 Huanhuan Zhao
Huanhuan Zhao Dan Liu
Dan Liu Qiumei Yan1
Qiumei Yan1 Jing Yu
Jing Yu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 05 January 2022
Sec. Integrative Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.736920
Zinc homeostasis has been known to play a role in myocardial ischemia/reperfusion (I/R) injury, but the precise molecular mechanisms regulating the expression of ZIP transporters during reperfusion are still unclear. The aim of this study was to determine whether ER Stress/CaMKII/STAT3 pathway plays a role in the regulation of cellular zinc homeostasis. Zinc deficiency increased mRNA and protein expressions of the ER stress relevant markers Chop and Bip, and STAT3 phosphorylation in H9c2 or HL-1 cells, an effect that was abolished by ZnCl2. ER calcium concentration [(Ca2+)ER] was decreased and cytosolic calcium concentration [(Ca2+)I] was increased at the condition of normoxia or ischemia/reperfusion, indicating that zinc deficiency triggers ER stress and Ca2+ leak. Further studies showed that upregulation of STAT3 phosphorylation was reversed by Ca2+ chelator, indicating that intracellular Ca2+ is important for zinc deficiency-induced STAT3 activation. In support, zinc deficiency enhanced ryanodine receptors (RyR), a channel in the ER that mediate Ca2+ release, and Ca2+-calmodulin-dependent protein kinase (CaMKII) phosphorylation, implying that zinc deficiency provoked Ca2+ leak from ER via RyR and p-CaMKII is involved in STAT3 activation. Moreover, inhibition of STAT3 activation blocked zinc deficiency induced ZIP9 expression, and resulted in increased Zn2+ loss in cardiomyocytes, further confirming that STAT3 activation during reperfusion promotes the expression of ZIP9 zinc transporter to correct the imbalance in zinc homeostasis. In addition, suppressed STAT3 activation aggravated reperfusion injury. These data suggest that the ER Stress/CaMKII/STAT3 axis may be an endogenous protective mechanism, which increases the resistance of the heart to I/R.
Myocardial ischemia/reperfusion (I/R) injury is a very common cardiovascular disease with a high mortality rate (Reed et al., 2017). It has been reported that zinc loss upon reperfusion contributes to myocardial ischemia/reperfusion (I/R) injury (McIntosh et al., 2010). Zinc is a trace element, which participates in the metabolism of many enzymes and it is also the basis for regulating nucleic acid metabolism, protein synthesis and the structure and function of transcription factors (Berg and Shi, 1996). The imbalance of zinc homeostasis will lead to immune deficiency, growth inhibition, neurological dysfunction, and susceptibility to infectious diseases (Prasad et al., 1961; Tapazoglou et al., 1985; Vallee and Falchuk, 1993). Intracellular zinc homeostasis is closely controlled by two families of zinc transporters: ZnT (SLC30) and ZIP (SLC39). So far, 10 ZnT and 14 ZIP transporters have been identified, which have the opposite effect in zinc homeostasis (Liuzzi and Cousins, 2004). ZnT family transporters promote zinc outflow from cytoplasm into organelles or across the plasma membrane to reduce cytosolic free zinc levels, while ZIP transporters facilitate zinc inflow from the extracellular space or organelles into cytoplasm to increase cytosolic free zinc levels. Our previous studies demonstrated that ZIP2 expression was increased at reperfusion in in vivo mouse hearts, an effect that was abolished by ZnCl2, indicating that the increased expression of ZIP2 during reperfusion was caused by the loss of zinc. In addition, ZIP2 gene knockout significantly exacerbated myocardial I/R injury, while ZIP2 overexpression could reduce I/R injury, indicating that ZIP2 is cardioprotective against I/R injury by correcting zinc dyshomeostasis (Du et al., 2019). Obviously, ZIP transporters play a significant role in the maintenance of zinc homeostasis in cardiomyocytes during reperfusion. However, the precise molecular mechanisms regulating the expression of ZIP transporters during reperfusion is still unclear.
The nuclear transcriptional factor signal transducer and activator of transcription 3 (STAT3) is important for cellular proliferation, differentiation, and survival. STAT3 is activated by tyrosine phosphorylation (Tyr705), leading to dimerization and subsequent translocation into the nucleus to promote gene expression through the interaction with regulatory elements (Fu et al., 1992; Schindler et al., 1992; Ihle, 1995). It has been reported that STAT3 plays a part in epithelial-mesenchymal transition in zebra fish by targeting ZIP6 (Yamashita et al., 2004). Proinflammatory cytokines, such as IL1β and IL6 can stimulate STAT3 to up-regulate ZIP14 and ZIP6 and facilitate cellular zinc influx (Cousins et al., 2006). Our latest research shows that STAT3 regulates ZIP2 expression in I/R. However, how zinc deficiency acts on STAT3 activation in the myocardial ischemia/reperfusion injury remain unknown.
As an important intracellular calcium reservoir, endoplasmic reticulum (ER) plays an important role in multiple key cellular functions, including calcium homeostasis, the synthesis of major structural lipids, and the folding of membrane and secreted proteins (Brodsky, 2012). The accumulation of misfolded proteins can lead to a variety of effects, such as ER stress and the subsequent activation of the unfolded protein response (UPR) (Hetz, 2012). Zinc is essential for normal ER function, as zinc deficiency can cause ER stress (Homma et al., 2013; Nguyen et al., 2013). ER stress has been proposed to activate STAT3 (Ahyi et al., 2013; Meares et al., 2014). Studies have reported that IRE1α and PERK can promote STAT3 activation to increase the expression of anti-apoptotic proteins, thus promoting cell survival (Cubillos-Ruiz et al., 2017). So it is possible that ER stress/STAT3 plays a role in regulating zinc transporter expression in response to cellular zinc deficiency. Ca2+/CaM-dependent protein kinase II (CaMKII) is a serine/threonine-specific protein kinase that is regulated by the Ca2+/calmodulin complex. Studies have shown that reperfusion can initiate ER stress, leading to increase in intracellular Ca2+ release and the activation of the CaMKII (Timmins et al., 2009; Wang et al., 2016; Sheng et al., 2018). Accumulating studies reveals that there is cross-talk between STAT3 and CaMKII (Ma et al., 2017). For example, a recent study defined CaMKII as an upstream effector, which showed that CaMKII activation during stress leads to releasing STAT3, and allows for its translocation to nucleus to alter gene expression (Unudurthi et al., 2018). However, whether CaMKII/STAT3 is involved in the regulation of zinc homeostasis has not yet been reported.
The purpose of this study was to identify whether ER Stress/CaMKII/STAT3 pathway plays a role in the regulation of cellular zinc homeostasis. Here we reveal that zinc deficiency activates STAT3 by ER stress-induced Ca2+ release and subsequent CaMKII activation, leading to enhancement of the transcriptional activity of the ZIP family zinc transporter genes. These data suggest that ER Stress/CaMKII/STAT3 pathway responses to decrease in intracellular free zinc and promotes the expression of the ZIP genes that are required for zinc import leading to the protection against cellular zinc deficiency.
N,N,N′,N′ -Tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) and thapsigargin (TG) were obtained from Sigma (St. Louis, MO, United States). BAPTA-AM, EGTA-AM, H89, 2APB, and stattic were purchased from MCE (NJ, United States). KN-93 and KN-92 were obtained from Selleck (Houston, TX, United States). Antibodies including anti-p-STAT3, -STAT3, -p-CaMKII, -CaMKII, -GAPDH, and the secondary antibody were obtained from Cell Signaling Technology (Beverly, MA, United States). Anti-IP3R,-p-RyR2 and -SERCA2 were purchased from Abcam (Cambridge, United Kingdom). Anti-RyR2 was purchased from Proteintech Group (Chicago, IL, United States). Anti-ZIP9 was obtained from Biorbyt (Cambridge, United Kingdom). Fluorescence dyes were obtained from Invitrogen (Carlsbad, CA, United States).
Rat heart tissue-derived H9c2 cardiac myoblast cell line and murine atrial tumor-derived HL-1 cardiomyocytes were purchased from ATCC. H9c2 cells were cultured and maintained with DMEM supplemented with 10% FBS and 100 U penicillin-streptomycin in a cell culture incubator at 37°C in a humidified 5% CO2-95% air atmosphere. HL-1 cells were grown at 37°C with 5% CO2 in claycomb medium (Sigma-Aldrich; Merck KGaA) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 0.1 mM norepinephrine.
Male mice (C57BL/6J, 8–10 weeks old) were obtained from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). All the treatments and subsequent analyses were conducted in a blind fashion and in accordance with the NIH Guide for the Care and Use of Laboratory Animals (Eighth Edition). The animal experiments have been approved by the Tianjin Medical University Animal Care and Use Committee.
To induce H/R injury, cells cultured in a 6-well plate filled with the hypoxia medium (1 g L–1 glucose without serum) were exposed to hypoxia (1% O2) by placing the plate in a humidified hypoxia glove box (Coy Laboratory Products Inc., Grass Lake, MI, United States) for 4 h. Then the normal DMEM (4.5 g L–1 glucose and 10% serum) replaced the hypoxia medium and cells were cultured in an incubator under normoxic conditions (room air with 5% CO2) for 2 h.
Male mice (8–10 weeks) were anesthetized with sodium pentobarbital (80mg/kg, i.p.), intubated through atracheotomy, and aerated with positive end-expiratory pressure of 3 cm H2O. The adequacy of anesthesia was monitored with the corneal and withdrawal reflexes. The ventilation frequency was 110 breaths per minute, and the tidal volume was 135–150 μl. After left thoracotomy, the left anterior descending coronary artery (LAD) was surrounded by 7-0 Prolene line, and then passed through a small plastic tube. After that, tighten the tubing against the heart surface in order to induce ischemia. At this time, myocardial pallor and ST-segment elevation of electrocardiogram can be observed with naked eyes, which is confirmed as local ischemia. Mice hearts were occluded LAD for 30 min and then reperfused for 30 min. However, the sham operation group was also sutured under LAD but not occluded. At the end of experiments, the mice were anesthetized with sodium pentobarbital again, then euthanized with cervical dislocation, and the left ventricle below ligation point were immediately collected for Western blotting analysis.
Myocardial infarct size was measured by Evans Blue and triphenyltetrazolium chloride (TTC) double staining methods. Mice hearts were occluded LAD for 30 min and then reperfused for 2 h. At the end reperfusion, Evans Blue was injected into the heart through the thoracic aorta. Hearts were excised and sliced. The slices were incubated in 1% TTC at 37°C for 20 min and fixed with 10% formalin at room temperature. Infarct size was measured with ImageJ in a single blind mode and was presented as a percentage of the risk zone.
Proteins were separated by SDS-polyacrylamide gel, and then transferred to a PVDF membrane. After blocking the non-specifc sites with non-fat milk, each membrane was incubated overnight at 4°C with a primary antibody (1:1,000). The membranes were washed and incubated for 90 min at room temperature with a secondary antibody (1:2,000). Protein bands were visualized by the ECL method.
Total mRNA were isolated from each of experimental groups using Trizol reagent. Reverse transcription (RT) was performed to convert RNA into cDNA by using reverse transcriptase. Primers used to determine the expression of ERS related genes and ZIP9 were described in Supplementary Table 1. mRNA expressions were evaluated by quantitative RT-PCR using EvaGreen 2X qPCR MasterMix (abm, Richmond, CA, United States) and Bio-Rad CFX96-Touch Real-Time PCR Systems.
Endoplasmic reticulum and free Ca2+ concentrations in H9c2 or HL-1 cells and mice hearts were detected by Mag-fluo-4 (5 μM) and Fluo-4 (5 μM), respectively. After treatment, H9c2 or HL-1 cells were stained with Mag-fluo-4 (5 μM) and Fluo-4 (5 μM) at 37°C for 30 min. Mice hearts were extracted, frozen, and cut into 7-μm sections. Cardiomyocytes were also stained with the fluorescence probes at 37°C for 30 min and washed twice with PBS. Then the fluorescence was detected by FACSVERSE Flow Cytometer (BD Biosciences, CA, United States) or a laser scanning confocal microscope (Olympus, Tokyo, Japan). A 488-nm line of helium-neon laser line excited the green fluorescence, which imaged through a 525-nm-long path filter.
After 30 min of ischemia and then 30 min of reperfusion, the cardiac tissue was pre-dried, weighed, and completely nitrated by 2 mL 65% HNO3 at 120°C for 30 min. Then the samples were dissolved in 4 mL mineral-free water. The concentration of zinc was measured by using inductively coupled plasma optical emission spectroscopy (ICPOES, Optima™ 8000, Perkin Elmer, CT, United States) at a wave-length of 206.200 nm. The detection limit is about 1 ppb. Tissue concentrations were expressed as μg/g dry weight.
H9c2 or HL-1 cells were seeded in 96-well plates (1 × 104 cells/ml). After treatment, culture medium was collected and Lactate dehydrogenase (LDH) release was detected using the CK12 kit (DOJINDO) according to the manufacturer’s instruction. Absorbance was measured at a wave-length of 490nm.
Serum myocardial injury marker, cardiac troponin I (cTnI), creatine kinase-MB (CK-MB), and lactate dehydrogenase (LDH) were detected by ELISA (Bio-Swamp, China).
H89 (10 mg/kg), KN93 (10 mg/kg), BAPTA-AM (10 mg/kg), or Stattic (3 mg/kg) were injected 5 min before reperfusion and continued for 30 min or 2 h through the tail vein. H9c2 or HL-1 cells were subjected to 4 h of hypoxia followed by 2 h of reoxygenation or exposed to TPEN for 2 h. KN93, KN92, H89, BAPTA-AM, EGTA-AM, Stattic (10 μM) or 2APB (100 μM) were applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. ZnCl2 (10 μM) and ionophore pyrithione (4 μM) was applied 1 h after exposure to TPEN for 1 h. Cells were exposed to 50 nM thapsigargin (TG) for 4 h.
Data are expressed as mean ± SEM and the number of experimental replications is indicated in the figure legends. Statistical difference was determined using Student t-test or ANOVA followed by Tukey’s test. Statistical significance was defined as p < 0.05. SigmaStat 3.5 was used for statistical analysis.
Our recent studies have shown that STAT3 is activated during reperfusion and protects the heart from I/R injury by up-regulating Zip2 protein expression (Du et al., 2019). In order to determine how zinc deficiency acts on STAT3 activation in the myocardial ischemia/reperfusion injury, RNA-seq analysis was performed. The pathway enrichment data suggest that zinc deficiency affects the protein processing in ER (Supplementary Figure 1). Zinc is required for normal ER function. To investigate whether zinc depletion can provoke ER stress, H9c2 or HL-1 cells were exposed to TPEN (a selective zinc chelator) for 4 h. TPEN increased mRNA expressions of the ER stress relevant markers CHOP and Bip in two cell lines (Figure 1A). TPEN has the highest affinity for Zn2+, it can also bind to other heavy metals such as iron (Jackson and Kodanko, 2010). To verify that ER stress was induced by the reduction of intracellular zinc levels, ZnCl2 (10 μM) and ionophore pyrithione (4 μM) were applied 1 h after the exposure to TPEN for 1 h. TPEN-induced CHOP, Bip protein expressions and STAT3 phosphorylation were inhibited by ZnCl2 (Figure 1B). Moreover, ER calcium concentration [(Ca2+)ER] was decreased and cytosolic calcium concentration [(Ca2+)I] was increased under the condition of normoxia (Figure 1C) or ischemia/reperfusion (Figure 1D), implying that the reduction of zinc levels in ER triggers ER stress and Ca2+ leak.
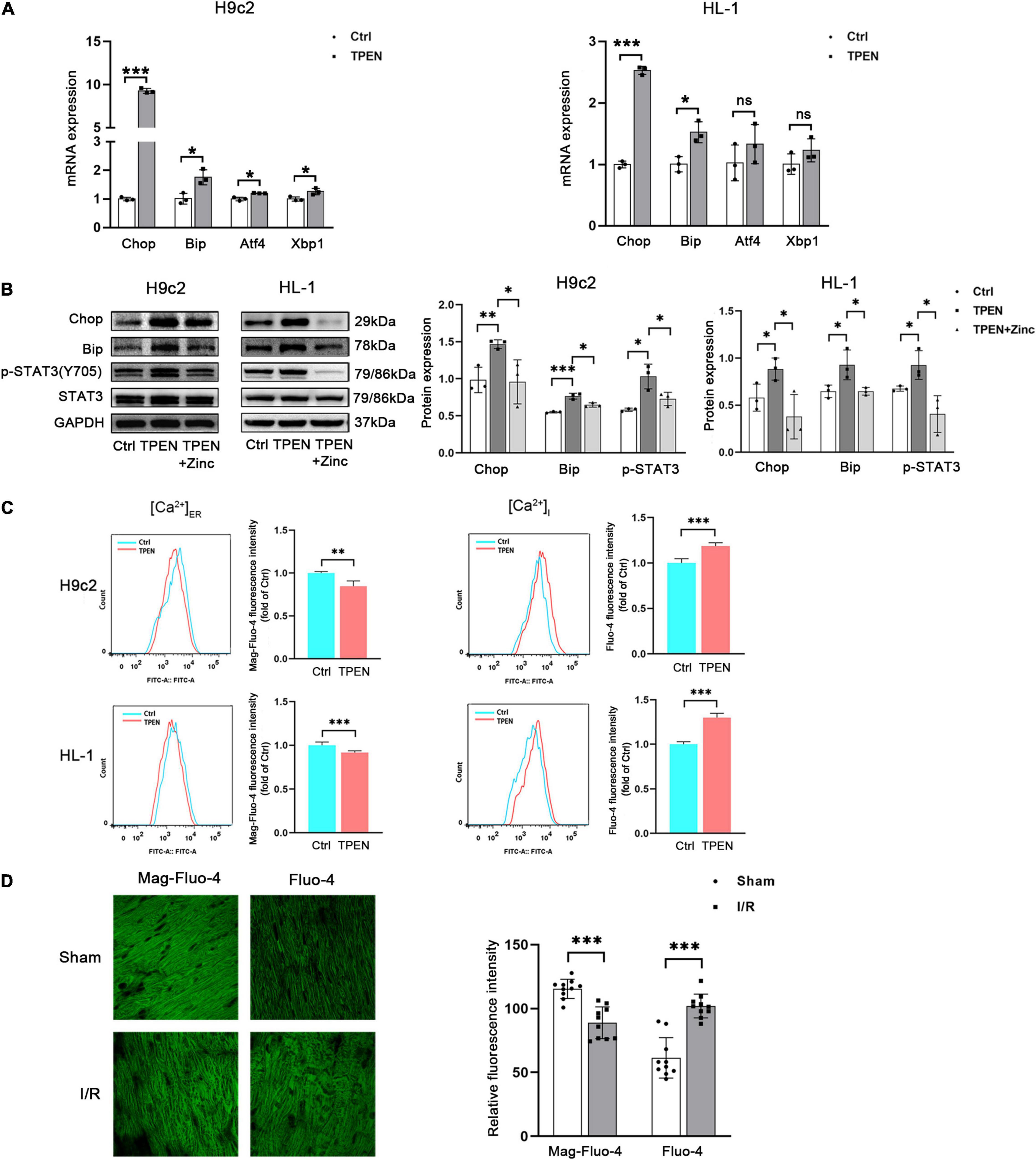
Figure 1. Zinc deficiency can induce ER Stress and Ca2+ leak. (A) Cells were exposed to TPEN (10 μM, n = 3) for 4 h. (B) TPEN was applied 1 h before exposure to ZnCl2 (10 μM, n = 3) and ionophore pyrithione (4 μM) for 1 h. (C) ER and free Ca2+ concentrations in cells were labeled with Mag-fluo4 (5 μM, n = 3) and Fluo-4 (5 μM, n = 3), respectively, and detected with confocal microscopy. (D) Mouse hearts were ischemic for 30 min and then reperfused for 30 min. Cardiomyocytes were labeled with Mag-fluo4 (5 μM) and Fluo-4 (5 μM), respectively (n = 10). *p < 0.05, **p < 0.01, and ***p < 0.001. ns, no significant difference P > 0.05.
Abnormal Zn2+ homeostasis has been reported to be associated with dysregulation of intracellular Ca2+ release (Reilly-O’Donnell et al., 2017). Since zinc deficiency can trigger both Ca2+ leak and STAT3 activation, it is possible that Ca2+ is involved in zinc deficiency-induced STAT3 activation. To test this hypothesis, we determined the effect of BAPTA-AM, an intracellular Ca2+ chelator, on zinc deficiency-induced STAT3 activation. BAPTA-AM (10 μM) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. As shown in Figures 2A,B, BAPTA-AM reduced TPEN or H/R-induced STAT3 phosphorylation. To confirm this observation, EGTA-AM, a membrane permeable form of EGTA, was applied as above. EGTA-AM also reversed TPEN or H/R-induced STAT3 phosphorylation (Figures 2C,D), indicating that intracellular Ca2+ is responsible for zinc deficiency-induced STAT3 activation.
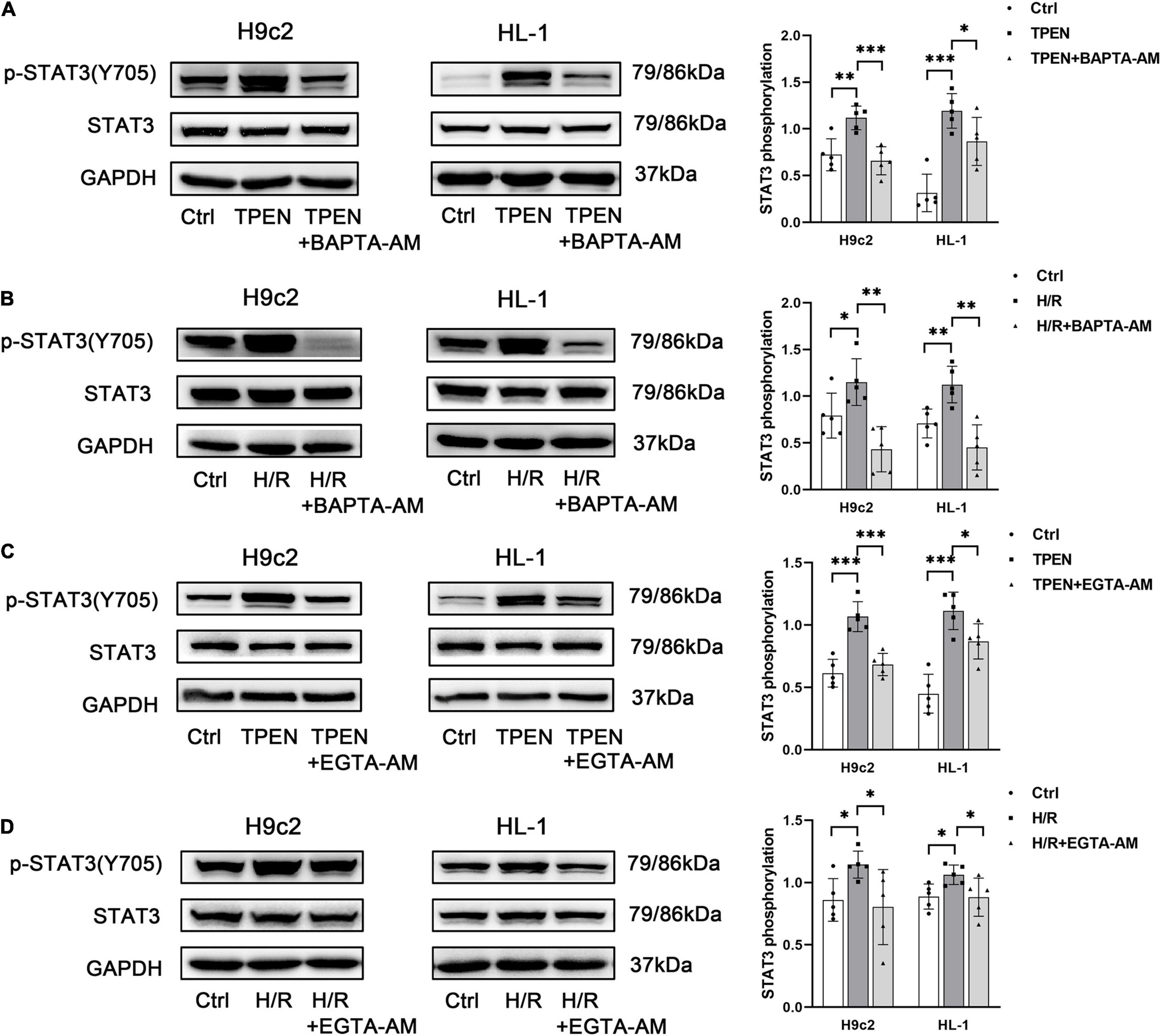
Figure 2. Calcium is responsible for zinc deficiency-induced STAT3 activation. BAPTA-AM (10 μM) (A,B) or EGTA-AM (10 μM) (C,D) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation (n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001.
The mechanism of zinc deficiency in regulating ER Ca2+ leak is poorly characterized. Ryanodine receptor (RyR) and inositol trisphosphate receptor (IP3R) are two channels in the ER that mediate Ca2+ release, while sarco(endo) plasmic reticulum Ca2+-ATPase (SERCA) is a pump that uptakes Ca2+ into SR(ER) (Popugaeva and Bezprozvanny, 2014; Zhai et al., 2020). Ca2+ release via the RyR and IP3R is facilitated by protein kinase A (PKA) (Leech et al., 2010). Then we examined how zinc deficiency shaped ER Ca2+ leak and further activated STAT3. H89 (PKA inhibitor, 10 μM) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. As shown in Figure 3A, the expression of IP3R and SERCA did not change in the setting of intracellular zinc depletion by TPEN, but the phosphorylation level of RyR2S2808 (PKA site) increased not only in the setting of intracellular zinc depletion by TPEN but also under hypoxia/reoxygen and ischemia/reperfusion conditions (Figures 3B,C). And H89 can inhibit TPEN, H/R or I/R-induced RyR2 and STAT3 phosphorylation. Moreover, H89 prevented the increase of cytoplasmic Ca2+ levels and the decrease of ER Ca2+ levels induced by TPEN or H/R (Figure 3D). Since PKA can also phosphorylate IP3R, 2-APB (a specific inhibitor of IP3 R, 10 μM) was applied. However, 2-APB treatment has no effect on TPEN induced STAT3 phosphorylation (Figure 3E). These results indicate that zinc deficiency can lead to RyR phosphorylation and increase its open probability, which result in an increased Ca2+ leakage from ER.
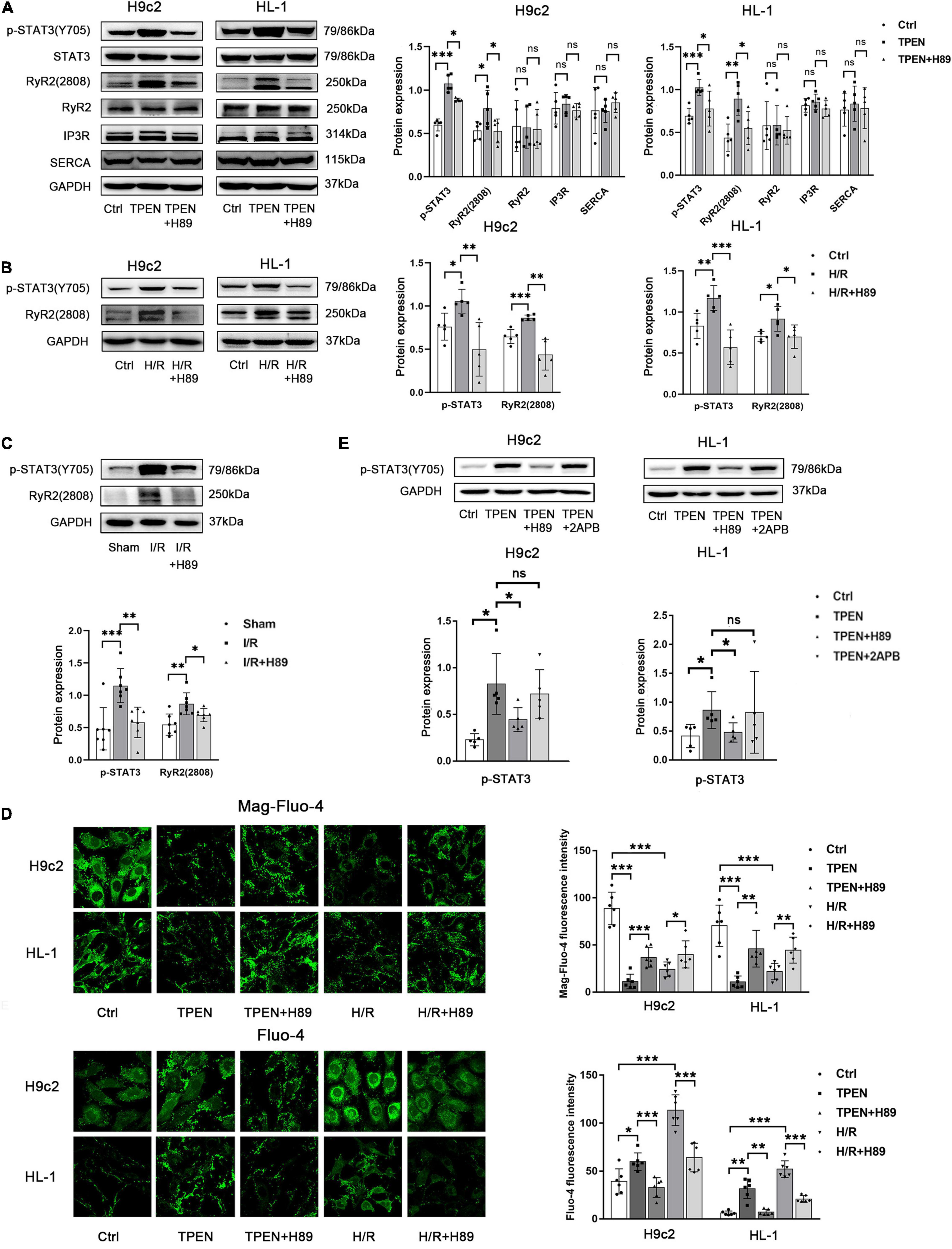
Figure 3. Zinc deficiency activates Ca2+ leak from ER via RyR2. (A,B) H89 (PKA inhibitor,10 μM, n = 5) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. (C) Mouse hearts were ischemic for 30 min and then reperfused for 30 min. H89 (10 mg/kg) was injected 5 min before reperfusion and continued for 30 min through the tail vein (n = 7). (D) H89 (10 μM, n = 6) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation and labeled with Mag-fluo-4 (5 μM, n = 6) and Fluo-4 (5 μM, n = 6), respectively. (E) H89 (10 μM, n = 5) and 2APB (100 μM, n = 5) were applied 2 h before exposure to TPEN for 2 h. *p < 0.05, **p < 0.01, and ***p < 0.001. ns, no significant difference P > 0.05.
CaMKII is a general integrator of Ca2+ signaling, which is activated when a large number of Ca2+ bind to calmodulin (CaM). To further determine whether CaMKII is involved in zinc deficiency-induced STAT3 activation, CaMKII inhibitor KN93 (10 uM) and its inactive analog KN92 (10 uM) were applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation (Figures 4A,B). Indeed, inhibition of CaMKII with KN-93, but not the inactive analog KN92, decreased zinc deficiency-induced STAT3 phosphorylation, pointing that CaMKII activation is vital to STAT3 activation. In addition, H89 and BAPTA-AM also reversed TPEN or H/R-induced CaMKII phosphorylation, implying that CaMKII phosphorylation does require the release of Ca2+ to participate in the activation of downstream STAT3 (Figures 4C,D).
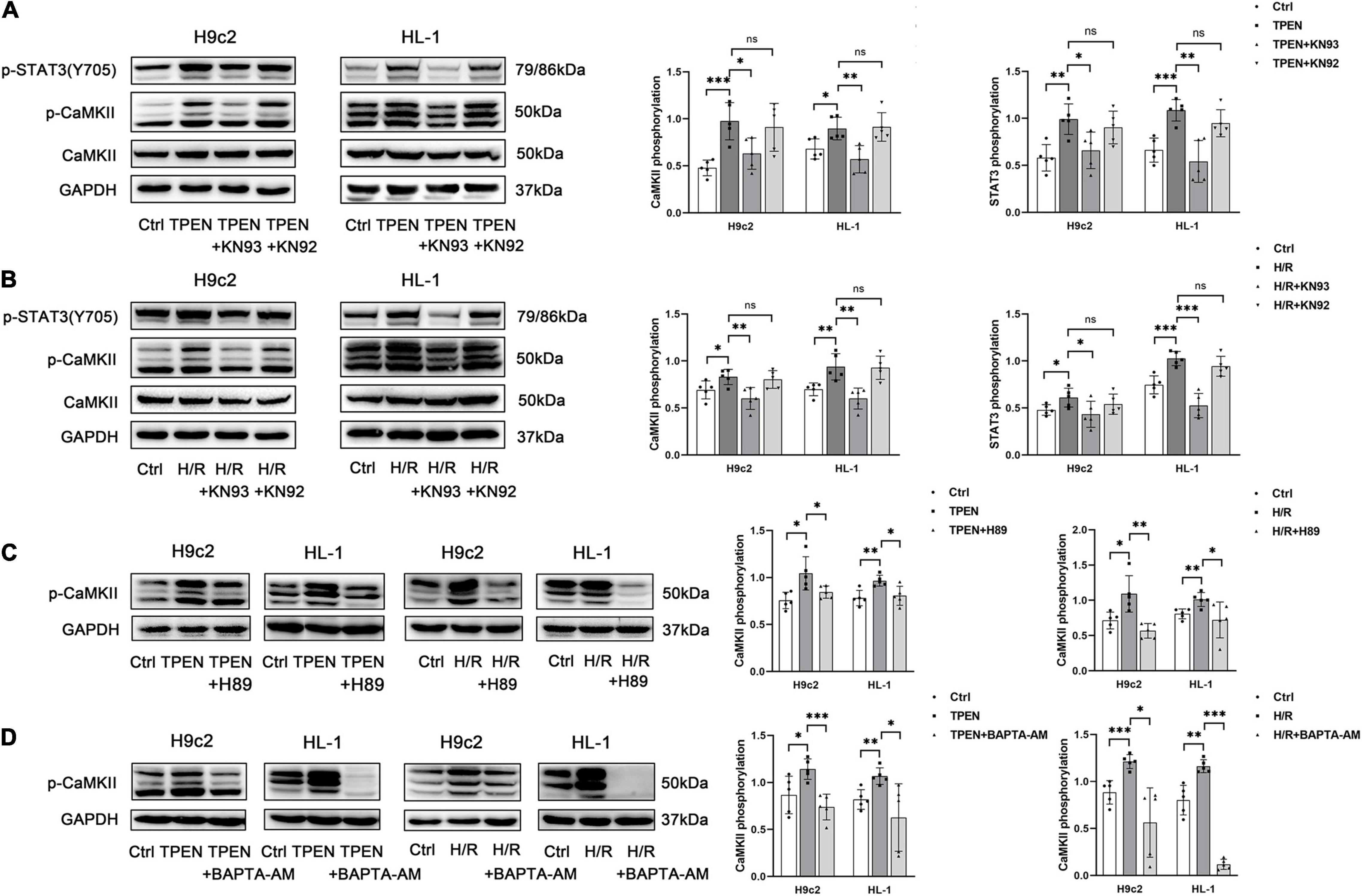
Figure 4. P-CaMKII is involved in zinc deficiency-induced STAT3 activation. KN93, KN92 (A,B), H89 (C), or BAPTA-AM (D) (10 μM, n = 5) were applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. *p < 0.05, **p < 0.01, and ***p < 0.001. ns, no significant difference P > 0.05.
Now more and more ZIP transporters have been found to play crucial roles in the maintenance of zinc homeostasis in cardiomyocytes during reperfusion. Our preliminary RNA sequencing data in H9c2 cells showed that ZIP9 expression is significantly increased after TPEN treatment (data not shown). Since STAT3 is activated upon zinc deficiency, it may play a role in the regulation of the Zip9 gene expression. Figures 5A,B showed that when STAT3 phosphorylation is inhibited by stattic, the expression of ZIP9 is also down-regulated. To test if cardiac zinc deficiency caused by ischemia/reperfusion can also provoke STAT3 activation and induce ZIP9 expression in vivo, mouse hearts were subjected to ischemia/reperfusion. Ischemia/reperfusion increased ZIP9 expression was reversed by stattic (Figure 5C), implying that zinc deficiency induced STAT3 activation leads to the upregulation of zinc importer ZIP9.
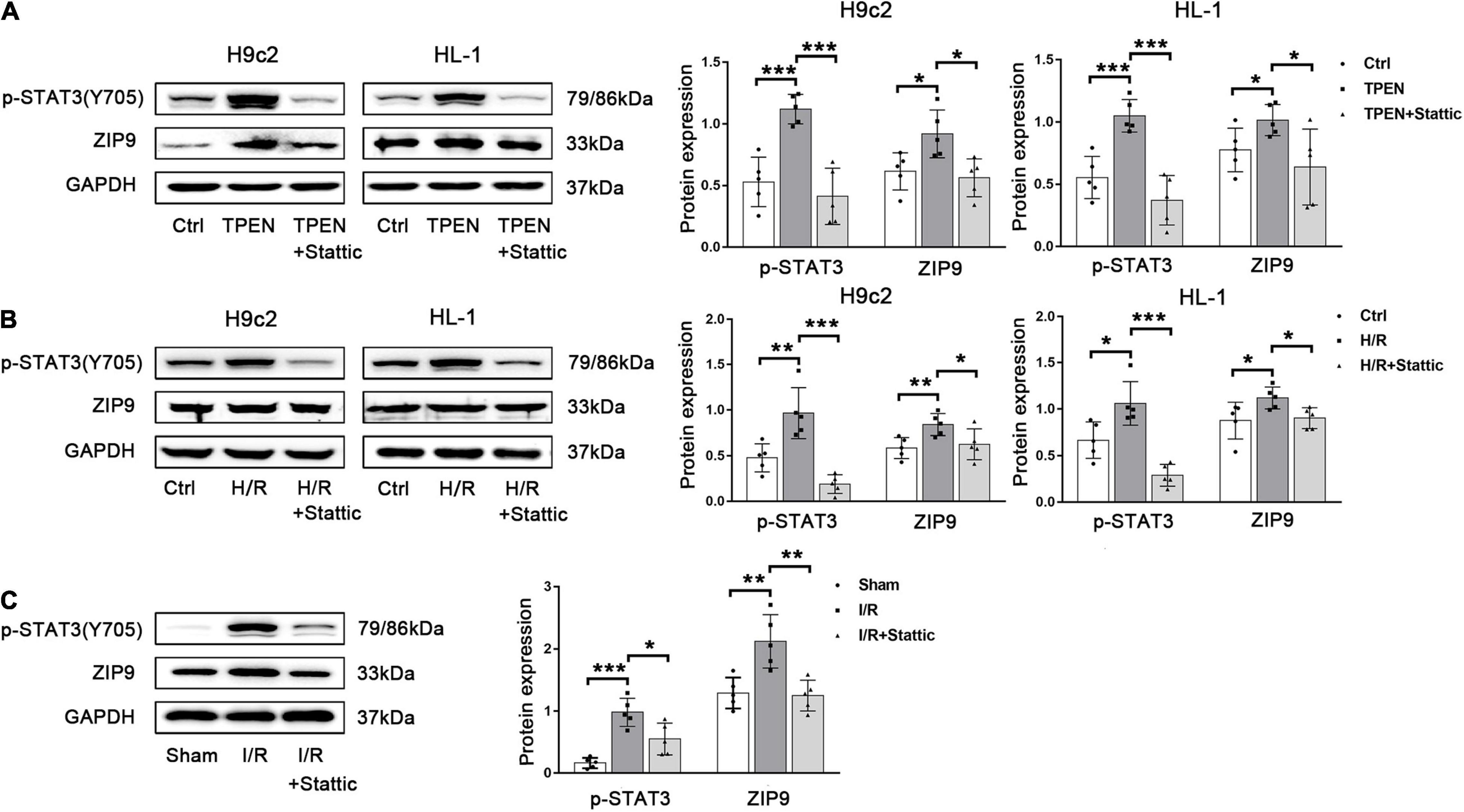
Figure 5. Zinc deficiency-induced STAT3 activation promotes ZIP9 expression. (A,B) Stattic (10 μM, n = 5) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. (C) Mouse hearts were ischemic for 30 min and then reperfused for 30 min. Stattic (3 mg/kg, n = 5) was injected 5 min before reperfusion and continued for 30 min through the tail vein. *p < 0.05, **p < 0.01, and ***p < 0.001.
To further assess whether ER Stress is involved in the regulation of ZIP9 by STAT3, ER stress inducer thapsigargin (TG) was applied. As shown in Supplementary Figure 2A, when TG induced strong ER stress, the mRNA expression of ZIP9 was also up-regulated. Besides, when STAT3 phosphorylation is inhibited by H89, KN93, BAPTA-AM, both the mRNA (Supplementary Figures 2B–D) and protein (Figures 6A–C) expression of ZIP9 are decreased, further confirming that ER Ca2+ leak and CaMKII activation act as upstream signals respond to zinc deficiency to activate STAT3 and promote zip9 expression.
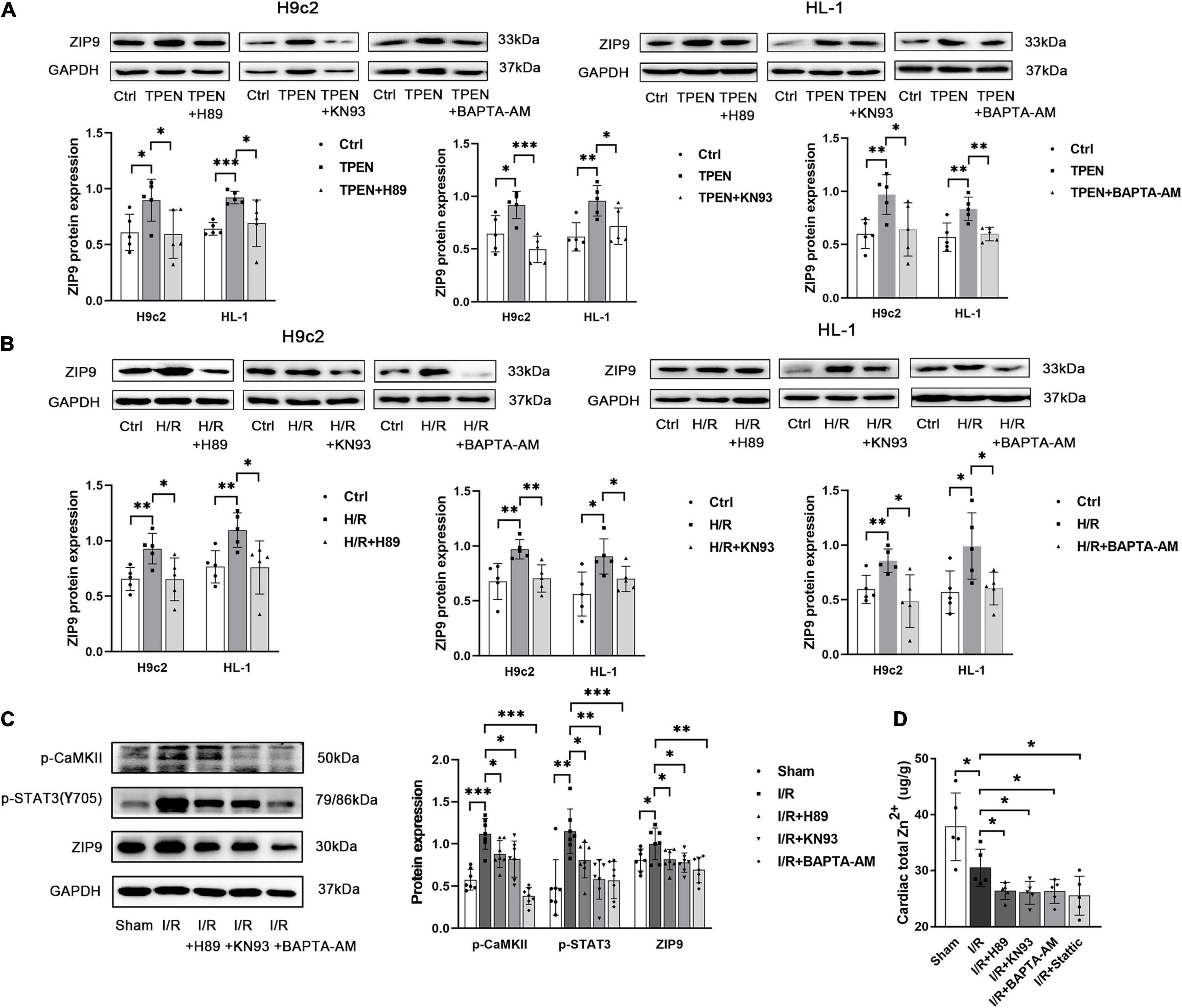
Figure 6. ER Stress/CaMKII is involved in the regulation of ZIP9 by STAT3 (A,B) H89, KN93, or BAPTA-AM (10 μM, n = 5) were applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. (C) Mouse hearts were ischemic for 30 min and then reperfused for 30 min. H89, KN93 or BAPTA-AM (10 mg/kg) were injected 5 min before reperfusion and continued for 30 min through the tail vein (n = 7). (D) Zn2+ levels were monitored with inductively coupled plasma optical emission spectroscopy (ICPOES, n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001.
In order to determine whether the ER stress/CaMKII/STAT3 pathway prevents myocardial Zn2+ loss, ICPOES was used to measure total Zn2+ levels in isolated rat hearts. Compared with the sham group, myocardial Zn2+ levels were significantly decreased after I/R. While the treatment of H89, KN93, BAPTA-AM, and Stattic resulted in increased Zn2+ loss in cardiomyocytes (Figure 6D). These results indicate the responding ER Stress/CaMKII/STAT3 pathway during reperfusion promotes the expression of ZIP9 zinc transporter to correct the imbalance in zinc homeostasis.
To clarify the relationship between STAT3 activation and reperfusion injury, we first detected the release of the lactate dehydrogenase LDH in H9c2 and HL-1 cells. As shown in Figure 7A, TPEN or H/R can increase the release of LDH, while after KN93 treatment, the release of LDH further increased, indicating that inhibition of STAT3 activation can aggravate cell injury, since KN93 can reverse zinc deficiency induced STAT3 activation. Next, we confirmed this result in in vivo myocardial ischemia-reperfusion models. After I/R, the serum LDH, CK-MB, and cTnl activities, as well as infarct size, were increased, but after administration of H89, KN93, BATPA-AM and Stattic, cardiac injury were further increased (Figures 7B,C). These results suggest that ER Stress/CaMKII/STAT3 axis during myocardial ischemia-reperfusion can alleviate reperfusion injury, which may be an endogenous myocardial protective mechanism.
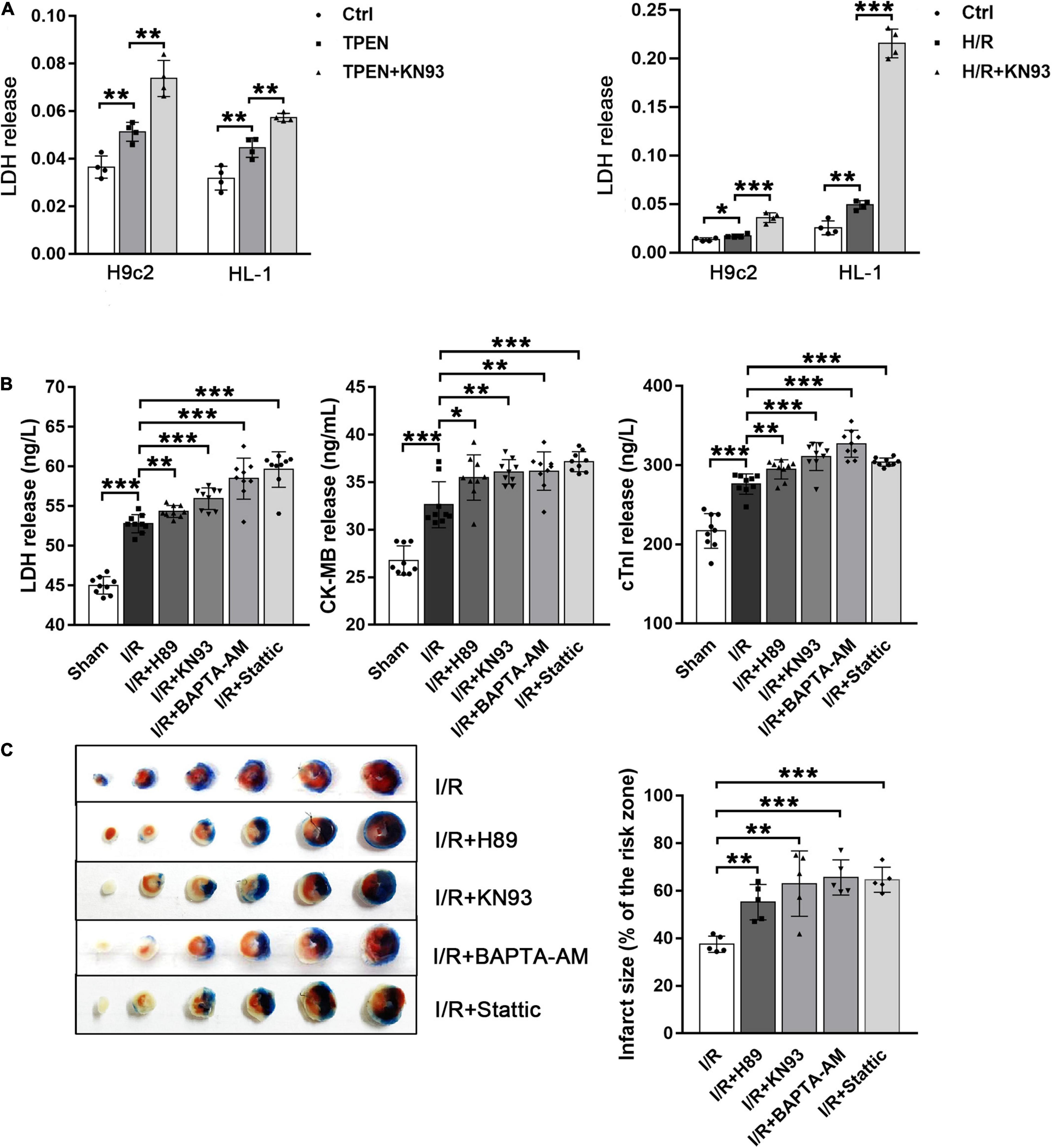
Figure 7. Activated STAT3 can reduce myocardial reperfusion injury. (A) KN93 (10 μM, n = 4) was applied 2 h before exposure to TPEN for 2 h or 30 min before the onset of reoxygenation. (B) Mouse hearts were ischemic for 30 min and then reperfused for 30 min. After I/R, the release of these myocardial injury markers were detected (n = 9). (C) Myocardial infarct size was assessed by TTC staining. Mouse hearts were subjected to 30 min ischemia followed by 2 h of reperfusion (n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001.
In this report, we demonstrate a new pathway response in the setting of intracellular zinc deficiency. Zinc deficiency activates STAT3 by ER stress-induced Ca2+ release and subsequent CaMKII activation, leading to enhancement of the transcriptional activity of the ZIP family zinc transporter genes. More importantly, the mechanism not only works in the setting of intracellular zinc depletion by TPEN but is also sensitive to zinc deficiency caused by hypoxia/reoxygen or ischemia/reperfusion.
Zinc has important structural, enzymatic, and regulatory functions (Ma et al., 2016). These functions of zinc require a tight control of zinc homeostasis. Zinc transporters strictly control intracellular zinc homeostasis in physiological conditions and zinc deficiency is related to many diseases (Hambidge and Krebs, 2007). Studies have demonstrated that ischemia/reperfusion in rat heart can lead to the decrease of intracellular zinc level, but supplementation of zinc can protect the heart, suggesting that the maintenance of zinc homeostasis is very important for the survival of heart during reperfusion (Karagulova et al., 2007). The cellular zinc homeostasis is maintained mainly by zinc transporters. Multiple ZIP family members, such as ZIP1, ZIP2, ZIP7, ZIP10, ZIP13, and ZIP14, were reported to response to zinc deficiency, with the mRNAs of these ZIPs accumulate when cells are exposed to zinc chelator TPEN (Cousins et al., 2003; Dietrich et al., 2017; Du et al., 2019; Thokala et al., 2019). Our preliminary RNA sequencing data in H9c2 cells showed that zinc deficiency can upregulate many ZIP transporters, ZIP9 is one of the most obvious ones. Many studies have reported that ZIP9 plays a critical role in regulating zinc homeostasis by transporting zinc across cell and organelle membranes into the cytoplasm (Matsuura et al., 2009; Taniguchi et al., 2013). In this study, we use ZIP9 as the representative of ZIP family to reduce work burden.
STAT proteins are part of the Janus tyrosine kinase (JAK)/STAT signal pathway (Darnell, 1997). Classical activation of STAT proteins occurs after cytokine bindings to cell surface receptors. The receptor dimerization induces activation of Janus tyrosine kinase (JAK) proteins and the activated JAK proteins activate STAT proteins through tyrosine phosphorylation (Tyr705for STAT3). Phosphorylated STAT proteins undergo dimerization and then STAT dimmers translocate into the nucleus and bind to specific DNA sequences of target genes to affect gene expression. STAT3 is an important protein in the signal transduction of cardioprotection. It has been reported that STAT3 activation protects the heart from I/R injury by reducing oxidative stress, inhibiting apoptosis, and inhibiting the inflammatory cascade (Wu et al., 2019, 2021; Yin et al., 2020). In addition to these results, we reveal that STAT3 activation can up-regulate the expression of ZIP family zinc transporters. In general, the ZIP family transporters transport zinc from cell exterior or intracellular organelles into the cytosol thereby increasing cytosolic free zinc. Therefore, STAT3 activation linking to ZIP transporter expression in the setting of cellular zinc deficiency is a cellular adaptive mechanism by which cells attempt to homeostatically compensate for zinc loss. In this study, STAT3 acts through its post translational phosphorylation at Tyr705 via ER stress-induced Ca2+ release and subsequent CaMKII activation (see below). This finding is very unique and provides a new STAT3 activation mechanism distinct from those previously proposed.
Zinc is required for ER function. Zinc deficiency can lead to an increase in the level of unfolded protein and ER stress because many proteins bind or acquire zinc in the ER (Ellis et al., 2004). Our preliminary RNA sequencing data suggest that zinc deficiency affects the protein processing in ER (Supplementary Figure 1). Further experimental data showed that zinc deficiency induced ER stress serves as an upstream signaling event leading to STAT3 activation and ZIP expression. ER Stress can activate STAT3 in several ways. A recent study has demonstrated that ER stress activated STAT3 in a PERK-dependent manner in glial cells (Meares et al., 2014). It has also been reported that ER transmembrane protein IRE1a can associate with STAT3 to enhancing STAT3 phosphorylation in mouse primary hepatocytes. (Liu et al., 2015). Besides, deletion of the ER resident thiol disulfide oxidoreductase ERp57 enhances STAT3 signaling (Coe et al., 2010). In this study, we reveal that ER stress induced Ca2+ release accounts for STAT3 activation.
Endoplasmic reticulum is an important intracellular calcium reservoir. The decrease of sarcoplasmic reticulum (SR) calcium content may help to protect myocardium from ischemia/reperfusion injury (Kumada et al., 1998). Ryanodine receptor (RyR) and inositol trisphosphate receptor (IP3R) in the ER can mediate Ca2+ release, and a sarco(endo) plasmic reticulum Ca2+-ATPase (SERCA) that uptakes Ca2+ into SR(ER) (Popugaeva and Bezprozvanny, 2014; Zhai et al., 2020). The signal network involved in the absorption and release of Ca2+ in the ER is very complex, and it has been suggested that protein kinase A (PKA) can regulate the uptake and release of Ca2+ in the ER by affecting IP3R, RyR and SERCA proteins, respectively (Leech et al., 2010). Sztretye et al. (2009) showed that zinc ion chelator TPEN could inhibit the increase of cytoplasmic calcium ion concentration by SERCA. Jung et al. (2007) showed that TPEN can activate the release of calcium ions by changing the activity of RyR. Lee et al. (2012) suggested that treatment of cells with IP3R inhibitor 2-APB could inhibit IL-31-induced STAT3 activation. In our study, we observed that the phosphorylation level of RyR2S2808 (PKA site), not the expression of IP3R, SERCA or total RyR, is increased in the setting of intracellular zinc depletion by TPEN. While PKA inhibitor H89 could reverse the up-regulation of p-RyR, the activation of CaMKII/STAT3 and the expression of downstream ZIP zinc transporter, indicating enhancing RyR phosphorylation is required for STAT3 activation. The different responses to TPEN can be attributed to various cell types, exposure time and/or threshold levels. TPEN is a specific cell-permeable heavy metal chelator. Although TPEN has the highest affinity for Zn2+, it can also chelate to other heavy metals (Jackson and Kodanko, 2010; Zhai et al., 2020). However, the enhanced phosphorylation of RyR may not be attributed to the chelation of other metal ions by TPEN, since p-RyR is also increased under pathophysiological hypoxia/reoxygen and ischemia/reperfusion conditions, and inhibition of RyR phosphorylation aggravates zinc ion loss during reperfusion (Karagulova et al., 2007; McIntosh et al., 2010). Obviously, the exact mechanism of zinc deficiency leading to RyR phosphorylation requires more researches.
It was shown that PKA-mediated phosphorylation of RyR2S2808 increases the Ca2+ binding affinity to RyR to enhance RyR opening probability (Houser, 2014). Increased ER Ca2+ leak activates CaMKII. In support, this study demonstrated that cytosolic Ca2+ chelated with BAPTA-AM significantly decreased CaMKII activation. CaMKII is composed of 4 isoforms (α, β, δ, and γ) which possesses expression patterns varying from tissues, mostly α and β in neurons, δ and some γ in cardiomyocytes (Gaertner et al., 2004; Kreusser et al., 2014; Hegyi et al., 2019). CaMKII activity is sustained by autophosphorylation at Thr286 (CaMK2α) or Thr-287 (β, δ, and γ) (Uniprot database). In this study, surprisingly, we detected a very significant increase in CaMKII phosphorylation at Thr286. Meanwhile, inhibition of CaMKII with KN-93, but not the inactive analog KN92, decreased STAT3 phosphorylation, pointing that CaMKII activation at Thr286 is vital to STAT3 activation. Contrary to our results, a large number of studies have shown that inhibiting CaMKII can reduce myocardial ischemia/reperfusion injury (Hong et al., 2010; Ling et al., 2013). Indeed, most groups detect phosphorylation at Thr287 CaMK2δ or CaMK2γ. It is possible that CaMK2α is the dominant isoform which autophosphorylation at Thr-286 that protects the heart from ischemia reperfusion injury. A lot more work are needed to study the mechanism of CaMKII in regulating STAT3.
In this study we have also demonstrated that ZIP9 is upregulated upon hypoxia/reoxygenation or ischemia-reperfusion in the heart or H9c2/HL-1 cells. One of our recent study has shown that ZIP2 was upregulated at reperfusion and protects the heart against ischemia-reperfusion injury (Du et al., 2019). Therefore, it is intriguing to explore the roles of ZIP9 in the setting of myocardial ischemia/reperfusion injury in future studies.
In conclusion (Figure 8), in this study, we demonstrated that zinc deficiency induced Ca2+ release can activate CaMKII at Thr286 which in turn leads to STAT3 activation. This is a new pathway which responses cellular zinc deficiency and promotes the transcription of the genes encoding for the zinc importers to compensate cellular zinc loss. The ER Stress/CaMKII/STAT3 axis provides new therapeutic targets for the treatment of diseases caused by zinc deficiency.

Figure 8. Cellular endogenous protective mechanism induced by zinc deficiency.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Tianjin Medical University Animal Care and Use Committee.
HZ, DL, and XB performed the experiments with cells. QY, JW, and JY conducted animal experiments. XC participated in data discussion and manuscript preparation. ZX designed and supervised the study. All authors approved the final version of the manuscript for publication.
This study was supported by Key Program of Tianjin Natural Science Foundation of Tianjin (2019KJ168).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.736920/full#supplementary-material
Ahyi, A. N., Quinton, L. J., Jones, M. R., Ferrari, J. D., Pepper-Cunningham, Z. A., Mella, J. R., et al. (2013). Roles of STAT3 in protein secretion pathways during the acute-phase response. Infect. Immun. 81, 1644–1653. doi: 10.1128/iai.01332-12
Berg, J. M., and Shi, Y. (1996). The galvanization of biology: a growing appreciation for the roles of zinc. Science 271, 1081–1085. doi: 10.1126/science.271.5252.1081
Brodsky, J. L. (2012). Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167. doi: 10.1016/j.cell.2012.11.012
Coe, H., Jung, J., Groenendyk, J., Prins, D., and Michalak, M. (2010). ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 285, 6725–6738. doi: 10.1074/jbc.M109.054015
Cousins, R. J., Blanchard, R. K., Popp, M. P., Liu, L., Cao, J., Moore, J. B., et al. (2003). A global view of the selectivity of zinc deprivation and excess on genes expressed in human THP-1 mononuclear cells. Proc. Natl. Acad. Sci. U.S.A. 100, 6952–6957. doi: 10.1073/pnas.0732111100
Cousins, R. J., Liuzzi, J. P., and Lichten, L. A. (2006). Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 281, 24085–24089. doi: 10.1074/jbc.R600011200
Cubillos-Ruiz, J. R., Bettigole, S. E., and Glimcher, L. H. (2017). Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168, 692–706. doi: 10.1016/j.cell.2016.12.004
Darnell, J. E. Jr. (1997). STATs and gene regulation. Science 277, 1630–1635. doi: 10.1126/science.277.5332.1630
Dietrich, N., Schneider, D. L., and Kornfeld, K. (2017). A pathway for low zinc homeostasis that is conserved in animals and acts in parallel to the pathway for high zinc homeostasis. Nucleic Acids Res. 45, 11658–11672. doi: 10.1093/nar/gkx762
Du, L., Zhang, H., Zhao, H., Cheng, X., Qin, J., Teng, T., et al. (2019). The critical role of the zinc transporter Zip2 (SLC39A2) in ischemia/reperfusion injury in mouse hearts. J. Mol. Cell Cardiol. 132, 136–145. doi: 10.1016/j.yjmcc.2019.05.011
Ellis, C. D., Wang, F., MacDiarmid, C. W., Clark, S., Lyons, T., and Eide, D. J. (2004). Zinc and the Msc2 zinc transporter protein are required for endoplasmic reticulum function. J. Cell Biol. 166, 325–335. doi: 10.1083/jcb.200401157
Fu, X. Y., Schindler, C., Improta, T., Aebersold, R., and Darnell, J. E. Jr. (1992). The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl. Acad. Sci. U.S.A. 89, 7840–7843. doi: 10.1073/pnas.89.16.7840
Gaertner, T. R., Kolodziej, S. J., Wang, D., Kobayashi, R., Koomen, J. M., Stoops, J. K., et al. (2004). Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 12484–12494. doi: 10.1074/jbc.M313597200
Hambidge, K. M., and Krebs, N. F. (2007). Zinc deficiency: a special challenge. J. Nutr. 137, 1101–1105. doi: 10.1093/jn/137.4.1101
Hegyi, B., Bers, D. M., and Bossuyt, J. (2019). CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J. Mol. Cell Cardiol. 127, 246–259. doi: 10.1016/j.yjmcc.2019.01.001
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Homma, K., Fujisawa, T., Tsuburaya, N., Yamaguchi, N., Kadowaki, H., Takeda, K., et al. (2013). SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol. Cell 52, 75–86. doi: 10.1016/j.molcel.2013.08.038
Hong, Y., Downey, T., Eu, K. W., Koh, P. K., and Cheah, P. Y. (2010). A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin. Exp. Metastasis 27, 83–90. doi: 10.1007/s10585-010-9305-4
Houser, S. R. (2014). Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ Res 114, 1320–1327; discussion1327. doi: 10.1161/CIRCRESAHA.114.300569
Jackson, C. S., and Kodanko, J. J. (2010). Iron-binding and mobilization from ferritin by polypyridyl ligands. Metallomics 2, 407–411. doi: 10.1039/c003414b
Jung, C., Zima, A. V., Szentesi, P., Jona, I., Blatter, L. A., and Niggli, E. (2007). Ca2+ release from the sarcoplasmic reticulum activated by the low affinity Ca2+ chelator TPEN in ventricular myocytes. Cell Calcium 41, 187–194. doi: 10.1016/j.ceca.2006.06.009
Karagulova, G., Yue, Y., Moreyra, A., Boutjdir, M., and Korichneva, I. (2007). Protective role of intracellular zinc in myocardial ischemia/reperfusion is associated with preservation of protein kinase C isoforms. J. Pharmacol. Exp. Ther. 321, 517–525. doi: 10.1124/jpet.107.119644
Kreusser, M. M., Lehmann, L. H., Keranov, S., Hoting, M. O., Oehl, U., Kohlhaas, M., et al. (2014). Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 130, 1262–1273. doi: 10.1161/CIRCULATIONAHA.114.006185
Kumada, Y., Yamamoto, F., Yamamoto, H., Ishikawa, T., Kagisaki, K., and Hirose, H. (1998). [Decreasing sarcoplasmic reticular calcium gives rise to myocardial protection–the effect of thapsigargin for myocardial protection under conditions of normothermia]. Jpn. J. Thorac. Cardiovasc. Surg. 46, 368–374. doi: 10.1007/BF03217757
Lee, C. H., Hong, C. H., Yu, W. T., Chuang, H. Y., Huang, S. K., Chen, G. S., et al. (2012). Mechanistic correlations between two itch biomarkers, cytokine interleukin-31 and neuropeptide beta-endorphin, via STAT3/calcium axis in atopic dermatitis. Br. J. Dermatol. 167, 794–803. doi: 10.1111/j.1365-2133.2012.11047.x
Leech, C. A., Chepurny, O. G., and Holz, G. G. (2010). Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam. Horm. 84, 279–302. doi: 10.1016/B978-0-12-381517-0.00010-2
Ling, H., Gray, C. B., Zambon, A. C., Grimm, M., Gu, Y., Dalton, N., et al. (2013). Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ. Res. 112, 935–944. doi: 10.1161/CIRCRESAHA.112.276915
Liu, Y., Shao, M., Wu, Y., Yan, C., Jiang, S., Liu, J., et al. (2015). Role for the endoplasmic reticulum stress sensor IRE1alpha in liver regenerative responses. J. Hepatol. 62, 590–598. doi: 10.1016/j.jhep.2014.10.022
Liuzzi, J. P., and Cousins, R. J. (2004). Mammalian zinc transporters. Annu. Rev. Nutr. 24, 151–172. doi: 10.1146/annurev.nutr.24.012003.132402
Ma, J., Zhao, N., and Zhu, D. (2016). Bioabsorbable zinc ion induced biphasic cellular responses in vascular smooth muscle cells. Sci. Rep. 6:26661. doi: 10.1038/srep26661
Ma, X., Meng, Z., Jin, L., Xiao, Z., Wang, X., Tsark, W. M., et al. (2017). CAMK2gamma in intestinal epithelial cells modulates colitis-associated colorectal carcinogenesis via enhancing STAT3 activation. Oncogene 36, 4060–4071. doi: 10.1038/onc.2017.16
Matsuura, W., Yamazaki, T., Yamaguchi-Iwai, Y., Masuda, S., Nagao, M., Andrews, G. K., et al. (2009). SLC39A9 (ZIP9) regulates zinc homeostasis in the secretory pathway: characterization of the ZIP subfamily I protein in vertebrate cells. Biosci. Biotechnol. Biochem. 73, 1142–1148. doi: 10.1271/bbb.80910
McIntosh, R., Lee, S., Ghio, A. J., Xi, J., Zhu, M., Shen, X., et al. (2010). The critical role of intracellular zinc in adenosine A(2) receptor activation induced cardioprotection against reperfusion injury. J. Mol. Cell Cardiol. 49, 41–47. doi: 10.1016/j.yjmcc.2010.02.001
Meares, G. P., Liu, Y., Rajbhandari, R., Qin, H., Nozell, S. E., Mobley, J. A., et al. (2014). PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell Biol. 34, 3911–3925. doi: 10.1128/mcb.00980-14
Nguyen, T. S., Kohno, K., and Kimata, Y. (2013). Zinc depletion activates the endoplasmic reticulum-stress sensor Ire1 via pleiotropic mechanisms. Biosci. Biotechnol. Biochem. 77, 1337–1339. doi: 10.1271/bbb.130130
Popugaeva, E., and Bezprozvanny, I. (2014). Can the calcium hypothesis explain synaptic loss in Alzheimer’s disease? Neurodegen. Dis. 13, 139–141. doi: 10.1159/000354778
Prasad, A. S., Halsted, J. A., and Nadimi, M. (1961). Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am. J. Med. 31, 532–546. doi: 10.1016/0002-9343(61)90137-1
Reed, G. W., Rossi, J. E., and Cannon, C. P. (2017). Acute myocardial infarction. Lancet 389, 197–210. doi: 10.1016/s0140-6736(16)30677-8
Reilly-O’Donnell, B., Robertson, G. B., Karumbi, A., McIntyre, C., Bal, W., Nishi, M., et al. (2017). Dysregulated Zn(2+) homeostasis impairs cardiac type-2 ryanodine receptor and mitsugumin 23 functions, leading to sarcoplasmic reticulum Ca(2+) leakage. J. Biol. Chem. 292, 13361–13373. doi: 10.1074/jbc.M117.781708
Schindler, C., Shuai, K., Prezioso, V. R., and Darnell, J. E. Jr. (1992). Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257, 809–813. doi: 10.1126/science.1496401
Sheng, M., Zhang, G., Wang, J., Yang, Q., Zhao, H., Cheng, X., et al. (2018). Remifentanil induces cardio protection against ischemia/reperfusion injury by inhibiting endoplasmic reticulum stress through the maintenance of zinc homeostasis. Anesth. Analg. 127, 267–276. doi: 10.1213/ANE.0000000000003414
Sztretye, M., Almassy, J., Deli, T., Szentesi, P., Jung, C., Dienes, B., et al. (2009). Altered sarcoplasmic reticulum calcium transport in the presence of the heavy metal chelator TPEN. Cell Calcium 46, 347–355. doi: 10.1016/j.ceca.2009.10.002
Taniguchi, M., Fukunaka, A., Hagihara, M., Watanabe, K., Kamino, S., Kambe, T., et al. (2013). Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS One 8:e58022. doi: 10.1371/journal.pone.0058022
Tapazoglou, E., Prasad, A. S., Hill, G., Brewer, G. J., and Kaplan, J. (1985). Decreased natural killer cell activity in patients with zinc deficiency with sickle cell disease. J. Lab. Clin. Med. 105, 19–22.
Thokala, S., Bodiga, V. L., Kudle, M. R., and Bodiga, S. (2019). Comparative response of cardiomyocyte ZIPs and ZnTs to extracellular zinc and TPEN. Biol. Trace Elem. Res. 192, 297–307. doi: 10.1007/s12011-019-01671-0
Timmins, J. M., Ozcan, L., Seimon, T. A., Li, G., Malagelada, C., Backs, J., et al. (2009). Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Invest. 119, 2925–2941. doi: 10.1172/JCI38857
Unudurthi, S. D., Nassal, D., Greer-Short, A., Patel, N., Howard, T., Xu, X., et al. (2018). betaIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J. Clin. Invest. 128, 5561–5572. doi: 10.1172/JCI99245
Vallee, B. L., and Falchuk, K. H. (1993). The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118. doi: 10.1152/physrev.1993.73.1.79
Wang, G., Huang, H., Zheng, H., He, Y., Zhang, Y., Xu, Z., et al. (2016). Zn(2+) and mPTP mediate endoplasmic reticulum stress inhibition-induced cardioprotection against myocardial ischemia/reperfusion injury. Biol. Trace Elem. Res. 174, 189–197. doi: 10.1007/s12011-016-0707-2
Wu, J. W., Hu, H., Li, D., and Ma, L. K. (2021). Hypoxia-inducible factor 2-alpha-dependent induction of IL-6 protects the heart from ischemia/reperfusion injury. Aging (Albany NY) 13, 3443–3458. doi: 10.18632/aging.202276
Wu, L., Tan, J. L., Chen, Z. Y., and Huang, G. (2019). Cardioprotection of post-ischemic moderate ROS against ischemia/reperfusion via STAT3-induced the inhibition of MCU opening. Basic Res. Cardiol. 114:39. doi: 10.1007/s00395-019-0747-9
Yamashita, S., Miyagi, C., Fukada, T., Kagara, N., Che, Y. S., and Hirano, T. (2004). Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 429, 298–302. doi: 10.1038/nature02545
Yin, Q., Zhao, B., Zhu, J., Fei, Y., Shen, W., Liang, B., et al. (2020). JLX001 improves myocardial ischemia-reperfusion injury by activating Jak2-Stat3 pathway. Life Sci. 257:118083. doi: 10.1016/j.lfs.2020.118083
Keywords: zinc deficiency, ischemia/reperfusion (I/R) injury, ER stress, CaMKII, STAT3
Citation: Zhao H, Liu D, Yan Q, Bian X, Yu J, Wang J, Cheng X and Xu Z (2022) Endoplasmic Reticulum Stress/Ca2+-Calmodulin-Dependent Protein Kinase/Signal Transducer and Activator of Transcription 3 Pathway Plays a Role in the Regulation of Cellular Zinc Deficiency in Myocardial Ischemia/Reperfusion Injury. Front. Physiol. 12:736920. doi: 10.3389/fphys.2021.736920
Received: 06 July 2021; Accepted: 29 November 2021;
Published: 05 January 2022.
Edited by:
Francisco Altamirano, Houston Methodist Research Institute, United StatesReviewed by:
Valeria Garrido Moreno, Centro Avanzado de Enfermedades Crónicas, ChileCopyright © 2022 Zhao, Liu, Yan, Bian, Yu, Wang, Cheng and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinxin Cheng, Y2hlbmd4aW54aW5AdG11LmVkdS5jbg==; Zhelong Xu, enh1QHRtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.