 Mira Hamze
Mira Hamze Igor Medina
Igor Medina Quentin Delmotte
Quentin Delmotte Christophe Porcher
Christophe Porcher
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 10 December 2021
Sec. Membrane Physiology and Membrane Biophysics
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.798066
This article is part of the Research Topic Chloride Homeostasis in Animal Cell Physiology View all 6 articles
In the early stages of the central nervous system growth and development, γ-aminobutyric acid (GABA) plays an instructive trophic role for key events including neurogenesis, migration, synaptogenesis, and network formation. These actions are associated with increased concentration of chloride ions in immature neurons [(Cl−)i] that determines the depolarizing strength of ion currents mediated by GABAA receptors, a ligand-gated Cl− permeable ion channel. During neuron maturation the (Cl−)i progressively decreases leading to weakening of GABA induced depolarization and enforcing GABA function as principal inhibitory neurotransmitter. A neuron restricted potassium-chloride co-transporter KCC2 is a key molecule governing Cl− extrusion and determining the resting level of (Cl−)i in developing and mature mammalian neurons. Among factors controlling the functioning of KCC2 and the maturation of inhibitory circuits, is Smoothened (Smo), the transducer in the receptor complex of the developmental protein Sonic Hedgehog (Shh). Too much or too little Shh-Smo action will have mirror effects on KCC2 stability at the neuron membrane, the GABA inhibitory strength, and ultimately on the newborn susceptibility to neurodevelopmental disorders. Both canonical and non-canonical Shh-Smo signal transduction pathways contribute to the regulation of KCC2 and GABAergic synaptic activity. In this review, we discuss the recent findings of the action of Shh-Smo signaling pathways on chloride ions homeostasis through the control of KCC2 membrane trafficking, and consequently on inhibitory neurotransmission and network activity during postnatal development.
Developing neuronal circuits generate primitive patterns of network activity, that are necessary to support more complex neuronal processes and future cognitive functions. These primitive activities require γ-aminobutyric acid (GABA) synaptic transmission and chloride flux through GABAA ionotropic receptor channels (GABAAR). The inhibitory strength of GABAAR transmission is dependent on intracellular neuronal chloride concentration [(Cl−)i], that is relatively high (15–25 mM) in immature neurons and decreases progressively to 4–6 mM in mature neurons. This developmental change of (Cl−)i is determined primarily by activity of two cation-chloride cotransporters sodium-potassium-chloride cotransporter type 1 (NKCC1) and potassium-chloride cotransporter type 2 (KCC2) (see reviews Medina et al., 2014; Virtanen et al., 2021). Several factors controlling the maturation of GABAergic transmission have been identified so far, including GABA itself, neurotrophic factors (Roussa et al., 2016; Porcher et al., 2018), pituitary neuropeptides (Tyzio et al., 2006; Leonzino et al., 2016; Spoljaric et al., 2017), peripheral metabolic and sex hormones (Galanopoulou and Moshé, 2003; Sawano et al., 2013; Dumon et al., 2018), and more recently the Sonic Hedgehog (Shh) peptide and its signal transducer Smoothened (Smo) receptor (Delmotte et al., 2020b). In this review, we discuss the recent achievements in Shh-Smo signaling contribution to developmental and functional maturation of GABAergic transmission (Figure 1) and chloride ion homeostasis in the postnatal rodent brain (Figure 2).
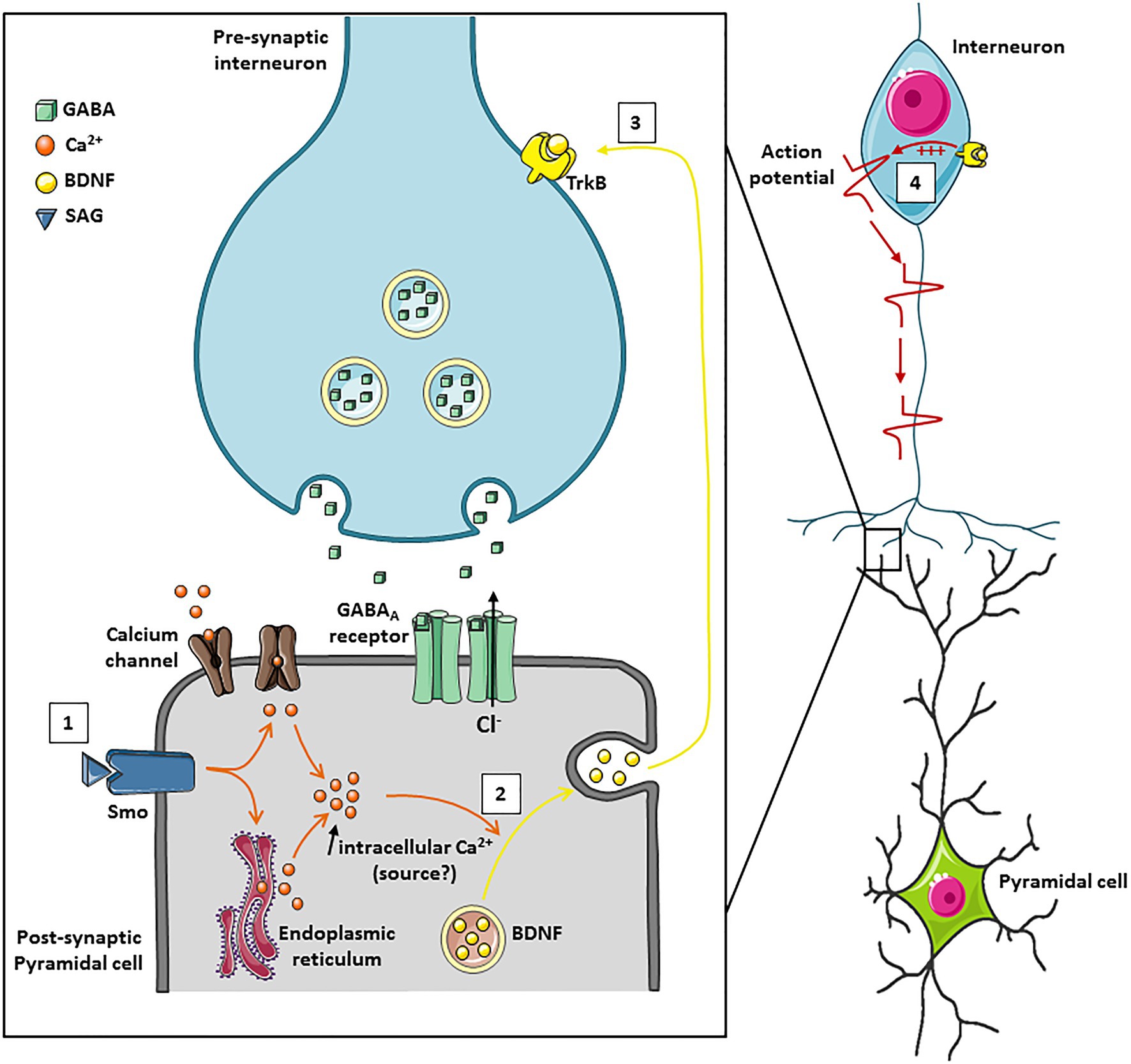
Figure 1. A schematic model of action of the Smoothened receptor signaling on GABAergic transmission in the immature hippocampus. (1) Activation of the non-canonical Smo signaling pathway by its ligand (SAG) leads to an intracellular calcium increase in the post-synaptic neuron which can be mediated either through calcium channels or by the release of intracellular stores from the endoplasmic reticulum. (2) The intracellular increase in calcium concentration triggers the exocytosis of BDNF. (3) BDNF binds to its receptor TrkB on neighboring synapses and (4) increases GABAergic synaptic transmission.
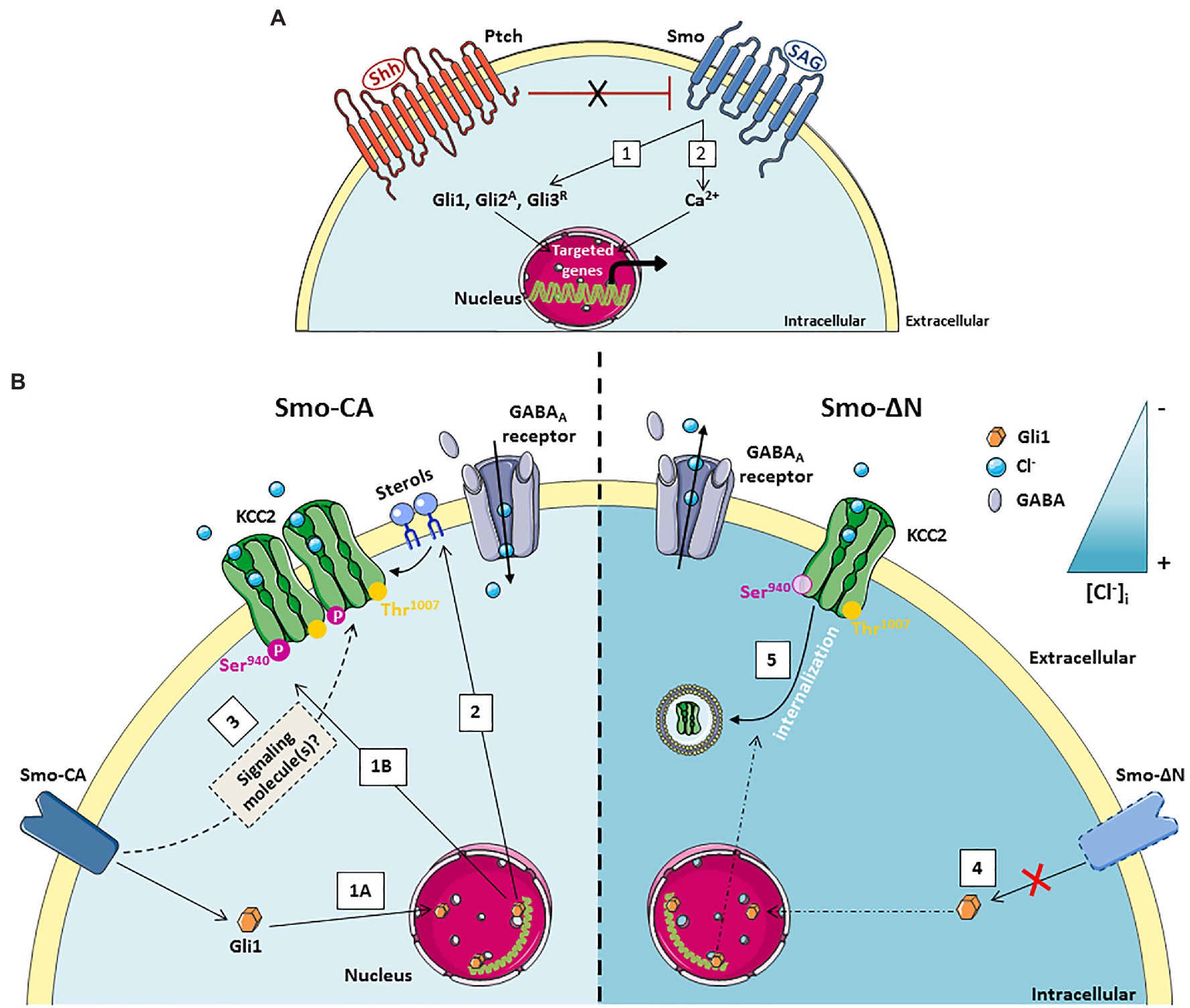
Figure 2. A schematic model of the Smoothened receptor signaling pathways controlling KCC2 activity. (A) In the absence of the ligand Shh, the Patched (Ptch) receptor maintains the Smoothened (Smo) receptor in an inactive state. Smo signaling can be activated either by the binding of Shh which inactivates Ptch and releases Smo or by the binding of its agonist SAG (Shh Signaling Agonist). Smo signaling can operate through: (1) canonical pathway which activates the transcription factors Gli allowing transcription of Gli target genes, (2) Non-canonical pathway that is Gli-independent but triggers transient calcium currents allowing transcription of targeted genes. (B) Left: (1a) Constitutive activity of the Smo-CA receptor promotes Gli1 downstream target genes expression. (1b) The Smo-Gli1 signaling pathways activate the phosphorylation of KCC2 at the residue Serine 940, increasing KCC2 function and stability at the cell surface, which renders GABAergic transmission less depolarizing. (2) The Smo/Gli1 signaling pathways may modify the synthesis/transport of sterols, impacting the composition of the plasma membrane and lipid rafts and the stability of KCC2 at the cell surface. (3) The Smo-Gli1 pathway may also regulate chloride ions homeostasis and KCC2 function through a second messenger. Right: (4) Dominant-negative activity of Smo-ΔN represses Gli1 expression. (5) Inhibition of Smo-Gli1 signaling pathway induces dephosphorylation of residue Serine 940 and KCC2 internalization.
Shh is a secreted glycoprotein preserved through the evolution and mostly known for its morphogenetic role during early phases of central nervous system development such as neural cell proliferation, neural progenitor cell fate, or neuronal differentiation (Ruat et al., 2012, 2014; Briscoe and Thérond, 2013; Belgacem et al., 2016). In the postnatal rodent brain the Shh signaling pathway, composed by the receptors complex Patched (Ptch) and Smo, is involved in neurogenesis (Breunig et al., 2008; Shqirat et al., 2021), growth of presynaptic terminals (Mitchell et al., 2012), connectivity of corticofugal projection neurons (Harwell et al., 2012) and functional maturation of GABAergic inhibitory neurotransmission (Delmotte et al., 2020a,b). In the hippocampus, Shh is produced in both neurons and glial cells distributed in the pyramidal layer (Rivell et al., 2019; Tirou et al., 2020), and Shh receptors Ptch and Smo are further described to be expressed mostly at the postsynaptic membranes of CA1 and CA3 pyramidal neurons and at the mossy fibers terminals, thus indicating that the Shh-Smo pathway may regulate the strengthening of synaptic connectivity (Masdeu et al., 2007; Sasaki et al., 2010; Petralia et al., 2011; Mitchell et al., 2012).
Shh signaling can operate through canonical and non-canonical pathways (Carballo et al., 2018; Figure 2A). In the presence of ligand, Shh receptor Ptch release its constitutive inhibition on its signal transducer Smo receptor, leading to the activation of Shh canonical signaling effector (Carballo et al., 2018). Among canonical Shh signaling downstream targets are Smo and activator (Gli1 and Gli2A) or repressor (Gli3R) forms of Glioma-associated (Gli) gene transcription factors (Briscoe and Thérond, 2013; Rimkus et al., 2016; Carballo et al., 2018). In the central nervous system (CNS), during the late embryonic stage of mouse cortical development, canonical Shh-Smo signaling, acting through the inactivation of Gli3R, is essential to regulate the lineage change of cortical neural stem cells for the generation of olfactory bulb GABAergic interneurons (Zhang et al., 2020a). In hippocampal neurons, Shh is involved in axonal elongation through Smo and the canonical transcription factor Gli (Yao et al., 2015). Concurrently, several non-canonical, Gli-independent, Shh pathways have been described (Riobo et al., 2006; Ayers and Thérond, 2010). For instance, an interaction between Shh signaling and Ca2+-dependent spike activity in the developing spinal cord has been identified for spinal neuron differentiation and GABAergic phenotype homeostatic specification (Belgacem and Borodinsky, 2011; Belgacem et al., 2016). Moreover, this non-canonical Ca2+-dependent Shh signaling pathway has been shown to inhibit the canonical Shh signaling as spinal cord development progresses (Belgacem and Borodinsky, 2015).
The developing hippocampus is characterized by spontaneous network oscillations called Giant Depolarizing Potentials (GDPs) that occur during the first postnatal weeks (Ben-Ari et al., 1989) and disappear when GABA currents became hyperpolarizing (Griguoli and Cherubini, 2017). In the developing human and mouse neocortex, Shh mRNA was identified in a subpopulation of glutamatergic and GABAergic neuronal cells (Komada et al., 2008; Memi et al., 2018). Interestingly, the gene expression profiles of both Ptch and Smo receptors in hippocampi of rat follow a similar pattern with a decreased expression levels when GABA became hyperpolarizing (Delmotte et al., 2020a). The GDPs could be modulated by several factors including neurotrophic factors such as brain-derived neurotrophic factor (BDNF) (Mohajerani et al., 2007), the metabolic hormone leptin (Dumon et al., 2019), the chemical stromal cell-derived factor-1-alpha (SDF-1; Kasyanov et al., 2006) and the hypothalamic neurohormone vasopressin (Spoljaric et al., 2017). More recently, Shh emerged as a new trophic factor in the functional maturation of GABAergic network (Delmotte et al., 2020a). In vitro experiments on hippocampal slices at postnatal days 5–7 showed that a non-canonical Ca2+-dependent Shh-Smo signaling increased the GDPs frequency and spontaneous GABAergic inhibitory postsynaptic currents without affecting glutamatergic synaptic activity (Delmotte et al., 2020a). The subcellular mechanism allowing the function of Shh-Smo pathway in GABAergic developmental plasticity depends on postsynaptic calcium-transients within the cytosol since the application of the Ca2+-chelator BAPTA in the recording pipette completely abolished this effect (Figure 2). This result suggests that Ca2+ serves as a second messenger in neuronal cells for the Smo receptor downstream pathway. However, the source for intracellular calcium signaling in neuronal cells following the binding of Shh or its agonist Shh Signaling Agonist (SAG; Chen et al., 2002) on their receptors remains to be determined (Figure 2). The interplay between Smo and Ca2+ activity has been also observed in embryonic spinal neurons where Smo receptor signaling leads to the recruitment of a heterotrimeric Gαi that in turn induces Inositol Triphosphate (IP3) oscillations apparent at the neuronal primary cilium synchronous with Ca2+ waves in the neuronal soma. The Shh-induced increase in Ca2+ spike activity depends on both intracellular Ca2+ stores and extracellular Ca2+ influx through voltage dependent calcium channels and transient receptor potential 1 channel (Belgacem and Borodinsky, 2011). Moreover, in embryonic spinal cord, the calcium increase generated by Smo signaling activates the PKA kinase which subsequently phosphorylates the cAMP response element-binding protein (CREB) transcription factor leading to the inhibition of Gli1 transcription factor (Belgacem and Borodinsky, 2015). These results are very similar to those observed during the first 2 weeks after birth in the rodent hippocampus where activation of the Smo signaling triggers phosphorylation of CREB as well as a decrease in Gli1 transcripts, suggesting an identical mechanism by a non-canonical Shh pathway (Delmotte et al., 2020a). Other Shh-Smo-mediated Ca2+ increases have been described under epileptic seizure discharges in hippocampal neurons through increased levels of extracellular glutamate and NMDA receptors activity (Feng et al., 2016). In this study, the authors suggest that Shh has no physiological function in adult hippocampal neuroplasticity. Interestingly, Delmotte et al. (2020a) reported that the application of SAG modulates the frequency of GDPs only during the first postnatal week of life and this action disappeared around P10. This divergence of the Shh-Smo effect between immature and mature hippocampal tissue may suggest that Shh-Smo signaling regulates the development of GABAergic neurotransmission only in immature neurons and disappears as soon as GABA becomes hyperpolarizing, thus reinforcing the hypothesis of a specific trophic role for Shh-Smo in the developing brain. This trophic function in the maturation of GABAergic neurotransmission acts through the regulated release of BDNF and activation of its high affinity tyrosine kinase receptor TrkB, suggesting that BDNF is a downstream target of Smo (Figure 1; Delmotte et al., 2020a). Other studies performed on different models of injury including cortical neurons, peripheral sciatic nerve, and cavernous nerve also showed an interplay between Shh-Smo and BDNF (Hashimoto et al., 2008; Dai et al., 2012; Bond et al., 2013). Although the subcellular mechanism remains to be demonstrated, a Shh-dependent increase of BDNF was found after SAG administration in lung organ cultures, and in hippocampal and spinal cord neuronal cells (Radzikinas et al., 2011; Liu et al., 2018; Delmotte et al., 2020a). In lung epithelial cells Shh promotes indirectly BDNF expression through a post-transcriptional mechanism, whereas in hippocampal neurons Shh-Smo triggered the dendritic release of BDNF via a calcium signal. These data indicate that Shh-Smo might acts through the BDNF–TrkB signaling pathway in both injured and healthy conditions.
The postnatal maturation of GABAergic inhibitory transmission is linked to the developmental sequence of GABA shift from depolarizing to inhibitory actions which results from a developmental decrease in (Cl−)i, brought about by the increased contribution of the Cl− extruder KCC2 (Rivera et al., 1999; Medina et al., 2014). Using molecular tools to manipulate Smo activity in the pyramidal neurons of the rat somatosensory cortex, Delmotte et al. (2020b) showed that Smo canonical signaling contributes to chloride ions homeostasis and KCC2 cell membrane stability (Delmotte et al., 2020b). Two Smo constructs were used, one mimicking Smo receptor in an activate state (Smo-CA) and a second blocking Smo signaling downstream pathway (Smo-ΔN). In this study, it has been demonstrated that Smo-CA accelerates the transition of GABA from depolarizing to hyperpolarizing, thus positioning the Shh-Smo pathway as a trigger of this GABAergic developmental sequence. This action on chloride ions homeostasis requires the activation of the Shh-Smo canonical signaling downstream target Gli1 as the inhibition of Gli1 abolished this effect (Delmotte et al., 2020b; Figure 1B). The abnormal GABAergic developmental sequence observed at the cellular level leads to behavioral consequences since electroporated rodents with the Smo-CA construct showed an increased susceptibility to seizures induced by the pentylenetrazol (PTZ), a chemical convulsive agent. In human, many studies illustrated compromised Cl− homeostasis and altered GABAergic inhibition in patients with temporal lobe epilepsiy (TLE). On the other hand, an alteration of the Shh-Smo pathway has also been identified in TLE (Fang et al., 2011) and hypothalamic hamartoma with gelastic epilepsy (Hildebrand et al., 2016; Saitsu et al., 2016). Although the (Cl−)i was not evaluated in the latter works, together above studies indicate the potential contribution of the Shh-Smo pathway to Cl− disruption in TLE patients.
The key molecule controlling the resting level of neuronal Cl− and directly linked to the switch of GABA from excitatory to inhibitory is KCC2 (Rivera et al., 1999; Virtanen et al., 2021 for recent comprehensive review). The dysfunctions of KCC2 are associated with large number of pathologies starting from pioneer observations of KCC2 change in different types of epilepsies (Miles et al., 2012) and ending with recent findings of KCC2 link to different neurological disorders (Tang, 2020). Naturally, the observations of Smo-dependent control on GABAergic polarity shift raised the question whether Smo transducer regulates the KCC2. The total expression level and the phosphorylated state of KCC2 are fundamental determinants of KCC2 function affecting its Cl− extrusion ability and hence controlling neuronal Cl− homeostasis (Box 1). Analysis of the phosphorylation state of Ser940 and Thr1007 residues of KCC2 revealed a strong and significant increase of Ser940 phosphorylation in neurons expressing Smo-CA, and significant decrease of Ser940 phosphorylation in neurons with Smo-ΔN (Delmotte et al., 2020b; Figure 2B). No effect of Smo was detected on the level of Thr1007 phosphorylation indicating on the high selectivity of Smo-dependent pathways controlling KCC2. Consistent with previous observations of Ser940 – dependent control of plasma membrane stabilization of KCC2 (Lee et al., 2007, 2011), the modulation of Smo pathway affected the surface expression of KCC2 construct harboring tag in extracellular domain and expressed in cultured hippocampal neurons (Delmotte et al., 2020b).
In the light of current knowledge on the regulation of KCC2 membrane trafficking, several hypotheses might be considered to explain the interplay between Smo and KCC2: (1) Activation of Smo downstream pathway with Smo-CA construct increases phosphorylation of KCC2 at Ser940 and consequently the stability of the co-transporter at the cell surface and its activity, thereby decreasing the intracellular chloride concentration and rendering GABA hyperpolarizing (Lee et al., 2007; Silayeva et al., 2015); (2) Complementary, the canonical Shh-Smo-Gli1 pathway may also regulate chloride ions homeostasis and KCC2 function through a second messenger including a Ca2+ transient frequency and/or an intermediary factor such as BDNF (Figure 1). As mentioned before, non-canonical Shh-Smo signaling pathway induce BDNF secretion and a previous study showed that the loss of canonical Shh-Smo signaling also decreased the levels of BDNF transcripts (Zhou et al., 2016). In line with this hypothesis, BDNF has been shown to modulate KCC2 expression and activity, but depending on the physiological context and maturation stage of neurons it can either upregulate (Ludwig et al., 2011) or down-regulate the KCC2 (Rivera et al., 2004); (3) A third hypothesis is based on the activation of Smo, which could affect the lipid organization of the membrane. Since the Smo binds sterols and endogenous cholesterol, a strong activation of the Smoothened receptor could prevent these bindings, leaving the unbound cholesterol free to move elsewhere in the membrane (Figure 1B). Cholesterol is an essential component of lipid rafts and is central to many mechanisms, including the stability of KCC2 at the membrane (Hartmann et al., 2009; Watanabe et al., 2009).
In this review, we highlight the significant potential role of Shh-Smo signaling in chloride homeostasis through the control of KCC2 cell surface expression and activity, which in turn fine-tune the strength of inhibitory synaptic transmission. A weak or an absence of Shh-Smo signaling delayed the GABA polarity shift, leading to an unbalanced excitation to inhibition ratio. A similar role of Shh-smo signaling has been shown on astrocytes in mature cortex (Hill et al., 2019). The conditional knockout of Smo results in an early increase in synapse number and in neuronal excitability, induced by a reduction in Kir 1.4 potassium inward rectifier currents, thus confirming an important role for the Shh pathway on synaptic transmission. It remains to determine whether modulation of Ptch receptor with Shh will have the same consequences as Smo activation on GABAergic developmental sequence. Indeed, activation of Smo by SAG or stimulation of Ptch with application of Shh results in the same outcome in a very large number of functions, both for regulation of axonal elongation and, activation of the ERK signaling pathway (Yao et al., 2015, 2019). Finally, these recent data on Shh-Smo showed that this pathway continues to have a very important role in development after birth, both in early network activity such as GDPs and in the maturation of inhibitory synaptic transmission with the developmental GABAergic sequence. This signaling pathway can trigger the release of a key neurotrophic factor in CNS development, BDNF, but also modulate the activity of a central protein in chloride homeostasis and GABAergic polarity control, KCC2.
These open new research avenues on the interplay between Shh and GABA in neurodevelopmental disorders like Autism Spectrum Disorders (ASD), Down Syndrome and, epilepsy (Belgacem et al., 2016; Feng et al., 2016; Kumar et al., 2019). As promising examples, treatments with SAG restored partially cerebellar morphology and behavioral deficits in a Down Syndrome mouse model (Roper et al., 2006; Das et al., 2013), and purmorphamine, a Smo agonist, ameliorated behavioral and cellular alterations in a rat model of ASD (Rahi et al., 2021).
BOX 1: Phosphorylation-dependent control of KCC2.
A large number of phosphorylatable residues were revealed on KCC2 using mass spectrometry analysis (Rinehart et al., 2011; Weber et al., 2014; Agez et al., 2017; Cordshagen et al., 2018; Zhang et al., 2020b). The mutation of some residues (e.g., Ser932, Thr934, Ser937, Ser940, Thr906, Thr1007) results in change of the ion-transport activity of KCC2 measured in heterologous expression system (Weber et al., 2014; Zhang et al., 2020b). The importance of other residues is not clear yet. Among above phosphorylatable sites with ion-transport importance only two group of residues (Ser940 and Thr906 plus Thr1007) were extensively studied for their role in native neuronal environment both in-vivo and in-vitro. Briefly, the protein kinase C (pkC) phosphorylation of Ser940 leads to stabilization of KCC2 on neuron’s surface leading to enhancement of ion-transport activity, whereas Ser940 dephosphorylation is associated with transporter internalization and reduction of the KCC2 activity (Lee et al., 2007, 2011). The transgenic mice harboring nonphosphorylatable mutation S940A showed reduced sociability that is compatible with autism-like behavior (Moore et al., 2019) and exhibited higher lethality when entering in kainate-induced status epilepticus (Silayeva et al., 2015). The phosphorylation of Thr906 and Thr1007 is associated with decrease of ion-transport ability of KCC2 (Rinehart et al., 2009; Friedel et al., 2015) due to enhancement of KCC2’s internalization rate (Friedel et al., 2015). In opposite, the dephosphorylation of Thr906 and Thr1007 results in increase of KCC2 activity and enhancement of its surface expression (Friedel et al., 2015). The mouse harboring phosphomimetic T906E/T1007E mutations in both alleles dies at birth (Watanabe et al., 2019). The heterozygous T906E/T1007E mouse is characterized by deficit in neuronal Cl− homeostasis, network activity and has altered social interaction behavior (Pisella et al., 2019). In opposite, the mouse with T906A/T1007A mutations, mimicking non-phosphorylated state of respective threonine residues, exhibited increased basal neuronal Cl − extrusion, showed delayed onset and severity of chemoconvulsant-induced seizure activity (Moore et al., 2018), and exhibited enhanced social behavior (Moore et al., 2019). The phosphorylation of both Thr906 and Thr1007 depend on activity of with no lysine kinase (WNK) including four members (WNK1–4) and the downstream SPAK/OSR1 (SPS1-related proline/alanine-rich kinase/oxidative stress responsive kinase-1) pathway (Alessi et al., 2014; Friedel et al., 2015), although the exact mechanisms of KCC2 phosphorylation by WNK/SPAK remain to be clarified.
The review was conceptualized, written, and edited by each of the authors. CP was the supervisor. All authors contributed to the article and approved the submitted version.
This work was supported by The National Institute of Health and Medical Research (INSERM), the National Center for Scientific Research (CNRS), the A*MIDEX project (ANR-11-IDEX-0001-02) funded by the “Investissements d’Avenir” French Government Program, managed by the Agence Nationale de la Recherche. IM was supported by French Foundation for Epilepsy Research. MH was supported by a Doctoral fellowship from the French Ministry of higher Education and Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agez, M., Schultz, P., Medina, I., Baker, D. J., Burnham, M. P., Cardarelli, R. A., et al. (2017). Molecular architecture of potassium chloride co-transporter KCC2. Sci. Rep. 7:16452. doi: 10.1038/s41598-017-15739-1
Alessi, D. R., Zhang, J., Khanna, A., Hochdörfer, T., Shang, Y., and Kahle, K. T. (2014). The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci. Signal. 7:re3. doi: 10.1126/scisignal.2005365
Ayers, K. L., and Thérond, P. P. (2010). Evaluating smoothened as a G-protein-coupled receptor for hedgehog signalling. Trends Cell Biol. 20, 287–298. doi: 10.1016/j.tcb.2010.02.002
Belgacem, Y. H., and Borodinsky, L. N. (2011). Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. PNAS 108, 4482–4487. doi: 10.1073/pnas.1018217108
Belgacem, Y. H., and Borodinsky, L. N. (2015). Inversion of sonic hedgehog action on its canonical pathway by electrical activity. PNAS 112, 4140–4145. doi: 10.1073/pnas.1419690112
Belgacem, Y. H., Hamilton, A. M., Shim, S., Spencer, K. A., and Borodinsky, L. N. (2016). The many hats of sonic hedgehog signaling in nervous system development and disease. J. Dev. Biol. 4:35. doi: 10.3390/jdb4040035
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. Lond. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Bond, C. W., Angeloni, N., Harrington, D., Stupp, S., and Podlasek, C. A. (2013). Sonic hedgehog regulates brain-derived neurotrophic factor in normal and regenerating cavernous nerves. J. Sex. Med. 10, 730–737. doi: 10.1111/jsm.12030
Breunig, J. J., Sarkisian, M. R., Arellano, J. I., Morozov, Y. M., Ayoub, A. E., Sojitra, S., et al. (2008). Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc. Natl. Acad. Sci. U. S. A. 105, 13127–13132. doi: 10.1073/pnas.0804558105
Briscoe, J., and Thérond, P. P. (2013). The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429. doi: 10.1038/nrm3598
Carballo, G. B., Honorato, J. R., de Lopes, G. P. F., and de Spohr, T. C. L. S. E. (2018). A highlight on sonic hedgehog pathway. Cell Commun. Signal 16, 1–15. doi: 10.1186/s12964-018-0220-7
Chen, J. K., Taipale, J., Young, K. E., Maiti, T., and Beachy, P. A. (2002). Small molecule modulation of smoothened activity. Proc. Natl. Acad. Sci. U. S. A. 99, 14071–14076. doi: 10.1073/pnas.182542899
Cordshagen, A., Busch, W., Winklhofer, M., Nothwang, H. G., and Hartmann, A.-M. (2018). Phosphoregulation of the intracellular termini of K+-cl- cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem. 293, 16984–16993. doi: 10.1074/jbc.RA118.004349
Dai, R., Xia, Y., Mao, L., Mei, Y., Xue, Y., and Hu, B. (2012). Involvement of PI3K/Akt pathway in the neuroprotective effect of sonic hedgehog on cortical neurons under oxidative stress. J. Sci. Technol. Med. Sci. 32, 856–860. doi: 10.1007/s11596-012-1047-x
Das, I., Park, J.-M., Shin, J. H., Jeon, S. K., Lorenzi, H., Linden, D. J., et al. (2013). Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci. Transl. Med. 5:201ra120. doi: 10.1126/scitranslmed.3005983
Delmotte, Q., Diabira, D., Belaidouni, Y., Hamze, M., Kochmann, M., Montheil, A., et al. (2020a). Sonic hedghog signaling agonist (SAG) triggers BDNF secretion and promotes the maturation of GABAergic networks in the postanatal rat hippocampus. Front. Cell. Neurosci. 14:98. doi: 10.3389/fncel.2020.00098
Delmotte, Q., Hamze, M., Medina, I., Buhler, E., Zhang, J., Belgacem, Y. H., et al. (2020b). Smoothened receptor signaling regulates the developmental shift of GABA polarity in rat somatosensory cortex. J. Cell Sci. 133, 1–18. doi: 10.1242/jcs.247700
Dumon, C., Diabira, D., Chudotvorova, I., Bader, F., Sahin, S., Zhang, J., et al. (2018). The adipocyte hormone leptin sets the emergence of hippocampal inhibition in mice. Elife 7:e36726. doi: 10.7554/eLife.36726
Dumon, C., Pisella, L., Diabira, D., Belaidouni, Y., Wayman, G. A., and Gaiarsa, J.-L. (2019). Developmental switch of Leptin action on network driven activity in the neonatal rat hippocampus. Front. Cell. Neurosci. 13:254. doi: 10.3389/fncel.2019.00254
Fang, M., Lu, Y., Chen, G.-J., Shen, L., Pan, Y.-M., and Wang, X.-F. (2011). Increased expression of sonic hedgehog in temporal lobe epileptic foci in humans and experimental rats. Neuroscience 182, 62–70. doi: 10.1016/j.neuroscience.2011.02.060
Feng, S., Ma, S., Jia, C., Su, Y., Yang, S., Zhou, K., et al. (2016). Sonic hedgehog is a regulator of extracellular glutamate levels and epilepsy. EMBO Rep. 17, 682–694. doi: 10.15252/embr.201541569
Friedel, P., Kahle, K. T., Zhang, J., Hertz, N., Pisella, L. I., Buhler, E., et al. (2015). WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 8:ra65. doi: 10.1126/scisignal.aaa0354
Galanopoulou, A. S., and Moshé, S. L. (2003). Role of sex hormones in the sexually dimorphic expression of KCC2 in rat substantia nigra. Exp. Neurol. 184, 1003–1009. doi: 10.1016/S0014-4886(03)00387-X
Griguoli, M., and Cherubini, E. (2017). Early correlated network activity in the hippocampus: its putative role in shaping neuronal circuits. Front. Cell. Neurosci. 11:255. doi: 10.3389/fncel.2017.00255
Hartmann, A.-M., Blaesse, P., Kranz, T., Wenz, M., Schindler, J., Kaila, K., et al. (2009). Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J. Neurochem. 111, 321–331. doi: 10.1111/j.1471-4159.2009.06343.x
Harwell, C. C., Parker, P. R. L., Gee, S. M., Okada, A., McConnell, S. K., Kreitzer, A. C., et al. (2012). Sonic hedgehog expression in corticofugal projection neurons directs cortical microcircuit formation. Neuron 73, 1116–1126. doi: 10.1016/j.neuron.2012.02.009
Hashimoto, M., Ishii, K., Nakamura, Y., Watabe, K., Kohsaka, S., and Akazawa, C. (2008). Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J. Neurochem. 107, 918–927. doi: 10.1111/j.1471-4159.2008.05666.x
Hildebrand, M. S., Griffin, N. G., Damiano, J. A., Cops, E. J., Burgess, R., Ozturk, E., et al. (2016). Mutations of the sonic hedgehog pathway underlie hypothalamic Hamartoma with Gelastic epilepsy. Am. J. Hum. Genet. 99, 423–429. doi: 10.1016/j.ajhg.2016.05.031
Hill, S. A., Blaeser, A. S., Coley, A. A., Xie, Y., Shepard, K. A., Harwell, C. C., et al. (2019). Sonic hedgehog signaling in astrocytes mediates cell-type-specific synaptic organization. Elife 8:e45545. doi: 10.7554/eLife.45545
Kasyanov, A., Tamamura, H., Fujii, N., and Xiong, H. (2006). HIV-1 gp120 enhances giant depolarizing potentials via chemokine receptor CXCR4 in neonatal rat hippocampus. Eur. J. Neurosci. 23, 1120–1128. doi: 10.1111/j.1460-9568.2006.04646.x
Komada, M., Saitsu, H., Kinboshi, M., Miura, T., Shiota, K., and Ishibashi, M. (2008). Hedgehog signaling is involved in development of the neocortex. Development 135, 2717–2727. doi: 10.1242/dev.015891
Kumar, S., Reynolds, K., Ji, Y., Gu, R., Rai, S., and Zhou, C. J. (2019). Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 11, 1–14. doi: 10.1186/s11689-019-9268-y
Lee, H. H. C., Deeb, T. Z., Walker, J. A., Davies, P. A., and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 14, 736–743. doi: 10.1038/nn.2806
Lee, H. H. C., Walker, J. A., Williams, J. R., Goodier, R. J., Payne, J. A., and Moss, S. J. (2007). Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride Cotransporter KCC2. J. Biol. Chem. 282, 29777–29784. doi: 10.1074/jbc.M705053200
Leonzino, M., Busnelli, M., Antonucci, F., Verderio, C., Mazzanti, M., and Chini, B. (2016). The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 15, 96–103. doi: 10.1016/j.celrep.2016.03.013
Liu, S., Yao, J.-L., Wan, X.-X., Song, Z.-J., Miao, S., Zhao, Y., et al. (2018). Sonic hedgehog signaling in spinal cord contributes to morphine-induced hyperalgesia and tolerance through upregulating brain-derived neurotrophic factor expression. J. Pain Res. 11, 649–659. doi: 10.2147/JPR.S153544
Ludwig, A., Uvarov, P., Soni, S., Thomas-Crusells, J., Airaksinen, M. S., and Rivera, C. (2011). Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 31, 644–649. doi: 10.1523/JNEUROSCI.2006-10.2011
Masdeu, C., Bernard, V., Faure, H., Traiffort, E., and Ruat, M. (2007). Distribution of smoothened at hippocampal mossy fiber synapses. Neuroreport 18, 395–399. doi: 10.1097/WNR.0b013e32801421ce
Medina, I., Friedel, P., Rivera, C., Kahle, K. T., Kourdougli, N., Uvarov, P., et al. (2014). Current view on the functional regulation of the neuronal K(+)-cl(−) cotransporter KCC2. Front. Cell. Neurosci. 8, 27. doi: 10.3389/fncel.2014.00027
Memi, F., Zecevic, N., and Radonjić, N. (2018). Multiple roles of sonic hedgehog in the developing human cortex are suggested by its widespread distribution. Brain Struct. Funct. 223, 2361–2375. doi: 10.1007/s00429-018-1621-5
Miles, R., Blaesse, P., Huberfeld, G., Wittner, L., and Kaila, K. (2012). “Chloride homeostasis and GABA signaling in temporal lobe epilepsy” in Jasper’s Basic Mechanisms of the Epilepsies. eds. J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda: National Center for Biotechnology Information).
Mitchell, N., Petralia, R. S., Currier, D. G., Wang, Y.-X., Kim, A., Mattson, M. P., et al. (2012). Sonic hedgehog regulates presynaptic terminal size, ultrastructure and function in hippocampal neurons. J. Cell Sci. 125, 4207–4213. doi: 10.1242/jcs.105080
Mohajerani, M. H., Sivakumaran, S., Zacchi, P., Aguilera, P., and Cherubini, E. (2007). Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3–CA1 connections in the hippocampus. Proc. Natl. Acad. Sci. U. S. A. 104, 13176–13181. doi: 10.1073/pnas.0704533104
Moore, Y. E., Deeb, T. Z., Chadchankar, H., Brandon, N. J., and Moss, S. J. (2018). Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. U. S. A. 115, 10166–10171. doi: 10.1073/pnas.1810134115
Moore, Y. E., Conway, L. C., Wobst, H. J., Brandon, N. J., Deeb, T. Z., and Moss, S. J. (2019). Developmental regulation of KCC2 phosphorylation has long-term impacts on cognitive function. Front. Mol. Neurosci. 12:173. doi: 10.3389/fnmol.2019.00173
Petralia, R. S., Schwartz, C. M., Wang, Y.-X., Mattson, M. P., and Yao, P. J. (2011). Subcellular localization of patched and smoothened, the receptors for sonic hedgehog signaling, in the hippocampal neuron. J. Comp. Neurol. 519, 3684–3699. doi: 10.1002/cne.22681
Pisella, L. I., Gaiarsa, J.-L., Diabira, D., Zhang, J., Khalilov, I., Duan, J., et al. (2019). Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 12:eaay0300. doi: 10.1126/scisignal.aay0300
Porcher, C., Medina, I., and Gaiarsa, J.-L. (2018). Mechanism of BDNF modulation in GABAergic synaptic transmission in healthy and disease brains. Front. Cell. Neurosci. 12:273. doi: 10.3389/fncel.2018.00273
Radzikinas, K., Aven, L., Jiang, Z., Tran, T., Paez-Cortez, J., Boppidi, K., et al. (2011). A Shh/miR-206/BDNF Cascade coordinates innervation and formation of airway smooth muscle. J. Neurosci. 31, 15407–15415. doi: 10.1523/JNEUROSCI.2745-11.2011
Rahi, S., Gupta, R., Sharma, A., and Mehan, S. (2021). SMO-SHH signaling activator purmorphamine ameliorates neurobehavioral, molecular, and morphological alterations in an intracerebroventricular propionic acid-induced experimental model of autism. Hum. Exp. Toxicol. :9603271211013456. doi: 10.1177/09603271211013456 [Epub Ahead of Print].
Rimkus, T. K., Carpenter, R. L., Qasem, S., Chan, M., and Lo, H.-W. (2016). Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers 8:22. doi: 10.3390/cancers8020022
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Hodson, C. A., Zhang, J., et al. (2009). Sites of regulated phosphorylation that control K-cl cotransporter activity. Cell 138, 525–536. doi: 10.1016/j.cell.2009.05.031
Rinehart, J., Vázquez, N., Kahle, K. T., Hodson, C. A., Ring, A. M., Gulcicek, E. E., et al. (2011). WNK2 kinase is a novel regulator of essential neuronal Cation-chloride Cotransporters. J. Biol. Chem. 286, 30171–30180. doi: 10.1074/jbc.M111.222893
Riobo, N. A., Saucy, B., Dilizio, C., and Manning, D. R. (2006). Activation of heterotrimeric G proteins by smoothened. Proc. Natl. Acad. Sci. U. S. A. 103, 12607–12612. doi: 10.1073/pnas.0600880103
Rivell, A., Petralia, R. S., Wang, Y.-X., Clawson, E., Moehl, K., Mattson, M. P., et al. (2019). Sonic hedgehog expression in the postnatal brain. Bio. Open 8:bio.040592. doi: 10.1242/bio.040592
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., et al. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-cl cotransporter KCC2. J. Neurosci. 24, 4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004
Roper, R. J., Baxter, L. L., Saran, N. G., Klinedinst, D. K., Beachy, P. A., and Reeves, R. H. (2006). Defective cerebellar response to mitogenic hedgehog signaling in Down’s syndrome mice. Proc. Natl. Acad. Sci. U. S. A. 103, 1452–1456. doi: 10.1073/pnas.0510750103
Roussa, E., Speer, J. M., Chudotvorova, I., Khakipoor, S., Smirnov, S., Rivera, C., et al. (2016). The membrane trafficking and functionality of the K+-cl− co-transporter KCC2 is regulated by TGF-β2. J. Cell Sci. 129, 3485–3498. doi: 10.1242/jcs.189860
Ruat, M., Hoch, L., Faure, H., and Rognan, D. (2014). Targeting of smoothened for therapeutic gain. Trends Pharmacol. Sci. 35, 237–246. doi: 10.1016/j.tips.2014.03.002
Ruat, M., Roudaut, H., Ferent, J., and Traiffort, E. (2012). Hedgehog trafficking, cilia and brain functions. Differentiation 83, S97–S104. doi: 10.1016/j.diff.2011.11.011
Saitsu, H., Sonoda, M., Higashijima, T., Shirozu, H., Masuda, H., Tohyama, J., et al. (2016). Somatic mutations in GLI3 and OFD1 involved in sonic hedgehog signaling cause hypothalamic hamartoma. Ann. Clin. Transl. Neurol. 3, 356–365. doi: 10.1002/acn3.300
Sasaki, N., Kurisu, J., and Kengaku, M. (2010). Sonic hedgehog signaling regulates actin cytoskeleton via Tiam1-Rac1 cascade during spine formation. Mol. Cell. Neurosci. 45, 335–344. doi: 10.1016/j.mcn.2010.07.006
Sawano, E., Takahashi, M., Negishi, T., and Tashiro, T. (2013). Thyroid hormone-dependent development of the GABAergic pre- and post-synaptic components in the rat hippocampus. Int. J. Dev. Neurosci. 31, 751–761. doi: 10.1016/j.ijdevneu.2013.09.007
Shqirat, M., Kinoshita, A., Kageyama, R., and Ohtsuka, T. (2021). Sonic hedgehog expands neural stem cells in the neocortical region leading to an expanded and wrinkled neocortical surface. Genes Cells 26, 399–410. doi: 10.1111/gtc.12847
Silayeva, L., Deeb, T. Z., Hines, R. M., Kelley, M. R., Munoz, M. B., Lee, H. H. C., et al. (2015). KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U. S. A. 112, 3523–3528. doi: 10.1073/pnas.1415126112
Spoljaric, A., Seja, P., Spoljaric, I., Virtanen, M. A., Lindfors, J., Uvarov, P., et al. (2017). Vasopressin excites interneurons to suppress hippocampal network activity across a broad span of brain maturity at birth. Proc. Natl. Acad. Sci. U. S. A. 114, E10819–E10828. doi: 10.1073/pnas.1717337114
Tang, B. L. (2020). The expanding therapeutic potential of neuronal KCC2. Cell 9:e240. doi: 10.3390/cells9010240
Tirou, L., Russo, M., Faure, H., Pellegrino, G., Sharif, A., and Ruat, M. (2020). C9C5 positive mature oligodendrocytes are a source of sonic hedgehog in the mouse brain. PLoS One 15:e0229362. doi: 10.1371/journal.pone.0229362
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hübner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792. doi: 10.1126/science.1133212
Virtanen, M. A., Uvarov, P., Mavrovic, M., Poncer, J. C., and Kaila, K. (2021). The multifaceted roles of KCC2 in cortical development. Trends Neurosci. 44, 378–392. doi: 10.1016/j.tins.2021.01.004
Watanabe, M., Wake, H., Moorhouse, A. J., and Nabekura, J. (2009). Clustering of neuronal K+-cl- cotransporters in lipid rafts by tyrosine phosphorylation. J. Biol. Chem. 284, 27980–27988. doi: 10.1074/jbc.M109.043620
Watanabe, M., Zhang, J., Mansuri, M. S., Duan, J., Karimy, J. K., Delpire, E., et al. (2019). Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 12:eaaw9315. doi: 10.1126/scisignal.aaw9315
Weber, M., Hartmann, A.-M., Beyer, T., Ripperger, A., and Nothwang, H. G. (2014). A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride Cotransporter KCC2 That interferes with N-Ethylmaleimide or Staurosporine-mediated activation. J. Biol. Chem. 289, 18668–18679. doi: 10.1074/jbc.M114.567834
Yao, Q., Liu, J., Xiao, L., and Wang, N. (2019). Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARγ stability. J. Biol. Chem. 294, 3284–3293. doi: 10.1074/jbc.RA118.004411
Yao, P. J., Petralia, R. S., Ott, C., Wang, Y.-X., Lippincott-Schwartz, J., and Mattson, M. P. (2015). Dendrosomatic sonic hedgehog signaling in hippocampal neurons regulates axon elongation. J. Neurosci. 35, 16126–16141. doi: 10.1523/JNEUROSCI.1360-15.2015
Zhang, J., Cordshagen, A., Medina, I., Nothwang, H. G., Wisniewski, J. R., Winklhofer, M., et al. (2020b). Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS One 15:e0232967. doi: 10.1371/journal.pone.0232967
Zhang, Y., Liu, G., Guo, T., Liang, X. G., Du, H., Yang, L., et al. (2020a). Cortical neural stem cell lineage progression is regulated by extrinsic signaling molecule sonic hedgehog. Cell Rep. 30, 4490–4504. doi: 10.1016/j.celrep.2020.03.027
Keywords: GABA, KCC2 activity, Smo, chloride homeostasis, Shh
Citation: Hamze M, Medina I, Delmotte Q and Porcher C (2021) Contribution of Smoothened Receptor Signaling in GABAergic Neurotransmission and Chloride Homeostasis in the Developing Rodent Brain. Front. Physiol. 12:798066. doi: 10.3389/fphys.2021.798066
Edited by:
Jiangtao Guo, Zhejiang University, ChinaReviewed by:
Werner Kilb, Johannes Gutenberg University Mainz, GermanyCopyright © 2021 Hamze, Medina, Delmotte and Porcher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Porcher, Y2hyaXN0b3BoZS5wb3JjaGVyQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.