 Cátia F. Lourenço
Cátia F. Lourenço João Laranjinha
João Laranjinha
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 22 October 2021
Sec. Redox Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.729201
This article is part of the Research TopicOxidative Stress Revisited - Major Role in Vascular Diseases, Volume IIView all 5 articles
The brain has impressive energy requirements and paradoxically, very limited energy reserves, implying its huge dependency on continuous blood supply. Aditionally, cerebral blood flow must be dynamically regulated to the areas of increased neuronal activity and thus, of increased metabolic demands. The coupling between neuronal activity and cerebral blood flow (CBF) is supported by a mechanism called neurovascular coupling (NVC). Among the several vasoactive molecules released by glutamatergic activation, nitric oxide (•NO) is recognized to be a key player in the process and essential for the development of the neurovascular response. Classically, •NO is produced in neurons upon the activation of the glutamatergic N-methyl-D-aspartate (NMDA) receptor by the neuronal isoform of nitric oxide synthase and promotes vasodilation by activating soluble guanylate cyclase in the smooth muscle cells of the adjacent arterioles. This pathway is part of a more complex network in which other molecular and cellular intervenients, as well as other sources of •NO, are involved. The elucidation of these interacting mechanisms is fundamental in understanding how the brain manages its energy requirements and how the failure of this process translates into neuronal dysfunction. Here, we aimed to provide an integrated and updated perspective of the role of •NO in the NVC, incorporating the most recent evidence that reinforces its central role in the process from both viewpoints, as a physiological mediator and a pathological stressor. First, we described the glutamate-NMDA receptor-nNOS axis as a central pathway in NVC, then we reviewed the link between the derailment of the NVC and neuronal dysfunction associated with neurodegeneration (with a focus on Alzheimer’s disease). We further discussed the role of oxidative stress in the NVC dysfunction, specifically by decreasing the •NO bioavailability and diverting its bioactivity toward cytotoxicity. Finally, we highlighted some strategies targeting the rescue or maintenance of •NO bioavailability that could be explored to mitigate the NVC dysfunction associated with neurodegenerative conditions. In line with this, the potential modulatory effects of dietary nitrate and polyphenols on •NO-dependent NVC, in association with physical exercise, may be used as effective non-pharmacological strategies to promote the •NO bioavailability and to manage NVC dysfunction in neuropathological conditions.
The brain is highly dependent on the continuous and dynamically regulated delivery of metabolic substrates to support ongoing neural function. The energy requirements of the brain are expressively high and paradoxically, it has very limited reserves which imply that the blood supply must be finely and timely adjusted to where it is needed the most, which are the areas of increased activity (Attwell and Laughlin, 2001). This process, namely, neurovascular coupling (NVC), is accomplished by a tight network communication between active neurons and vascular cells that involves the cooperation of the other cells from the neurovascular unit (namely, astrocytes, and pericytes) (Attwell et al., 2010; Iadecola, 2017). Despite the extensive investigations and huge advances in the field over the last decades, a clear definition of the mechanisms underlying this process and particularly, the underlying cross-interactions and balance, is still elusive. This is accounted for by the difficulties in measuring the process dynamically in vivo, allied with the intrinsic complexity of the process, likely enrolling diverse signaling pathways that reflect the specificities of the neuronal network of different brain regions and the diversity of the neurovascular unit along the cerebrovascular tree (from pial arteries to capillaries). Within such complexity, there is a prevailing common assumption that points to glutamate, the main excitatory neurotransmitter in the brain, as the trigger for NVC in the feed-forward mechanisms elicited by activated neurons. The pathways downstream glutamate may then involve multiple vasoactive molecules released by neurons (via activation of ligand-gated cationic channels – iGluRs) and/or astrocytes (via G-coupled receptors activation – mGluRs) (Attwell et al., 2010; Iadecola, 2017; Lourenço et al., 2017a). Among them, nitric oxide (•NO) is widely recognized to be an ubiquitous key player in the process and essential for the development of the neurovascular response, as will be discussed in a later section (Figure 1).
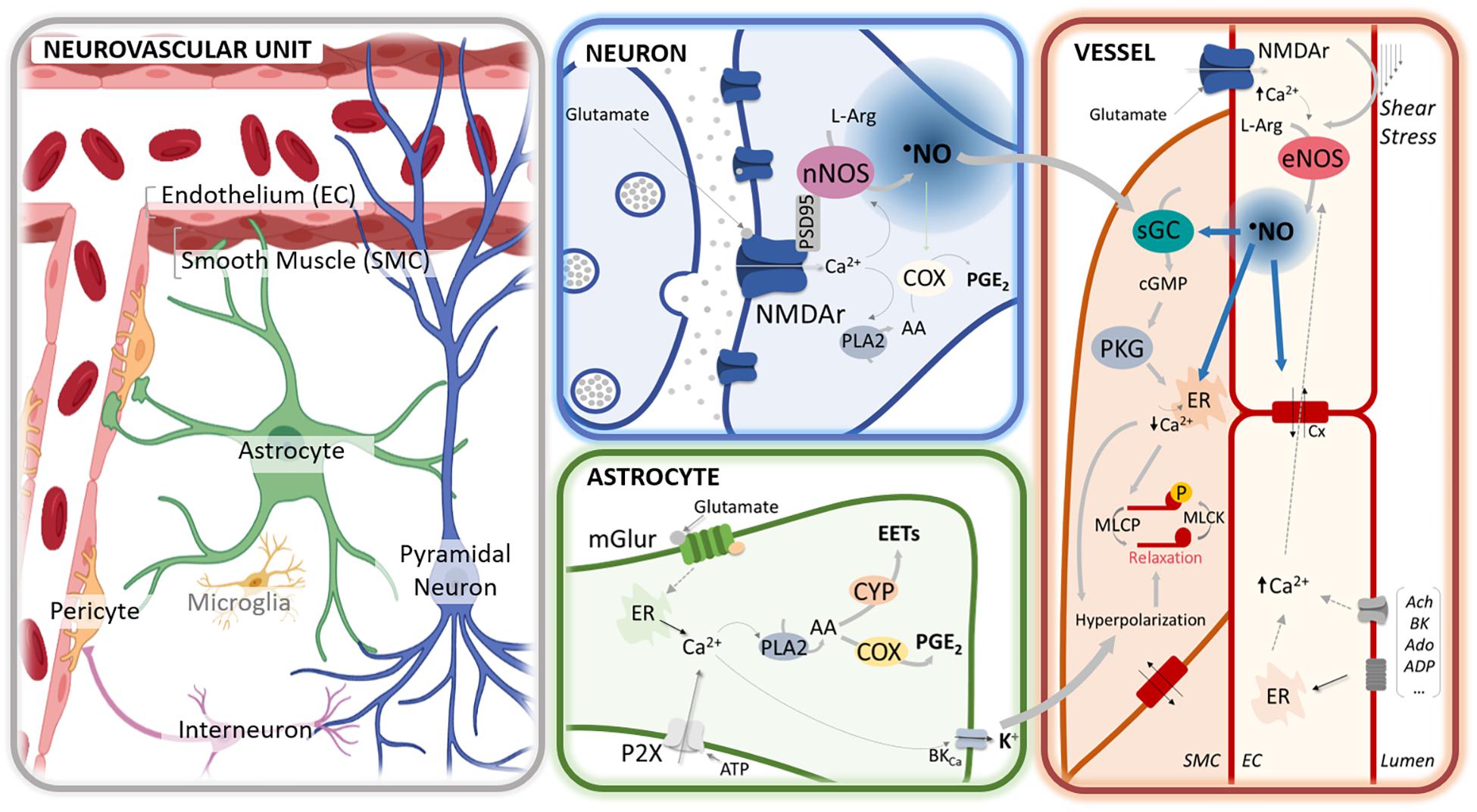
Figure 1. •NO-mediated regulation of neurovascular coupling at different cellular compartments of the neurovascular unit. In neurons, glutamate release activates the N-methyl-D-aspartate (NMDA) receptors (NMDAr), leading to an influx of calcium cation (Ca2+) that activates the neuronal nitric oxide synthase (nNOS), physically anchored to the receptor via the scaffold protein PSD95. The influx of Ca2+ may further activate phospholipase A2 (PLA2), leading to the synthesis of prostaglandins (PGE) via cyclooxygenase (COX) activation. In astrocytes, the activation of mGluR by glutamate by rising Ca2+ promotes the synthesis of PGE via COX and epoxyeicosatrienoic acids (EETs) via cytochrome P450 epoxygenase (CYP) activation and leads to the release of K + via the activation of BKCa. At the capillary level, glutamate may additionally activate the NMDAr in the endothelial cells (EC), thereby eliciting the activation of endothelial NOS (eNOS). The endothelial-dependent nitric oxide (•NO) production can be further elicited via shear stress or the binding of different agonists (e.g., acetylcholine, bradykinin, adenosine, ATP). Additionally, erythrocytes may contribute to •NO release (via nitrosated hemoglobin or hemoglobin-mediated nitrite reduction). At the smooth muscle cells (SMC), paracrine •NO activates the sGC to produce cGMP and activate the cGMP-dependent protein kinase (PKG). The PKG promotes a decrease of Ca2+ [e.g., by stimulating its reuptake by sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA)] that leads to the dephosphorylation of the myosin light chain through the associated phosphatase (MLCP) and, ultimately to vasorelaxation. Additionally, PKG triggers the efflux of K+ by the large-conductance Ca2+-sensitive potassium channel (BKCa) that leads to cell hyperpolarization. Hyperpolarization is additionally triggered via the activation of the inward rectifier potassium channels (Kir) and spread rapidly to adjacent cells via gap junctions (Cx). Further, •NO can regulate vasodilation via the stimulation of SERCA, modulation of the synthesis of arachidonic acid (AA) derivatives, and regulation of potassium channels and connexins.
A full understanding of the mechanisms underlying NVC is fundamental to know how the brain manages its energy requirements under physiological conditions and how the failure in regulating this process is associated with neurodegeneration. The connection between NVC dysfunction and neurodegeneration is nowadays well-supported by a range of neurological conditions, including Alzheimer’s disease (AD), vascular cognitive impairment and dementia (VCID), traumatic brain injury (TBI), multiple sclerosis (MS), among others (Iadecola, 2004, 2017; Lourenço et al., 2017a; Iadecola and Gottesman, 2019). In line with this, the advancing of our understanding of the mechanisms through which the brain regulates, like no other organ, its blood perfusion may provide relevant cues to forward new therapeutic strategies targeting neurodegeneration and cognitive decline. A solid understanding of NVC is also relevant, considering that the hemodynamic responses to neural activity underlie the blood-oxygen-level-dependent (BOLD) signal used in functional MRI (fMRI) (Attwell and Iadecola, 2002).
In the next sections, the status of the current knowledge on the involvement of •NO in regulating the NVC will be discussed. Furthermore, we will explore how the decrease in •NO bioavailability may support the link between NVC impairment and neuronal dysfunction in some neurodegenerative conditions. Finally, we will discuss some strategies that can be used to counteract NVC dysfunction, and thus, to improve cognitive function.
The classical pathway for •NO synthesis involves a family of enzymes – nitric oxide synthase (NOS) – that catalyzes the oxidation of L-arginine to L-citrulline and •NO, provided that oxygen (O2) and several other cofactors are available [nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), heme and tetrahydrobiopterin (BH4)]. For this to occur, the enzyme must be in a homodimeric form that results from the assembly of two monomers through the oxygenase domains and allows the electrons released by the NADPH in the reductase domain to be transferred through the FAD and FMN to the heme group of the opposite subunit. At this point, in the presence of the substrate L-arginine and the cofactor BH4, the electrons enable the reduction of O2 and the formation of •NO and L-citrulline. Under conditions of disrupted dimerization, ensured by different factors (e.g., BH4 bioavailability), the enzyme catalyzes the uncoupled oxidation of NADPH with the consequent production of superoxide anion (O2–•) instead of •NO (Knowles and Moncada, 1994; Stuehr, 1999). There are three major members of the NOS family which may diverge in terms of the cellular/subcellular localization, regulation of their enzymatic activity, and physiological function: type I neuronal NOS (nNOS), type II inducible NOS (iNOS), and type III endothelial NOS (eNOS) (Stuehr, 1999). The nNOS and eNOS are constitutively expressed enzymes that rely on Ca2+-calmodulin binding for activation. The nNOS and eNOS activity is further regulated both at the transcriptional and post-translational levels and via protein-protein interactions (Forstermann and Sessa, 2012). While not exclusively, the nNOS is mainly expressed in neurons where it is intimately associated with glutamatergic neurotransmission. The dominant splice variant of this isoform (nNOSα) possesses an N-terminal PDZ motif that allows the enzyme to bind other PDZ-containing proteins, such as the synaptic density scaffold protein PSD-95. This allows the enzyme to anchor itself to the synaptic membrane by forming a supramolecular complex with the N-methyl-D-aspartate receptors (NMDAr), whose activation upon glutamate binding results in Ca2+ influx, and ultimately, •NO production. The eNOS isoform is mainly expressed at the endothelium and is critically involved in vascular homeostasis. In the endothelial cells, the eNOS is predominantly localized within the caveolae, forming a complex with caveolin-1 that inhibits its activity. The stretching of the vascular wall, induced by shear stress, results in the dissociation of this complex and allows the enzyme to be activated, either by Ca2+-calmodulin binding and/or by PI3K/Akt-mediated phosphorylation of specific serine residues (e.g., 1,177) (Forstermann and Sessa, 2012). Unlike the other two isoforms, iNOS does not rely on Ca2+ increases for activation but on the de novo synthesis, which occurs predominantly in glial cells following an immunological or inflammatory stimulation. Because iNOS has much lower Ca2+ requirements (calmodulin binds with very high affinity to the enzyme even at basal Ca2+ levels), it produces •NO for as long as the enzyme remains from being degraded (Knott and Bossy-Wetzel, 2009).
In recent years, studies have supported •NO production independent of NOS activity, through the stepwise reduction of nitrate (NO3–) and nitrite (NO2–) via the so-called nitrate-nitrite-nitric oxide pathway. Viewed as stable end products of •NO metabolism, both NO3– and NO2– are now recognized to be able to be recycled back into •NO, thereby acting as important •NO reservoirs in vivo. NO3– and NO2– can be consumed in the regular vegetable components of a diet, fueling the nitrate-nitrite-nitric oxide pathway (Rocha et al., 2011; Lundberg et al., 2018). NO3– can be reduced to NO2– by the commensal bacteria in the gastrointestinal tract and/or by the mammalian enzymes that can acquire a nitrate reductase activity under acidic and hypoxic environments. In turn, the reduction of NO2– to •NO can be achieved non-enzymatically via a redox interaction with one-electron reductants (e.g., ascorbate and polyphenols) or can be catalyzed by different enzymes (e.g., hemoglobin, xanthine oxidoreductase, and cytochrome P450 reductase). All these reactions are favored by low O2 and decreased pH, thereby ensuring the generation of •NO under conditions of limited synthesis by the canonical NOS-mediated pathways which require O2 as a substrate (Lundberg et al., 2008). It is also worth mentioning that S-nitrosothiols might serve as downstream •NO-carrying signaling molecules regulating protein expression/function (Chen et al., 2008).
The transduction of •NO signaling may involve several reactions that reflect, among other factors, the high diffusion of •NO, the relative spatial and temporal abundance of the targets, and the relative rate constants with the potential targets. Most of the physiological actions of •NO are promoted by the chemical modification of relevant proteins either via nitrosylation or nitrosation [reviewed in Picon-Pages et al. (2019)]. Nitrosylation refers to the reversible binding of •NO to inorganic protein moieties (e.g., iron in heme groups), while nitrosation involves the modification of organic moieties (e.g., thiol groups in cysteine residues), not directly, but intermediated by the species produced upon •NO autoxidation, namely N2O3. In addition, •NO can react with superoxide anion (O2–•), yielding peroxynitrite (ONOO–), a potent oxidant and nitrating species that conveys the main deleterious actions associated with the •NO signaling (e.g., oxidation and/or nitration of proteins, lipids and nucleic acids) (Radi, 2018).
The best characterized molecular target for the physiological action of •NO is the soluble guanylate cyclase (sGC), a hemeprotein that is frequently and controversially tagged as the classical “•NO receptor.” The activation of the sGC by •NO involves the nitrosylation of heme moiety of the enzyme that induces a conformational change, enabling it to catalyze the conversion of guanosine triphosphate (GTP) to the second messenger cyclic guanosine monophosphate (cGMP) (Martin et al., 2005). Nitric oxide may additionally regulate the catalytic activity of sGC by promoting its inhibition via nitrosation of critical cysteine residues (Beuve, 2017).
After being recognized as the endothelial-derived relaxing factor (EDRF) in the late 80s, it did not take long for •NO to be implicated in NVC (Iadecola, 1993). This is not unexpected if we consider that •NO is well suited for such function: it is produced upon glutamate stimulation in the brain, is highly diffusible, and is a potent vasodilator involved in the regulation of the vascular tone.
The conventional pathway for •NO- mediated NVC involves the activation of the glutamate-NMDAr-nNOS pathway in neurons. The binding of glutamate to the NMDAr stimulates the influx of [Ca2+] through the channel that, upon binding calmodulin, promotes the activation of nNOS and the synthesis of •NO. Being hydrophobic and highly diffusible, the •NO produced in neurons can diffuse intercellularly and reach the smooth muscle cells (SMC) of adjacent arterioles, there inducing the activation of sGC and promoting the formation of cGMP. The subsequent activation of the cGMP-dependent protein kinase (PKG) leads to a decrease [Ca2+] that results in the dephosphorylation of the myosin light chain and consequent SMC relaxation [reviewed by Iadecola (1993) and Lourenço et al. (2017a)]. Additionally, •NO may promote vasodilation via the stimulation of the sarco/endoplasmic reticulum calcium ATPase (SERCA), via activation of the Ca2+-dependent K+ channels, or via modulation of the synthesis of other vasoactive molecules [reviewed by Lourenço et al. (2017a)]. Specifically, the ability of •NO to regulate the activity of critical heme-containing enzymes involved in the metabolism of arachidonic acid to vasoactive compounds suggests the complementary role of •NO as a modulator of NVC via the modulation of the signaling pathways linked to mGLuR activation at the astrocytes. •NO has been demonstrated to play a permissive role in PGE2-dependent vasodilation by regulating cyclooxygenase activity (Fujimoto et al., 2004) and eliciting ATP release from astrocytes (Bal-Price et al., 2002).
The notion of •NO as a key intermediate in NVC was initially grounded by a large set of studies describing the blunting of NVC responses by the pharmacological NOS inhibition under different experimental paradigms [reviewed (Lourenço et al., 2017a)]. A recent meta-analysis, covering studies on the modulation of different signaling pathways in NVC, found that a specific nNOS inhibition produced a larger blocking effect than any other individual target (e.g., prostanoids, purines, and K+). In particular, the nNOS inhibition promoted an average reduction of 2/3 in the NVC response (Hosford and Gourine, 2019). It is recognized that the dominance of the glutamate-NMDAr-NOS pathway in NVC likely reflects the specificities of the neuronal networks, particularly concerning the heterogenic pattern of nNOS expression/activity in the brain. Although nNOS is ubiquitously expressed in different brain areas, the pattern of nNOS immunoreactivity in the rodent telencephalon has been pointed to a predominant expression in the cerebellum, olfactory bulb, and hippocampus and scarcely in the cerebral cortex (Bredt et al., 1990; Lourenço et al., 2014a). Coherently, there is a prevalent consensus for the role of •NO as the direct mediator of the neuron-to-vessels signaling in the hippocampus and cerebellum. In the hippocampus of anesthetized rats, it was demonstrated that the •NO production and hemodynamic changes evoked by the glutamatergic activation in dentate gyrus (DG) are temporally correlated and both dependent on the glutamate-NMDAr-nNOS pathway (Lourenço et al., 2014b). The blockage of either the NMDAr or nNOS also showed to blunt the •NO production and vessels dilation to mossy fiber stimulation in the cerebellar slices (Mapelli et al., 2017).
In the cerebral cortex, •NO has been suggested to act as a modulator rather than a direct mediator of the NVC responses, but this view has been challenged in recent years. Emergent evidence from ex vivo approaches indicates that the regulation of vasodilation may diverge along the cerebrovascular tree: at the capillary level, vasodilation seems to be mainly controlled by pericytes via an ATP-dependent astrocytic pathway while at the arteriolar level it involves neuronal •NO-NMDAr signaling (Mishra et al., 2016).
Recent data support that the optogenetic stimulation of nNOS positive interneurons can promote central blood flow (CBF) changes in the somatosensory cortex comparable to those evoked by whiskers stimulation on awake and behaving rodents (Krawchuk et al., 2020; Lee et al., 2020). The implication of the GABAergic interneurons in NVC has been previously demonstrated, both in the cerebellum and somatosensory cortex (Cauli et al., 2004; Rancillac et al., 2006). Also, in the hippocampus, parvalbumin GABAergic interneurons are suggested to drive, via •NO signaling, the NVC response to hippocampus-engaged exploration activities with impact in the neurogenesis in the dentate gyrus (Shen et al., 2019). The involvement of GABAergic interneurons in neurovascular regulation is not unexpected as some of them have extended projections in close contact with arterial vessels and secrete diverse molecules with vasoactive properties which are able to modulate the vascular tone (e.g., •NO, vasopressin, and NPY) (Hamel, 2006). A novel and striking hypothesis suggest that nNOS-expressing neurons can control vasodilation independent of neural activities. The optogenetic activation of NOS-positive interneurons regulates CBF without detectable changes in the activity of other neurons (Echagarruga et al., 2020; Lee et al., 2020). The activation of GABAergic interneurons has further been shown to promote vasodilation while decreasing neuronal activity; this occurring independently of ionotropic glutamatergic or GABAergic synaptic transmission (Scott and Murphy, 2012; Anenberg et al., 2015). The hypothesis stating that evoked CBF is dynamically regulated by different subsets of neurons, some independently of neuronal activity, calls into question the linearity of the correlation between the net ongoing neuronal activity and CBF changes and raises concerns regarding the interpretation of functional MRI (fMRI) data.
As for the systemic vascular network, endothelial-derived •NO has also been implicated in the regulation of CBF. Endothelial cells are able to respond to diverse chemical and physical stimuli by producing, via Ca2+-dependent signaling pathways, a myriad of vasoactive compounds (e.g., •NO), thereby modulating the vascular tone. Additionally, Ca2+ may directly induce the hyperpolarization of the endothelial membrane and adjacent SMC through the activation of Ca2+-dependent K+ channels (Chen et al., 2014; Guerra et al., 2018). Despite this, the critical requirement of endothelium for the development of a full neurovascular response to neuronal activity only recently started to be valued. Specifically, endothelial-mediated signaling stands to be essential for the retrograde propagation of NVC-associated vasodilation. The discrete ablation of the endothelium was demonstrated to halt the retrograde dilation of pial arteries in response to hindpaw stimulation (Chen et al., 2014). Additionally, in the somatosensory cortex, NVC was shown to be regulated via eNOS upon the activation of the purinergic receptors at the endothelium in a mechanism involving a glio-endothelial coupling (Toth et al., 2015). Recent data further pointed to the ability of endothelial cells to directly sense neuronal activity via the NMDAr expressed in the basolateral endothelial membranes, thereby eliciting vasodilation via eNOS activation (Stobart et al., 2013; Hogan-Cann et al., 2019; Lu et al., 2019). While the precise mechanisms by which the eNOS-derived •NO shape NVC response is still to be defined, eNOS activation is suggested to contribute to the local but not to the conducted vasodilation, the latter being associated with K+-mediated hyperpolarization (Lu et al., 2019). Yet, it is proposed that •NO-dependent vasodilation may be also involved in a slower and shorter-range retrograde propagation cooperating with the faster and long-range propagation mediated by endothelial hyperpolarization (Chen et al., 2014; Tran et al., 2018). Of note, •NO can modulate the activity of connexins at the gap junctions to favor the propagation of the hyperpolarizing current upstream to the feeding vessels (Kovacs-Oller et al., 2020). Additionally, vascular-derived •NO has been pointed to facilitate Ca2+ astrocytic signal and was forwarded as an explanation for the late endfoot Ca2+ signaling (Tran et al., 2018).
Despite the extensive accumulated evidence for the involvement of •NO in the NVC in animal models, these studies have only been applied to humans recently. By addressing the hemodynamic response to visual stimulation, Hoiland and coworkers provided the first demonstration for the involvement of •NO in the NVC in humans via modulation by a systemic intravenous infusion of the non-selective competitive NOS inhibitor L-NMMA (Hoiland et al., 2020). The authors proposed a two-step signaling mechanism for the NVC in humans translated in a biphasic response with the first component being attributed to the NOS activation elicited by glutamatergic activation. They hypothesized that •NO may be further involved in the second component of the hemodynamic response via erythrocyte-mediated signaling (either by releasing •NO from nitrosated hemoglobin or by mediating NO2– reduction) (Hoiland et al., 2020).
The tight coupling between neuronal activity and CBF is crucial in supporting the functional integrity of the brain, by both providing the essential metabolic substrates for ongoing neuronal activities and by contributing to the clearance of the metabolic waste byproducts. Disturbances of the mechanisms that regulate CBF, both under resting and activated conditions, can therefore critically impair neural function. Coherently, a robust amount of data support neurovascular dysfunction implicated in the mechanisms of neurodegeneration and cognitive decline associated with several conditions, including aberrant brain aging, AD, VCID, and TBI, among others [reviewed by Zlokovic (2011), Lourenço et al. (2017a), Sweeney et al. (2018), and Moretti and Caruso (2020)]. A large amount of clinical studies has been focused on AD, for which the regional CBF changes were described to follow a stepwise pattern along the clinical stages of the disease in connection with a cognitive decline (Wierenga et al., 2012; Leeuwis et al., 2017; Mokhber et al., 2021). Alongside, both patients with mild cognitive impairment and AD displayed decreased hemodynamic responses to neuronal activation (memory encoding tasks) (Small et al., 1999; Xu et al., 2007). Interestingly, a retrospective neuroimaging analysis of healthy subjects and patients with mild cognitive impairment and AD suggested that vascular abnormalities are early events, preceding the changes in Aβ deposition, functional impairment, and cerebral atrophy (Iturria-Medina et al., 2016). These and other clinical data are strongly supported by an extensive portfolio of studies in animal models of AD that recapitulate the NVC dysfunction observed in patients [(Mueggler et al., 2003; Shin et al., 2007; Rancillac et al., 2012; Lourenço et al., 2017b; Tarantini et al., 2017), reviewed by Nicolakakis and Hamel (2011)]. The latter has also proved to be valuable in providing insights on the mechanisms underpinning NVC dysfunction and their correlation with AD classical pathological hallmarks, namely, Aβ accumulation, tau hyperphosphorylation, and neuronal loss. For instance, both in vitro and in vivo studies demonstrated that Aβ can reduce the CBF changes in response to vasodilators and neuronal activation (Price et al., 1997; Thomas et al., 1997; Niwa et al., 2000). In turn, hypoperfusion has been demonstrated to foster both the Aβ production and accumulation (Koike et al., 2010; Park et al., 2019; Shang et al., 2019). Simplistically, this points to a vicious cycle that may sustain the progression of the disease. In this cycle, CBF alterations stand out as important prompters. For instance, in the 3xTgAD mice model of AD, the impairment of the NVC in the hippocampus was demonstrated to precede an obvious cognitive dysfunction or altered neuronal-derived •NO signaling, suggestive of an altered cerebrovascular dysfunction (Lourenço et al., 2017b). Also, the suppression of NVC to whiskers stimulation reported in the tau-expressing mice was described to precede tau pathology and cognitive impairment. In this case, the NVC dysfunction was attributed to the specific uncoupling of the nNOS from the NMDAr and the consequent disruption of •NO production in response to neuronal activation (Park et al., 2020). Overall, these studies point to dysfunctional NVC as a trigger event of the toxic cascade leading to neurodegeneration and dementia.
The mechanisms underpinning the NVC dysfunction in AD and other pathologies are expectedly complex and likely enroll several intervenients through a myriad of pathways, that may reflect both the specificities of neuronal networks (as the NVC itself) and that of the neurodegenerative pathways. Yet, oxidative stress (nowadays conceptually denoted by Sies and Jones as oxidative distress) is recognized as an important and ubiquitous contributor to the dysfunctional cascades that culminate in the NVC deregulation in several neurodegenerative conditions (Hamel et al., 2008; Carvalho and Moreira, 2018). Oxidative distress is generated when the production of oxidants [traditionally referred to as reactive oxygen species (ROS)], outpace the control of the cellular antioxidant enzymes or molecules [e.g., superoxide dismutase (SOD), peroxidases, and catalase] reaching toxic steady-state concentrations (Sies and Jones, 2020). While ROS are assumed to be critical signaling molecules for maintaining brain homeostasis, an unbalanced redox environment toward oxidation is recognized to play a pivotal role in the development of cerebrovascular dysfunction in different pathologies. In the context of AD, Aβ has been demonstrated to induce excessive ROS production in the brain, this occurring earlier in the vasculature than in parenchyma (Park et al., 2004).
At the cerebral vasculature, ROS can be produced by different sources, including NADPH oxidase (NOX), mitochondria respiratory chain, uncoupled eNOS, and cyclooxygenase (COXs), among others. In this list, the NOX family has been reported to produce more ROS [essentially O2–• but also hydrogen peroxide (H2O2)] than any other enzyme. Interestingly, the NOX activity in the cerebral vasculature is much higher than in the peripheral arteries (Miller et al., 2006) and is further increased by aging, AD, and VCID (Choi and Lee, 2017; Ma et al., 2017). Also, both the NOX enzyme activity level and protein levels of the different subunits (p67phox, p47phox, and p40phox) were reported to be elevated in the brains of patients with AD (Ansari and Scheff, 2011) and AD transgenic mice in correlation with a cognitive decline (Park et al., 2008; Bruce-Keller et al., 2011; Han et al., 2015; Lin et al., 2016). As mentioned earlier, NOS enzymes may produce O2–• themselves in their uncoupled state, critically contributing to the decreased BH4 bioavailability. Of note, the BH4 metabolism is described to be deregulated in AD (Foxton et al., 2007).
The reaction of O2–• with •NO proceeds at diffusion-controlled rates and is favored by an increased steady-state concentration of O2–•, providing that •NO diffuses to the sites of O2–• formation. This radical-radical interaction has two important consequences for cerebrovascular dysfunction: (1) reduced •NO bioavailability and ensued diminished •NO- mediated vasodilation, and (2) increased peroxynitrite formation and fostering of oxidative distress (Figure 2).
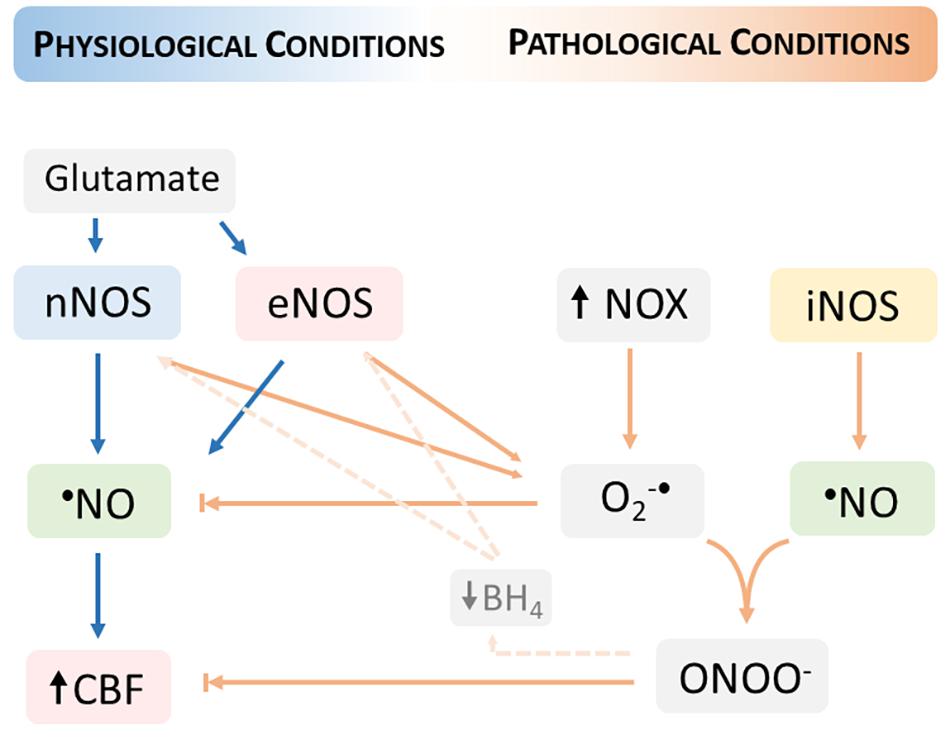
Figure 2. Neurovascular coupling dysfunction in pathological conditions fostered by oxidative distress. The increase in the steady-state concentration of oxygen (O2–•) produced by the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) promotes a decrease in the •NO bioavailability and thus limiting the •NO- mediated cerebral blood flow (CBF) increase. The reaction of •NO with O2–• produces peroxynitrite (ONOO)– which additionally contributes to the dysfunction of neurovascular coupling, including by reducing the levels of tetrahydrobiopterin (BH4) and thus promoting the uncoupling of nitric oxide synthases (nNOS and eNOS). The uncoupled NOS produces O2–• contributing to increase its steady-state concentration. Under oxidative distress conditions, the inducible NOS (iNOS) supports a sustained •NO production that further reacts with O2–•.
The decrease of •NO bioavailability is translated into inefficient signaling to support the hemodynamic response to neuronal activation and might occur either due to increased scavenging (e.g., by O2–•) or limited •NO synthesis (e.g., in hypoxia). Thus, the fast reaction of •NO with O2–• might be a persuasive reaction at the origins of the •NO decreased bioavailability and NVC dysfunction.
The impact of O2–•-mediated •NO scavenging in the efficiency of NVC has been addressed in different animal models. In healthy young rats, we were able to mimic the impairment of •NO-mediated NVC observed in the hippocampus of 3xTg-AD mice and aged Fisher 344 rats by using 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), a redox-cycle quinone that triggers the intracellular O2–• generation (Lourenço et al., 2017b, 2018). Park and coworkers demonstrated that the attenuation of the CBF changes to whisker stimulation, as observed in aged and AD mice, could be abrogated by both a SOD mimetic (MnTBAP) and a NOX inhibitor (peptide gp91ds-tat) (Park et al., 2007, 2008). In the Tg2576 mice, it was further shown that the genetic ablation of the NOX2 subunit of NADPH oxidase was able to prevent both the NVC dysfunction and spatial memory decline (Park et al., 2008). More recently, the mitochondria-targeted overexpression of catalase has been shown to hamper the age-related NVC dysfunction by preserving the •NO-mediated component of the hemodynamic response (Csiszar et al., 2019).
The •NO synthesis by the NOS enzymes involves the oxidation of L-arginine to L-citrulline, dependent on O2. Under conditions of limited O2 concentration (e.g., ischemic conditions) and going lower than the KM for NOS, the synthesis of •NO by the canonical pathway became limited, and expectedly, the •NO concentration decreases (Adachi et al., 2000).
As mentioned earlier, the reaction of •NO with O2–•, yielding ONOO–, conveys the major pathway underlying the deleterious actions of •NO, that eventually culminates into neurodegeneration (Radi, 2018). This pathway is largely fueled by the activity of iNOS, an isoform much less dependent on Ca2+ concentration and capable to sustain a continuous •NO production, thereby producing a much larger amount of •NO relative to the constitutive isoforms (Pautz et al., 2010). The ONOO– formed can oxidize and nitrate several biomolecules, including proteins. Specifically, the nitration of the tyrosine residues of proteins, resulting in the formation of 3-nitrotyrosine (3-NT), may irreversibly impact signaling pathways (either by promoting a loss or a gain of function of the target protein) (Radi, 2018).
A large body of evidence supports the enhanced 3-NT immunoreactivity in the brains of AD patients and rodent models, as well as the nitration and oxidation of several relevant proteins [reviewed in Butterfield et al. (2011) and Butterfield and Boyd-Kimball (2019)]. Among them, the mitochondrial isoform of SOD (MnSOD) was reported to occur nitrated in AD (Aoyama et al., 2000), a modification associated with enzyme inactivation (Radi, 2004) and expected increased oxidative distress. Also, tau protein has been demonstrated to be a target for nitration, a modification linked to increased aggregation (Horiguchi et al., 2003). In the 3xTgAD mice with impaired NVC, we detected increased levels of 3-NT and iNOS of the hippocampus (Lourenço et al., 2017b). Peroxynitrite can further impair NVC by altering the mechanisms for vasodilation (e.g., oxidizing BH4, inhibiting sGC expression/activity, inactivating prostacyclin) and by promoting structural alterations in the blood vessels [reviewed by Chrissobolis and Faraci (2008) and Lee and Griendling (2008)].
From the abovementioned notions, we infer that sustaining •NO bioavailability may have a relevant impact in the NVC, and thus in the neuronal function. Strategies capable to enhance •NO bioavailability may therefore configure attractive approaches to be explored both from a therapeutic standpoint in patients with impaired NVC function (and cognitive decline) or from a prophylactic standpoint in individuals at risk of developing cognitive dysfunctions linked to neurovascular alterations. In line with this, several strategies have been tested, both in animal models and humans, targeting the promotion of •NO synthesis and/or the limitation of its scavenging by the ROS [reviewed by Lundberg et al. (2015)]. Some of the most promising strategies involving non-pharmacological intervention are discussed below (Figure 3).
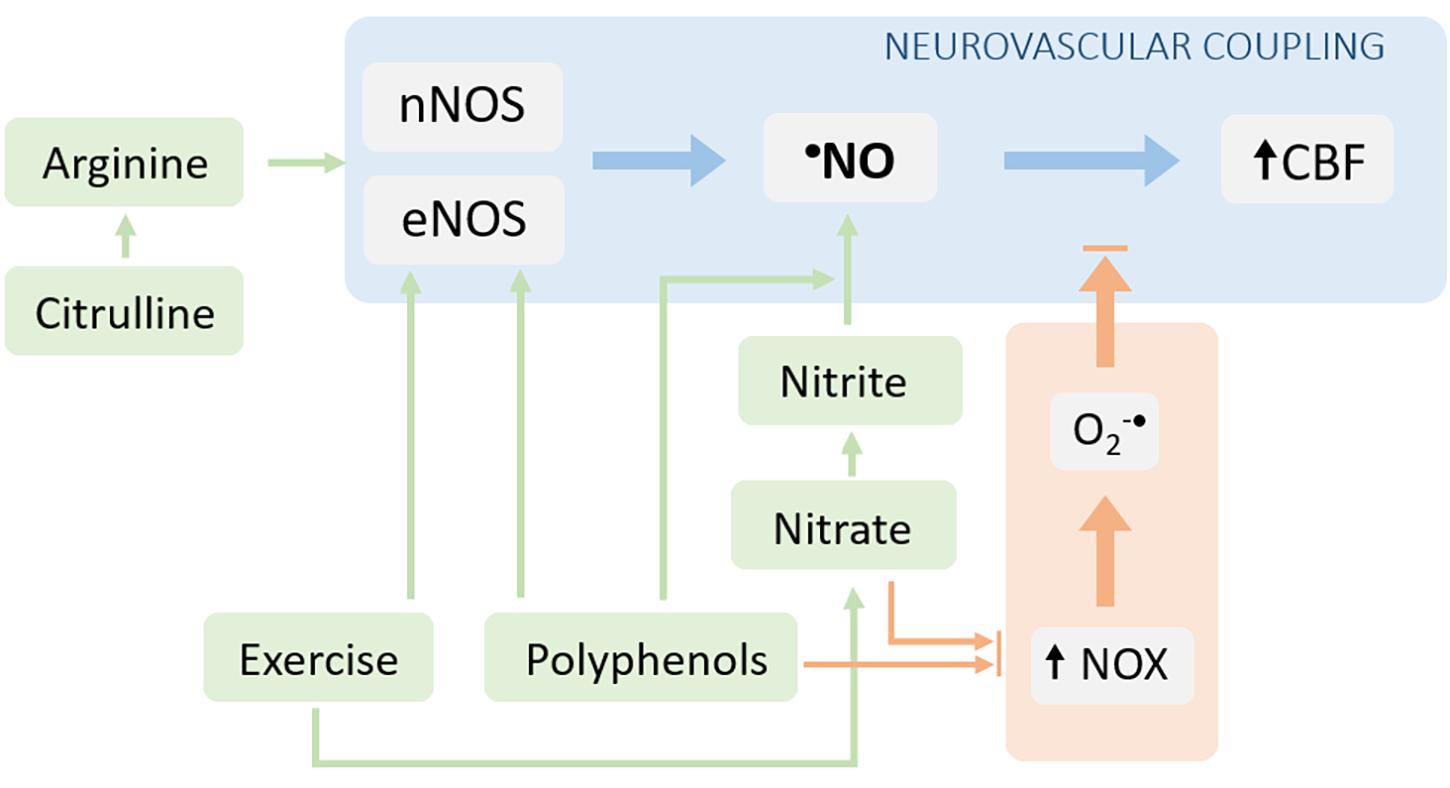
Figure 3. Interventions aiming to target neurovascular coupling dysfunction via reestablishment of •NO bioavailability. The •NO bioavailability can be increased either by stimulating its production (blue arrows) or by decreasing its degradation (orange arrows). The NOS-dependent •NO production can be increased by providing substrates for the enzyme (arginine or its precursor, citrulline) and via the post-translational regulation of the enzyme (e.g., signaling pathways stimulated by polyphenols and/or exercise). Additionally, the •NO synthesis can be enhanced via the alternative nitrate-nitrite-•NO pathway. The limitation of •NO scavenging can be achieved by decreasing the production of superoxide radical (both nitrate/nitrite supplementation and exercise can decrease the activity/expression of NOX enzymes).
In the past two decades, both arginine (NOS substrate) and its precursor, citrulline, received a lot of attention for their potential to increase •NO-dependent signaling, being currently accepted that citrulline is more efficient than arginine as a precursor for •NO synthesis (Bahadoran et al., 2021). Citrulline, but not arginine, has been demonstrated to improve the acetylcholine-induced retinal vasodilation in diabetic rats (Kurauchi et al., 2017). It also has been shown to mitigate the cognitive dysfunction linked to transient brain ischemia in mice (Yabuki et al., 2013) and to improve the CBF response associated with spreading depolarization events in rats (Kurauchi et al., 2017). Yet, in patients with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome, oral treatment with L-Arginine was able to restore the NVC response to the visual cortex stimulus (Rodan et al., 2020).
More recently, much focus has been paid to both NO2– and NO3– anions as relevant biological precursors of •NO, capable to support •NO-dependent mechanisms, including vasodilation, via the NO3–-NO2–-•NO pathway (Lundberg et al., 2008). In line with this, different pieces of evidence provide support for the benefits of NO2– and NO3–, particularly in the cardiovascular system (Lundberg et al., 2015). Additionally, acute nitrite was demonstrated to enhance basal CBF in rats (Rifkind et al., 2007) and to improve the hemodynamic response to somatosensory activation under conditions of NOS inhibition (Piknova et al., 2011). In essence, NO2– is now recognized as a regulator of hypoxic signaling in mammalian physiology through the formation of •NO (Rocha et al., 2011; Lundberg et al., 2018), and human studies support the application of NO2– to alleviate cardiovascular dysfunctions and as a supplement for healthy arterial aging (Lundberg et al., 2018). NO3–, which can be obtained from diets, salt supplementation, or endogenous •NO oxidation, is also proposed to increase both cerebral blood perfusion and cognitive performance in humans, but the overall evidence is still not conclusive (Clifford et al., 2019). This may be justified by the short duration of the interventions and the health status of the subjects. Magnetic resonance imaging studies in an elderly human population exposed to an increased intake of NO3–-rich foods showed that a 2-day intervention with dietary NO3– had no effect on the global CBF but enhanced the CBF in the deep white matter in the frontal lobe, suggested to be involved in executive functioning (Presley et al., 2011). Moreover, in healthy adults, a single dose of beetroot juice (containing 5.5 mmol NO3–) was demonstrated to modulate their CBF response to task performance in the frontal cortex assessed by near-infrared spectroscopy and to improve their performance to a serial 3 s subtraction task (Wightman et al., 2015). Also, intrathecal levels of NO3– in patients with vascular dementia are inversely correlated to the degree of intellectual impairment (Tarkowski et al., 2000). In the cardiovascular and renal systems, dietary NO3– was able to attenuate oxidative distress by inhibiting NOX expression/activity, conveying an additional mechanism by which NO3– supplementation could improve •NO bioavailability (Carlstrom and Montenegro, 2019).
Found predominantly in fruits and vegetables, polyphenols are another set of bioactive compounds recognized to improve •NO bioavailability, thereby providing positive health benefits (Oak et al., 2018). For decades, explored for their antioxidant capacity associated with the free radical scavenging, polyphenols are recognized to directly modulate the redox-sensitive processes in vivo (Fraga et al., 2019). Moreover, polyphenols are known to stimulate •NO production in the endothelial cells via the phosphorylation of eNOS mediated by both PI3K/Akt and p38 MAPK signaling pathways. Several polyphenols were also described to modulate the redox status of the cellular environment via the activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a transcription factor needed for anti-oxidant response element gene (ARE) expression, leading to the expression of several antioxidant enzymes (e.g., SOD). Interestingly, they have also been demonstrated to downregulate the expression of NOX enzymes (Forte et al., 2016; Fraga et al., 2018). Furthermore, as mentioned above, polyphenols are also capable to promote the reduction of NO2– to •NO, particularly under acidic environments (Rocha et al., 2009). Polyphenols accompany NO2– and NO3– in the vegetables consumed in the human diet and mounting evidence from epidemiological and randomized controlled trials support their beneficial effects in preventing age-associated cognitive decline (Fraga et al., 2019). The ability of some polyphenols to improve NVC has also been demonstrated by some clinical and pre-clinical studies. For instance, resveratrol, a polyphenol found in grape seeds, was able to rescue the hemodynamic responses to neuronal and endothelial activation in aged mice, occurring along with downregulation of the NADPH oxidase and decreased 3-NT levels (Toth et al., 2014). In humans, a similar beneficial effect was demonstrated in type 2 diabetic patients and postmenopausal women. In diabetic patients, a single dose of resveratrol enhanced their performance on a multi-tasking test battery and the blood flow velocity in the middle cerebral artery to this cognitive stimulus (Wong et al., 2016). In postmenopausal women, a long-term treatment with resveratrol significantly increased their cognitive performance and NVC response as compared with their basal condition (Thaung Zaw et al., 2020). Cocoa flavonoids were also demonstrated to improve NVC in humans, increasing the BOLD fMRI response to cognitive tasks in healthy subjects (Decroix et al., 2016) and type 1 diabetic patients (Decroix et al., 2019).
Regular physical exercise is widely recognized to have a substantial neuroprotective impact on brain function, contributing to restore and maintain cognitive performance (Alkadhi, 2018), and amending several cardiovascular risk factors (e.g., diabetes, hypertension) (Kokkinos and Myers, 2010). In particular, exercise is suggested to promote the upregulation of eNOS expression and activity via activation of Akt-dependent signaling by temporally increasing shear stress (Chistiakov et al., 2017). Shear stress has also been shown to stimulate the expression of Nrf2 and thus, the upregulation of antioxidant defenses (Warabi et al., 2007). Also, exercise is suggested to enhance NO2– plasmatic levels similar to dietary NO3– (Lundberg et al., 2015). The evidence for the beneficial effect of exercise in the CBF and brain functioning is overwhelmingly convincing both in humans and experimental animal models (Ogoh and Ainslie, 2009; Smith and Ainslie, 2017; Trigiani and Hamel, 2017). Recently, physical exercise has been demonstrated to alter the white matter pathology and hemodynamic response to whiskers stimulation in a VCID mouse model (Trigiani et al., 2020). Also, in an AD mouse model, exercise training was demonstrated to reduce cerebrovascular dysfunction via enhancing the P2Y2-eNOS signaling (Hong et al., 2020).
The NVC is a crucial pathway supporting both the structural and functional integrity of the brain. The diffusible vasodilator, •NO, is widely recognized to be a key player in the intricate communication between the neurovascular unit cells required to supply energetic substrates. The canonical pathway for •NO-mediated NVC involves the NMDAr-mediated activation of the nNOS at the neurons and the stimulation of the sGC at the SMC. Additionally, the •NO produced by different sources (interneurons, endothelial cells, erythrocytes) and modulating other signaling pathways likely converge to assure the proper development of the neurovascular response. The failure in this process is linked to neurodegeneration in different pathological conditions and may be fostered by the changes in the redox environment toward oxidation that decreases the •NO bioavailability and subverts its bioactivity. Diet is established among the most relevant adjustable variables of human health in modern societies. Considering that WHO recognizes cognitive impairment and dementia associated with aging as one of the major public health challenges of our time (World Health Organization [WHO], 2012), a more comprehensive understanding of how the different aspects of lifestyle and diet affect neural function and consequent cognitive performance is an imperative need. In line with this, the potential modulatory effects of dietary NO3– and polyphenols on •NO-dependent NVC, in association with physical exercise, may be used as effective non-pharmacological strategies to promote the •NO bioavailability and thereby to manage NVC dysfunction in neuropathological conditions.
CL and JL discussed the content of the review and approved the final version of the manuscript. CL preformed the literature research, draft the manuscript, and draw the figures. JL edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the European Regional Development Fund (ERDF) through the COMPETE 2020 – Operational Program for Competitiveness and Internationalization and the Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, under the projects POCI-01-0145-FEDER-029099 and UIDB/04539/2020.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adachi, N., Lei, B., Soutani, M., and Arai, T. (2000). Different roles of neuronal and endothelial nitric oxide synthases on ischemic nitric oxide production in gerbil striatum. Neurosci. Lett. 288, 151–154. doi: 10.1016/S0304-3940(00)01222-2
Alkadhi, K. A. (2018). Exercise as a Positive Modulator of Brain Function. Mol. Neurobiol. 55, 3112–3130. doi: 10.1007/s12035-017-0516-4
Anenberg, E., Chan, A. W., Xie, Y., Ledue, J. M., and Murphy, T. H. (2015). Optogenetic stimulation of GABA neurons can decrease local neuronal activity while increasing cortical blood flow. J. Cereb. Blood Flow Metab. 35, 1579–1586. doi: 10.1038/jcbfm.2015.140
Ansari, M. A., and Scheff, S. W. (2011). NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol. Med. 51, 171–178. doi: 10.1016/j.freeradbiomed.2011.03.025
Aoyama, K., Matsubara, K., Fujikawa, Y., Nagahiro, Y., Shimizu, K., and Umegae, N. (2000). Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol. 47, 524–527. doi: 10.1002/1531-8249(200004)47:4<524::AID-ANA19>3.0.CO;2-5
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Attwell, D., and Iadecola, C. (2002). The neural basis of functional brain imaging signals. Trends Neurosci. 25, 621–625. doi: 10.1016/S0166-2236(02)02264-6
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bahadoran, Z., Mirmiran, P., Kashfi, K., and Ghasemi, A. (2021). Endogenous flux of nitric oxide: Citrulline is preferred to Arginine. Acta Physiol. 231:e13572. doi: 10.1111/apha.13572
Bal-Price, A., Moneer, Z., and Brown, G. C. (2002). Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 40, 312–323. doi: 10.1002/glia.10124
Beuve, A. (2017). Thiol-Based Redox Modulation of Soluble Guanylyl Cyclase, the Nitric Oxide Receptor. Antioxid. Redox. Signal. 26, 137–149. doi: 10.1089/ars.2015.6591
Bredt, D. S., Hwang, P. M., and Snyder, S. H. (1990). Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347, 768–770. doi: 10.1038/347768a0
Bruce-Keller, A. J., Gupta, S., Knight, A. G., Beckett, T. L., Mcmullen, J. M., and Davis, P. R. (2011). Cognitive impairment in humanized APPxPS1 mice is linked to Abeta(1–42) and NOX activation. Neurobiol. Dis. 44, 317–326. doi: 10.1016/j.nbd.2011.07.012
Butterfield, D. A., and Boyd-Kimball, D. (2019). Redox proteomics and amyloid beta-peptide: insights into Alzheimer disease. J. Neurochem. 151, 459–487. doi: 10.1111/jnc.14589
Butterfield, D. A., Reed, T., and Sultana, R. (2011). Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 45, 59–72. doi: 10.3109/10715762.2010.520014
Carlstrom, M., and Montenegro, M. F. (2019). Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J. Intern. Med. 285, 2–18. doi: 10.1111/joim.12818
Carvalho, C., and Moreira, P. I. (2018). Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 9:806. doi: 10.3389/fphys.2018.00806
Cauli, B., Tong, X. K., Rancillac, A., Serluca, N., Lambolez, B., Rossier, J., and Hamel, E. (2004). Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J. Neurosci. 24, 8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004
Chen, B. R., Kozberg, M. G., Bouchard, M. B., Shaik, M. A., and Hillman, E. M. (2014). A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 3:e000787. doi: 10.1161/JAHA.114.000787
Chen, K., Pittman, R. N., and Popel, A. S. (2008). Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid. Redox. Signal. 10, 1185–1198. doi: 10.1089/ars.2007.1959
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2017). Effects of shear stress on endothelial cells: go with the flow. Acta Physiol. 219, 382–408. doi: 10.1111/apha.12725
Choi, D. H., and Lee, J. (2017). A Mini-Review of the NADPH oxidases in Vascular Dementia: Correlation with NOXs and Risk Factors for VaD. Int. J. Mol. Sci. 18:2500 doi: 10.3390/ijms18112500
Chrissobolis, S., and Faraci, F. M. (2008). The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol. Med. 14, 495–502. doi: 10.1016/j.molmed.2008.09.003
Clifford, T., Babateen, A., Shannon, O. M., Capper, T., Ashor, A., and Stephan, B. (2019). Effects of inorganic nitrate and nitrite consumption on cognitive function and cerebral blood flow: A systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Food Sci. Nutr. 59, 2400–2410. doi: 10.1080/10408398.2018.1453779
Csiszar, A., Yabluchanskiy, A., Ungvari, A., Ungvari, Z., and Tarantini, S. (2019). Overexpression of catalase targeted to mitochondria improves neurovascular coupling responses in aged mice. Geroscience 41, 609–617. doi: 10.1007/s11357-019-00111-0
Decroix, L., Tonoli, C., Soares, D. D., Tagougui, S., Heyman, E., and Meeusen, R. (2016). Acute cocoa flavanol improves cerebral oxygenation without enhancing executive function at rest or after exercise. Appl. Physiol. Nutr. Metab. 41, 1225–1232. doi: 10.1139/apnm-2016-0245
Decroix, L., Van Schuerbeek, P., Tonoli, C., Van Cutsem, J., Soares, D. D., Heyman, E., Vanderhasselt, T., Verrelst, R., Raeymaekers, H., De Mey, J., and Meeusen, R. (2019). The effect of acute cocoa flavanol intake on the BOLD response and cognitive function in type 1 diabetes: a randomized, placebo-controlled, double-blinded cross-over pilot study. Psychopharmacology 236, 3421–3428. doi: 10.1007/s00213-019-05306-z
Echagarruga, C. T., Gheres, K. W., Norwood, J. N., and Drew, P. J. (2020). nNOS-expressing interneurons control basal and behaviorally evoked arterial dilation in somatosensory cortex of mice. Elife 9:e60533. doi: 10.7554/eLife.60533
Forstermann, U., and Sessa, W. C. (2012). Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837. doi: 10.1093/eurheartj/ehr304
Forte, M., Conti, V., Damato, A., Ambrosio, M., Puca, A. A., Sciarretta, S., Frati, G., Vecchione, C., and Carrizzo, A. (2016). Targeting Nitric Oxide with Natural Derived Compounds as a Therapeutic Strategy in Vascular Diseases. Oxid. Med. Cell Longev. 2016:7364138. doi: 10.1155/2016/7364138
Foxton, R. H., Land, J. M., and Heales, S. J. (2007). Tetrahydrobiopterin availability in Parkinson’s and Alzheimer’s disease; potential pathogenic mechanisms. Neurochem. Res. 32, 751–756. doi: 10.1007/s11064-006-9201-0
Fraga, C. G., Croft, K. D., Kennedy, D. O., and Tomas-Barberan, F. A. (2019). The effects of polyphenols and other bioactives on human health. Food Funct. 10, 514–528. doi: 10.1039/C8FO01997E
Fraga, C. G., Oteiza, P. I., and Galleano, M. (2018). Plant bioactives and redox signaling: (-)-Epicatechin as a paradigm. Mol. Aspects Med. 61, 31–40. doi: 10.1016/j.mam.2018.01.007
Fujimoto, Y., Uno, E., and Sakuma, S. (2004). Effects of reactive oxygen and nitrogen species on cyclooxygenase-1 and -2 activities. Prostaglandins Leukot Essent Fatty Acids 71, 335–340. doi: 10.1016/j.plefa.2004.06.002
Guerra, G., Lucariello, A., Perna, A., Botta, L., De Luca, A., and Moccia, F. (2018). The Role of Endothelial Ca(2+) Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 19:938. doi: 10.3390/ijms19040938
Hamel, E. (2006). Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 100, 1059–1064. doi: 10.1152/japplphysiol.00954.2005
Hamel, E., Nicolakakis, N., Aboulkassim, T., Ongali, B., and Tong, X. K. (2008). Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp. Physiol. 93, 116–120. doi: 10.1113/expphysiol.2007.038729
Han, B. H., Zhou, M. L., Johnson, A. W., Singh, I., Liao, F., Vellimana, A. K., Nelson, J. W., Milner, E., Cirrito, J. R., Basak, J., Yoo, M., Dietrich, H. H., Holtzman, D. M., and Zipfel, G. J. (2015). Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. U.S.A. 112, E881–E890. doi: 10.1073/pnas.1414930112
Hogan-Cann, A. D., Lu, P., and Anderson, C. M. (2019). Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. U.S.A. 116, 10229–10231. doi: 10.1073/pnas.1902647116
Hoiland, R. L., Caldwell, H. G., Howe, C. A., Nowak-Fluck, D., Stacey, B. S., and Bailey, D. M. (2020). Nitric oxide is fundamental to neurovascular coupling in humans. J. Physiol. 598, 4927–4939. doi: 10.1113/JP280162
Hong, J., Hong, S. G., Lee, J., Park, J. Y., Eriksen, J. L., Rooney, B. V., and Park, Y. (2020). Exercise training ameliorates cerebrovascular dysfunction in a murine model of Alzheimer’s disease: role of the P2Y2 receptor and endoplasmic reticulum stress. Am. J. Physiol. Heart Circ. Physiol. 318, H1559–H1569. doi: 10.1152/ajpheart.00129.2020
Horiguchi, T., Uryu, K., Giasson, B. I., Ischiropoulos, H., Lightfoot, R., Bellmann, C., Richter-Landsberg, C., Lee, V. M., and Trojanowski, J. Q. (2003). Nitration of tau protein is linked to neurodegeneration in tauopathies. Am. J. Pathol. 163, 1021–1031. doi: 10.1016/S0002-9440(10)63462-1
Hosford, P. S., and Gourine, A. V. (2019). What is the key mediator of the neurovascular coupling response? Neurosci. Biobehav. Rev. 96, 174–181. doi: 10.1016/j.neubiorev.2018.11.011
Iadecola, C. (1993). Regulation of the cerebral microcirculation during neural activity: is nitric oxide the missing link? Trends Neurosci. 16, 206–214. doi: 10.1016/0166-2236(93)90156-G
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iadecola, C. (2017). The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Iadecola, C., and Gottesman, R. F. (2019). Neurovascular and Cognitive Dysfunction in Hypertension. Circ. Res. 124, 1025–1044. doi: 10.1161/CIRCRESAHA.118.313260
Iturria-Medina, Y., Sotero, R. C., Toussaint, P. J., Mateos-Perez, J. M., and Evans, A. C. (2016). Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 7:11934. doi: 10.1038/ncomms11934
Knott, A. B., and Bossy-Wetzel, E. (2009). Nitric oxide in health and disease of the nervous system. Antioxid. Redox. Signal. 11, 541–554. doi: 10.1089/ars.2008.2234
Knowles, R. G., and Moncada, S. (1994). Nitric oxide synthases in mammals. Biochem. J. 298, 249–258. doi: 10.1042/bj2980249
Koike, M. A., Green, K. N., Blurton-Jones, M., and Laferla, F. M. (2010). Oligemic hypoperfusion differentially affects tau and amyloid-{beta}. Am. J. Pathol. 177, 300–310. doi: 10.2353/ajpath.2010.090750
Kokkinos, P., and Myers, J. (2010). Exercise and physical activity: clinical outcomes and applications. Circulation 122, 1637–1648. doi: 10.1161/CIRCULATIONAHA.110.948349
Kovacs-Oller, T., Ivanova, E., Bianchimano, P., and Sagdullaev, B. T. (2020). The pericyte connectome: spatial precision of neurovascular coupling is driven by selective connectivity maps of pericytes and endothelial cells and is disrupted in diabetes. Cell Discov. 6:39. doi: 10.1038/s41421-020-0180-0
Krawchuk, M. B., Ruff, C. F., Yang, X., Ross, S. E., and Vazquez, A. L. (2020). Optogenetic assessment of VIP, PV, SOM and NOS inhibitory neuron activity and cerebral blood flow regulation in mouse somato-sensory cortex. J. Cereb. Blood Flow Metab. 40, 1427–1440. doi: 10.1177/0271678X19870105
Kurauchi, Y., Mokudai, K., Mori, A., Sakamoto, K., Nakahara, T., Morita, M., Kamimura, A., and Ishii, K. (2017). l-Citrulline ameliorates cerebral blood flow during cortical spreading depression in rats: Involvement of nitric oxide- and prostanoids-mediated pathway. J. Pharmacol. Sci. 133, 146–155. doi: 10.1016/j.jphs.2017.02.004
Lee, L., Boorman, L., Glendenning, E., Christmas, C., Sharp, P., and Redgrave, P. (2020). Key Aspects of Neurovascular Control Mediated by Specific Populations of Inhibitory Cortical Interneurons. Cereb. Cortex 30, 2452–2464. doi: 10.1093/cercor/bhz251
Lee, M. Y., and Griendling, K. K. (2008). Redox signaling, vascular function, and hypertension. Antioxid. Redox. Signal. 10, 1045–1059. doi: 10.1089/ars.2007.1986
Leeuwis, A. E., Benedictus, M. R., Kuijer, J. P. A., Binnewijzend, M. A. A., Hooghiemstra, A. M., and Verfaillie, S. C. J. (2017). Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement 13, 531–540. doi: 10.1016/j.jalz.2016.08.013
Lin, B., Hasegawa, Y., Takane, K., Koibuchi, N., Cao, C., and Kim-Mitsuyama, S. (2016). High-Fat-Diet Intake Enhances Cerebral Amyloid Angiopathy and Cognitive Impairment in a Mouse Model of Alzheimer’s Disease, Independently of Metabolic Disorders. J. Am. Heart Assoc. 5:e003154. doi: 10.1161/JAHA.115.003154
Lourenço, C.F., Ferreira, N. R., Santos, R. M., Lukacova, N., Barbosa, R. M., and Laranjinha, J. (2014a). The pattern of glutamate-induced nitric oxide dynamics in vivo and its correlation with nNOS expression in rat hippocampus, cerebral cortex and striatum. Brain Res. 1554, 1–11. doi: 10.1016/j.brainres.2014.01.030
Lourenço, C.F., Ledo, A., Barbosa, R. M., and Laranjinha, J. (2017a). Neurovascular-neuroenergetic coupling axis in the brain: master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic. Biol. Med. 108, 668–682. doi: 10.1016/j.freeradbiomed.2017.04.026
Lourenço, C.F., Ledo, A., Barbosa, R. M., and Laranjinha, J. (2017b). Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp. Neurol. 291, 36–43. doi: 10.1016/j.expneurol.2017.01.013
Lourenço, C.F., Ledo, A., Caetano, M., Barbosa, R. M., and Laranjinha, J. (2018). Age-Dependent Impairment of Neurovascular and Neurometabolic Coupling in the Hippocampus. Front. Physiol. 9:913. doi: 10.3389/fphys.2018.00913
Lourenço, C.F., Santos, R. M., Barbosa, R. M., Cadenas, E., Radi, R., and Laranjinha, J. (2014b). Neurovascular coupling in hippocampus is mediated via diffusion by neuronal-derived nitric oxide. Free Radic. Biol. Med. 73, 421–429. doi: 10.1016/j.freeradbiomed.2014.05.021
Lu, L., Hogan-Cann, A. D., Globa, A. K., Lu, P., Nagy, J. I., Bamji, S. X., and Anderson, C. M. (2019). Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 39, 481–496. doi: 10.1177/0271678X17734100
Lundberg, J. O., Carlstrom, M., and Weitzberg, E. (2018). Metabolic Effects of Dietary Nitrate in Health and Disease. Cell Metab. 28, 9–22. doi: 10.1016/j.cmet.2018.06.007
Lundberg, J. O., Gladwin, M. T., and Weitzberg, E. (2015). Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 14, 623–641. doi: 10.1038/nrd4623
Lundberg, J. O., Weitzberg, E., and Gladwin, M. T. (2008). The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 7, 156–167. doi: 10.1038/nrd2466
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., and Brann, D. W. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 12:7. doi: 10.1186/s13024-017-0150-7
Mapelli, L., Gagliano, G., Soda, T., Laforenza, U., Moccia, F., and D’angelo, E. U. (2017). Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO- Dependent System. J. Neurosci. 37, 1340–1351. doi: 10.1523/JNEUROSCI.2025-16.2016
Martin, E., Berka, V., Tsai, A. L., and Murad, F. (2005). Soluble guanylyl cyclase: the nitric oxide receptor. Methods Enzymol. 396, 478–492. doi: 10.1016/S0076-6879(05)96040-0
Miller, A. A., Drummond, G. R., and Sobey, C. G. (2006). Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol. Ther. 111, 928–948. doi: 10.1016/j.pharmthera.2006.02.005
Mishra, A., Reynolds, J. P., Chen, Y., Gourine, A. V., Rusakov, D. A., and Attwell, D. (2016). Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 19, 1619–1627. doi: 10.1038/nn.4428
Mokhber, N., Shariatzadeh, A., Avan, A., Saber, H., Babaei, G. S., Chaimowitz, G., and Azarpazhooh, M. R. (2021). Cerebral blood flow changes during aging process and in cognitive disorders: A review. Neuroradiol. J. 34, 300–307. doi: 10.1177/19714009211002778
Moretti, R., and Caruso, P. (2020). Small Vessel Disease-Related Dementia: An Invalid Neurovascular Coupling? Int. J. Mol. Sci. 21:1095 doi: 10.3390/ijms21031095
Mueggler, T., Baumann, D., Rausch, M., Staufenbiel, M., and Rudin, M. (2003). Age-dependent impairment of somatosensory response in the amyloid precursor protein 23 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 23, 8231–8236. doi: 10.1523/JNEUROSCI.23-23-08231.2003
Nicolakakis, N., and Hamel, E. (2011). Neurovascular function in Alzheimer’s disease patients and experimental models. J. Cereb. Blood Flow Metab. 31, 1354–1370. doi: 10.1038/jcbfm.2011.43
Niwa, K., Younkin, L., Ebeling, C., Turner, S. K., Westaway, D., Younkin, S., Ashe, K. H., Carlson, G. A., and Iadecola, C. (2000). Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. U.S.A. 97, 9735–9740. doi: 10.1073/pnas.97.17.9735
Oak, M. H., Auger, C., Belcastro, E., Park, S. H., Lee, H. H., and Schini-Kerth, V. B. (2018). Potential mechanisms underlying cardiovascular protection by polyphenols: Role of the endothelium. Free Radic. Biol. Med. 122, 161–170. doi: 10.1016/j.freeradbiomed.2018.03.018
Ogoh, S., and Ainslie, P. N. (2009). Cerebral blood flow during exercise: mechanisms of regulation. J. Appl. Physiol. 107, 1370–1380. doi: 10.1152/japplphysiol.00573.2009
Park, J. H., Hong, J. H., Lee, S. W., Ji, H. D., Jung, J. A., and Yoon, K. W. (2019). The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: A positron emission tomography study in rats. Sci. Rep. 9:14102. doi: 10.1038/s41598-019-50681-4
Park, L., Anrather, J., Forster, C., Kazama, K., Carlson, G. A., and Iadecola, C. (2004). Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J. Cereb. Blood Flow Metab. 24, 334–342. doi: 10.1097/01.WCB.0000105800.49957.1E
Park, L., Anrather, J., Girouard, H., Zhou, P., and Iadecola, C. (2007). Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J. Cereb. Blood Flow Metab. 27, 1908–1918. doi: 10.1038/sj.jcbfm.9600491
Park, L., Hochrainer, K., Hattori, Y., Ahn, S. J., Anfray, A., and Wang, G. (2020). Tau induces PSD95-neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nat. Neurosci. 23, 1079–1089. doi: 10.1038/s41593-020-0686-7
Park, L., Zhou, P., Pitstick, R., Capone, C., Anrather, J., Norris, E. H., Younkin, L., Younkin, S., Carlson, G., Mcewen, B. S., and Iadecola, C. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 105, 1347–1352. doi: 10.1073/pnas.0711568105
Pautz, A., Art, J., Hahn, S., Nowag, S., Voss, C., and Kleinert, H. (2010). Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 23, 75–93. doi: 10.1016/j.niox.2010.04.007
Picon-Pages, P., Garcia-Buendia, J., and Munoz, F. J. (2019). Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1949–1967. doi: 10.1016/j.bbadis.2018.11.007
Piknova, B., Kocharyan, A., Schechter, A. N., and Silva, A. C. (2011). The role of nitrite in neurovascular coupling. Brain Res. 1407, 62–68. doi: 10.1016/j.brainres.2011.06.045
Presley, T. D., Morgan, A. R., Bechtold, E., Clodfelter, W., Dove, R. W., and Jennings, J. M. (2011). Acute effect of a high nitrate diet on brain perfusion in older adults. Nitric. Oxide 24, 34–42. doi: 10.1016/j.niox.2010.10.002
Price, J. M., Sutton, E. T., Hellermann, A., and Thomas, T. (1997). beta-Amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol. Res. 19, 534–538. doi: 10.1080/01616412.1997.11740853
Radi, R. (2004). Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 101, 4003–4008. doi: 10.1073/pnas.0307446101
Radi, R. (2018). Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. U.S.A. 115, 5839–5848. doi: 10.1073/pnas.1804932115
Rancillac, A., Geoffroy, H., and Rossier, J. (2012). Impaired neurovascular coupling in the APPxPS1 mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 9, 1221–1230. doi: 10.2174/156720512804142859
Rancillac, A., Rossier, J., Guille, M., Tong, X. K., Geoffroy, H., Amatore, C., Arbault, S., Hamel, E., and Cauli, B. (2006). Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the Cerebellum. J. Neurosci. 26, 6997–7006. doi: 10.1523/JNEUROSCI.5515-05.2006
Rifkind, J. M., Nagababu, E., Barbiro-Michaely, E., Ramasamy, S., Pluta, R. M., and Mayevsky, A. (2007). Nitrite infusion increases cerebral blood flow and decreases mean arterial blood pressure in rats: a role for red cell NO. Nitric Oxide 16, 448–456. doi: 10.1016/j.niox.2007.04.002
Rocha, B. S., Gago, B., Barbosa, R. M., and Laranjinha, J. (2009). Dietary polyphenols generate nitric oxide from nitrite in the stomach and induce smooth muscle relaxation. Toxicology 265, 41–48. doi: 10.1016/j.tox.2009.09.008
Rocha, B. S., Gago, B., Pereira, C., Barbosa, R. M., Bartesaghi, S., Lundberg, J. O., Radi, R., and Laranjinha, J. (2011). Dietary nitrite in nitric oxide biology: a redox interplay with implications for pathophysiology and therapeutics. Curr. Drug Targets 12, 1351–1363. doi: 10.2174/138945011796150334
Rodan, L. H., Poublanc, J., Fisher, J. A., Sobczyk, O., Mikulis, D. J., and Tein, I. (2020). L-arginine effects on cerebrovascular reactivity, perfusion and neurovascular coupling in MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) syndrome. PLoS One 15:e0238224. doi: 10.1371/journal.pone.0238224
Scott, N. A., and Murphy, T. H. (2012). Hemodynamic responses evoked by neuronal stimulation via channelrhodopsin-2 can be independent of intracortical glutamatergic synaptic transmission. PLoS One 7:e29859. doi: 10.1371/journal.pone.0029859
Shang, J., Yamashita, T., Tian, F., Li, X., Liu, X., and Shi, X. (2019). Chronic cerebral hypoperfusion alters amyloid-beta transport related proteins in the cortical blood vessels of Alzheimer’s disease model mouse. Brain Res. 1723:146379. doi: 10.1016/j.brainres.2019.146379
Shen, J., Wang, D., Wang, X., Gupta, S., Ayloo, B., Wu, S., Prasad, P., Xiong, Q., Xia, J., and Ge, S. (2019). Neurovascular Coupling in the Dentate Gyrus Regulates Adult Hippocampal Neurogenesis. Neuron 103, 878–890 e873. doi: 10.1016/j.neuron.2019.05.045
Shin, H. K., Jones, P. B., Garcia-Alloza, M., Borrelli, L., Greenberg, S. M., and Bacskai, B. J. (2007). Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130, 2310–2319. doi: 10.1093/brain/awm156
Sies, H., and Jones, D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383. doi: 10.1038/s41580-020-0230-3
Small, S. A., Perera, G. M., Delapaz, R., Mayeux, R., and Stern, Y. (1999). Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer’s disease. Ann. Neurol. 45, 466–472. doi: 10.1002/1531-8249(199904)45:4<466::AID-ANA8>3.0.CO;2-Q
Smith, K. J., and Ainslie, P. N. (2017). Regulation of cerebral blood flow and metabolism during exercise. Exp. Physiol. 102, 1356–1371. doi: 10.1113/EP086249
Stobart, J. L., Lu, L., Anderson, H. D., Mori, H., and Anderson, C. M. (2013). Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 110, 3149–3154. doi: 10.1073/pnas.1215929110
Stuehr, D. J. (1999). Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1411, 217–230. doi: 10.1016/S0005-2728(99)00016-X
Sweeney, M. D., Kisler, K., Montagne, A., Toga, A. W., and Zlokovic, B. V. (2018). The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 21, 1318–1331. doi: 10.1038/s41593-018-0234-x
Tarantini, S., Fulop, G. A., Kiss, T., Farkas, E., Zolei-Szenasi, D., Galvan, V., Toth, P., Csiszar, A., Ungvari, Z., and Yabluchanskiy, A. (2017). Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience 39, 465–473. doi: 10.1007/s11357-017-9980-z
Tarkowski, E., Ringqvist, A., Blennow, K., Wallin, A., and Wennmalm, A. (2000). Intrathecal release of nitric oxide in Alzheimer’s disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 11, 322–326. doi: 10.1159/000017261
Thaung Zaw, J. J., Howe, P. R. C., and Wong, R. H. X. (2020). Sustained Cerebrovascular and Cognitive Benefits of Resveratrol in Postmenopausal Women. Nutrients 12:828 doi: 10.3390/nu12030828
Thomas, T., Mclendon, C., Sutton, E. T., and Thomas, G. (1997). Cerebrovascular endothelial dysfunction mediated by beta-amyloid. Neuroreport 8, 1387–1391. doi: 10.1097/00001756-199704140-00014
Toth, P., Tarantini, S., Davila, A., Valcarcel-Ares, M. N., Tucsek, Z., Varamini, B., Ballabh, P., Sonntag, W. E., Baur, J. A., Csiszar, A., and Ungvari, Z. (2015). Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am. J. Physiol. Heart Circ. Physiol. 309, H1837–H1845. doi: 10.1152/ajpheart.00463.2015
Toth, P., Tarantini, S., Tucsek, Z., Ashpole, N. M., Sosnowska, D., Gautam, T., Ballabh, P., Koller, A., Sonntag, W. E., Csiszar, A., and Ungvari, Z. (2014). Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 306, H299–H308. doi: 10.1152/ajpheart.00744.2013
Tran, C. H. T., Peringod, G., and Gordon, G. R. (2018). Astrocytes Integrate Behavioral State and Vascular Signals during Functional Hyperemia. Neuron 100, 1133–1148 e1133. doi: 10.1016/j.neuron.2018.09.045
Trigiani, L. J., and Hamel, E. (2017). An endothelial link between the benefits of physical exercise in dementia. J. Cereb. Blood Flow Metab. 37, 2649–2664. doi: 10.1177/0271678X17714655
Trigiani, L. J., Lacalle-Aurioles, M., Bourourou, M., Li, L., Greenhalgh, A. D., Zarruk, J. G., David, S., Fehlings, M. G., and Hamel, E. (2020). Benefits of physical exercise on cognition and glial white matter pathology in a mouse model of vascular cognitive impairment and dementia. Glia 68, 1925–1940. doi: 10.1002/glia.23815
Warabi, E., Takabe, W., Minami, T., Inoue, K., Itoh, K., Yamamoto, M., Ishii, T., Kodama, T., and Noguchi, N. (2007). Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 42, 260–269. doi: 10.1016/j.freeradbiomed.2006.10.043
World Health Organization [WHO] (2012). Dementia: A Public Health Priority. Geneva: World Health Organization.
Wierenga, C. E., Dev, S. I., Shin, D. D., Clark, L. R., Bangen, K. J., Jak, A. J., Rissman, R. A., Liu, T. T., Salmon, D. P., and Bondi, M. W. (2012). Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J. Cereb. Blood Flow Metab. 32, 1589–1599. doi: 10.1038/jcbfm.2012.58
Wightman, E. L., Haskell-Ramsay, C. F., Thompson, K. G., Blackwell, J. R., Winyard, P. G., Forster, J., Jones, A. M., and Kennedy, D. O. (2015). Dietary nitrate modulates cerebral blood flow parameters and cognitive performance in humans: A double-blind, placebo-controlled, crossover investigation. Physiol. Behav. 149, 149–158. doi: 10.1016/j.physbeh.2015.05.035
Wong, R. H., Raederstorff, D., and Howe, P. R. (2016). Acute Resveratrol Consumption Improves Neurovascular Coupling Capacity in Adults with Type 2 Diabetes Mellitus. Nutrients 8:425 doi: 10.3390/nu8070425
Xu, G., Antuono, P. G., Jones, J., Xu, Y., Wu, G., Ward, D., and Li, S. J. (2007). Perfusion fMRI detects deficits in regional CBF during memory-encoding tasks in MCI subjects. Neurology 69, 1650–1656. doi: 10.1212/01.wnl.0000296941.06685.22
Yabuki, Y., Shioda, N., Yamamoto, Y., Shigano, M., Kumagai, K., Morita, M., and Fukunaga, K. (2013). Oral L-citrulline administration improves memory deficits following transient brain ischemia through cerebrovascular protection. Brain Res. 1520, 157–167. doi: 10.1016/j.brainres.2013.05.011
Keywords: nitric oxide, neurovascular coupling (NVC), cerebral blood flow (CBF), neurodegeneration, oxidative stress, diet, exercise
Citation: Lourenço CF and Laranjinha J (2021) Nitric Oxide Pathways in Neurovascular Coupling Under Normal and Stress Conditions in the Brain: Strategies to Rescue Aberrant Coupling and Improve Cerebral Blood Flow. Front. Physiol. 12:729201. doi: 10.3389/fphys.2021.729201
Received: 22 June 2021; Accepted: 20 September 2021;
Published: 22 October 2021.
Edited by:
George Perry, University of Texas at San Antonio, United StatesReviewed by:
Stephen Black, Florida International University, United StatesCopyright © 2021 Lourenço and Laranjinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cátia F. Lourenço, Y2F0aWFmbG1hcnF1ZXNAZ21haWwuY29t; Y2ZsbWFycXVlc0B1Yy5wdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.