 Lale A. Ertuglu
Lale A. Ertuglu Fernando Elijovich
Fernando Elijovich Cheryl L. Laffer
Cheryl L. Laffer Annet Kirabo
Annet Kirabo- 1Division of Nephrology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
Salt sensitivity of blood pressure (SSBP) is an independent risk factor for cardiovascular morbidity and mortality that is seen in both hypertensive and normotensive populations. Insulin resistance (IR) strongly correlates with SSBP and affects nearly 50% of salt sensitive people. While the precise mechanism by which IR and SSBP relate remains elusive, several common pathways are involved in the genesis of both processes, including vascular dysfunction and immune activation. Vascular dysfunction associated with insulin resistance is characterized by loss of nitric oxide (NO)-mediated vasodilation and heightened endothelin-1 induced vasoconstriction, as well as capillary rarefaction. It manifests with increased blood pressure (BP) in salt sensitive murine models. Another common denominator in the pathogenesis of insulin resistance, hypertension, and salt sensitivity (SS) is immune activation involving pro-inflammatory cytokines like tumor necrosis factor (TNF)-α, IL-1β, and IL-6. In the last decade, a new understanding of interstitial sodium storage in tissues such as skin and muscle has revolutionized traditional concepts of body sodium handling and pathogenesis of SS. We have shown that interstitial Na+ can trigger a T cell mediated inflammatory response through formation of isolevuglandin protein adducts in antigen presenting cells (APCs), and that this response is implicated in salt sensitive hypertension. The peroxisome proliferator-activated receptor γ (PPARγ) is a transcription factor that modulates both insulin sensitivity and BP. PPARγ agonists increase insulin sensitivity and ameliorate salt sensitivity, whereas deficiency of PPARγ results in severe insulin resistance and hypertension. These findings suggest that PPARγ plays a role in the common pathogenesis of insulin sensitivity and salt sensitivity, perhaps via effects on the immune system and vascular function. The goal of this review is to discuss those mechanisms that may play a role in both SSBP and in insulin resistance.
Introduction
Salt sensitivity (SS) of blood pressure (BP) is a phenotype characterized by changes of BP that parallel changes in dietary salt intake. SS affects more than half of all hypertensive subjects as well as a quarter of normotensive individuals in the United States (Weinberger et al., 1986) and is a cardiovascular risk factor for both normotensive and hypertensive humans (Morimoto et al., 1997; Weinberger et al., 2001). As a polygenic trait normally distributed in human populations, SS is well-known to be associated with insulin resistance (IR; Zavaroni et al., 1995; Fuenmayor et al., 1998; Suzuki et al., 2000; Giner et al., 2001). In fact, epidemiological studies suggest that approximately 50% of salt sensitive individuals are insulin resistant (Reaven, 2003), independent of confounding factors such as age, obesity, and glucose intolerance (Galletti et al., 1997). Furthermore, the degree of salt sensitivity seems to correlate with the severity of IR (Zavaroni et al., 1995; Giner et al., 2001), suggesting a causal relationship between the two states. However, the mechanisms underlying this association remain unclear. This article reviews the existing evidence on the interplay between salt sensitivity and insulin resistance and propose mechanisms to explain their relationship.
Pathogenesis of Salt Sensitivity
Since SSBP was described, its underlying pathophysiologic mechanisms have been a matter of controversy. The traditional view follows the classic concept of Guyton (1991), who postulated that SS individuals must have an intrinsic defect in renal sodium handling. According to this view, a salt load expands plasma volume to reach isosmotic balance, which in turn activates systemic and renal natriuretic mechanisms, leading to renal salt excretion without change in arterial pressure (Guyton, 1991). Thus, only an impairment in some renal natriuretic system could lead to hypertension. While defects in renin angiotensin system (Giner et al., 2000), renal sodium transport (Lluch et al., 1996), and sympathetic system (de la Sierra et al., 1996) among others, have been implicated, the exact pathogenesis has not been established. Impaired natriuresis should induce salt retention and plasma volume expansion in SS individuals. However, several studies showed that there is no difference in sodium balance, plasma volume, and cardiac output in response to salt loading or depletion between SS and salt resistant (SR) individuals (Schmidlin et al., 2007; Laffer et al., 2016). Instead, hemodynamic measurements in humans and animals revealed an absence of the normal vasodilator response to salt or even a paradoxical vasoconstriction in SS (Ganguli et al., 1979; Sullivan and Ratts, 1983; Simchon et al., 1989; Qi et al., 1999; Schmidlin et al., 2007; Laffer et al., 2016). This would imply a shift of the paradigm from one of renal excretory defects to one of extrarenal mechanisms producing vascular dysfunction. Indeed, while cardiac output increases after a salt load in both SR and SS subjects, SS subjects lack the concomitant decrease in total peripheral resistance (TPR) that is seen in SR subjects. Similarly, TPR is also unchanged after salt depletion in SS subjects. These findings suggested that BP elevation in SS was mediated by abnormalities in vasoconstrictor/vasodilator responses.
Our understanding of whole-body Na+ distribution has been recently expanded by new knowledge about the storage of Na+ in the interstitial compartment. Previous literature on salt sensitivity regarded body salt balance in terms of the traditional isoosmolar sodium distribution, including the intravascular, interstitial, and intracellular compartments. However, recent studies showed that Na+ may accumulate in the interstitium without commensurate water retention but in association with glycosaminoglycans instead (Titze et al., 2004; Wenstedt et al., 2021). Whether this Na+ is or is not hyperosmolar is controversial (Rossitto et al., 2020), but irrelevant in terms of activation of immune cells, which is due to the Na+ concentration, not to its osmolality (Barbaro et al., 2017). In any case, the finding sheds doubt on the traditional model of sodium-water balance (Machnik et al., 2009; Wiig et al., 2018). In murine models, extrusion of Na+ from this compartment involves a macrophage salt-sensitive tonicity-responsive enhancer binding protein (TonEBP) and stimulation of vascular endothelial growth factor C (VEGFc) leading to lymphangiogenesis (Machnik et al., 2010; Wiig et al., 2013). Blocking this pathway by pharmacological or genetic means results in salt-sensitive hypertension.
Studies using 23Na MRI confirmed that Na+ is stored in the interstitia of skin and skeletal muscle in humans, indicating that this may also occur in other organs less accessible for measurement, and that this storage may provide a buffering system for excess salt intake (Kopp et al., 2013). Others have shown that skin Na+ positively correlates with BP. We have made a similar observation with muscle Na+, which positively correlated with systolic and diastolic blood pressures in patients studied either on their usual diets or during the sodium loading and depletion phases of a research protocol (Sahinoz et al., 2021). Also, skin and skeletal muscle Na+ increase with aging and hypertension (Rossitto et al., 2020), suggesting a direct role of these stores in BP regulation. Some evidence for differential regulation of this Na+ storage between SS and SR individuals has been obtained (Laffer et al., 2016), but this remains to be established with certainty in future studies.
Insulin Resistance, Hypertension, and Salt Sensitivity
Insulin resistance, or reduced insulin sensitivity, is a key component of the metabolic syndrome, which includes hypertension, obesity, and dyslipidemia. Research in the last three decades has proved a strong association between hypertension and insulin resistance, and this relationship is stronger in salt sensitive hypertension (Yatabe et al., 2010).
Salt intake has a close relationship with hypertension (Grillo et al., 2019) and may be a determinant of the pathogenetic link between salt sensitivity and insulin resistance, because it impairs insulin sensitivity in normotensive and hypertensive patients with salt sensitivity but not in those with salt resistance (Sharma et al., 1991; Zavaroni et al., 1995). A high-salt diet exaggerated the insulin response to an oral glucose load in SS but not SR patients (Ferri et al., 1998; Fuenmayor et al., 1998). This suggests that in salt sensitive states, high salt intake may exacerbate insulin resistance. In turn, insulin resistance seems to heighten the blood pressure response to sodium intake (Zhou et al., 2014). Whether the effect of aldosterone, many times inappropriately secreted in SS subjects, plays a role in determining insulin resistance via hybridization of the insulin and insulin growth factor receptors (Sherajee et al., 2012) is not known.
Several mechanisms have been postulated to explain the association between insulin resistance, salt sensitivity, and hypertension. In the following sections, we will describe renal vascular and immune mechanisms, including novel findings about a role for endothelin (Liu et al., 2013) and for interstitial Na+ storage (Nikpey et al., 2017).
Renal Sodium Retention
Insulin resistance is primarily due to downregulation or inactivation of insulin signaling, which is mainly mediated by insulin receptor substrates (IRSs) in effector organs. IRSs exhibit tissue specific distribution. Whereas IRS1 facilitates insulin dependent glucose transport in adipose and skeletal muscle tissue, IRS2 primarily mediates the effects of insulin on the kidney (Kido et al., 2000; Hookham et al., 2013). The latter increase Na+ reabsorption in multiple nephron segments via stimulation of Na+/H+ exchanger type 3 (Klisic et al., 2002), Na+/K+ ATPase (Ewart and Klip, 1995), Na-K-2CL cotransporter (Tsimaratos et al., 2003; Horita et al., 2011), sodium-bicarbonate cotransporter (NBCe1; Horita et al., 2011; Pavlov et al., 2013), and the epithelial sodium channel (ENaC; Loffing and Korbmacher, 2009). It is now known that insulin resistance affects IRSs’ expression differently in each target tissue (Soleimani, 2015). For example, insulin resistant humans have significantly reduced IRS1 and IRS2 expression in metabolically active tissues such as muscle, while IRS2 expression in the kidney is exceptionally preserved (Sechi, 1999; Nakamura et al., 2015). Thus, the compensatory hyperinsulinemia required to maintain normoglycemia can aggravate insulin sodium-retaining actions (Miyazaki et al., 1996), which may potentially lead to hypertension (Figure 1). This is supported by the observation that mice lacking the insulin receptor specifically in the collecting duct principal cells showed significantly lower ENaC activity, while ENaC subunit expression was not changed compared to wild type mice. Also, wild type mice had enhancement in ENaC activity following insulin treatment, indicating insulin mediated channel opening in renal tubules (Pavlov et al., 2013). Thus, selective insulin resistance, through decreasing sodium excretion, may increase the effective sodium load in response to high oral sodium intake, which could aggravate the blood pressure response in salt sensitive patients.
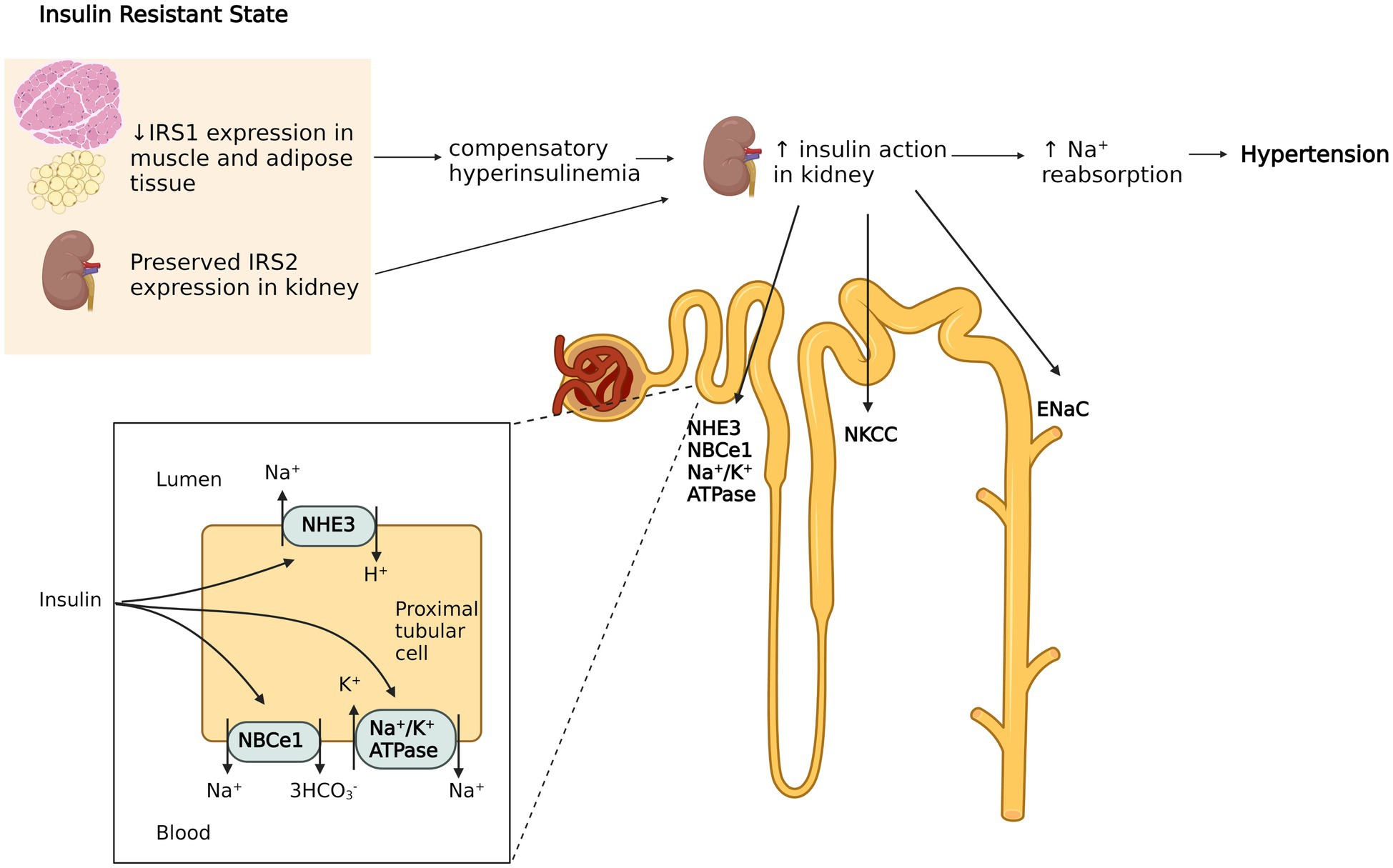
Figure 1. Hyperinsulinemia induced sodium retention in the kidney. Insulin resistance (IR) in humans selectively affects insulin receptor substrate (IRS) 1 in muscle and adipose tissue, while the function of IRS2 in kidney is preserved. The compensatory hyperinsulinemia of insulin resistant states increases insulin’s effects in renal tubules by activating of Na+/H+ exchanger type 3, Na+/K+ ATPase, Na-K-2CL cotransporter, sodium-bicarbonate cotransporter (NBCe1) and the epithelial sodium channel (ENaC). This leads to increased sodium retention which contributes to hypertension. The figure created with BioRender.com.
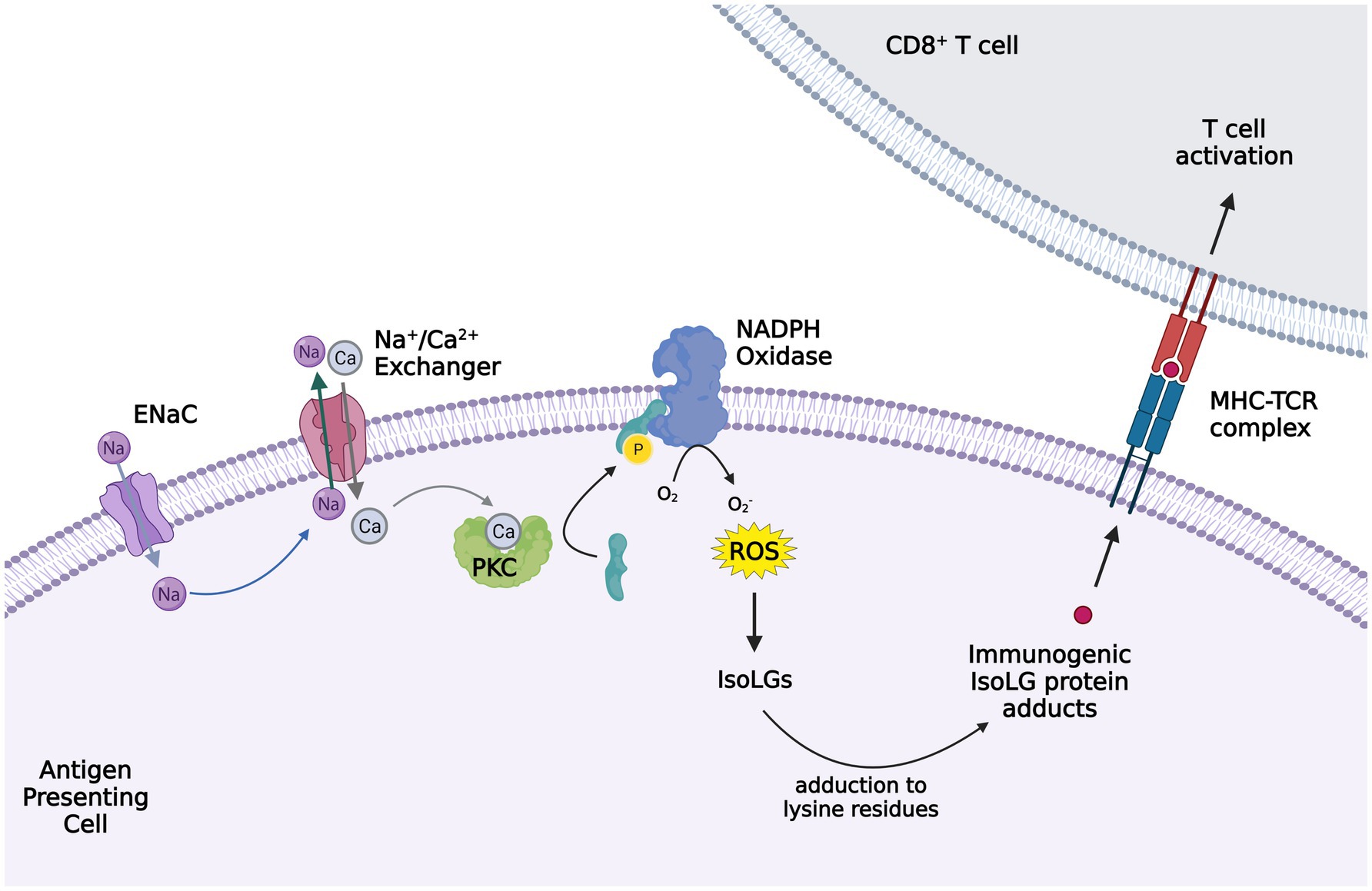
Figure 2. Activation of ENaC in antigen presenting cells (APC) leading to isoleuvoglandin (IsoLG) formation and activation of CD8+ T cells. Elevated interstitial sodium (Na+) enters APCs, in particular dendritic cells, through ENaC and triggers intracellular calcium entry via the sodium calcium exchanger. The increased intracellular calcium activates protein kinase C (PKC), which in turn activates NAPDH oxidase via phosphorylation of p47phox. Superoxide and ROS formation by NAPDH oxidase leads to IsoLG production, which adduct to the lysine residues in proteins and form IsoLG adducts that are presented as neoantigens and trigger CD8+ T cells activation. The figure created with BioRender.com.
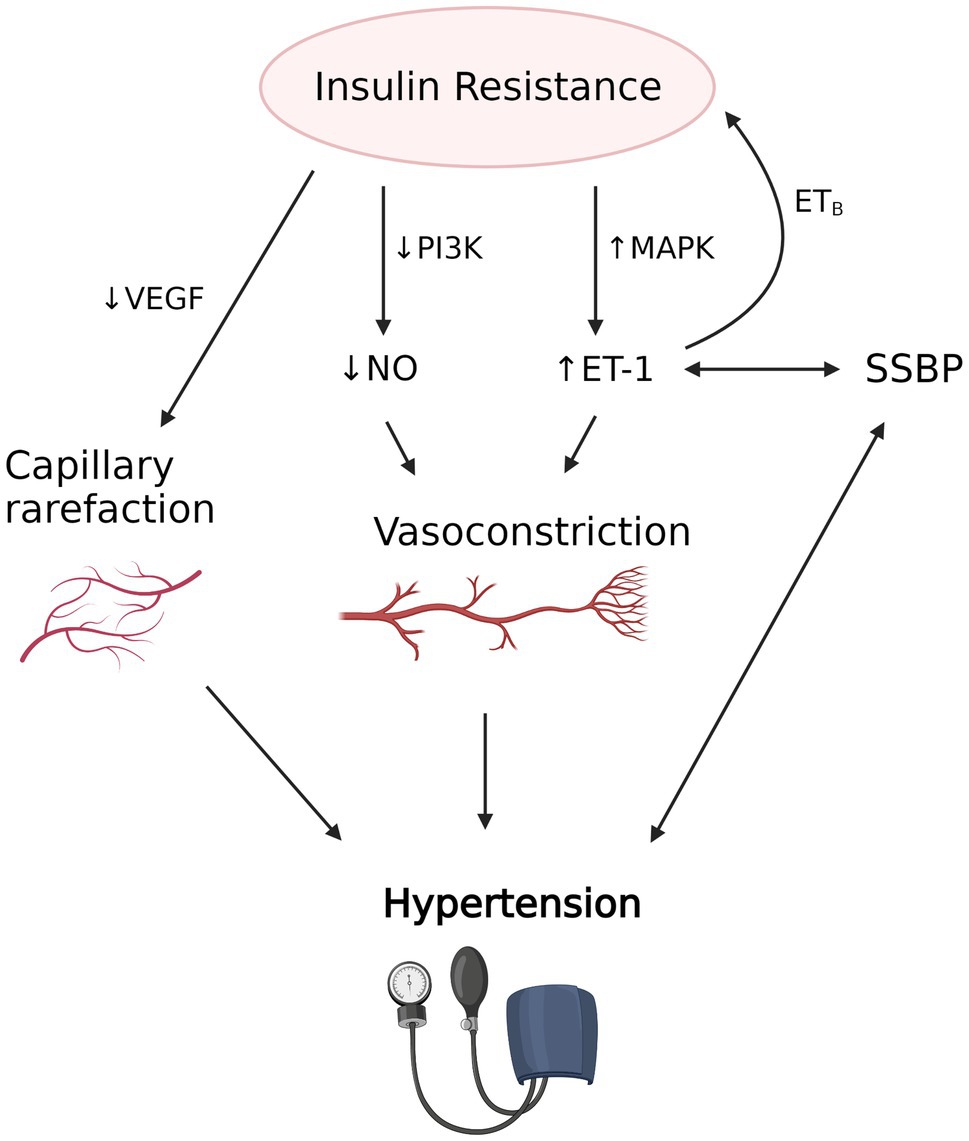
Figure 3. The proposed interplay between insulin resistance induced vascular dysfunction, salt sensitivity of blood pressure (SSBP) and hypertension. Insulin resistance is associated with decreased nitric oxide (NO) production due to decreased activity of PI3K and increased endothelin-1 (ET-1) production due to increased mitogen-activated protein kinase (MAPK) activity, which results in vasoconstriction in precapillary arterioles. Decreased vascular endothelial growth factor (VEGF) and angiogenesis also causes capillary rarefaction. Evidence suggests that SSBP, which strongly correlates with hypertension, is associated with increased availability of ET-1. Increased ET-1, in turn, may contribute to the development of insulin resistance via the action of ETB receptor in adipose tissue. Vascular dysfunction caused by insulin resistance and possibly SSBP leads to hypertension and its clinical manifestations. The figure created with BioRender.com.
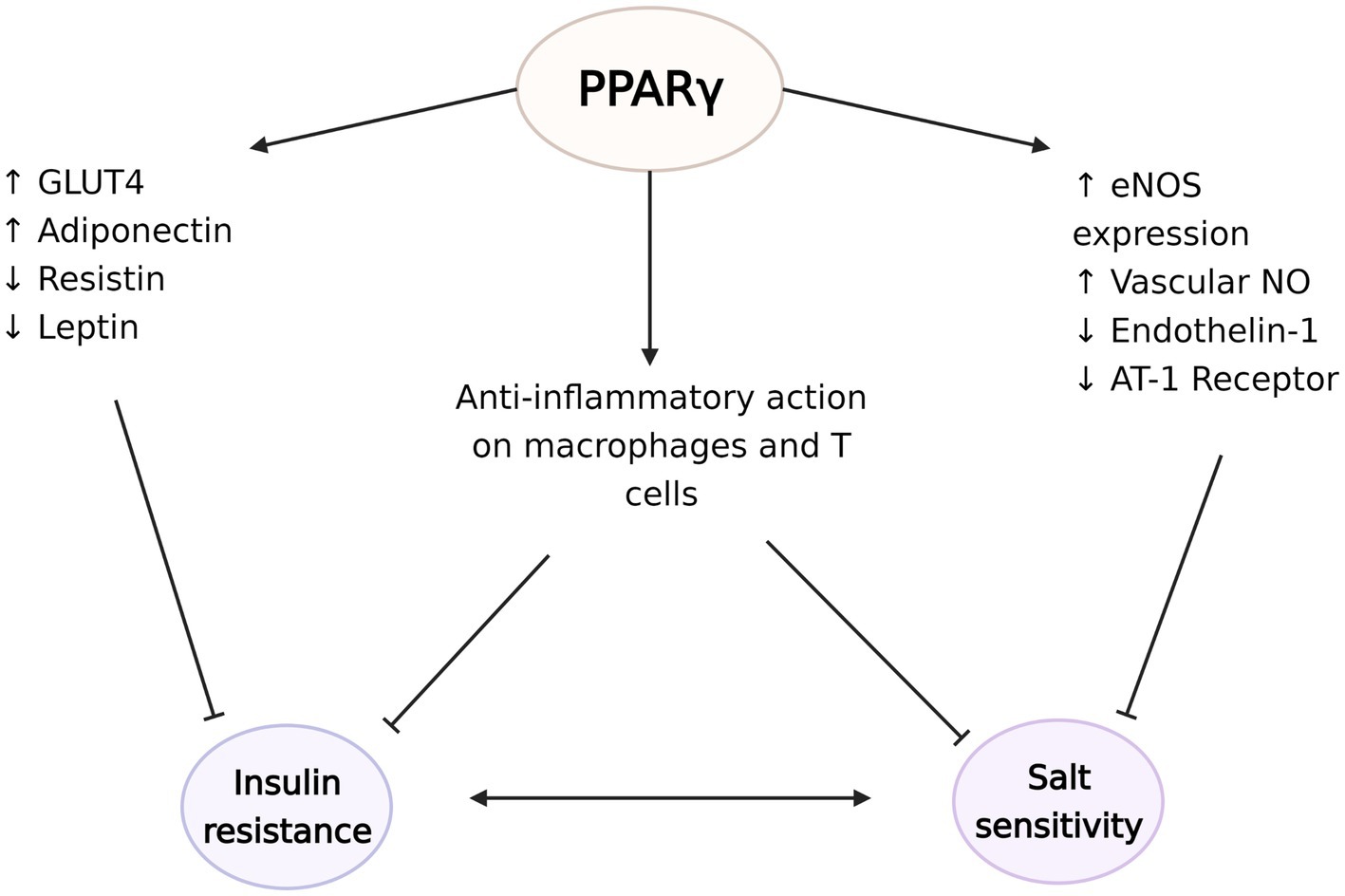
Figure 4. The protective effects of peroxisome proliferator-activated receptor γ (PPARγ) against the development of insulin resistance and salt sensitivity. PPARγ plays a critical role in regulation of the expression of glucose transporter type 4 (GLUT4), adiponectin, resistin, and leptin to increase insulin sensitivity. At the same time, PPARγ induces endothelial nitric oxide synthase (eNOS) expression and NO synthesis and decreases endothelin-1 and angiotensin II type 1 (AT-1) receptor availabilities. These vasodilatory actions potentially ameliorate the vascular dysfunction seen in salt sensitivity. PPARγ activation also has anti-inflammatory effects on macrophages and regulatory T cells in the visceral adipose tissue, which possibly counteract the development of insulin resistance and salt sensitivity. The figure created with BioRender.com.
Vascular Dysfunction
Another mechanism by which IR is postulated to induce hypertension is through vascular insulin resistance (Yki-Järvinen and Utriainen, 1998). Insulin has important vasodilatory effects in skeletal muscle to augment muscle blood flow and glucose transport (Steinberg et al., 1996) and these are impaired in insulin resistant states (Mather et al., 2013). Insulin relaxes precapillary arterioles to facilitate transcapillary insulin transport, increasing microvascular blood flow by a nitric oxide (NO) dependent process termed microvascular recruitment (Herkner et al., 2003; Vincent et al., 2004). Insulin-induced NO production is due to activation of nitric oxide synthase via the phosphatidylinositol 3-kinase (PI3K) pathway, similar to other metabolic insulin signaling (Muniyappa et al., 2020).
In contrast to its vasodilatory effects, insulin also triggers vasoconstriction via mitogen-activated protein kinase (MAPK)-dependent production of vasoconstrictor endothelin 1 (ET-1). This mechanism is involved in fine-tuning of vascular tone (Muniyappa et al., 2020). Selective resistance to the action of insulin on the PI3K-dependent pathway depresses nitric oxide synthesis, whereas the unaffected MAPK pathway maintains endothelin-1 production. This results in a shift of the normally delicate balance between the two opposing vascular actions of insulin toward vasoconstriction and hypertension (Ko et al., 2010). Supporting the involvement of insulin resistance-associated vascular dysfunction in SSBP are observations in Dahl salt sensitive rats, which after a high salt diet exhibit hypertension, metabolic insulin resistance, impaired insulin-dependent activation of PI3K/endothelial NO synthase (eNOS), and impaired NO-mediated vasorelaxation (Zhou et al., 2009, 2010). Other studies of mice have also found high salt diet induced reduction in NO synthesis, through Rho kinase-dependent inhibitory phosphorylation of eNOS by circulating interleukin-17 (IL-17; Faraco et al., 2018).
Insulin’s vasodilatory effects are also crucial to ensure adequate transendothelial transport, a rate limiting step of insulin’s metabolic action (Yang et al., 1989). There is evidence that microvascular IR precedes metabolic IR (Zhao et al., 2015), suggesting an important role for endothelial dysfunction in the pathogenesis of metabolic IR. IR also causes capillary rarefaction in the skeletal muscle, a process of reduced capillary density due to impairment in the angiogenesis mediated by the vascular endothelial growth factor (VEGF; Chung et al., 2006; Frisbee, 2007). This contributes further to insulin-induced vascular dysfunction.
The evidence above suggests that vascular dysfunction induced by insulin resistance may play a role in the pathogenesis of hypertension. Conversely, hypertension associated increased vascular resistance may contribute to the development of metabolic insulin resistance. Indeed, a relationship between hypertension and insulin resistance is further suggested by the finding that monotherapies with either enalapril (vasodilator ACE inhibitor) or rosiglitazone (insulin sensitizer), effectively reduce metabolic insulin resistance, plasma levels of ET-1 and blood pressure in spontaneously hypertensive rats (Potenza et al., 2006). Similar trends have also been reported in nondiabetic hypertensive patients treated with insulin sensitizers and vasodilator antihypertensive agents (Raji et al., 2003; Geng et al., 2013).
Interplay Between Endothelin-1 and Ir in Hypertension
Endothelin-1 is a potent vasoconstrictor secreted by vascular endothelial cells and has been long thought to play a role in the development of the hypertension component of the metabolic syndrome (Dhaun et al., 2008). As discussed above, selective insulin resistance enhances the prohypertensive action of ET-1 by disrupting its physiological balance with NO. This is supported by observations in rodents and humans. In rats, a continuous insulin infusion leads to development of insulin resistance and hypertension along with higher plasma ET-1 levels (Juan et al., 2004). In humans, (a) insulin stimulates ET-1 production during an euglycemic hyperinsulinemic clamp, (b) insulin resistant patients have elevated plasma ET-1 concentrations (Wolpert et al., 1993), (c) plasma insulin and ET-1 levels correlate in essential hypertensive patients (Zaporowska-Stachowiak et al., 1997), and (d) a mixed ET-1 type A (ETA) and type B (ETB) receptor antagonist produces vasodilation in insulin resistant but not insulin sensitive humans (Shemyakin et al., 2006). All these findings suggest a role of insulin-induced ET-1 predominance in the development of hypertension.
Conversely, ET-1 may play a causal role in the development of insulin resistance, since it inhibits insulin mediated glucose uptake in adipocytes through ETB receptors (Chou et al., 1994). It is via stimulation of this receptor that ET-1 regulates adiponectin expression and promotes adipose tissue deposition and insulin resistance (Juan et al., 2007; Rivera-Gonzalez et al., 2020).
Interplay Between Endothelin-1 and Salt in Hypertension
Murine studies have also shown that high salt intake upregulates vascular ET-1 expression independent of changes in blood pressure (Tsai et al., 2006). Also, if the endothelin-1 gene is knocked out from the endothelium (the VEET KO mouse), a high salt diet fails to increase mean arterial pressure to the same extent as in control mice (Speed et al., 2015).
In normotensive and hypertensive humans, high salt intake increases plasma ET-1 levels (Liu et al., 2013). We have discussed above how tissue Na+ storage is involved in salt sensitivity of blood pressure. It is therefore noteworthy that skin Na+ accumulation in response to high salt diet is accompanied by increased ET-1 mRNA expression in vascular tissues (Speed et al., 2015). Therefore, ET-1 may modulate the blood pressure response to salt intake through a direct vasoconstrictive effect as well as an effect on tissue Na+ accumulation. Actually, the interaction between ET-1 and salt in hypertension may have a third player, because tissue Na+ also associates with insulin resistance, with the caveat that the latter observation has only been made in patients with end stage kidney disease (Sahinoz et al., 2020).
Several observations suggest that participation of ET-1 in salt-induced hypertension may be different in SS vs. SR subjects. For example, ET-1 levels are higher in salt sensitive than in salt resistant essential hypertensive patients (Ferri et al., 1998). Also, while receiving an intermediate salt diet, salt sensitive patients have a higher plasma ET-1 response to oral glucose load than their salt resistant counterparts (Ferri et al., 1998). Finally, in a study of 155 normotensive and mild hypertensive patients, a 7-day high salt diet increased plasma ET-1 levels and arterial stiffness much more in SS than in SR patients (Liu et al., 2013). All these studies show that exaggerated ET-1 responses to salt are a feature of SS hypertension.
It is well-known that SS hypertension is associated with more severe target organ damage than SR hypertension and that this may be explained by worse endothelial dysfunction in the former (Franco and Oparil, 2006). It is therefore highly conceivable that ET-1-dependent and IR-dependent endothelial dysfunction are important determinants of the early and severe target organ damage of SS hypertension, and thus explain the poor prognosis associated with this phenotype.
Adipokines in Insulin Resistance and Hypertension
As a key element of metabolic syndrome, obesity closely relates with both hypertension and insulin resistance. Increased adiposity is proposed to contribute to both blood pressure and glucose homeostasis through secretion of various adipokines. One such is chemerin, an adipocyte-derived factor responsible for the development and differentiation of adipocytes (Goralski et al., 2007). Chemerin has vasoconstrictive effects through its actions on vascular smooth muscle (Ferland et al., 2017) and endothelium (Lobato et al., 2012) in both human and animal vasculature (Kennedy et al., 2016) and decrease in whole body chemerin by chemerin antisense oligonucleotides reduces blood pressure in normal-fed rats (Ferland et al., 2018) as well as in Dahl salt-sensitive (DS) rats fed high-salt and high fat diets (Ferland et al., 2019). In humans, chemerin has been found to associate with higher risk of hypertension in a large population-based study including 3,986 subjects (Zylla et al., 2017).
In addition, chemerin regulates glucose metabolism and its actions in skeletal and cardiac muscles have been linked with insulin resistance in several studies (Sell et al., 2009; Becker et al., 2010; Zhang et al., 2014). Rodent models of obesity/diabetes demonstrated significantly elevated serum levels of chemerin; exogenous chemerin administration further exacerbated glucose intolerance (Ernst et al., 2010). Nevertheless, human studies have not shown a consistent relationship between insulin sensitivity and serum chemerin levels and its exact role in whole body insulin resistance remains controversial (Takahashi et al., 2008; Alfadda et al., 2012; Helfer and Wu, 2018; Karczewska-Kupczewska et al., 2020).
Another factor produced by adipose tissue that has overlapping roles in obesity, insulin resistance, and hypertension is leptin. Data from human as well as animal studies suggest that leptin plays a major role in the neurohormonal mechanisms of obesity-induced hypertension (Simonds et al., 2014). Obese subjects display high circulating levels of leptin and an insensitivity to the anorexigenic effects of exogeneous leptin, known as leptin resistance (Izquierdo et al., 2019). Leptin is proposed to affect blood pressure through its sympathetic activity and leptin levels directly associated with blood pressure changes in rodent models of obesity, which is not seen in models of leptin deficiency. Humans with loss-of-function mutation in leptin or leptin receptor have been found to be protected from hypertension despite obesity (Simonds et al., 2014), nevertheless, exogenous leptin administration does not increase blood pressure in leptin deficient states including congenital leptin deficiency and lipodystrophy (Brown et al., 2015). Importantly, obesity secondary to leptin resistance was found to result in increased salt sensitive blood pressure response to high salt in SHHF rat, a model of spontaneous hypertension. The increase in salt sensitivity was driven by endothelin and was obliterated by bosentan (Radin et al., 2003). In another study, high salt diet has been demonstrated to cause leptin resistance, obesity as well as high blood pressure and insulin resistance in mice, further implying a link between leptin resistance, high salt-associated hypertension, and insulin resistance (Lanaspa et al., 2018).
Leptin also has important pro-inflammatory effects, which may play a role in insulin resistance and hypertension. Leptin activates monocytes to produce inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-6, and IL-12(Gainsford et al., 1996), while suppressing the anti-inflammatory Th2 response (Fernández-Riejos et al., 2010). The chronic inflammatory state observed in high adiposity, whether partially driven by leptin or not, seems to play a major role in the interplay between insulin resistance and hypertension.
Immunity in Obesity and IR
Obesity is characterized by high levels of plasma inflammatory cytokines (Villarreal-Calderón et al., 2019) and a low-grade inflammatory state in visceral adipose tissue, which is a critical contributor to the development of insulin resistance (Winer and Winer, 2012). The innate immune system plays an important role in the proinflammatory state seen in obesity (Patel et al., 2013). Macrophages of the inflamed adipose tissue are polarized to the M1 state with enhanced antigen-presenting and proinflammatory cytokine-producing properties, to the detriment of M2 macrophages that produce anti-inflammatory mediators such as IL-10 and TGF-β (Orliaguet et al., 2020). M1 production of TNF-α, IL-1β, and IL-6, IL-12, and iNOS activates serine kinases that phosphorylate IRS proteins and insulin receptors, thus hindering insulin signaling (Mantovani et al., 2002; Olefsky and Glass, 2010). This paracrine process in the adipose tissue has local and also systemic effects due to leakage of cytokines into circulation, which can be detected with plasma measurements (Olefsky and Glass, 2010).
The adaptive immune system is also activated in obesity. T cells accumulate in the adipose tissue and interact with macrophages to enhance the inflammatory response (Nishimura et al., 2009). Indeed, obesity and insulin resistance are associated with predominance of CD8+ and CD4+ Th1 T cells with IFN-γ secretion in visceral adipose tissue that promotes the M1 polarization of macrophages described above (Winer and Winer, 2012). In mice fed a high-fat diet, depletion of CD8+ T cells reduces local adipose tissue inflammation and insulin resistance, whereas conversely, transfer of CD8+ cells into CD8 deficient mice induces insulin resistance (Nishimura et al., 2009).
Immunity in Hypertension
Hypertension is also strongly associated with inflammation. T cells and macrophages infiltrate the perivascular space and kidneys in animal models of hypertension (Kirabo et al., 2014; Caillon et al., 2017) and relate to hypertensive end organ damage. Mice lacking T cells or the T cell cytokine IL-17A are protected from angiotensin II-induced hypertension and related vascular dysfunction (Crowley et al., 2010; Madhur et al., 2010). Moreover, immunosuppression has antihypertensive effects in animal studies (McMaster et al., 2015).
Hypertension is associated with T cell clonal expansion, a hallmark of antigen-presentation found in various immune pathologies including autoimmune disease (Acha-Orbea et al., 1998), atherosclerosis (Paulsson et al., 2000), and obesity (Yang et al., 2010). By examining T cell receptor (TCR) usage, we showed that accumulation of an oligoclonal CD8+ T cell population in the kidney contributes to hypertension by inducing endothelial dysfunction and vascular rarefaction as well as sodium and volume retention (Trott et al., 2014). More recently, we found that the CD8+ T cell populations in the adipose tissue of hypertensive high-fat fed mice are more clonal and demonstrate enrichment for positively charged amino acids, particularly arginine, which is a characteristic previously reported in TCRs associated with obesity and insulin resistance (McDonnell et al., 2018). Furthermore, two of the TCR clonotypes found in abundance in adipose tissue of high-fat fed mice were previously identified diabetogenic clonotypes (McDonnell et al., 2018).
In multiple hypertensive models, we showed that hypertension is associated with production of isolevuglandins (IsoLGs; alternatively named Isoketals or γ-ketoaldehydes) in murine dendritic cells (DCs) and human monocytes (Figure 2). IsoLGs are highly reactive products of lipid peroxidation and adduct to lysine residues on proteins, leading to alteration of protein function and formation of neoantigens (Kirabo et al., 2014). These immunogenic proteins lead to T cell activation and hypertension (Kirabo et al., 2014), and contribute to the oligoclonal T cell expansion in the adipose tissue of high-fat induced obesity (McDonnell et al., 2018). These protein adducts, owing to their negative charge (McDonnell et al., 2018) particularly associate with TCRs found in obese adipose tissue, which are enriched with positively charged amino acids. IsoLGs represent a potential interventional target, since administration of isoLG scavengers prevents the inflammatory responses and the development of hypertension (Kirabo et al., 2014).
Moreover, we reported that elevated interstitial Na+ is a potent stimulus for IsoLG-adduct formation in murine DCs. Na+ enters DCs through amiloride-sensitive ENaC channels and promotes calcium (Ca2+) entry into the cell via the Na+/Ca2+ exchanger. Increased intracellular Ca2+ activates protein kinase C (PKC), which phosphorylates and activates the NADPH oxidase. The increased superoxide and ROS formation by NADPH oxidase promote IsoLG formation and subsequent DC activation (Barbaro et al., 2017). These high-salt treated DCs produce IL-1β and induce T cell production of pro-hypertensive cytokines IL-17A and IFN-γ (Barbaro et al., 2017). Other studies of mice have also shown increased circulating IL-17 levels in response to high salt diet, leading to endothelial dysfunction (Faraco et al., 2018). These findings indicate that elevated tissue Na+ may contribute to the development of hypertension and salt sensitivity through immune cell activation. Indeed, previous studies suggested altered regulation of the hyperosmolar Na+ storage in tissue in the pathogenesis of salt sensitivity (Laffer et al., 2016). Whether differences in tissue Na+ or differences in the immune response to tissue Na+ play a role in the salt sensitive phenotype is yet to be discovered.
The relationship between the immune system and interstitial Na+ stores is complex. It is pretty clear that interstitial Na+ storage triggers immune system activation. Alternatively, the immune system regulates interstitial Na+ storage. As discussed earlier, interstitial Na+ storage in tissue, primarily in muscle and skin, may be an important buffer system for high salt intake that may determine SSBP. Innate immune cells have been suggested to play a role in the regulation of the amount of Na+ that can be stored in the interstitium, which in turn may activate adaptive immune cells through ENaC mediated Na+ entry into antigen presenting cells (APCs) as described above. The mononuclear phagocyte system (MPS) and macrophages have been shown to regulate tissue Na+ accumulation through modulating interstitial Na+ clearance. The high salt diet-induced increase in skin Na+ relates with hyperplasia of the lymphatic capillary network, through activation of TonEBP and consequent secretion of VEGF-C by macrophages (Machnik et al., 2009). This lymphangiogenesis provides a buffer for extra sodium and water in tissue as shown by the fact that either depletion of MPS or inhibition of VEGF action augmented interstitial hypertonic water retention and salt-induced blood pressure elevation (Machnik et al., 2009). The MPS/TonEBP/VEGF-C pathway belongs to the physiological anti-inflammatory phenotype of M2 subtype of macrophage polarization (Machnik et al., 2010). Shifting the balance of macrophage polarization into the pro-inflammatory M1 subtype associated with insulin resistant states, may disrupt MPS modulation of interstitial Na+. This most likely converted the salt resistant rats into a salt sensitive phenotype (Machnik et al., 2010) in the experiments above.
Role of the Inflammasome
The inflammatory responses seen in both hypertension and obesity induced insulin resistance include inflammasome activation. NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, is a member of the nucleotide-binding oligomerization domain leucine-rich repeat (NLR) PRR family and drives sterile inflammation in response to damaged cell derived “danger-associated molecular patterns,” leading to caspase-1 activation and subsequent IL-1β and IL-18 secretion (Chen and Nunez, 2010; Van Tassell et al., 2013; Lamkanfi and Dixit, 2014).
NLRP3 has also been suggested to produce inflammatory responses in adipose tissue, since they can be triggered by ceramides, a product of fatty acid metabolism, as well as oxidized LDL and cholesterol (Duewell et al., 2010). The common end point of NLRP3 inflammasome activation is a chronic systemic inflammatory state because the locally secreted inflammatory cytokines are released into blood with subsequent development of global insulin resistance (Xu et al., 2003; Zhou et al., 2014). In animal models, elimination of NLRP3 inflammasome protects from high-fat induced insulin resistance, while in obese diabetic individuals, adipose tissue expression of NLRP3 inversely correlates with the increase in insulin sensitivity after weight loss (Vandanmagsar et al., 2011). Analysis of UKBiobank revealed that individuals with a gene variant increasing NLRP3 mRNA expression was associated with higher prevalence of diabetes (Schunk et al., 2021).
Hypertension has also been related with the activation of NLRP3 inflammasome. IL-1β and IL-18, the main products of NLRP3 activation, are elevated in plasma and monocytes of hypertensive individuals (Dörffel et al., 1999; Li et al., 2005; Rabkin, 2009) and associate with renal and vascular dysfunction (De Miguel et al., 2021). Hypertensive patients also have high concentrations of NF-κB, a crucial intracellular trigger of NLRP3 activation, in tissue and inflammatory cells (De Miguel et al., 2021). NF-κB inhibition ameliorates hypertension and prevents hypertension-induced organ damage in mice (Zambom et al., 2019). NF-κB may also play a role in the interplay between salt sensitivity and insulin resistance, since the association between these two traits in Dahl salt sensitive rats was partially NFkB dependent. Pyrrolidine dithiocarbamate (PDTC), an inhibitor of NFκB, significantly improved blood pressure as well as insulin sensitivity and insulin mediated vasorelaxation in this salt sensitive rat model (Zhou et al., 2010). Indeed, NLRP3 (Sun et al., 2017) and NALP3 (Fu et al., 2018) inflammasomes mediate endothelial dysfunction, main contributors to both salt sensitivity and insulin resistance as proposed earlier. Moreover, eNOS and NO pathways, which are disrupted in salt sensitivity, are in part regulated by the NLRP3 inflammasome (Sogawa et al., 2018). Importantly, NLRP3 inflammasome is activated by NADPH oxidase and ROS (Abais et al., 2014; Cui et al., 2014; Banoth and Cassel, 2018), which are crucial mediators of IsoLG formation.
Immunity and the Link Between SSBP and IR
While inflammation has been shown to separately contribute to the pathogenesis of both insulin resistance and salt sensitive hypertension, is not known whether it has a causal role in the link between these two traits (Figure 3). Animal models extensively used as a paradigm for salt-sensitive hypertension in humans provide some evidence in this regard. In DS rats, inflammation and oxidative stress were found to associate with both hypertension and insulin resistance (Zhou et al., 2010). Furthermore, treatment with tempol, an intracellular antioxidant, improved inflammatory markers, vascular insulin signaling, endothelium-dependent relaxation, and insulin sensitivity. Similarly, the angiotensin receptor blocker candesartan, a commonly used antihypertensive, also improved inflammatory markers and insulin sensitivity (Zhou et al., 2009).
The potential role of inflammation in the pathogenesis of SS may also have further implications regarding the increased cardiovascular risk in individuals with SS. Considering that pro-inflammatory status is consistently associated with increased risk of cardiovascular disease (Sorriento and Iaccarino, 2019), it is plausible that inflammatory activation related to SS development may also link with higher cardiovascular risk. Furthermore, if elevated, interstitial Na+ may indeed lead to a systemic immune activation through infiltration of T cells in various organs, an altered tissue Na+ storage may potentially predispose the salt sensitive population to poor cardiovascular outcomes.
PPARγ, Insulin Resistance, and Immunity
The peroxisome proliferator-activated receptors are a nuclear receptor superfamily of ligand-inducible transcription factors found in humans and includes three subtypes: PPARα, PPARβ/δ, and peroxisome proliferator-activated receptor γ (PPARγ; Ahmadian et al., 2013). PPARγ is well-known as the master regulator of adipogenesis and lipid metabolism and is predominantly expressed in white and brown adipose tissue. PPARγ is bound and activated by fatty acids and their derivatives endogenously and by thiazolidinediones (TZDs), highly specific synthetic ligands, exogenously. PPARγ activation has a robust insulin sensitizing effect, making TZDs potent insulin sensitizers and highly effective oral medications for type 2 diabetes, although, their clinical use is limited by major adverse effects including weight gain, fluid retention, and osteoporosis (Ahmadian et al., 2013).
PPARγ acts through various gene networks to modulate glucose homeostasis. Its activation is directly related with increased expression of glucose transporter type 4, while also controlling the expression of many factors that affect insulin sensitivity, such as adiponectin, resistin, leptin, and TNF-α. Therefore, PPARγ agonists’ insulin sensitizing effects can be explained by various mechanisms including modulation of cellular glucose update, hepatic glucose release, systemic inflammation, or food intake (Iwaki et al., 2003; Tomaru et al., 2009; Ahmadian et al., 2013). Indeed, PPARγ’s regulatory roles are not limited to adipocytes. PPARγ also has crucial functions in immune cells, especially in macrophages and APCs (Tontonoz et al., 1998; Wahli and Michalik, 2012). PPARγ regulates lipid metabolism and exerts anti-inflammatory effects in macrophages, including polarization into the anti-inflammatory M2 subtype (Odegaard et al., 2007). Mice with macrophage specific deletion of PPARγ exhibit disruption of M2 macrophage activation, and development of diet induced obesity and whole-body insulin resistance (Odegaard et al., 2007). In turn, evidence suggest that TZDs downregulate the expression of M1 mediators (Xu et al., 2016). Moreover, PPARγ also regulates the accumulation and function of regulator T cells in the visceral adipose tissue and expression of PPARγ in this unique T cell population is essential for the complete insulin-sensitizing activity of TZDs in obese mice (Cipolletta et al., 2012). PPARγ-axis dependent insulin signaling has also shown to drive state transition between adipose regulatory T cell subsets (Li et al., 2021). PPARγ agonists have also been reported to suppress the progression of atherosclerosis and aortic aneurysmal changes by reducing the expression of TNF-α and other markers of inflammation (Zinn et al., 2008; Motoki et al., 2015). These anti-inflammatory actions of PPARγ can potentially explain some of its beneficial effects on insulin sensitivity as well as blood pressure (Figure 4).
PPARγ and Hypertension
Compelling evidence suggest that PPARγ is also an important regulator of blood pressure. Various mutations in PPARγ, causing loss of function, have been shown to strongly associate not only with insulin resistance and diabetes but also with severe hypertension (Barroso et al., 1999; Fang et al., 2021). In addition to these rare mutations, common variants of PPARγ may also play a role. For example, in a meta-analysis of more than 5,500 Asian hypertensives (Zhang et al., 2019), the P12A polymorphism was associated with the risk of hypertension. Furthermore, treatment with PPARγ agonists is associated with decreased blood pressure (Nolan et al., 1994).
A well-known side effect of TZD is fluid retention. TZDs increase renal sodium reabsorption without modulation of RAS system, in people with diabetes and hypertension (Zanchi et al., 2010) and in healthy subjects (Zanchi et al., 2004). Despite this, large scale clinical studies consistently show that TZDs, including pioglitazone (Dormandy et al., 2005; Yoshii et al., 2014), rosiglitazone (Komajda et al., 2008), and troglitazone (Nolan et al., 1994), lead to modest decreases in blood pressure (Qayyum and Adomaityte, 2006). Various cardiovascular actions of TZD could explain its effect on blood pressure, including its anti-inflammatory and endothelium-mediated vasodilatory actions. PPARγ activation upregulates eNOS expression and availability of vascular NO (Sartori-Valinotti et al., 2010; Nagao and Yamaguchi, 2012), while it downregulates the synthesis of vasoconstrictor endothelin-1 (Satoh et al., 1999; Kang et al., 2017) and the expression of Angiotensin II type 1 receptor [AT (1)-R], thus promoting vasodilation in the vascular smooth muscle (Ketsawatsomkron et al., 2012; Pelham et al., 2012). Mice lacking endothelial-specific PPARγ have deficient NO production and increased oxidative stress markers (Kleinhenz et al., 2009), and show augmented Ang-II induced endothelial dysfunction, a response dependent on superoxide, NADPH oxidase, and Rho kinase (Silva et al., 2017). PPARγ further inhibits pro-inflammatory gene expression in vascular smooth muscle through inhibition of NF-κB activity (Mukohda et al., 2017) and protects against IL-1β-induced oxidative stress in endothelium (Mukohda et al., 2016).
Considering the impaired vasodilatory response observed in salt sensitive patients and the beneficial effects of PPARγ on the vasodilatory system, PPARγ agonists would be expected to ameliorate salt sensitivity of blood pressure (SSBP). Indeed, despite increased sodium retention, clinical studies demonstrated that TZD abolished the blood pressure response to salt in salt-sensitive individuals (Zanchi et al., 2010). In animal models, TZDs also attenuated salt sensitivity, a result observed along with increased NO availability and decreased renal macrophage infiltration and inflammation (Sartori-Valinotti et al., 2010). Furthermore, mice models of PPARγ impairment develop salt sensitive hypertension. Salt sensitive humans and animals demonstrate decreased renal blood flow and increased renal vascular resistance, and these abnormalities are attenuated by TZDs (Fink et al., 1980; Campese et al., 1991; Wu et al., 2021). Mice with dominant negative mutation of PPARγ in vascular smooth muscle start showing salt-induced impairment of vasodilation along with blunted NO responsiveness after 3 days of high salt diet, and this is followed by the development of salt sensitive hypertension (Wu et al., 2021). Regarding its actions to increase insulin sensitivity, PPARγ agonists can also counteract the effects of insulin resistance on blood pressure. In insulin resistant fatty rat models, rosiglitazone treatment prevents the development of hypertension and partially restores the vasodilatory effects of insulin, thus ameliorating endothelial dysfunction associated with insulin resistance (Walker et al., 1999). Therefore, mounting evidence points to PPARγ as a common regulator of both glucose and blood pressure homeostasis. As numerous mutations of PPARγ have been shown to result in both severe insulin resistance and hypertension, deficiency of physiologic PPARγ activation may be a pathogenetic factor linking insulin resistance and salt sensitive hypertension, a question that remains to be answered in definitive manner.
Oxidative Metabolites, IR, and SSBP
Methylglyoxal (MGO), a metabolite of the glycolysis pathway that is increased in diabetes, has been proposed to contribute to the development of salt sensitivity as well as insulin resistance through oxidative stress and advanced glycation endproducts (AGE; Guo et al., 2009). Increased levels of MGO along with increased levels of ROS have been found in vascular tissue of hypertensive animals (Chang et al., 2005; Jia and Wu, 2007) and in plasma of diabetic patients. High fructose induced accumulation of endogenous MGO in plasma and tissue induces the development of hypertension and insulin resistance in rats (Wang et al., 2008).
The mechanisms by which MGO produces insulin resistance are multiple. Accumulation of endogenous MGO reduces IRS-1/PI3K association and alters PI3K activity in adipose tissue, leading to decreased insulin stimulated glucose uptake and insulin resistance (Jia and Wu, 2007). Other studies have also shown that MGO disrupts insulin signaling pathways by formation of AGEs and ROS (Unoki et al., 2007) and by direct binding to IRS1 protein. This inhibits IRS1 association with other proteins in cultured muscle cells and in adipose tissue of fructose-induced hypertensive rats independent of formation of ROS (Riboulet-Chavey et al., 2006). Furthermore, MGO directly produces a dose-dependent decrease in adipose tissue capillarization and blood flow, which associate with systemic and muscle insulin resistance (Rodrigues et al., 2017). Finally, MGO also alters insulin structure by binding to its arginine residue and decreasing its activity (Jia et al., 2006).
In addition to its role in the pathogenesis of insulin resistance, MGO contributes to the development of salt sensitivity of blood pressure by mechanisms not yet fully understood. In normotensive rats, MGO treatment induces both the development of insulin resistance and salt sensitivity. Although MGO or salt, given separately did not induce hypertension, co-administration of both significantly increased blood pressure. N-acetyl cysteine, a MGO scavenger, or an AGE inhibitor completely improved MGO-induced insulin resistance in this model (Guo et al., 2009), whereas metformin improved both, the increase in endogenous MGO and in blood pressure observed in a high fructose diet model (Wang et al., 2008).
Conclusion
Salt sensitivity of blood pressure, a significant cardiovascular risk factor, strongly associates with insulin resistance. Salt sensitivity and insulin resistance share several pathogenetic factors. They include vascular dysfunction, particularly caused by endothelin-1 overproduction, and immune activation primarily driven by CD8+ T cells action. The transcription factor PPARγ also modulates insulin sensitivity and hypertension through its anti-inflammatory and vasodilatory actions. Impairment of PPARγ action results in insulin resistance and hypertension in both animal models and humans, and perhaps is a common denominator linking insulin resistance and salt sensitive hypertension in the population. The oxidative metabolite of the glycolysis pathway MGO, also contributes to the development of both salt sensitivity and insulin resistance through yet to be explored mechanisms. The presence of these shared pathways underlying the mechanisms of both, insulin resistance and salt sensitivity of blood pressure, is very suggestive of a possible causal bidirectional relationship between these two cardiovascular risk factors, a contention that remains to be proven in definitive manner.
Author Contributions
LE wrote the draft. FE, CL, and AK revised and approved the manuscript. AK obtained funding for the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Vanderbilt CTSA grant UL1TR002243 from NCATS/NIH and the National Institutes of Health grants K01HL13049, R03HL155041, and R01HL144941 to AK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abais, J. M., Xia, M., Li, G., Gehr, T. W. B., Boini, K. M., and Li, P. L. (2014). Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic. Biol. Med. 67, 211–220. doi: 10.1016/j.freeradbiomed.2013.10.009
Acha-Orbea, H., Mitchell, D. J., Timmermann, L., Wraith, D. C., Tausch, G. S., Waldor, M. K., et al. (1998). Limited heterogeneity of T cell receptors from lymphocytes mediating autoimmune encephalomyelitis allows specific immune intervention. Cell 54, 263–273. doi: 10.1016/0092-8674(88)90558-2
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi: 10.1038/nm.3159
Alfadda, A. A., Sallam, R. M., Chishti, M. A., Moustafa, A. S., Fatma, S., Alomaim, W. S., et al. (2012). Differential patterns of serum concentration and adipose tissue expression of chemerin in obesity: adipose depot specificity and gender dimorphism. Mol. Cell 33, 591–596. doi: 10.1007/s10059-012-0012-7
Banoth, B., and Cassel, S. L. (2018). Mitochondria in innate immune signaling. Transl. Res. 202, 52–68. doi: 10.1016/j.trsl.2018.07.014
Barbaro, N. R., Foss, J. D., Kryshtal, D. O., Tsyba, N., Kumaresan, S., Xiao, L., et al. (2017). Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 21, 1009–1020. doi: 10.1016/j.celrep.2017.10.002
Barroso, I., Gurnell, M., Crowley, V. E., Agostini, M., Schwabe, J. W., Soos, M. A., et al. (1999). Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402, 880–883. doi: 10.1038/47254
Becker, M., Rabe, K., Lebherz, C., Zugwurst, J., Göke, B., Parhofer, K. G., et al. (2010). Expression of human chemerin induces insulin resistance in the skeletal muscle but does not affect weight, lipid levels, and atherosclerosis in LDL receptor knockout mice on high-fat diet. Diabetes 59, 2898–2903. doi: 10.2337/db10-0362
Brown, R. J., Meehan, C. A., and Gorden, P. (2015). Leptin does not mediate hypertension associated with human obesity. Cell 162, 465–466. doi: 10.1016/j.cell.2015.07.007
Caillon, A., Mian, M. O. R., Fraulob-Aquino, J. C., Huo, K. G., Barhoumi, T., Ouerd, S., et al. (2017). γδ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation 135, 2155–2162. doi: 10.1161/CIRCULATIONAHA.116.027058
Campese, V. M., Parise, M., Karubian, F., and Bigazzi, R. (1991). Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension 18, 805–812. doi: 10.1161/01.HYP.18.6.805
Chang, T., Wang, R., and Wu, L. (2005). Methylglyoxal-induced nitric oxide and peroxynitrite production in vascular smooth muscle cells. Free Radic. Biol. Med. 38, 286–293. doi: 10.1016/j.freeradbiomed.2004.10.034
Chen, G. Y., and Nunez, G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837. doi: 10.1038/nri2873
Chou, Y. C., Perng, J. C., Juan, C. C., Jang, S. Y., Kwok, C. F., Chen, W. L., et al. (1994). Endothelin-1 inhibits insulin-stimulated glucose uptake in isolated rat adipocytes. Biochem. Biophys. Res. Commun. 202, 688–693. doi: 10.1006/bbrc.1994.1985
Chung, A. W., Hsiang, Y. N., Matzke, L. A., McManus, B. M., van Breemen, C., and Okon, E. B. (2006). Reduced expression of vascular endothelial growth factor paralleled with the increased angiostatin expression resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in human type 2 diabetic arterial vasculature. Circ. Res. 99, 140–148. doi: 10.1161/01.RES.0000232352.90786.fa
Cipolletta, D., Feuerer, M., Li, A., Kamei, N., Lee, J., Shoelson, S. E., et al. (2012). PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553. doi: 10.1038/nature11132
Crowley, S. D., Song, Y. S., Lin, E. E., Griffiths, R., Kim, H. S., and Ruiz, P. (2010). Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am. J. Phys. Regul. Integr. Comp. Phys. 298, R1089–R1097. doi: 10.1152/ajpregu.00373.2009
Cui, J., Chen, Y., Wang, H. Y., and Wang, R. F. (2014). Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccin. Immunother. 10, 3270–3285. doi: 10.4161/21645515.2014.979640
de la Sierra, A., Lluch, M. M., Coca, A., Aguilera, M. T., Giner, V., Bragulat, E., et al. (1996). Fluid, ionic and hormonal changes induced by high salt intake in salt-sensitive and salt-resistant hypertensive patients. Clin. Sci. 91, 155–161. doi: 10.1042/cs0910155
De Miguel, C., Pelegrín, P., Baroja-Mazo, A., and Cuevas, S. (2021). Emerging role of the inflammasome and pyroptosis in hypertension. Int. J. Mol. Sci. 22:1064. doi: 10.3390/ijms22031064
Dhaun, N., Goddard, J., Kohan, D. E., Pollock, D. M., Schiffrin, E. L., and Webb, D. J. (2008). Role of endothelin-1 in clinical hypertension. Hypertension 52, 452–459. doi: 10.1161/HYPERTENSIONAHA.108.117366
Dörffel, Y., Lätsch, C., Stuhlmüller, B., Schreiber, S., Scholze, S., Burmester, G. R., et al. (1999). Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension 34, 113–117. doi: 10.1161/01.HYP.34.1.113
Dormandy, J. A., Charbonnel, B., Eckland, D. J. A., Erdmann, E., Massi-Benedetti, M., Moules, I. K., et al. (2005). Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (prospective pioglitazone clinical trial in macrovascular events): a randomised controlled trial. Lancet 366, 1279–1289. doi: 10.1016/S0140-6736(05)67528-9
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Ernst, M. C., Issa, M., Goralski, K. B., and Sinal, C. J. (2010). Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 151, 1998–2007. doi: 10.1210/en.2009-1098
Ewart, H. S., and Klip, A. (1995). Hormonal regulation of the Na(+)-K(+)-ATPase: mechanisms underlying rapid and sustained changes in pump activity. Am. J. Phys. 269, C295–C311. doi: 10.1152/ajpcell.1995.269.2.C295
Fang, S., Livergood, M. C., Nakagawa, P., Wu, J., and Sigmund, C. T. (2021). Role of the peroxisome proliferator activated receptors in hypertension. Circ. Res. 128, 1021–1039. doi: 10.1161/CIRCRESAHA.120.318062
Faraco, G., Brea, D., Garcia-Bonilla, L., Wang, G., Racchumi, G., Chang, H., et al. (2018). Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 21, 240–249. doi: 10.1038/s41593-017-0059-z
Ferland, D. J., Darios, E. S., Neubig, R. R., Sjögren, B., Truong, N., Torres, R., et al. (2017). Chemerin-induced arterial contraction is G(i)- and calcium-dependent. Vasc. Pharmacol. 88, 30–41. doi: 10.1016/j.vph.2016.11.009
Ferland, D. J., Flood, E. D., Garver, H., Yeh, S. T., Riney, S., Mullick, A. E., et al. (2019). Different blood pressure responses in hypertensive rats following chemerin mRNA inhibition in dietary high fat compared to dietary high-salt conditions. Physiol. Genomics 51, 553–561. doi: 10.1152/physiolgenomics.00050.2019
Ferland, D. J., Seitz, B., Darios, E. S., Thompson, J. M., Yeh, S. Y., Mullick, A. E., et al. (2018). Whole-body but not hepatic knockdown of chemerin by antisense oligonucleotide decreases blood pressure in rats. J. Pharmacol. Exp. Ther. 365, 212–218. doi: 10.1124/jpet.117.245456
Fernández-Riejos, P., Najib, S., Santos-Alvarez, J., Martín-Romero, C., Pérez-Pérez, A., González-Yanes, C., et al. (2010). Role of leptin in the activation of immune cells. Mediat. Inflamm. 2010:568343. doi: 10.1155/2010/568343
Ferri, C., Bellini, C., Desideri, G., Giuliani, E., De Siati, L., Cicogna, S., et al. (1998). Clustering of endothelial markers of vascular damage in human salt-sensitive hypertension: influence of dietary sodium load and depletion. Hypertension 32, 862–868. doi: 10.1161/01.HYP.32.5.862
Fink, G. D., Takeshita, A., Mark, A. L., and Brody, M. J. (1980). Determinants of renal vascular resistance in the dahl strain of genetically hypertensive rat. Hypertension 2, 274–280. doi: 10.1161/01.HYP.2.3.274
Franco, V., and Oparil, S. (2006). Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J. Am. Coll. Nutr. 25, 247s–255s. doi: 10.1080/07315724.2006.10719574
Frisbee, J. C. (2007). Obesity, insulin resistance, and microvessel density. Microcirculation 14, 289–298. doi: 10.1080/10739680701282945
Fu, H., Chen, J. K., Lu, W. J., Jiang, Y. J., Wang, Y. Y., Li, D. J., et al. (2018). Inflammasome-independent NALP3 contributes to high-salt induced endothelial dysfunction. Front. Pharmacol. 9:968. doi: 10.3389/fphar.2018.00968
Fuenmayor, N., Moreira, E., and Cubeddu, L. X. (1998). Salt sensitivity is associated with insulin resistance in essential hypertension. Am. J. Hypertens. 11, 397–402. doi: 10.1016/S0895-7061(97)00490-1
Gainsford, T., Willson, T. A., Metcalf, D., Handman, E., McFarlane, C., Ng, A., et al. (1996). Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. U. S. A. 93, 14564–14568. doi: 10.1073/pnas.93.25.14564
Galletti, F., Strazzullo, P., Ferrara, I., Annuzzi, G., Rivellese, A., Gatto, S., et al. (1997). NaCl sensitivity of essential hypertensive patients is related to insulin resistance. J. Hypertens. 15, 1485–1491. doi: 10.1097/00004872-199715120-00017
Ganguli, M., Tobian, L., and Iwai, J. (1979). Cardiac output and peripheral resistance in strains of rats sensitive and resistant to NaCl hypertension. Hypertension 1, 3–7. doi: 10.1161/01.HYP.1.1.3
Geng, D. F., Jin, D. M., Wu, W., Liang, Y. D., and Wang, J. F. (2013). Angiotensin converting enzyme inhibitors for prevention of new-onset type 2 diabetes mellitus: a meta-analysis of 72,128 patients. Int. J. Cardiol. 167, 2605–2610. doi: 10.1016/j.ijcard.2012.06.125
Giner, V., Coca, A., and de la Sierra, A. (2001). Increased insulin resistance in salt sensitive essential hypertension. J. Hum. Hypertens. 15, 481–485. doi: 10.1038/sj.jhh.1001216
Giner, V., Poch, E., Bragulat, E., Oriola, J., González, D., Coca, A., et al. (2000). Renin-angiotensin system genetic polymorphisms and salt sensitivity in essential hypertension. Hypertension 35, 512–517. doi: 10.1161/01.HYP.35.1.512
Goralski, K. B., McCarthy, T. C., Hanniman, E. A., Zabel, B. A., Butcher, E. C., Parlee, S. D., et al. (2007). Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 282, 28175–28188. doi: 10.1074/jbc.M700793200
Grillo, A., Salvi, L., Coruzzi, P., Salvi, P., and Parati, G. (2019). Sodium intake and hypertension. Nutrients 11:1970. doi: 10.3390/nu11091970
Guo, Q., Mori, T., Jiang, Y., Hu, C., Osaki, Y., Yoneki, Y., et al. (2009). Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague-dawley rats. J. Hypertens. 27, 1664–1671. doi: 10.1097/HJH.0b013e32832c419a
Guyton, A. C. (1991). Blood pressure control--special role of the kidneys and body fluids. Science 252, 1813–1816. doi: 10.1126/science.2063193
Helfer, G., and Wu, Q.-F. (2018). Chemerin: a multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 238, R79–R94. doi: 10.1530/JOE-18-0174
Herkner, H., Klein, N., Joukhadar, C., Lackner, E., Langenberger, H., Frossard, M., et al. (2003). Transcapillary insulin transfer in human skeletal muscle. Eur. J. Clin. Investig. 33, 141–146. doi: 10.1046/j.1365-2362.2003.01106.x
Hookham, M. B., O'Donovan, H. C., Church, R. H., Mercier-Zuber, A., Luzi, L., Curran, S. P., et al. (2013). Insulin receptor substrate-2 is expressed in kidney epithelium and up-regulated in diabetic nephropathy. FEBS J. 280, 3232–3243. doi: 10.1111/febs.12305
Horita, S., Seki, G., Yamada, H., Suzuki, M., Koike, K., and Fujita, T. (2011). Insulin resistance, obesity, hypertension, and renal sodium transport. Int. J. Hypertens. 2011:391762. doi: 10.4061/2011/391762
Iwaki, M., Matsuda, M., Maeda, N., Funahashi, T., Matsuzawa, Y., Makishima, M., et al. (2003). Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 52, 1655–1663. doi: 10.2337/diabetes.52.7.1655
Izquierdo, A. G., Crujeiras, A. B., Casanueva, F. F., and Carreira, M. C. (2019). Leptin, obesity, and leptin resistance: where are we 25 years later? Nutrients 11:2704. doi: 10.3390/nu11112704
Jia, X., Olson, D. J. H., Ross, A. R. S., and Wu, L. (2006). Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 20, 1555–1557. doi: 10.1096/fj.05-5478fje
Jia, X., and Wu, L. (2007). Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 306:133. doi: 10.1007/s11010-007-9563-x
Juan, C. C., Chuang, T. Y., Chang, C. L., Huang, S. W., and Ho, L. T. (2007). Endothelin-1 regulates adiponectin gene expression and secretion in 3T3-L1 adipocytes via distinct signaling pathways. Endocrinology 148, 1835–1842. doi: 10.1210/en.2006-0654
Juan, C.-C., Shen, Y. W., Chien, Y., Lin, Y. J., Chang, S. F., and Ho, L. T. (2004). Insulin infusion induces endothelin-1-dependent hypertension in rats. Am. J. Phys. 287, E948–E954. doi: 10.1152/ajpendo.00536.2003
Kang, B.-Y., Park, K., Kleinhenz, J. M., Murphy, T. C., Sutliff, R. L., Archer, D., et al. (2017). Peroxisome proliferator-activated receptor γ regulates the V-Ets avian erythroblastosis virus E26 oncogene homolog 1/microRNA-27a axis to reduce endothelin-1 and endothelial dysfunction in the sickle cell mouse lung. Am. J. Respir. Cell Mol. Biol. 56, 131–144. doi: 10.1165/rcmb.2016-0166OC
Karczewska-Kupczewska, M., Nikołajuk, A., Stefanowicz, M., Matulewicz, N., Kowalska, I., and Strączkowski, M. (2020). Serum and adipose tissue chemerin is differentially related to insulin sensitivity. Endocr. Connect. 9, 360–369. doi: 10.1530/EC-20-0084
Kennedy, A. J., Yang, P., Read, C., Kuc, R. E., Yang, L., Taylor, E. J. A., et al. (2016). Chemerin elicits potent constrictor actions via chemokine-like receptor 1 (CMKLR1), not G-protein-coupled receptor 1 (GPR1), in human and rat vasculature. J. Am. Heart Assoc. 5:e004421. doi: 10.1161/JAHA.116.004421
Ketsawatsomkron, P., Lorca, R. A., Keen, H. L., Weatherford, E. T., Liu, X., Pelham, C. J., et al. (2012). PPARγ; regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ. Res. 111, 1446–1458. doi: 10.1161/CIRCRESAHA.112.271577
Kido, Y., Burks, D. J., Withers, D., Bruning, J. C., Kahn, C. R., White, M. F., et al. (2000). Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105, 199–205. doi: 10.1172/JCI7917
Kirabo, A., Fontana, V., de Faria, A. P. C., Loperena, R., Galindo, C. L., Wu, J., et al. (2014). DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Invest. 124, 4642–4656. doi: 10.1172/JCI74084
Kleinhenz, J. M., Kleinhenz, D. J., You, S., Ritzenthaler, J. D., Hansen, J. M., Archer, D. R., et al. (2009). Disruption of endothelial peroxisome proliferator-activated receptor-γ reduces vascular nitric oxide production. Am. J. Physiol. Heart Circ. Physiol. 297, H1647–H1654. doi: 10.1152/ajpheart.00148.2009
Klisic, J., Hu, M. C., Nief, V., Reyes, L., Fuster, D., Moe, O. W., et al. (2002). Insulin activates Na(+)/H(+) exchanger 3: biphasic response and glucocorticoid dependence. Am. J. Physiol. Ren. Physiol. 283, F532–F539. doi: 10.1152/ajprenal.00365.2001
Ko, S.-H., Cao, W., and Liu, Z. (2010). Hypertension management and microvascular insulin resistance in diabetes. Curr. Hypertens. Rep. 12, 243–251. doi: 10.1007/s11906-010-0114-6
Komajda, M., Curtis, P., Hanefeld, M., Beck-Nielsen, H., Pocock, S. J., Zambanini, A., et al. (2008). Effect of the addition of rosiglitazone to metformin or sulfonylureas versus metformin/sulfonylurea combination therapy on ambulatory blood pressure in people with type 2 diabetes: a randomized controlled trial (the RECORD study). Cardiovasc. Diabetol. 7:10. doi: 10.1186/1475-2840-7-10
Kopp, C., Linz, P., Dahlmann, A., Hammon, M., Jantsch, J., Müller, D. N., et al. (2013). 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 61, 635–640. doi: 10.1161/HYPERTENSIONAHA.111.00566
Laffer, C. L., Scott, R. C., Titze, J. M., Luft, F. C., and Elijovich, F. (2016). Hemodynamics and salt-and-water balance link sodium storage and vascular dysfunction in salt-sensitive subjects. Hypertension 68, 195–203. doi: 10.1161/HYPERTENSIONAHA.116.07289
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Lanaspa, M. A., Kuwabara, M., Andres-Hernando, A., Li, N., Cicerchi, C., Jensen, T., et al. (2018). High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc. Natl. Acad. Sci. U. S. A. 115, 3138–3143. doi: 10.1073/pnas.1713837115
Li, Q.-Z., Deng, Q., Li, J. Q., Yi, G. H., and Zhao, S. P. (2005). Valsartan reduces interleukin-1β secretion by peripheral blood mononuclear cells in patients with essential hypertension. Clin. Chim. Acta 355, 131–136. doi: 10.1016/j.cccn.2004.12.006
Li, Y., Lu, Y., Lin, S. H., Li, N., Han, Y., Huang, Q., et al. (2021). Insulin signaling establishes a developmental trajectory of adipose regulatory T cells. Nat. Immunol. 22, 1175–1185. doi: 10.1038/s41590-021-01010-3
Liu, Z., Peng, J., Lu, F., Zhao, Y., Wang, S., Sun, S., et al. (2013). Salt loading and potassium supplementation: effects on ambulatory arterial stiffness index and endothelin-1 levels in normotensive and mild hypertensive patients. J. Clin. Hypertens. 15, 485–496. doi: 10.1111/jch.12109
Lluch, M. M., de la Sierra, A., Poch, E., Coca, A., Aguilera, M. T., Compte, M., et al. (1996). Erythrocyte sodium transport, intraplatelet pH, and calcium concentration in salt-sensitive hypertension. Hypertension 27, 919–925. doi: 10.1161/01.HYP.27.4.919
Lobato, N. S., Neves, K. B., Filgueira, F. P., Fortes, Z. B., Carvalho, M. H. C., Webb, R. C., et al. (2012). The adipokine chemerin augments vascular reactivity to contractile stimuli via activation of the MEK-ERK1/2 pathway. Life Sci. 91, 600–606. doi: 10.1016/j.lfs.2012.04.013
Loffing, J., and Korbmacher, C. (2009). Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch. 458, 111–135. doi: 10.1007/s00424-009-0656-0
Machnik, A., Dahlmann, A., Kopp, C., Goss, J., Wagner, H., van Rooijen, N., et al. (2010). Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension 55, 755–761. doi: 10.1161/HYPERTENSIONAHA.109.143339
Machnik, A., Neuhofer, W., Jantsch, J., Dahlmann, A., Tammela, T., Machura, K., et al. (2009). Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 15, 545–552. doi: 10.1038/nm.1960
Madhur, M. S., Lob, H. E., McCann, L. A., Iwakura, Y., Blinder, Y., Guzik, T. J., et al. (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55, 500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094
Mantovani, A., Sozzani, S., Locati, M., Allavena, P., and Sica, A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555. doi: 10.1016/S1471-4906(02)02302-5
Mather, K. J., Steinberg, H. O., and Baron, A. D. (2013). Insulin resistance in the vasculature. J. Clin. Invest. 123, 1003–1004. doi: 10.1172/JCI67166
McDonnell, W. J., Koethe, J. R., Mallal, S. A., Pilkinton, M. A., Kirabo, A., Ameka, M. K., et al. (2018). High CD8 T-cell receptor clonality and altered CDR3 properties are associated with elevated isolevuglandins in adipose tissue during diet-induced obesity. Diabetes 67, 2361–2376. doi: 10.2337/db18-0040
McMaster, W. G., Kirabo, A., Madhur, M. S., and Harrison, D. G. (2015). Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 116, 1022–1033. doi: 10.1161/CIRCRESAHA.116.303697
Miyazaki, Y., Shimamoto, K., Ise, T., Shiiki, M., Higashiura, K., Hirata, A., et al. (1996). Effects of hyperinsulinaemia on renal function and the pressor system in insulin-resistant obese adolescents. Clin. Exp. Pharmacol. Physiol. 23, 287–290. doi: 10.1111/j.1440-1681.1996.tb02825.x
Morimoto, A., Uzu, T., Fujii, T., Nishimura, M., Kuroda, S., Nakamura, S., et al. (1997). Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet 350, 1734–1737. doi: 10.1016/S0140-6736(97)05189-1
Motoki, T., Kurobe, H., Hirata, J., Nakayama, T., Kinoshita, H., Rocco, K. A., et al. (2015). PPAR-γ agonist attenuates inflammation in aortic aneurysm patients. Gen. Thorac. Cardiovasc. Surg. 63, 565–571. doi: 10.1007/s11748-015-0576-1
Mukohda, M., Lu, K. T., Guo, D. F., Wu, J., Keen, H. L., Liu, X., et al. (2017). Hypertension-causing mutation in peroxisome proliferator-activated receptor γ impairs nuclear export of nuclear factor-κB p65 in vascular smooth muscle. Hypertension 70, 174–182. doi: 10.1161/HYPERTENSIONAHA.117.09276
Mukohda, M., Stump, M., Ketsawatsomkron, P., Hu, C., Quelle, F. W., and Sigmund, C. D. (2016). Endothelial PPAR-γ provides vascular protection from IL-1β-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 310, H39–H48. doi: 10.1152/ajpheart.00490.2015
Muniyappa, R., Chen, H., Montagnani, M., Sherman, A., and Quon, M. J. (2020). Endothelial dysfunction due to selective insulin resistance in vascular endothelium: insights from mechanistic modeling. Am. J. Phys. 319, E629–E646. doi: 10.1152/ajpendo.00247.2020
Nagao, S., and Yamaguchi, T. (2012). PPAR-γ agonists in polycystic kidney disease with frequent development of cardiovascular disorders. Curr. Mol. Pharmacol. 5, 292–300. doi: 10.2174/1874467211205020292
Nakamura, M., Yamazaki, O., Shirai, A., Horita, S., Satoh, N., Suzuki, M., et al. (2015). Preserved Na/HCO3 cotransporter sensitivity to insulin may promote hypertension in metabolic syndrome. Kidney Int. 87, 535–542. doi: 10.1038/ki.2014.351
Nikpey, E., Karlsen, T. V., Rakova, N., Titze, J. M., Tenstad, O., and Wiig, H. (2017). High-salt diet causes osmotic gradients and hyperosmolality in skin without affecting interstitial fluid and lymph. Hypertension 69, 660–668. doi: 10.1161/HYPERTENSIONAHA.116.08539
Nishimura, S., Manabe, I., Nagasaki, M., Eto, K., Yamashita, H., Ohsugi, M., et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15, 914–920. doi: 10.1038/nm.1964
Nolan, J. J., Ludvik, B., Beerdsen, P., Joyce, M., and Olefsky, J. (1994). Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 331, 1188–1193. doi: 10.1056/NEJM199411033311803
Odegaard, J. I., Ricardo-Gonzalez, R. R., Goforth, M. H., Morel, C. R., Subramanian, V., Mukundan, L., et al. (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. doi: 10.1038/nature05894
Olefsky, J. M., and Glass, C. K. (2010). Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246. doi: 10.1146/annurev-physiol-021909-135846
Orliaguet, L., Dalmas, E., Drareni, K., Venteclef, N., and Alzaid, F. (2020). Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front. Endocrinol. 11:62. doi: 10.3389/fendo.2020.00062
Patel, P. S., Buras, E. D., and Balasubramanyam, A. (2013). The role of the immune system in obesity and insulin resistance. J. Obes. 2013:616193. doi: 10.1155/2013/616193
Paulsson, G., Zhou, X., Törnquist, E., and Hansson, G. K. (2000). Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 20, 10–17. doi: 10.1161/01.ATV.20.1.10
Pavlov, T. S., Ilatovskaya, D. V., Levchenko, V., Li, L., Ecelbarger, C. M., and Staruschenko, A. (2013). Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J. 27, 2723–2732. doi: 10.1096/fj.12-223792
Pelham, C. J., Ketsawatsomkron, P., Groh, S., Grobe, J. L., de Lange, W. J., Ibeawuchi, S. R. C., et al. (2012). Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/rho-kinase. Cell Metab. 16, 462–472. doi: 10.1016/j.cmet.2012.08.011
Potenza, M. A., Marasciulo, F. L., Tarquinio, M., Quon, M. J., and Montagnani, M. (2006). Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes 55, 3594–3603. doi: 10.2337/db06-0667
Qayyum, R., and Adomaityte, J. (2006). A meta-analysis of the effect of thiazolidinediones on blood pressure. J. Clin. Hypertens. 8, 19–28. doi: 10.1111/j.1524-6175.2005.04784.x
Qi, N., Rapp, J. P., Brand, P. H., Metting, P. J., and Britton, S. L. (1999). Body fluid expansion is not essential for salt-induced hypertension in SS/Jr rats. Am. J. Phys. 277, R1392–R1400. doi: 10.1152/ajpregu.1999.277.5.R1392
Rabkin, S. W. (2009). The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat. Rev. Cardiol. 6, 192–199. doi: 10.1038/ncpcardio1453
Radin, M. J., Holycross, B. J., Hoepf, T. M., and McCune, S. A. (2003). Increased salt sensitivity secondary to leptin resistance in SHHF rats is mediated by endothelin. Mol. Cell. Biochem. 242, 57–63. doi: 10.1023/A:1021181527060
Raji, A., Seely, E. W., Bekins, S. A., Williams, G. H., and Simonson, D. C. (2003). Rosiglitazone improves insulin sensitivity and lowers blood pressure in hypertensive patients. Diabetes Care 26, 172–178. doi: 10.2337/diacare.26.1.172
Reaven, G. M. (2003). Insulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular disease. J. Clin. Endocrinol. Metab. 88, 2399–2403. doi: 10.1210/jc.2003-030087
Riboulet-Chavey, A., Pierron, A., Durand, I., Murdaca, J., Giudicelli, J., and Van Obberghen, E. (2006). Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 55, 1289–1299. doi: 10.2337/db05-0857
Rivera-Gonzalez, O. J., Kasztan, M., Johnston, J. G., Hyndman, K. A., and Speed, J. S. (2020). Loss of endothelin type B receptor function improves insulin sensitivity in rats. Can. J. Physiol. Pharmacol. 98, 604–610. doi: 10.1139/cjpp-2019-0666
Rodrigues, T., Matafome, P., Sereno, J., Almeida, J., Castelhano, J., Gamas, L., et al. (2017). Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep. 7:1698. doi: 10.1038/s41598-017-01730-3
Rossitto, G., Mary, S., Chen, J. Y., Boder, P., Chew, K. S., Neves, K. B., et al. (2020). Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nat. Commun. 11:4222. doi: 10.1038/s41467-020-17820-2
Sahinoz, M., Elijovich, F., Laffer, C., Pitzer, A., Ikizler, T., and Kirabo, A. (2021). Reduction in monocyte isolevuglandins associated with high interstitial sodium mirrors salt-sensitivity of blood pressure in patients with essential hypertension. FASEB J. 35, doi: 10.1096/fasebj.2021.35.S1.02022
Sahinoz, M., Tintara, S., Deger, S. M., Alsouqi, A., Crescenzi, R. L., Mambungu, C., et al. (2020). Tissue sodium stores in peritoneal dialysis and hemodialysis patients determined by 23-sodium magnetic resonance imaging. Nephrol. Dial. Transplant. 36, 1307–1317. doi: 10.1093/ndt/gfaa350
Sartori-Valinotti, J. C., Venegas-Pont, M. R., Lamarca, B. B., Romero, D. G., Yanes, L. L., Racusen, L. C., et al. (2010). Rosiglitazone reduces blood pressure in female Dahl salt-sensitive rats. Steroids 75, 794–799. doi: 10.1016/j.steroids.2009.10.010
Satoh, H., Tsukamoto, K., Hashimoto, Y., Hashimoto, N., Togo, M., Hara, M., et al. (1999). Thiazolidinediones suppress endothelin-1 secretion from bovine vascular endothelial cells: a new possible role of PPARgamma on vascular endothelial function. Biochem. Biophys. Res. Commun. 254, 757–763. doi: 10.1006/bbrc.1998.0126
Schmidlin, O., Sebastian, A. F., and Morris, R. C. (2007). What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension 49, 1032–1039. doi: 10.1161/HYPERTENSIONAHA.106.084640
Schunk, S. J., Kleber, M. E., März, W., Pang, S., Zewinger, S., Triem, S., et al. (2021). Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur. Heart J. 42, 1742–1756. doi: 10.1093/eurheartj/ehab107
Sechi, L. A. (1999). Mechanisms of insulin resistance in rat models of hypertension and their relationships with salt sensitivity. J. Hypertens. 17, 1229–1237. doi: 10.1097/00004872-199917090-00001
Sell, H., Laurencikiene, J., Taube, A., Eckardt, K., Cramer, A., Horrighs, A., et al. (2009). Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes 58, 2731–2740. doi: 10.2337/db09-0277
Sharma, A. M., Ruland, K., Spies, K. P., and Distler, A. (1991). Salt sensitivity in young normotensive subjects is associated with a hyperinsulinemic response to oral glucose. J. Hypertens. 9, 329–335. doi: 10.1097/00004872-199104000-00004
Shemyakin, A., Böhm, F., Wagner, H., Efendic, S., Båvenholm, P., and Pernow, J. (2006). Enhanced endothelium-dependent vasodilatation by dual endothelin receptor blockade in individuals with insulin resistance. J. Cardiovasc. Pharmacol. 47, 385–390. doi: 10.1097/01.fjc.0000210070.47205.16
Sherajee, S. J., Fujita, Y., Rafiq, K., Nakano, D., Mori, H., Masaki, T., et al. (2012). Aldosterone induces vascular insulin resistance by increasing insulin-like growth factor-1 receptor and hybrid receptor. Arterioscler. Thromb. Vasc. Biol. 32, 257–263. doi: 10.1161/ATVBAHA.111.240697
Silva, T. M. D., Hu, C., Kinzenbaw, D. A., Modrick, M. L., Sigmund, C. D., and Faraci, F. M. (2017). Genetic interference with endothelial PPAR-γ (peroxisome proliferator-activated receptor-γ) augments effects of angiotensin II while impairing responses to angiotensin 1-7. Hypertension 70, 559–565. doi: 10.1161/HYPERTENSIONAHA.117.09358
Simchon, S., Manger, W. M., Carlin, R. D., Peeters, L. L., Rodriguez, J., Batista, D., et al. (1989). Salt-induced hypertension in dahl salt-sensitive rats. Hemodynamics and renal responses. Hypertension 13, 612–621. doi: 10.1161/01.hyp.13.6.612
Simonds, S. E., Pryor, J. T., Ravussin, E., Greenway, F. L., Dileone, R., Allen, A. M., et al. (2014). Leptin mediates the increase in blood pressure associated with obesity. Cell 159, 1404–1416. doi: 10.1016/j.cell.2014.10.058
Sogawa, Y., Chen, J. K., Lu, W. J., Jiang, J., Wang, Y. Y., Li, D. J., et al. (2018). The eNOS-NO pathway attenuates kidney dysfunction via suppression of inflammasome activation in aldosterone-induced renal injury model mice. PLoS One 13:e0203823. doi: 10.1371/journal.pone.0203823
Soleimani, M. (2015). Insulin resistance and hypertension: new insights. Kidney Int. 87, 497–499. doi: 10.1038/ki.2014.392
Sorriento, D., and Iaccarino, G. (2019). Inflammation and cardiovascular diseases: the most recent findings. Int. J. Mol. Sci. 20:3879. doi: 10.3390/ijms20163879
Speed, J. S., Heimlich, J. B., Hyndman, K. A., Fox, B. M., Patel, V., Yanagisawa, M., et al. (2015). Endothelin-1 as a master regulator of whole-body Na+ homeostasis. FASEB J. 29, 4937–4944. doi: 10.1096/fj.15-276584
Steinberg, H. O., Chaker, H., Leaming, R., Johnson, A., Brechtel, G., and Baron, A. D. (1996). Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J. Clin. Invest. 97, 2601–2610. doi: 10.1172/JCI118709
Sullivan, J. M., and Ratts, T. E. (1983). Hemodynamic mechanisms of adaptation to chronic high sodium intake in normal humans. Hypertension 5, 814–820. doi: 10.1161/01.HYP.5.6.814
Sun, H.-J., Ren, X. S., Xiong, X. Q., Chen, Y. Z., Zhao, M. Z., Wang, J. J., et al. (2017). NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 8:e3074. doi: 10.1038/cddis.2017.470
Suzuki, M., Kimura, Y., Tsushima, M., and Harano, Y. (2000). Association of insulin resistance with salt sensitivity and nocturnal fall of blood pressure. Hypertension 35, 864–868. doi: 10.1161/01.HYP.35.4.864
Takahashi, M., Takahashi, Y., Takahashi, K., Zolotaryov, F. N., Hong, K. S., Kitazawa, R., et al. (2008). Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett. 582, 573–578. doi: 10.1016/j.febslet.2008.01.023
Titze, J., Shakibaei, M., Schafflhuber, M., Schulze-Tanzil, G., Porst, M., Schwind, K. H., et al. (2004). Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 287, H203–H208. doi: 10.1152/ajpheart.01237.2003
Tomaru, T., Steger, D. J., Lefterova, M. I., Schupp, M., and Lazar, M. A. (2009). Adipocyte-specific expression of murine resistin is mediated by synergism between peroxisome proliferator-activated receptor gamma and CCAAT/enhancer-binding proteins. J. Biol. Chem. 284, 6116–6125. doi: 10.1074/jbc.M808407200
Tontonoz, P., Nagy, L., Alvarez, J. G., Thomazy, V. A., and Evans, R. M. (1998). PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93, 241–252. doi: 10.1016/S0092-8674(00)81575-5
Trott, D. W., Thabet, S. R., Kirabo, A., Saleh, M. A., Itani, H., Norlander, A. E., et al. (2014). Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 64, 1108–1115. doi: 10.1161/HYPERTENSIONAHA.114.04147
Tsai, Y. H., Ohkita, M., and Gariepy, C. E. (2006). Chronic high-sodium diet increases aortic wall endothelin-1 expression in a blood pressure-independent fashion in rats. Exp. Biol. Med. 231, 813–817. doi: 10.3181/00379727-231-2310813
Tsimaratos, M., Roger, F., Chabardès, D., Mordasini, D., Hasler, U., Doucet, A., et al. (2003). C-peptide stimulates Na+, K+-ATPase activity via PKC alpha in rat medullary thick ascending limb. Diabetologia 46, 124–131. doi: 10.1007/s00125-002-0996-1
Unoki, H., Bujo, H., Yamagishi, S. I., Takeuchi, M., Imaizumi, T., and Saito, Y. (2007). Advanced glycation end products attenuate cellular insulin sensitivity by increasing the generation of intracellular reactive oxygen species in adipocytes. Diabetes Res. Clin. Pract. 76, 236–244. doi: 10.1016/j.diabres.2006.09.016
Vandanmagsar, B., Youm, Y. H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188. doi: 10.1038/nm.2279
Van Tassell, B. W., Toldo, S., Mezzaroma, E., and Abbate, A. (2013). Targeting interleukin-1 in heart disease. Circulation 128, 1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199
Villarreal-Calderón, J. R., Cuéllar, R. X., Ramos-González, M. R., Rubio-Infante, N., Castillo, E. C., Elizondo-Montemayor, L., et al. (2019). Interplay between the adaptive immune system and insulin resistance in weight loss induced by bariatric surgery. Oxid. Med. Cell. Longev. 2019:3940739. doi: 10.1155/2019/3940739
Vincent, M. A., Clerk, L. H., Lindner, J. R., Klibanov, A. L., Clark, M. G., Rattigan, S., et al. (2004). Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53, 1418–1423. doi: 10.2337/diabetes.53.6.1418
Wahli, W., and Michalik, L. (2012). PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 23, 351–363. doi: 10.1016/j.tem.2012.05.001
Walker, A. B., Chattington, P. D., Buckingham, R. E., and Williams, G. (1999). The thiazolidinedione rosiglitazone (BRL-49653) lowers blood pressure and protects against impairment of endothelial function in Zucker fatty rats. Diabetes 48, 1448–1453. doi: 10.2337/diabetes.48.7.1448
Wang, X., Jia, X., Chang, T., Desai, K., and Wu, L. (2008). Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J. Hypertens. 26, 765–772. doi: 10.1097/HJH.0b013e3282f4a13c
Weinberger, M. H., Fineberg, N. S., Fineberg, S. E., and Weinberger, M. (2001). Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension 37, 429–432. doi: 10.1161/01.HYP.37.2.429
Weinberger, M. H., Miller, J. Z., Luft, F. C., Grim, C. E., and Fineberg, N. S. (1986). Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 8, 127–134. doi: 10.1161/01.hyp.8.6_pt_2.ii127
Wenstedt, E. F. E., Oppelaar, J. J., Besseling, S., Rorije, N. M. G., Engberink, R. H. G. O., Oosterhof, A., et al. (2021). Distinct osmoregulatory responses to sodium loading in patients with altered glycosaminoglycan structure: a randomized cross-over trial. J. Transl. Med. 19:38. doi: 10.1186/s12967-021-02700-0
Wiig, H., Luft, F. C., and Titze, J. M. (2018). The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol. 222:e13006. doi: 10.1111/apha.13006
Wiig, H., Schröder, A., Neuhofer, W., Jantsch, J., Kopp, C., Karlsen, T. V., et al. (2013). Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Invest. 123, 2803–2815. doi: 10.1172/JCI60113
Winer, S., and Winer, D. A. (2012). The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol. Cell Biol. 90, 755–762. doi: 10.1038/icb.2011.110
Wolpert, H. A., Steen, S. N., Istfan, N. W., and Simonson, D. C. (1993). Insulin modulates circulating endothelin-1 levels in humans. Metabolism 42, 1027–1030. doi: 10.1016/0026-0495(93)90018-J
Wu, J., Agbor, L. N., Fang, S., Mukohda, M., Nair, A. R., Nakagawa, P., et al. (2021). Failure to vasodilate in response to salt loading blunts renal blood flow and causes salt-sensitive hypertension. Cardiovasc. Res. 117, 308–319. doi: 10.1093/cvr/cvaa147
Xu, H., Barnes, G. T., Yang, Q., Tan, G., Yang, D., Chou, C. J., et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830. doi: 10.1172/JCI200319451
Xu, Y., Romero, R., Miller, D., Kadam, L., Mial, T. N., Plazyo, O., et al. (2016). An M1-like macrophage polarization in decidual tissue during spontaneous preterm labor that is attenuated by rosiglitazone treatment. J. Immunol. 196, 2476–2491. doi: 10.4049/jimmunol.1502055
Yang, Y. J., Hope, I. D., Ader, A., and Bergman, R. N. (1989). Insulin transport across capillaries is rate limiting for insulin action in dogs. J. Clin. Invest. 84, 1620–1628. doi: 10.1172/JCI114339
Yang, H., Youm, Y. H., Vandanmagsar, B., Ravussin, A., Gimble, J. F., Greenway, F., et al. (2010). Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J. Immunol. 185, 1836–1845. doi: 10.4049/jimmunol.1000021
Yatabe, M. S., Yatabe, J., Yoneda, M., Watanabe, T., Otsuki, M., Felder, R. A., et al. (2010). Salt sensitivity is associated with insulin resistance, sympathetic overactivity, and decreased suppression of circulating renin activity in lean patients with essential hypertension. Am. J. Clin. Nutr. 92, 77–82. doi: 10.3945/ajcn.2009.29028
Yki-Järvinen, H., and Utriainen, T. (1998). Insulin-induced vasodilatation: physiology or pharmacology? Diabetologia 41, 369–379. doi: 10.1007/s001250050919
Yoshii, H., Onuma, T., Yamazaki, T., Watada, H., Matsuhisa, M., Matsumoto, M., et al. (2014). Effects of pioglitazone on macrovascular events in patients with type 2 diabetes mellitus at high risk of stroke: the PROFIT-J study. J. Atheroscler. Thromb. 21, 563–573. doi: 10.5551/jat.21626
Zambom, F. F. F., Oliveira, K. C., Foresto-Neto, O., Faustino, V. D., Ávila, V. F., Albino, A. H., et al. (2019). Pathogenic role of innate immunity in a model of chronic NO inhibition associated with salt overload. Am. J. Physiol. Ren. Physiol. 317, F1058–F1067. doi: 10.1152/ajprenal.00251.2019
Zanchi, A., Chiolero, A., Maillard, M., Nussberger, J., Brunner, H. R., and Burnier, M. (2004). Effects of the peroxisomal proliferator-activated receptor-γ agonist pioglitazone on renal and hormonal responses to salt in healthy men. J. Clin. Endocrinol. Metab. 89, 1140–1145. doi: 10.1210/jc.2003-031526
Zanchi, A., Maillard, M., Jornayvaz, F. R., Vinciguerra, M., Deleaval, P., Nussberger, J., et al. (2010). Effects of the peroxisome proliferator-activated receptor (PPAR)-gamma agonist pioglitazone on renal and hormonal responses to salt in diabetic and hypertensive individuals. Diabetologia 53, 1568–1575. doi: 10.1007/s00125-010-1756-2
Zaporowska-Stachowiak, I., Gluszek, J., and Chodera, A. (1997). Comparison of the serum insulin and endothelin level in patients with essential and renovascular hypertension. J. Hum. Hypertens. 11, 795–800. doi: 10.1038/sj.jhh.1000552
Zavaroni, I., Coruzzi, P., Bonini, L., Mossini, G. L., Musiari, L., Gasparini, P., et al. (1995). Association between salt sensitivity and insulin concentrations in patients with hypertension. Am. J. Hypertens. 8, 855–858. doi: 10.1016/0895-7061(95)00152-F
Zhang, R., Liu, S., Guo, B., Chang, L., and Li, Y. (2014). Chemerin induces insulin resistance in rat cardiomyocytes in part through the ERK1/2 signaling pathway. Pharmacology 94, 259–264. doi: 10.1159/000369171
Zhang, M., Zhang, J., Li, L., Wang, Q., and Feng, L. (2019). Association between peroxisome proliferator-activated receptor γ-2 gene Pro12Ala polymorphisms and risk of hypertension: an updated meta-analysis. Biosci. Rep. 39:BSR20190022. doi: 10.1042/BSR20190022
Zhao, L., Fu, Z., Wu, J., Aylor, K. W., Barrett, E. J., Cao, W., et al. (2015). Inflammation-induced microvascular insulin resistance is an early event in diet-induced obesity. Clin. Sci. 129, 1025–1036. doi: 10.1042/CS20150143
Zhou, M. S., Schulman, I. H., and Raij, L. (2009). Role of angiotensin II and oxidative stress in vascular insulin resistance linked to hypertension. Am. J. Physiol. Heart Circ. Physiol. 296, H833–H839. doi: 10.1152/ajpheart.01096.2008
Zhou, M. S., Schulman, I. H., and Raij, L. (2010). Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: role of nuclear factor kappa B activation. J. Hypertens. 28, 527–535. doi: 10.1097/HJH.0b013e3283340da8
Zhou, M.-S., Wang, A., and Yu, H. (2014). Link between insulin resistance and hypertension: what is the evidence from evolutionary biology? Diabetol. Metab. Syndr. 6:12. doi: 10.1186/1758-5996-6-12
Zinn, A., Felson, S., Fisher, E., and Schwartzbard, A. (2008). Reassessing the cardiovascular risks and benefits of thiazolidinediones. Clin. Cardiol. 31, 397–403. doi: 10.1002/clc.20312
Keywords: salt-sensitivity, blood pressure, insulin resistance, immune activation, PPARγ
Citation: Ertuglu LA, Elijovich F, Laffer CL and Kirabo A (2021) Salt-Sensitivity of Blood Pressure and Insulin Resistance. Front. Physiol. 12:793924. doi: 10.3389/fphys.2021.793924
Edited by:
Jing Wu, Medical College of Wisconsin, United StatesReviewed by:
Thiago Bruder Do Nascimento, University of Pittsburgh, United StatesMonica M. Santisteban, Cornell University, United States
Tao Yang, University of Toledo, United States
Copyright © 2021 Ertuglu, Elijovich, Laffer and Kirabo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annet Kirabo, YW5uZXQua2lyYWJvQHZ1bWMub3Jn