 Nouf J. Althali
Nouf J. Althali Kathryn E. Hentges
Kathryn E. Hentges
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 06 October 2022
Sec. Cell Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.1012665
This article is part of the Research TopicCell Signalling in Heart Development, Disease and RegenerationView all 5 articles
Congenital heart defects (CHD) include structural abnormalities of the heart or/and great vessels that are present at birth. CHD affects around 1% of all newborns worldwide. Tetralogy of Fallot (TOF) is the most prevalent cyanotic congenital cardiac abnormality, affecting three out of every 10,000 live infants with a prevalence rate of 5–10% of all congenital cardiac defects. The four hallmark characteristics of TOF are: right ventricular hypertrophy, pulmonary stenosis, ventricular septal defect, and overriding aorta. Approximately 20% of cases of TOF are associated with a known disease or chromosomal abnormality, with the remaining 80% of TOF cases being non-syndromic, with no known aetiology. Relatively few TOF patients have been studied, and little is known about critical causative genes for non-syndromic TOF. However, rare genetic variants have been identified as significant risk factors for CHD, and are likely to cause some cases of TOF. Therefore, this review aims to provide an update on well-characterized genes and the most recent variants identified for non-syndromic TOF.
Congenital heart defects (CHD) are structural malformations of the heart or/and great vessels which affect about 1% of live born infants (Hoffman and Kaplan, 2002). Globally, CHDs are the greatest cause of death from non-communicable diseases in individuals under 30 years of age, and are also the leading cause of mortality from congenital abnormalities (Su et al., 2022). In a study conducted between 2000 and 2005 in Europe, it was reported that approximately 88% of CHD cases are not associated with chromosomal alterations (Dolk et al., 2011), suggesting other genetic defects are responsible for the majority of CHD in this population. Within CHD, Tetralogy of Fallot (TOF) is the most common cyanotic congenital heart defect, affecting 3 per 10,000 live births, and comprising 5–10% of all congenital heart abnormalities (Suzuki et al., 1996; Bailliard and Anderson, 2009; Postoev et al., 2014); and (Park and Thayer, 2014). The hallmark characteristics of TOF include four structural alterations to the heart and great vessels (Figure 1), which are: pulmonary stenosis, ventricular septal defect (VSD), overriding aorta, and right ventricular (RV) hypertrophy (Shinebourne et al., 2006; Bailliard and Anderson, 2009); and reviewed in (Bedair and Iriart, 2019).
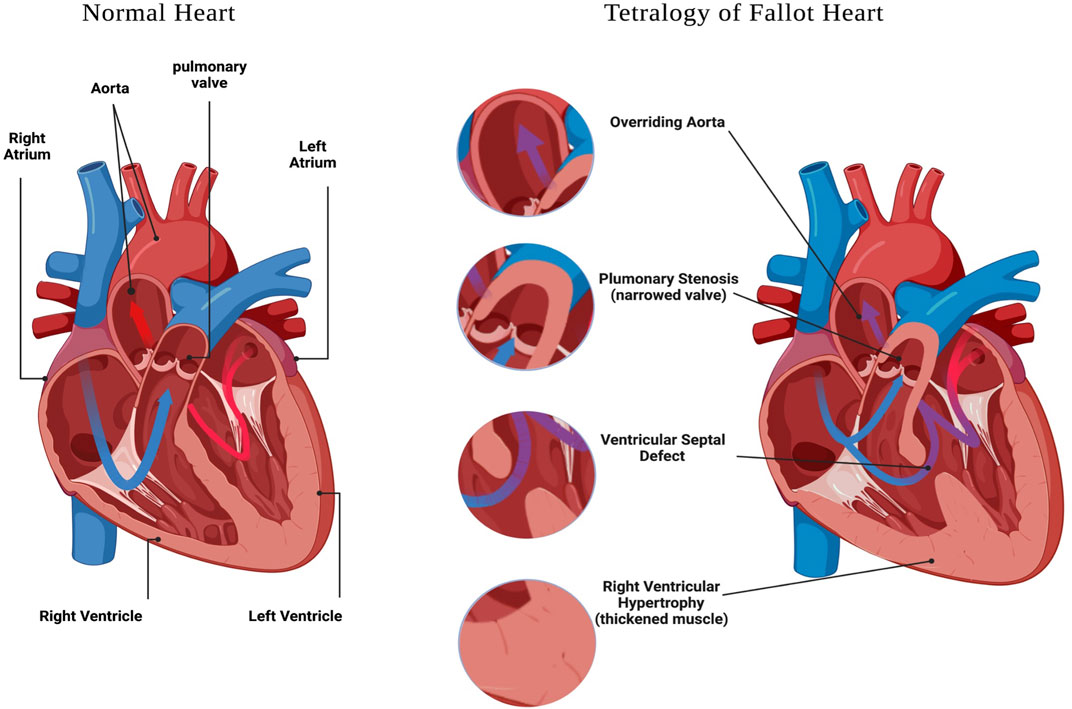
FIGURE 1. Tetralogy of Fallot features. A schematic representation of a heart with: Ventricular Septal Defect, Overriding Aorta, Pulmonary Stenosis and Right Ventricular Hypertrophy. Created with BioRender.com.
The TOF heart anatomy permits blood to be mixed between the pulmonary and systemic circulations. This mixture commonly happens at the VSD, where a right-to-left shunt introduces deoxygenated blood into the systemic circulation, resulting in cyanosis. The differential pressure gradient between the RV and LV determines the right-to-left shunt across the VSD. The degree of the right ventricular outflow tract obstrucrtion (or pulmonary stenosis) determines the quantity of pulmonary blood flow (the RV stroke volume) (Wilson et al., 2019). Understanding the dynamical variables that aggravate or improve the right-to-left shunt is critical to understanding the essential care of disease or preoperative neonates with TOF (Wilson et al., 2019).
An irregular blood flow pattern, chamber hypoplasia, obstructive lesions and aberrant venous and arterial connections found in individuals with CHD results in some of the most interesting and complicated abnormalities in normal heart physiology (Škorić-Milosavljević et al., 2021). The physiological changes surrounding hyper cyanotic incidents consist of either a reduction in systemic vascular resistance or a rise in pulmonary resistance, both of which paticipate to a right-to-left shunt across the VDS (Mcleod et al., 2018; Ho et al., 2019); and (Senst et al., 2022). Due to the right ventricle to left ventricle flow shunt and high flow levels through the overriding aorta, the endocardium encompassing the VSD faces higher wall shear stresses. In addition, the TOF right ventricle and left ventricle have higher pressures than normal hearts, but only the TOF right ventricle has higher wall shear stresses and thickening (Wiputra et al., 2018). Understanding the blood flow dynamics in TOF hearts, most likely due to anatomical differences and their different illnesses, could aid in future minimally invasive foetal heart therapies.
Surgical repair of the morphological and physiological alterations in the TOF heart has markedly increased survival rates for infants with TOF. Repair in the first year of life is preferable, and earlier repair improves survival outcomes (Peck et al., 2021). Although patients with repaired TOF have a high chance of surviving into adulthood, the transition into middle life is accompanied by severe morbidity and increased mortality rates (Dennis et al., 2017). Both atrial and ventricular arrhythmias are a common source of morbidity and mortality following TOF repair (Pinsker et al., 2022). Additional causes of mortality for individuals with TOF are heart failure and sudden cardiac death (Norgaard et al., 1996; Oechslin et al., 2000); and (Dennis et al., 2017). Sudden mortality in the repaired TOF population is widely documented, with multiple risk factors, which are assumed to be related to chronic RV volume overload over time.
A higher incidence of CHD in first-degree relatives and children of TOF patients (Postoev et al., 2014) suggests a genetic aetiology to this disorder. The genetic analysis of syndromic TOF patients has offered valuable insights into causal genes in specific individuals. Approximately 20% of cases are linked to a recognised condition or chromosomal abnormality (Bailliard and Anderson, 2009). These TOF cases are found in patients with genetic syndromes including 22q11 microdeletions, trisomy 21, Alagille’s disorder, Cat Eye syndrome, or CHARGE and VATER/VACTERL syndromes (Goldmuntz et al., 1998; Goldmuntz, 2001; McElhinney et al., 2002; Michielon et al., 2006); and (Marino et al., 2012). About 80% of TOF cases are non-syndromic, and display no indication of familial Mendelian CHD segregation, with no identified aetiology (Goodship et al., 2012; Palomino Doza et al., 2013). Rare, de novo genetic variations have been found as substantial factors in TOF (Jin et al., 2017). Notably, variants in the key cardiac transcription factor NKX2.5 were the first to be identified as a genetic cause of TOF (Goldmuntz et al., 1998; Benson et al., 1999). Subsequent studies have further extended our knowledge of genetic alterations resulting in TOF. Therefeore, this mini review aims to offer an update on the genes and variants reported to be associated with non-syndromic TOF (Table 1). As highlighted in this review, disruption of the balanced interaction of transcription factors which are crucial for cardiogenesis plays a substantial role in the aetiology of non-syndromic TOF.

TABLE 1. Non-syndromic TOF genetic variants.
Whole Exome Sequencing (WES) has been used effectively to find new candidate genes for CHD (Zaidi et al., 2013; Al Turki et al., 2014; Homsy et al., 2015); and (Sifrim et al., 2016). Previous research revealed that ultra-rare non-synonymous variants play a major role in the genetic aetiology of CHD, particularly TOF (Jin et al., 2017; Page et al., 2019); and (Reuter et al., 2019). Although familial cases of TOF are rare, likely due to the severity of the disease, inherited variants in the genes GATA4, NKX2.5, JAG1, FOXC2, TBX5 and TBX1 have been reported (reviewed in (Morgenthau and Frishman, 2018). A recent study of the largest group of non-syndromic TOF patients observed to date, using WES of 829 TOF patients, has found that the NOTCH1 locus, followed by FLT4, is the most common site of genetic variants leading to non-syndromic TOF. Variants in these genes are detected in about 7% of TOF cases (Page et al., 2019). Moreover, rare variants in some specific transcription factors, such as FOXC1 and HAND2, have been identified in non-syndromic TOF patients (Töpf et al., 2014). Because there are links between normal heart formation and multiple transcription factors, variants in transcription factors required during cardiac development can result in aberrant cardiac morphology or function, thus resulting in CHD.
NOTCH family proteins 1-4 are evolutionarily conserved transmembrane proteins that have a crucial impact on cell fate decisions (Andersson et al., 2011). The sequential expression of Notch components and related genes are required during vascular and cardiac development (Luxán et al., 2016). The NOTCH pathway is necessary for the developing heart for the regulation of vital pathways involving the formation of cardiac trabeculae and sequential activation of many NOTCH genes promotes myocardial development, formation, and compaction (Luxán et al., 2016).
Since NOTCH signalling is so significant for cardiac and vascular development, variants in genes encoding NOTCH signalling components cause a variety of cardiovascular abnormalities (Eldadah et al., 2001; McElhinney et al., 2002; Tomita-Mitchell et al., 2007; Garg et al., 2005; Stittrich et al., 2014; Kerstjens-Frederikse et al., 2016); and (Luxán et al., 2016). A recent study conducted by (Page et al., 2019) found five new de novo NOTCH1 variants in the TOF cohort; three of these were missense variations, whereas the remaining two were truncating variants (Table 1): p. G200V, p. R448X, p. C1549Y, p. W1638X and p. N1875S. The differences in NOTCH1 signalling observed were not due to the variants’ reduced mRNA expression. As a result, two variants, the p. C607Y and p. N1875S, recognised in TOF patients who underwent functional testing, were found to affect canonical NOTCH1 signalling (Page et al., 2019). Furthermore, eight ultra-rare missense NOTCH1 variants were found in the investigation of a 175-member TOF cohort in a study performed by (Manshaei et al., 2020). Their analysis confirmed that those variants were ultra-rare deleterious and involved in the pathogenesis of TOF, suggesting abnormal vascular development and related signalling defects as probable causes of TOF.
Vascular endothelial growth factor receptors (VEGFR1-3) are crucial in the development and long-term maintenance of the cardiovascular and lymphovascular systems. VEGFR3 (Vascular Endothelial Growth Factor 3), encoded by the FLT4 gene, plays an essential and well-studied role in the development and formation of the circulatory vessels (Olsson et al., 2006; Karaman et al., 2022). Statistical analysis revealed that FLT4 was among the most common genes predisposing individuals to non-syndromic TOF (Matos-Nieves et al., 2019). Likewise, exome sequence analysis revealed that FLT4 and NOTCH1 genes were the most significant genes that caused non-syndromic TOF in a cohort of 829 TOF patients (Page et al., 2019). Recent analysis of exome sequencing data from 811 TOF probands has found 14 FLT4 loss of function variants (Reuter et al., 2019). Additionally, (Page et al., 2019); found that several FLT4 variants in TOF patients are unique and severe, due to causing premature protein truncation and frameshifts located in the FLT4 immunoglobulin (Ig) and VEGFR3 protein kinase domains. Taken together, these findings highlight the role of FLT4 in TOF and CHD; however, the precise mechanism of how FLT4 variants provoke the cardiac structural alterations of TOF remains to be elucidated.
The human transcription factor NKX2.5 is the first gene associated with human cardiac development. It sits at the 5q34-q35 chromosome five region and is abundantly expressed in the human foetal heart (Shinebourne et al., 2006). An essential role of NKX2.5 during secondary heart field development is its interactions with the broader network of transcriptional factors that regulate cardiac differentiation and fate (Clark et al., 2013). Moreover, deletion of Nkx2.5 has been linked to abnormalities in the structure of the embryonic heart, growth delay, and embryo mortality in mice (Lyons et al., 1995). Several studies have also reported that variants in NKX2.5 cause a wide range of CHDs, including ASD, VSD, TOF, HLH, CoA, TGA, Double Outlet Right Ventricle (DORV), IAA, and cardiac outflow tract (OFT) anomalies; reviewed in (Chung and Rajakumar, 2016). Recently, new NKX2.5 variants were discovered in a Moroccan cohort with non-syndromic TOF; a missense variant R25C and a synonymous variant E21E (EL Bouchikhi et al., 2021). The R25C variant is expected to impair NKX2.5 dimerisation and impede activation of downstream target genes (Kasahara et al., 2000; Dentice et al., 2006). Bioinformatic predictions indicate that the E21E variant might impair NKX2.5 transcript splicing by removing a whole exon from the transcript, resulting in translation of a shortened protein that is most likely inactive. Further in vitro functional tests are needed to validate this finding (EL Bouchikhi et al., 2021).
In mammals, the master cardiac transcription factors (TFs) such as GATA4, TBX5, HAND2, KNX2.5, and MESP1 control the intricate and complicated process of heart development (Olsson et al., 2006). Pathological variants in essential cardiac transcription factor genes such as GATA4, GATA5 and GATA6 are associated with CHD (Kodo et al., 2009); Jiang et al., 2013; and (Granados-Riveron et al., 2011). Moreover (Töpf et al., 2014), reported that rare variants in the transcription factors of the second heart field, such as HAND2, FOXC1, FOXC2, FOXH1, and TBX1, can be found in non-syndromic TOF patients. These variants affect transcription activity; thereby disrupting the vital role these proteins play in the development of the outflow tract and right-sided cardiovascular components.
Via the general process of combinatorial interactions, a significant subset of cardiac transcription factors can regulate the intricate spatiotemporal control of heart morphogenesis and intermolecular interactions (Durocher and Nemer, 1998; Bruneau, 2008); and (Pradhan et al., 2017). For instance, physical interaction between GATA4, NKX2.5, and TBX5 activates downstream targets in a synergistic manner (Bruneau et al., 2001; Hiroi et al., 2001). Thus, mutations in these transcription factors may damage their interactions, thereby affecting protein function. Interestingly, according to a semi-quantitative study in the TOF cohort, (Qian et al., 2017); found that the interaction between the ZFPM2/FOG2 E1148K mutant protein with GATA4 was significantly damaged and was reduced by 50 per cent compared to the wild-type ZFPM2 protein (Qian et al., 2017). On the other hand, this result is not consistent with the previous findings whereby (Zhang et al., 2014) demonstrated that although the p.M703L and p. T843M ZFPM2 missense variants were assumed to be damaging, they had only a minor effect, if any, on the protein interaction with GATA4. Therefore, the impacts of individual variants may disrupt protein interactions to differing extents, or different variants may involve other unknown mechanisms to provoke CHD.
Moreover, multiple new potential candidate genes for causing TOF, that are linked to the function of FLT4, were identified, such as CLDN9, CCDC168, SN2, L1TD, TACC3, CCDC36 and TTC5 (Manshaei et al., 2020). More recently, it has been noted that twenty-three confirmed TOF genes form an interaction network with KDR/VEGFR2 and NOTCH1, acting as key nodes in the protein interaction network, each directly linking to 11 additional proteins via functional biological connections (Reuter et al., 2019). Reanalysis of the accuracy of these protein interactions may help to understand the interaction mechanisms and might inform therapeutic targets of TOF patients.
De novo variants have a major role in the development of early-onset genetic diseases such as CHD. A screen of a CHD cohort was performed by (Bauer et al., 2010); and found that JAG1 variants, producing functionally relevant sequence changes, were detected in 3% (2/94) of TOF cases. These individuals did not show signs of any other recognised syndromes. A recent study has concluded that a JAG1 variant: c.765C>T, is strongly linked to TOF in Iranian patients with a ratio of 60.2% versus 5.7% in controls (Safari-Arababadi et al., 2018); this variant may affect gene function. However, additional investigations need to be performed to understand the mechanisms of JAG1 alterations in TOF.
Furthermore, despite the limited sample size in a study conducted by (Manshaei et al., 2020); burden test findings indicated ultra-rare truncating variants in novel candidate genes, such as ZFAND5 and WNT5A, that might be involved in the genesis of TOF. A novel FLNA variant, p. L1139F, was discovered in two Iranian brothers diagnosed with X-linked recessive TOF. This variant was heterozygous in their mother, whilst their father had the wild type reference allele. Hemizygosity for this variant is therefore predicted to have a role in CHD, particularly TOF (Kalayinia et al., 2021). Nevertheless, further research is required to demonstrate the role of this variant or other FLNA variants in TOF individuals and their frequency in different populations. These new gene associations reveal the need for further investigation of the role of the contribution of rare variants to TOF in a larger patient cohort, which might give new insights into additional genetic causes of non-syndromic TOF.
In addition, it has also been identified that the loss of function of three emerging candidate genes, KDR, IQGAP1, and GDF1 may cause CHD and TOF (Reuter et al., 2019), but further clinical confirmation is needed. Highlighting the genetic variability of TOF; 16 additional patients have been identified to have one pathogenic/likely pathogenic variant (11 loss of function and five missense) in 16 CHD genes (ARHGAP31, ATRX, CACNA1C, CHD7, CSNK2A1, DLL4, EP300, GATAD2B, KAT6A, LZTR1, NF1, NODAL, PIK3CA, RAF1, RASA1, and SMAD2) and one patient has been identified with loss of function variants in two genes (ASXL1 and PSMD12) (Reuter et al., 2019). In addition, recent studies have shown that new variants found in TOF cases in the NDRG4 gene cause multiple heart abnormalities (Peng et al., 2021). Evidence suggests that SMARCC2 is required for cardiac development by promoting cardiac myocyte differentiation and controlling temporal steps in cardiac differentiation (Hota et al., 2019). More recently, in 2022, Sun et al. found a de novo SMARCC2 variant in TOF patients, suggesting there may be a link between CHD and deleterious SMARCC2 variants (Sun et al., 2022). Further research into the pathogenic mechanisms linked with these new variants, and genotype-phenotype associations is required, which may give new therapeutic insights for treating TOF and CHD.
Tetralogy of Fallot is the most common type of cyanotic congenital heart defect and a serious condition that includes four heart abnormalities: a ventricular septal defect, pulmonary stenosis/right ventricular outflow tract obstruction, the aorta overriding the VSD, and right ventricular hypertrophy (Hota et al., 2019). Despite advanced medical interventions, morbidity and mortality for individuals with TOF remain high due to the severe developmental cardiac abnormalities present in this condition. Additionally, the critical causative genes associated with non-syndromic TOF are poorly understood.
Recent progress in identifying the genetic basis for TOF has shown that deleterious variants in NOTCH1 and FLT4 play a significant role in TOF and CHD; however, the precise mechanism of these gene variations in TOF remains unknown. In addition, variants in additional cardiac transcription factors were connected to cardiac developmental defects and non-syndromic TOF (Granados-Riveron et al., 2011); Jiang et al., 2013, and (Töpf et al., 2014), suggesting the impacts of these mutants disrupt protein interactions which disrupts the crucial role these proteins play in the evolution of the cardiovascular components.
Recent research findings showed that several rare variants in many different genes might be associated with TOF in individual patients. Additionally, multiple variants in a single individual may act together to perturb cardiac development and cause TOF. Therefore, there may be a large number of genes that cause TOF in only a small number of individuals. These genes may have links to each other in protein interactions or signalling pathways needed for cardiac development. This finding suggests that more research is needed into the pathophysiological mechanisms associated with these new variants and genotype-phenotype associations. This research may be a promising aspect to provide new therapeutic insights for treating TOF and CHD.
NA: Performed Research, Wrote Manuscript KH: Wrote Manuscript, Edited Manuscript, Obtained Funding.
NA was supported by a PhD studentship from the Saudi Cultural Bureau in London and King Khalid Univesity. Work in KH’s laboratory is supported by British Heart Foundation grant PG/20/14/35030.
We thank Areej Alenazy for assistance with the creation of Figure 1 in BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al Turki S., Manickaraj A. K., Mercer C. L., Gerety S. S., Hitz M.-P., Lindsay S., et al. 2014). Rare variants in NR2F2 cause congenital heart defects in humans. Am. J. Hum. Genet., 95(1), p.126. doi:10.1016/j.ajhg.2014.06.013
Andersson E. R., Sandberg R., Lendahl U. (2011) Notch signaling: Simplicity in design, versatility in function, 138. Development, (17), pp.3593–3612. doi:10.1242/dev.063610
Bailliard F., Anderson R. H. (2009). Tetralogy of Fallot. Orphanet J. Rare Dis. 4, 2. doi:10.1186/1750-1172-4-2
Bauer R. C., Laney A. O., Smith R., Gerfen J., Morrissette J. J. D., Woyciechowski S., et al. (2010). Jagged1 (Jag1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat., 31(5), Pp.594–601. doi:10.1002/Humu.21231
Bedair R., Iriart X. (2019). Educational series in congenital heart disease: Tetralogy of Fallot: Diagnosis to long-term follow-up. Echo Res. Pract., 6(1), pp.R10–R24. doi:10.1530/erp-18-0049
Benson D. W., Silberbach G. M., Kavanaugh-McHugh A., Cottrill C., Zhang Y., Riggs S., et al. (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Invest., 104(11), pp.1567–1573. doi:10.1172/jci8154
Bruneau B. G., Nemer G., Schmitt J. P., Charron F., Robitaille L., Caron S., et al. (2001). A murine model of holt-oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell, 106(6), pp.709–721. doi:10.1016/s0092-8674(01)00493-7
Bruneau B. G. (2008). The developmental genetics of congenital heart disease. Nature, 451(7181), pp.943–948. doi:10.1038/nature06801
Chung I.-M., Rajakumar G. (2016). Genetics of congenital heart defects: The NKX2-5 gene, a key player. Genes, 7(2), p.6. doi:10.3390/genes7020006
Clark C. D., Zhang B., Lee B., Evans S. I., Lassar A. B., Lee K. H. (2013). Evolutionary conservation of Nkx2.5 autoregulation in the second heart field. Dev. Biol. 374 (1), 198–209. doi:10.1016/j.ydbio.2012.11.007
De Luca A., Sarkozy A., Ferese R., Consoli F., Lepri F., Dentici M., Vergara P., De Zorzi A., Versacci P., Digilio M., Marino B., Dallapiccola B. (2010). New mutations in zfpm2/fog2 gene in tetralogy of Fallot and Double outlet right ventricle. Clin. Genet., 80(2), Pp.184–190. doi:10.1111/J.1399-0004.2010.01523.X
Dennis M., Moore B., Kotchetkova I., Pressley L., Cordina R., Celermajer D. S. (2017). Adults with repaired tetralogy: Low mortality but high morbidity up to middle age. Open Heart, 4(1), p.e000564. doi:10.1136/openhrt-2016-000564
Dentice M., Cordeddu V., Rosica A., Ferrara A. M., Santarpia L., Salvatore D., et al. (2006). Missense mutation in the transcription factor NKX2-5: A novel molecular event in the pathogenesis of thyroid dysgenesis. J. Clin. Endocrinol. Metabolism, 91(4), pp.1428–1433. doi:10.1210/jc.2005-1350
Dolk H., Loane M., Garne E. Congenital heart defects in Europe. (2011) congenital heart defects in Europe: Prevalence and perinatal mortality, 2000 to 2005. Circulation. 123(8):841–849. doi:10.1161/CIRCULATIONAHA.110.958405
Durocher D., Nemer M. (1998). Combinatorial interactions regulating cardiac transcription. Dev. Genet., 22(3), pp.250–262. doi:10.1002/(sici)1520-
El Bouchikhi I., Belhassan K., Moufid F. Z., Bouguenouch L., Samri I., Iraqui Houssaïni M., et al. (2021). Screening of NKX2.5 gene in Moroccan tetralogy of Fallot (TOF) patients: Worldwide mutation rate comparisons show a significant association between R25C variant and TOF phenotype. Egypt J. Med. Hum. Genet., 22(1). doi:10.1186/s43042-021-00136-1
Eldadah Z. A., Hamosh A., Biery N. J., Montgomery R. A., Duke M., Elkins R., et al. (2001). Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum. Mol. Genet. 10 (2), 163–169. [online]. doi:10.1093/hmg/10.2.163
Garg V., Muth A. N., Ransom J. F., Schluterman M. K., Barnes R., King I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature. 437 (7056), 270–274. doi:10.1038/nature03940
Goldmuntz E., Clark B. J., Mitchell L. E., Jawad A. F., Cuneo B. F., Reed L., et al. (1998). Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol., 32(2), pp.492–498. doi:10.1016/s0735-1097(98)00259-9
Goldmuntz E. (2001). The epidemiology and genetics of congenital heart disease. Clin. Perinatology, 28(1), pp.1–10. doi:10.1016/s0095-5108(05)70067-1
Goodship J. A., Hall D., Topf A., Mamasoula C., Griffin H., Rahman T. J., et al. 2012). A common variant in the PTPN11 gene contributes to the risk of tetralogy of Fallot. Circ. Cardiovasc Genet., 5(3), pp.287–292. doi:10.1161/circgenetics.111.962035
Granados-Riveron J. T., Pope M., Bu'Lock F. A., Thornborough C., Eason J., Setchfield K., et al. (2011). Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: Multiple heterozygosity and novel mutations. Congenit. Heart Dis., 7(2), pp.151–159. doi:10.1111/j.1747-0803.2011.00573.x
Hiroi Y., Kudoh S., Monzen K., Ikeda Y., Yazaki Y., Nagai R., et al. (2001). Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet., 28(3), pp.276–280. doi:10.1038/90123
Ho C. Y., Day S. M., Ashley E. A., Michels M., Pereira A. C., Jacoby D., et al. (2019). Response by Ho et al to Letter Regarding Article, "Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights From the Sarcomeric Human Cardiomyopathy Registry (SHaRe)"’. Circulation, 139(12), pp.1559–1560. doi:10.1161/circulationaha.118.039069
Hoffman J. I. E., Kaplan S. (2002). The incidence of congenital heart disease. J. Am. Coll. Cardiol., 39(12), pp.1890–1900. doi:10.1016/s0735-1097(02)01886-7
Homsy J., Zaidi S., Shen Y., Ware J. S., Samocha K. E., Karczewski K. J., et al. 2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science, 350(6265), pp.1262–1266. doi:10.1126/science.aac9396
Hota S. K., Johnson J. R., Verschueren E., Thomas R., Blotnick A. M., Zhu Y., et al. (2019), Dynamic BAF chromatin remodeling complex subunit inclusion promotes temporally distinct gene expression programs in cardiogenesis. Development. 146, dev174086. doi:10.1242/Dev.174086
Huang X., Niu W., Zhang Z., Zhou C., Xu Z., Liu J., et al. (2014). Identification of novel significant variants of zfpm2/fog2 in non-syndromic tetralogy of Fallot and Double outlet right ventricle in A Chinese han population. Mol. Biol. Rep., 41(4), Pp.2671–2677. doi:10.1007/S11033-014-3126-5
Jiang J. Q., Li R. G., Wang J., Liu X. Y., Xu Y. J., Fang W. Y., et al. (2013). Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int. J. Cardiol. 165 (3), 570–573. doi:10.1016/j.ijcard.2012.09.039
Jin J. Y., Wu L. P., Dong Y., Pi H. C., Wu H. F., Xiang R. (2022). Identification of a novel GATA binding protein 5 variant (c.830C>T/p.P277L) damaging the nuclear translocation and causing tetralogy of Fallot. Qjm,115(4):256–258. doi:10.1093/Qjmed/Hcab317.Pmid:34904678
Jin S. C., Homsy J., Zaidi S., Lu Q., Morton S., DePalma Sr., et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet., 49(11):1593–1601. doi:10.1038/Ng.3970
Kalayinia S., Maleki M., Mahdavi M., Mahdieh N. (2021) Whole-exome sequencing reveals a novel mutation of FLNA gene in an Iranian family with nonsyndromic tetralogy of Fallot. Lab. Med., 52, 614, 618. doi:10.1093/Labmed/Lmab018
Karaman S., Paavonsalo S., Heinolainen K., Lackman M. H., Ranta A., Hemanthakumar K. A., et al. (2022). Interplay of vascular endothelial growth factor receptors in organ-specific vessel maintenance. J. Exp. Med., 219(3). doi:10.1084/jem.20210565
Kasahara H., Lee B., Schott J.-J., Benson D. W., Seidman J. G., Seidman C. E., et al. (2000). Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J. Clin. Invest., 106(2), pp.299–308. doi:10.1172/jci9860
Kassab K., Hariri H., Gharibeh L., Fahed A. C., Zein M., El‐Rassy I., et al. (2015). Gata5 mutation homozygosity linked to A Double outlet right ventricle phenotype in A Lebanese patient. Mol. Genet. Genomic Med., 4(2), Pp.160–171. doi:10.1002/Mgg3.190
Kerstjens-Frederikse W. S., van de Laar I. M. B. H., Vos Y. J., Verhagen J. M. A., Berger R. M. F., Lichtenbelt K. D., et al. (2016). Cardiovascular malformations caused by NOTCH1 mutations do not keep left: Data on 428 probands with left-sided CHD and their families. Genet. Med., 18(9), pp.914–923. doi:10.1038/gim.2015.193
Kodo K., Nishizawa T., Furutani M., Arai S., Yamamura E., Joo K., et al. (2009). GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. U.S.A, 106(33), pp.13933–13938. doi:10.1073/pnas.0904744106
Luxán G., D’Amato G., MacGrogan D., de la Pompa J. L. (2016). Endocardial Notch signaling in cardiac development and disease. Circ. Res., 118(1). e1-e18. doi:10.1161/circresaha.115.305350
Lyons I., Parsons L. M., Hartley L., Li R., Andrews J. E., Robb L., et al. (1995). Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 9 (13), 1654–1666. doi:10.1101/gad.9.13.1654
Manshaei R., Merico D., Reuter M. S., Engchuan W., Mojarad B. A., Chaturvedi R., et al. 2020). Genes and pathways implicated in tetralogy of Fallot revealed by ultra-rare variant burden analysis in 231 genome sequences. Front. Genet., 11. 957. doi:10.3389/Fgene.2020.00957
Marino B. S., Lipkin P. H., Newburger J. W., Peacock G., Gerdes M., Gaynor J. W., et al. (2012). Neurodevelopmental outcomes in children with congenital heart disease: Evaluation and management. Circulation, 126(9), pp.1143–1172. doi:10.1161/cir.0b013e318265ee8a
Matos-Nieves A., Yasuhara J., Garg V. (2019). Another Notch in the genetic puzzle of tetralogy of Fallot. Circ. Res., 124(4), pp.462–464. doi:10.1161/circresaha.118.314520
McElhinney D. B., Krantz I. D., Bason L., Piccoli D. A., Emerick K. M., Spinner N. B., et al. (2002). Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation, 106(20), pp.2567–2574. doi:10.1161/01.cir.0000037221.45902.69
Mcleod G., Shum K., Gupta T., Chakravorty S., Kachur S., Bienvenu L., et al. (2018). Echocardiography in congenital heart disease. Prog. Cardiovasc. Dis. 61(5-6):468–475. doi:10.1016/j.pcad.2018.11.004
Michielon G., Marino B., Formigari R., Gargiulo G., Picchio F., Digilio M. C., et al. (2006). Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann. Thorac. Surg., 81(3), pp.968–975. doi:10.1016/j.athoracsur.2005.09.033
Morgenthau A., Frishman W. H. (2018). Genetic origins of tetralogy of Fallot, Cardiology in review: 26(2), pp 86–92. doi:10.1097/CRD.0000000000000170
Norgaard M. A., Olsen P. S., Svendsen U. G., Pettersson G. (1996). Revascularization of the bronchial arteries in lung transplantation: An overview. Ann. Thorac. Surg., 62(4), pp.1215–1221. doi:10.1016/0003-4975(96)00539-5
Oechslin E. N., Attenhofer Jost C. H., Rojas J. R., Kaufmann P. A., Jenni R. (2000). Long-term follow-up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. J. Am. Coll. Cardiol., 36(2), pp.493–500. doi:10.1016/s0735-1097(00)00755-5
Olsson A.-K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006). VEGF receptor signalling ? In control of vascular function. Nat. Rev. Mol. Cell Biol, 7(5), pp.359–371. doi:10.1038/nrm1911
Page D. J., Miossec M. J., Williams S. G., Monaghan R. M., Fotiou E., Cordell H. J., et al. 2019). Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ. Res., 124(4), Pp.553–563. doi:10.1161/Circresaha.118.313250
Palomino Doza J., Topf A., Bentham J., Bhattacharya S., Cosgrove C., Brook J. D., et al. (2013). Low-frequency intermediate penetrance variants in the ROCK1 gene predispose to Tetralogy of Fallot. BMC Genet., 14(1). 57. doi:10.1186/1471-2156-14-57
Park G., Thayer J. F. (2014). From the heart to the mind: Cardiac vagal tone modulates top-down and bottom-up visual perception and attention to emotional stimuli. Front. Psychol., 5. 278. doi:10.3389/fpsyg.2014.00278
Peck D., Tretter J., Possner M., Yutzey K., Zafar F., Morales D., et al. (2021). Timing of repair in tetralogy of Fallot: Effects on outcomes and myocardial health. Cardiol. Rev., 29,(2), Pp.62–67. doi:10.1097/Crd.0000000000000293
Peng J., Wang Q., Meng Z., Wang J., Zhou Y., Zhou S., Song W., Chen S., Chen A. F., Sun K. (2021). A loss‐of‐function mutation p.T256M in NDRG4 is implicated in the pathogenesis of pulmonary atresia with ventricular septal defect (PA/VSD) and tetralogy of Fallot (TOF). Febs Open Bio, 11(2), Pp.375–385. doi:10.1002/2211-5463.13044
Pinsker B. L., Serfas J. D., Krasuski R. A. (2022). Burden and impact of arrhythmias in repaired tetralogy of Fallot. Curr. Cardiol. Rep., 24(3), Pp.225–234. doi:10.1007/S11886-022-01638-Z
Postoev V. A., Talykova L. V., Vaktskjold A. (2014). Epidemiology of cardiovascular malformations among newborns in monchegorsk (North-West Russia): A register-based study. J. Public Health Res. ;3(2):270. doi:10.4081/jphr.2014.270.PMID:25343136
Pradhan A., Zeng X.-X. I., Sidhwani P., Marques S. R., George V., Targoff K. L., et al. (2017). FGF signaling enforces cardiac chamber identity in the developing ventricle. Development, 144(7), pp.1328–1338. doi:10.1242/dev.143719
Qian Y., Xiao D., Guo X., Chen H., Hao L., Ma X., Huang G., Ma D., Wang H. (2017). Multiple gene variations contributed to congenital heart disease via gata family transcriptional regulation. J. Transl. Med., 15(1), 69. doi:10.1186/S12967-017-1173-0
Reuter M. S., Jobling R., Chaturvedi R. R., Manshaei R., Costain G., Heung T., et al. (2019). Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med., 21(4), Pp.1001–1007. doi:10.1038/S41436-018-0260-9
Safari-Arababadi A., Behjati-Ardakani M., Kalantar S. M., Jaafarinia M. (2018). Silencing mutations in JAG1 gene may play crucial roles in the pathogenesis of tetralogy of Fallot. Cell Mol. Biol. (Noisy-le-grand), 64 (4), P.103. doi:10.14715/Cmb/2018.64.4.17
Senst B., Kumar A., Diaz R. R. Cardiac surgery. In: Treasure island (FL), Tampa, Florida, United States: StatPearls Publishing. 2022, StatPearls [internet].
Shi L.-M., Tao J.-W., Qiu X.-B., Wang J., Yuan F., Xu L., et al. (2014). Gata5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Of Mol. Med., 33(5), Pp.1219–1226. doi:10.3892/Ijmm.2014.1700
Shinebourne E. A., Babu-Narayan S. V., Carvalho J. S. (2006) Tetralogy of Fallot: From fetus to adult. Heart. 92(9):1353–1359. doi:10.1136/hrt.2005.061143
Sifrim A., Hitz M.-P., Hitz A., Wilsdon J., Breckpot S. H. A., Turki B., et al. 2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet., 48(9), pp.1060–1065. doi:10.1038/ng.3627
Škorić-Milosavljević D., Lahrouchi N., Bosada F. M., Dombrowsky G., Williams S. G., Lesurf R., et al. (2021). Rare variants in kdr, encoding vegf receptor 2, are associated with tetralogy of Fallot. Genet. Med., 23(10), Pp.1952–1960. doi:10.1038/S41436-021-01212-Y
Stittrich A.-B., Lehman A., Bodian Dale L., Ashworth J., Zong Z., Li H., et al. (2014). Mutations in NOTCH1 cause adams-oliver syndrome. Am. J. Hum. Genet., 95, (3), pp.275–284. doi:10.1016/j.ajhg.2014.07.011
Su Z., Zou Z., Hay S. I., Liu Y., Li S., Chen H., et al. (2022). Global, regional, and national time trends in mortality for congenital heart disease, 1990-2019: An age-period-cohort analysis for the Global Burden of Disease 2019 study. Eclinicalmedicine, 43, P.101249. doi:10.1016/J.Eclinm.2021.101249
Sun H., Zhang S., Wang J., Zhou X., Zhang H., Yang H., et al. (2022). Expanding the phenotype Associated with Smarcc2 variants: A fetus with tetralogy of Fallot. BMC Med. Genomics, 15(1). 40. doi:10.1186/S12920-022-01185-0
Suzuki N., Svensson K., Eriksson U. J. (1996). High glucose concentration inhibits migration of rat cranial neural crest cells in vitro. Diabetologia, 39(4), pp.401–411. doi:10.1007/bf00400671
Tan Z-P., Huang C., Xu Z-B., Yang J-F., Yang Y-F. (2011). Novelzfpm2/Fog2variants in patients with Double outlet right ventricle. Clin. Genet., 82(5), Pp.466–471. doi:10.1111/J.1399-0004.2011.01787.X
Tomita-Mitchell A., Maslen C. L., Morris C. D., Garg V., Goldmuntz E. (2007). GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet., 44(12), pp.779–783. doi:10.1136/jmg.2007.052183
Töpf A., Griffin H. R., Glen E., Soemedi R., Brown D. L., Hall D., et al. (2014). Functionally significant, rare transcription factor variants in tetralogy of Fallot. Plos One, 9(8), P.E95453. doi:10.1371/Journal.Pone.0095453
Wei D., Bao H., Liu X.-Y., Zhou N., Wang Q., Li R.-G., et al. (2013). Gata5 loss-of-function mutations underlie tetralogy of Fallot. Int. J. Med. Sci., 10(1), Pp.34–42. doi:10.7150/Ijms.5270
Wilson R., Ross O., Griksaitis M. J. (2019, Tetralogy of Fallot, BJA Educ., 19). (11), Pp.362–369. doi:10.1016/J.Bjae.2019.07.003
Wiputra H., Chen C. K., Talbi E., Lim G. L., Soomar S. M., Biswas A., et al. (2018). Human fetal hearts with tetralogy of Fallot have altered fluid dynamics and forces. Am. J. Of Physiology-Heart Circulatory Physiology, 315(6), Pp.H1649–H1659. doi:10.1152/Ajpheart.00235.2018
Zaidi S., Choi M., Wakimoto H., Ma L., Jiang J., Overton J. D., et al. 2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature, 498(7453), pp.220–223. doi:10.1038/nature12141
Keywords: Tetralogy of Fallot, non-syndromic, genetics, congenital heart disease, NOTCH1, FLT4, transcription factors
Citation: Althali NJ and Hentges KE (2022) Genetic insights into non-syndromic Tetralogy of Fallot. Front. Physiol. 13:1012665. doi: 10.3389/fphys.2022.1012665
Received: 05 August 2022; Accepted: 13 September 2022;
Published: 06 October 2022.
Edited by:
Diego Franco, University of Jaén, SpainReviewed by:
Cathy J. Hatcher, Philadelphia College of Osteopathic Medicine (PCOM), United StatesCopyright © 2022 Althali and Hentges. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn E. Hentges, a2F0aHJ5bi5oZW50Z2VzQG1hbmNoZXN0ZXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.