 Rudolf Lucas1,2,3*Yalda Hadizamani4,5Perenlei Enkhbaatar6Gabor Csanyi1,2Robert W. Caldwell2Harald Hundsberger7,8Supriya Sridhar1Alice Ann Lever1,3Martina Hudel9Dipankar Ash1Masuko Ushio-Fukai1,3Tohru Fukai1,2,10Trinad Chakraborty9Alexander Verin1,3Douglas C. Eaton11Maritza Romero1,2,12Jürg Hamacher4,5,13,14*
Rudolf Lucas1,2,3*Yalda Hadizamani4,5Perenlei Enkhbaatar6Gabor Csanyi1,2Robert W. Caldwell2Harald Hundsberger7,8Supriya Sridhar1Alice Ann Lever1,3Martina Hudel9Dipankar Ash1Masuko Ushio-Fukai1,3Tohru Fukai1,2,10Trinad Chakraborty9Alexander Verin1,3Douglas C. Eaton11Maritza Romero1,2,12Jürg Hamacher4,5,13,14*
- 1Vascular Biology Center, Augusta University, Augusta, GA, United States
- 2Department of Pharmacology and Toxicology, Augusta University, Augusta, GA, United States
- 3Department of Medicine, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 4Lungen-und Atmungsstiftung Bern, Bern, Switzerland
- 5Pneumology, Clinic for General Internal Medicine, Lindenhofspital Bern, Bern, Switzerland
- 6Department of Anesthesiology, University of Texas Medical Branch, Galveston, TX, United States
- 7Department of Medical Biotechnology, University of Applied Sciences, Krems, Austria
- 8Department of Dermatology, University Hospital of the Paracelsus Medical University, Salzburg, Austria
- 9Institute for Medical Microbiology, Justus-Liebig University, Giessen, Germany
- 10Charlie Norwood Veterans Affairs Medical Center, Augusta, GA, United States
- 11Department of Medicine, School of Medicine, Emory University, Atlanta, GA, United States
- 12Department of Anesthesiology and Perioperative Medicine, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 13Medical Clinic V-Pneumology, Allergology, Intensive Care Medicine, and Environmental Medicine, Faculty of Medicine, University Medical Centre of the Saarland, Saarland University, Homburg, Germany
- 14Institute for Clinical & Experimental Surgery, Faculty of Medicine, Saarland University, Homburg, Germany
Alveolar-capillary leak is a hallmark of the acute respiratory distress syndrome (ARDS), a potentially lethal complication of severe sepsis, trauma and pneumonia, including COVID-19. Apart from barrier dysfunction, ARDS is characterized by hyper-inflammation and impaired alveolar fluid clearance (AFC), which foster the development of pulmonary permeability edema and hamper gas exchange. Tumor Necrosis Factor (TNF) is an evolutionarily conserved pleiotropic cytokine, involved in host immune defense against pathogens and cancer. TNF exists in both membrane-bound and soluble form and its mainly -but not exclusively- pro-inflammatory and cytolytic actions are mediated by partially overlapping TNFR1 and TNFR2 binding sites situated at the interface between neighboring subunits in the homo-trimer. Whereas TNFR1 signaling can mediate hyper-inflammation and impaired barrier function and AFC in the lungs, ligand stimulation of TNFR2 can protect from ventilation-induced lung injury. Spatially distinct from the TNFR binding sites, TNF harbors within its structure a lectin-like domain that rather protects lung function in ARDS. The lectin-like domain of TNF -mimicked by the 17 residue TIP peptide- represents a physiological mediator of alveolar-capillary barrier protection. and increases AFC in both hydrostatic and permeability pulmonary edema animal models. The TIP peptide directly activates the epithelial sodium channel (ENaC) -a key mediator of fluid and blood pressure control- upon binding to its α subunit, which is also a part of the non-selective cation channel (NSC). Activity of the lectin-like domain of TNF is preserved in complexes between TNF and its soluble TNFRs and can be physiologically relevant in pneumonia. Antibody- and soluble TNFR-based therapeutic strategies show considerable success in diseases such as rheumatoid arthritis, psoriasis and inflammatory bowel disease, but their chronic use can increase susceptibility to infection. Since the lectin-like domain of TNF does not interfere with TNF’s anti-bacterial actions, while exerting protective actions in the alveolar-capillary compartments, it is currently evaluated in clinical trials in ARDS and COVID-19. A more comprehensive knowledge of the precise role of the TNFR binding sites versus the lectin-like domain of TNF in lung injury, tissue hypoxia, repair and remodeling may foster the development of novel therapeutics for ARDS.
TNF via TNF Receptors Induces Cell Death or Inflammation
About a century ago the New York surgeon William Coley observes that vaccination of cancer patients with a mixture of bacterial toxins (Coley’s toxins) induces tumor remission (Coley, 1910). Six decades later the group of Dr. Lloyd Old identifies an LPS-induced protein with profound hemorrhagic necrotic activity against tumors (mainly mediated by apoptosis in tumor-associated vasculature), as the active principle of Coley’s toxins and named it Tumor Necrosis Factor (TNF) (Carswell et al., 1975). As such, TNF was discovered at the interface between infection and cancer. Systemic treatment with high bolus doses of TNF in cancer patients is not possible, due to severe hypotension and hepatotoxicity [reviewed in Brouckaert et al. (1992)]. This problem can be partially circumvented in patients with melanoma and soft tissue sarcoma by local administration of TNF in a closed circuit, in combination with melphalan and Interferon-γ (Lejeune et al., 2006; Verhoef et al., 2007).
In the 1980s, the TNF gene is cloned by several groups in Belgium, Japan and the United States (Fransen et al., 1985; Pennica et al., 1985; Shirai et al., 1985) and is found to encode a protein that assembles into a homo-trimeric cytokine, harboring three receptor binding sites which are situated at the interface between neighboring subunits (Bodmer et al., 2002). TNF is expressed in two alternative forms (Horiuchi et al., 2010; Wajant and Beilhack, 2019) - as a surface membrane-anchored precursor, named transmembrane TNF (mTNF) (Kriegler et al., 1988), which consists of 233 residues (26 kDa), and as a soluble form (sTNF), composed of 157 amino acid residues (17 kDa), secreted upon the action of the TNF-Alpha Converting Enzyme [TACE, a.k.a A Disintegrin And Metalloprotease 17 (ADAM17)] on transmembrane TNF (Horiuchi et al., 2010; Wajant and Beilhack, 2019). Mice deficient in the natural inhibitor of TACE -Tissue Inhibitor of Metalloproteinases 3 (TIMP3)- display significantly elevated levels of TNF and severe liver inflammation (Black, 2004; Mohammed et al., 2004), highlighting the physiological importance of sTNF as a pro-inflammatory cytokine.
Tumor necrosis factor is a key mediator for the activation and recruitment of inflammatory cells, including polymorphonuclear neutrophils (PMNs) and macrophages, to sites of infection, and is as such involved in infection control, tissue repair and healing (Gardam et al., 2003). However, when generation of the cytokine is too high, this can promote organ injury.
Tumor necrosis factor expression is induced upon contact with pathogen- or danger-associated molecular patterns (PAMPs and DAMPs) in immune cells, as well as in numerous types of non-immune cells, including endothelial and epithelial cells, neurons, fibroblasts, microglia and cardiomyocytes (Medler and Wajant, 2019). As summarized in Figure 1, TNF signals through two distinct membrane-associated receptors - TNF receptor 1 (TNFR1), which contains a death domain and TNF receptor 2 (TNFR2), which lacks a death domain (Wajant and Beilhack, 2019). Numerous biological functions -ranging from apoptosis and necroptosis to survival and inflammation- are mediated upon binding of TNF ligands to their receptors (Chen and Goeddel, 2002; Saperstein et al., 2009).
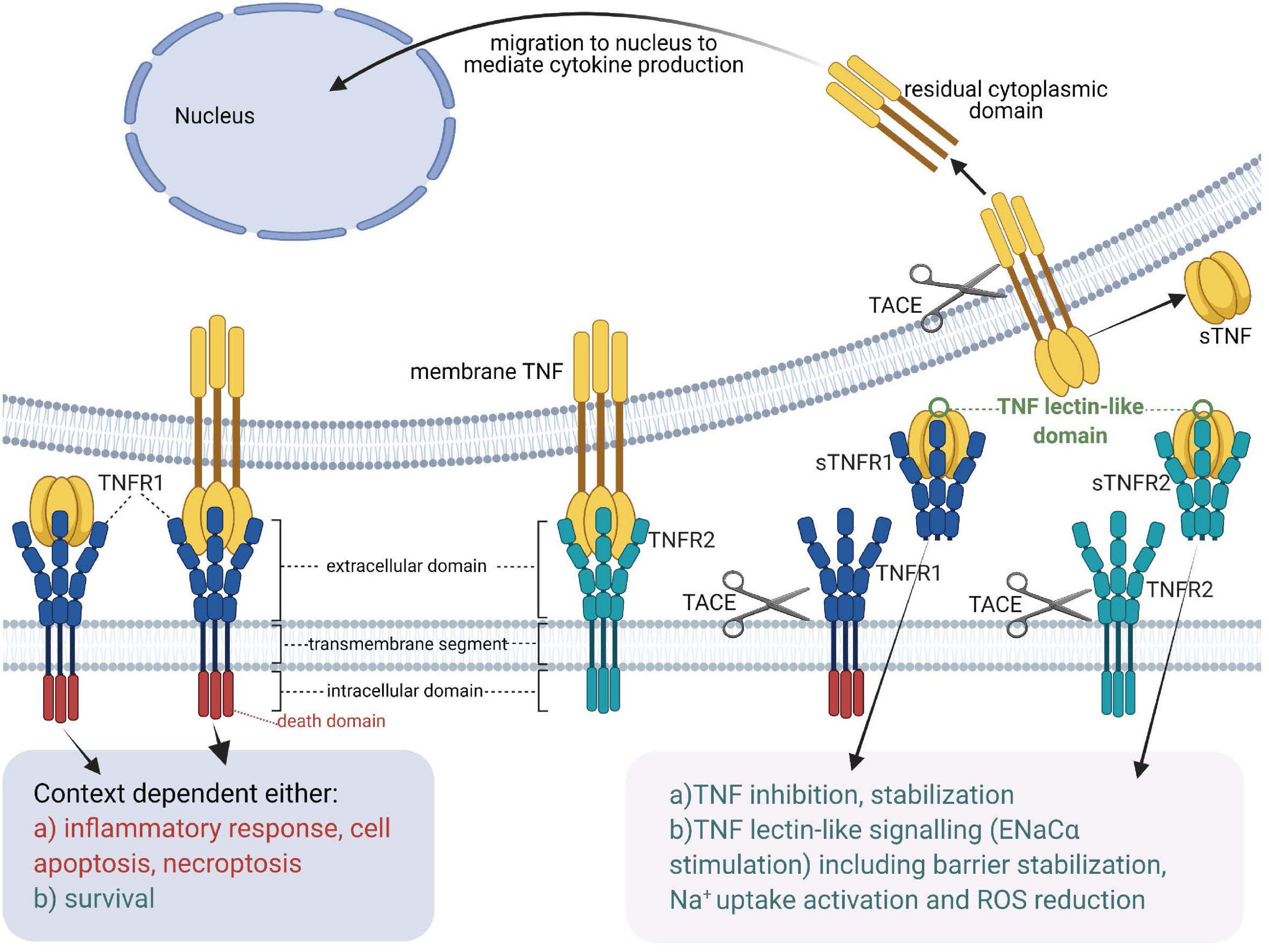
Figure 1. Tumor necrosis factor receptors, TNF ligands, TNF-soluble TNF receptor complexes and TNF lectin-like activity [Adapted from Horiuchi et al. (2010)]. sTNF, soluble TNF; mTNF, transmembrane TNF; sTNFR, soluble TNF receptor; TACE, TNF Alpha Converting Enzyme; TNF, tumor necrosis factor; TNFR1, membrane-associated TNF receptor 1; TNFR2, membrane-associated TNF receptor 2.
Whereas both transmembrane TNF (mTNF) and soluble TNF (sTNF) can bind to both TNF receptors, TNFR2, which is typically expressed in the membrane of immune cells, only becomes fully activated by transmembrane TNF (Grell et al., 1995). Both mTNF and sTNF can induce inflammatory responses (Horiuchi et al., 2010). Through cell-cell contact mTNF can also mediate juxtacrine signaling in the local medium, while sTNF launches paracrine and systemic signaling via TNFR1 even in remote locations from its source cells (Richter et al., 2012). A casein kinase I (CKI) consensus sequence in the cytoplasmic domain of mTNF was suggested to perform “reverse signaling,” which means that it can transmit signals both as a ligand and as a receptor upon interacting with TNFRs (Watts et al., 1999; Horiuchi et al., 2010).
Tumor necrosis factor receptor 1 (also referred to a p55, p60, CD120a, or TNFRSF1A) (Tartaglia et al., 1991) is a prototypical, 55 kDa type 1 transmembrane glycoprotein (Tartaglia et al., 1993) with an N-terminal extracellular domain (ECD) comprising four cysteine-rich domains (CRD1, −2, −3, −4), an α-helical transmembrane domain, a transmembrane segment and a cytoplasmic intracellular death domain (Bodmer et al., 2002). TNFR1 is expressed by most cell types, except erythrocytes (Martínez-Reza et al., 2017). Upon binding to TNF and depending on the ubiquitination status of receptor interacting protein kinase 1 (RIPK1), TNFR1 activates several pro-inflammatory signaling pathways, including nuclear factor kappa B (NF-κB), p38 MAP kinase or JNK. Alternatively, TNFR1 induces cell death, either in the form of apoptosis or necroptosis, depending on the activity of caspase 8 (Wajant et al., 2003). These pathways are summarized in Figure 2.
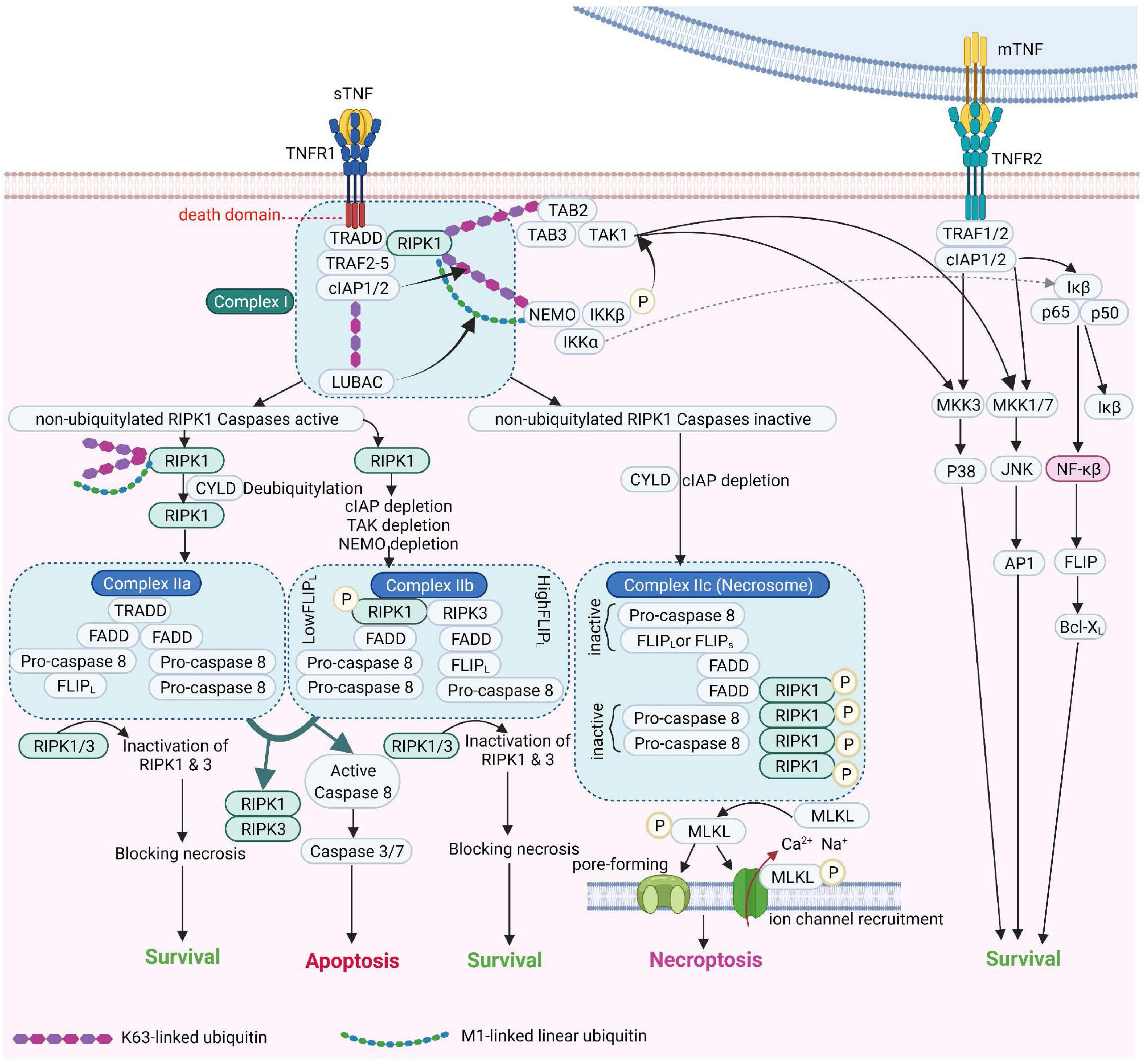
Figure 2. TNF Receptor-1 and TNF Receptor-2 signaling pathways [Adapted from Brenner et al. (2015) and Conrad et al. (2016)]. cIAP, Cellular Inhibitor of Apoptosis Protein; FADD, Fas-associated protein with death domain; FLIPL, FLICE-like inhibitory protein; IKK, IκB kinase; JNK, c-Jun NH2-terminal kinase; LUBAC, linear ubiquitin chain assembly complex; MLKL, mixed lineage kinase domain-like; mTNF, membrane TNF; NEMO, NF-κB essential modulator; NF-κB, Nuclear factor kappa B; NIK, NF-κB-inducing kinase; RIPK1, Receptor-interacting serine/threonine-protein kinase 1; sTNF, soluble TNF; TAK1, transforming growth factor-β Transforming growth factor beta-activated kinase 1; TNF, tumor necrosis factor; TNFR1, TNF receptor 1; TNFR2, TNF receptor 2; TRAF2, TNF receptor-associated factor 2; TRADD, TNFRSF1A Associated via death domain.
Tumor necrosis factor receptor 2 (also known as p75, p80, CD120b, or TNFRSF1B) (Tartaglia et al., 1991) is a transmembrane glycoprotein with a molecular weight of 75kDa (Tartaglia et al., 1993). Similar to TNFR1, TNFR2 has an N-terminal ECD made by four cysteine-rich domains (CRDs) as well as an α-helical transmembrane domain. In contrast to TNFR1, the cytoplasmic domain of TNFR2 harbors no death domain (Martínez-Reza et al., 2017; Yang S. et al., 2018). TNFR2 is expressed mainly in immune cells such as regulatory T cells (Tregs), hematopoietic lineage cells, neurological tissues and endothelial cells (Richter et al., 2012; Martínez-Reza et al., 2017), some solid tumors and hematopoietic malignancies (Wajant and Beilhack, 2019). Somewhat surprisingly, TNFR2, but not TNFR1, upon interacting with mTNF or with lymphotoxin α is a crucial mediator of ICAM-1 upregulation, an adhesion molecule important in the binding of Plasmodium-infected erythrocytes and leukocytes to microvessels in the brain (Lucas et al., 1997b; Favre et al., 1999; Lou et al., 2001).
Although TNFR2 cannot form a death-inducing signaling complex (DISC), it nevertheless promotes TNFR1-mediated apoptosis, upon degrading anti-apoptotic factors, such as Bcl-xL in lung microvascular endothelial cells (MVEC) (Lucas et al., 1998) or TRAF-2, which in turn prevents the binding of apoptosis inhibitory proteins to TNFR1 (Shu et al., 1996; Li et al., 2002).
Like TNF, also the TNF receptors can be cleaved from the membrane by TACE (Black, 2004). In addition, soluble TNF receptors not only neutralize, but also stabilize TNF (Aderka et al., 1992).
The Lectin-Like Domain of TNF
Spatially distinct from the TNF receptor 1 and 2 binding sites, TNF harbors a functional domain with lectin-like activity for specific oligosaccharides, such as N,N′-diacetylchitobiose and branched tri-mannoses (Lucas et al., 1994). The lectin-like activity of TNF was originally discovered by its capacity to bind to uromodulin, a glycoform of Tamm-Horsfall protein expressed in the loops of Henle in the kidneys (Hession et al., 1987; Sherblom et al., 1988). Whereas TNF is able to directly kill long slender bloodstream forms of the protozoan parasite Trypanosoma brucei brucei causing sleeping sickness, by means of lysosomal lysis (Lucas et al., 1994; Magez et al., 1997; Daulouède et al., 2001) upon lectin-like recognition of their variant surface glycoprotein, lymphotoxin-α, which has a similar conformation as TNF and which can bind to TNF receptors, failed to do so. The domain responsible for the lectin-like activity was mapped by comparing the tertiary structures of TNF and lymphotoxin-α (Lucas et al., 1994). Moreover, soluble TNFR1, which blunts binding of TNF ligand to both TNF receptors, failed to inhibit the trypanolytic activity. The TIP peptide (a.k.a. AP301, Solnatide) is a 17 amino acid cyclic synthetic peptide (sequence CGQRETPEGAEAKPWYC) that can mimic the lectin-like domain of human TNF (Lucas et al., 1994). Mutation of the lectin-like domain of TNF does not interfere with the cytokine’s anti-bacterial activities in a cecal ligation and puncture model (Lucas et al., 1997a). There is neutralizing cross-reactivity of monoclonal antibodies raised against the lectin-like (TIP) domain of TNF and those generated against coelomic cytolytic protein 1 (CCF-1), an invertebrate analog of TNF in Eisenia foetida, which -like TNF- can also lyse African trypanosomes through a similar lectin-like interaction (Beschin et al., 1999, 2004). Intriguingly, although there is a high conformational similarity between both molecules, the residues making up the active site for the lectin-like activity in CCF-1 and TNF are different, as such suggesting a convergent evolution of these cytokines (Beschin et al., 1999).
The Acute Respiratory Distress Syndrome: A Potentially Lethal Complication of Acute Lung Injury and Severe Pneumonia
The combination of predominantly type 1 and fewer tpe 2 alveolar epithelial (AT1/2) cells (make up a total surface area around 140 m2 in an adult human lung) and capillary endothelial cells (represent a surface area around 90 m2) provides a large barrier that ordinarily prevents serum proteins and fluid from leaving the capillaries and entering the alveoli. While preventing the movement of serum proteins and liquid into the alveolar space, the barriers must allow breathing, i.e. facilitate the movement of O2 from the alveoli into the blood and CO2 from the blood into the alveoli. Disruption of the alveolar epithelial barrier results in pneumonia, but additional compromise of the endothelial barrier can lead to the acute respiratory distress syndrome (ARDS) -an underdiagnosed potentially lethal complication of severe pneumonia. COVID-19, sepsis and trauma (Hariri and Hardin, 2020). The resulting accumulation of fluid, proteins and cells in the alveolar space significantly reduces breathing efficacy and patients often require mechanical ventilation. Pulmonary edema resolution is often hampered in ARDS patients and alveolar fluid clearance (AFC) capacity even inversely correlates with mortality in ARDS patients (Ware and Matthay, 2001). AFC is mediated by vectorial Na+ transport, with Na+ uptake mainly via the apically expressed epithelial sodium channel (ENaC) and Na+ release into the interstitial space via basolateral Na+-K+-ATPase in AT1/2 cells (Matthay et al., 2005). At least four pathological parameters must be addressed in ARDS: (i) hyperinflammation, (ii) alveolar epithelial barrier dysfunction, (iii) impaired AFC and (iv) capillary leak.
Despite recent advances in the treatment of ARDS i.e., low tidal volume ventilation (Amato et al., 1998), ventilation in prone position (Guérin et al., 2013), neuromuscular blockade (Papazian et al., 2010) and corticosteroid use (Villar et al., 2020), mortality remains as high as 30-40% (Bellani et al., 2016), at least partially due to the lack of major pharmacologic treatments for ARDS refractory to conventional therapy.
Deleterious Outcomes of TNF-TNF Receptor Interactions in ARDS
TNF is intimately involved in inflammatory pulmonary disease, including severe pneumonia and ARDS (Mukhopadhyay et al., 2006). TNF-TNF receptor signaling has an essential impact on alveolar-capillary barrier integrity, on the physiopathology of neutrophilic alveolitis and on AFC (Lipke et al., 2010). Death signaling, AFC impairment and hyperpermeability triggered by TNF-TNFR signaling are of considerable importance in the pathogenesis of ARDS (Patel et al., 2013). Accordingly, TNF inhibitory treatments have been proposed in order to attenuate the degree of lung injury (Mazzon and Cuzzocrea, 2007).
Numerous studies have investigated the role of TNF in the regulation of capillary barrier function, but these results have to be interpreted with caution, as the reaction of cells to TNF depends on the relative expression of both TNF receptor types, the tissue, the local concentration of the ligand and its soluble TNF receptors as well as on the context, like the type of flow [laminar (high shear stress) versus turbulent (disturbed)] (Rangamani and Sirovich, 2007). Depending on its concentration, TNF can act either as a regulator of survival or cell death (Rangamani and Sirovich, 2007). Moreover, functional and phenotypical features of the lung microvasculature are distinct from most other vascular beds (Doerschuk, 2001) and from large pulmonary vessels, at least in part due to differential basal and TNF-induced expression of adhesion molecules (such as ICAM-1 and VCAM-1), varying requirements for leukocyte extravasation and relative susceptibility to bacterial toxins, e.g., LPS (Doerschuk, 2001). Thus, there are potential risks of erroneous extrapolation of results gained in in vitro studies in pulmonary large vessel endothelial cells to lung MVEC or to endothelial cells from other tissues (Bertok et al., 2011). Moreover, lung MVEC isolated from patients who developed ARDS were shown to be functionally and phenotypically different from their counterparts isolated from patients who had undergone a lobectomy for lung carcinoma. As such, constitutive expression of the adhesion molecule ICAM-1 and of TNFR2 was found to be significantly increased in pulmonary MVEC from ARDS patients, as well as their basal generation of the proinflammatory cytokine IL-6 and of the chemokine IL-8 (Grau et al., 1996). The paragraphs below provide a short overview of TNF/TNFR-mediated effects in capillary endothelial cells.
Tumor Necrosis Factor-Induced Endothelial Cell Death and Capillary Barrier Dysfunction in the Lungs
Although TNFR1 is sufficient to induce sensitized apoptosis (in the presence of transcriptional or translational inhibitors) in primary lung mouse MVEC, the presence of both receptors is required for direct TNF-induced cell death (Lucas et al., 1998). While TNF-induced apoptosis in lung endothelium involves myosin light chain (MLC) phosphorylation, TNF-induced capillary barrier dysfunction does not (Petrache et al., 2001). Elevated plasma levels of soluble TNFRs are associated with morbidity and mortality in ALI patients (Parsons et al., 2005). Bronchoalveolar lavage fluid from early ARDS patients contains significantly elevated levels of both TNF and soluble TNF receptors, as compared to lavages from control patients investigated for chronic cough, hilar masses or pulmonary nodules (Hamacher et al., 2002).
TNF-TNFR2 interactions can be either destructive or preventive (Tartaglia et al., 1993; Lucas et al., 1997c; Wilson et al., 2007). On the one hand, TNF-TNFR2 signaling inhibits edema formation in a murine model of ventilator-induced lung injury, since mice lacking TNFR2 are more prone to the formation of edema, whereas mice not expressing TNFR1 are protected (Wilson et al., 2007). On the other hand, as indicated above, TNFR2 may increase TNFR1-mediated apoptosis upon impairing anti-apoptotic pathways (Tartaglia et al., 1993; Lucas et al., 1998; Li et al., 2002). As both TNF receptor types can mediate divergent signaling pathways, their relative expression in lung endothelial and alveolar epithelial cells can profoundly affect TNF signaling during ALI (Bertok et al., 2011).
Receptor interacting protein kinase 1(RIPK1)–dependent endothelial necroptosis underlies the TNF-induced systemic inflammatory response syndrome in mice (Zelic et al., 2018) and the blocking of connexin 43 hemichannels, which promote Ca2+ influx, with TAT-Gap19 protected mice against TNF-induced vascular leakage and mortality (Delvaeye et al., 2019). Laminar flow fosters pro-survival signaling, at least partially by impairing TNF-induced caspase-3 activity (Garin et al., 2007).
TNF not only decreases barrier function in ARDS by inducing cell death, but it can also affect junction protein status. Increased endothelial Ca2+-influx is the initial pivotal signal preceding pathways leading to contraction, including myosin light chain (MLC)-dependent mechanisms and MLC-independent microtubule rearrangement (Sandoval et al., 2001). TNF induces F-actin depolymerization in bovine pulmonary artery endothelial cell monolayers (Goldblum et al., 1993; Matsumura et al., 1997) to promote intracellular gap formation and barrier dysfunction. Across human pulmonary artery endothelial cells, TNF decreases trans-endothelial electrical resistance. This process occurs independently of MLC phosphorylation, which is catalyzed by either MLC kinase or Rho-kinase (Petrache et al., 2003). Rather, TNF causes microtubule destabilization and actin rearrangement in human pulmonary artery endothelial cells (Petrache et al., 2003). TNF-induced disassembly of the peripheral microtubule network involves activation of the p38 mitogen-activated protein kinase pathway in endothelial cells of human pulmonary arteries, which leads to an increase in permeability of endothelial cell layers (Petrache et al., 2003). Depolymerization of microtubules causes disassembly of adherens junction proteins they associate with, such as VE-cadherin, which increases permeability (Petrache et al., 2003). TNF can increase tyrosine phosphorylation of VE-cadherin (Angelini et al., 2006) and degrade the endothelial glycocalyx (Nieuwdorp et al., 2009). The Ca2+–dependent protein kinase C-α isoform can also play an important role in initiating endothelial cell contraction and disassembly of VE-cadherin–mediated cell–cell contacts (Petrache et al., 2003).
Tumor necrosis factor can also interact with other pathways promoting vascular leak. As such membrane-bound TNF (mTNF) and VEGF signaling pathways converge at the level of the secondary messenger, SAPK-2/p38, with continuous autocrine signaling by mTNF sensitizing endothelial cells to respond to VEGF by increasing their vascular permeability (Petrache et al., 2003).
Tumor Necrosis Factor-Induced Oxidative and Nitrosative Stress in Pulmonary Endothelium
Although pulmonary venous and capillary endothelial cells and are at times highly exposed to O2-rich environments, they use the glycolytic pathway, rather than mitochondrial respiration as the main ATP-generating pathway in quiescent and proliferating states. They likely do so in order to maximize O2 delivery to tissues and perivascular cells. This prevents the generation of high levels of reactive oxygen species in this cell compartment (Xiao et al., 2021). The endothelium, however, also provides the first line of defense against inflammatory or infectious insults in the vasculature. Upon inflammatory stimulation by TNF, otherwise quiescent endothelial cells are activated to produce significantly increased levels of pro-inflammatory molecules and NADPH oxidase-generated ROS (Gertzberg et al., 2004), and they moreover increase PFKFB3-mediated glycolysis (Cao et al., 2019). ROS can in turn combine with nitric oxide (NO) to generate barrier-disruptive peroxynitrite (Neumann et al., 2006). Although endothelial cells were shown to express NOX-1, -2, -4 and -5, especially NOX-2 was demonstrated to contribute to TNF-induced ROS generation (Deng et al., 2012; Drummond and Sobey, 2014).
Reduction of endothelial nitric oxide synthase (eNOS)-mediated NO generation was shown to promote pulmonary microvessel leakage in eNOS–/– mice (Predescu et al., 2005), suggesting a barrier-protective effect of eNOS-derived NO in the pulmonary microvasculature (Di Lorenzo et al., 2013). Increased ROS generation was shown to impair the expression and activity of eNOS (Neumann et al., 2004) and to increase the activity of arginase 1, an enzyme competing with eNOS for the common substrate arginine (Romero et al., 2008; Chandra et al., 2012; Lucas et al., 2013). TNF was shown to be critical for arginase 1 induction in a murine model of ischemia and reperfusion, since TNF–/– mice had significantly reduced endothelial arginase activity (Gao et al., 2007). All of these events involve increased endothelial oxidative stress and can disrupt the vascular integrity by reducing adherens and tight junction protein expression and/or by inducing endothelial cell death. ROS can moreover increase expression of adhesion molecules in endothelium and as such foster transendothelial migration of circulating leukocytes (Wang and Doerschuk, 2000). A stop of blood flow, as can occur in TNF-induced multiple organ failure, was moreover shown to activate a mechano-sensing machinery that generates ROS and that subsequently drives neutrophil influx (Noel et al., 2013). By contrast, laminar shear stress exerts a profound anti-apoptotic effect on endothelial cells treated with TNF, via the increased NO synthesis and post-transcriptional activation of eNOS (Cicha et al., 2009).
Tumor Necrosis Factor-Induced Upregulation of Adhesion Molecule Expression in Endothelial Cells
Apart from directly inducing barrier impairment, TNF can also indirectly induce hyperpermeability. TNFR1 activation increases neutrophil and monocyte adhesion to endothelium (Zhao et al., 2014), through upregulation of cell surface expression of endothelial cell adhesion molecules such as E-selectin, ICAM-1 and VCAM-1 in activated human pulmonary microvascular endothelial monolayers in vitro (Zhou et al., 2007; Su et al., 2008; Proudfoot et al., 2018). The induction of VCAM expression is associated with ROS generation, and endothelial NOX-2 is a major source of ROS associated with pulmonary inflammation (Frey et al., 2006). In patients and mice with critical limb ischemia, increased expression of the Cu2+ transport protein antioxidant-1 (Atox1) stimulates transcription of the NADPH oxidase organizer p47phox, thereby increasing VCAM-1/ICAM-1 expression and promoting monocyte adhesion in endothelial cells inflamed with TNF (Chen et al., 2015; Das et al., 2019).
Tumor Necrosis Factor-Induced Release of Pro-inflammatory Markers in Endothelial Cells
The TNF-TNFR1/p38 MAPK/MK2 signaling axis induces secretion of pro-inflammatory chemokines, such as the neutrophil attractant IL-8 in human lung MVEC (Su et al., 2008). Moreover, TNF-induced activation of PI3Kγ in endothelial cells in turn induces PKC-ζ-dependent activation of NOX, leading to the generation of ROS and thereby degradation of the proteasome-dependent inhibitor of NF-κBα –IκBα- which further activates NF-κB-dependent inflammation (Frey et al., 2006).
Tumor Necrosis Factor-Induced Thrombotic Events
TNF can promote thrombosis by inducing luminal endothelial expression of tissue factor. Tissue factor interacts with circulating coagulation factor VII and as such triggers the extrinsic coagulation pathway. This process involves NADPH oxidase activation (Clauss et al., 1996; Mechtcheriakova et al., 2001). Selective inhibitors of either TNFR1 or TNFR2-induced signaling pathways were shown to inhibit tissue factor expression (Clauss et al., 1996; Mechtcheriakova et al., 2001). However, TNFR1 is the predominant receptor responsible for the synergistic interaction between VEGF and TNF in endothelial tissue factor expression (Clauss et al., 1996; Mechtcheriakova et al., 2001).
Tumor Necrosis Factor in COVID-19-Associated Acute Respiratory Distress Syndrome
Acute Respiratory Distress Syndrome develops in about 20% of COVID-19 patients with severe symptoms (Matthay et al., 2020; Du et al., 2021) and is influenced by age-, sex-, the presence of co-morbidities like diabetes, obesity and arterial hypertension, immunodeficiency and vaccination status. The development of COVID-19 associated ARDS is highly age-dependent and represents the main cause of COVID-19 related morbidity and mortality. As a matter of fact, COVID-19-related ARDS has increased the prevalence of ARDS in most highly industrialized countries by a factor of three (Matthay et al., 2020), leading to immense problems in terms of human resources, including excessive work load for physicians, nurses and other health care workers, with a profound impact on patients and their families, including social isolation and reduced patient care for other diseases. Severe COVID-19 is characterized by increased permeability of the alveolar-capillary barriers combined with diffuse alveolar damage, as shown in autopsies (Zhang et al., 2020). Human lung capillary endothelial cells express the main receptor for SARS-CoV2, i.e., angiotensin converting enzyme 2 (ACE-2) (Gheblawi et al., 2020), as well as of the cellular proteases, furin (Mayer et al., 2004) and TMPRSS2, necessary to mature the spike glycoprotein for subsequent receptor binding and membrane fusion (Hoffmann et al., 2020). The combination of these post-translational events causes endothelial cells to be vulnerable to SARS-CoV2. Some groups are detecting SARS-CoV2 within the pulmonary endothelium of COVID-19 patients (Ackermann et al., 2020; Varga et al., 2020) and in infected human blood vessel organoids (Monteil et al., 2020). Although therapeutic strategies to blunt host damage in COVID-19 mainly focus on the alveolar epithelium, COVID-19, particularly in the later complicated stages, is a disease of endothelial cells, particularly in the pulmonary microvasculature (Libby and Lüscher, 2020).
COVID-19-associated ARDS is accompanied by a significant imbalance in the cytokine/chemokine profile (Pedersen and Ho, 2020), with increased plasma levels of pro-inflammatory cytokines and chemokines (Pedersen and Ho, 2020). The severity of the inflammatory response, combined with T cell lymphopenia significantly contributes to pulmonary devastation and respiratory distress (Pedersen and Ho, 2020). An excessive inflammatory response during SARS-CoV-2 infection, which includes high levels of TNF, can provoke death of resident lung cells, which can further amplify barrier dysfunction. Elevated BAL and plasma levels of TNF are a hallmark manifestation of COVID-19, and can be associated with significant morbidity and mortality rates (Del Valle et al., 2020; Robinson et al., 2020). Increased soluble TNF levels can be caused by SARS-CoV2-induced activation of TACE (Haga et al., 2008; Atzeni et al., 2021). Mast cells activated by SARS-Cov-2 infection in respiratory tracts are also an important source of TNF (Kritas et al., 2020; Motta Junior et al., 2020) and together with the virus they can activate macrophages (Kritas et al., 2020). TNF is a major mediator of pulmonary edema and multi-organ failure in cases of severe COVID-19 (Eisenhut and Shin, 2020). TNF also promotes viral entry and dissemination since it induces adhesion molecule expression in endothelial cells and promotes VEGF generation, all of which contribute to increased permeability of the endothelial layer and thereby dissemination of SARS-CoV2 in patients with COVID-19 (Polidoro et al., 2020). Hyperpermeability of pulmonary blood vessels also results in improper accumulation of inflammatory monocytes, macrophages and neutrophils in the alveolar space (Channappanavar et al., 2016). Lung biopsies from patients who succumbed to severe COVID-19 show remarkable pathological injuries, with alveolar exudates, proliferation of alveolar epithelium, interstitial inflammation, and formation of hyaline membranes (Tan et al., 2020).
Activation of the coagulation pathway is another known feature of severe COVID-19 with potential development of disseminated intravascular coagulation (Teuwen et al., 2020). This is associated with pulmonary endothelial cell activation and dysfunction as cell death and the disruption of vascular endothelial integrity leads to exposure of the thrombogenic basement membrane and further in the activation of the coagulation cascade (Nachman and Rafii, 2008). In addition, IL-1β and TNF activate endothelial cells, which causes activation of coagulation cascade by expressing P-selectin, von Willebrand factor and fibrinogen, that are required for attaching platelets (Pober and Sessa, 2007).
COVID-19 endothelialitis is associated with platelet aggregation and thrombocoagulatory and fibrinolysis-inhibitory (D’Agnillo et al., 2021) features that can obstruct vessels, as manifested by the important frequency of thromboses, including pulmonary thromboembolism. This has been shown particularly in the pulmonary microvasculature, especially in vessels with a cross-sectional area between 1.25 and 5 mm2 (Libby and Lüscher, 2020; Morris et al., 2021), containing highly activated and functionally distinct neutrophils, which generate neutrophil extracellular traps (NETs) (Leppkes et al., 2020; Zuo et al., 2020; Panda et al., 2021), which may stimulate alternative treatment strategies besides corticosteroids and standard care (Panda et al., 2021). One has to question whether virus-associated endothelial cell injury, quite frequently co-localized with NETs, or rather the collateral of other inflammatory processes in plasma, represents the key step to neutrophil activation. The high pro-coagulation context in COVID-19 in part motivates therapeutic anticoagulation approaches especially with heparins in hospitalized patients who are not critically ill (Leppkes et al., 2020; Houston et al., 2020). Such patients seem to profit and seem less in danger of anticoagulation than sicker and frailer poly-monitorized ventilated ARDS patients, in whom both risk and benefit need to be more closely examined. To circumvent progressive respiratory failure, prone positioning of the patient, which has high preventative and therapeutic value, is recommended since it improves ventilation and perfusion mismatch (Gattinoni et al., 2020). Conceptually, the high frequency of the above-mentioned pulmonary embolism, including the site of peripheral arterial large vessel occlusions like coronary, cerebral, limb, and other arterial occlusions, may have a similar pathogenetic background, which may also be the case for COVID-19 pneumonia and makes it a more privileged respiratory disease, as compared to non-COVID ARDS in terms of prognosis.
Tumor Necrosis Factor Neutralizing Agents
To counteract excessive deleterious TNF signaling leading to tissue damage, barrier dysfunction and hyperinflammation, several types of molecular-based therapeutic approaches have been developed over the past decades (Fischer et al., 2020). These include monoclonal antibodies such as infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi) and soluble TNF receptor constructs, such as etanercept (Enbrel) (Russell et al., 2020). TNF inhibitors may directly compete with the TNF ligand, or deactivate its receptors or the enzyme generating the soluble TNF ligand, i.e., TACE, resulting in inhibition of downstream signaling pathways leading to cell death or inflammation. Figure 3 illustrates some of the different targets associated with TNF and TNFRs that can be inhibited by natural or pharmacological TNF blockers. In addition, data obtained from various studies and clinical trials on the impact of natural or pharmaceutically designed TNF blockers are summarized in Tables 1A,B.
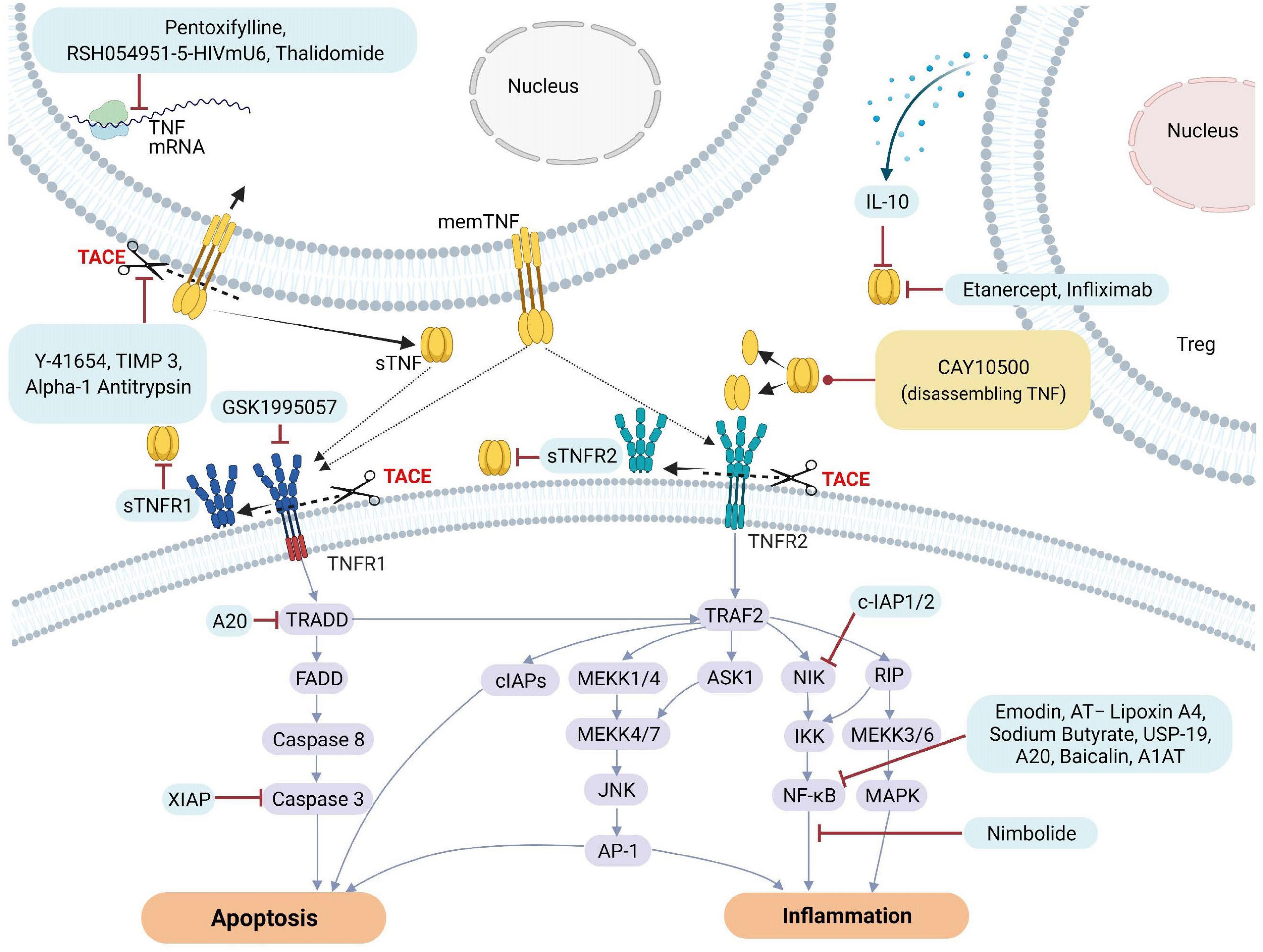
Figure 3. Natural and pharmacological inhibitors of TNF and TNF receptor expression and bioactivity. AP-1, Activator protein 1; AQP5, Aquaporin 5; ASK1, Apoptosis signal-regulating kinase 1; ASM, acid sphingomyelinase; cIAP1, Cellular Inhibitor of Apoptosis Protein 1; ENaC, epithelial sodium channel; FADD, Fas-associated protein with death domain; ICAM-1, intercellular adhesion molecule-1; IL, Interleukin; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MEKK, mitogen-activated protein Kinase/ERK kinase kinase; mRNA, messenger RNA; NF-κB, Nuclear factor kappa B; NIK, NF-κB-inducing kinase; sTNF, soluble TNF; TACE, TNF converting enzyme; TJs, tight junctions; TNF, tumor necrosis factor; TNFR1, TNF receptor 1; TNFR2, TNF receptor 2; TRAF2, TNF receptor-associated factor 2; TRADD, TNFRSF1A Associated via death domain; XIAP, X-Linked inhibitor of apoptosis.
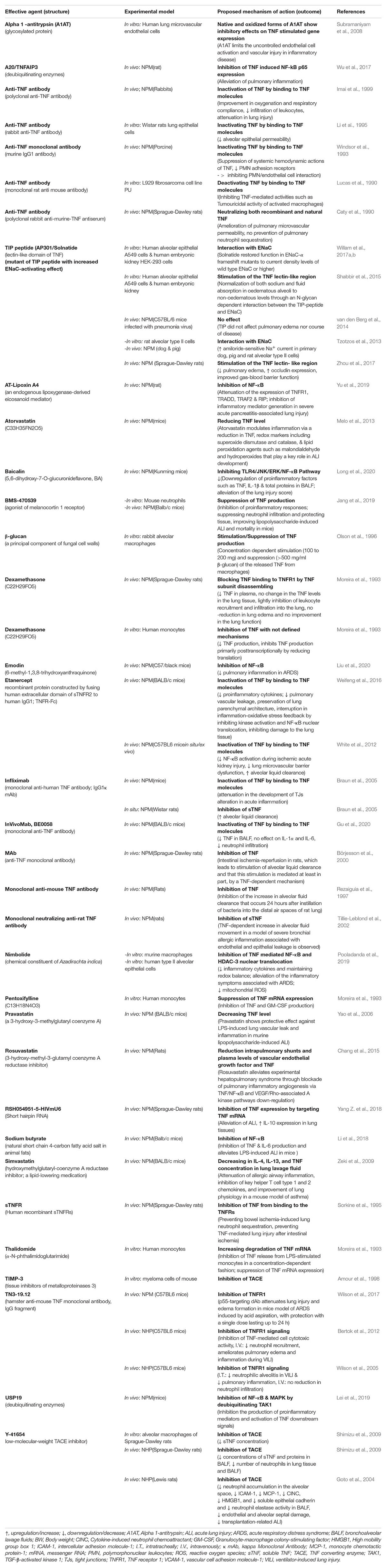
Table 1A. Impact of TNF inhibitors/stimulators in experimental settings using non-human models including non-human primates (NHP) and non-primates mammals (NPM)
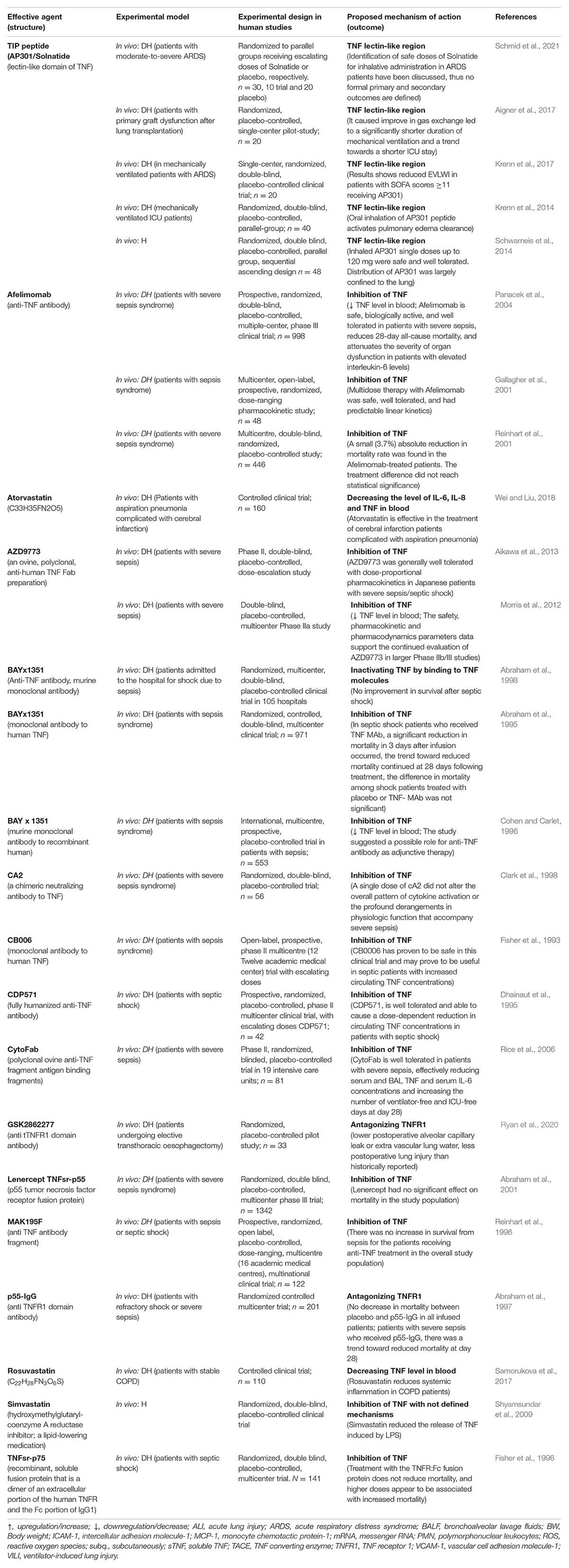
Table 1B. Impact of TNF inhibitor/stimulators or lectin-like domain stimulators in experimental settings using human trials with diseased human beings (DH) or volunteer human beings (H).
Despite the involvement of a vast variety of pro-inflammatory mediators, exclusive blockage of TNF seems to result in better clinical outcomes in a broad range of diseases associated with inflammation and autoimmunity and clinical trials assessing TNF inhibitors for treatment of ARDS in severe COVID-19 patients have been proposed (Feldmann et al., 2020). Drugs with anti-inflammatory and/or anti-viral effects can be considered as add-on therapy against acute lung injury- (Gallelli et al., 2020) and COVID-19-associated hyper-inflammation (Robinson et al., 2020). Indeed, genetic or pharmacological inhibition of TNF and TNFRs abrogates production of TNF in pleural exudates and lung tissues and reduces infiltration of inflammatory cells into the pleural cavity and lung tissues, alveolar-capillary barrier dysfunction and resident lung cell apoptosis (Mazzon and Cuzzocrea, 2007). TNF neutralization has shown protection against SARS-CoV2 infection in animal models (Jamilloux et al., 2020).
As a note of caution, the observation of higher rates of, sometimes fatal, bacterial infection in patients with rheumatoid arthritis and psoriasis treated with TNF neutralizing antibodies and soluble receptors (Keane et al., 2001) clearly documents the danger of chronically suppressing these sometimes crucial bactericidal pathways. Taken together, these findings demonstrate limitations of chronically using TNF neutralizing agents that blunt binding of the ligand to its receptors, since this approach can inhibit both deleterious and beneficial TNF-TNFR signaling. The search for alternative treatments that selectively impair TNF-mediated vascular dysfunction, AFC impairment and hyper-inflammation in cells of the alveolar-capillary compartment, without impairing the cytokine’s beneficial actions in host response to infection, is therefore therapeutically significant.
The Yin and Yang of the Tumor Necrosis Factor Molecule in the Lungs – the Lectin-Like Domain Versus the Receptor Binding Sites of Tumor Necrosis Factor
Divergent Roles of Tumor Necrosis Factor’s Receptor Binding Sites and Lectin-Like Domain in Alveolar Fluid Clearance
Alveolar fluid clearance is essential to keep the alveolar space relatively dry, to assure efficient gas exchange. AFC is mediated by vectorial Na+ transport through the apically expressed epithelial sodium channel (ENaC) and basolateral Na+-K+-ATPase in type I and II alveolar epithelial cells (Matthay et al., 2005; Vadász et al., 2007). The amiloride-sensitive ENaC in its native configuration consists of three subunits of α, β, and γ and controls the overall rate of transapical Na+ transport (Dagenais et al., 2004). A fourth δ subunit was also identified and can replace the α subunit to form an alternative active ENaC configuration (Ji et al., 2012). Na+ absorption by ENaC is the critical driving force of AFC at birth and is crucially involved in pulmonary edema clearance in adulthood (Dagenais et al., 2004). Hence, mice lacking ENaC-α die from alveolar flooding at birth (Hummler et al., 1996).
Epithelial sodium channel activity is regulated by subunit maturation upon cleavage of the extracellular loops of the α and γ subunits by proteases such as furin and prostasin (Kleyman and Eaton, 2020), by its cell surface expression N and by its open probability time Po, the latter of which is at least partially controlled by complex formation of ENaC with the protein MARCKS and with PIP2 (Alli et al., 2012, 2015; Archer et al., 2020).
Conflicting study outcomes are found with regard to the effect of TNF on AFC. Whereas TNF generation is increased in lavage from acute ARDS patients (Hamacher et al., 2002), whose AFC capacity inversely correlates with mortality (Ware and Matthay, 2001), a TNF-dependent increase in AFC is found in a number of animal models, three of which are presented in more detail (Rezaiguia et al., 1997; Börjesson et al., 2000; Tillie-Leblond et al., 2002; Hamacher et al., 2018). Further studies show that TNF can either stimulate (Fukuda et al., 2001) or inhibit ENaC in alveolar epithelial cells (Johnson et al., 2002, 2006; Dagenais et al., 2004; Eaton et al., 2004) and that, accordingly, the cytokine has a substantial impact on the capacity of alveolar epithelial cells to transport Na+ (Dagenais et al., 2004). In the following paragraphs we will discuss the opposing actions of TNF-TNFR1 signaling and activities of the lectin-like domain of TNF on ENaC.
The inhibitory effect of TNF on ENaC is mainly mediated by TNFR1 (Dagenais et al., 2004; Braun et al., 2005). TNF decreases the mRNA expression levels of α, β, and γ subunits of ENaC in alveolar epithelial cells isolated from male Sprague-Dawley rats (Dagenais et al., 2004), which correlates with a significant reduction in amiloride-sensitive current in these cells (Dagenais et al., 2004). TNF moreover affects the stability of ENaC mRNA (Dagenais et al., 2004). In addition, TNF downregulates ENaC-α mRNA involving a complex process that requires the transcription of multiple genes (Dagenais et al., 2004). Along with the abovementioned results, direct exposure of rat type II alveolar epithelial cells to TNF inhibits mRNA and protein expression of the α- and γ-, but not of the β subunit of ENaC (Yamagata et al., 2009) and impairs ENaC function, as revealed by reduced amiloride-sensitive currents in these cells (Yamagata et al., 2009). These results document the substantial negative influence TNF can exert on the capacity of alveolar epithelial cells to transport sodium (Dagenais et al., 2004).
One of the mechanisms through which TNF can impair ENaC activity is through the TNFR1-mediated activation of acid sphingomyelinase (ASMase), an enzyme located in lysosomes and in the extracellular space, which catalyzes the conversion of sphingomyelin, a major component of membranes, into ceramide and phosphocholine (Gorelik et al., 2016). Low levels of ASMase maintain an equilibrium within the alveolar space between ceramides and sphingomyelin substrate (Ryan et al., 2003), but during inflammation this balance can be altered, resulting in increased generation of injurious sphingolipids, that can in turn accelerate pulmonary edema formation (Schissel et al., 1996; Haimovitz-Friedman et al., 1997; Ryan et al., 2003). Ceramide is a crucial mediator of TNF/TNFR1-induced inhibition of ENaC, via a pathway associated with PKC-dependent externalization of phosphatidyl serine (Bao et al., 2007).
Apart from inhibiting ENaC, TNF/TNFR1 signaling can also affect mRNA and/or protein expression of other channels involved in AFC. As such, TNF impairs expression of the major water channel aquaporin 5, expressed in alveolar, tracheal, and upper bronchial epithelium, through a mechanism involving activated NF-κB (Towne et al., 2000, 2001). Moreover, TNF downregulates the steady-state mRNA level of the cystic fibrosis transmembrane conductance regulator (CFTR) in HT-29 cells (Nakamura et al., 1992). Depending on the direction of the chloride transport, CFTR can either activate (inward) or inhibit (outward) ENaC activity. Similar to the impact of TNF on ENaC, its inhibitory action on CFTR occurs at the post-transcriptional level in a dose- and time-dependent manner (Nakamura et al., 1992), by reducing CFTR mRNA half-life (Nakamura et al., 1992).
In a Pseudomonas aeruginosa (P. aeruginosa)-induced pneumonia model in rats, Rezaiguia et al., measured alveolar protein and liquid clearance, 24 h after intratracheal instillation of bacteria, by instilling 5% bovine albumin solution with 125I-labeled human albumin into the airspaces (Rezaiguia et al., 1997). The concentration of unlabeled and labelled protein in the distal airspaces over 1h is used as an index of net AFC (Rezaiguia et al., 1997). Surprisingly, the investigators detect a P. aeruginosa pneumonia-induced increase in AFC over 1h by about 48% in experiments with blood flow, and by 43% in those without blood flow, compared with control groups (Rezaiguia et al., 1997). In both experiments, this AFC increase is inhibited by amiloride, which blunts ENaC activity. As propranolol has no inhibitory effect on AFC, a catecholamine-dependent mechanism can be excluded (Rezaiguia et al., 1997). Intratracheal instillation of an anti-TNF neutralizing antibody into the lungs 5 min before bacterial instillation is shown to prevent the increase in AFC in rats with pneumonia (Rezaiguia et al., 1997) and instillation of 5 mg TNF in normal rats increased AFC by 43% over 1h compared to control rats receiving vehicle (Rezaiguia et al., 1997), an effect not blunted by propranolol. As such, TNF at early time points following G– pneumonia can upregulate net AFC, partly by a TNF-dependent mechanism (Rezaiguia et al., 1997). This study thus provides evidence of a positive role of TNF in AFC, in a catecholamine-independent manner.
Studies by Börjesson et al. (2000) in ventilated, anesthetized rats with 45 min intestinal ischemia followed by 3 h reperfusion show development of remote organ injury, including the lungs. A 76% increase in AFC as compared to controls, measured as the increase in alveolar protein concentration over 45 min, is at least partially mediated by a TNF-dependent mechanism, since systemic administration of a neutralizing polyclonal anti-TNF antibody before induction of intestinal ischemia fully inhibits the rise in AFC. Similar to the results above (Rezaiguia et al., 1997), the stimulated AFC after intestinal ischemia-reperfusion cannot be inhibited by propranolol and is therefore assumed to be catecholamine-independent (Börjesson et al., 2000). Intracellular cAMP levels are not increased in this model and amiloride inhibits similar fractions of AFC in animals subjected to ischemia-reperfusion or in control animals (Börjesson et al., 2000). In conclusion, the rat intestinal ischemia-reperfusion model leads to an AFC increase that is TNF-dependent.
Evaluation of AFC and epithelial and endothelial permeability in a model of chronic allergic inflammation in Brown-Norway rats challenged with ovalbumin by Tillie-Leblond et al., demonstrates lung sections with inflammatory infiltrates associated with perivascular and peribronchiolar edema in the ovalbumin group (Tillie-Leblond et al., 2002). To measure AFC in this study, a 5% bovine albumin solution with 125I-labeled human albumin is instilled into their air spaces and alveolar-capillary barrier permeability is evaluated in another group of animals by intravenous injection of 1 μCi of 131I-labeled albumin (Tillie-Leblond et al., 2002). Endothelial permeability in this study significantly increased in the ovalbumin group but the final-to-initial protein ratio reaches significantly higher levels in the ovalbumin group, as compared to the control group (Tillie-Leblond et al., 2002). Administration of neutralizing rat anti-TNF antibodies in the instillation significantly decreases this ratio. As such, also in this rat model of severe bronchial allergic inflammation associated with increased endothelial and epithelial permeability, a TNF-dependent increase in AFC is observed.
These three in vivo studies of G- pneumonia, intestinal ischemia and reperfusion or ovalbumin-induced inflammation give clear evidence of an increase in AFC involving a crucial role of TNF (Hamacher et al., 2018).
One of the explanations for the observed AFC-promoting actions of TNF discussed above can come from either positive actions of TNFR2 signaling or from a role for the lectin-like domain of TNF. Indeed, in sharp contrast to the TNFR1 binding site, the lectin-like domain of TNF, mimicked by the TIP peptide (a.k.a. Solnatide, AP301), can activate amiloride-sensitive Na+ uptake in alveolar epithelial cells, as well as in lung and glomerular MVEC, which also express all ENaC subunits (Hribar et al., 1999; Elia et al., 2003; Braun et al., 2005; Madaio et al., 2019). This effect of the lectin-like domain of TNF occurs even in cells lacking both TNF receptors (Hribar et al., 1999) thus clearly documenting that the TIP peptide binds to partners different from the TNF receptors. Actually, TIP peptide binds to the α subunit of ENaC and as such directly increases the channel’s activity. This occurs by affecting both the open probability (Po) and the cell surface (N) expression of ENaC (Czikora et al., 2014; Shabbir et al., 2015; Lucas et al., 2016). Treatment of cells with N-glycosidase F, which removes N-linked glycosylation from ENaC subunits, completely abrogates channel activation by the TIP peptide (Czikora et al., 2014). This interaction mainly affects the promotion of ENaC’s cell surface expression and involves the Asn511 residue in the extracellular loop of ENaC-α (Shabbir et al., 2015). The effects of the peptide on ENaC open probability rather involve a different interaction site within the second transmembrane domain of ENaC-α in which Val567 and Glu568 are crucial (Lucas et al., 2016). The TIP peptide even activates ENaC in the presence of bacterial toxins like pneumococcal pneumolysin, a pore-forming toxin which impairs both the Po (Lucas et al., 2012a) and N of ENaC (Lucas et al., 2016), both in vitro and in vivo (Czikora et al., 2014; Hamacher et al., 2018).
Shifting the Balance Between the Tumor Necrosis Factor Receptor- and Lectin-Like Domain-Mediated Effects of Tumor Necrosis Factor
Despite of the negative effects of TNFR1 on ENaC, the lower affinity of TNF for oligosaccharides in glycoproteins than for its membrane-associated TNF receptors (Hession et al., 1987; Sherblom et al., 1988) and the much higher expression level of TNF receptors than of ENaC in alveolar epithelial cells, an intriguing observation is that inhibition of TNF with a neutralizing antibody worsened edema formation in a rat pneumonia model (Rezaiguia et al., 1997). An important question to ask is under what conditions can the beneficial activity of the lectin-like domain of TNF on ENaC predominate the deleterious one exerted by TNF-TNFR1 signaling? In an isolated perfused rat flooded lung model, which mimics cardiogenic (hydrostatic) edema, human TNF significantly decreases lung liquid clearance. Whereas the neutralizing anti-TNF monoclonal antibody Infliximab restores basal liquid clearance in this model, complex formation of TNF with its soluble TNF receptor 1 increases fluid clearance above basal levels, comparable to treatment of the lungs with TIP peptide (Braun et al., 2005). Whereas Infliximab likely interferes with both the receptor binding sites and the lectin-like domain of TNF, the soluble TNFR1 construct only blunts the former (Lucas et al., 1994; Braun et al., 2005). As such, it is tempting to hypothesize that the lectin-like domain of TNF can play a physiologically relevant role especially when present in complexes between TNF and its soluble TNF receptors, which blunt the receptor binding sites but leave the lectin-like domain active. This condition can occur in pneumonia, as such providing a potential explanation for the observed positive effects of TNF in a rat model of severe bacterial pneumonia (Rezaiguia et al., 1997). Triple mutant TNF mice expressing a mutant TNF lacking a functional lectin-like domain exhibit increased lung injury upon pneumolysin instillation as compared to wild type mice (Czikora et al., 2014). These findings indicate that the lectin-like domain can exert its protective role within the context of the entire TNF molecule and suggests that it represents an intramolecular negative feedback mechanism to keep a brake on TNF’s potent inflammatory activities.
Barrier-Protective Activities of the Lectin-Like Domain of Tumor Necrosis Factor
Important progress in the understanding of the biological role of the lectin-like domain of TNF comes from in vivo pneumonia and ARDS models evaluating the potential therapeutic potential of the TIP peptide. As shown in Figure 4, both ENaC (in its native configuration α, β, γ) and the non-selective cation channel (NSC, consisting of an ASIC1a and an ENaC-α subunit), can be expressed in lung MVEC (Czikora et al., 2017). TIP peptide, which binds to ENaC-α, can strengthen barrier function in cells treated with the pore-forming bacterial toxins listeriolysin-O (LLO, from Listeria monocytogenes) and pneumolysin (PLY, from Streptococcus pneumoniae). Both LLO and PLY induce Ca2+-influx, which in turn activates Ca2+-dependent PKC isozymes, such as PKC-α, involved in capillary barrier dysfunction (Xiong et al., 2010; Lucas et al., 2012b). Both LLO and PLY impair ENaC activity (Lucas et al., 2012a; Yang G. et al., 2018). Our results show a profound protective effect of TIP peptide toward both LLO and PLY-induced barrier dysfunction, assessed by electrical cell substrate impedance sensing (ECIS), in human lung microvascular endothelial cell (MVEC) monolayers in vitro and in mice in vivo, the latter assessed by Evans Blue Dye incorporation (Xiong et al., 2010; Lucas et al., 2012b). A crucial role for ENaC-α in this process is documented by the significantly elevated sensitivity to PLY-induced barrier dysfunction in human lung MVEC in which ENaC-α is depleted by specific siRNA. Moreover, the protective effect of both the TIP peptide and of the NSC activator MitTx is lost under these conditions. TIP peptide blunts PLY-induced phosphorylation of calmodulin-dependent kinase II (CaMKII), as well as of its substrate filamin A (FLN-A) (Czikora et al., 2017). Non-phosphorylated FLN-A both promotes ENaC open probability and blunts stress fiber formation, thus providing a mechanism and a link for the ENaC activating and barrier protective role of the lectin-like domain of TNF.
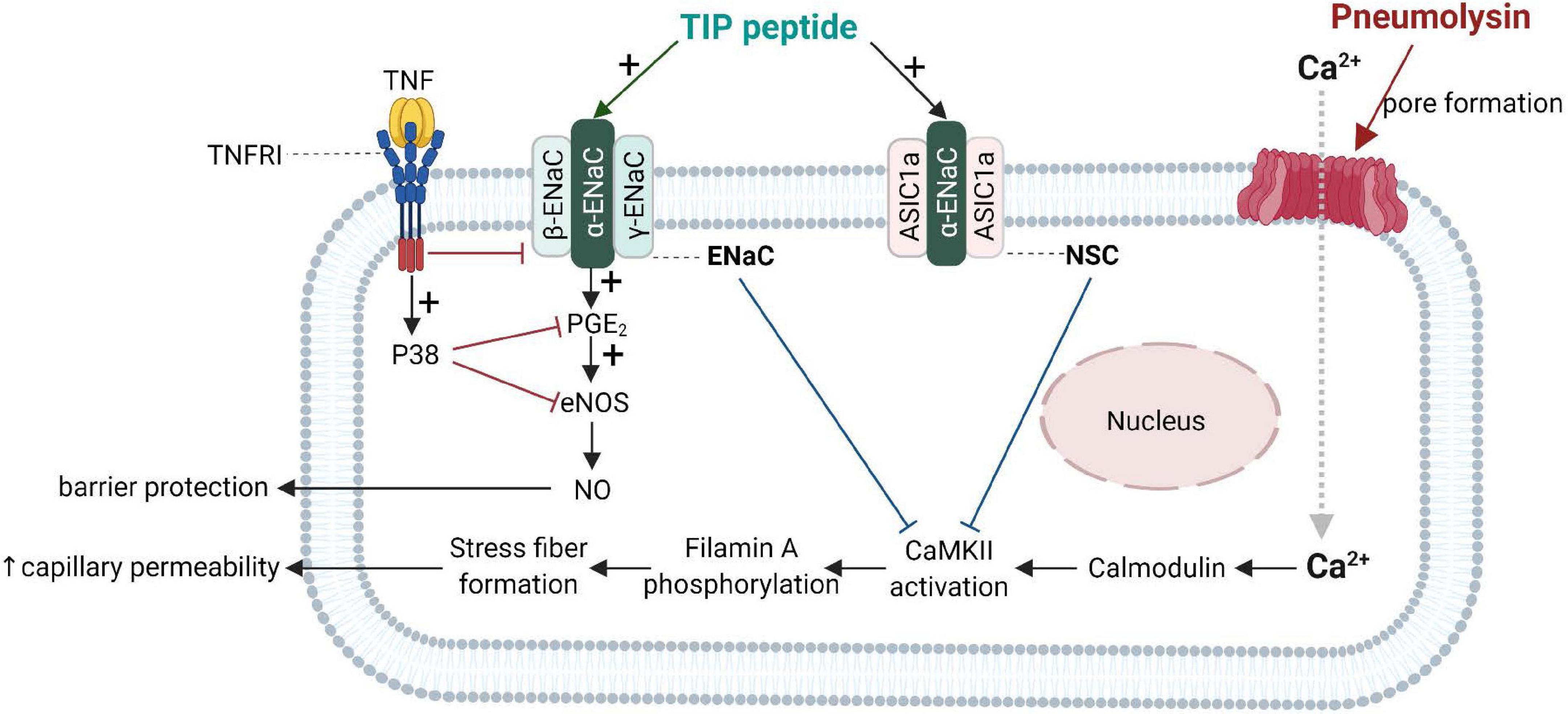
Figure 4. Barrier-protective mechanism mediated by the TNF lectin-like domain in pneumolysin-treated human lung microvascular endothelial cells [adapted from Czikora et al. (2017)]. Pneumolysin forms pores in cholesterol-containing cell membranes, which increases Ca2+ -influx (Lucas et al., 2012b), which mobilizes calmodulin to activate CaMKII, which in turn phosphorylates its substrate filamin A. Phosphorylated filamin A (Alli et al., 2015) then promotes stress fiber formation and enhances permeability of capillaries. Activation of ENaC by TIP peptide or of the non-selective cation channel, (NSC) either by TIP peptide (binding to ENaC-α) or MitTx (binding to ASIC1a), inhibits pneumolysin-mediated CaMKII activation and subsequent hyperpermeability. ENaC activation moreover promotes PGE2 generation and eNOS activation, resulting in increased barrier-protective NO production. ASIC1a, acid-sensing ion channel 1a; CaMKII, Calcium/calmodulin- dependent protein kinase II; ENaC, epithelial sodium channel.
Transplanted lungs often experience severe alveolar epithelial and endothelial dysfunction immediately following the procedure, due to ischemia-reperfusion injury. In the rat left-sided lung transplantation model of severe acute lung injury due to 20 h of cold ischemia and 24 h of reimplantation response, TIP peptide given intratracheally immediately before reimplantation into the left lung increases the left lung PaO2/FiO2 (P/F) ratio from about 70 mm Hg -indicating a condition similar to severe ARDS- to more than 400 mm Hg, which rather corresponds to a mild hypoxia-induced lung injury where a patient can typically walk up even stairs (Hamacher et al., 2010, 2018). Patient with a similar drop in P/F ratio as the one observed in the rat transplantation study group not treated with TIP peptide need to be tracheally intubated and are completely dependent on both the ventilator’s respiratory work and on a high fraction of oxygen supplementation. When applied together with the pharmacological ENaC inhibitor amiloride, the protective effect of the TIP peptide is abrogated. Complex formation of TIP peptide with N, N′-diacetylchitobiose, a selective oligosaccharide inhibitor of the lectin-like domain of TNF, or use of an inactive mutant TIP peptide, in which three crucial residues (1 Threonine and 2 glutamic acids) are changed for alanine also fail to restore oxygenation. These data strongly suggest that the significant protective effect of the TIP peptide in a severe ischemia-reperfusion lung injury model involves the lectin-like activity of TNF and is due to the activation of Na+ uptake via ENaC (Hamacher et al., 2010). The reduced infiltration of neutrophils in the bronchoalveolar lavage fluid, as well as the reduced generation of reactive oxygen species (ROS, discussed in the next paragraph), further underlines that the lectin-like domain of TNF can modify important inflammatory pathways in lung ischemia-reperfusion injury.
Anti-oxidant and Anti-inflammatory Actions of the TIP Peptide
One of the main reasons for endothelial dysfunction following lung transplantation is the alteration in blood flow, which results in shear stress that can be sensed by a mechanosensitive complex -the mechanosome- in the endothelial cell membrane. The components of this mechanosome include caveolae, the adhesion molecule PECAM, vascular endothelial growth factor receptor 2 (VEGFR2) and the adherens junction protein VE-cadherin (Chatterjee and Fisher, 2014; Chatterjee et al., 2015). The shear signal can be transduced by components of the cell surface membrane, resulting in the activation of NADPH oxidases and the generation of ROS. In a rat iso-transplantation model, a significantly increased ROS generation occurs in the transplanted lung 24h after the procedure, but this is prevented in by pretreatment of the transplanted lungs with TIP peptide, but not with an inactive mutant peptide. This ROS-reducing activity of the TIP peptide correlates with by a significantly reduced infiltration of neutrophils in the bronchoalveolar lavage fluid (Hamacher et al., 2010). Both of these effects can be blunted by the ENaC inhibitor amiloride. TIP peptide also blunts ROS generation in vitro in ovine pulmonary artery endothelial cells undergoing hypoxia-reoxygenation. The results from these preclinical studies suggest that the TIP peptide is a therapeutic candidate to treat ischemia-reperfusion injury associated with lung transplantation, since it can blunt both oxidative stress and inflammation in endothelium.
Another study documenting the anti-inflammatory capacity of the TIP peptide comes from a study using a rat model of high-altitude pulmonary edema (HAPE). In this study, treatment with TIP peptide (a.k.a. Solnatide, AP301) reduces leukocyte infiltration and pro-inflammatory cytokine levels in lavage, while improving occludin expression in alveolar epithelial cells and reducing edema formation (Zhou et al., 2017).
The TIP peptide also has an anti-inflammatory action in other organs than the lungs, as shown in a mouse glomerulonephritis model, in which animals were treated with nephrotoxic serum. In this model, local immune deposits in the glomeruli generate TNF, which activates pro-inflammatory pathways in both glomerular endothelial cells and podocytes. Intraperitoneal treatment with TIP peptide in this model significantly reduces inflammation (reduced systemic and local pro-inflammatory cytokine generation, and reduced renal leukocyte infiltration), as well as proteinuria and blood urea nitrogen (Madaio et al., 2019). TIP peptide also increases PGE2 generation in glomerular endothelial cells, which in turn promotes eNOS-dependent NO generation and barrier function (Figure 4). Despite the capacity of TIP peptide to activate ENaC, it does not increase mean arterial blood pressure in mice, possibly because it is captured in the loops of Henle by Tamm-Horsfall protein (Hession et al., 1987; Sherblom et al., 1988) before it can reach the distal tubules of the kidney. Figure 5 and Table 2 give an overview of the divergent actions of the TNF receptor binding sites versus the lectin-like domain in models of acute lung injury.
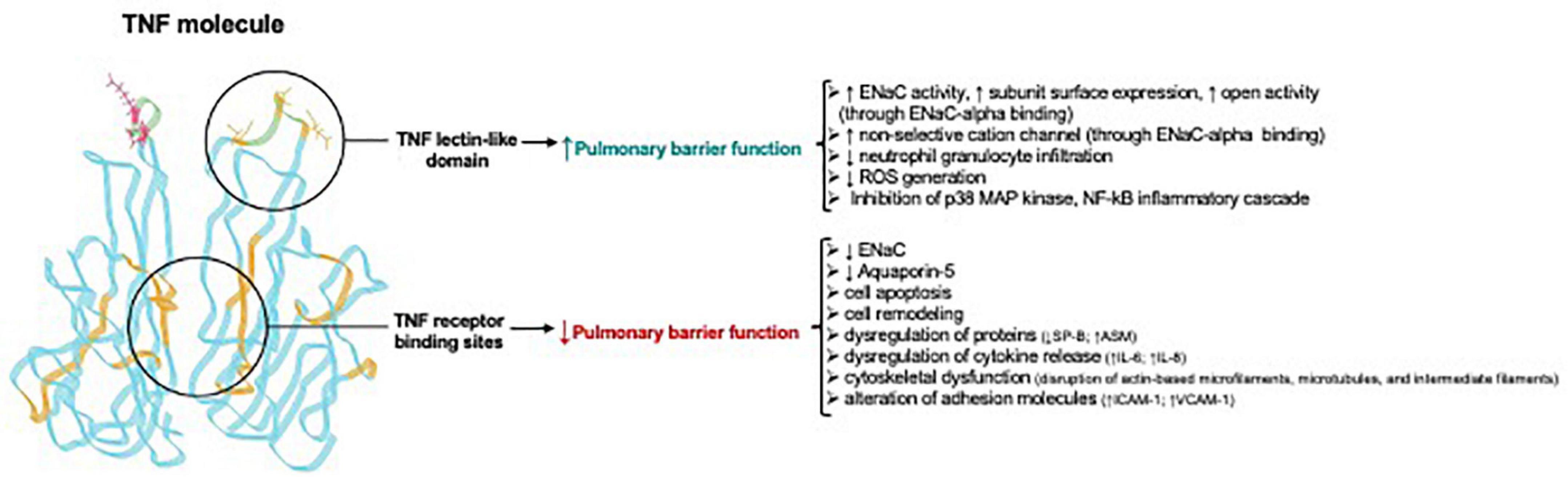
Figure 5. Divergent actions of TNF receptor binding sites versus the lectin-like domain in acute lung injury Adapted from Hamacher et al. (2018). Note that the TNF receptor binding sites for TNF receptor type 1 and TNF receptor type 2 are in the regions between two TNF homotrimers and are not identical, but in part overlapping. ↑, upregulation; ↓, downregulation; ASM, acid sphingomyelinase; ENaC, Epithelial sodium channel; ICAM-1, intercellular adhesion molecule 1; Na+/K+ -ATPase, sodium–potassium adenosine triphosphatase; VCAM-1, vascular cell adhesion molecule-1; ROS, reactive oxygen species.

Table 2. Divergent actions of TNF via its receptor binding sites versus its lectin-like domain in the lungs
Clinical Trials With the Tip Peptide in Patients With Acute Respiratory Distress Syndrome and Following Lung Transplantation
In view of promising data resulting from preclinical studies of TIP peptide performed by others and our group in several animal species, including mice, rats, rabbits and pigs (Elia et al., 2003; Braun et al., 2005; Hamacher et al., 2010; Madaio et al., 2019), clinical trials assessing safety and efficacy of TIP peptide (a.k.a. Solnatide, AP301) organized by APEPTICO, Vienna, Austria have been conducted at the Hospital of the Medical University Vienna, Austria. A double-blind placebo-controlled phase I study evaluating spirometry, exhaled NO, vital signs, ECG, safety laboratory and quantification of peptide in plasma, associated with six ascending inhaled doses of the TIP peptide in 48 healthy male subjects (Schwameis et al., 2014) demonstrates no serious, local, or dose-limiting adverse events with very low systemic exposure levels to the peptide. Subsequently, two double-blind, placebo-controlled phase 2a clinical trials in 40 patients with acute lung injury (ALI) (Krenn et al., 2017) and in 20 lung transplantation patients (Hamacher et al., 2010; Aigner et al., 2017; Ware, 2017) further document safety and efficacy. The main parameters in these studies are extravascular lung water index (EVLWI) -indicative of pulmonary edema and measured with the Picco method- (ALI trial) and days on the ventilator in the latter. In both the acute lung injury trial and the lung transplantation trial, patients receive 2 doses of the peptide versus placebo in the mechanical ventilator over seven days. Although no significant reduction in EVLWI occurs with TIP peptide inhalation over the entire patient population, it is interesting to note that there is a positive effect in a sub-group of patients with a sequential organ failure assessment (SOFA) score >11 (representing more than half of the study population), accompanied by a reduced ventilation pressure (Krenn et al., 2017). In the lung transplant patient group, a significant reduction in days on the ventilator is observed in patients receiving the peptide (Aigner et al., 2017). In 2016, TIP peptide obtains orphan drug designation for acute lung injury (from EMA), for lung transplantation (FDA and EMA) and for high-altitude pulmonary edema (HAPE) (FDA and EMA) (Hamacher et al., 2018). A phase 2b dose escalation study with TIP peptide in ARDS patients (a.k.a. Solnatide, AP301) is currently being conducted in eight university hospitals in Germany and Austria (ClinicalTrials.gov Identifier: NCT03567577](Schmid et al., 2021). Moreover, a phase 2a clinical trial in moderate to severe COVID-19 patients is being conducted at the Medical University Vienna (EudraCT 2020-001244-26).
Results obtained in the clinical trials so far document the translational and therapeutic potential of the TIP peptide -mimicking the lectin-like domain of TNF- for the treatment of alveolar-capillary dysfunction in ARDS and ischemia reperfusion injury after transplantation.
Conclusion
Tumor necrosis factor is a highly complex dual mediator, harboring within its structure spatially distinct domains that can act either in a deleterious or a protective manner in ARDS (Braun et al., 2005; Czikora et al., 2014; Hamacher et al., 2018).
At high concentrations, the role of TNF in the regulation of lung function during pneumonia, ARDS and lung transplantation is traditionally thought to be mainly deleterious, mediated by TNF-TNFR1 signaling and characterized by increased alveolar epithelial and capillary endothelial cell demise, impaired AFC, barrier dysfunction, oxidative stress and hyper-inflammation. The situation is, however, far more complex. First, in certain pathological conditions, such as in ventilator-induced lung injury, the two TNF receptor types can mediate divergent effects upon binding TNF, with TNFR1 being deleterious and TNFR2 rather protective. Second, the lectin-like domain of TNF which is spatially distinct from the receptor binding sites exerts largely protective activities in ARDS and severe pneumonia upon binding to ENaC-α. These protective activities include activation of alveolar fluid clearance, strengthening of alveolar-capillary barrier function, and reduction of inflammation including a lesser production of tissular reactive oxygen species. The lectin-like domain of TNF is with high probability physiologically relevant, since it is clearly active in complexes between TNF and its soluble TNFRs (Braun et al., 2005). Moreover, mice expressing a mutant TNF lacking a functional lectin-like activity have increased lung injury upon bacterial toxin instillation (Czikora et al., 2014).
How the TNF-TNF receptor 1 and TNF receptor 2 actions on the one hand and the TNF lectin-like domain actions on the other hand interact with one another within the TNF homotrimer remains largely unknown and represents an important field of research in the forthcoming years. Not much is known with regard to the contribution of the receptor binding sites versus the lectin-like domain during the evolution of lung injury, its repair or remodeling and concerning mediator co-signaling, alveolar oncotic pressure increase, tissue hypoxia, and many others (Hamacher et al., 2018). Our data suggest that TNF contains within its tertiary structure a negative feedback mechanism able to put a brake on and to even counteract TNF-TNFR-mediated inflammation, as well as barrier and AFC dysfunction in the lungs. The concept to compare outcomes on lung function of clinical trials with neutralizing anti-TNF antibodies versus soluble TNF receptor constructs seems us to be of high interest, since only the latter approach would not interfere with the activities of the protective lectin-like domain of TNF.
The TNF lectin-like region, mimicked by the TIP peptide (a.k.a. Solnatide, AP-301), is showing promising outcomes in recent clinical trials in ARDS and lung transplantation and may represent a prototype for alternative therapeutic strategies, apart from TNF inhibitory antibodies or soluble receptor constructs, to blunt the harmful, but retain the potentially beneficial activities of TNF required in host defense in acute lung injury and ARDS, as well as in bacterial and viral pneumonia, including COVID-19 pneumonia. It should probably further be studied in important clinical settings where thus far no specific treatment exists, such as in idiopathic lung fibrosis.
Much experimental and clinical work has to be done for scientists and clinicians to better understand the role of TNF in lung diseases such as ARDS, in order to be able to therapeutically modulate the cytokine and its associated inflammatory disorders and in order to improve patient outcome and as such health care. Novel mediators and pathways -like those related to the lectin-like domain of TNF- might contribute to this endeavor.
Author Contributions
JH, YH, DE, MR, and RL wrote the manuscript. RL, GC, RC, HH, SS, AL, MH, DA, MU-F, TF, TC, AV, DE, MR, and JH contributed to studies reported in the review. PE, DA, MU-F, TF and TC assisted with redaction of the manuscript for clarity and with the figures. All authors contributed to the article and approved the submitted version.
Funding
Funding was obtained from R01HL138410 (to RL and DE), R01HL135584 and R01HL160014 (to MU-F), R01HL147550 (to MU-F and TF), R01HL116976 (to TF and MU-F), R01HL139562 (to GC), Veterans Administration Merit Review Award 2I01BX001232 (to TF) and by the German Scientific Research Foundation (DFG) through the Collaborative Research Center TRR84 (Project A4, to TC). The work of YH and JH was funded by the Lungen-und Atmungsstiftung, Bern, Switzerland. RL also received funding from this foundation for the preparation of this review.
Conflict of Interest
RL is inventor of several patents relating to the use of the TNF-derived TIP peptide in pulmonary edema reabsorption.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All co-authors dedicate this article to the hardworking nurses and the irreplaceable caregivers at this stressful time: Be it in hospitals, care homes, or home care. JH would also like to encourage those in governmental, insurance, institutional, and related positions to better recognize the current extraordinary burden on nurses, and the need for better pay and improved working conditions for these members of the healthcare system. JH thanks his family for their support in his quest to improve healthcare.
References
Abraham, E., Anzueto, A., Gutierrez, G., Tessler, S. San Pedro, G., Wunderink, R., et al. (1998). Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet, 351, 929–933. doi: 10.1016/S0140-6736(05)60602-2
Abraham, E., Glauser, M. P., Butler, T., Garbino, J., Gelmont, D., Laterre, P. F., et al. (1997). p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 Study Group. JAMA 277, 1531–1538. doi: 10.1001/jama.1997.03540430043031
Abraham, E., Laterre, P. F., Garbino, J., Pingleton, S., Butler, T., Dugernier, T., et al. (2001). Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit. Care Med. 29, 503–510. doi: 10.1097/00003246-200103000-00006
Abraham, E., Wunderink, R., Silverman, H., Perl, T. M., Nasraway, S., Levy, H., et al. (1995). Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome: a randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb sepsis study group. JAMA 273, 934–941. doi: 10.1001/jama.1995.03520360048038
Ackermann, M., Verleden, S. E., Kuehnel, M., Haverich, A., Welte, T., Laenger, F., et al. (2020). Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. New Engl. J. Med. 383, 120–128. doi: 10.1056/NEJMoa2015432
Aderka, D., Engelmann, H., Maor, Y., Brakebusch, C., and Wallach, D. (1992). Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 175, 323–329. doi: 10.1084/jem.175.2.323
Aigner, C., Slama, A., Barta, M., Mitterbauer, A., Lang, G., Taghavi, S., et al. (2017). Treatment of primary graft dysfunction after lung transplantation with orally inhaled AP301: A prospective, randomized pilot study. J. Heart Lung. Transplant 2017:21. doi: 10.1016/j.healun.2017.09.021
Aikawa, N., Takahashi, T., Fujimi, S., Yokoyama, T., Yoshihara, K., Ikeda, T., et al. (2013). A phase II study of polyclonal anti-TNF-α (AZD9773) in Japanese patients with severe sepsis and/or septic shock. J. Infect. Chemother. 19, 931–940. doi: 10.1007/s10156-013-0612-y
Alli, A. A., Bao, H. F., Alli, A. A., Aldrugh, Y., Song, J. Z., Ma, H. P., et al. (2012). Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am. J. Phys. Renal Physiol. 303, F800–F811. doi: 10.1152/ajprenal.00703.2011
Alli, A. A., Bao, H. F., Liu, B. C., Yu, L., Aldrugh, S., Montgomery, D. S., et al. (2015). Calmodulin and CaMKII modulate ENaC activity by regulating the association of MARCKS and the cytoskeleton with the apical membrane. Am. J. Phys. Renal Phys. 309, F456–F463. doi: 10.1152/ajprenal.00631.2014
Amato, M. B., Barbas, C. S., Medeiros, D. M., Magaldi, R. B., Schettino, G. P., Lorenzi-Filho, G., et al. (1998). Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. New Engl. J. Med. 338, 347–354. doi: 10.1056/NEJM199802053380602
Amour, A., Slocombe, P. M., Webster, A., Butler, M., Knight, C. G., Smith, B. J., et al. (1998). TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39–44. doi: 10.1016/s0014-5793(98)01031-x
Angelini, D. J., Hyun, S. W., Grigoryev, D. N., Garg, P., Gong, P., Singh, I. S., et al. (2006). TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mole. Phys. 291, L1232–L1245. doi: 10.1152/ajplung.00109.2006
Archer, C. R., Enslow, B. T., Carver, C. M., and Stockand, J. D. (2020). Phosphatidylinositol 4,5-bisphosphate directly interacts with the β and γ subunits of the sodium channel ENaC. J. Biol. Chem. 295, 7958–7969. doi: 10.1074/jbc.RA120.012606
Atzeni, F., Masala, I. F., Rodríguez-Carrio, J., Ríos-Garcés, R., Gerratana, E., La Corte, L., et al. (2021). The Rheumatology Drugs for COVID-19 Management: Which and When? J. Clin. Med. 10:783. doi: 10.3390/jcm10040783
Bao, H. F., Zhang, Z. R., Liang, Y. Y., Ma, J. J., Eaton, D. C., and Ma, H. P. (2007). Ceramide mediates inhibition of the renal epithelial sodium channel by tumor necrosis factor-alpha through protein kinase C. Am. J. Phys. Renal Phys. 293, F1178–F1186. doi: 10.1152/ajprenal.00153.2007
Bellani, G., Laffey, J. G., Pham, T., Fan, E., Brochard, L., Esteban, A., et al. (2016). Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. Jama 315, 788–800. doi: 10.1001/jama.2016.0291
Bertok, S., Wilson, M. R., Dorr, A. D., Dokpesi, J. O., O’Dea, K. P., Marczin, N., et al. (2011). Characterization of TNF receptor subtype expression and signaling on pulmonary endothelial cells in mice. Am. J. Phys. Lung Cell. Mole Phys. 300, L781–L789. doi: 10.1152/ajplung.00326.2010
Bertok, S., Wilson, M. R., Morley, P. J., de Wildt, R., Bayliffe, A., and Takata, M. (2012). Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax 67, 244–251. doi: 10.1136/thoraxjnl-2011-200590
Beschin, A., Bilej, M., Brys, L., Torreele, E., Lucas, R., Magez, S., et al. (1999). Convergent evolution of cytokines. Nature 400, 627–628. doi: 10.1038/23164
Beschin, A., Bilej, M., Magez, S., Lucas, R., and De Baetselier, P. (2004). Functional convergence of invertebrate and vertebrate cytokine-like molecules based on a similar lectin-like activity. Prog. Mol. Subcell Biol. 34, 145–163. doi: 10.1007/978-3-642-18670-7_6
Bodmer, J.-L., Schneider, P., and Tschopp, J. (2002). The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 27, 19–26. doi: 10.1016/s0968-0004(01)01995-8
Börjesson, A., Norlin, A., Wang, X., Andersson, R., and Folkesson, H. G. (2000). TNF-alpha stimulates alveolar liquid clearance during intestinal ischemia-reperfusion in rats. Am. J. Physiol. Lung Cell. Mole. Physiol. 278, L3–L12. doi: 10.1152/ajplung.2000.278.1.L3
Braun, C., Hamacher, J., Morel, D. R., Wendel, A., and Lucas, R. (2005). Dichotomal role of TNF in experimental pulmonary edema reabsorption. J. Immunol. 175, 3402–3408. doi: 10.4049/jimmunol.175.5.3402
Brenner, D., Blaser, H., and Mak, T. W. (2015). Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15, 362–374. doi: 10.1038/nri3834
Brouckaert, P. G., Fiers, W., and Lejeune, F. J. (1992). Coley’s vaccine and TNF therapy. Nature 358:630. doi: 10.1038/358630a0
Burke-Gaffney, A., and Hellewell, P. G. (1996). Tumour necrosis factor-alpha-induced ICAM-1 expression in human vascular endothelial and lung epithelial cells: modulation by tyrosine kinase inhibitors. Br. J. Pharmacol. 119, 1149–1158. doi: 10.1111/j.1476-5381.1996.tb16017.x
Cao, Y., Zhang, X., Wang, L., Yang, Q., Ma, Q., Xu, J., et al. (2019). PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. U S A. 116, 13394–13403. doi: 10.1073/pnas.1821401116
Carswell, E. A., Old, L. J., Kassel, R. L., Green, S., Fiore, N., and Williamson, B. (1975). An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U S A. 72, 3666–3670. doi: 10.1073/pnas.72.9.3666
Caty, M. G., Guice, K. S., Oldham, K. T., Remick, D. G., and Kunkel, S. I. (1990). Evidence for tumor necrosis factor-induced pulmonary microvascular injury after intestinal ischemia-reperfusion injury. Ann. Surg. 212, 694–700. doi: 10.1097/00000658-199012000-00007
Chandra, S., Romero, M. J., Shatanawi, A., Alkilany, A. M., Caldwell, R. B., and Caldwell, R. W. (2012). Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 165, 506–519. doi: 10.1111/j.1476-5381.2011.01584.x
Chang, C. C., Wang, S. S., Hsieh, H. G., Lee, W. S., Chuang, C. L., Lin, H. C., et al. (2015). Rosuvastatin improves hepatopulmonary syndrome through inhibition of inflammatory angiogenesis of lung. Clin. Sci. 129, 449–460. doi: 10.1042/cs20140622
Channappanavar, R., Fehr, A. R., Vijay, R., Mack, M., Zhao, J., Meyerholz, D. K., et al. (2016). Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 19, 181–193. doi: 10.1016/j.chom.2016.01.007
Chatterjee, S., and Fisher, A. B. (2014). Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal. 20, 899–913. doi: 10.1089/ars.2013.5624
Chatterjee, S., Fujiwara, K., Pérez, N. G., Ushio-Fukai, M., and Fisher, A. B. (2015). Mechanosignaling in the vasculature: emerging concepts in sensing, transduction and physiological responses. Am. J. Phys. Heart Circul. Phys. 308, H1451–H1462. doi: 10.1152/ajpheart.00105.2015
Chen, G., and Goeddel, D. V. (2002). TNF-R1 Signaling: A Beautiful Pathway. Science 296:1634. doi: 10.1126/science.1071924
Chen, G.-F., Sudhahar, V., Youn, S.-W., Das, A., Cho, J., Kamiya, T., et al. (2015). Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 5:14780. doi: 10.1038/srep14780
Cicha, I., Beronov, K., Ramirez, E. L., Osterode, K., Goppelt-Struebe, M., Raaz, D., et al. (2009). Shear stress preconditioning modulates endothelial susceptibility to circulating TNF-alpha and monocytic cell recruitment in a simplified model of arterial bifurcations. Atherosclerosis 207, 93–102. doi: 10.1016/j.atherosclerosis.2009.04.034
Clark, M. A., Plank, L. D., Connolly, A. B., Streat, S. J., Hill, A. A., Gupta, R., et al. (1998). Effect of a chimeric antibody to tumor necrosis factor-alpha on cytokine and physiologic responses in patients with severe sepsis–a randomized, clinical trial. Crit. Care Med. 26, 1650–1659. doi: 10.1097/00003246-199810000-00016
Clauss, M., Grell, M., Fangmann, C., Fiers, W., Scheurich, P., and Risau, W. (1996). Synergistic induction of endothelial tissue factor by tumor necrosis factor and vascular endothelial growth factor: functional analysis of the tumor necrosis factor receptors. FEBS Lett. 390, 334–338. doi: 10.1016/0014-5793(96)00690-4
Cohen, J., and Carlet, J. (1996). INTERSEPT: an international, multicenter, placebocontrolled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International sepsis trial study group. Crit. Care Med. 24, 1431–1440. doi: 10.1097/00003246-199609000-00002
Coley, W. B. (1910). The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 3, 1–48.
Conrad, M., Angeli, J. P. F., Vandenabeele, P., and Stockwell, B. R. (2016). Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Dis. 15, 348–366. doi: 10.1038/nrd.2015.6
Czikora, I., Alli, A., Bao, H. F., Kaftan, D., Sridhar, S., Apell, H. J., et al. (2014). A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am. J. Respirat. Crit. Care Med. 190, 522–532. doi: 10.1164/rccm.201405-0833OC
Czikora, I., Alli, A. A., Sridhar, S., Matthay, M. A., Pillich, H., Hudel, M., et al. (2017). Epithelial Sodium Channel-alpha Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 8:842. doi: 10.3389/fimmu.2017.00842
D’Agnillo, F., Walters, K. A., Xiao, Y., Sheng, Z. M., Scherler, K., Park, J., et al. (2021). Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 13:eabj7790. doi: 10.1126/scitranslmed.abj7790
Dagenais, A., Fréchette, R., Yamagata, Y., Yamagata, T., Carmel, J. F., Clermont, M. E., et al. (2004). Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mole. Physiol. 286, L301–L311. doi: 10.1152/ajplung.00326.2002
Das, A., Sudhahar, V., Ushio-Fukai, M., and Fukai, T. (2019). Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. Am. J. Physiol. Cell Physiol. 317, C1161–C1171. doi: 10.1152/ajpcell.00264.2019
Daulouède, S., Bouteille, B., Moynet, D., De Baetselier, P., Courtois, P., Lemesre, J. L., et al. (2001). Human macrophage tumor necrosis factor (TNF)-alpha production induced by Trypanosoma brucei gambiense and the role of TNF-alpha in parasite control. J. Infect Dis. 183, 988–991. doi: 10.1086/319257
Del Valle, D. M., Kim-Schulze, S., Huang, H. H., Beckmann, N. D., Nirenberg, S., Wang, B., et al. (2020). An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643. doi: 10.1038/s41591-020-1051-9
Delvaeye, T., De Smet, M. A. J., Verwaerde, S., Decrock, E., Czekaj, A., Vandenbroucke, R. E., et al. (2019). Blocking connexin43 hemichannels protects mice against tumour necrosis factor-induced inflammatory shock. Sci. Rep. 9:16623. doi: 10.1038/s41598-019-52900-4
Deng, B., Xie, S., Wang, J., Xia, Z., and Nie, R. (2012). Inhibition of protein kinase C β(2) prevents tumor necrosis factor-α-induced apoptosis and oxidative stress in endothelial cells: the role of NADPH oxidase subunits. J. Vasc. Res. 49, 144–159. doi: 10.1159/000332337
Dhainaut, J. F., Vincent, J. L., Richard, C., Lejeune, P., Martin, C., Fierobe, L., et al. (1995). CDP571, a humanized antibody to human tumor necrosis factor-alpha: safety, pharmacokinetics, immune response, and influence of the antibody on cytokine concentrations in patients with septic shock. CPD571 sepsis study group. Crit. Care Med. 23, 1461–1469. doi: 10.1097/00003246-199509000-00004
Di Lorenzo, A., Lin, M. I., Murata, T., Landskroner-Eiger, S., Schleicher, M., Kothiya, M., et al. (2013). eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 126(Pt 24), 5541–5552. doi: 10.1242/jcs.115972
Doerschuk, C. M. (2001). Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation 8, 71–88. doi: 10.1111/j.1549-8719.2001.tb00159.x
Drummond, G. R., and Sobey, C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25, 452–463. doi: 10.1016/j.tem.2014.06.012
Du, Z. S., Xu, H., and Liu, M. C. (2021). A retrospective analysis of medication in children with SARS-CoV-2 infection in Wuhan. Chinese J. Contemp. Pediat. 23, 61–66. doi: 10.7499/j.issn.1008-8830.2007212
Eaton, D. C., Chen, J., Ramosevac, S., Matalon, S., and Jain, L. (2004). Regulation of Na+ channels in lung alveolar type II epithelial cells. Proc. Am. Thorac. Soc. 1, 10–16. doi: 10.1513/pats.2306008
Eisenhut, M., and Shin, J. I. (2020). Pathways in the Pathophysiology of Coronavirus 19 Lung Disease Accessible to Prevention and Treatment. Front. Physiol. 11:872. doi: 10.3389/fphys.2020.00872
Elia, N., Tapponnier, M., Matthay, M. A., Hamacher, J., Pache, J. C., Brundler, M. A., et al. (2003). Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am. J. Respir. Crit. Care Med. 168, 1043–1050. doi: 10.1164/rccm.200206-618OC
Favre, N., Da Laperousaz, C., Ryffel, B., Weiss, N. A., Imhof, B. A., Rudin, W., et al. (1999). Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microb. Infect. 1, 961–968. doi: 10.1016/s1286-4579(99)80513-9
Feldmann, M., Maini, R. N., Woody, J. N., Holgate, S. T., Winter, G., Rowland, M., et al. (2020). Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395, 1407–1409. doi: 10.1016/S0140-6736(20)30858-8
Fisher, C. J. Jr., Agosti, J. M., Opal, S. M., Lowry, S. F., Balk, R. A., Sadoff, J. C., et al. (1996). Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The soluble TNF receptor sepsis study group. N. Engl. J. Med. 334, 1697–1702. doi: 10.1056/nejm199606273342603
Fischer, R., Kontermann, R. E., and Pfizenmaier, K. (2020). Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 8:401. doi: 10.3389/fcell.2020.00401
Fisher, C. J. Jr., Opal, S. M., Dhainaut, J. F., Stephens, S., Zimmerman, J. L., Nightingale, P., et al. (1993). Influence of an anti-tumor necrosis factor monoclonal antibody on cytokine levels in patients with sepsis. The CB0006 sepsis syndrome study group. Crit. Care Med. 21, 318–327. doi: 10.1097/00003246-199303000-00006
Fransen, L., Müller, R., Marmenout, A., Tavernier, J., Van der Heyden, J., Kawashima, E., et al. (1985). Molecular cloning of mouse tumour necrosis factor cDNA and its eukaryotic expression. Nucleic Acids Res. 13, 4417–4429. doi: 10.1093/nar/13.12.4417
Frey, R. S., Gao, X., Javaid, K., Siddiqui, S. S., Rahman, A., and Malik, A. B. (2006). Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-kappaB activation in endothelial cells. J. Biol. Chem. 281, 16128–16138. doi: 10.1074/jbc.M508810200
Fukuda, N., Jayr, C., Lazrak, A., Wang, Y., Lucas, R., Matalon, S., et al. (2001). Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am. J. Physiol. Lung Cell. Mole. Physiol. 280, L1258–L1265. doi: 10.1152/ajplung.2001.280.6.L1258
Gallagher, J., Fisher, C., Sherman, B., Munger, M., Meyers, B., Ellison, T., et al. (2001). A multicenter, open-label, prospective, randomized, dose-ranging pharmacokinetic study of the anti-TNF-alpha antibody afelimomab in patients with sepsis syndrome. Intensive Care Med. 27, 1169–1178. doi: 10.1007/s001340100973
Gallelli, L., Zhang, L., Wang, T., and Fu, F. (2020). Severe Acute Lung Injury Related to COVID-19 Infection: A Review and the Possible Role for Escin. J. Clin. Pharm. 60, 815–825. doi: 10.1002/jcph.1644
Gao, X., Xu, X., Belmadani, S., Park, Y., Tang, Z., Feldman, A. M., et al. (2007). TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 27, 1269–1275. doi: 10.1161/ATVBAHA.107.142521
Gardam, M. A., Keystone, E. C., Menzies, R., Manners, S., Skamene, E., Long, R., et al. (2003). Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155. doi: 10.1016/s1473-3099(03)00545-0
Garin, G., Abe, J., Mohan, A., Lu, W., Yan, C., Newby, A. C., et al. (2007). Flow antagonizes TNF-alpha signaling in endothelial cells by inhibiting caspase-dependent PKC zeta processing. Circul. Res. 101, 97–105. doi: 10.1161/CIRCRESAHA.107.148270
Gertzberg, N., Neumann, P., Rizzo, V., and Johnson, A. (2004). NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am. J. Physiol. Lung Cell. Mole. Phys. 286, L37–L48. doi: 10.1152/ajplung.00116.2003
Gattinoni, L., Chiumello, D., and Rossi, S. (2020). COVID-19 pneumonia: ARDS or not? Crit. Care 24:154. doi: 10.1186/s13054-020-02880-z
Gheblawi, M., Wang, K., Viveiros, A., Nguyen, Q., Zhong, J. C., Turner, A. J., et al. (2020). Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circul. Res. 126, 1456–1474. doi: 10.1161/circresaha.120.317015
Goldblum, S. E., Ding, X., and Campbell-Washington, J. (1993). TNF-alpha induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am. J. Physiol. 264(4 Pt 1), C894–C905. doi: 10.1152/ajpcell.1993.264.4.C894
Gorelik, A., Illes, K., Heinz, L. X., Superti-Furga, G., and Nagar, B. (2016). Crystal structure of mammalian acid sphingomyelinase. Nat. Comm. 7:12196. doi: 10.1038/ncomms12196
Goto, T., Ishizaka, A., Kobayashi, F., Kohno, M., Sawafuji, M., Tasaka, S., et al. (2004). Importance of tumor necrosis factor-alpha cleavage process in post-transplantation lung injury in rats. Am. J. Respir. Crit. Care Med. 170, 1239–1246. doi: 10.1164/rccm.200402-146OC
Grau, G. E., Mili, N., Lou, J. N., Morel, D. R., Ricou, B., Lucas, R., et al. (1996). Phenotypic and functional analysis of pulmonary microvascular endothelial cells from patients with acute respiratory distress syndrome. Lab. Investig. 74, 761–770.
Grell, M., Douni, E., Wajant, H., Löhden, M., Clauss, M., Maxeiner, B., et al. (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83, 793–802. doi: 10.1016/0092-8674(95)90192-2
Gu, T., Zhao, S., Jin, G., Song, M., Zhi, Y., Zhao, R., et al. (2020). Cytokine Signature Induced by SARS-CoV-2 Spike Protein in a Mouse Model. Front. Immunol. 11:621441. doi: 10.3389/fimmu.2020.621441
Guérin, C., Reignier, J., Richard, J. C., Beuret, P., Gacouin, A., Boulain, T., et al. (2013). Prone positioning in severe acute respiratory distress syndrome. New Engl. J. Med. 368, 2159–2168.
Haga, S., Yamamoto, N., Nakai-Murakami, C., Osawa, Y., Tokunaga, K., Sata, T., et al. (2008). Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. U S A. 105, 7809–7814. doi: 10.1073/pnas.0711241105
Haimovitz-Friedman, A., Cordon-Cardo, C., Bayoumy, S., Garzotto, M., McLoughlin, M., Gallily, R., et al. (1997). Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J. Exp. Med. 186, 1831–1841. doi: 10.1084/jem.186.11.1831
Hamacher, J., Hadizamani, Y., Borgmann, M., Mohaupt, M., Männel, D. N., Moehrlen, U., et al. (2018). Cytokine–Ion Channel Interactions in Pulmonary Inflammation. Front. Imm. 2018:8.
Hamacher, J., Lucas, R., Lijnen, H. R., Buschke, S., Dunant, Y., Wendel, A., et al. (2002). Tumor necrosis factor-alpha and angiostatin are mediators of endothelial cytotoxicity in bronchoalveolar lavages of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 166, 651–656. doi: 10.1164/rccm.2109004
Hamacher, J., Stammberger, U., Roux, J., Kumar, S., Yang, G., Xiong, C., et al. (2010). The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation–potential role for a reduction in reactive oxygen species generation. Crit. Care Med. 38, 871–878. doi: 10.1097/CCM.0b013e3181cdf725
Hardyman, M. A., Wilkinson, E., Martin, E., Jayasekera, N. P., Blume, C., Swindle, E. J., et al. (2013). TNF-α-mediated bronchial barrier disruption and regulation by src-family kinase activation. J. Allergy Clin. Immunol. 132, 665.e–675.e. doi: 10.1016/j.jaci.2013.03.005
Hariri, L., and Hardin, C. C. (2020). Covid-19, Angiogenesis, and ARDS Endotypes. New Engl. J. Med. 383, 182–183. doi: 10.1056/NEJMe2018629
Hartmann, E. K., Boehme, S., Duenges, B., Bentley, A., Klein, K. U., Kwiecien, R., et al. (2013). An inhaled tumor necrosis factor-alpha-derived TIP peptide improves the pulmonary function in experimental lung injury. Acta Anaesthesiol. Scand. 57, 334–341. doi: 10.1111/aas.12034
Hartmann, E. K., Ziebart, A., Thomas, R., Liu, T., Schad, A., Tews, M., et al. (2015). Inhalation therapy with the synthetic TIP-like peptide AP318 attenuates pulmonary inflammation in a porcine sepsis model. BMC Pulm. Med. 15:7. doi: 10.1186/s12890-015-0002-6
Hazemi, P., Tzotzos, S. J., Fischer, B., Andavan, G. S. B., Fischer, H., Pietschmann, H., et al. (2010). Essential structural features of TNF-α lectin-like domain derived peptides for activation of amiloride-sensitive sodium current in A549 cells. J. Med. Chem. 53, 8021–8029. doi: 10.1021/jm100767p
Hession, C., Decker, J. M., Sherblom, A. P., Kumar, S., Yue, C. C., Mattaliano, R. J., et al. (1987). Uromodulin (Tamm-Horsfall glycoprotein): a renal ligand for lymphokines. Science 237, 1479–1484. doi: 10.1126/science.3498215
Hocking, D. C., Ferro, T. J., and Johnson, A. (1991). Dextran sulfate inhibits PMN-dependent hydrostatic pulmonary edema induced by tumor necrosis factor. J. Appl. Physiol. 70, 1121–1128. doi: 10.1152/jappl.1991.70.3.1121
Hocking, D. C., Phillips, P. G., Ferro, T. J., and Johnson, A. (1990). Mechanisms of pulmonary edema induced by tumor necrosis factor-alpha. Circ. Res. 67, 68–77. doi: 10.1161/01.res.67.1.68
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271.e–280.e.
Horgan, M. J., Palace, G. P., Everitt, J. E., and Malik, A. B. (1993). TNF-alpha release in endotoxemia contributes to neutrophil-dependent pulmonary edema. Am. J. Physiol. 264(4 Pt 2), H1161–H1165. doi: 10.1152/ajpheart.1993.264.4.H1161
Horiuchi, T., Mitoma, H., Harashima, S., Tsukamoto, H., and Shimoda, T. (2010). Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology 49, 1215–1228. doi: 10.1093/rheumatology/keq031
Houston, B. L., Lawler, P. R., Goligher, E. C., Farkouh, M. E., Bradbury, C., Carrier, M., et al. (2020). Anti-thrombotic therapy to ameliorate complications of COVID-19 (ATTACC): study design and methodology for an international, adaptive Bayesian randomized controlled trial. Clin. Trials 17, 491–500. doi: 10.1177/1740774520943846
Hribar, M., Bloc, A., van der Goot, F. G., Fransen, L., De Baetselier, P., Grau, G. E., et al. (1999). The lectin-like domain of tumor necrosis factor-alpha increases membrane conductance in microvascular endothelial cells and peritoneal macrophages. Eur. J. Immunol. 29, 3105–3111. doi: 10.1002/(SICI)1521-4141(199910)29:10<3105::AID-IMMU3105>3.0.CO;2-A
Hummler, E., Barker, P., Gatzy, J., Beermann, F., Verdumo, C., Schmidt, A., et al. (1996). Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 12, 325–328. doi: 10.1038/ng0396-325
Imai, Y., Kawano, T., Iwamoto, S., Nakagawa, S., Takata, M., and Miyasaka, K. (1999). Intratracheal anti-tumor necrosis factor-alpha antibody attenuates ventilator-induced lung injury in rabbits. J. Appl. Physiol. 87, 510–515. doi: 10.1152/jappl.1999.87.2.510
Jamilloux, Y., Henry, T., Belot, A., Viel, S., Fauter, M., El Jammal, T., et al. (2020). Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 19:102567. doi: 10.1016/j.autrev.2020.102567
Jang, E. A., Kim, J. Y., Tin, T. D., Song, J. A., Lee, S. H., and Kwak, S. H. (2019). The effects of BMS-470539 on lipopolysaccharide-induced acute lung injury. Acute Crit. Care 34, 133–140. doi: 10.4266/acc.2019.00507
Ji, H. L., Zhao, R. Z., Chen, Z. X., Shetty, S., Idell, S., and Matalon, S. (2012). δ ENaC: a novel divergent amiloride-inhibitable sodium channel. Am. J. Physiol. Lung Cell. Mole. Physiol. 303, L1013–L1026. doi: 10.1152/ajplung.00206.2012
Johnson, M. D., Bao, H.-F., Helms, M. N., Chen, X.-J., Tigue, Z., Jain, L., et al. (2006). Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc. Natl. Acad. Sci. U S A. 103, 4964–4969. doi: 10.1073/pnas.0600855103
Johnson, M. D., Widdicombe, J. H., Allen, L., Barbry, P., and Dobbs, L. G. (2002). Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc. Natl. Acad. Sci. U S A. 99, 1966–1971. doi: 10.1073/pnas.042689399
Keane, J., Gershon, S., Wise, R. P., Mirabile-Levens, E., Kasznica, J., Schwieterman, W. D., et al. (2001). Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. New Engl. J. Med. 345, 1098–1104. doi: 10.1056/NEJMoa011110
Kim, H., Hwang, J. S., Woo, C. H., Kim, E. Y., Kim, T. H., Cho, K. J., et al. (2008). TNF-alpha-induced up-regulation of intercellular adhesion molecule-1 is regulated by a Rac-ROS-dependent cascade in human airway epithelial cells. Exp. Mole. Med. 40, 167–175. doi: 10.3858/emm.2008.40.2.167
Kleyman, T. R., and Eaton, D. C. (2020). Regulating ENaC’s gate. Am. J. Physiol. Cell Physiol. 318, C150–C162. doi: 10.1152/ajpcell.00418.2019
Koh, Y., Hybertson, B. M., Jepson, E. K., and Repine, J. E. (1996). Tumor necrosis factor induced acute lung leak in rats: less than with interleukin-1. Inflammation 20, 461–469. doi: 10.1007/bf01487039
Koss, M., Pfeiffer, G. R. II, Wang, Y., Thomas, S. T., Yerukhimovich, M., Gaarde, W. A., et al. (2006). Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J. Immunol. 176, 1218–1227. doi: 10.4049/jimmunol.176.2.1218
Krenn, K., Croize, A., Klein, K. U., Böhme, S., Markstaller, K., Ullrich, R., et al. (2014). Oral inhalation of AP301 peptide activates pulmonary oedema clearance: initial results from a phase IIa clinical trial in mechanically ventilated ICU patients. Eur. Respir. J. 44(Suppl. 58):1386.
Krenn, K., Lucas, R., Croizé, A., Boehme, S., Klein, K. U., Hermann, R., et al. (2017). Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: a phase IIa randomized placebo-controlled trial. Crit. Care 21:194. doi: 10.1186/s13054-017-1795-x
Kriegler, M., Perez, C., DeFay, K., Albert, I., and Lu, S. D. (1988). A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell 53, 45–53. doi: 10.1016/0092-8674(88)90486-2
Kritas, S. K., Ronconi, G., Caraffa, A., Gallenga, C. E., Ross, R., and Conti, P. (2020). Mast cells contribute to coronavirus-induced inflammation: new anti-inflammatory strategy. J. Biol. Regul. Homeost. Agents 34, 9–14. doi: 10.23812/20-Editorial-Kritas
Kwon, O. J., Au, B. T., Collins, P. D., Adcock, I. M., Mak, J. C., Robbins, R. R., et al. (1994). Tumor necrosis factor-induced interleukin-8 expression in cultured human airway epithelial cells. Am. J. Physiol. 267(4 Pt 1), L398–L405. doi: 10.1152/ajplung.1994.267.4.L398
Lei, C. Q., Wu, X., Zhong, X., Jiang, L., Zhong, B., and Shu, H. B. (2019). USP19 Inhibits TNF-α- and IL-1β-Triggered NF-κB Activation by Deubiquitinating TAK1. J. Imm. 203, 259–268. doi: 10.4049/jimmunol.1900083
Lejeune, F. J., Liénard, D., Matter, M., and Rüegg, C. (2006). Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. 6:6. doi: 10.1016/0165-4608(89)90205-7
Leppkes, M., Knopf, J., Naschberger, E., Lindemann, A., Singh, J., Herrmann, I., et al. (2020). Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 58:102925. doi: 10.1016/j.ebiom.2020.102925
Li, N., Liu, X. X., Hong, M., Huang, X. Z., Chen, H., Xu, J. H., et al. (2018). Sodium butyrate alleviates LPS-induced acute lung injury in mice via inhibiting HMGB1 release. Internat. Imm. 56, 242–248. doi: 10.1016/j.intimp.2018.01.017
Li, X., Yang, Y., and Ashwell, J. D. (2002). TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416, 345–347. doi: 10.1038/416345a
Li, X. Y., Donaldson, K., Brown, D., and MacNee, W. (1995). The role of tumor necrosis factor in increased airspace epithelial permeability in acute lung inflammation. Am. J. Respir. Cell Mol. Biol. 13, 185–195. doi: 10.1165/ajrcmb.13.2.7626286
Libby, P., and Lüscher, T. (2020). COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 41, 3038–3044. doi: 10.1093/eurheartj/ehaa623
Lipke, A. B., Matute-Bello, G., Herrero, R., Kurahashi, K., Wong, V. A., Mongovin, S. M., et al. (2010). Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis. J. Imm. 184, 3801–3813. doi: 10.4049/jimmunol.0903191
Liu, B., Cheng, Y., Wu, Y., Zheng, X., Li, X., Yang, G., et al. (2020). Emodin improves alveolar hypercoagulation and inhibits pulmonary inflammation in LPS-provoked ARDS in mice via NF-κB inactivation. Intern. Imm. 88:107020. doi: 10.1016/j.intimp.2020.107020
Long, Y., Xiang, Y., Liu, S., Zhang, Y., Wan, J., Yang, Q., et al. (2020). Baicalin Liposome Alleviates Lipopolysaccharide-Induced Acute Lung Injury in Mice via Inhibiting TLR4/JNK/ERK/NF-κB Pathway. Med. Inflamm. 2020:8414062. doi: 10.1155/2020/8414062
Lo, S. K., Everitt, J., Gu, J., and Malik, A. B. (1992). Tumor necrosis factor mediates experimental pulmonary edema by ICAM-1 and CD18-dependent mechanisms. J. Clin. Invest. 89, 981–988. doi: 10.1172/JCI115681
Lou, J., Lucas, R., and Grau, G. E. (2001). Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin. Microb. Rev. 14, 810–820. doi: 10.1128/CMR.14.4.810-820.2001
Lucas, R., Czikora, I., Sridhar, S., Zemskov, E. A., Oseghale, A., Circo, S., et al. (2013). Arginase 1: an unexpected mediator of pulmonary capillary barrier dysfunction in models of acute lung injury. Front Immunol. 4:228. doi: 10.3389/fimmu.2013.00228
Lucas, R., Echtenacher, B., Sablon, E., Juillard, P., Magez, S., Lou, J., et al. (1997a). Generation of a mouse tumor necrosis factor mutant with antiperitonitis and desensitization activities comparable to those of the wild type but with reduced systemic toxicity. Infect Immun. 65, 2006–2010. doi: 10.1128/iai.65.6.2006-2010.1997
Lucas, R., Garcia, I., Donati, Y. R., Hribar, M., Mandriota, S. J., Giroud, C., et al. (1998). Both TNF receptors are required for direct TNF-mediated cytotoxicity in microvascular endothelial cells. Eur. J. Imm. 28, 3577–3586. doi: 10.1002/(SICI)1521-4141(199811)28:11<3577::AID-IMMU3577>3.0.CO;2-#
Lucas, R., Heirwegh, K., Neirynck, A., Remels, L., Van Heuverswyn, H., and De Baetselier, P. (1990). Generation and characterization of a neutralizing rat anti-rmTNF-alpha monoclonal antibody. Immunology 71, 218–223.
Lucas, R., Juillard, P., Decoster, E., Redard, M., Burger, D., Donati, Y., et al. (1997b). Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur. J. Imm. 27, 1719–1725. doi: 10.1002/eji.1830270719
Lucas, R., Lou, J., Morel, D. R., Ricou, B., Suter, P. M., and Grau, G. E. (1997c). TNF receptors in the microvascular pathology of acute respiratory distress syndrome and cerebral malaria. J. Leukocyte Biol. 61, 551–558. doi: 10.1002/jlb.61.5.551
Lucas, R., Magez, S., De Leys, R., Fransen, L., Scheerlinck, J. P., Rampelberg, M., et al. (1994). Mapping the lectin-like activity of tumor necrosis factor. Science 263, 814–817. doi: 10.1126/science.8303299
Lucas, R., Sridhar, S., Rick, F. G., Gorshkov, B., Umapathy, N. S., Yang, G., et al. (2012a). Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc. Natl. Acad. Sci. U S A. 109, 2084–2089. doi: 10.1073/pnas.1121075109
Lucas, R., Yang, G., Gorshkov, B. A., Zemskov, E. A., Sridhar, S., Umapathy, N. S., et al. (2012b). Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mole. Biol. 47, 445–453. doi: 10.1165/rcmb.2011-0332OC
Lucas, R., Yue, Q., Alli, A., Duke, B. J., Al-Khalili, O., Thai, T. L., et al. (2016). The Lectin-like Domain of TNF Increases ENaC Open Probability through a Novel Site at the Interface between the Second Transmembrane and C-terminal Domains of the α-Subunit. J. Biol. Chem. 291, 23440–23451. doi: 10.1074/jbc.M116.718163
Madaio, M. P., Czikora, I., Kvirkvelia, N., McMenamin, M., Yue, Q., Liu, T., et al. (2019). The TNF-derived TIP peptide activates the epithelial sodium channel and ameliorates experimental nephrotoxic serum nephritis. Kidney Internat. 95, 1359–1372. doi: 10.1016/j.kint.2018.12.022
Magez, S., Geuskens, M., Beschin, A., del Favero, H., Verschueren, H., Lucas, R., et al. (1997). Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J. Cell Biol. 137, 715–727. doi: 10.1083/jcb.137.3.715
Martínez-Reza, I., Díaz, L., and García-Becerra, R. (2017). Preclinical and clinical aspects of TNF-α and its receptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 24:90. doi: 10.1186/s12929-017-0398-9
Matsumura, T., Wolff, K., and Petzelbauer, P. (1997). Endothelial cell tube formation depends on cadherin 5 and CD31 interactions with filamentous actin. J. Immunol. 158, 3408–3416.
Matthay, M. A., Leligdowicz, A., and Liu, K. D. (2020). Biological Mechanisms of COVID-19 Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 202, 1489–1491. doi: 10.1164/rccm.202009-3629ED
Matthay, M. A., Robriquet, L., and Fang, X. (2005). Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc. Am. Thorac. Soc. 2, 206–213. doi: 10.1513/pats.200501-009AC
Mayer, G., Boileau, G., and Bendayan, M. (2004). Sorting of furin in polarized epithelial and endothelial cells: expression beyond the Golgi apparatus. J. Histochem. Cytochem. 52, 567–579. doi: 10.1177/002215540405200502
Mazzon, E., and Cuzzocrea, S. (2007). Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir. Res. 8:75. doi: 10.1186/1465-9921-8-75
Mechtcheriakova, D., Schabbauer, G., Lucerna, M., Clauss, M., De Martin, R., Binder, B. R., et al. (2001). Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor up-regulation via EGR-1 in endothelial cells. FASEB J. 15, 230–242. doi: 10.1096/fj.00-0247com
Medler, J., and Wajant, H. (2019). Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Exp. Opin. Ther. Targets 23, 295–307. doi: 10.1080/14728222.2019.1586886
Melo, A. C., Valença, S. S., Gitirana, L. B., Santos, J. C., Ribeiro, M. L., Machado, M. N., et al. (2013). Redox markers and inflammation are differentially affected by atorvastatin, pravastatin or simvastatin administered before endotoxin-induced acute lung injury. Int. Immunopharmacol. 17, 57–64. doi: 10.1016/j.intimp.2013.05.016
Meyrick, B., Christman, B., and Jesmok, G. (1991). Effects of recombinant tumor necrosis factor-alpha on cultured pulmonary artery and lung microvascular endothelial monolayers. Am. J. Pathol. 138, 93–101.
Mohammed, F. F., Smookler, D. S., Taylor, S. E., Fingleton, B., Kassiri, Z., Sanchez, O. H., et al. (2004). Abnormal TNF activity in Timp3-/- mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat. Genet. 36, 969–977. doi: 10.1038/ng1413
Monteil, V., Kwon, H., Prado, P., Hagelkrüys, A., Wimmer, R. A., Stahl, M., et al. (2020). Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 181, 905.e–913.e. doi: 10.1016/j.cell.2020.04.004
Moreira, A. L., Sampaio, E. P., Zmuidzinas, A., Frindt, P., Smith, K. A., and Kaplan, G. (1993). Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 177, 1675–1680. doi: 10.1084/jem.177.6.1675
Morris, G., Bortolasci, C. C., Puri, B. K., Olive, L., Marx, W. O’Neil, A., et al. (2021). Preventing the development of severe COVID-19 by modifying immunothrombosis. Life Sci. 264:118617. doi: 10.1016/j.lfs.2020.118617
Morris, P. E., Zeno, B., Bernard, A. C., Huang, X., Das, S., Edeki, T., et al. (2012). A placebo-controlled, double-blind, dose-escalation study to assess the safety, tolerability and pharmacokinetics/pharmacodynamics of single and multiple intravenous infusions of AZD9773 in patients with severe sepsis and septic shock. Crit. Care 16:R31. doi: 10.1186/cc11203
Motta Junior, J. D. S., Miggiolaro, A., Nagashima, S., de Paula, C. B. V., Baena, C. P., Scharfstein, J., et al. (2020). Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front. Immunol. 11:574862. doi: 10.3389/fimmu.2020.574862
Mukhopadhyay, S., Hoidal, J. R., and Mukherjee, T. K. (2006). Role of TNFalpha in pulmonary pathophysiology. Respir. Res. 7:125. doi: 10.1186/1465-9921-7-125
Nachman, R. L., and Rafii, S. (2008). Platelets, petechiae, and preservation of the vascular wall. New Engl. J. Med. 359, 1261–1270. doi: 10.1056/NEJMra0800887
Nakamura, H., Yoshimura, K., Bajocchi, G., Trapnell, B. C., Pavirani, A., and Crystal, R. G. (1992). Tumor necrosis factor modulation of expression of the cystic fibrosis transmembrane conductance regulator gene. FEBS Lett. 314, 366–370. doi: 10.1016/0014-5793(92)81507-i
Neumann, P., Gertzberg, N., and Johnson, A. (2004). TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell. Mole. Physiol. 286, L452–L459. doi: 10.1152/ajplung.00378.2002
Neumann, P., Gertzberg, N., Vaughan, E., Weisbrot, J., Woodburn, R., Lambert, W., et al. (2006). Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am. J. Physiol. Lung Cell. Mole. Physiol. 290, L674–L684. doi: 10.1152/ajplung.00391.2005
Nieuwdorp, M., Meuwese, M. C., Mooij, H. L., van Lieshout, M. H., Hayden, A., Levi, M., et al. (2009). Tumor necrosis factor-alpha inhibition protects against endotoxin-induced endothelial glycocalyx perturbation. Atherosclerosis 202, 296–303. doi: 10.1016/j.atherosclerosis.2008.03.024
Noel, J., Wang, H., Hong, N., Tao, J. Q., Yu, K., Sorokina, E. M., et al. (2013). PECAM-1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium. Am. J. Physiol. Lung Cell. Mole. Physiol. 305, L805–L818. doi: 10.1152/ajplung.00123.2013
Olson, E. J., Standing, J. E., Griego-Harper, N., Hoffman, O. A., and Limper, A. H. (1996). Fungal beta-glucan interacts with vitronectin and stimulates tumor necrosis factor alpha release from macrophages. Infect. Immun. 64, 3548–3554. doi: 10.1128/iai.64.9.3548-3554.1996
Panacek, E. A., Marshall, J. C., Albertson, T. E., Johnson, D. H., Johnson, S., MacArthur, R. D., et al. (2004). Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit. Care Med. 32, 2173–2182. doi: 10.1097/01.ccm.0000145229.59014.6c
Panda, R., Castanheira, F. V., Schlechte, J. M., Surewaard, B. G., Shim, H. B., Zucoloto, A. Z., et al. (2021). A functionally distinct neutrophil landscape in severe COVID-19 reveals opportunities for adjunctive therapies. JCI Insight 7:e152291. doi: 10.1172/jci.insight.152291
Papazian, L., Forel, J. M., Gacouin, A., Penot-Ragon, C., Perrin, G., Loundou, A., et al. (2010). Neuromuscular blockers in early acute respiratory distress syndrome. New Engl. J. Med. 363, 1107–1116. doi: 10.1056/NEJMoa1005372
Parsons, P. E., Matthay, M. A., Ware, L. B., and Eisner, M. D. (2005). Elevated plasma levels of soluble TNF receptors are associated with morbidity and mortality in patients with acute lung injury. Am. J. Phys. Lung Cell. Mole. Physiol. 288, L426–L431. doi: 10.1152/ajplung.00302.2004
Patel, B. V., Wilson, M. R., O’Dea, K. P., and Takata, M. (2013). TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J. Immunol. 190, 4274–4282. doi: 10.4049/jimmunol.1202437
Pedersen, S. F., and Ho, Y. C. (2020). SARS-CoV-2: a storm is raging. J. Clin. Investig. 130, 2202–2205. doi: 10.1172/JCI137647
Pennica, D., Hayflick, J. S., Bringman, T. S., Palladino, M. A., and Goeddel, D. V. (1985). Cloning and expression in Escherichia coli of the cDNA for murine tumor necrosis factor. Proc. Natl. Acad. Sci. U S A. 82, 6060–6064. doi: 10.1073/pnas.82.18.6060
Petrache, I., Birukova, A., Ramirez, S. I., Garcia, J. G., and Verin, A. D. (2003). The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 28, 574–581. doi: 10.1165/rcmb.2002-0075OC
Petrache, I., Verin, A. D., Crow, M. T., Birukova, A., Liu, F., and Garcia, J. G. (2001). Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am. J. Physiol. Lung Cell. Mole. Physiol. 280, L1168–L1178. doi: 10.1152/ajplung.2001.280.6.L1168
Pober, J. S., and Sessa, W. C. (2007). Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815. doi: 10.1038/nri2171
Polidoro, R. B., Hagan, R. S., de Santis Santiago, R., and Schmidt, N. W. (2020). Overview: Systemic Inflammatory Response Derived From Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Imm. 2020:1626. doi: 10.3389/fimmu.2020.01626
Pooladanda, V., Thatikonda, S., Bale, S., Pattnaik, B., Sigalapalli, D. K., Bathini, N. B., et al. (2019). Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death Dis. 10:81. doi: 10.1038/s41419-018-1247-9
Predescu, D., Predescu, S., Shimizu, J., Miyawaki-Shimizu, K., and Malik, A. B. (2005). Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J. Physiol. Lung Cell. Mole. Physiol. 289, L371–L381. doi: 10.1152/ajplung.00175.2004
Proudfoot, A., Bayliffe, A., O’Kane, C. M., Wright, T., Serone, A., Bareille, P. J., et al. (2018). Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 73, 723–730. doi: 10.1136/thoraxjnl-2017-210305
Rangamani, P., and Sirovich, L. (2007). Survival and apoptotic pathways initiated by TNF-alpha: modeling and predictions. Biotechnol. Bioeng. 97, 1216–1229. doi: 10.1002/bit.21307
Reinhart, K., Menges, T., Gardlund, B., Harm Zwaveling, J., Smithes, M., Vincent, J. L., et al. (2001). Randomized, placebo-controlled trial of the anti-tumor necrosis factor antibody fragment afelimomab in hyperinflammatory response during severe sepsis: the RAMSES study. Crit. Care Med. 29, 765–769. doi: 10.1097/00003246-200104000-00015
Reinhart, K., Wiegand-Löhnert, C., Grimminger, F., Kaul, M., Withington, S., Treacher, D., et al. (1996). Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK 195F, in patients with sepsis and septic shock: a multicenter, randomized, placebo-controlled, doseranging study. Crit. Care Med. 24, 733–742. doi: 10.1097/00003246-199605000-00003
Rezaiguia, S., Garat, C., Delclaux, C., Meignan, M., Fleury, J., Legrand, P., et al. (1997). Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J. Clin. Invest. 99, 325–335. doi: 10.1172/JCI119161
Rice, T. W., Wheeler, A. P., Morris, P. E., Paz, H. L., Russell, J. A., Edens, T. R., et al. (2006). Safety and efficacy of affinity-purified, anti-tumor necrosis factor-alpha, ovine fab for injection (CytoFab) in severe sepsis. Crit. Care Med. 34, 2271–2281. doi: 10.1097/01.Ccm.0000230385.82679.34
Richter, C., Messerschmidt, S., Holeiter, G., Tepperink, J., Osswald, S., Zappe, A., et al. (2012). The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mole. Cell. Biol. 32, 2515–2529. doi: 10.1128/MCB.06458-11
Robinson, P. C., Liew, D. F. L., Liew, J. W., Monaco, C., Richards, D., Shivakumar, S., et al. (2020). The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med 1, 90–102. doi: 10.1016/j.medj.2020.11.005
Romero, M. J., Platt, D. H., Tawfik, H. E., Labazi, M., El-Remessy, A. B., Bartoli, M., et al. (2008). Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circul. Res. 102, 95–102. doi: 10.1161/CIRCRESAHA.107.155028
Russell, B., Moss, C., George, G., Santaolalla, A., Cope, A., Papa, S., et al. (2020). Associations between immune-suppressive and stimulating drugs and novel COVID-19-a systematic review of current evidence. Ecancermedicalscience 14:1022. doi: 10.3332/ecancer.2020.1022
Ryan, A. J., McCoy, D. M., McGowan, S. E., Salome, R. G., and Mallampalli, R. K. (2003). Alveolar sphingolipids generated in response to TNF-alpha modifies surfactant biophysical activity. J. Appl. Physiol. 94, 253–258. doi: 10.1152/japplphysiol.00184.2002
Samorukova, E., Malyavin, A., Adasheva, T., and Zadionchenko, V. (2017). Rosuvastatin influence on inflammatory and anti-inflammatory cytokines in blood in patients with chronic obstructive pulmonary disease. Eur. Respir. J. 50(Suppl. 61):PA1074. doi: 10.1183/1393003.congress-2017.PA1074
Sandoval, R., Malik, A. B., Minshall, R. D., Kouklis, P., Ellis, C. A., and Tiruppathi, C. (2001). Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J. Physiol. 533(Pt 2), 433–445. doi: 10.1111/j.1469-7793.2001.0433a.x
Saperstein, S., Chen, L., Oakes, D., Pryhuber, G., and Finkelstein, J. (2009). IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J. Interf. Cytok. Res. 29, 273–284. doi: 10.1089/jir.2008.0076
Schissel, S. L., Schuchman, E. H., Williams, K. J., and Tabas, I. (1996). Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J. Biol. Chem. 271, 18431–18436. doi: 10.1074/jbc.271.31.18431
Schmid, B., Kredel, M., Ullrich, R., Krenn, K., Lucas, R., Markstaller, K., et al. (2021). Safety and preliminary efficacy of sequential multiple ascending doses of solnatide to treat pulmonary permeability edema in patients with moderate-to-severe ARDS-a randomized, placebo-controlled, double-blind trial. Trials 22:643. doi: 10.1186/s13063-021-05588-9
Schwameis, R., Eder, S., Pietschmann, H., Fischer, B., Mascher, H., Tzotzos, S., et al. (2014). A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J. Clin. Pharm. 54, 341–350. doi: 10.1002/jcph.203
Schwingshackl, A., Teng, B., Ghosh, M., Lim, K. G., Tigyi, G., Narayanan, D., et al. (2013). Regulation of interleukin-6 secretion by the two-pore-domain potassium channel Trek-1 in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mole. Physiol. 304, L276–L286. doi: 10.1152/ajplung.00299.2012
Shabbir, W., Scherbaum-Hazemi, P., Tzotzos, S., Fischer, B., Fischer, H., Pietschmann, H., et al. (2013). Mechanism of action of novel lung edema therapeutic AP301 by activation of the epithelial sodium channel. Mole. Pharm. 84, 899–910. doi: 10.1124/mol.113.089409
Shabbir, W., Tzotzos, S., Bedak, M., Aufy, M., Willam, A., Kraihammer, M., et al. (2015). Glycosylation-dependent activation of epithelial sodium channel by solnatide. Biochem. Pharm. 98, 740–753. doi: 10.1016/j.bcp.2015.08.003
Sherblom, A. P., Decker, J. M., and Muchmore, A. V. (1988). The lectin-like interaction between recombinant tumor necrosis factor and uromodulin. J. Biol. Chem. 263, 5418–5424.
Shimizu, M., Hasegawa, N., Nishimura, T., Endo, Y., Shiraishi, Y., Yamasawa, W., et al. (2009). Effects of TNF-alpha-converting enzyme inhibition on acute lung injury induced by endotoxin in the rat. Shock 32, 535–540. doi: 10.1097/SHK.0b013e3181a2adb7
Shirai, T., Yamaguchi, H., Ito, H., Todd, C. W., and Wallace, R. B. (1985). Cloning and expression in Escherichia coli of the gene for human tumour necrosis factor. Nature 313, 803–806. doi: 10.1038/313803a0
Shu, H. B., Takeuchi, M., and Goeddel, D. V. (1996). The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. U S A. 93, 13973–13978. doi: 10.1073/pnas.93.24.13973
Shyamsundar, M., McKeown, S. T., O’Kane, C. M., Craig, T. R., Brown, V., Thickett, D. R., et al. (2009). Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am. J. Respir. Crit. Care Med. 179, 1107–1114. doi: 10.1164/rccm.200810-1584OC
Sorkine, P., Setton, A., Halpern, P., Miller, A., Rudick, V., Marmor, S., et al. (1995). Soluble tumor necrosis factor receptors reduce bowel ischemia-induced lung permeability and neutrophil sequestration. Crit. Care Med. 23, 1377–1381. doi: 10.1097/00003246-199508000-00011
Standiford, T. J., Kunkel, S. L., Basha, M. A., Chensue, S. W., Lynch, J. P. III, Toews, G. B., et al. (1990). Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J. Clin. Investig. 86, 1945–1953. doi: 10.1172/JCI114928
Su, X., Ao, L., Zou, N., Song, Y., Yang, X., Cai, G.-Y., et al. (2008). Post-transcriptional regulation of TNF-induced expression of ICAM-1 and IL-8 in human lung microvascular endothelial cells: an obligatory role for the p38 MAPK-MK2 pathway dissociated with HSP27. Biochim. et Biophys. Acta 1783, 1623–1631. doi: 10.1016/j.bbamcr.2008.04.009
Subramaniyam, D., Virtala, R., Pawłowski, K., Clausen, I. G., Warkentin, S., Stevens, T., et al. (2008). TNF-alpha-induced self expression in human lung endothelial cells is inhibited by native and oxidized alpha1-antitrypsin. Int. J. Biochem. Cell Biol. 40, 258–271. doi: 10.1016/j.biocel.2007.07.016
Tan, B. H., Zhang, Y., Gui, Y., Wu, S., and Li, Y. C. (2020). The possible impairment of respiratory-related neural loops may be associated with the silent pneumonia induced by SARS-CoV-2. J. Med. Virol. 92, 2269–2271. doi: 10.1002/jmv.26158
Tartaglia, L. A., Pennica, D., and Goeddel, D. V. (1993). Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J. Biol. Chem. 268, 18542–18548. doi: 10.1016/s0021-9258(17)46661-0
Tartaglia, L. A., Weber, R. F., Figari, I. S., Reynolds, C., Palladino, M. A. Jr., and Goeddel, D. V. (1991). The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. U S A. 88, 9292–9296. doi: 10.1073/pnas.88.20.9292
Teuwen, L. A., Geldhof, V., Pasut, A., and Carmeliet, P. (2020). COVID-19: the vasculature unleashed. Nat. Rev. Immunol. 20, 389–391. doi: 10.1038/s41577-020-0343-0
Tillie-Leblond, I., Guery, B. P., Janin, A., Leberre, R., Just, N., Pittet, J. F., et al. (2002). Chronic bronchial allergic inflammation increases alveolar liquid clearance by TNF-alpha -dependent mechanism. Am. J. Phys. Lung Cell. Mole. Physiol. 283, L1303–L1309. doi: 10.1152/ajplung.00147.2002
Towne, J. E., Harrod, K. S., Krane, C. M., and Menon, A. G. (2000). Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection. Am. J. Respir. Cell Mol. Biol. 22, 34–44. doi: 10.1165/ajrcmb.22.1.3818
Towne, J. E., Krane, C. M., Bachurski, C. J., and Menon, A. G. (2001). Tumor necrosis factor-alpha inhibits aquaporin 5 expression in mouse lung epithelial cells. J. Biol. Chem. 276, 18657–18664. doi: 10.1074/jbc.M100322200
Tzotzos, S., Fischer, B., Fischer, H., Pietschmann, H., Lucas, R., Dupré, G., et al. (2013). AP301, a synthetic peptide mimicking the lectin-like domain of TNF, enhances amiloride-sensitive Na(+) current in primary dog, pig and rat alveolar type II cells. Pulm. Pharm. Ther. 26, 356–363. doi: 10.1016/j.pupt.2012.12.011
Tzotzos, S., Fischer, B., Pietschmann, H., Fischer, H., Lucas, R., Shabbir, W., et al. (2011). Characterisation of TNF-alpha lectin-like domain derived peptides associated with improved alveolar fluid clearance in pulmonary oedema. Eur. Respirat. J. 38, (Suppl. 55):4688.
Vadász, I., Raviv, S., and Sznajder, J. I. (2007). Alveolar epithelium and Na,K-ATPase in acute lung injury. Intens. Care Med. 33, 1243–1251. doi: 10.1007/s00134-007-0661-8
Vadasz, I., Schermuly, R. T., Ghofrani, H. A., Rummel, S., Wehner, S., Muhldorfer, I., et al. (2008). The lectin-like domain of tumor necrosis factor-alpha improves alveolar fluid balance in injured isolated rabbit lungs. Crit. Care Med. 36, 1543–1550. doi: 10.1097/CCM.0b013e31816f485e
van den Berg, E., Bem, R. A., Bos, A. P., Lutter, R., and van Woensel, J. B. (2014). The effect of TIP on pneumovirus-induced pulmonary edema in mice. PLoS One 9:e102749. doi: 10.1371/journal.pone.0102749
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418. doi: 10.1016/S0140-6736(20)30937-5
Verhoef, C., de Wilt, J. H., Grünhagen, D. J., van Geel, A. N., Hagen, T. L., and Eggermont, A. M. (2007). Isolated limb perfusion with melphalan and TNF-alpha in the treatment of extremity sarcoma. Curr. Treat Opt. Oncol. 8, 417–427. doi: 10.1007/s11864-007-0044-y
Villar, J., Ferrando, C. Martínez, D. Ambrós, A. Muñoz, T., Soler, J. A., et al. (2020). Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir. Med. 8, 267–276. doi: 10.1016/S2213-2600(19)30417-5
Wajant, H., and Beilhack, A. (2019). Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front. Immunol. 10:2040. doi: 10.3389/fimmu.2019.02040
Wajant, H., Pfizenmaier, K., and Scheurich, P. (2003). Tumor necrosis factor signaling. Cell Death Diff. 10, 45–65.
Wang, Q., and Doerschuk, C. M. (2000). Neutrophil-induced changes in the biomechanical properties of endothelial cells: roles of ICAM-1 and reactive oxygen species. J. Imm. 164, 6487–6494. doi: 10.4049/jimmunol.164.12.6487
Ware, L. B. (2017). Targeting resolution of pulmonary edema in primary graft dysfunction after lung transplantation: Is inhaled AP301 the answer? J. Heart Lung. Transplant 2017:9. doi: 10.1016/j.healun.2017.11.013
Ware, L. B., and Matthay, M. A. (2001). Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 163, 1376–1383. doi: 10.1164/ajrccm.163.6.2004035
Watts, A. D., Hunt, N. H., Wanigasekara, Y., Bloomfield, G., Wallach, D., Roufogalis, B. D., et al. (1999). A casein kinase I motif present in the cytoplasmic domain of members of the tumour necrosis factor ligand family is implicated in ‘reverse signalling’. Embo J. 18, 2119–2126. doi: 10.1093/emboj/18.8.2119
Wei, B., and Liu, Y. (2018). Clinical significance and efficacy analysis of atorvastatin in the treatment of patients with cerebral infarction and aspiration pneumonia. Exp. Ther. Med. 16, 5144–5148. doi: 10.3892/etm.2018.6829
Weifeng, Y., Li, L., Yujie, H., Weifeng, L., Zhenhui, G., and Wenjie, H. (2016). Inhibition of Acute Lung Injury by TNFR-Fc through Regulation of an Inflammation-Oxidative Stress Pathway. PLoS One 11:e0151672. doi: 10.1371/journal.pone.0151672
White, L. E., Santora, R. J., Cui, Y., Moore, F. A., and Hassoun, H. T. (2012). TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury. Am. J. Physiol. Lung Cell. Mole. Physiol. 303, L449–L459. doi: 10.1152/ajplung.00301.2011
Willam, A., Aufy, M., Tzotzos, S., El-Malazi, D., Poser, F., Wagner, A., et al. (2017a). TNF Lectin-Like Domain Restores Epithelial Sodium Channel Function in Frameshift Mutants Associated with Pseudohypoaldosteronism Type 1B. Front. Imm. 2017:601. doi: 10.3389/fimmu.2017.00601
Willam, A., Aufy, M., Tzotzos, S., Evanzin, H., Chytracek, S., Geppert, S., et al. (2017b). Restoration of Epithelial Sodium Channel Function by Synthetic Peptides in Pseudohypoaldosteronism Type 1B Mutants. Front. Pharmacol. 8:85. doi: 10.3389/fphar.2017.00085
Wilson, M. R., Choudhury, S., and Takata, M. (2005). Pulmonary inflammation induced by high-stretch ventilation is mediated by tumor necrosis factor signaling in mice. Am. J. Physiol. Lung. Cell. Mol. Physiol. 288, L599–L607. doi: 10.1152/ajplung.00304.2004
Wilson, M. R., Goddard, M. E., O’Dea, K. P., Choudhury, S., and Takata, M. (2007). Differential roles of p55 and p75 tumor necrosis factor receptors on stretch-induced pulmonary edema in mice. Am. J. Physiol. Lung Cell. Mole. Physiol. 293, L60–L68. doi: 10.1152/ajplung.00284.2006
Wilson, M. R., Wakabayashi, K., Bertok, S., Oakley, C. M., Patel, B. V., O’Dea, K. P., et al. (2017). Inhibition of TNF receptor p55 by a domain antibody attenuates the initial phase of acid-induced lung injury in mice. Front. Immunol. 8:128. doi: 10.3389/fimmu.2017.00128
Windsor, A. C., Walsh, C. J., Mullen, P. G., Cook, D. J., Fisher, B. J., Blocher, C. R., et al. (1993). Tumor necrosis factor-alpha blockade prevents neutrophil CD18 receptor upregulation and attenuates acute lung injury in porcine sepsis without inhibition of neutrophil oxygen radical generation. J. Clin. Investig. 91, 1459–1468. doi: 10.1172/JCI116351
Wu, D.-Q., Wu, H.-B., Zhang, M., and Wang, J.-A. (2017). Effects of Zinc Finger Protein A20 on Lipopolysaccharide (LPS)-Induced Pulmonary Inflammation/Anti-Inflammatory Mediators in an Acute Lung Injury/Acute Respiratory Distress Syndrome Rat Model. Med. Sci. Monit. 23, 3536–3545. doi: 10.12659/msm.901700
Xiao, W., Oldham, W. M., Priolo, C., Pandey, A. K., and Loscalzo, J. (2021). Immunometabolic Endothelial Phenotypes: Integrating Inflammation and Glucose Metabolism. Circul. Res. 129, 9–29. doi: 10.1161/CIRCRESAHA.120.318805
Xiong, C., Yang, G., Kumar, S., Aggarwal, S., Leustik, M., Snead, C., et al. (2010). The lectin-like domain of TNF protects from listeriolysin-induced hyperpermeability in human pulmonary microvascular endothelial cells - a crucial role for protein kinase C-alpha inhibition. Vasc. Pharm. 52, 207–213. doi: 10.1016/j.vph.2009.12.010
Yamagata, T., Yamagata, Y., Nishimoto, T., Hirano, T., Nakanishi, M., Minakata, Y., et al. (2009). The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-alpha in injured lungs and alveolar type II cells. Respir. Phys. Neurobiol. 166, 16–23. doi: 10.1016/j.resp.2008.12.008
Yang, G., Pillich, H., White, R., Czikora, I., Pochic, I., Yue, Q., et al. (2018). Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins 10:79. doi: 10.3390/toxins10020079
Yang, S., Wang, J., Brand, D. D., and Zheng, S. G. (2018). Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immun. 9:784. doi: 10.3389/fimmu.2018.00784
Yang, Z., Zhang, X.-R., Zhao, Q., Wang, S.-L., Xiong, L.-L., Zhang, P., et al. (2018). Knockdown of TNF-α alleviates acute lung injury in rats with intestinal ischemia and reperfusion injury by upregulating IL-10 expression. Int. J. Mole. Med. 42, 926–934. doi: 10.3892/ijmm.2018.3674
Yao, H. W., Mao, L. G., and Zhu, J. P. (2006). Protective effects of pravastatin in murine lipopolysaccharideinduced acute lung injury. Clin. Exp. Pharmacol. Physiol. 33, 793–797. doi: 10.1111/j.1440-1681.2006.04440.x
Yu, J., Ma, Z., Shetty, S., Ma, M., and Fu, J. (2016). Selective HDAC6 inhibition prevents TNF-α-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am. J. Physiol. Lung. Cell. Mol. Physiol. 311, L39–L47. doi: 10.1152/ajplung.00051.2016
Yu, S., Xie, J., Xiang, Y., Dai, S., Yu, D., Sun, H., et al. (2019). Downregulation of TNFα/TNF-R1 Signals by AT-Lipoxin A4 May Be a Significant Mechanism of Attenuation in SAP-Associated Lung Injury. Mediat. Inflamm. 2019:9019404. doi: 10.1155/2019/9019404
Zeki, A. A., Franzi, L., Last, J., and Kenyon, N. J. (2009). Simvastatin inhibits airway hyperreactivity: implications for the mevalonate pathway and beyond. Am. J. Respir. Crit. Care Med. 180, 731–740. doi: 10.1164/rccm.200901-0018OC
Zelic, M., Roderick, J. E., O’Donnell, J. A., Lehman, J., Lim, S. E., Janardhan, H. P., et al. (2018). RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clin. Invest. 128, 2064–2075. doi: 10.1172/JCI96147
Zhang, H., Liu, Y., Li, H., Li, J., Luo, Y., and Yan, X. (2019). Novel insights into the role of LRRC8A in ameliorating alveolar fluid clearance in LPS induced acute lung injury. Eur. J. Pharmacol. 861:172613. doi: 10.1016/j.ejphar.2019.172613
Zhang, H., Penninger, J. M., Li, Y., Zhong, N., and Slutsky, A. S. (2020). Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intens. Care Med. 46, 586–590. doi: 10.1007/s00134-020-05985-9
Zhao, Y., Zhang, C.-W., Zhou, W.-J., Chen, J., Luo, N.-F., Gong, L.-N., et al. (2014). Is there a role of TNFR1 in acute lung injury cases associated with extracorporeal circulation? J. Zhejiang Univ. Sci. B. 15, 281–288. doi: 10.1631/jzus.B1300147
Zhou, Q., Wang, D., Liu, Y., Yang, X., Lucas, R., and Fischer, B. (2017). Solnatide Demonstrates Profound Therapeutic Activity in a Rat Model of Pulmonary Edema Induced by Acute Hypobaric Hypoxia and Exercise. Chest 151, 658–667. doi: 10.1016/j.chest.2016.10.030
Zhou, Z., Connell, M. C., and MacEwan, D. J. (2007). TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal. 19, 1238–1248. doi: 10.1016/j.cellsig.2006.12.013
Keywords: TNF receptor, TNF lectin-like domain, acute respiratory distress syndrome, COVID-19, epithelial sodium channel
Citation: Lucas R, Hadizamani Y, Enkhbaatar P, Csanyi G, Caldwell RW, Hundsberger H, Sridhar S, Lever AA, Hudel M, Ash D, Ushio-Fukai M, Fukai T, Chakraborty T, Verin A, Eaton DC, Romero M and Hamacher J (2022) Dichotomous Role of Tumor Necrosis Factor in Pulmonary Barrier Function and Alveolar Fluid Clearance. Front. Physiol. 12:793251. doi: 10.3389/fphys.2021.793251
Received: 11 October 2021; Accepted: 30 November 2021;
Published: 21 February 2022.
Edited by:
Sabah Hussain, McGill University, CanadaReviewed by:
Ulrich Herbert Thome, Leipzig University, GermanyEmilia Lecuona, Northwestern University, United States
Copyright © 2022 Lucas, Hadizamani, Enkhbaatar, Csanyi, Caldwell, Hundsberger, Sridhar, Lever, Hudel, Ash, Ushio-Fukai, Fukai, Chakraborty, Verin, Eaton, Romero and Hamacher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rudolf Lucas, rlucas@augusta.edu; Jürg Hamacher, hamacher@greenmail.ch