F291
Satoh M., Nagasu H., Morita Y., Yamaguchi T. P., Kanwar Y. S., Kashihara N. (2012). Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am. J. Physiol. Ren. Physiol. 303 (12), F1641–F1651. doi:10.1152/ajprenal.00460.2012
Scheel O., Zdebik A. A., Lourdel S., Jentsch T. J. (2005). Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436 (7049), 424–427. doi:10.1038/nature03860
Schelling J. R. (2016). Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatr. Nephrol. 31 (5), 693–706. doi:10.1007/s00467-015-3169-4
Schenk L. K., Buchholz B., Henke S. F., Michgehl U., Daniel C., Amann K., et al. (2018). Nephron-specific knockout of TMEM16A leads to reduced number of glomeruli and albuminuria. Am. J. Physiol. Ren. Physiol. 315 (6), F1777–F86. doi:10.1152/ajprenal.00638.2017
Schild L., Giebisch G., Green R. (1988). Chloride transport in the proximal renal tubule. Annu. Rev. physiology 50, 97–110. doi:10.1146/annurev.ph.50.030188.000525
Schilling T., Miralles F., Eder C. (2014). TRPM7 regulates proliferation and polarisation of macrophages. J. Cell Sci. 127 (21), 4561–4566. doi:10.1242/jcs.151068
Schilling T., Quandt F. N., Cherny V. V., Zhou W., Heinemann U., Decoursey T. E., et al. (2000). Upregulation of Kv1.3 K(+) channels in microglia deactivated by TGF-beta. Am. J. Physiol. Cell Physiol. 279 (4), C1123–C1134. doi:10.1152/ajpcell.2000.279.4.C1123
Schlichter L. C., Mertens T., Liu B. (2011). Swelling activated Cl- channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels (Austin) 5 (2), 128–137. doi:10.4161/chan.5.2.14310
Schmick M., Bastiaens P. I. H. (2014). The interdependence of membrane shape and cellular signal processing. Cell 156 (6), 1132–1138. doi:10.1016/j.cell.2014.02.007
Scott R. I., Markus D., Clapham David E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi:10.1146/annurev.physiol.68.040204.100431
Seino S., Miki T. (2003). Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 81 (2), 133–176. doi:10.1016/s0079-6107(02)00053-6
Serrano-Albarras A., Estadella I., Cirera-Rocosa S., Navarro-Perez M., Felipe A. (2018). Kv1.3: A multifunctional channel with many pathological implications. Expert Opin. Ther. Targets 22 (2), 101–105. doi:10.1080/14728222.2017.1420170
Shao P. P., Liu C. J., Xu Q., Zhang B., Li S. H., Wu Y., et al. (2018). Eplerenone reverses cardiac fibrosis via the suppression of tregs by inhibition of Kv1.3 channel. Front. Physiol. 9, 899. doi:10.3389/fphys.2018.00899
Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., Arbel N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 31 (3), 227–285. doi:10.1016/j.mam.2010.03.002
Shukla A., Edwards R., Yang Y., Hahn A., Folkers K., Ding J., et al. (2014). CLIC4 regulates TGF-beta-dependent myofibroblast differentiation to produce a cancer stroma. Oncogene 33 (7), 842–850. doi:10.1038/onc.2013.18
Shukla A., Malik M., Cataisson C., Ho Y., Friesen T., Suh K. S., et al. (2009). TGF-beta signalling is regulated by Schnurri-2-dependent nuclear translocation of CLIC4 and consequent stabilization of phospho-Smad2 and 3. Nat. Cell Biol. 11 (6), 777–784. doi:10.1038/ncb1885
Shukla A., Yuspa S. H. (2010). CLIC4 and schnurri-2: A dynamic duo in TGF-beta signaling with broader implications in cellular homeostasis and disease. Nucl. (Austin, Tex) 1 (2), 144–149. doi:10.4161/nucl.1.2.10920
Simons M., Gault W. J., Gotthardt D., Rohatgi R., Klein T. J., Shao Y., et al. (2009). Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat. Cell Biol. 11 (3), 286–294. doi:10.1038/ncb1836
Solanki A. K., Arif E., Morinelli T., Wilson R. C., Hardiman G., Deng P., et al. (2018). A novel CLCN5 mutation associated with focal segmental glomerulosclerosis and podocyte injury. Kidney Int. Rep. 3 (6), 1443–1453. doi:10.1016/j.ekir.2018.06.003
Solini A., Iacobini C., Ricci C., Chiozzi P., Amadio L., Pricci F., et al. (2005). Purinergic modulation of mesangial extracellular matrix production: Role in diabetic and other glomerular diseases. Kidney Int. 67 (3), 875–885. doi:10.1111/j.1523-1755.2005.00152.x
Solini A., Menini S., Rossi C., Ricci C., Santini E., Blasetti Fantauzzi C., et al. (2013). The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: Possible role of NLRP3 inflammasome activation. J. Pathol. 231 (3), 342–353. doi:10.1002/path.4237
Sontia B., Montezano A. C., Paravicini T., Tabet F., Touyz R. M. (2008). Downregulation of renal TRPM7 and increased inflammation and fibrosis in aldosterone-infused mice: Effects of magnesium. Hypertension 51 (4), 915–921. doi:10.1161/HYPERTENSIONAHA.107.100339
Staruschenko A., Spires D., Palygin O. (2019). Role of TRPC6 in progression of diabetic kidney disease. Curr. Hypertens. Rep. 21 (7), 48. doi:10.1007/s11906-019-0960-9
Stauber T., Weinert S., Jentsch T. J. (2012). Cell biology and physiology of CLC chloride channels and transporters. Compr. Physiol. 2 (3), 1701–1744. doi:10.1002/cphy.c110038
Steinmeyer K., Schwappach B., Bens M., Vandewalle A., Jentsch T. J. (1995). Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J. Biol. Chem. 270 (52), 31172–31177. doi:10.1074/jbc.270.52.31172
Stienstra R., Van Diepen J. A., Tack C. J., Zaki M. H., Van De Veerdonk F. L., Perera D., et al. (2011). Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 108 (37), 15324–15329. doi:10.1073/pnas.1100255108
Stocker M. (2004). Ca(2+)-activated K+ channels: Molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5 (10), 758–770. doi:10.1038/nrn1516
Sudarikova A. V., Vasileva V. Y., Sultanova R. F., Ilatovskaya D. V. (2021). Recent advances in understanding ion transport mechanisms in polycystic kidney disease. Clin. Sci. 135 (21), 2521–2540. doi:10.1042/CS20210370
Sun H., Li H., Yan J., Wang X., Xu M., Wang M., et al. (2022). Loss of CLDN5 in podocytes deregulates WIF1 to activate WNT signaling and contributes to kidney disease. Nat. Commun. 13 (1), 1600. doi:10.1038/s41467-022-29277-6
Sun H., Wang Y., Zhang J., Chen Y., Liu Y., Lin Z., et al. (2018). CFTR mutation enhances Dishevelled degradation and results in impairment of Wnt-dependent hematopoiesis. Cell Death Dis. 9 (3), 275. doi:10.1038/s41419-018-0311-9
Sun L., Dong Y., Zhao J., Yin Y., Zheng Y. (2016). The CLC-2 chloride Channel modulates ECM synthesis, differentiation, and migration of human conjunctival fibroblasts via the PI3K/akt signaling pathway. Int. J. Mol. Sci. 17 (6), E910. doi:10.3390/ijms17060910
Suzuki M., Morita T., Iwamoto T. (2006). Diversity of Cl(-) channels. Cell. Mol. Life Sci. 63 (1), 12–24. doi:10.1007/s00018-005-5336-4
Suzuki S., Penner R., Fleig A. (2020). TRPM7 contributes to progressive nephropathy. Sci. Rep. 10 (1), 2333. doi:10.1038/s41598-020-59355-y
Syeda R., Qiu Z., Dubin A. E., Murthy S. E., Florendo M. N., Mason D. E., et al. (2016). LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell 164 (3), 499–511. doi:10.1016/j.cell.2015.12.031
Takezawa R., Schmitz C., Demeuse P., Scharenberg A. M., Penner R., Fleig A. (2004). Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc. Natl. Acad. Sci. U. S. A. 101 (16), 6009–6014. doi:10.1073/pnas.0307565101
Tan T. K., Zheng G., Hsu T. T., Wang Y., Lee V. W., Tian X., et al. (2010). Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am. J. Pathol. 176 (3), 1256–1270. doi:10.2353/ajpath.2010.090188
Tarjus A., Gonzalez-Rivas C., Amador-Martinez I., Bonnard B., Lopez-Marure R., Jaisser F., et al. (2019). The absence of endothelial sodium channel α (αENaC) reduces renal ischemia/reperfusion injury. Int. J. Mol. Sci. 20 (13), E3132. doi:10.3390/ijms20133132
Therkildsen J. R., Christensen M. G., Tingskov S. J., Wehmoller J., Norregaard R., Praetorius H. A. (2019). Lack of P2X7 receptors protects against renal fibrosis after pyelonephritis with alpha-hemolysin-producing Escherichia coli. Am. J. Pathol. 189 (6), 1201–1211. doi:10.1016/j.ajpath.2019.02.013
Tinker A., Aziz Q., Li Y., Specterman M. (2018). ATP-sensitive potassium channels and their physiological and pathophysiological roles. Compr. Physiol. 8 (4), 1463–1511. doi:10.1002/cphy.c170048
Toyama K., Wulff H., Chandy K. G., Azam P., Raman G., Saito T., et al. (2008). The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest. 118 (9), 3025–3037. doi:10.1172/JCI30836
Turner C. M., Vonend O., Chan C., Burnstock G., Unwin R. J. (2003). The pattern of distribution of selected ATP-sensitive P2 receptor subtypes in normal rat kidney: An immunohistological study. Cells Tissues Organs 175 (2), 105–117. doi:10.1159/000073754
Ulmasov B., Bruno J., Gordon N., Hartnett M. E., Edwards J. C. (2009). Chloride intracellular channel protein-4 functions in angiogenesis by supporting acidification of vacuoles along the intracellular tubulogenic pathway. Am. J. Pathol. 174 (3), 1084–1096. doi:10.2353/ajpath.2009.080625
Urrego D., Tomczak A. P., Zahed F., Stuhmer W., Pardo L. A. (2014). Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369 (1638), 20130094. doi:10.1098/rstb.2013.0094
Vallon V., Unwin R., Inscho E. W., Leipziger J., Kishore B. K. (2020). Extracellular nucleotides and P2 receptors in renal function. Physiol. Rev. 100 (1), 211–269. doi:10.1152/physrev.00038.2018
Vasko R., Xavier S., Chen J., Lin C. H., Ratliff B., Rabadi M., et al. (2014). Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: Relevance to fibrosis of vascular senescence. J. Am. Soc. Nephrol. 25 (2), 276–291. doi:10.1681/ASN.2013010069
Voets T., Droogmans G., Raskin G., Eggermont J., Nilius B. (1999). Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc. Natl. Acad. Sci. U. S. A. 96 (9), 5298–5303. doi:10.1073/pnas.96.9.5298
Voss F. K., Ullrich F., Munch J., Lazarow K., Lutter D., Mah N., et al. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344 (6184), 634–638. doi:10.1126/science.1252826
Vriens J., Nilius B., Vennekens R. (2008). Herbal compounds and toxins modulating TRP channels. Curr. Neuropharmacol. 6 (1), 79–96. doi:10.2174/157015908783769644
Wang B., Xie J., He H. Y., Huang E. W., Cao Q. H., Luo L., et al. (2017). Suppression of CLC-3 chloride channel reduces the aggressiveness of glioma through inhibiting nuclear factor-κB pathway. Oncotarget 8 (38), 63788–63798. doi:10.18632/oncotarget.19093
Wang H., Gao M., Li J., Sun J., Wu R., Han D., et al. (2019). MMP-9-positive neutrophils are essential for establishing profibrotic microenvironment in the obstructed kidney of UUO mice. Acta Physiol. 227 (2), e13317. doi:10.1111/apha.13317
Wang J., Morishima S., Okada Y. (2003). Ik channels are involved in the regulatory volume decrease in human epithelial cells. Am. J. Physiol. Cell Physiol. 284 (1), C77–C84. doi:10.1152/ajpcell.00132.2002
Wang J., Ullah S. H., Li M., Zhang M., Zhang F., Zheng J., et al. (2019). DR region specific antibody ameliorated but ouabain worsened renal injury in nephrectomized rats through regulating Na, K-ATPase mediated signaling pathways. Aging 11 (4), 1151–1162. doi:10.18632/aging.101815
Wang X., Li G., Guo J., Zhang Z., Zhang S., Zhu Y., et al. (2019). Kv1.3 channel as a key therapeutic target for neuroinflammatory diseases: State of the art and beyond. Front. Neurosci. 13, 1393. doi:10.3389/fnins.2019.01393
Wang Y., Babankova D., Huang J., Swain G. M., Wang D. H. (2008). Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension 52 (2), 264–270. doi:10.1161/HYPERTENSIONAHA.108.110197
Wang Y., Wang D. H. (2011). Protective effect of TRPV1 against renal fibrosis via inhibition of TGF-β/Smad signaling in DOCA-salt hypertension. Mol. Med. 17 (11-12), 1204–1212. doi:10.2119/molmed.2011.00063
Wang Y., Wang M., Ning F., Ren D., Tao J., Xie W., et al. (2022). A novel role of BK potassium channel activity in preventing the development of kidney fibrosis. Kidney Int. 101 (5), 945–962. doi:10.1016/j.kint.2021.11.033
Warntges S., Grone H. J., Capasso G., Lang F. (2001). Cell volume regulatory mechanisms in progression of renal disease. J. Nephrol. 14 (5), 319–326.
Wei A. D., Gutman G. A., Aldrich R., Chandy K. G., Grissmer S., Wulff H. (2005). International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 57 (4), 463–472. doi:10.1124/pr.57.4.9
Wei X., Gao P., Pu Y., Li Q., Yang T., Zhang H., et al. (2017). Activation of TRPV4 by dietary apigenin antagonizes renal fibrosis in deoxycorticosterone acetate (DOCA)-salt-induced hypertension. Clin. Sci. 131 (7), 567–581. doi:10.1042/CS20160780
Wei X., Wei X., Lu Z., Li L., Hu Y., Sun F., et al. (2020). Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism. 105, 154182. doi:10.1016/j.metabol.2020.154182
Welling P. A. (2016). Roles and regulation of renal K channels. Annu. Rev. Physiol. 78, 415–435. doi:10.1146/annurev-physiol-021115-105423
Wu K. L., Khan S., Lakhe-Reddy S., Jarad G., Mukherjee A., Obejero-Paz C. A., et al. (2004). The NHE1 Na+/H+ exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J. Biol. Chem. 279 (25), 26280–26286. doi:10.1074/jbc.M400814200
Wu K. L., Khan S., Lakhe-Reddy S., Wang L., Jarad G., Miller R. T., et al. (2003). Renal tubular epithelial cell apoptosis is associated with caspase cleavage of the NHE1 Na+/H+ exchanger. Am. J. Physiol. Ren. Physiol. 284 (4), F829–F839. doi:10.1152/ajprenal.00314.2002
Wu L. J., Sweet T. B., Clapham D. E. (2010). International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol. Rev. 62 (3), 381–404. doi:10.1124/pr.110.002725
Wu P., Ren Y., Ma Y., Wang Y., Jiang H., Chaudhari S., et al. (2017). Negative regulation of Smad1 pathway and collagen IV expression by store-operated Ca(2+) entry in glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 312 (6), F1090–F100. doi:10.1152/ajprenal.00642.2016
Wu P., Wang Y., Davis M. E., Zuckerman J. E., Chaudhari S., Begg M., et al. (2015). Store-operated Ca2+ channels in mesangial cells inhibit matrix protein expression. J. Am. Soc. Nephrol. 26 (11), 2691–2702. doi:10.1681/ASN.2014090853
Wu Y. L., Xie J., An S. W., Oliver N., Barrezueta N. X., Lin M. H., et al. (2017). Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 91 (4), 830–841. doi:10.1016/j.kint.2016.09.039
Wu Y. S., Liang S., Li D. Y., Wen J. H., Tang J. X., Liu H. F. (2021). Cell cycle dysregulation and renal fibrosis. Front. Cell Dev. Biol. 9, 714320. doi:10.3389/fcell.2021.714320
Wu Z., Yin W., Sun M., Si Y., Wu X., Chen M. (2020). BKCa mediates dysfunction in high glucose induced mesangial cell injury via TGF-β1/smad2/3 signaling pathways. Int. J. Endocrinol. 2020, 3260728. doi:10.1155/2020/3260728
Wulff H., Castle N. A., Pardo L. A. (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8 (12), 982–1001. doi:10.1038/nrd2983
Xie J. X., Zhang S., Cui X., Zhang J., Yu H., Khalaf F. K., et al. (2018). Na/K-ATPase/src complex mediates regulation of CD40 in renal parenchyma. Nephrol. Dial. Transpl. 33 (7), 1138–1149. doi:10.1093/ndt/gfx334
Xu T., Wu B. M., Yao H. W., Meng X. M., Huang C., Ni M. M., et al. (2015). Novel insights into TRPM7 function in fibrotic diseases: A potential therapeutic target. J. Cell. Physiol. 230 (6), 1163–1169. doi:10.1002/jcp.24801
Xu W., Hong S. J., Zeitchek M., Cooper G., Jia S., Xie P., et al. (2015). Hydration status regulates sodium flux and inflammatory pathways through epithelial sodium channel (ENaC) in the skin. J. Invest. Dermatol. 135 (3), 796–806. doi:10.1038/jid.2014.477
Xue H., Zhang Y. L., Liu G. S., Wang H. (2005). A new ATP-sensitive potassium channel opener protects the kidney from hypertensive damage in spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 315 (2), 501–509. doi:10.1124/jpet.105.089722
Yan Y., Shapiro A. P., Haller S., Katragadda V., Liu L., Tian J., et al. (2013). Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 288 (47), 34249–34258. doi:10.1074/jbc.M113.461020
Yang D., Yang D. (2013). Role of intracellular Ca2+ and Na+/Ca2+ exchanger in the pathogenesis of contrast-induced acute kidney injury. Biomed. Res. Int. 2013, 678456. doi:10.1155/2013/678456
Yang S. X., Zhang Z. C., Bai H. L. (2019). ClC-5 alleviates renal fibrosis in unilateral ureteral obstruction mice. Hum. Cell 32 (3), 297–305. doi:10.1007/s13577-019-00253-5
Yang Y. D., Cho H., Koo J. Y., Tak M. H., Cho Y., Shim W. S., et al. (2008). TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455 (7217), 1210–1215. doi:10.1038/nature07313
Yang Z., He L. J., Sun S. R. (2019). Role of endothelial cells in renal fibrosis. Adv. Exp. Med. Biol. 1165, 145–163. doi:10.1007/978-981-13-8871-2_8
Yin Z., Tong Y., Zhu H., Watsky M. A. (2008). ClC-3 is required for LPA-activated Cl- current activity and fibroblast-to-myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 294 (2), C535–C542. doi:10.1152/ajpcell.00291.2007
Zhang J. T., Wang Y., Chen J. J., Zhang X. H., Dong J. D., Tsang L. L., et al. (2017). Defective CFTR leads to aberrant beta-catenin activation and kidney fibrosis. Sci. Rep. 7 (1), 5233. doi:10.1038/s41598-017-05435-5
Zhang M., Chen J., Lai L., You L., Lin S., Hao C., et al. (2010). Aldosterone promotes fibronectin synthesis in rat mesangial cells via ERK1/2-stimulated Na-H+ exchanger isoform 1. Am. J. Nephrol. 31 (1), 75–82. doi:10.1159/000256665
Zhang Y., Yin N., Sun A., Wu Q., Hu W., Hou X., et al. (2020). Transient receptor potential channel 6 knockout ameliorates kidney fibrosis by inhibition of epithelial-mesenchymal transition. Front. Cell Dev. Biol. 8, 602703. doi:10.3389/fcell.2020.602703
Zhao H., Carney K. E., Falgoust L., Pan J. W., Sun D., Zhang Z. (2016). Emerging roles of Na⁺/H⁺ exchangers in epilepsy and developmental brain disorders. Prog. Neurobiol. 138-140, 19–35. doi:10.1016/j.pneurobio.2016.02.002
Zheng J. (2013). Molecular mechanism of TRP channels. Compr. Physiol. 3 (1), 221–242. doi:10.1002/cphy.c120001
Zitka O., Kukacka J., Krizkova S., Huska D., Adam V., Masarik M., et al. (2010). Matrix metalloproteinases. Curr. Med. Chem. 17 (31), 3751–3768. doi:10.2174/092986710793213724
Keywords: ion channels, renal fibrosis, kidney, Cl− channels, Ca2+ signaling, K+ channels, Na+ transport
Citation: Yan P, Ke B and Fang X (2022) Ion channels as a therapeutic target for renal fibrosis. Front. Physiol. 13:1019028. doi: 10.3389/fphys.2022.1019028
Received: 14 August 2022; Accepted: 20 September 2022;
Published: 05 October 2022.
Edited by:
Susumu Ohya, Nagoya City University, JapanReviewed by:
Anastasia Sudarikova, Institute of Cytology (RAS), RussiaYoshiaki Suzuki, Nagoya City University, Japan
Copyright © 2022 Yan, Ke and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangdong Fang, eGlhbmdkb25nZmFuZzgxOEBzaW5hLmNvbQ==