 Qiping Wang
Qiping Wang Dafei Chai2†
Dafei Chai2† Navid Sobhani
Navid Sobhani Praveen Neeli
Praveen Neeli Junnian Zheng
Junnian Zheng Hui Tian
Hui Tian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 17 November 2022
Sec. Mitochondrial Research
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.1012112
This article is part of the Research TopicCurrent Challenges in Complement DiagnosticsView all 27 articles
Mitochondrial plasticity including mitochondrial dynamics, metabolic flexibility, and mitochondrial quality control, impact tumor cells’ progression and determine immune cells’ fate. Complement C1q binding protein (C1QBP) plays an indispensable role through regulating mitochondrial morphology, metabolism, and autophagy. C1QBP promotes mitochondrial plasticity to impact tumor metastasis and their therapeutic response. At the same time, C1QBP is involved in regulating immune cells’ maturation, differentiation, and effector function through the enhancement of mitochondrial function. In this regard, manipulation of C1QBP has been shown to adjust the competitive balance between tumor cells and immune cells. In the course of evolution, mitochondrial plasticity has endowed numerous advantages against the relentless microenvironment of tumors. In this current review, we summarize the current knowledge of the mechanism of C1QBP regulation of cancer and immunity. We explain this process in vision of potentially new anticancer therapies.
Mitochondria can rapidly sense energy availability and generate precursors necessary for the biosynthesis of amino acids, lipids, and nucleotides to meet cellular requirements or respond to adverse cellular stimulation (Mishra and Chan, 2016; Ahola et al., 2019). Most importantly, mitochondria function as the hub of numerous signal pathways, including the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), and fatty acid oxidation (FAO), which endow cells with the considerable adaptation to face a variety of challenging stresses (Vyas et al., 2016; Schvartzman et al., 2018).
Complement C1q binding protein (C1QBP), also known as p32 or hyaluronic acid-binding protein (HABP1), is a multicompartmental protein. In other words, C1QBP exists in multiple subcellular compartments, including the nucleus, endoplasmic reticulum, Golgi, and on the cell surface (Ghebrehiwet et al., 2001; Fogal et al., 2008). For example, C1QBP located on the cell surface interacts with tumor homing peptide, Lyp-1, which specifically recognizes an epitope in tumor cells (Fogal et al., 2008). Moreover, it binds with the pre-mRNA splicing factor (SF2/ASF) in the nucleus, which controls RNA splicing by sequestering an essential RNA splicing factor into an inhibitory complex (Petersen-Mahrt et al., 1999). However, increasing evidence has demonstrated that C1QBP predominantly resides in the mitochondria through its 33-residue N-terminal mitochondria-targeting signal (MTS) sequence and is essential for embryonic development, which is dependent on mitochondrial translation and OXPHOS (Muta et al., 1997; Fogal et al., 2010; Yagi et al., 2012; Zhai et al., 2021). It has been reported that C1qbp deletion induces embryos’ mid-gestation lethality and triggers severe dysfunction of the mitochondrial respiratory chain due to failure of mitochondrial protein synthesis. The association of C1QBP with mitochondrial RNA and mitochondrial ribosomes is required for some mitochondrial protein translation. Besides, C1QBP is also involved in mitochondrial autophagy to remove the dysfunctional mitochondria to maintain healthy cellular homeostasis (Jiao et al., 2015; Jiao and You, 2016). Consequently, C1QBP is integral to mitochondrial fitness, cell metabolism, and survival.
Given that tumor cells and T cells share the metabolic similarities of proliferation, expansion as well as activation, manipulation of mitochondrial plasticity would impact the metabolic competition between tumor cells and T cells in the context of the tumor microenvironment (TME). Tumor cells depend on C1QBP for progression and therapeutic resistance, while immune cells also depend on it for a persistent and powerful antitumor response. This review discusses the dual roles of C1QBP for mitochondrial function in tumor malignancy and immune cells. Finally, we give our expert opinion on potential new mitochondrial-dependent immune stimulation mechanism based-therapies targeting C1QBP.
Mitochondria are highly dynamic organelles constantly undergoing fission and fusion. When they are too damaged, they go through mitophagy, which is the selective degradation of mitochondria by autophagy. It has been elucidated that C1QBP plays a vital role in maintaining mitochondrial homeostasis through regulating mitochondrial morphology, metabolism, and mitophagy. Thereby this protein is fundamental for modulating mitochondrial plasticity and fitness.
Mitochondria undergo constant fission and fusion cycles according to the cellular bioenergetic situation. This fine-tuning of mitochondria is vital to give cells the optimal energy supply and survival. The mitochondrial architecture principally consists of optical atrophy 1 (OPA1), mitofusin 1/2 (Mfn1/2), the mitochondrial fission controlled by dynamin-related protein 1 (Drp1), and its mitochondrial adaptor fission protein 1 (Fis1) (Santel and Fuller, 2001; Chen and Chan, 2005; Loson et al., 2013).
C1QBP modulates mitochondrial morphology and integrity. C1qbp silencing not only promoted mitochondrial fragmentation but also repressed mitochondrial fusion (Fogal et al., 2010; Li et al., 2011; Hu et al., 2013; Noh et al., 2020). For example, Solhee Noh et al. found that genetic ablation of C1QBP activates the overlapping activity with m-AAA protease (OMA1) to cleave OPA1, which leads to mitochondrial fragmentation and swelling (Noh et al., 2020). C1QBP regulated mitochondrial morphology by regulating OMA1-dependent proteolytic processing of OPA1. On the other hand, MengJie Hu et al. revealed that overexpression of C1QBP increased mitochondrial fibrils (Hu et al., 2013). At the same time, C1QBP siRNA-treated cells contained predominantly small spherical mitochondria compared with the mainly normal elongated mitochondria in control cells. To further explore the underlying mechanism, they examined the expression of Mfn1, Mfn2, and mitochondrial Drp-1 and found that the C1QBP siRNA treatment resulted in a detectable loss of Mfn1 and Mfn2 and decreased Drp-1 levels. Here, siRNA-mediated C1qbp knockdown induced a more punctate mitochondrial morphology, which was associated with the reduction of the mitochondrial fusion mediator proteins, a sign of potential subsequent degradation.
Some tumor cells significantly depend on mitochondrial-mediated oxidative phosphorylation (Funes et al., 2007; Moreno-Sanchez et al., 2007). Since oxidative metabolism may recycle lactate, mitochondrial metabolism is advantageous for tumor cells that are highly glycolytic (Sonveaux et al., 2008). Moreover, glycolytic pathway oncogenes may upregulate some genes encoding mitochondrial proteins or those involved in mitochondrial biogenesis (Li et al., 2005). Additionally, mitochondria regulate cellular bioenergetic and biosynthetic processes of important cancer signaling pathways, such as proliferation, metastasis and self-renewal (Viale et al., 2014; Liu and Shi, 2020). Collectively, these findings suggest that oncogene-driven glycolytic metabolism should be balanced, at least in part, by concomitant changes to mitochondria. Mitochondrial metabolism may potentiate cellular plasticity and metabolic flexibility to deal with the relentless microenvironment.
C1QBP plays an indispensable role in mitochondrial function and metabolic regulation. For example, C1qbp-deficient mouse embryonic fibroblasts (MEFs) cells exhibit reduced OXPHOS function (Yagi et al., 2012). A mutation of C1qbp has also been suggested as a cause of mitochondrial respiratory chain disorder. In this regard, C1QBP is a crucial regulator of mitochondria-mediated OXPHOS.
A functional mitochondrial network responding to physiological adaptations and stress conditions is critical for a healthy cellular condition. Given that mitochondria are exposed to high levels of reactive oxygen species, they have evolved multiple systems of quality control to ensure that the requisite number of functional mitochondria are present to meet cellular demands. Mitochondria eliminate the damaged mitochondrial proteins through autophagy. Thus, understanding how C1QBP orchestrates mitophagy to maintain mitochondrial homeostasis could provide critical insights.
Jiao et al. (2015) reported that C1QBP controlled mitochondrial autophagy to help cells adapt better to the challenging microenvironment (Jiao and You, 2016). Serine/threonine kinase Unc-51-like kinase-1 (ULK1) is crucial in inducing mitophagy. This protein is susceptible to proteasome-mediated degradation. C1QBP regulates ULK1 stability by forming a protein complex with ULK1. The interaction between ULK1 and C1QBP is indispensable to maintaining steady-state levels of ULK1, thereby preventing its polyubiquitylation. Moreover, it has been shown that mitophagy defects can be recovered by re-introducing ULK1 into C1QBP-deficient cells, which suggested that C1QBP protected mitophagy through the prevention of ULK1 degradation. Considering that mitochondrial homeostasis and oxidative phosphorylation might be mutually beneficial (Wrighton, 2013), mitochondrial quality control may serve as an important mechanism for maintaining cellular energy homeostasis. Therefore, C1QBP exerts a protective effect under starvation conditions by both inducing Ulk1-dependent autophagy and maintaining oxidative phosphorylation. In this regard, C1QBP-Ulk1-mitophagy axis may provide a survival advantage when nutrients are scarce, which ultimately promotes tumorigenesis.
Taken together, mitochondrial plasticity including mitochondrial dynamics, metabolism, and autophagy is tightly linked to mitochondrial structure, function and homeostasis. C1QBP overexpression promotes mitochondrial plasticity to endow tumor cells with proliferation and malignant progression. On the contrast, C1qbp knocking down dampens mitochondrial adaptation and metabolic flexibility, which is responsible for tumor repression.
C1QBP is expressed at high levels in a significant number of tumor types, including melanoma, colon, ovarian, gastric, prostate, brain, and breast, compared with their nonmalignant counterparts (Chen et al., 2009; Amamoto et al., 2011; Yu et al., 2013; Niu et al., 2015; Wang et al., 2015; Gao et al., 2016; Rousso-Noori et al., 2021; Sinha et al., 2021). Various studies involving patients have revealed that C1QBP is positively linked to the tumor stage and poor prognosis, as shown in Table 1.

TABLE 1. C1QBP overexpressed in a variety of tumor cells is implicated in tumor malignant progression and tumor therapeutic treatment.
As expected, high expression levels of C1QBP are inversely correlated with tumor patients’ prognosis, making C1QBP a valuable independent prognostic marker of outcomes in tumor patients. For example, upregulation of C1qbp mRNA has been detected in low and high-grade gliomas, especially recurrent glioblastoma (GBM) (Rousso-Noori et al., 2021). This protein significantly is associated with the extent of the disease progression. It might also serve as a tumor-associated antigen (TAA) in gliomas.
Furthermore, C1QBP modulates tumor proliferation and metastasis. Its expression level in breast cancer was closely linked with distant metastasis and tumor node metastasis (TNM) stages (Wang et al., 2015). X Zhang et al. (2013) reported that during metastasis, the C1QBP up-modulation was confined to metastatic islands, which could have significant implications if further experiments could show it can regulate tumor metastasis. On this note, the authors showed that C1QBP held the activity of protein kinase C and modulated EGF-induced cancer cell chemotaxis, which thus impacted tumor cell migration. In contrast, C1QBP silencing attenuated tumor progression and metastasis in vivo by modulating the tumor microenvironment through inhibiting tumor angiogenesis and macrophage infiltration (Sinha et al., 2021). Based on such premises, C1QBP can be a target of a novel therapeutic approaches in the regulation of tumor progression and metastasis. Additionally, C1QBP has been observed to adhere to the cell surface and interact with integrin αvβ3, a regulatory molecule of cell migration. C1QBP-integrin interaction results in transcriptional upregulation of MT1-MMP expression and finally MMP-2 activation, leading to enhanced tumorigenicity and cell migration in B16F10 melanoma cell line (Prakash et al., 2011). Besides, C1qbp knocking down exhibits the disruption of lamellipodia formation and a concomitant decrease in focal adhesion kinase (FAK) kinase and receptor kinase, inhibiting cellular migration and tumorigenesis (Kim et al., 2011).
On the other hand, increasing evidence has shown that C1QBP-mediated tumorigenesis is tightly associated with mitochondrial metabolism maintenance. Fogal et al. (2010) found that knocking down C1QBP expression in human cancer cells strongly shifted their metabolism from oxidative OXPHOS to glycolysis. C1QBP deficiency resulted in less in vivo tumorigenicity, probably due to the reduced synthesis of the mitochondrial-DNA-encoded OXPHOS polypeptides. Thus, C1QBP is implicated in mitochondrial protein translation and affects several electron transport complexes’ functions. On the other hand, recovering C1QBP reestablished their animal tumorigenicity in these knockdown cells.
Notably, C1QBP was also reported to be involved in glutamine oxidation (Fogal et al., 2015). It is commonly known that glutamine is essential for mammalian cell proliferation. It is therefore associated with tumor progression of any type of cancer. Higher glutamine oxidation positively correlates with a higher degree of tumor progression in vivo. Myc can upregulate C1qbp transcription, resulting in the regulation of glutamine metabolism, which suggest that a high level of Myc in malignant brain cancers is accompanied by increased expression of C1QBP. On the other hand, attenuation of C1QBP expression stunted glioma cells’ growth. C1QBP silencing in glutamine-addicted glioma cells induced resistance to glutamine deprivation. In other words, C1QBP is involved in mitochondrial metabolism and plays an integral role in Myc-induced glutamine addiction in cancer cells.
Additionally, C1QBP has been shown implicated in mitochondrial-dependent apoptosis of tumor cells. Hu et al. reported that siRNA-mediated C1qbp knockdown enhanced mitochondrial fragmentation, which was accompanied by a loss of detectable levels of the mitochondrial fusion mediator proteins Mfn 1/2. Furthermore, siRNA-mediated C1QBP knockdown enhanced tumor cell death in response to cisplatin treatment (Hu et al., 2013).
Overall, C1QBP regulates mitochondrial metabolism and dynamics to potentiate mitochondrial plasticity, thereby endowing tumor cells with a bioenergetic advantage for proliferation, metastasis, and resistance to therapies, as shown in Figure 1.
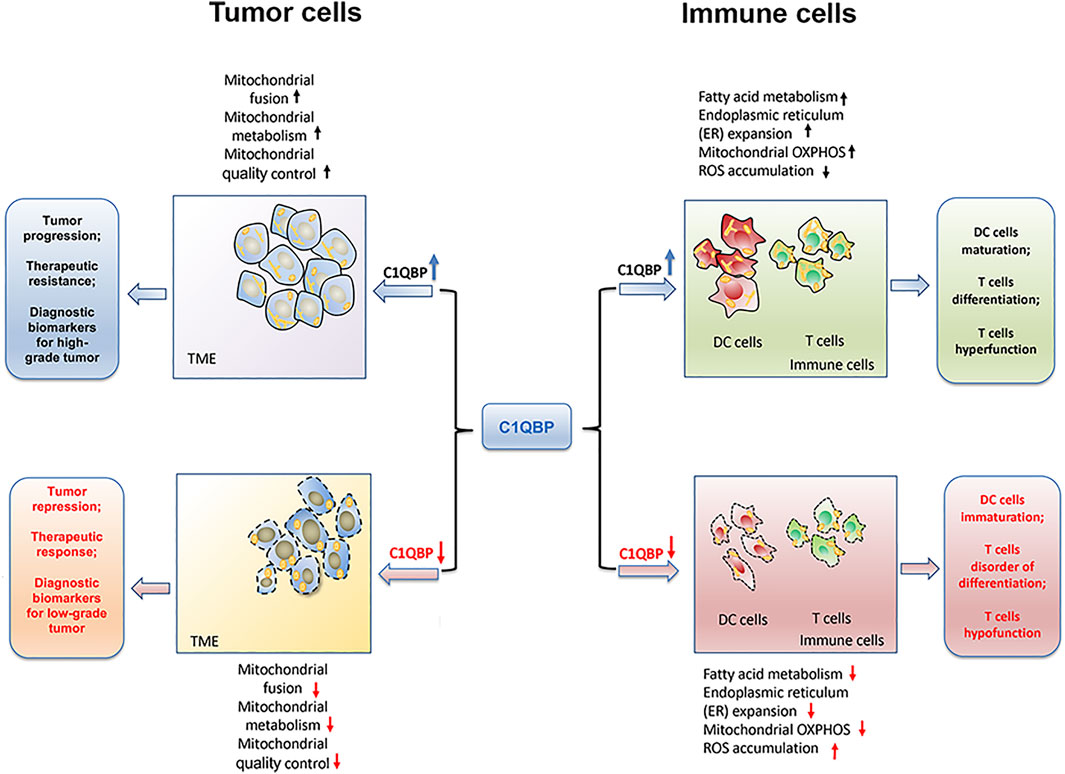
FIGURE 1. C1QBP regulation of mitochondrial plasticity to impact tumor progression and antitumor immunity. C1QBP regulates mitochondrial fusion, mitochondrial OXPHOS, and mitochondrial quality control to impact tumor malignant progression and therapeutic response. On the other hand, C1QBP also changes fatty acid metabolism, ER expansion, and ROS accumulation, thus modulating DCs’ maturation as well as T cells’ differentiation and their effector function.
Warburg hypothesized that the shift to aerobic glycolysis was a cause of cancer. Aerobic glycolysis supports macromolecule synthesis, rapid proliferation and heightened ATP demand, which are necessary for tumor rapid progression. However, even though tumor cells have high rates of glucose consumption via glycolysis, mitochondrial function was later found to be essential in several types of tumor (Wallace, 2012; Ward and Thompson, 2012). A significant proportion of their ATP is still produced via OXPHOS, again suggesting that at least some mitochondrial function is preserved in tumor cells. Similar to tumor cells, immune cells always shift to glycolysis to support their rapid growth and production of biosynthetic factors for differentiation into effector cells (Kouidhi et al., 2017). However, mitochondria generate metabolic intermediates that are important for their growth and proliferation as well as for their cytotoxicity and cytokine production. Several mitochondrial energy metabolism pathways have been implicated in shaping T-cells function and differentiation (Sena et al., 2013). Besides, memory CD8+ T cells require mitochondria-mediated FAO to support their long-term survival (Araki et al., 2009). Therefore, it is evident that mitochondrial function not only affects tumor onset, progression and distant metastasis but also controls immune cells’ maturation, differentiation, and long-term maintenance. A body of literature indicates that C1QBP plays a vital role in mitochondrial-dependent metabolic flexibility of critical immune cells of the immune systems (Gotoh et al., 2018; Zhai et al., 2021; Tian et al., 2022).
Dendritic cells (DCs) have different functions, and among them are the following ones: 1) constitute the most potent antigen-presenting cells; 2) induce primary immune responses; 3) capture antigens in peripheral tissues; 4) recognize pathogen-associated molecular patterns of invading microbes post-Toll-like receptors (TLRs) ligation, which leads to cellular activation and changes in gene expression and cellular metabolism in DCs (Banchereau et al., 2000; Kawai and Akira, 2011). Gotoh et al. (2018) reported that C1QBP was indispensable in DCs metabolism and maturation. Their study found that mitochondrial C1QBP and pyruvate dehydrogenase (PDH) activity are necessary for DC maturation both in vitro and in vivo. Although pyruvate and lactate were similarly produced by both W.T. and C1qbp−/− DCs, after lipopolysaccharide (LPS) stimulation, LPS-induced citrate production was impaired C1qbp−/− DCs. Generally, glucose is processed by the cytosolic glycolytic pathway to generate pyruvate. Acetyl-coenzyme A (acetyl-CoA) is produced from pyruvate by the PDH complex and subsequently reacts with oxaloacetate to form citrate in mitochondria. Given that after LPS stimulation, the upstream metabolites of citrate in wild type (WT) and C1qbp−/− DCs were produced at similar levels, the authors focused on the activity of PDH in both DCs types. They revealed that C1QBP could bind to PDH-E2 in DCs by immunoprecipitation experiments. Furthermore, the PDH activity in WT was significantly higher than that in C1qbp−/− DCs at the basal condition, which means C1QBP may control PDH activity by binding to PDH-E2 to regulate citrate production and further support DC. maturation. Notably, PDH-mediated citrate production from pyruvate is involved in fat acid synthesis (FAS). This mechanism impacts the endoplasmic reticulum (ER) expansion. Interestingly, ER expansion was observed in WT DCs but not in C1qbp−/− DCs after LPS stimulation.
Therefore, C1QBP bound to PDH-E2 promoted the activity of PDH, facilitating citrate production and enhancing FAS and ER expansion. In this regard, C1QBP involvement in regulating DCs maturation by controlling mitochondrial metabolism other than the electron transport chain (ETC) has many consequences to help fine-tune mitochondria expression, and alterations could lead to the decreased immune function of DCs.
Besides DCs maturation, C1QBP was also reported to be implicated in the regulation of T cells differentiation. Zhai et al. (2021) revealed that C1QBP was involved in the modulation of effector CD8+ T cells differentiation to affect their antiviral and antitumor immune responses further. Firstly, C1qbp deficiency in T cells intrinsically impaired the differentiation of effector CD8+ T cells, which is attributed to a failure to increase their mitochondrial respiratory capacities. They found that activated CD8+ T cells had greater glycolytic and OXPHOS requirements than activated CD4+ T cells and were more sensitive to OXPHOS impairment. In other words, C1QBP-mediated augmentation of OXPHOS was necessary for the differentiation of effector CD8+ T cells. Moreover, C1QBP promoted the epigenetic and transcriptional programs of effector CD8+ T cells. It achieved it by controlling the production of metabolites, including acetyl-CoA, fumarate, and 2-HG. Their study revealed that C1qbp deficiency mediated OXPHOS impairment. This, in turn, capitulated in decreased acetyl-CoA but increased fumarate and 2-HG. Moreover, downregulation of acetyl-CoA subsequently dampens histone protein H3K27 acetylation, which enhances CpG methylation to reduce some genes expression encoding several master transcription factors that drive effector CD8+ T cell differentiation such as Id2, Prdm1, and Tbx21.
Consequently, CD8+ T cell differentiation becomes retarded. C1QBP facilitates the epigenetic and transcriptional programs of effector CD8+ T cells differentiation by controlling metabolites coupled to OXPHOS. In this regard, they provide a mechanistic association between mitochondrial metabolism and CD8+ T cell differentiation, which means these metabolite supplementations or targeting OXPHOS may have significant application in T cell therapies for cancer.
Tumor-infiltrating T cells encounter not only persistent tumor antigen but also experience metabolic stress from the tumor microenvironment such as glucose competition, metabolite accumulation, and hypoxia (Vardhana et al., 2020; Yu et al., 2020). Scharping et al. (2016) revealed that continuous antigen stimulation, coordinated with hypoxia, impaired mitochondrial functions to drive T cells terminal exhaustion. Although hypoxia alone cannot induce T cells dysfunction, its association with persistent antigen stimulation plays a fundamental role in T cells exhaustion. While on the one hand, the tumor microenvironment makes tumor-infiltrating T cells with dysfunctional mitochondria have a lower mass and activity (measured as spare respiratory capacity), driving T cells’ metabolic insufficiency. On the other hand, the continuous activation of tumor-infiltrating cells further increases mitochondrial reactive oxygen species (ROS) to exacerbate the exhausted phenotype of tumor-specific T cells. Of note, mitigating hypoxia effectively alleviates these tumor-infiltrating T cells exhaustion. Mitochondria has a fundamental role in modulating T cells’ development, fate, and function. Fine-tuning mitochondria is critical for altering the different metabolic preferences and activities within cells. Recently it was shown that impaired ATP production and accumulation of mitochondrial ROS in dysfunctional T cells within the suppressive tumor microenvironment aggravated T cells exhaustion (Li et al., 2020; Vardhana et al., 2020). However, the relationship between metabolic stress and T cell exhaustion remains still unclear.
The question of whether the mitochondrial protein C1QBP is implicated with T cells exhaustion is interesting. Our previous study found that C1qbp knockdown modulated T cells with the accumulation of ROS and the loss of mitochondrial membrane potential, thus impairing T cell mitochondrial fitness (Tian et al., 2022). At the same time, C1QBP insufficiency aggravated exhausted T cells. We assessed exhaustion markers, such as PD-1, Tim-3, and LAG-3 in these tumor-specific T cells. We found that these inhibitory receptors exhibited higher expression levels in tumor-infiltrating C1qbp+/− T cells than the corresponding WT cells. In this regard, C1qbp knocking down exacerbated the tumor-infiltrating T cells exhaustion, leading to a reduced antitumor immune response.
Chronic antigen stimulation drives the defective oxidative phosphorylation and promotes ROS generation in activated T cells, so C1qbp could dampen T cells proliferation while remaining susceptible to cellular apoptosis. To detect the underlying molecular mechanism, we noticed AKT/mTOR proliferation signaling pathway and Bcl-2 anti-apoptotic signaling pathway. Our previous results demonstrated that C1qbp insufficiency inhibited the AKT/mTOR signaling pathway, which conferred T cells with relatively weaker proliferation capacity (Tian et al., 2022). At the same time, C1qbp knocking down reduced the anti-apoptotic proteins recruitment, such as Bcl-2 and Bcl-XL, but aggravated caspase-3 activation and poly (ADP-ribose) polymerase (PARP) cleavage, which thus accelerated T cells apoptotic process. Therefore, C1qbp knockdown induces T cells exhaustion and pre-apoptosis but retarded their proliferation as well as antitumor immune function.
Overall, the potentiation of immune cells’ mitochondrial plasticity through C1QBP promotes fatty acid metabolism, ER expansion, and mitochondrial OXPHOS, but relieves the accumulation of ROS, which could further enhance the DCs maturation, T cells differentiation as well as the tumor-infiltration T cells durability (Figure 1).
C1QBP is involved in tumor progression and metastasis, making it an attractive, actionable target against cancer, bringing to the development of monoclonal antibodies (mAbs). Such therapies were successful in inhibiting tumor growth and migration (Sanchez-Martin et al., 2011). The unique upregulation of C1QBP in certain tumors, and targeting it, in various ways, in such tumors could be a valuable strategy (Yenugonda et al., 2017; Rousso-Noori et al., 2021). C1QBP can also be found at the cell surface of angiogenic endothelial cells besides tumor cells. In nature, C1QBP acts as a receptor bound by the CGKRK peptide. Based on such premises, Agemy et al. devised a tumor-targeted nanosystem, through which CGKRK pentapeptide delivers a proapoptotic peptide into the mitochondria of tumor blood vessel endothelial cells and tumor cells (Agemy et al., 2013). The treatment was highly effective, especially in the refractory glioblastoma mouse models, compared to other antiangiogenic therapies.
Moreover, another tumor-homing peptide, LyP-1 (CGNKRTRGC), capable of targeting the cell surface of localized C1QBP, has been used to deliver nanoparticles to breast tumors overexpressing C1QBP and has shown efficacy in vivo (Luo et al., 2010). However, both the anti-C1QBP antibody and the LyP-1 peptide targeted the C1QBP subset that was exposed on the cell surface. Yenugonda et al. (2017) developed C1QBP-directed therapeutics to inhibit the protein in all subcellular compartments, including mitochondria. A primary screening assay employing a glutamine-addicted glioma cell line identified a hit compound called M36, which could bind and inhibit C1QBP located on the cellular membrane and in the mitochondria. In addition, they further demonstrated that M36 impedes the growth of glioma cells and patient-derived glioma stem cells in culture due to C1QBP genetic knockdown.
In addition, antibody neutralization of C1QBP inhibits lamellipodia formation, cell migration and focal adhesion kinase activation (Kim et al., 2016). At the same time, this antigen-antibody reaction inactivates receptor tyrosine kinases (RTKs) activation in various cancer cells and prevents even angiogenesis, resulting in the decreased tumorigenesis in vivo. Notably, A novel tumor penetrating peptide, linTT1 (AKRGARSTA), as a tumor targeting ligand for nanoparticles (Sharma et al., 2017). C1QBP as the primary homing receptor for linTT1, is expressed on the cell surface of peritoneal carcinoma cell lines (Paasonen et al., 2016). Iron oxide nanoworms (NWs) functionalized with the linTT1 peptide in tandem with a pro-apoptotic peptide showed C1QBP-dependent cytotoxicity (Hunt et al., 2017). At the same time, the linTT1-NWs predominantly accumulates in CD31-positive blood vessels, in LYVE-1-positive lymphatic structures, and in CD11b-positive tumor macrophages, thus resulting in significant reduction of weight of peritoneal tumors and significant decrease in the number of metastatic tumor nodules. In this regard, C1QBP-directed intraperitoneal targeting of other anticancer agents may represent a potential strategy to improve their therapeutic index.
Considering that C1QBP is be specifically expressed on the surface of glioma cells, it is regarded as a suitable tumor associated antigen (TAA) for redirected chimeric antigen receptor (CAR) T cell therapy. Rousso-Noori et al. (2021) reported that C1QBP-specific CAR T cells exerted a dual antitumor and antiangiogenic therapeutic benefit in gliomas. Their study focused on designing CAR T cells targeting C1QBP positive glioma cells. They found that the CAR T cells targeting C1QBP reduced tumor vascularization and extended the overall survival of mice bearing gliomas, thus providing proof-of-principle evidence that C1QBP-CAR T cells can recognize and eliminate not only glioma cells but also tumor endothelial cells. Thus, CAR T cells targeting C1QBP may serve as a therapeutic option for glioblastoma patients through specifically recognizing and eliminating C1QBP expressing glioma cells and tumor derived endothelial cells, finally controlling tumor growth in orthotopic syngeneic and xenograft mouse models.
On the other hand, upregulation of mitochondrial plasticity may effectively reinvigorate T cell’s robust and persistent immune function. This could alter the metabolic competition between tumor cells and immune systems. Thus, it is worth noting that C1QBP modified CAR T cells’ immune function and their long-term maintenance. Our previous study demonstrated that C1qbp knockdown attenuated CAR T cells’ antitumor immune response (Tian et al., 2022). Specifically, we constructed the C1qbp+/− and C1qbp+/+ CAR T cells targeting B7-H3. We found that C1qbp knocking down reduced CAR T cells’ release of IFN-γ and TNF-α cytokines, which reduced the corresponding immunotherapeutic efficacy of CAR T cells in vivo. In contrast, sufficient C1qbp potentiated CAR T cells’ mitochondrial adaptation to the relentless TME by ameliorating their exhaustion, thereby improving their durable antitumor immune function.
Since the mitochondrial function and metabolic flexibility imply the fate and function of immune cells, metabolic interventions manipulating immunity are rare. Using new proteins to achieve this purpose represents a great opportunity that remains untapped. Therefore, developing strategies for potentiation of mitochondrial-dependent metabolic flexibility could improve the efficacy of current immune-based anticancer therapy.
Mitochondrial plasticity endows cells with superior adaptation to the relentless microenvironment. This adaptation is regulated spatially and temporally by regulating gene expression and metabolic signatures, associated mitochondrial dynamics, metabolism, and quality control.
On the one hand, for tumor cells to survive, they are forced to remodel their signaling pathways by altering transcription, translation, and post-translational modifications to acquire phenotypic heterogeneity. Such heterogeneity, in turn, determines whether tumor cells resist environmental stress, enter dormancy or metastasize. Notably, it is imperative to know that mitochondrial plasticity plays a critical role in aiding cancer cells in acquiring metastatic traits under adverse environmental conditions. Cancer stem cells (CSCs) as a population of cells with stem cell-like properties are considered to be the root cause of tumor heterogeneity, which is also responsible for metastatic dissemination and therapeutic resistance (Clevers, 2011; Pattabiraman and Weinberg, 2014). CSCs are characterized by the increased mitochondrial mass and the elevated mitochondrial biogenesis (Luo and Wicha, 2015; Song et al., 2015; Peiris-Pages et al., 2016). In this scenario, mitochondrial biogenesis accompanied with increased membrane potential, and higher generation of mitochondria-derived ROS, has recently been regarded as a regulatory instigator of tumor stemness. Thus, the question whether C1QBP would regulate mitochondrial biogenesis to further impact tumor stemness will be of interest.
On the other hand, we must also stress that potentiation of immune cells, through enhancement of mitochondrial plasticity, contributes to preventing immune exhaustion and durable antitumor immunity. Improving T cells’ mitochondrial fitness can result in persistent immune function. It can cope with a stressful microenvironment. Consequently, in our opinion, an enhancement of mitochondrial function could be a promising strategy for tumor immunotherapy that could be used in combination with the current checkpoint blockade therapies available, such as anti-PD-L1, PD-1, and CTLA-4. Besides, a successful immune response depends on the ability of T cells not only to proliferate extensively and attain effector functions but also to generate long-lived memory T cells. FAO engagement is critical for the generation of memory T cells (Raud et al., 2018). C1QBP was reported to be involved in regulation of lipid metabolism and homeostasis (Liu et al., 2017; Liu et al., 2018). Therefore, the question whether metabolic reprogramming such as FAO through modulation of C1QBP would probably impact the generation of long-lived memory T cells requires further evaluation.
In conclusion, C1QBP is vital in maintaining mitochondrial plasticity. Therefore, inhibition of C1QBP in tumor cells and promotion of C1QBP in immune cells would beneficially adjust the competitive balance between tumor and immune cells, providing a very reasonable therapeutic opportunity by finding a way to correctly fine-tune mitochondrial-based metabolic flexibility (Figure 2).
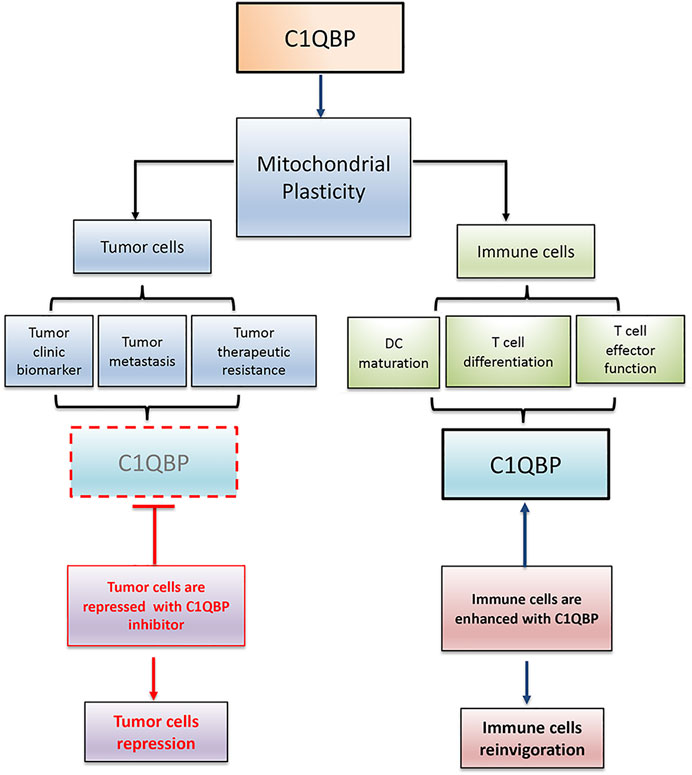
FIGURE 2. C1QBP regulation of mitochondrial plasticity alters the competitive balance between tumor cells and immune cells. Tumor cells disarmed from C1QBP exhibit reduced tumor proliferation, metastasis, and therapeutic resistance, while immune cells potentiated with C1QBP perform the active and durable antitumor immunity. In this regard, strategic manipulation of C1QBP would repress tumor progression but optimize antitumor immune function.
Conception and Writing: QW, DC, JZ, and HT; Revision of the manuscript: NSo, NSu, PN, and HT. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (Nos. 82073012, 81871869, and 32200124), Jiangsu Universtiy Clinical Medicine Technology Development Fund, Jiangsu Young Medical Talents Project, Jiangsu Provincial Key Medical Discipline, the Project of Invigorating Health Care through Science, Technology and Education (No. ZDXKA2016014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agemy L., Kotamraju V. R., Friedmann-Morvinski D., Sharma S., Sugahara K. N., Ruoslahti E. (2013). Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32. Mol. Ther. 21 (12), 2195–2204. doi:10.1038/mt.2013.191
Ahola S., Langer T., MacVicar T. (2019). Mitochondrial proteolysis and metabolic control. Cold Spring Harb. Perspect. Biol. 11 (7), a033936. doi:10.1101/cshperspect.a033936
Amamoto R., Yagi M., Song Y., Oda Y., Tsuneyoshi M., Naito S., et al. (2011). Mitochondrial p32/C1QBP is highly expressed in prostate cancer and is associated with shorter prostate-specific antigen relapse time after radical prostatectomy. Cancer Sci. 102 (3), 639–647. doi:10.1111/j.1349-7006.2010.01828.x
Araki K., Turner A. P., Shaffer V. O., Gangappa S., Keller S. A., Bachmann M. F., et al. (2009). mTOR regulates memory CD8 T-cell differentiation. Nature 460 (7251), 108–112. doi:10.1038/nature08155
Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y. J., et al. (2000). Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811. doi:10.1146/annurev.immunol.18.1.767
Chen H., Chan D. C. (2005). Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 142, R283–R289. doi:10.1093/hmg/ddi270
Chen Y. B., Jiang C. T., Zhang G. Q., Wang J. S., Pang D. (2009). Increased expression of hyaluronic acid binding protein 1 is correlated with poor prognosis in patients with breast cancer. J. Surg. Oncol. 100 (5), 382–386. doi:10.1002/jso.21329
Clevers H. (2011). The cancer stem cell: Premises, promises and challenges. Nat. Med. 17 (3), 313–319. doi:10.1038/nm.2304
Fogal V., Babic I., Chao Y., Pastorino S., Mukthavaram R., Jiang P., et al. (2015). Mitochondrial p32 is upregulated in Myc expressing brain cancers and mediates glutamine addiction. Oncotarget 6 (2), 1157–1170. doi:10.18632/oncotarget.2708
Fogal V., Richardson A. D., Karmali P. P., Scheffler I. E., Smith J. W., Ruoslahti E. (2010). Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 30 (6), 1303–1318. doi:10.1128/MCB.01101-09
Fogal V., Zhang L., Krajewski S., Ruoslahti E. (2008). Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 68 (17), 7210–7218. doi:10.1158/0008-5472.CAN-07-6752
Funes J. M., Quintero M., Henderson S., Martinez D., Qureshi U., Westwood C., et al. (2007). Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc. Natl. Acad. Sci. U. S. A. 104 (15), 6223–6228. doi:10.1073/pnas.0700690104
Gao H., Yao Q., Lan X., Li S., Wu J., Zeng G., et al. (2016). Elevated HABP1 protein expression correlates with progression and poor survival in patients with gastric cancer. Onco. Targets. Ther. 9, 6711–6718. doi:10.2147/OTT.S114756
Ghebrehiwet B., Lim B. L., Kumar R., Feng X., Peerschke E. I. (2001). gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol. Rev. 180, 65–77. doi:10.1034/j.1600-065x.2001.1800106.x
Gotoh K., Morisaki T., Setoyama D., Sasaki K., Yagi M., Igami K., et al. (2018). Mitochondrial p32/C1qbp is a critical regulator of dendritic cell metabolism and maturation. Cell Rep. 25 (7), 1800–1815. doi:10.1016/j.celrep.2018.10.057
Hu M., Crawford S. A., Henstridge D. C., Ng I. H., Boey E. J., Xu Y., et al. (2013). p32 protein levels are integral to mitochondrial and endoplasmic reticulum morphology, cell metabolism and survival. Biochem. J. 453 (3), 381–391. doi:10.1042/BJ20121829
Hunt H., Simon-Gracia L., Tobi A., Kotamraju V. R., Sharma S., Nigul M., et al. (2017). Targeting of p32 in peritoneal carcinomatosis with intraperitoneal linTT1 peptide-guided pro-apoptotic nanoparticles. J. Control. Release 260, 142–153. doi:10.1016/j.jconrel.2017.06.005
Jiao H., Su G. Q., Dong W., Zhang L., Xie W., Yao L. M., et al. (2015). Chaperone-like protein p32 regulates ULK1 stability and autophagy. Cell Death Differ. 22 (11), 1812–1823. doi:10.1038/cdd.2015.34
Jiao H., You H. (2016). p32: A new player in autophagy. Mol. Cell. Oncol. 3 (3), e1061097. doi:10.1080/23723556.2015.1061097
Kawai T., Akira S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34 (5), 637–650. doi:10.1016/j.immuni.2011.05.006
Kim B. C., Hwang H. J., An H. T., Lee H., Park J. S., Hong J., et al. (2016). Antibody neutralization of cell-surface gC1qR/HABP1/SF2-p32 prevents lamellipodia formation and tumorigenesis. Oncotarget 7 (31), 49972–49985. doi:10.18632/oncotarget.10267
Kim K. B., Yi J. S., Nguyen N., Lee J. H., Kwon Y. C., Ahn B. Y., et al. (2011). Cell-surface receptor for complement component C1q (gC1qR) is a key regulator for lamellipodia formation and cancer metastasis. J. Biol. Chem. 286 (26), 23093–23101. doi:10.1074/jbc.M111.233304
Kouidhi S., Elgaaied A. B., Chouaib S. (2017). Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front. Immunol. 8, 270. doi:10.3389/fimmu.2017.00270
Li F., Wang Y., Zeller K. I., Potter J. J., Wonsey D. R., O'Donnell K. A., et al. (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 25 (14), 6225–6234. doi:10.1128/MCB.25.14.6225-6234.2005
Li W., Cheng H., Li G., Zhang L. (2020). Mitochondrial damage and the road to exhaustion. Cell Metab. 32 (6), 905–907. doi:10.1016/j.cmet.2020.11.004
Li Y., Wan O. W., Xie W., Chung K. K. (2011). p32 regulates mitochondrial morphology and dynamics through parkin. Neuroscience 199, 346–358. doi:10.1016/j.neuroscience.2011.10.003
Liu Y., Leslie P. L., Jin A., Itahana K., Graves L. M., Zhang Y. (2017). p32 heterozygosity protects against age- and diet-induced obesity by increasing energy expenditure. Sci. Rep. 7 (1), 5754. doi:10.1038/s41598-017-06209-9
Liu Y., Leslie P. L., Jin A., Itahana K., Graves L. M., Zhang Y. (2018). p32 regulates ER stress and lipid homeostasis by down-regulating GCS1 expression. FASEB J. 32 (7), 3892–3902. doi:10.1096/fj.201701004RR
Liu Y., Shi Y. (2020). Mitochondria as a target in cancer treatment. MedComm 1 (2), 129–139. doi:10.1002/mco2.16
Loson O. C., Song Z., Chen H., Chan D. C. (2013). Fis1, mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24 (5), 659–667. doi:10.1091/mbc.E12-10-0721
Luo G., Yu X., Jin C., Yang F., Fu D., Long J., et al. (2010). LyP-1-conjugated nanoparticles for targeting drug delivery to lymphatic metastatic tumors. Int. J. Pharm. 385 (1-2), 150–156. doi:10.1016/j.ijpharm.2009.10.014
Luo M., Wicha M. S. (2015). Metabolic plasticity of cancer stem cells. Oncotarget 6 (34), 35141–35142. doi:10.18632/oncotarget.6177
Mishra P., Chan D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212 (4), 379–387. doi:10.1083/jcb.201511036
Moreno-Sanchez R., Rodriguez-Enriquez S., Marin-Hernandez A., Saavedra E. (2007). Energy metabolism in tumor cells. FEBS J. 274 (6), 1393–1418. doi:10.1111/j.1742-4658.2007.05686.x
Muta T., Kang D., Kitajima S., Fujiwara T., Hamasaki N. (1997). p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 272 (39), 24363–24370. doi:10.1074/jbc.272.39.24363
Niu M., Sun S., Zhang G., Zhao Y., Pang D., Chen Y. (2015). Elevated expression of HABP1 is correlated with metastasis and poor survival in breast cancer patients. Am. J. Cancer Res. 5 (3), 1190–1198.
Noh S., Phorl S., Naskar R., Oeum K., Seo Y., Kim E., et al. (2020). p32/C1QBP regulates OMA1-dependent proteolytic processing of OPA1 to maintain mitochondrial connectivity related to mitochondrial dysfunction and apoptosis. Sci. Rep. 10 (1), 10618. doi:10.1038/s41598-020-67457-w
Paasonen L., Sharma S., Braun G. B., Kotamraju V. R., Chung T. D., She Z. G., et al. (2016). New p32/gC1qR ligands for targeted tumor drug delivery. Chembiochem 17 (7), 570–575. doi:10.1002/cbic.201500564
Pattabiraman D. R., Weinberg R. A. (2014). Tackling the cancer stem cells - what challenges do they pose? Nat. Rev. Drug Discov. 13 (7), 497–512. doi:10.1038/nrd4253
Peiris-Pages M., Martinez-Outschoorn U. E., Pestell R. G., Sotgia F., Lisanti M. P. (2016). Cancer stem cell metabolism. Breast Cancer Res. 18 (1), 55. doi:10.1186/s13058-016-0712-6
Petersen-Mahrt S. K., Estmer C., Ohrmalm C., Matthews D. A., Russell W. C., Akusjarvi G. (1999). The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J. 18 (4), 1014–1024. doi:10.1093/emboj/18.4.1014
Prakash M., Kale S., Ghosh I., Kundu G. C., Datta K. (2011). Hyaluronan-binding protein 1 (HABP1/p32/gC1qR) induces melanoma cell migration and tumor growth by NF-kappa B dependent MMP-2 activation through integrin α(v)β(3) interaction. Cell. Signal. 23 (10), 1563–1577. doi:10.1016/j.cellsig.2011.04.009
Raud B., McGuire P. J., Jones R. G., Sparwasser T., Berod L. (2018). Fatty acid metabolism in CD8(+) T cell memory: Challenging current concepts. Immunol. Rev. 283 (1), 213–231. doi:10.1111/imr.12655
Rousso-Noori L., Mastandrea I., Talmor S., Waks T., Globerson Levin A., Haugas M., et al. (2021). P32-specific CAR T cells with dual antitumor and antiangiogenic therapeutic potential in gliomas. Nat. Commun. 12 (1), 3615. doi:10.1038/s41467-021-23817-2
Sanchez-Martin D., Cuesta A. M., Fogal V., Ruoslahti E., Alvarez-Vallina L. (2011). The multicompartmental p32/gClqR as a new target for antibody-based tumor targeting strategies. J. Biol. Chem. 286 (7), 5197–5203. doi:10.1074/jbc.M110.161927
Santel A., Fuller M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114 (5), 867–874. doi:10.1242/jcs.114.5.867
Scharping N. E., Menk A. V., Moreci R. S., Whetstone R. D., Dadey R. E., Watkins S. C., et al. (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45 (2), 374–388. doi:10.1016/j.immuni.2016.07.009
Schvartzman J. M., Thompson C. B., Finley L. W. S. (2018). Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217 (7), 2247–2259. doi:10.1083/jcb.201803061
Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38 (2), 225–236. doi:10.1016/j.immuni.2012.10.020
Sharma S., Kotamraju V. R., Molder T., Tobi A., Teesalu T., Ruoslahti E. (2017). Tumor-penetrating nanosystem strongly suppresses breast tumor growth. Nano Lett. 17 (3), 1356–1364. doi:10.1021/acs.nanolett.6b03815
Sinha S., Singh S. K., Jangde N., Ray R., Rai V. (2021). p32 promotes melanoma progression and metastasis by targeting EMT markers, Akt/PKB pathway, and tumor microenvironment. Cell Death Dis. 12 (11), 1012. doi:10.1038/s41419-021-04311-5
Song I. S., Jeong J. Y., Jeong S. H., Kim H. K., Ko K. S., Rhee B. D., et al. (2015). Mitochondria as therapeutic targets for cancer stem cells. World J. Stem Cells 7 (2), 418–427. doi:10.4252/wjsc.v7.i2.418
Sonveaux P., Vegran F., Schroeder T., Wergin M. C., Verrax J., Rabbani Z. N., et al. (2008). Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 118 (12), 3930–3942. doi:10.1172/JCI36843
Tian H., Wang G., Wang Q., Zhang B., Jiang G., Li H., et al. (2022). Complement C1q binding protein regulates T cells' mitochondrial fitness to affect their survival, proliferation, and anti-tumor immune function. Cancer Sci. 113 (3), 875–890. doi:10.1111/cas.15261
Vardhana S. A., Hwee M. A., Berisa M., Wells D. K., Yost K. E., King B., et al. (2020). Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21 (9), 1022–1033. doi:10.1038/s41590-020-0725-2
Viale A., Pettazzoni P., Lyssiotis C. A., Ying H., Sanchez N., Marchesini M., et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514 (7524), 628–632. doi:10.1038/nature13611
Vyas S., Zaganjor E., Haigis M. C. (2016). Mitochondria and cancer. Cell 166 (3), 555–566. doi:10.1016/j.cell.2016.07.002
Wallace D. C. (2012). Mitochondria and cancer. Nat. Rev. Cancer 12 (10), 685–698. doi:10.1038/nrc3365
Wang J., Song Y., Liu T., Shi Q., Zhong Z., Wei W., et al. (2015). Elevated expression of HABP1 is a novel prognostic indicator in triple-negative breast cancers. Tumour Biol. 36 (6), 4793–4799. doi:10.1007/s13277-015-3131-x
Ward P. S., Thompson C. B. (2012). Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 21 (3), 297–308. doi:10.1016/j.ccr.2012.02.014
Wrighton K. H. (2013). Metabolism: Putting energy into mitophagy. Nat. Rev. Mol. Cell Biol. 14 (6), 324. doi:10.1038/nrm3586
Yagi M., Uchiumi T., Takazaki S., Okuno B., Nomura M., Yoshida S., et al. (2012). p32/gC1qR is indispensable for fetal development and mitochondrial translation: importance of its RNA-binding ability. Nucleic Acids Res. 40 (19), 9717–9737. doi:10.1093/nar/gks774
Yenugonda V., Nomura N., Kouznetsova V., Tsigelny I., Fogal V., Nurmemmedov E., et al. (2017). A novel small molecule inhibitor of p32 mitochondrial protein overexpressed in glioma. J. Transl. Med. 15 (1), 210. doi:10.1186/s12967-017-1312-7
Yu H., Liu Q., Xin T., Xing L., Dong G., Jiang Q., et al. (2013). Elevated expression of hyaluronic acid binding protein 1 (HABP1)/P32/C1QBP is a novel indicator for lymph node and peritoneal metastasis of epithelial ovarian cancer patients. Tumour Biol. 34 (6), 3981–3987. doi:10.1007/s13277-013-0986-6
Yu Y. R., Imrichova H., Wang H., Chao T., Xiao Z., Gao M., et al. (2020). Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 21 (12), 1540–1551. doi:10.1038/s41590-020-0793-3
Zhai X., Liu K., Fang H., Zhang Q., Gao X., Liu F., et al. (2021). Mitochondrial C1qbp promotes differentiation of effector CD8(+) T cells via metabolic-epigenetic reprogramming. Sci. Adv. 7 (49), eabk0490. doi:10.1126/sciadv.abk0490
Keywords: C1QBP, mitochondrial plasticity, metabolic flexibility, tumor progression, antitumor immune response, immunotherapy
Citation: Wang Q, Chai D, Sobhani N, Sun N, Neeli P, Zheng J and Tian H (2022) C1QBP regulates mitochondrial plasticity to impact tumor progression and antitumor immune response. Front. Physiol. 13:1012112. doi: 10.3389/fphys.2022.1012112
Received: 15 August 2022; Accepted: 02 November 2022;
Published: 17 November 2022.
Edited by:
David Sebastián, Institute for Research in Biomedicine, SpainReviewed by:
Juliana Camacho-Pereira, Federal University of Rio de Janeiro, BrazilCopyright © 2022 Wang, Chai, Sobhani, Sun, Neeli, Zheng and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junnian Zheng, am56aGVuZ0B4emhtdS5lZHUuY24=; Hui Tian, aHVpdGlhbkB4emhtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.