 An-Jin Yang
An-Jin Yang Ning-Na Yin
Ning-Na Yin Dan-Lu Chen
Dan-Lu Chen Yu-Ruo Guo
Yu-Ruo Guo Yu-Jie Zhao
Yu-Jie Zhao Nai-Yong Liu
Nai-Yong Liu- 1Key Laboratory of Forest Disaster Warning and Control of Yunnan Province, Southwest Forestry University, Kunming, China
- 2Key Laboratory of National Forestry and Grassland Administration on Biodiversity Conservation in Southwest China, Southwest Forestry University, Kunming, China
The wood-boring beetles, including the majority of Cerambycidae, have developed the ability to metabolize a variety of toxic compounds derived from host plants and the surrounding environment. However, detoxification mechanisms underlying the evolutionary adaptation of a cerambycid beetle Pharsalia antennata to hosts and habitats are largely unexplored. Here, we characterized three key gene families in relation to detoxification (cytochrome P450 monooxygenases: P450s, carboxylesterases: COEs and glutathione-S-transferases: GSTs), by combinations of transcriptomics, gene identification, phylogenetics and expression profiles. Illumina sequencing generated 668,701,566 filtered reads in 12 tissues of P. antennata, summing to 100.28 gigabases data. From the transcriptome, 215 genes encoding 106 P450s, 77 COEs and 32 GSTs were identified, of which 107 relatives were differentially expressed genes. Of the identified 215 genes, a number of relatives showed the orthology to those in Anoplophora glabripennis, revealing 1:1 relationships in 94 phylogenetic clades. In the trees, P. antennata detoxification genes mainly clustered into one or two subfamilies, including 64 P450s in the CYP3 clan, 33 COEs in clade A, and 20 GSTs in Delta and Epsilon subclasses. Combining transcriptomic data and PCR approaches, the numbers of detoxification genes expressed in abdomens, antennae and legs were 188, 148 and 141, respectively. Notably, some genes exhibited significantly sex-biased levels in antennae or legs of both sexes. The findings provide valuable reference resources for further exploring xenobiotics metabolism and odorant detection in P. antennata.
Introduction
The cerambycid beetles are members of the most diverse groups, with over 30,000 described extant species to date. The majority of cerambycid larvae feed on wood tissues of trees and thus are typical representatives of wood-boring pests (Slipinski et al., 2011). Similar to other herbivorous insects, each cerambycid beetle has evolved specialized feeding host plant(s). Therefore, they are most likely to develop various detoxification mechanisms to adapt the hosts. Apart from that, due to the differences of habitats, toxic compounds (like insecticides and herbicides) from the external environment further shape the evolution of detoxification-related enzyme genes, such as cytochrome P450 monooxygenases (P450s), carboxylesterases (COEs), and glutathione-S-transferases (GSTs) (Allison et al., 2004; Despres et al., 2007).
Insect P450 is the most extensively studied detoxification gene family, emphasizing the roles in insecticide resistance (Zhu et al., 2010; Zimmer et al., 2014; Dermauw et al., 2020). Beyond the degradation of P450s to synthetic insecticides, studies have demonstrated their involvement in detoxifying plant-derived toxic compounds (Amezian et al., 2022). Hence, this gene family is of paramount important for the adaption of herbivorous insects to host plants and habitats. For wood-boring pests including the majority of cerambycid beetles, they spend most of the life history in the trunk of trees. To defense plant secondary metabolites (often defensive compounds), some members of the P450 gene repertoire regulate their expression or generate many gene copies via gene duplications or alternative splicing, particularly for the CYP3 and CYP4 clans. In Anoplophora glabripennis, the expression of 25 P450 genes in guts was modulated after larvae fed on sugar maple (McKenna et al., 2016). Similarly, the genes of CYP4 and CYP9 clades in Ips paraconfusus were induced by feeding on the host plant Pinus ponderosa (Huber et al., 2007). Beyond the detoxification, P450 genes in Ips pini (IpinCYP9T2) and Dendroctonus ponderosae (DponCYP345E2) were associated with the pheromone production and odorant detection, respectively (Sandstrom et al., 2006; Keeling et al., 2013a).
The COE is another critical component of the insect defense system and is capable of hydrolyzing a variety of esters of carboxylic acids (Wheelock et al., 2005; Oakeshott et al., 2019). Similar to P450s, the transcription of COE genes is also induced by xenobiotic compounds, including host plant and man-made chemicals. In A. glabripennis, a number of COE genes had inducible expression in sugar maple-fed larvae, suggesting their putative roles in the detoxification of plant allelochemicals (McKenna et al., 2016). For the roles of COEs in metabolizing insecticides, several studies showed a particular focus on their resistance to organophosphates, including Laodelphax striatellus (Zhang et al., 2012), Locusta migratoria manilensis (Zhang et al., 2011), and Nilaparvata lugens (Small and Hemingway, 2000). In some cases, insect COEs specifically or highly expressed in antennae are responsible for the clearance of redundant plant odorants or sex pheromones, so as to recover the sensitivity of olfactory sensory neurons to semiochemicals (Durand et al., 2010; He et al., 2020; Wang et al., 2021).
In comparison to the above two gene families, the numbers of GSTs in most insects were relatively small, ranging from 28 genes in A. glabripennis to 43 in Aethina tumida for the Coleoptera, and 22 genes in Plutella xylostella to 45 in Spodoptera litura for the Lepidoptera (You et al., 2015; McKenna et al., 2016; Cheng et al., 2017; Evans et al., 2018; Zhao et al., 2020b). In the two other cerambycid beetles Xylotrechus quadripes and Rhaphuma horsfieldi, GST numbers identified from the transcriptomes were 18 and 20, respectively (Zhao et al., 2020b). As a multifunctional gene family presented ubiquitously in insects, GSTs, mainly members of insect-specific Delta and Epsilon classes, become well-known detoxification enzymes due to their involvement in insecticide resistance. Such the resistance of COEs to insecticides occurred in Coleoptera (Kostaropoulos et al., 2001), Lepidoptera (Liu et al., 2017), Diptera (Ranson et al., 2001) and Hemiptera (Zhou et al., 2013). Outside their association in insecticide resistance, GSTs also responded to plant defensive compounds as a positive feedback for adapting feeding host plants (Dai et al., 2016; Liu et al., 2021).
The cerambycid beetle, Pharsalia antennata Gahan (Coleoptera: Cerambycidae), is a wood-boring pest and feeds mainly on the Juglandaceae family. To date, information on this beetle is focused on its morphological characteristics, including the ultrastructure of sensilla on antennae and tarsi of both sexes (Yin et al., 2020). Like other cerambycids, P. antennata utilizes a detoxification system to adapt diverse host plants and complex habitats. Previously, the phase II enzyme uridine diphosphate (UDP)-glycosyltransferase (UGT) in P. antennata was surveyed, summing to a comparable number of 59 transcripts (Yin et al., 2022). Such the number raises the possibility that the other gene families in relation to detoxification may also have lineage-specific expansions in this beetle, deserving further studies. In the present study, we identified and characterized three other detoxification enzyme families (P450s, COEs and GSTs) in P. antennata, based on the transcriptomic data of 12 adult tissues. This study identifies candidate molecular targets for detoxifying toxic compounds and greatly improves the detoxification gene repertoires in P. antennata.
Materials and methods
Insects and tissue samples
P. antennata adults were collected from the trunks of Juglans sigillata at Santai Village, Dayao County, Chuxiong City, Yunnan Province (26°00′01.6″ N, 101°04′04.7″ E). The damaged walnut trees were cut and brought to the laboratory. Rearing conditions were described in a previous study (Yin et al., 2022). After adult emergence, female and male beetles were distinguished by sex, following morphological characteristics of abdomens and the lengths of antennae (Yin et al., 2020). To construct cDNA libraries of various tissues and investigate expression profiles of detoxification genes, we dissected 12 tissues from 3– to 5–days–old beetles of both sexes, including 10 antennae, five heads without antennae, three thoraxes, three abdomens, 10 legs and 15 wings per biological replicate. In the expression profiling analyses, three biological pools were collected. All the tissues were immediately frozen and then stored at –80°C.
Total ribonucleic acid preparation and cDNA synthesis
Total RNA of each tissue was extracted using TRIzol reagent (Ambion, Life Technologies, Carlsbad, CA, United States), following the manufacturer’s instructions. Briefly, tissues were homogenized with 1 ml of TRIzol. We used a NanoDrop 1,000 Spectrophotometer (Thermo Fisher Scientific, San Jose, CA, United States) and agarose gel electrophoresis (1%, w/v) to measure the quality and concentration of purified RNA samples. Genomic DNA in RNA samples was digested with DNase I (Amplification Grade; Invitrogen Life Technologies, Carlsbad, CA, United States). For de novo transcriptome sequencing, at least 1 μg of total RNA for each tissue was sent to the Novogene Bioinformatics Technology Co. Ltd. (Tianjing, China). In the expression profiles of genes, cDNA synthesis was conducted with 1 μg of total RNA, according to the protocols of a PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, Liaoning, China). Briefly, the reverse transcription reaction was performed at 37°C for 15 min, and stopped at 85°C for 5 s. Three biological templates for each tissue were prepared.
Sequencing, assembly and annotation of the transcriptome
After assessing the quality and quantity of RNA samples by using a Qubit 2.0 Fluorometer (Invitrogen Life Technologies, Waltham, MA, United States) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States), cDNA libraries were produced via NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB, United States), along with the manufacturer’s recommendations. The libraries were sequenced on an Illumina Hiseq–PE150 platform using a paired-end sequencing strategy. The initially generating sequences (raw reads) were filtered after removing adapter sequences, reads with poly-N and low quality reads. Next, the resulting reads (clean reads) were assembled into the trinity transcriptome using Trinity v2.5.1 (Grabherr et al., 2011), or used for subsequent analyses. After the transcripts were clustered by Corset v1.05 with default settings (Davidson and Oshlack, 2014), the longest transcript in each cluster was picked and pooled into a unigene database (defined as the unigene transcriptome). Using the unigenes as queries, we aligned them into seven functional databases (GO, Gene Ontology; KO, KEGG Ortholog; KOG/COG, EuKaryotic Orthologous Groups/Clusters of Orthologous Groups; NR, National Center for Biotechnology Information (NCBI) non-redundant (NR) protein sequences; NT, NCBI non-redundant nucleotide sequences; PFAM, Protein family and SwissProt). The annotated information of unigenes in each database was summarized.
Estimation of gene expression levels
To obtain the expression levels of each gene in various tissues, clean reads from each tissue were mapped to the reference unigene transcriptome using Bowtie2 (Langmead and Salzberg, 2012). Next, reads of each gene in different tissues were counted by RSEM v1.2.15 (Li and Dewey, 2011). Expression levels of genes were calculated using a FPKM (fragments per kilobase of transcript per million mapped reads) method (Trapnell et al., 2010). For the analysis of differentially expressed genes (DEGs), the edgeR package (version 3.0.8) was used to adjust read counts via one scaling normalized factor (Robinson et al., 2010). Differential expression between two samples was determined using the DEGSeq R package (version 1.12.0) (Wang et al., 2009). The statistical significance of DEGs was set at a corrected p value <0.005 and |log2(foldchange)| >1.
Identification of candidate detoxification genes
Considering that some short sequences were possibly lost during the process of Corset clusters, we queried two different transcriptomic data, i.e., the trinity transcriptome and the unigene transcriptome. A sequence query file was set using amino acid sequences of detoxification genes (P450s, COEs and GSTs) from three cerambycid beetles (A. glabripennis, X. quadripes, and R. horsfieldi) (McKenna et al., 2016; Zhao et al., 2020b) and a model coleopteran organism Tribolium castaneum (Oakeshott et al., 2010; Shi et al., 2012; Zhu et al., 2013). Three detoxification gene families in P. antennata were identified using amino acid sequences collected above as queries, by blasting the above two transcriptomes. The searching parameters were as follows: maximum hit of 20, BLOSUM62 matrix and a max E-value of 1e−5. The homology and conserved domains of detoxification enzymes were determined by blasting the NCBI NR protein sequence database. Further, we used identified genes as queries to iteratively search the transcriptomes until no novel candidate genes were found.
Phylogenetics
Due to the size differences of three detoxification gene repertoires, we selected the corresponding protein sequences from different species to build the trees. In the P450 tree, P450 sequences from A. glabripennis (McKenna et al., 2016) and P. antennata were included. The COE tree was constructed with the protein sequences from A. glabripennis (McKenna et al., 2016), P. antennta, T. castaneum (Oakeshott et al., 2010) and Drosophila melanogaster (Claudianos et al., 2006). In the GST tree, seven coleopteran species were chosen, including A. glabripennis (McKenna et al., 2016), D. ponderosae (Keeling et al., 2013b), Leptinotarsa decemlineata (Schoville et al., 2018), R. horsfieldi, X. quadripes (Zhao et al., 2020b), P. antennata and T. castaneum (Shi et al., 2012). The protein sequences were aligned using MAFFT v7.450 with the L–INS–I algorithm and default parameters (scoring matrix: BLOSUM62, offset value: 0.123 and gap open penalty: 1.53) (Katoh and Standley, 2013). The aligned sequences were used to infer maximum likelihood trees under the Whelan–And–Goldman (WAG) model with FastTree v2.1.11 (Price et al., 2010). The tree was visualized and edited using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Expression profiling analysis
Based on the results by RNA sequencing (RNA–Seq), we randomly selected 48 P450s, 38 COEs and 15 GSTs to validate and investigate their expression in 12 tissues of both sexes. These genes were mainly composed of antenna-, thorax- or abdomen-enriched relatives. A reverse transcription PCR (RT–PCR) strategy was first employed to determine their tissue expression characteristics. To measure the quality and reliability of templates, we used a control gene in P. antennata (ribosomal protein S3, PantRPS3) to detect their usability (Yin et al., 2022). PCR reactions were conducted with a 25 μL mixture, containing 1 μL of cDNA, 1.5 μL of forward or reverse primers (10 μM). 2 μL of dNTP mixture (each 2.5 mM), 2.5 μL of 10×PCR buffer (Mg2+), 0.15 μL of TaKaRa Taq (5 U/μL) and 15.35 μL of sterile water. According to the manufacturer’s protocols of a TaKaRa Taq™ Kit (TaKaRa, Dalian, Liaoning, China), an annealing temperature of 55°C was applied with 35 cycles. If the results were inconsistent between RNA–Seq and RT–PCR, an additional replicate was performed using the third biological pool as the template. PCR products were analyzed using 1.2% (w/v) agarose gels. The primer sequences are listed in Supplementary Table S1.
To further determine the relative expression of detoxification genes in tissues and their putative roles in chemoreception, we picked 25 genes to examine their expression in antennae, legs and other tissues of female and male beetles using real-time quantitative PCR (qPCR). A reaction mixture of 20 μL was prepared, comprising 2 μL of cDNA, 10 μL of Bestar® SybrGreen qPCR mastermix (DBI® Bioscience, Germany), 0.5 μL of forward or reverse primers (10 μM), and 7 μL of sterile water. The specificity of PCR products was validated by melting curve analysis from 60 to 95°C. The amplification efficiencies of primers designed by Beacon Designer 8.14 (PREMIER Biosoft International, CA, United States) were checked using gradient dilutions of cDNA templates. The qPCR was run on qTOWER 2.2 (Analytic Jena AG, Germany) under the conditions: 2 min at 95°C, 40 cycles of 10 s at 95°C, 31 s at 58°C, and 30 s at 72°C. Fluorescence readings were taken at the elongation step of 72°C. Expression levels of detoxification genes were computed relative to two control genes (PantRPS3 and ribosomal protein L10, PantRPL10), using a Q–Gene method (Muller et al., 2002; Simon, 2003). In order to ensure the reproducibility and reliability of qPCR data, the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines were followed (Bustin et al., 2009). Three biological templates for each tissue were used. The primers are listed in Supplementary Table S1. Significant differences between female and male tissues were compared by Student’s t–test in GraphPad Prism 7.00 (GraphPad Software Inc., San Diego, CA, United States).
Results
Transcriptome sequencing and assembly
Through the Illumina sequencing technology, we constructed and sequenced 12 cDNA libraries, representing 12 different tissues of female and male adults. In total, 669,610,108 raw reads were produced, comprising 327,705,854 sequences in males and 341,904,254 in females. All raw data of 12 tissues have been deposited in the NCBI Sequence Read Archive (SRA) database with the BioProject accession number PRJNA821002. The numbers of clean reads were variable in tissues, ranging from 44,598,376 sequences in male abdomens to 66,321,758 in female antennae. The clean data were summed to 100.28 gigabases (G), containing 668,701,566 sequences, high Q20 (97.36–97.90%) and Q30 (92.26–93.75%), and an average of 43.33% GC content (Table 1). All clean reads of 12 tissues were merged into a reference transcriptome (i.e., the unigene transcriptome), with a size of 76.9 megabytes (Mb). Such the assembly generated 196,717 transcripts and 81,744 unigenes, of which 41.83% (82,290) of the former were less than 301 bp in length and over half of unigenes (55.74%, 45,567) varied from 301 to 1,000 bp (Figure 1A).
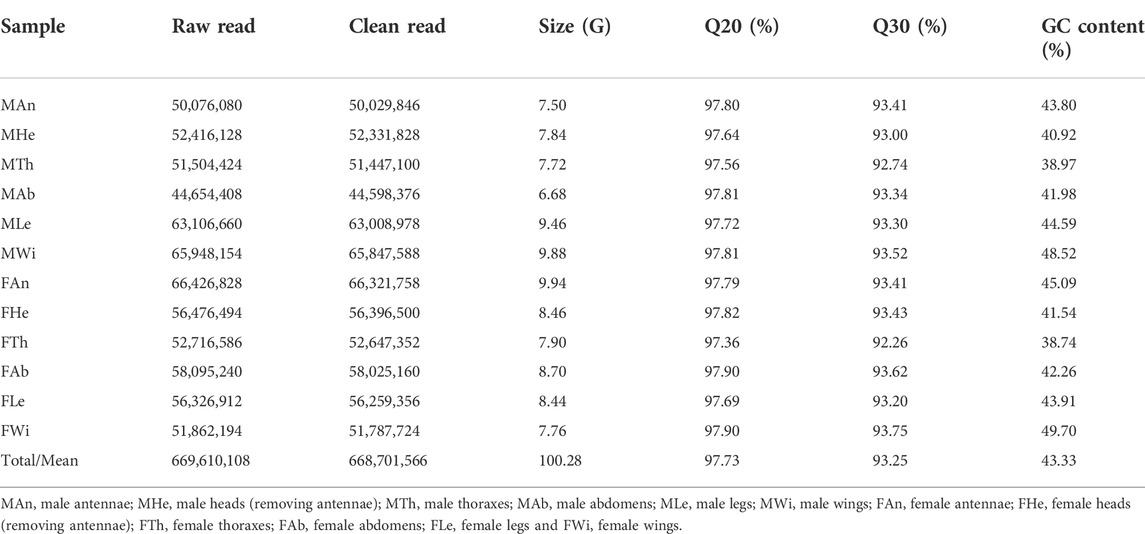
TABLE 1. Summary statistics of the transcriptome in various tissues of P. antennata.
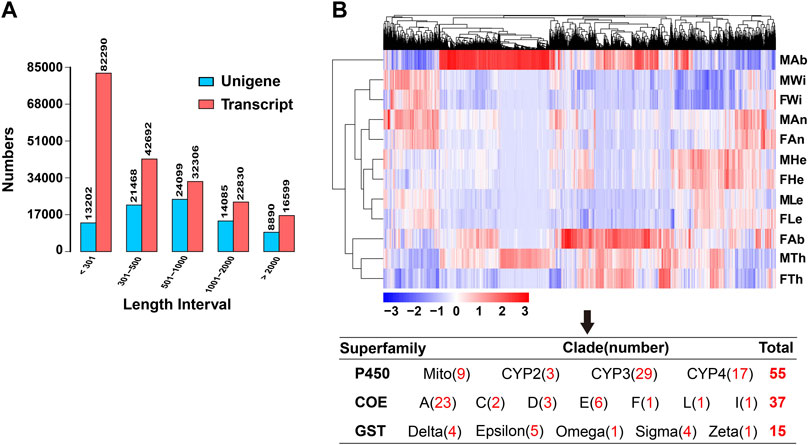
FIGURE 1. Summary of the transcriptome in P. antennata. (A) Length distribution and comparison of unigenes and transcripts. (B) The clustering analysis of differentially expressed genes. Differentially expressed detoxification genes (55 P450s, 37 COEs and 15 GSTs) in various clades are indicated.
Transcriptome analysis and gene functional annotation
A comparative analysis of unigenes among 12 tissues revealed 7,998 DEGs, accounting for 9.78% of all the unigenes. Clustering analyses showed that the majority of DEGs were enriched in female and male abdomens. In contrast, a number of DEGs were found to have relatively low expression in wings of both sexes. Among these DEGs, a comparable number of differentially expressed detoxification genes were identified, including 55 P450s, 37 COEs and 15 GSTs. Of these, CYP3 (29 DEGs) and CYP4 (17 DEGs) members contributed to the majority of differentially expressed P450s, differential COEs mainly presented in clade A (23 DEGs) and differential GSTs mainly presented in Epsilon (five DEGs), Delta (four DEGs) and Sigma (four DEGs) subclasses (Figure 1B).
Out of the 81,744 unigenes, most of them were distributed in two regions of E-values by blasting the genes against the NCBI NR protein sequence database, accounting for 38.9% of the genes for 0–1e−60 and 36.7% for 1e−30–1e−5 (Figure 2A). Further, 38.49% (31,471), 30.33% (24,795), 30.33% (24,795) and 25.27% (20,663) of the genes were annotated separately in NR, GO, PFAM and SwissProt databases, representing the most abundant presence among seven databases. The genes annotated in at least one database contributed to 44.82% (36,643) of the unigenes, but only 3.54% (2,901) of the genes were mapped into all seven databases (Figure 2B). In the NR annotation, P. antennata unigenes were mainly aligned to those in T. castaneum, accounting for 46.5% of the genes (Figure 2C). Approximately 72.2% of unigenes in P. antennata shared more than 60% similarities with those in the NCBI NR protein sequence database (Figure 2D).
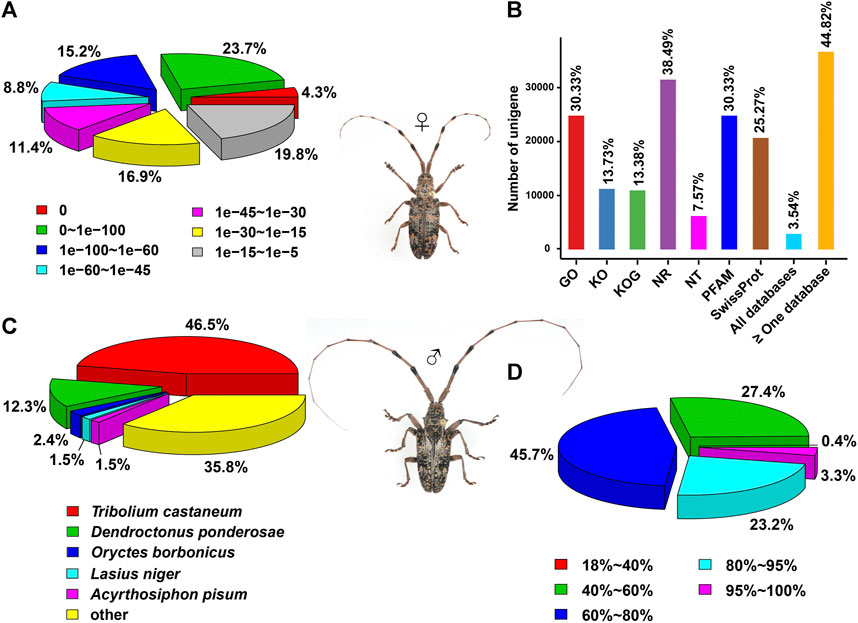
FIGURE 2. Annotation of the transcriptome in P. antennata illustrating the E-value distribution (A), database annotation (B), species classification (C), and similarity distribution (D).
Identification of candidate genes involving detoxification
Based on the trinity transcriptome and the unigene transcriptome, a blasted-based homology search led to the identification of a total of 215 transcripts encoding 106 P450s, 77 COEs, and 32 GSTs. The P450 sequences in P. antennata were sent to the Cytochrome P450 Nomenclature Committees for the nomenclature. COEs were numbered 1 to 77 (COE1–COE77), and GST names were assigned with a combination of a lowercase of the GST subclass and numbers (like GSTe1). Four P450s (CYP307B1/CYP6BJ9/CYP6BJ67/CYP9Z200) and four COEs (COE2/COE60/COE74/COE76) were absent in the unigene transcriptome. The remaining 207 detoxification genes were presented in two transcriptomes. Of the identified genes, 129 relatives were predicted to have complete open reading frames (ORFs), with the protein sizes of 427–591 amino acids for 63 full-length P450s, 401–1,292 amino acids for 41 full-length COEs, and 204–244 amino acids for 25 full-length GSTs. Other 86 genes were partial sequences, including 43 P450s (99–542 amino acids), 36 COEs (106–554 amino acids) and seven GSTs (114–190 amino acids) (Supplementary Table S2). All identified genes have been submitted to the NCBI GenBank database, under the accession numbers OP314595–OP314809.
In the analyses of NCBI blast hits, approximately 90% (193/215) of detoxification genes in P. antennata were aligned to the corresponding genes from A. glabripennis, with relatively high conservation (>50% amino acid identities). In particular, a comparable number of genes (100/193) shared over 80% amino acid identities with their respective orthologs in A. glabripennis (Supplementary Table S2).
Phylogeny and expression profile of candidate cytochrome P450 monooxygenase genes
With an aligned protein sequence of 104 A. glabripennis P450s and 106 P. antennata P450s, a maximum likelihood tree was inferred. The phylogeny classified cerambycid P450s into four clades, representing 11 mitochondrial (Mito) members, six CYP2s, 64 CYP3s and 25 CYP4s in P. antennata. Between P. antennata and A. glabripennis, the former harbored more members in the CYP3 clade, but relatively fewer CYP4s compared to A. glabripennis (CYP3: 55 genes; CYP4: 31 genes). Other two clades Mito and CYP2 had similar gene numbers between the two cerambycid beetles. The tree revealed 52 orthologous pairs between the two species, showing 1:1 relationships in eight Mito, six CYP2, 25 CYP3 and 13 CYP4 pairs. Hence, in most cases, P450s between the beetles shared a high degree of conservation. Exceptionally, we detected a small cluster in P. antennata, representing PantCYP9Z-F4/CYP9Z-F6/CYP9Z-F9/CYP9Z199/CYP9Z200 in the CYP3 clan (Figure 3).
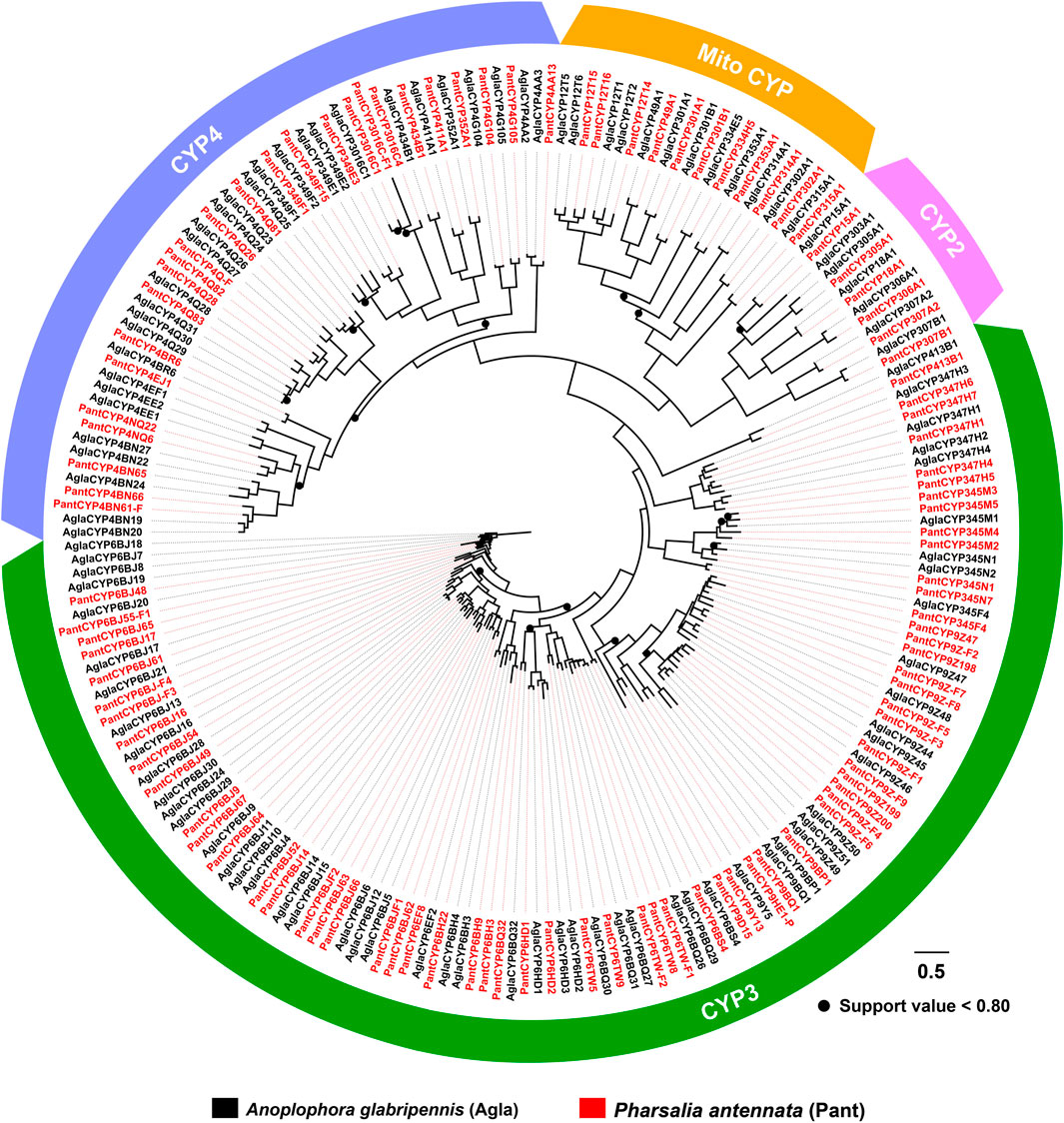
FIGURE 3. Maximum-likelihood tree of P450s in two cerambycid beetles. The P450 proteins in P. antennata are highlighted in red.
Based on FPKM values of RNA–Seq, 98 out of 102 P450 genes had detectable expression in at least one tissue (FPKM >1). The majority of the genes displayed a broad tissue expression profile, some of which were enriched in each tissue such as CYP12T14 in Mito, CYP18A1 in CYP2, CYP347H1/CYP6HD1/CYP9Z198 in CYP3, and CYP4G104/CYP4Q82/CYP4Q83 in CYP4. Four genes had extremely low levels in tissues (0 < FPKM <1), including CYP307A2/CYP9Z-F6/CYP9Z-F7/CYP434B1. In antennae and abdomens, there were 80 and 91 genes, respectively, with FPKM values of above 1. Of these, some genes appeared to have obviously sex-biased levels in the two tissues. For instance, CYP4Q28 showed particularly high expression in male antennae, with a 464.98–fold difference relative to females. CYP334H5 was a male-abdomen-dominant gene, showing 86.25-fold higher expression than females (Supplementary Table S3).
To validate the expression of P450 genes in tissues, we mainly focused on the genes highly expressed in antennae, thoraxes or abdomens, and further investigated their tissue presence by RT–PCR. Accordingly, a total of 48 relatives were selected, representing five Mitos, five CYP2s, 24 CYP3s and 14 CYP4s. In agreement with RNA–Seq results, most of the genes were widely transcribed in various tissues. As shown in RT–PCR assays, as many as 31 genes were presented in all 12 tissues, including three, two, 10 and 16 genes in Mito, CYP2, CYP3 and CYP4 clans, respectively. Notably, molecular evidence of CYP4Q28 and CYP334H5 further supported their preferential expression in male antennae and male abdomens, respectively. In addition, the expression of CYP349F15 and CYP6BJ64 was female-antenna-biased, whereas CYP6HD2 had a stronger band in male antennae compared to females. CYP345N1 exhibited the highest expression in antennae of both sexes among 12 tissues (Figure 4).
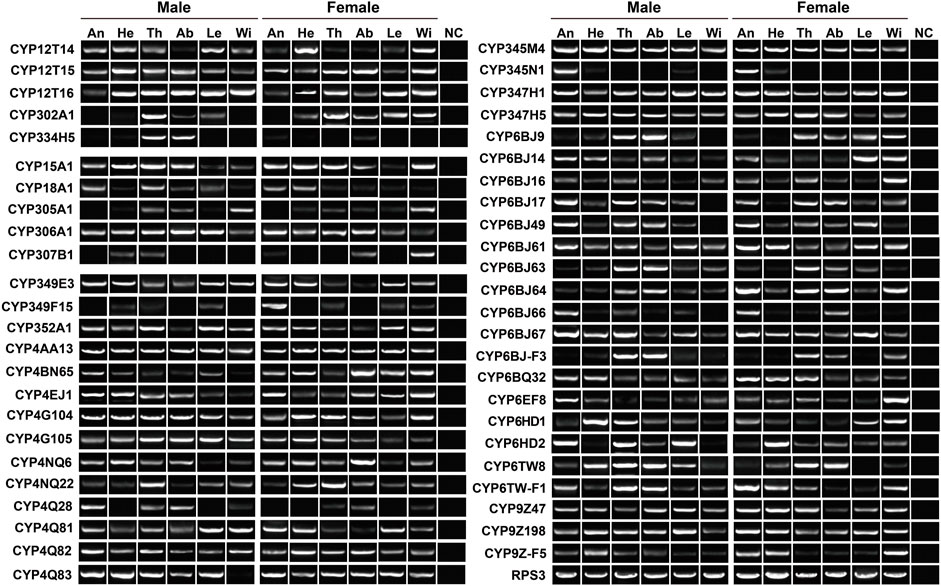
FIGURE 4. Expression pattern of 48 P450 genes in various tissues of P. antennata. The abbreviation of tissues is seen in Table 1. NC, negative control using sterile water as the templates.
Phylogeny and expression profile of candidate carboxylesterase genes
The phylogenetic tree built by 79 AglaCOEs, 77 PantCOEs, 63 TcasCOEs and 35 DmelCOEs revealed 10 clades in Coleoptera (clades A–F/H–M), excluding clades B and H containing COEs solely from D. melanogaster. P. antennata COEs were distributed in each of 10 clades, with the majority of clade A (33 relatives). Other nine clades were composed of relatively few genes, ranging from one to 13 members. Similar to A. glabripennis, P. antennata possessed two lineage-specific expansions in clades A and E, i.e., COE39–COE41/COE47/COE52/COE75 and COE6/COE19/COE35/COE38/COE49/COE54. Between A. glabripennis and P. antennata, there were 31 orthologous pairs dispersed into eight clades, except for clades D and K. Among three coleopteran species, two cerambycid beetles had a similar proportion of genes in each clade, with the exception of clade E where a large species-specific expansion (15 genes) was observed in A. glabripennis. In comparison, T. castaneum had contracted genes in clade A (17 genes), but an obvious expansion was observed in clade C (20 genes) (Figure 5).
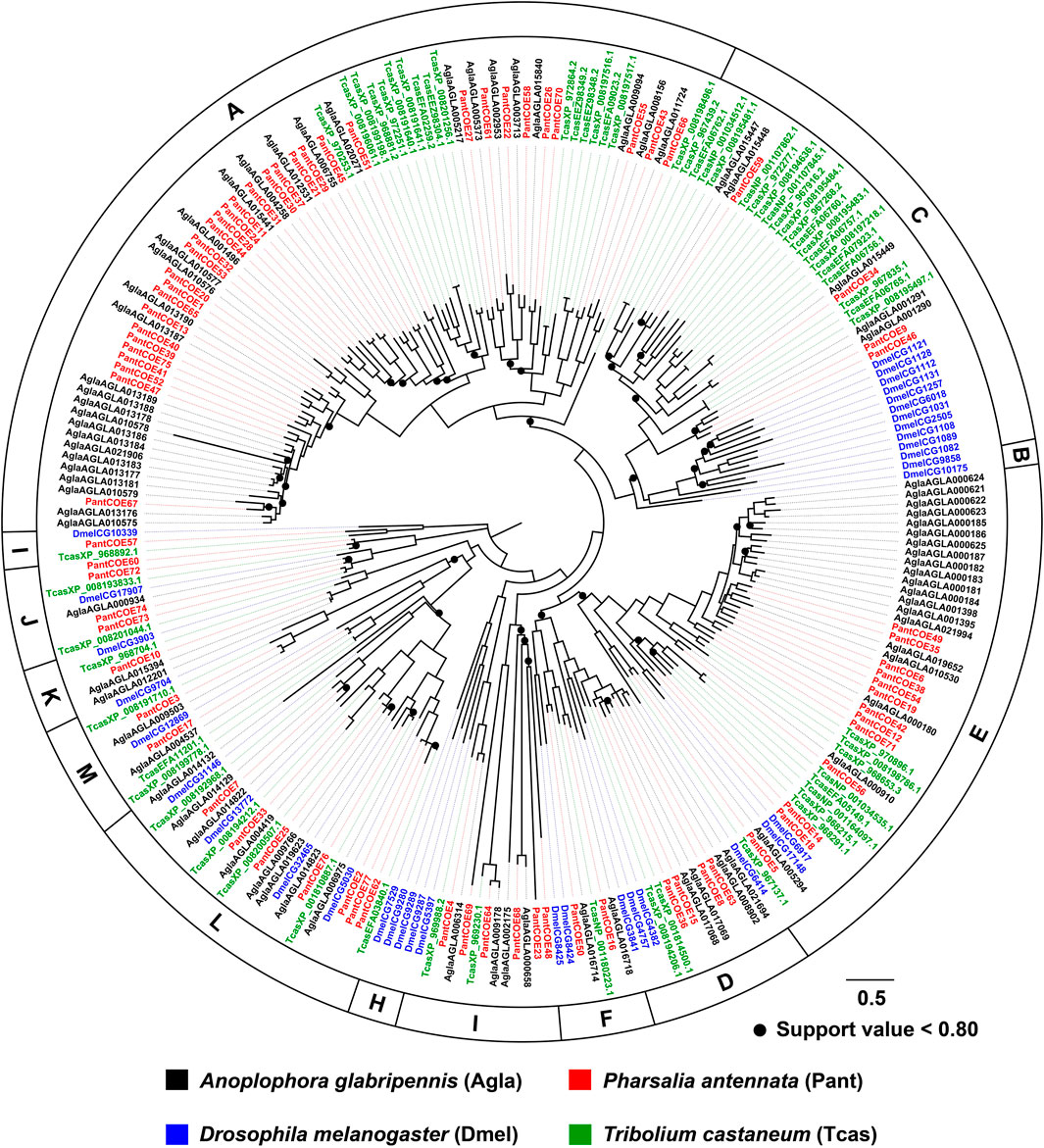
FIGURE 5. Maximum-likelihood tree of COEs in three coleopteran species and D. melanogaster. The P450 proteins in P. antennata are highlighted in red.
Except for COE2/COE60/COE74/COE76, the remaining 73 COEs had available FPKM values in tissues. However, four genes had an extremely low expression in tested tissues (0 < FPKM <1), including COE61/COE62/COE70/COE73. Three COE genes (COE15/COE34/COE64) were broadly expressed in each tissue at a high level (FPKM >10). The numbers of COEs presented in antennae, thoraxes and abdomens (FPKM >1) were 32, 60 and 63, respectively. Of the tested tissues, some genes were preferentially transcribed in males or females with over 10–fold differences. For instance, COE20 and COE51 separately displayed 22.17– and 17.11–fold higher expression in female thoraxes than males. In contrast, the expression level of COE67 in male thoraxes was 10.46–fold higher relative to females. Such the sex-biased expression was also obtained in abdominal tissues, including COE51 (17.63–fold higher) and COE53 (38.85–fold higher) in female abdomens, as well as COE56 in males (140.20–fold higher) (Supplementary Table S3).
RT–PCR was employed to further check the expression of 38 COEs highly transcribed in antennae, thoraxes or abdomens. Outside COE55, the remaining 37 genes were detected in thoraxes or abdomens. In the antennae, 18 COEs were found to have the expression. Overall, molecular results with RT–PCR assays well support the existence of 38 COEs in tissues (Figure 6).
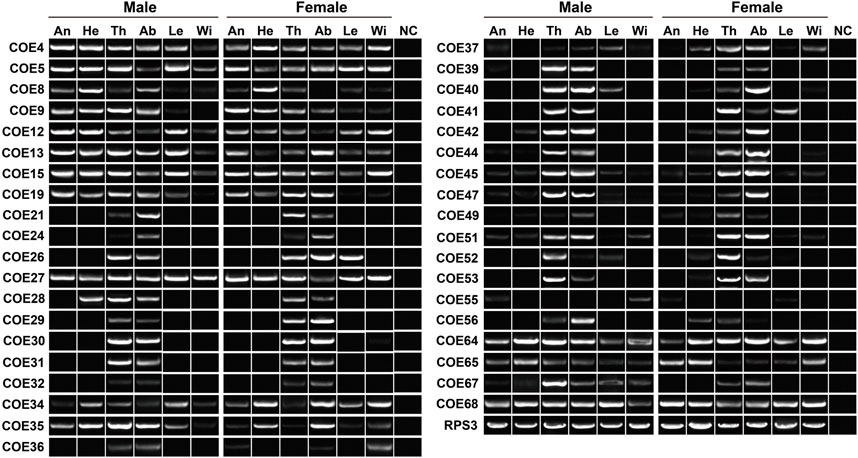
FIGURE 6. Expression pattern of 38 COE genes in various tissues of P. antennata. The abbreviation of tissues is seen in Table 1. NC, negative control using sterile water as the templates.
Phylogeny and expression profile of candidate glutathione-S-transferase genes
The tree of 194 GSTs from seven coleopteran species was inferred, including 98 sequences in four cerambycids A. glabripennis, P. antennata, R. horsfieldi and X. quadripes. In six subclasses, the Delta clade comprised the largest number of P. antennata GSTs (11 relatives), followed by Epsilon (nine relatives) and Sigma (seven relatives) clades. This feature on the largest proportion of GSTs in Delta and Epsilon subclasses was observed in the other three cerambycid beetles. As indicated in the tree, non-cerambycid species harbored at least one species-specific cluster. Differing from that, GSTs from the longhorned beetles mainly clustered together in Cerambycidae-specific patterns. Among four cerambycids, only two orthologous groups were detected, representing AglaGSTo1/PantGSTo2/RhorGSTo3/XquaGSTo2 and AglaGSTs1/PantGSTs2/RhorGSTs1/XquaGSTs2. Between A. glabripennis and P. antennata, there were 1:1 orthologs in 11 clades distributed into five GST subclasses outside Zeta (Figure 7).
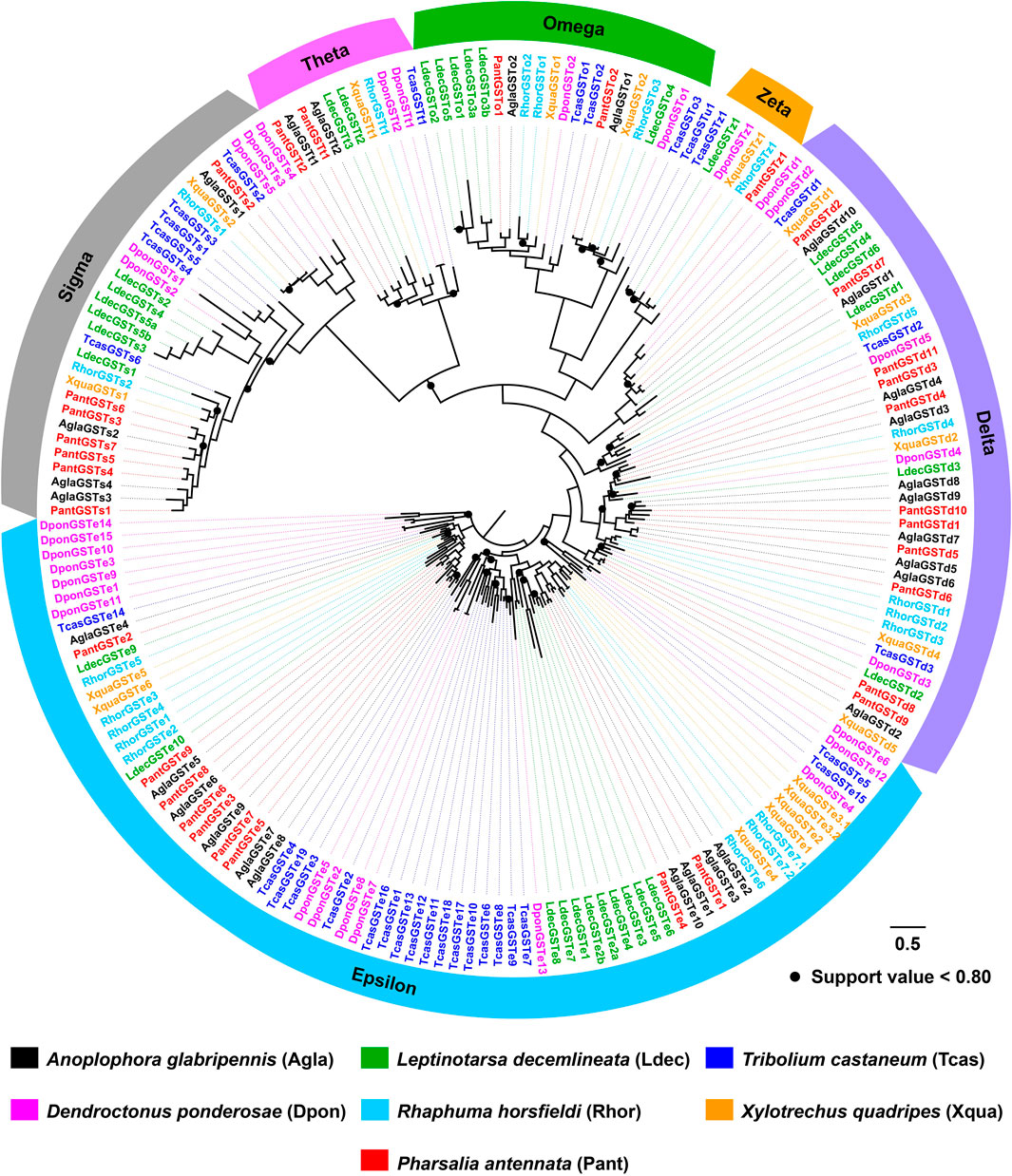
FIGURE 7. Maximum-likelihood tree of GSTs in seven coleopteran species. The GST proteins in P. antennata are highlighted in red.
In the expression profiling analyses of GSTs, all 32 genes had the expression in at least one tissue, with FPKM values of above 1. In particular, nine genes were enriched in all 12 tissues (FPKM >10). Following the standard of FPKM >1, we identified 24, 29 and 30 GSTs in antennae, abdomens and thoraxes, respectively. A Delta gene, GSTd2, was highly expressed in female abdomens with a 10.88–fold difference relative to males. The GSTe3 expression was restricted to female abdomens and thoraxes (Supplementary Table S3). In RT–PCR analyses of 15 GSTs, eight genes exhibited a wide tissue expression, consistent with RNA–Seq results. Except for GSTd5/GSTe7/GSTs5, other 12 genes were detected in antennae. Nearly all the genes had the expression in thoraxes (14 relatives) or abdomens (14 relatives) (Figure 8).
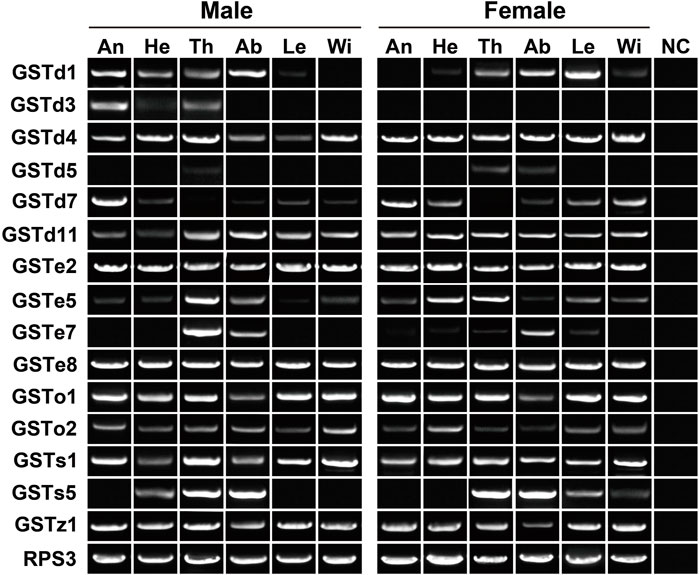
FIGURE 8. Expression patterns of 15 GST genes in various tissues of P. antennata. The abbreviation of tissues is seen in Table 1. NC, negative control using sterile water as the templates.
Candidate detoxification genes involving chemoreception
The antenna and leg are important chemosensory organs in insects, mainly serving as smell and taste (Hansson and Stensmyr, 2011). Combining the results of RNA–Seq (FPKM >1) and RT–PCR, 148 and 141 out of 215 genes had the expression in antennae and legs, respectively, representing 87 and 77 P450s, 36 and 38 COEs, and 25 and 26 GSTs. Here, we further used qPCR to investigate the relative expression of 25 genes specifically or highly expressed in the two chemosensory tissues. As a result, all 25 genes could be detected in the antennae with the majority of them being antenna-enriched relatives. Of 14 P450 genes, 12 relatives were enriched in the antennae, of which four genes (CYP18A1/CYP345N1/CYP6EF8/CYP4Q28) were significantly male-biased relatives while CYP347H1 showed a significant level in females. Notably, CYP4Q28 displayed a 159.64–fold higher expression in male antennae compared to females, further supporting RNA–Seq and RT–PCR results. Some P450 genes, like CYP349E3/CYP349F15/CYP4EJ1, showed relatively high expression in legs. However, most P450s had relatively low expression in bodies (removing antennae and legs). A significantly sex-biased expression was also obtained in legs and bodies, such as four P450s in male legs, five in female legs, 11 in male bodies and one in female bodies (Figure 9).
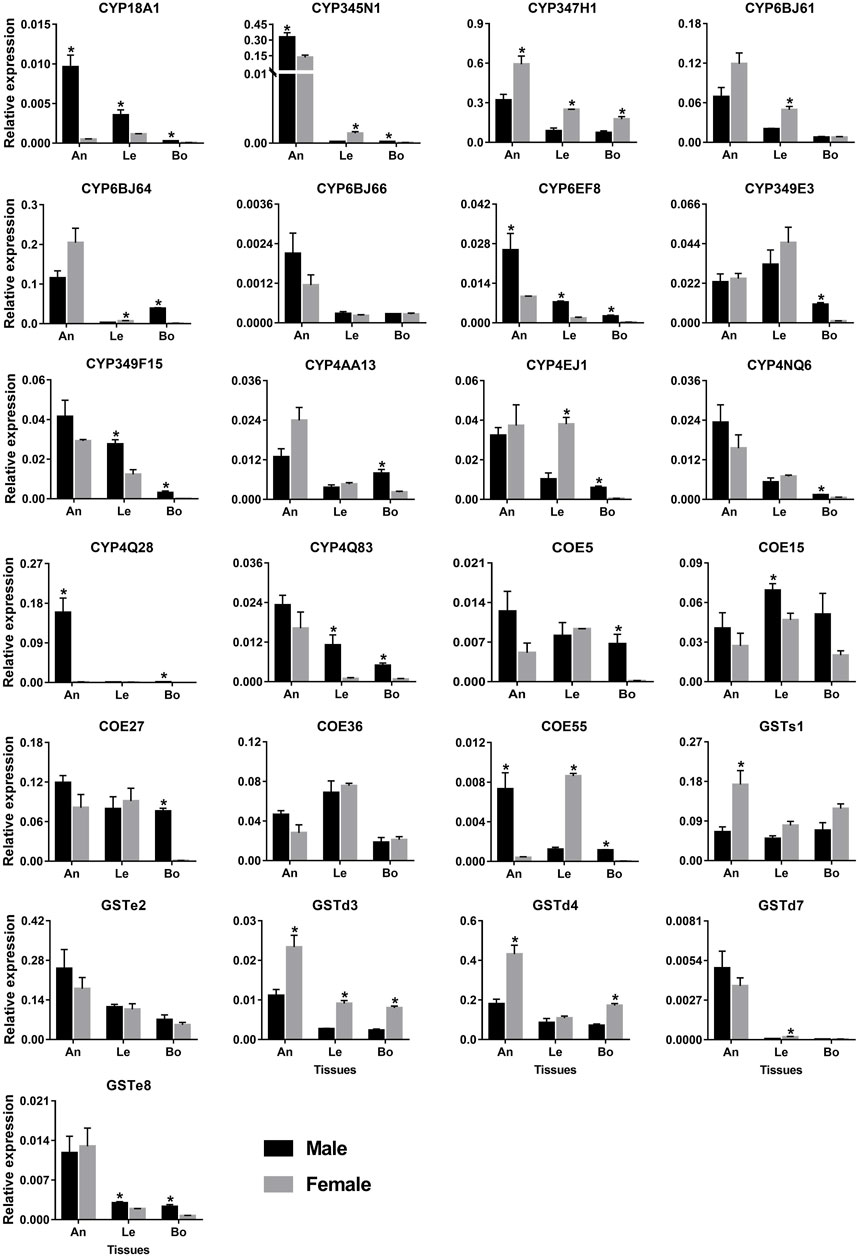
FIGURE 9. qPCR analysis of 25 detoxification genes in various tissues of P. antennata. Data denote mean ± standard errors. Asterisks represent significant differences between male and female tissues (ANOVA, Student’s t–test, p < 0.05). An, antennae; Le, legs and Bo, bodies without antennae and legs.
Five COE genes were abundantly expressed in antennae and legs. Of these, the expression of COE15 was significantly higher in male legs than females, whereas COE55 was a female-leg-biased gene. Besides, COE55 also showed a significant level in male antennae. Similar to P450s, GST genes were expressed in the antennae at a high level. Three GST genes (GSTd3/GSTd4/GSTs1) exhibited significantly higher expression in female antennae compared to males. In legs, GSTd3 was a female-biased gene while GSTe8 was significantly enriched in males (Figure 9).
Discussion
The wood-boring beetles, including P. antennata of the Cerambycidae family, have developed complex detoxification mechanisms to adapt specialized host plants (Allison et al., 2004; McKenna et al., 2016). Considering the differences of host plants, as well as special habitats underneath the bark for wood-borers, it is hypothesized that these beetles, particularly for generalists, are possibly subjected to the defences of more plant secondary metabolites. Such the defenses of plants to herbivorous insects may drive the expansions of P450s, COEs or GSTs via gene duplications or alternative splicing in insects. As implied in Coleoptera, the generalists (polyphagous species) in general possessed more P450s and COEs compared to the specialists (stenophagous and oligophagous species) (Zhao et al., 2020b). Correspondingly, a large size of the detoxification enzyme gene repertoire, to some extent, possibly reflects the host plant range of one coleopteran species. Our study identified a comparable number of P450s (106 genes) and COEs (77 genes) from the P. antennata transcriptome. Although host plants of this beetle remain to be largely unknown to date, relatively large detoxification gene repertoires are regarded as an implication on a broad range of host plants in P. antennata.
Among four cerambycid beetles P. antennata, A. glabripennis, R. horsfieldi and X. quadripes, the Asian longhorned beetle A. glabripennis was the only species with available genome sequences (accession number: GCA_000390,285.2) (McKenna et al., 2016). In comparison, this beetle harbored more P450s (125 genes) and COEs (107 genes), as well as nearly equal GST numbers (28 genes) with P. antennata. This result indicated that some genes encoding P450s and COEs in P. antennata remained to be identified, while GSTs were most likely to be completely found. For detoxification genes in three cerambycids derived from the transcriptomes, P. antennata had more relatives in each family, probably attributed to RNA–Seq of multiple tissues (X. quadripes: mixed female and male antennae (Ji et al., 2018); R. horsfieldi: six tissues (Zhao et al., 2020a); P. antennata: 12 tissues in this study). Furthermore, a wide tissue expression profile of most detoxification genes in P. antennata further supported the notion that the transcriptome of multiple tissues led to the identification of more detoxification genes. On the other hand, it was also possible that the breadth and/or differences of host plants among these beetles resulted in different sizes of detoxification genes. For example, A. glabripennis was a generalist with the larvae feeding on over 100 host plants (Meng et al., 2015).
Insect P450 is known as its role in insecticide resistance and thus is the most extensively studied detoxification gene family (Feyereisen, 1999; Dermauw et al., 2020). Among four P450 clans, both CYP3 and CYP4 commonly have species- or family-specific expansions, mainly contributing to the size differences of this gene family across insects. In contrast, Mito and CYP2 members are highly conserved among species (Feyereisen, 2006; Zhao et al., 2020b). Our phylogenetic analyses of P450s revealed a high degree of orthology between P. antennata and A. glabripennis (52 orthologous pairs), especially Mito and CYP2 clades associated with cuticle formation (Sztal et al., 2012), circadian rhythm (Sathyanarayanan et al., 2008), and the biosynthesis of juvenile hormones and 20-hydroxyecdysone (Feyereisen, 2011; Iga and Kataoka, 2012). However, no obvious clusters were found in the tree. These results suggest functional conservation of orthologous P450s between the two beetles.
In Coleoptera, members of CYP3 and CYP4 families mainly respond to insecticides and plant secondary metabolites. In three Dendroctonus bark beetles D. valens, D. rhizophagus and D. armandi, some CYP3 and CYP4 genes enriched in antennae and guts had inducible expression by host monoterpenes (Cano-Ramirez et al., 2013; Lopez et al., 2013; Dai et al., 2021). Similarly, A. glabripennis P450s expressed in larval guts were induced by feeding host plant sugar maple, as most of the genes belonged to CYP3 and CYP4 clans (McKenna et al., 2016). In the two CYP families, our study detected a number of genes expressed in antennae and abdomens by combinations of RNA–Seq and PCR approaches, possibly associated with the detoxification of host plant chemicals. In particular, some P450s specifically or highly expressed in the antennae of P. antennata may play important roles in the degradation of host volatiles. Focusing on PantCYP4Q28 significantly enriched in male antennae, it was regarded as a candidate pheromone degrading enzyme (PDE), coupled with pheromone degrading functions of a male-antenna-specific gene PdivCYP4AW1 in Phyllopertha diversa (Maibeche-Coisne et al., 2004). Outside the ability of P450s to metabolize plant chemicals, CYP3 and CYP4 enzymes that were broadly expressed in tissues have been demonstrated to have the involvement in insecticide resistance (Zhu et al., 2010; Zimmer et al., 2014). The majority of CYP3 and CYP4 genes in P. antennata also presented a wide tissue expression profile including abdomens with detoxification tissues, suggestive of their putative roles in the detoxification of insecticides.
In the COE gene family, the species-specific expansion mainly occurred in clades A, C and E (Zhao et al., 2020b). For example, 13 A. glabripennis COEs clustered into a group of clade A (coleopteran xenobiotic metabolizing enzymes), of which the majority of them were distributed on scaffold 498 with high conservation (69.40% mean amino acid identity) (McKenna et al., 2016). Six P. antennata COEs phylogenetically close to the A. glabripennis cluster formed a relatively small clade, and shared an average of 72.51% amino acid identity. Such the cluster and conservation were obtained in the two cerambycid beetles of clade E (β- and pheromone esterases). These findings suggest that cerambycid species possibly utilize gene duplications to generate a number of gene copies so as to adapt feeding host plants and the changeable habitats.
Previously, PjapPDE from Popillia japonica belonging to clade E was exclusive to male antennae and was capable of degrading a sex pheromone (R)-japonilure (Ishida and Leal, 2008). Focusing on 13 PantCOEs in this clade, we found that PantCOE71 exhibited 8.36-fold higher expression in male antennae relative to females, suggesting its putative roles in inactivating female-specific compounds (e.g., sex pheromones). Except for clade E, esterases from other clades were involved in the degradation of odorant molecules, including clades D (integument esterases) and G (Lepidopteran juvenile hormone esterases) where some antenna-enriched esterases in Lepidoptera could degrade sex pheromones or plant odorants (He et al., 2014; He et al., 2015). In Coleoptera, clade F replaced clade G as candidate non-lepidopteran juvenile hormone esterases (Zhao et al., 2020b). In clades D and F, we sought to identify candidate odorant degrading enzymes (ODEs) in P. antennata. As shown in expression profiles, PantCOE15/COE16/COE36 highly expressed in antennae were possibly associated with the removal of odorants and thus were identified as candidate ODEs. In addition, we noticed that most PantCOEs were enriched in thoraxes or abdomens, similar to the results in X. quadripes and R. horsfieldi (Zhao et al., 2020b). This could be explained by the fact that insect abdomen contains three important detoxification tissues (midgut, fat body and Malpighian tubules). Although thorax-enriched COEs were also found in other species (Zhao et al., 2020b), their putative functions in this tissue remain unknown and deserve to be further addressed.
As two major subclasses of Delta and Epsilon involved in the detoxification of xenobiotics including plant allelochemicals and insecticides, they accounted for a large proportion of GSTs in most insects (Zhao et al., 2020b; Gao et al., 2020). Meanwhile, due to the absence of Delta and Epsilon members in non-insect species (Sheehan et al., 2001), this further reflects their importance for survival and reproduction of insects. P. antennata possessed a similar proportion of GSTs in the two clades (62.50%) with the other three cerambycid beetles A. glabripennis (71.43%) (Mason et al., 2016), R. horsfieldi (65.00%) and X. quadripes (66.67%) (Zhao et al., 2020b), suggesting the requirements for detoxifying a variety of toxic compounds. The extensive expression of Delta and Epsilon genes in abdomens of P. antennata further conferred their resistance to xenobiotics, similar to the GST genes in Tenebrio molitor (Liu et al., 2015), A. glabripennis (Scully et al., 2013) and X. quadripes (Zhao et al., 2020b). Beyond the detoxification, insect GSTs have been also implied to have olfactory roles responsible for the degradation of odorants (Rogers et al., 1999). In P. antennata, the number (25 genes) of GSTs expressed in antennae was fewer compared to that in the antennal transcriptome of D. melanogaster (31 GSTs) (Younus et al., 2014), but more than those derived from antennal transcriptomes in X. quadripes (18 relatives) (Zhao et al., 2020b) and Agrilus planipennis (13 relatives) (Praveen et al., 2013). Hence, it is possible that antenna-enriched GSTs in P. antennata may be correlated with odorant degradation.
In conclusion, we sequenced the transcriptome of 12 tissues in P. antennata, generating 327,263,716 clean reads in males and 341,437,850 in females with a total of 100.28 G data. Transcriptome analysis combined with the bioinformatics-based search led to the yields of 215 detoxification genes, 107 of which were identified as DEGs. Phylogenetic analyses revealed that P. antennata detoxification genes shared high conservation with those in A. glabripennis, representing 52 P450, 31 COE and 11 GST orthologous pairs. Expression profiles identified a number of detoxification genes in abdomens, suggesting putative roles in xenobiotics metabolism. On the other hand, a certain number of detoxification genes were enriched in antennae, as important molecular candidates for odorant detection. Taken together, this study provides valuable resources for further investigating the adaptation of P. antennata to host plants and habitats, and identifies potential molecular targets involved in the detoxification and chemoreception.
Data availability statement
Raw data of the transcriptome presented in this study were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) repository, with the BioProject accession number PRJNA821002. All nucleotide sequences of detoxification genes from Pharsalia antennata were deposited in the NCBI GenBank repository, with accession numbers OP314595–OP314700 for P450s, OP314701–OP314777 for COEs and OP314778–OP314809 for GSTs.
Author contributions
N-YL conceived and designed the experiments. A-JY, D-LC and Y-JZ performed the experiments. A-JY, N-NY, Y-RG and N-YL analyzed the data. A-JY and N-YL drafted the manuscript. A-JY, N-NY and N-YL revised the manuscript. All authors contributed to the article and approved the final version of the manuscript.
Funding
This work was funded by the Key Project of Applied Basic Research of Yunnan Province of China (202201AS070017), the National Natural Science Foundation of China (31960115), and the Yunnan Provincial Support Plan for the Cultivation of High-level Talents Young Top-notch Talents (YNWR_ QNBJ_2019_057).
Acknowledgments
We are grateful to David Nelson for the nomenclature of P450 genes in Pharsalia antennata.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.1015793/full#supplementary-material
References
Allison J. D., Borden J. H., Seybold S. J. (2004). A review of the chemical ecology of the Cerambycidae (Coleoptera). Chemoecology 14, 123–150. doi:10.1007/s00049-004-0277-1 |
Amezian D., Mehlhorn S., Vacher-Chicane C., Nauen R., Le Goff G. (2022). Spodoptera frugiperda Sf9 cells as a model system to investigate the role of detoxification gene expression in response to xenobiotics. Curr. Res. Insect Sci. 2, 100037. doi:10.1016/j.cris.2022.100037 | |
Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi:10.1373/clinchem.2008.112797 | |
Cano-Ramirez C., Lopez M. F., Cesar-Ayala A. K., Pineda-Martinez V., Sullivan B. T., Zuniga G. (2013). Isolation and expression of cytochrome P450 genes in the antennae and gut of pine beetle Dendroctonus rhizophagus (Curculionidae: Scolytinae) following exposure to host monoterpenes. Gene 520, 47–63. doi:10.1016/j.gene.2012.11.059 | |
Cheng T., Wu J., Wu Y., Chilukuri R. V., Huang L., Yamamoto K., et al. (2017). Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat. Ecol. Evol. 1, 1747–1756. doi:10.1038/s41559-017-0314-4 | |
Claudianos C., Ranson H., Johnson R. M., Biswas S., Schuler M. A., Berenbaum M. R., et al. (2006). A deficit of detoxification enzymes: Pesticide sensitivity and environmental response in the honeybee. Insect Mol. Biol. 15, 615–636. doi:10.1111/j.1365-2583.2006.00672.x | |
Dai L., Gao H., Chen H. (2021). Expression levels of detoxification enzyme genes from Dendroctonus armandi (Coleoptera: Curculionidae) fed on a solid diet containing pine phloem and terpenoids. Insects 12, 926. doi:10.3390/insects12100926 | |
Dai L., Ma J., Ma M., Zhang H., Shi Q., Zhang R., et al. (2016). Characterisation of GST genes from the Chinese white pine beetle Dendroctonus armandi (Curculionidae: Scolytinae) and their response to host chemical defence. Pest Manag. Sci. 72, 816–827. doi:10.1002/ps.4059 | |
Davidson N. M., Oshlack A. (2014). Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 15, 410. doi:10.1186/s13059-014-0410-6 | |
Dermauw W., Van Leeuwen T., Feyereisen R. (2020). Diversity and evolution of the P450 family in arthropods. Insect Biochem. Mol. Biol. 127, 103490. doi:10.1016/j.ibmb.2020.103490 | |
Despres L., David J. P., Gallet C. (2007). The evolutionary ecology of insect resistance to plant chemicals. Trends Ecol. Evol. 22, 298–307. doi:10.1016/j.tree.2007.02.010 | |
Durand N., Carot-Sans G., Chertemps T., Montagne N., Jacquin-Joly E., Debernard S., et al. (2010). A diversity of putative carboxylesterases are expressed in the antennae of the noctuid moth Spodoptera littoralis. Insect Mol. Biol. 19, 87–97. doi:10.1111/j.1365-2583.2009.00939.x | |
Evans J. D., McKenna D., Scully E., Cook S. C., Dainat B., Egekwu N., et al. (2018). Genome of the small hive beetle (Aethina tumida, Coleoptera: Nitidulidae), a worldwide parasite of social bee colonies, provides insights into detoxification and herbivory. Gigascience 7, giy138. doi:10.1093/gigascience/giy138 | |
Feyereisen R. (2011). Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Biochim. Biophys. Acta 1814, 19–28. doi:10.1016/j.bbapap.2010.06.012 | |
Feyereisen R. (2006). Evolution of insect P450. Biochem. Soc. Trans. 34, 1252–1255. doi:10.1042/BST0341252 | |
Feyereisen R. (1999). Insect P450 enzymes. Annu. Rev. Entomol. 44, 507–533. doi:10.1146/annurev.ento.44.1.507 | |
Gao H., Dai L., Fu D., Sun Y., Chen H. (2020). Isolation, expression profiling, and regulation via host allelochemicals of 16 glutathione S-transferases in the Chinese white pine beetle, Dendroctonus armandi. Front. Physiol. 11, 546592. doi:10.3389/fphys.2020.546592 | |
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA–Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi:10.1038/nbt.1883 | |
Hansson B. S., Stensmyr M. C. (2011). Evolution of insect olfaction. Neuron 72, 698–711. doi:10.1016/j.neuron.2011.11.003 | |
He P., Li Z. Q., Liu C. C., Liu S. J., Dong S. L. (2014). Two esterases from the genus Spodoptera degrade sex pheromones and plant volatiles. Genome 57, 201–208. doi:10.1139/gen-2014-0041 | |
He P., Mang D. Z., Wang H., Wang M. M., Ma Y. F., Wang J., et al. (2020). Molecular characterization and functional analysis of a novel candidate of cuticle carboxylesterase in Spodoptera exigua degradating sex pheromones and plant volatile esters. Pestic. Biochem. Physiol. 163, 227–234. doi:10.1016/j.pestbp.2019.11.022 | |
He P., Zhang Y. N., Yang K., Li Z. Q., Dong S. L. (2015). An antenna-biased carboxylesterase is specifically active to plant volatiles in Spodoptera exigua. Pestic. Biochem. Physiol. 123, 93–100. doi:10.1016/j.pestbp.2015.03.009 | |
Huber D. P., Erickson M. L., Leutenegger C. M., Bohlmann J., Seybold S. J. (2007). Isolation and extreme sex-specific expression of cytochrome P450 genes in the bark beetle, Ips paraconfusus, following feeding on the phloem of host ponderosa pine, Pinus ponderosa. Insect Mol. Biol. 16, 335–349. doi:10.1111/j.1365-2583.2007.00731.x | |
Iga M., Kataoka H. (2012). Recent studies on insect hormone metabolic pathways mediated by cytochrome P450 enzymes. Biol. Pharm. Bull. 35, 838–843. doi:10.1248/bpb.35.838 | |
Ishida Y., Leal W. S. (2008). Chiral discrimination of the Japanese beetle sex pheromone and a behavioral antagonist by a pheromone-degrading enzyme. Proc. Natl. Acad. Sci. U. S. A. 105, 9076–9080. doi:10.1073/pnas.0802610105 | |
Ji S. S., Zhuang X. L., Liu N. Y. (2018). Identification and expression profile analysis of odorant binding protein genes from Xylotrechus quadripes Chevrolat (Coleoptera: Cerambycidae). J. Sichuan Agric. Univ. 36, 193–202. doi:10.16036/j.issn.1000-2650.2018.02.010 |
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010 | |
Keeling C. I., Henderson H., Li M., Dullat H. K., Ohnishi T., Bohlmann J. (2013a). CYP345E2, an antenna-specific cytochrome P450 from the mountain pine beetle, Dendroctonus ponderosae Hopkins, catalyses the oxidation of pine host monoterpene volatiles. Insect Biochem. Mol. Biol. 43, 1142–1151. doi:10.1016/j.ibmb.2013.10.001 | |
Keeling C. I., Yuen M. M., Liao N. Y., Docking T. R., Chan S. K., Taylor G. A., et al. (2013b). Draft genome of the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major forest pest. Genome Biol. 14, R27. doi:10.1186/gb-2013-14-3-r27 | |
Kostaropoulos I., Papadopoulos A. I., Metaxakis A., Boukouvala E., Papadopoulou-Mourkidou E. (2001). The role of glutathione S-transferases in the detoxification of some organophosphorus insecticides in larvae and pupae of the yellow mealworm, Tenebrio molitor (Coleoptera: Tenebrionidae). Pest Manag. Sci. 57, 501–508. doi:10.1002/ps.323 | |
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923 | |
Li B., Dewey C. N. (2011). Rsem: Accurate transcript quantification from RNA–seq data with or without a reference genome. BMC Bioinforma. 12, 323. doi:10.1186/1471-2105-12-323 | |
Liu H., Tang Y., Wang Q., Shi H., Yin J., Li C. (2021). Identification and characterization of an antennae-specific glutathione S-transferase from the Indian meal moth. Front. Physiol. 12, 727619. doi:10.3389/fphys.2021.727619 | |
Liu S., Shi X. X., Jiang Y. D., Zhu Z. J., Qian P., Zhang M. J., et al. (2015). De novo analysis of the Tenebrio molitor (Coleoptera: Tenebrionidae) transcriptome and identification of putative glutathione S-transferase genes. Appl. Entomol. Zool. 50, 63–71. doi:10.1007/s13355-014-0305-8 |
Liu S., Zhang Y. X., Wang W. L., Zhang B. X., Li S. G. (2017). Identification and characterisation of seventeen glutathione S-transferase genes from the cabbage white butterfly Pieris rapae. Pestic. Biochem. Physiol. 143, 102–110. doi:10.1016/j.pestbp.2017.09.001 | |
Lopez M. F., Cano-Ramirez C., Cesar-Ayala A. K., Ruiz E. A., Zuniga G. (2013). Diversity and expression of P450 genes from Dendroctonus valens LeConte (Curculionidae: Scolytinae) in response to different kairomones. Insect Biochem. Mol. Biol. 43, 417–432. doi:10.1016/j.ibmb.2013.02.004 | |
Maibeche-Coisne M., Nikonov A. A., Ishida Y., Jacquin-Joly E., Leal W. S. (2004). Pheromone anosmia in a scarab beetle induced by in vivo inhibition of a pheromone-degrading enzyme. Proc. Natl. Acad. Sci. U. S. A. 101, 11459–11464. doi:10.1073/pnas.0403537101 | |
Mason C. J., Scully E. D., Geib S. M., Hoover K. (2016). Contrasting diets reveal metabolic plasticity in the tree-killing beetle, Anoplophora glabripennis (Cerambycidae: Lamiinae). Sci. Rep. 6, 33813. doi:10.1038/srep33813 | |
McKenna D. D., Scully E. D., Pauchet Y., Hoover K., Kirsch R., Geib S. M., et al. (2016). Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle-plant interface. Genome Biol. 17, 227. doi:10.1186/s13059-016-1088-8 | |
Meng P. S., Hoover K., Keena M. A. (2015). Asian longhorned beetle (Coleoptera: Cerambycidae), an introduced pest of maple and other hardwood trees in North America and Europe. J. Integr. Pest Manag. 6, 4. doi:10.1093/jipm/pmv003 |
Muller P. Y., Janovjak H., Miserez A. R., Dobbie Z. (2002). Processing of gene expression data generated by quantitative real-time RT–PCR. BioTechniques 32, 1372–1374. 1376, 1378-1379. |
Oakeshott J. G., Claudianos C., Campbell P. M., Newcomb R. D., Russell R. J. (2019). Biochemical genetics and genomics of insect esterases, reference module in life sciences. Elsevier. doi:10.1016/B978-0-12-809633-8.04063-2 |
Oakeshott J. G., Johnson R. M., Berenbaum M. R., Ranson H., Cristino A. S., Claudianos C. (2010). Metabolic enzymes associated with xenobiotic and chemosensory responses in Nasonia vitripennis. Insect Mol. Biol. 19, 147–163. doi:10.1111/j.1365-2583.2009.00961.x | |
Praveen M., Wijeratne A., Wijeratne S., Poland T., Qazi S., Doucet D., et al. (2013). Identification of odor-processing genes in the emerald ash borer, Agrilus planipennis. PLoS One 8, e56555. doi:10.1371/journal.pone.0056555 | |
Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. doi:10.1371/journal.pone.0009490 | |
Ranson H., Rossiter L., Ortelli F., Jensen B., Wang X., Roth C. W., et al. (2001). Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem. J. 359, 295–304. doi:10.1042/0264-6021:3590295 | |
Robinson M. D., McCarthy D. J., Smyth G. K. (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi:10.1093/bioinformatics/btp616 | |
Rogers M. E., Jani M. K., Vogt R. G. (1999). An olfactory-specific glutathione-S-transferase in the sphinx moth Manduca sexta. J. Exp. Biol. 202, 1625–1637. doi:10.1242/jeb.202.12.1625 | |
Sandstrom P., Welch W. H., Blomquist G. J., Tittiger C. (2006). Functional expression of a bark beetle cytochrome P450 that hydroxylates myrcene to ipsdienol. Insect Biochem. Mol. Biol. 36, 835–845. doi:10.1016/j.ibmb.2006.08.004 | |
Sathyanarayanan S., Zheng X., Kumar S., Chen C. H., Chen D., Hay B., et al. (2008). Identification of novel genes involved in light-dependent CRY degradation through a genome-wide RNAi screen. Genes Dev. 22, 1522–1533. doi:10.1101/gad.1652308 | |
Schoville S. D., Chen Y. H., Andersson M. N., Benoit J. B., Bhandari A., Bowsher J. H., et al. (2018). A model species for agricultural pest genomics: The genome of the Colorado potato beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Sci. Rep. 8, 1931. doi:10.1038/s41598-018-20154-1 | |
Scully E. D., Hoover K., Carlson J. E., Tien M., Geib S. M. (2013). Midgut transcriptome profiling of Anoplophora glabripennis, a lignocellulose degrading cerambycid beetle. BMC Genomics 14, 850. doi:10.1186/1471-2164-14-850 | |
Sheehan D., Meade G., Foley V. M., Dowd C. A. (2001). Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360, 1–16. doi:10.1042/0264-6021:3600001 | |
Shi H., Pei L., Gu S., Zhu S., Wang Y., Zhang Y., et al. (2012). Glutathione S-transferase (GST) genes in the red flour beetle, Tribolium castaneum, and comparative analysis with five additional insects. Genomics 100, 327–335. doi:10.1016/j.ygeno.2012.07.010 | |
Simon P. (2003). Q–Gene: Processing quantitative real-time RT–PCR data. Bioinformatics 19, 1439–1440. doi:10.1093/bioinformatics/btg157 | |
Slipinski S. A., Leschen R. A. B., Lawrence J. F. (2011). “Order Coleoptera Linnaeus, 1758,” in Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Editor Z. Q. Zhang, 3148, 203–208. |
Small G. J., Hemingway J. (2000). Molecular characterization of the amplified carboxylesterase gene associated with organophosphorus insecticide resistance in the brown planthopper, Nilaparvata lugens. Insect Mol. Biol. 9, 647–653. doi:10.1046/j.1365-2583.2000.00229.x | |
Sztal T., Chung H., Berger S., Currie P. D., Batterham P., Daborn P. J. (2012). A cytochrome P450 conserved in insects is involved in cuticle formation. PLoS ONE 7, e36544. doi:10.1371/journal.pone.0036544 | |
Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., et al. (2010). Transcript assembly and quantification by RNA–Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi:10.1038/nbt.1621 | |
Wang L., Feng Z., Wang X., Wang X., Zhang X. (2009). DEGseq: An R package for identifying differentially expressed genes from RNA–seq data. Bioinformatics 26, 136–138. doi:10.1093/bioinformatics/btp612 | |
Wang M. M., Long G. J., Guo H., Liu X. Z., Wang H., Dewer Y., et al. (2021). Two carboxylesterase genes in Plutella xylostella associated with sex pheromones and plant volatiles degradation. Pest Manag. Sci. 77, 2737–2746. doi:10.1002/ps.6302 | |
Wheelock C. E., Shan G., Ottea J. (2005). Overview of carboxylesterases and their role in the metabolism of insecticides. J. Pestic. Sci. 30, 75–83. doi:10.1584/jpestics.30.75 |
Yin N. N., Wang Z. Q., Xiao H. Y., Lu T. T., Liu N. Y. (2022). Identification and characterization of UDP-glycosyltransferase genes in a cerambycid beetle, Pharsalia antennata Gahan, 1894 (Coleoptera: Cerambycidae). Diversity 14, 348. doi:10.3390/d14050348 |
Yin N. N., Zhao Y. J., Zhao N., Liu N. Y. (2020). Morphological characteristics of Pharsalia antennata and its sensilla ultrastructure. Plant Prot. 46, 30–40. doi:10.16688/j.zwbh.2019493 |
You Y., Xie M., Ren N., Cheng X., Li J., Ma X., et al. (2015). Characterization and expression profiling of glutathione S-transferases in the diamondback moth, Plutella xylostella (L.). BMC Genomics 16, 152. doi:10.1186/s12864-015-1343-5 | |
Younus F., Chertemps T., Pearce S. L., Pandey G., Bozzolan F., Coppin C. W., et al. (2014). Identification of candidate odorant degrading gene/enzyme systems in the antennal transcriptome of Drosophila melanogaster. Insect Biochem. Mol. Biol. 53, 30–43. doi:10.1016/j.ibmb.2014.07.003 | |
Zhang J., Zhang J., Yang M., Jia Q., Guo Y., Ma E., et al. (2011). Genomics-based approaches to screening carboxylesterase-like genes potentially involved in malathion resistance in oriental migratory locust (Locusta migratoria manilensis). Pest Manag. Sci. 67, 183–190. doi:10.1002/ps.2049 | |
Zhang Y., Wang L., Guo H., Li G., Zhang Z., Xie L., et al. (2012). A transcriptome-based screen of carboxylesterase-like genes that are involved in chlorpyrifos resistance in Laodelphax striatellus (Fallén). Anal. Chem. 104, 224–231. doi:10.1021/ac202405q |
Zhao Y. J., Li G. C., Zhu J. Y., Liu N. Y. (2020a). Genome-based analysis reveals a novel SNMP group of the Coleoptera and chemosensory receptors in Rhaphuma horsfieldi. Genomics 112, 2713–2728. doi:10.1016/j.ygeno.2020.03.005 | |
Zhao Y. J., Wang Z. Q., Zhu J. Y., Liu N. Y. (2020b). Identification and characterization of detoxification genes in two cerambycid beetles, Rhaphuma horsfieldi and Xylotrechus quadripes (Coleoptera: Cerambycidae: Clytini). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 243–244, 110431. doi:10.1016/j.cbpb.2020.110431 | |
Zhou W. W., Liang Q. M., Xu Y., Gurr G. M., Bao Y. Y., Zhou X. P., et al. (2013). Genomic insights into the glutathione S-transferase gene family of two rice planthoppers, Nilaparvata lugens (stål) and Sogatella furcifera (horváth) (Hemiptera: Delphacidae). PLoS ONE 8, e56604. doi:10.1371/journal.pone.0056604 | |
Zhu F., Moural T. W., Shah K., Palli S. R. (2013). Integrated analysis of cytochrome P450 gene superfamily in the red flour beetle, Tribolium castaneum. BMC Genomics 14, 174. doi:10.1186/1471-2164-14-174 | |
Zhu F., Parthasarathy R., Bai H., Woithe K., Kaussmann M., Nauen R., et al. (2010). A brain-specific cytochrome P450 responsible for the majority of deltamethrin resistance in the QTC279 strain of Tribolium castaneum. Proc. Natl. Acad. Sci. U. S. A. 107, 8557–8562. doi:10.1073/pnas.1000059107 | |
Zimmer C. T., Bass C., Williamson M. S., Kaussmann M., Wolfel K., Gutbrod O., et al. (2014). Molecular and functional characterization of CYP6BQ23, a cytochrome P450 conferring resistance to pyrethroids in European populations of pollen beetle, Meligethes aeneus. Insect Biochem. Mol. Biol. 45, 18–29. doi:10.1016/j.ibmb.2013.11.008 | |
Keywords: Pharsalia antennata, cytochrome P450 monooxygenase, carboxylesterase, glutathione-S-transferase, detoxification, olfaction
Citation: Yang A-J, Yin N-N, Chen D-L, Guo Y-R, Zhao Y-J and Liu N-Y (2022) Identification and characterization of candidate detoxification genes in Pharsalia antennata Gahan (Coleoptera: Cerambycidae). Front. Physiol. 13:1015793. doi: 10.3389/fphys.2022.1015793
Received: 10 August 2022; Accepted: 31 August 2022;
Published: 16 September 2022.
Edited by:
Ya-Nan Zhang, Huaibei Normal University, ChinaReviewed by:
Guan-Heng Zhu, School of Agriculture, Sun Yat-sen University, ChinaPeng He, Guizhou University, China
Copyright © 2022 Yang, Yin, Chen, Guo, Zhao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nai-Yong Liu, bmFpeW9uZy5saXVAc3dmdS5lZHUuY24=