 Hirohito Shimizu1†Simon Huber2†Adam D. Langenbacher1Lauren Crisman1Jie Huang1Kevin Wang1Fabiola Wilting2Thomas Gudermann2,3Johann Schredelseker2,3*‡Jau-Nian Chen1*‡
Hirohito Shimizu1†Simon Huber2†Adam D. Langenbacher1Lauren Crisman1Jie Huang1Kevin Wang1Fabiola Wilting2Thomas Gudermann2,3Johann Schredelseker2,3*‡Jau-Nian Chen1*‡- 1Department of Molecular, Cell and Developmental Biology, University of California, Los Angeles, Los Angeles, CA, United States
- 2Walther Straub Institute of Pharmacology and Toxicology, Faculty of Medicine, LMU Munich, Munich, Germany
- 3German Centre for Cardiovascular Research (DZHK), Partner Site Munich Heart Alliance, Munich, Germany
Mitochondria critically regulate a range of cellular processes including bioenergetics, cellular metabolism, apoptosis, and cellular Ca2+ signaling. The voltage-dependent anion channel (VDAC) functions as a passageway for the exchange of ions, including Ca2+, across the outer mitochondrial membrane. In cardiomyocytes, genetic or pharmacological activation of isoform 2 of VDAC (VDAC2) effectively potentiates mitochondrial Ca2+ uptake and suppresses Ca2+ overload-induced arrhythmogenic events. However, molecular mechanisms by which VDAC2 controls mitochondrial Ca2+ transport and thereby influences cardiac rhythmicity remain elusive. Vertebrates express three highly homologous VDAC isoforms. Here, we used the zebrafish tremblor/ncx1h mutant to dissect the isoform-specific roles of VDAC proteins in Ca2+ handling. We found that overexpression of VDAC1 or VDAC2, but not VDAC3, suppresses the fibrillation-like phenotype in zebrafish tremblor/ncx1h mutants. A chimeric approach showed that moieties in the N-terminal half of VDAC are responsible for their divergent functions in cardiac biology. Phylogenetic analysis further revealed that a glutamate at position 73, which was previously described to be an important regulator of VDAC function, is sevolutionarily conserved in VDAC1 and VDAC2, whereas a glutamine occupies position 73 (Q73) of VDAC3. To investigate whether E73/Q73 determines VDAC isoform-specific anti-arrhythmic effect, we mutated E73 to Q in VDAC2 (VDAC2E73Q) and Q73 to E in VDAC3 (VDAC3Q73E). Interestingly, VDAC2E73Q failed to restore rhythmic cardiac contractions in ncx1 deficient hearts, while the Q73E conversion induced a gain of function in VDAC3. In HL-1 cardiomyocytes, VDAC2 knockdown diminished the transfer of Ca2+ from the SR into mitochondria and overexpression of VDAC2 or VDAC3Q73E restored SR-mitochondrial Ca2+ transfer in VDAC2 deficient HL-1 cells, whereas this rescue effect was absent for VDAC3 and drastically compromised for VDAC2E73Q. Collectively, our findings demonstrate a critical role for the evolutionary conserved E73 in determining the anti-arrhythmic effect of VDAC isoforms through modulating Ca2+ cross-talk between the SR and mitochondria in cardiomyocytes.
Introduction
Mitochondria govern a variety of cellular processes including bioenergetics, metabolism, reactive oxygen species production, cell survival, and Ca2+ homeostasis. In particular, a tight cross-talk between mitochondria and the endoplasmic reticulum (ER) or sarcoplasmic reticulum (SR) was associated with important physiological and pathophysiological processes (Rizzuto et al., 1998; Gomez et al., 2016; Paillusson et al., 2016; Dia et al., 2020). In the heart, the close proximity of the SR and mitochondria facilitates Ca2+ cross-talk between these cellular organelles and provides a mechanism for the regulation of cardiac Ca2+ handling and adaptation to higher workload. This SR-mitochondria cross-talk depends on an array of inner and outer mitochondrial membrane channels, including the highly abundant voltage-dependent anion channels (VDACs) in the outer mitochondrial membrane, which critically regulate transport of ions, substrates, and metabolites across the outer mitochondrial membrane. VDAC dysfunction has previously been linked to pathophysiological outcomes (Feng et al., 2019; Santin et al., 2020), but the roles of VDAC proteins in cardiac function and disease are still poorly understood.
The vertebrate VDAC family consists of three paralogs, VDAC1, VDAC2, and VDAC3. While these proteins are highly similar in their sequences, structures, and biophysical properties, different biological functions have been noted among these isoforms (Naghdi and Hajnóczky, 2016; Caterino et al., 2017). In particular, there is a distinct requirement for VDAC2 during mammalian embryogenesis: Mice without functional VDAC1 or VDAC3 can survive to adulthood (Anflous et al., 2001; Sampson et al., 2001), but VDAC2 deficiency leads to embryonic lethality (Cheng et al., 2003). Furthermore, conditional ablation of VDAC2 specifically in cardiomyocytes causes cardiomyopathy suggesting an essential role for VDAC2 in maintaining the physiological function of the heart (Raghavan et al., 2012).
In cardiomyocytes, VDAC2 serves as a gatekeeper for transferring Ca2+ into mitochondria under both normal physiological and stressed conditions (Rosencrans et al., 2021; Sander et al., 2021). For example, knockdown of VDAC2 significantly extended Ca2+ sparks, supporting a physiological role for VDAC2 in Ca2+ cycling (Subedi et al., 2011; Min et al., 2012). Our laboratories previously showed that enhancing mitochondrial Ca2+ uptake with efsevin, a gating modifier of VDAC2 (Wilting et al., 2020), can suppress arrhythmogenesis in cardiomyocytes (Shimizu et al., 2015; Schweitzer et al., 2017). Efsevin efficiently restored rhythmic cardiac contractions in zebrafish tremblor/ncx1h mutant embryos, which are characterized by cardiomyocyte Ca2+ overload and chaotic cardiac contractions (Ebert et al., 2005; Langenbacher et al., 2005; Shimizu et al., 2015) and suppressed erratic, diastolic Ca2+ events in both a murine model for human catecholaminergic polymorphic ventricular tachycardia (CPVT) and in iPSC-derived cardiomyocytes from a CPVT patient (Schweitzer et al., 2017).
Overexpression of VDAC2 allows tremblor/ncx1h mutant zebrafish hearts to establish coordinated cardiac contractions and to preserve the integrity of myofibrils (Shimizu et al., 2015, 2017), indicating that tremblor/ncx1h can serve as an animal model for the study of cardiac Ca2+ regulation by VDAC. In this study, we investigate the potential of the three VDAC isoforms to regulate Ca2+ signaling in the heart, with the aim of identifying molecular moieties which confer isoform specificity. We show that overexpression of VDAC1, but not VDAC3, restored regular contractions in tremblor/ncx1h mutants comparable to VDAC2. We further show that VDAC1 and VDAC2 possess a glutamate at position 73 (E73), whereas VDAC3 contains a glutamine (Q73), a residue that was previously reported to be involved in VDAC regulation. Strikingly, substitution of VDAC3 Q73 for a glutamate confers a gain of function ability to restore rhythmic cardiac contractions in tremblor/ncx1h mutants while the converse exchange in VDAC2 abolishes its phenotype rescue ability. Finally, using cellular assays, we show that the glutamate at position 73 of VDAC proteins is essential for mediating mitochondrial Ca2+ uptake.
Materials and Methods
Phylogenetic Analysis
Protein sequences used for phylogenetic analyses were downloaded from the NCBI database.1 A full list of the NCBI accession IDs for each sequence used is found in Supplementary File 1. For analysis of vertebrate VDACs, full-length VDAC protein sequences were aligned using the MUSCLE algorithm in the MEGA7 application (Kumar et al., 2016), producing a 284 position alignment. For expanded analysis of vertebrate and non-vertebrate VDACs, full-length protein sequences were aligned using the ClustalW algorithm in MEGA7, producing a 397 position alignment. Phylogenetic analyses were performed with the software RAxML using a maximum likelihood method, the JTT substitution matrix, and empirical frequencies (Stamatakis, 2014). RAxML software was accessed using the CIPRES Science Gateway (Miller et al., 2010) and trees were visualized using the Interactive Tree of Life website (Letunic and Bork, 2007).
Pairwise Comparison of VDAC Proteins
Human and zebrafish VDAC protein sequences were downloaded from the NCBI database and their percent identity was determined using a standard protein BLAST (blastp). Pairwise comparisons were visualized in R v3.6.3 using the ggcorrplot package.
Cloning
Plasmids containing the full-length zebrafish VDAC1, 2, and 3 cDNA were purchased from Open Biosystems and cloned into pCS2+ or pCS2+3xFLAG plasmid for mRNA synthesis. Point mutations VDAC2E73Q and VDAC3Q73E were inserted by SOE-PCR.
For HeLa cell transfections, coding regions of VDAC genes were cloned into the pCS2+ vector together with the NLS-EGFP fragments and the viral T2A sequence. The T2A sequence enables bicistronic expression of VDAC proteins and NLS-EGFP for the identification of VDAC-overexpressing cells.
Plasmids for the generation of transgenic lines were generated using the Tol2Kit (Kwan et al., 2007). Plasmids shLenti2.4G-mVDAC2 and shLenti2.4G-Ctrl for VDAC2 knockdown in HL-1 cells were generously provided by Dr. Yeon Soo Kim from Inje University, Gimhae, South Korea. Plasmids pCCLc-CMV, pCMVΔ8.91, and pCAGGS-VSV-G for lentivirus production were obtained as a gift from Dr. Donald Kohn, University of California Los Angeles, United States. For production of lentiviruses, the IRES-nlsEGFP element from p3E-IRES-nlsEGFPpA (Kwan et al., 2007) was fused to VDAC elements by subcloning into pCS2+ constructs, before the entire VDAC-eGFP element was fused into pCCLc-CMV using the In-Fusion HD Cloning Kit (TaKaRa; Wilting et al., 2020).
Zebrafish Husbandry, Generation of Transgenic Fish Lines, and Chemical Induction
Zebrafish were raised and maintained under standard laboratory conditions. The zebrafish tremblor (tretc318d) mutant line (ZDB-ALT-980203-1756) was bred and maintained as previously described (Langenbacher et al., 2005). To induce transgene expression in Tg(myl7:Gal4EcR-EGFP-UAS-vdac1-FLAG), Tg(myl7:Gal4EcR-EGFP-UAS-vdac2-FLAG), and Tg(myl7:Gal4EcR-EGFP-UAS-vdac3-FLAG), 1 μM tebufenozide (TBF) was added to the embryo media at 1 day post-fertilization (dpf; Lu et al., 2017). Cardiac phenotypes were assessed at 2 dpf (Shimizu et al., 2015, 2017).
Zebrafish Injections
mRNA was synthesized from pCS2+-constructs using the SP6 mMESSAGE mMACHINE kit (Life Technologies). mRNA was injected into one-cell stage embryos collected from crosses of tretc318 heterozygotes. Cardiac performance was analyzed by visual inspection of cardiac contractions on 1 dpf. and genotypes were confirmed at 2–3 dpf.
Zebrafish Expression Profiling
Total RNA of embryos was isolated using Trizol Reagent (Life Technologies) and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Sequences for forward and reverse primers used for RT-PCR are provided in Supplementary File 2.
In-Situ Hybridization
Whole-mount in situ hybridization was performed as previously described (Chen and Fishman, 1996) using VDAC1, 2, and 3 probes spanning the entire coding sequences. For riboprobe synthesis, plasmids were linearized and in vitro transcription was performed using the DIG RNA labeling kit (Roche).
HeLa Ca2+ Uptake Assay
HeLa cells were transfected with expression plasmids using Lipofectamine2000 (Life Technologies). 24 h after transfection, cells were loaded with 5 μM Rhod2-AM (Life Technologies) in loading buffer (5 mM HEPES, 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 10 mM glucose, and pH adjusted to 7.4 with NaOH) for 1 h at 15°C followed by an additional 30 min incubation in wash buffer (10 mM HEPES, 140 mM TEA-Cl, 1 mM MgCl2, 2 mM Na-EGTA, 10 mM glucose, and pH adjusted to 7.4 with Trizma base) at 37°C to allow de-esterification of cytosolic AM esters. Prior to imaging, cells were permeabilized with 100 μM digitonin for 1 min at room temperature. After approximately 10 s of baseline recording, cells were exposed to an external Ca2+ pulse (final free Ca2+ concentration is calculated to be approximately 10 μM using WEBMAXC at https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/webmaxc/webmaxcS.htm). Confocal images were recorded in internal buffer (5 mM K-EGTA, 20 mM HEPES, 100 mM K-aspartate, 40 mM KCl, 1 mM MgCl2, 2 mM maleic acid, 2 mM glutamic acid, 5 mM pyruvic acid, 0.5 mM KH2PO4, 5 mM MgATP, and pH adjusted to 7.2 with Trizma base) every 0.6 s (Nikon Eclipse Ti microscope) at 545 nm excitation using 20x objective to monitor mitochondrial Ca2+ dynamics. Confocal images were analyzed and quantified using ImageJ (National Institutes of Health, Bethesda, MD).
Stable HL-1 Cell Lines
HL-1 cells (RRID:CVCL_0303) were a gift from William Claycomb (Louisiana State University) and were cultured as previously described (Claycomb et al., 1998). To knock down expression of the endogenous murine VDAC2 cells were lentivirally transduced with a construct expressing shRNA directed against murine VDAC2 (Min et al., 2012; Wilting et al., 2020) followed by selection using 3 μg/ml puromycin. This cell line was then again lentivirally transduced to overexpress zVDAC2, zVDAC2E73Q, zVDAC3, and zVDAC3Q73E using respective constructs. Stable cell lines were established by selecting for nlsGFP expression by FACS sorting.
SR-Mitochondria Ca2+ Transfer
Ca2+ transfer from the SR into mitochondria was measured as described previously (Schweitzer et al., 2017). In brief, HL-1 cardiomyocytes plated in a 96-well plate were loaded with 6 μM Rhod-2 AM (Life technologies) and 0.12% (w/v) Pluronic® F-127 for 30 min at 37°C. After permeabilizing the cells with 100 mM digitonin in internal solution (in mM: 1 BAPTA, 20 HEPES, 100 L-Aspartic acid potassium salt, 40 KCl, 0.5 MgCl2, 2 maleic acid, 2 glutamic acid, 5 pyruvic acid, 0.5 KH2PO4, 5 MgATP, and 0.46 CaCl2; pH = 7.2 with KOH), cells were washed with internal solution before measurements. Rhod-2 fluorescence was monitored at excitation wavelength 540 ± 9 nm and emission wavelength 580 ± 20 nm with an Infinite® 200 PRO multimode reader (Tecan, Maennedorf, Switzerland). After recording 30 s of baseline fluorescence, 10 mM caffeine was added to release Ca2+ from the SR.
Statistical Analysis
Data are expressed as mean ± s.e.m. Statistical testing was carried out using student t-test unless otherwise specified. *Significance levels are expressed as *p < 0.05; **p < 0.01; ***p < 0.001; and NS not significant.
Results
Identification of Zebrafish VDAC Genes
In vertebrates, the VDAC gene family consists of three paralogs: VDAC1, VDAC2, and VDAC3 (Naghdi and Hajnóczky, 2016). We examined the zebrafish genome and found three genes encoding VDAC proteins with high sequence identity to human VDAC1, VDAC2, and VDAC3 (Figure 1A). To investigate whether these three zebrafish proteins represent homologs of the mammalian VDACs and examine the evolutionary relationships among VDAC homologs, we constructed phylogenetic trees of VDAC amino acid sequences using a maximum likelihood approach. Consistent with the findings of previous studies (Young et al., 2007; Wojtkowska et al., 2012), vertebrate VDACs clustered in three distinct clades representing homologs of VDAC1, VDAC2, and VDAC3 (Figure 1B). Within each VDAC clade, zebrafish VDACs clustered tightly with the VDACs of other fish species (medaka and pufferfish). Importantly, one zebrafish VDAC protein was a member in each VDAC clade based on strong bootstrap support, indicating that zebrafish VDAC1, VDAC2, and VDAC3 are the bona fide homologs of their mammalian equivalents (Figure 1B).
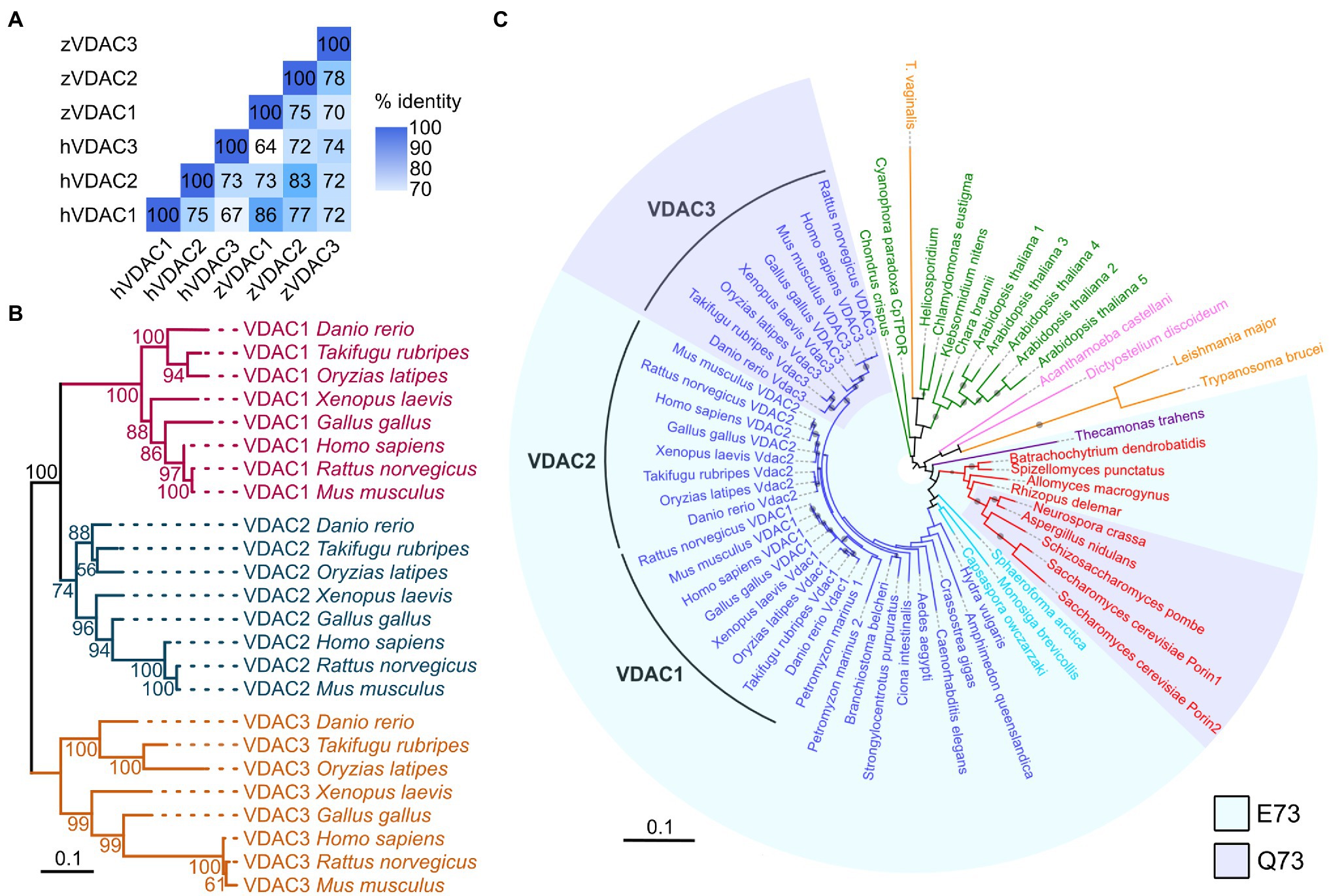
Figure 1. VDACs are highly conserved proteins. (A) Pairwise comparison of sequence identity of human (h) and zebrafish (z) VDAC protein sequences. (B) Maximum likelihood phylogenetic analysis of 24 vertebrate VDAC proteins. Branches are colored based on VDAC gene families (VDAC1: magenta, VDAC2: blue, and VDAC3: gold). Nodes are labeled with bootstrap values in units of percentage. The scale bar for branch lengths indicates the mean number of inferred substitutions per site. (C) Maximum likelihood phylogenetic analysis of 63 VDAC homologs from 41 species. Branches and labels are colored based on phylogenetic groupings of species: blue – animals, light blue – Holozoa (excluding animals), red – fungi, purple – Apusomonadida, orange – Excavata, pink – Amoebozoa, and green – Archaeplastida. Clades are shaded to indicate if proteins contain glutamate (E, light blue) or glutamine (Q, light purple) at amino acid position 73 (with respect to human VDAC1) or are unshaded if this region of the protein lacks high homology with human VDAC1. The size of circles on branches represents bootstrap values and is only displayed for values of 50% and greater. The scale bar for branch lengths indicates the mean number of inferred substitutions per site.
Interestingly, while some plants and fungi possess multiple VDAC paralogs, only a single VDAC protein is present in the majority of the invertebrate animal species surveyed (Figure 1C). This suggests that the three VDAC proteins present in zebrafish likely arose from duplication events occurring in a common ancestor of teleost and tetrapod animals (amphibians, reptiles, birds, and mammals). The retention of the three VDAC paralogs throughout vertebrate evolution may indicate that they have acquired differing and crucial functional roles in mitochondrial biology.
All Three VDAC Isoforms Are Expressed in the Hearts of Embryonic and Adult Zebrafish
We have previously shown that injecting wild-type VDAC2 mRNA in tremblor/ncx1h mutant embryos at the 1-cell stage allows tremblor/ncx1h mutant hearts to establish rhythmic Ca2+ transients and consequently preserves the integrity of myofibrils and maintains coordinated cardiac contractions (Shimizu et al., 2015, 2017). To investigate whether all three VDAC genes share this cardioprotective activity, we first examined if zebrafish VDAC1, 2, and 3 are expressed in the developing zebrafish heart. Whole-mount in situ hybridization revealed that all three VDACs are expressed in the embryonic heart (Figure 2A). Strong signals of VDAC transcripts are also detected in adult zebrafish hearts (both atrium and ventricle; Figure 2B), suggesting a role for VDAC proteins in the maintenance of cardiac physiology.
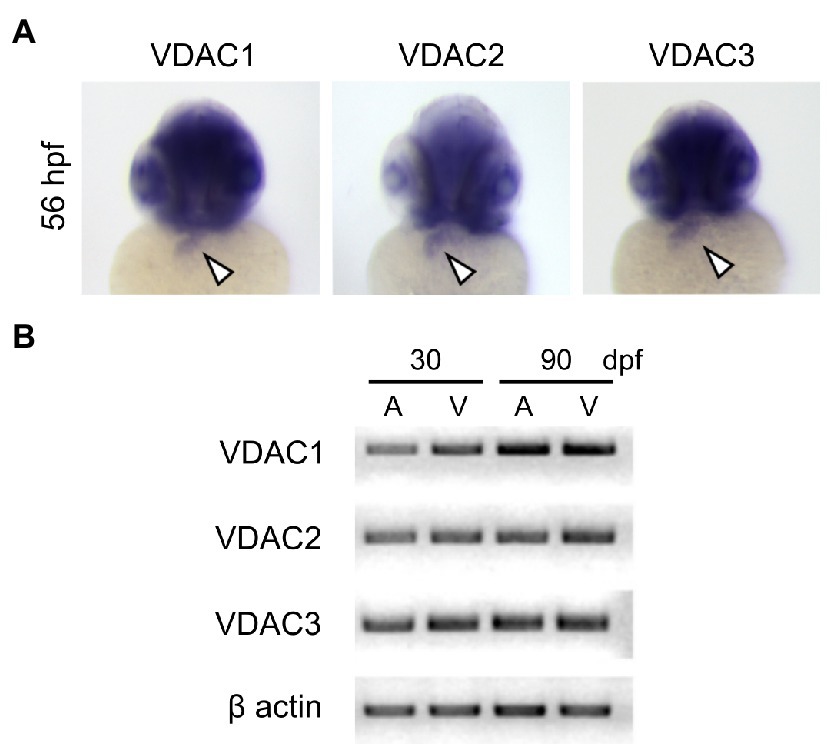
Figure 2. Expression of VDAC isoforms during zebrafish embryonic development. (A) Whole-mount in situ hybridization analysis using full-length riboprobes reveals that all VDAC isoforms are expressed in the embryonic heart (arrowheads). (B) RT-PCR analysis of VDAC mRNA from zebrafish hearts at 30 and 90 days post-fertilization (dpf). Expression of all VDAC isoforms is maintained to adulthood in both the atrium (A) and the ventricle (V).
Diverged Cardioprotective Effects Among VDAC Genes
To evaluate the cardioprotective potential of VDAC1 and VDAC3, we injected FLAG-tagged VDAC1, 2, or 3 RNA into tremblor/ncx1h at the 1-cell stage (Figure 3). As expected, the majority of uninjected tremblor/ncx1h mutant hearts fibrillates where each individual cardiomyocytes contracts spontaneously but fails to coordinate with other cardiomyocytes within the same chamber to support a heartbeat (Langenbacher et al., 2005). However, a significantly higher number of FLAG-tagged VDAC2 RNA injected mutant embryos establish persistent and coordinated cardiac contractions as previous described (Shimizu et al., 2015, 2017), confirming a cardioprotective effect of VDAC2 against aberrant Ca2+ handling-induced arrhythmia. Interestingly, we observed that overexpression of VDAC1 and VDAC3 resulted in different effects on tremblor/ncx1h hearts. Similar to VDAC2, tremblor/ncx1h mutant embryos receiving VDAC1 mRNA established persistent cardiac contractions but the hearts of tremblor/ncx1h mutant embryos receiving VDAC3 RNA continued to fibrillate despite comparable expression levels as assessed by Western blot analysis against the flag epitope. These data demonstrate divergent cardioprotective effects among VDAC isoforms.
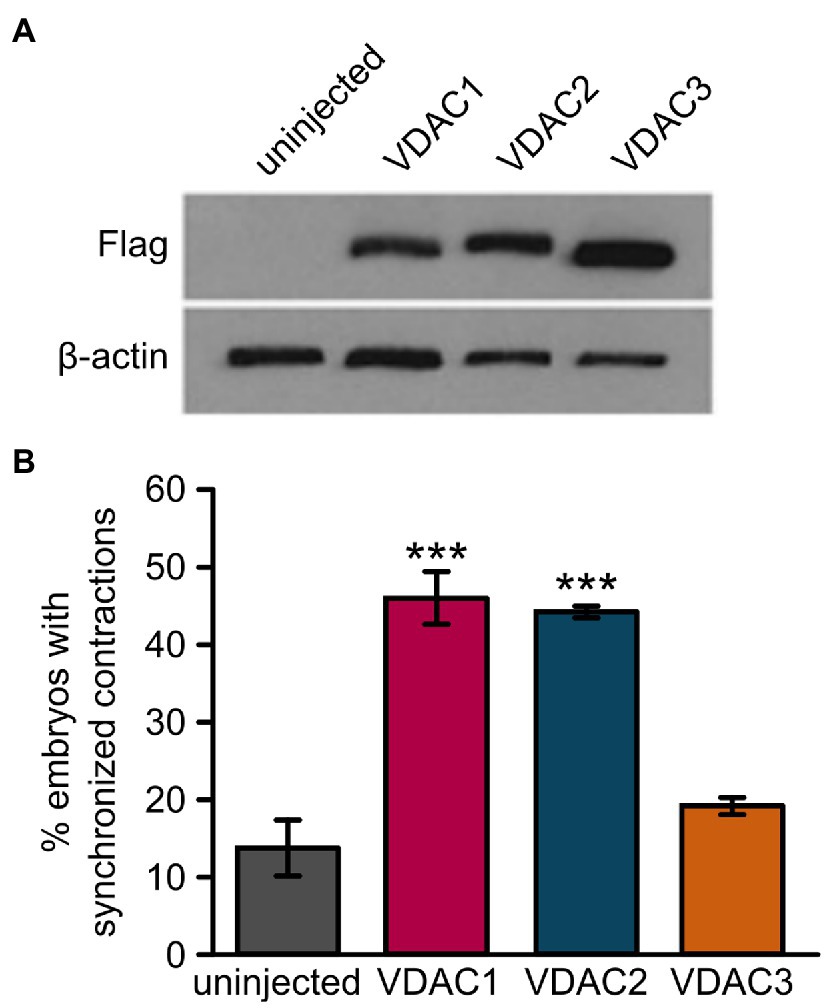
Figure 3. Overexpression of VDAC1 or VDAC2 but not VDAC3 restores rhythmic cardiac contractions in tremblor/ncx1h mutants. (A) Western blot with anti-FLAG antibody after SDS-PAGE with lysate from 32 hpf uninjected embryos or embryos injected with 25 pg. of C-terminally FLAG-tagged VDAC mRNA demonstrates that all VDAC mRNAs are expressed at comparable levels in embryos. β-actin was used as a loading control. (B) While injection of 25 pg. VDAC1 or VDAC2 mRNA significantly restored rhythmic cardiac contractions in 1-day-old tremblor/ncx1h mutant embryos (46.5 ± 3.4%, N = 3, n = 120, and 44.8 ± 0.4%, N = 3, n = 134, respectively, as opposed to 14 ± 3.5%, N = 3, n = 175 in uninjected siblings), injection of 25 pg. VDAC3 failed to recapitulate this effect (19.4 ± 1.1%, N = 3, n = 114). Overall rescue percentages represent the mean rescue percentage ± s.e.m. from N independent experiments, using a total of n embryos.
Cardiomyocyte-Specific Expression of VDAC1 and VDAC2 Is Sufficient to Restore Cardiac Contractions in ncx1 Deficient Mutants
RNA injection into newly fertilized eggs results in a global upregulation of VDAC proteins prior to the onset of Ca2+ handling defects in tremblor/ncx1h mutant embryos. This approach lacks cell type specificity and does not address the question of whether VDAC activation in cardiomyocytes alone is sufficient to suppress aberrant Ca2+ handling-induced cardiac dysfunction. To gain temporal- and cell type-specific resolution of the divergent effects of VDAC proteins on ncx1 deficient hearts, we created transgenic lines Tg:VDAC1, Tg:VDAC2, and Tg:VDAC3, where the expression of FLAG-tagged VDAC isoforms as well as the GFP reporter is regulated by the cardiac-specific promoter myl7 driven Gal4-ecdysone receptor fusion protein (Gal4-EcR; Figure 4A). We then crossed these transgenic fish into the tremblor/ncx1h mutant background, subjected the embryos to either vehicle (DMSO) or tebufenozide (TBF) treatment after 1 day of development, at which time the primitive heart has already formed and the tremblor mutant hearts are fibrillating (Langenbacher et al., 2005).
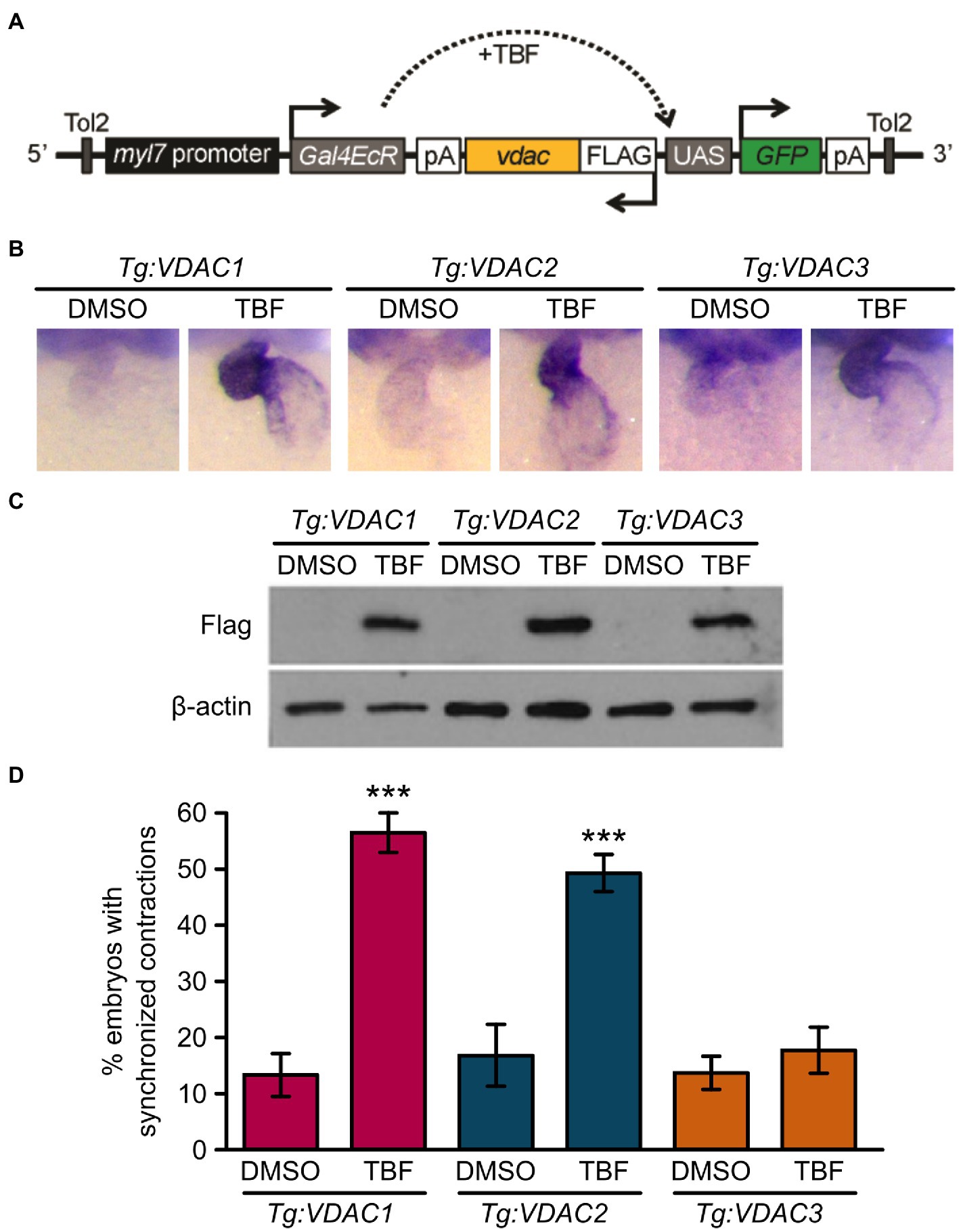
Figure 4. Induction of VDAC1 and VDAC2 but not VDAC3 expression restores rhythmic cardiac contraction in transgenic tremblor/ncx1h lines. (A) Schematic diagram of VDAC transgenic construct. The cardiomyocytes-specific promoter myl7 drives Gal4-ecdysone receptor fusion protein (Gal4EcR), which becomes transcriptionally activated in response to tebufenozide (TBF), an ecdysone receptor agonist and binds to the upstream activating sequence (UAS), resulting in the simultaneous expression of both FLAG-tagged VDAC and EGFP. Transgenic lines were bred in the tremblor/ncx1h background. (B) Whole-mount in situ hybridization analysis demonstrating that VDAC expression is induced specifically in the heart upon TBF treatment in transgenic zebrafish (Tg:VDAC). Embryos were treated with either DMSO or TBF from 24 hpf until they were fixed for in situ hybridization at 48 hpf. (C) Western blotting of 32 hpf transgenic embryo lysate with an anti-FLAG antibody showing that VDAC protein expression is induced after embryos are treated with TBF. β-actin was used as a loading control. (D) While only 13.5 ± 3.6% of DMSO-treated Tg:VDAC1; tremblor/ncx1h embryos exhibit rhythmic cardiac contraction (N = 3, n = 233), 56.72 ± 3.5% of TBF-treated Tg:VDAC1; tremblor/ncx1h embryos established rhythmic contraction (N = 3, n = 238). Similarly, as opposed to only 16.8 ± 5.7% of DMSO-treated Tg:VDAC2; tremblor/ncx1h embryos (N = 3, n = 161), 49.6 ± 3.1% of TBF-treated Tg:VDAC2; tremblor/ncx1h embryos showed cardiac contraction (N = 3, n = 227). In contrast, the effect of TBF-induced overexpression is minimal in Tg:VDAC3; tremblor/ncx1h (13.7 ± 2.9%, N = 3, n = 283 in DMSO-treated embryos compared to 17.9 ± 4.0%, N = 3, n = 373 in TBF-treated embryos). Overall rescue percentages represent the mean rescue percentage ± s.e.m. from N independent experiments, using a total of n embryos.
In the absence of TBF, Gal4-EcR is inactive and the transgenes are not expressed (Figures 4B,C; Lu et al., 2017). In situ hybridization analysis showed that TBF treatment effectively induced VDAC expression in the heart (Figure 4B). Western blotting further confirmed the induction of VDAC proteins by TBF treatment and detected comparable levels of induced VDAC proteins among three transgenic lines (Figure 4C). As expected, the majority of DMSO-treated transgenic ncx1 deficient hearts exhibited fibrillation-like chaotic cardiac movement; coordinated and persistent cardiac contractions were observed in only approximately 14% of these embryos. Excitingly, about half of the hearts of Tg:VDAC1;tremblor/ncx1h and Tg:VDAC2; and tremblor/ncx1h embryos stopped fibrillating after TBF induction and began to contract. The hearts of Tg:VDAC3; tremblor/ncx1h embryos, on the other hand, continued to fibrillate after TBF treatment (Figure 4D). These findings parallel the divergent cardioprotective effects of VDAC1, 2, and 3 revealed by our global overexpression approach. In addition, because the induction of VDAC proteins in tremblor/ncx1h embryos occurs after the manifestation of cardiac fibrillation, our results demonstrate a cardiomyocyte autonomous suppressive effect for VDAC1 and VDAC2 on aberrant Ca2+ handling-induced cardiac dysfunction.
The N-Terminal Domain of VDAC Proteins Is Responsible for Their Divergent Cardioprotective Effect
Mechanisms by which VDAC proteins regulate cardiac contraction remain elusive at the molecular level. The differing abilities of VDAC isoforms to restore of cardiac contractions in tremblor/ncx1h mutant hearts provide a reliable and robust platform to dissect critical domains for VDAC’s ability to suppress aberrant Ca2+ handling-induced arrhythmogenic effects in cardiomyocytes. We created FLAG-tagged chimeric constructs consisting of the N-terminal half of VDAC2 and the C-terminal half of VDAC3 (VDACN2C3) and vice-versa (VDACN3C2; Figure 5A). We then injected synthetic RNAs made from these chimeric constructs into tremblor/ncx1h embryos. While Western blots showed that both VDACN2C3 and VDACN3C2 were expressed at comparable levels (Figure 5B), only those tremblor/ncx1h mutant embryos receiving VDACN2C3 mRNA manifested rhythmic cardiac contractions. The hearts of those mutant embryos receiving VDACN3C2 RNA continued to fibrillate (Figure 5C). These findings indicate that the anti-arrhythmic activity of VDAC proteins is determined by amino acids located in their N-terminal half.
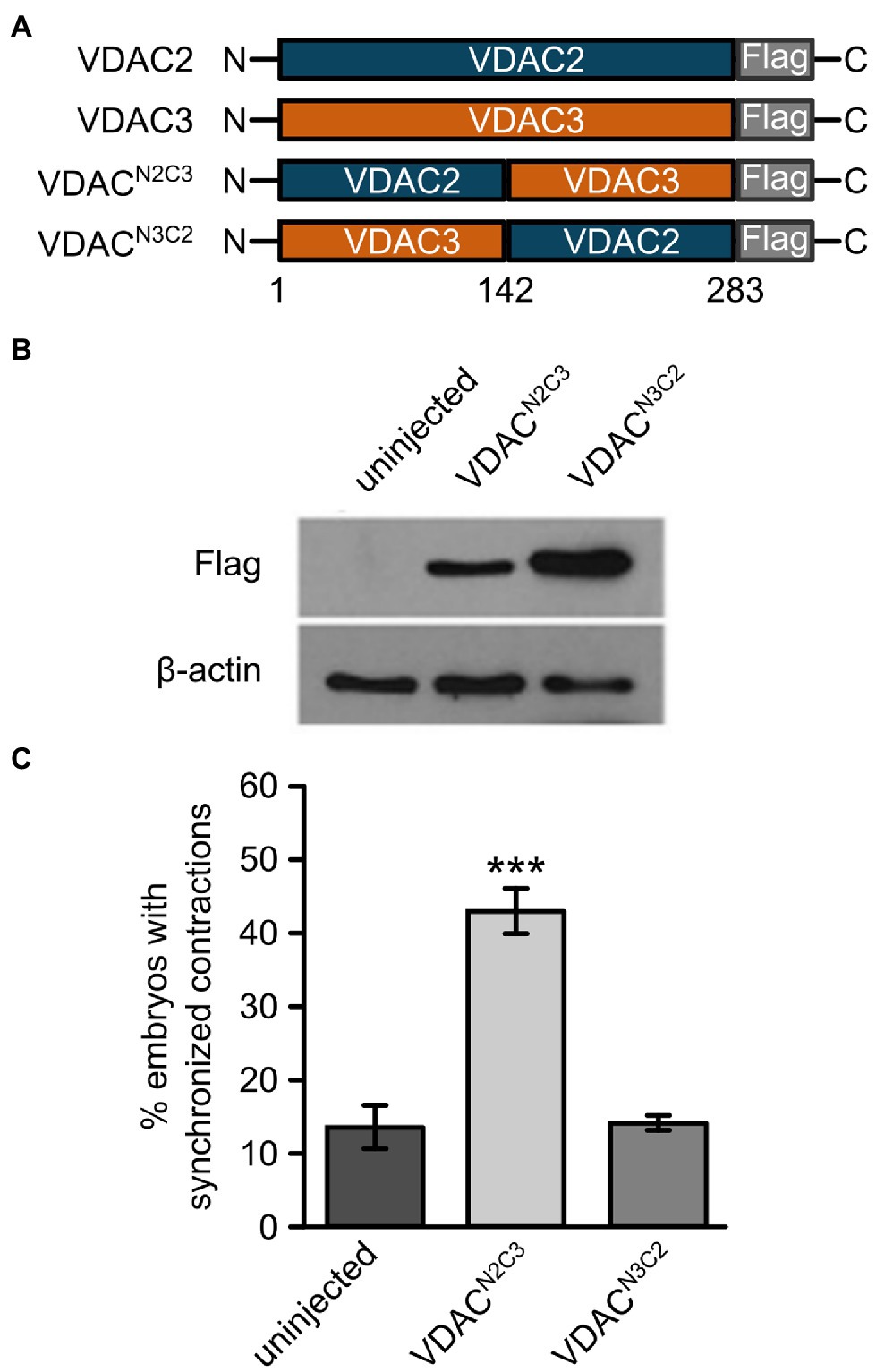
Figure 5. The N-terminal domain of VDAC2 contains critical elements for its cardioprotective activity. (A) Schematic representation of VDAC2/3 chimeric constructs. Reciprocal chimeric constructs were generated by swapping the N- and C-terminal halves of VDAC2 and VDAC3 and tagging a FLAG epitope at the C-terminal end. VDACN2C3: The N-terminal half of VDAC2 (amino acids 1 to 142) was fused to the C-terminal half of VDAC3. VDACN3C2: The N-terminal half of VDAC3 was fused to the C-terminal half of VDAC2. (B) Western blot with anti-FLAG antibody after SDS-PAGE with lysates from 30 hpf uninjected embryos or embryos injected with 60 pg. FLAG-tagged chimeric VDAC mRNA. β-actin was used as a loading control. (C) Injection of VDACN2C3 mRNA restored synchronized cardiac contractions in 1-day-old tremblor/ncx1h embryos (43.4 ± 2.8%, N = 3, n = 168 compared to 13.7 ± 3.1%, N = 3, n = 156 in uninjected siblings), whereas injection of VDACN3C2 mRNA fails to produce this effect (14.2 ± 1.2%, N = 3, n = 162). Overall rescue percentages represent the mean rescue percentage ± s.e.m. from N independent experiments, using a total of n embryos.
E73 Is Evolutionary Conserved in VDAC1 and VDAC2
While the N-terminal half of zebrafish VDAC1, 2, and 3 are highly similar (67–74% similarity at the amino acid level), the amino acid at position 73 (with respect to zebrafish VDAC2) differs between VDAC1 and 2, which exhibit anti-arrhythmic activity, and VDAC3, which does not (Figures 6A,B). Residue 73 is located in β-sheet 4 of the channel facing the outside of the barrel (Figure 6B) and a glutamate is present at this position in zebrafish VDAC2 (E73), while a glutamine is present at the corresponding position in zebrafish VDAC3 (Q73). E73 is highly conserved among animals; it is present in all animal VDAC proteins we examined excluding the VDAC3 family (Figure 1C).
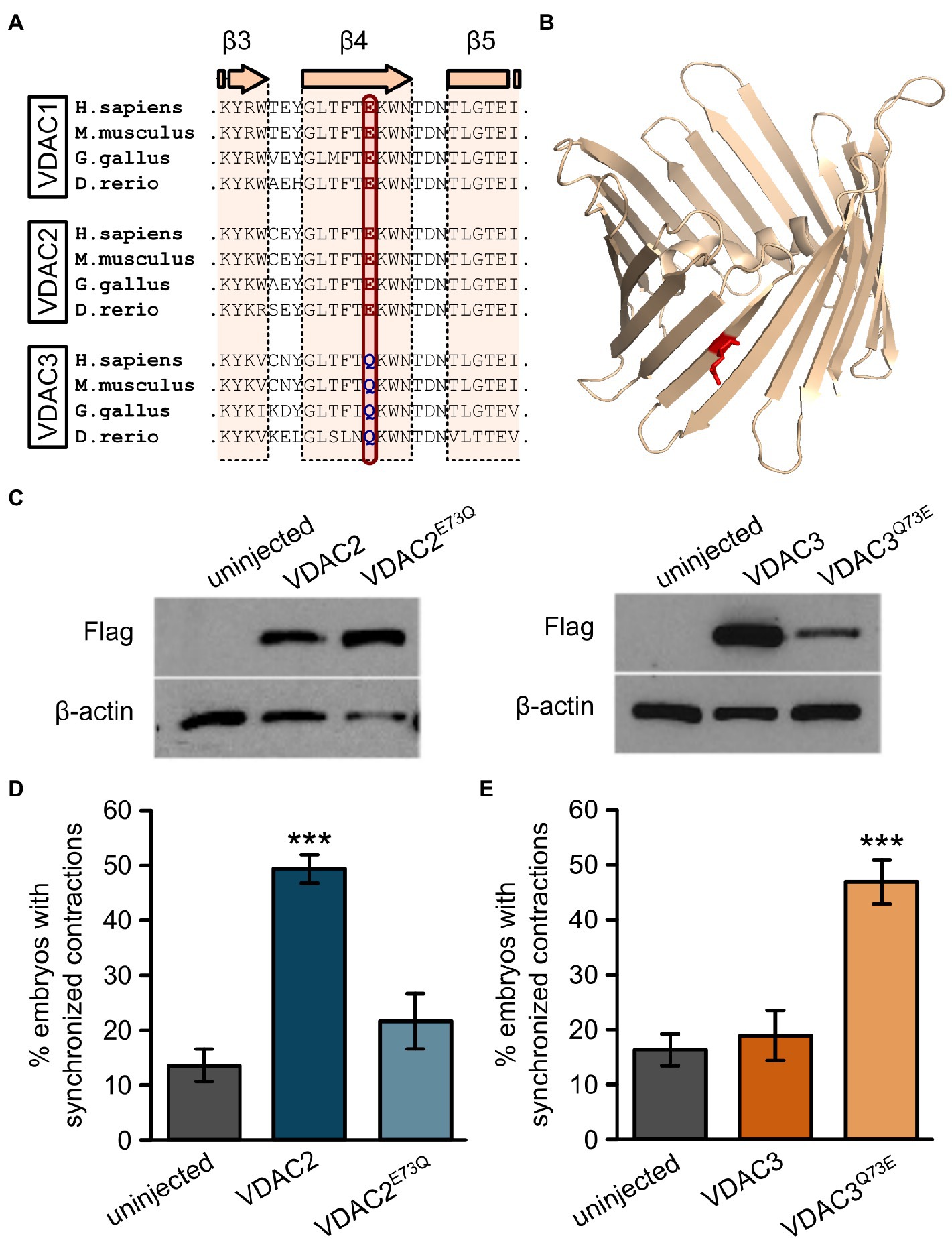
Figure 6. E73 is the critical amino acid residue that determines the ability of VDAC2 to suppress cardiac fibrillation in the tremblor/ncx1h mutant. (A) Alignment of protein sequences of VDAC1, 2, and 3 from different species. In all the species examined the position corresponding to zebrafish residue 73 is consistently occupied by a glutamate (E) in VDAC1 and VDAC2, whereas this position is occupied by glutamine (Q) in VDAC3. (B) Three-dimensional model of the VDAC2 protein (pdb: 4bum) showing the location of amino acid 73 in β-sheet 4 in red. (C) Western blot analysis of lysates from 30 hpf uninjected embryos or embryos injected with 25 pg. FLAG-tagged wild type and point mutant VDAC mRNA. β-actin was used as a loading control. (D) Mutation of E73 to Q in VDAC2 abrogates its ability to suppress cardiac fibrillation in tremblor/ncx1h mutants (49.7 ± 2.8%, N = 3, n = 144 with VDAC2 in contrast to 21.7 ± 5.1%, N = 3, n = 155 with VDAC2E73Q). (E) Vice-versa, by mutating Q73 to E, VDAC3 gained the ability to restore rhythmic cardiac contraction in tre mutants (19.0 ± 4.4%, N = 3, n = 182 with VDAC3 in contrast to 47.2 ± 4.3%, N = 3, n = 145 with VDAC3Q73E). Overall rescue percentages represent the mean rescue percentage ± s.e.m. from N independent experiments, using a total of n embryos.
Interestingly, E73 also appears in some fungi and in several protists (including choanoflagellates) that are believed to be closely related to animals (Figure 1C), suggesting an early evolutionary origin and subsequent conservation of this residue. While E73 is present in early evolutionary branches of fungi (Blastocladiomycota, Chytridiomycota, Chytridiomycota, and Mucoromycota), Q73 is instead present in fungi from the subkingdom Dikarya. Therefore, Q73 appears to have evolved twice, once in animals (VDAC3) and once in a subset of fungi (Dikarya). Intriguingly, like E73, Q73 also appears to be highly conserved within the taxa that it evolved (Figure 1C). Together, the simultaneous conservation of E73 and Q73 suggests that these two residues may impart significant functional differences to the VDAC protein that in turn created an evolutionary pressure for their retention.
Given that upregulation of mitochondrial Ca2+ uptake restores cardiac contraction in tremblor/ncx1h mutant embryos (Shimizu et al., 2015) and that E73 was previously reported to have an essential role in Ca2+ handling (Israelson et al., 2007; Peng et al., 2020), we suspected that E73 may be a critical residue that determines the isoform-specific rescue effect of VDAC genes on tremblor/ncx1h’s arrhythmia phenotype. To investigate this possibility, we created two point mutants: VDAC2E73Q by replacing E73 in VDAC2 with a glutamine and VDAC3Q73E by substituting Q73 of VDAC3 with a glutamate. We then injected VDAC2E73Q and VDAC3Q73E RNA into tremblor/ncx1h mutant embryos. Western blots showed that both VDAC2E73Q and VDAC3Q73E are stably expressed in embryos (Figure 6C). Interestingly, opposite to wild-type VDAC2 and VDAC3 overexpression effects, the hearts that received VDAC2E73Q RNA continued to fibrillate, whereas VDAC3Q73E overexpression suppressed cardiac fibrillation and restored rhythmic cardiac contraction in tremblor/ncx1h mutant embryos (Figures 6D,E).
E73 Controls VDAC-Dependent Mitochondrial Ca2+ Uptake
VDAC2 activation suppresses cardiac fibrillation in tremblor/ncx1h mutants by buffering excess Ca2+ into the mitochondria (Shimizu et al., 2015). To test whether VDAC’s differential cardioprotective activities on tremblor/nxc1h mutant hearts are rooted in their differential Ca2+ transporting activity, we transfected HeLa cells with VDAC isoforms and measured mitochondrial Ca2+ uptake using Rhod-2 (Figure 7A). Cells transfected with VDAC2 showed increased mitochondrial Ca2+ levels compared to empty vector transfected control cells upon stimulation, whereas no significant differences in mitochondrial Ca2+ uptake between control and VDAC3 transfected cells were observed (Figure 7B). Given the previously observed correlation between position 73 and the cardioprotective activity of VDAC2 and VDAC3, we next examined whether E73 might determine the differential mitochondrial Ca2+ uptake activities between VDAC isoforms. Indeed, wild-type VDAC2 enhanced mitochondrial Ca2+ uptake, while VDAC2E73Q showed a mitochondrial Ca2+ uptake profile comparable to control cells. In contrast, VDAC3 did not enhance mitochondrial Ca2+ uptake above control, while introduction of E73 into VDAC3 (VDAC3Q73E) significantly enhanced mitochondrial Ca2+ uptake, demonstrating that E73 is a key determinant of VDAC2’s mitochondrial Ca2+ uptake activity (Figures 7B–D).
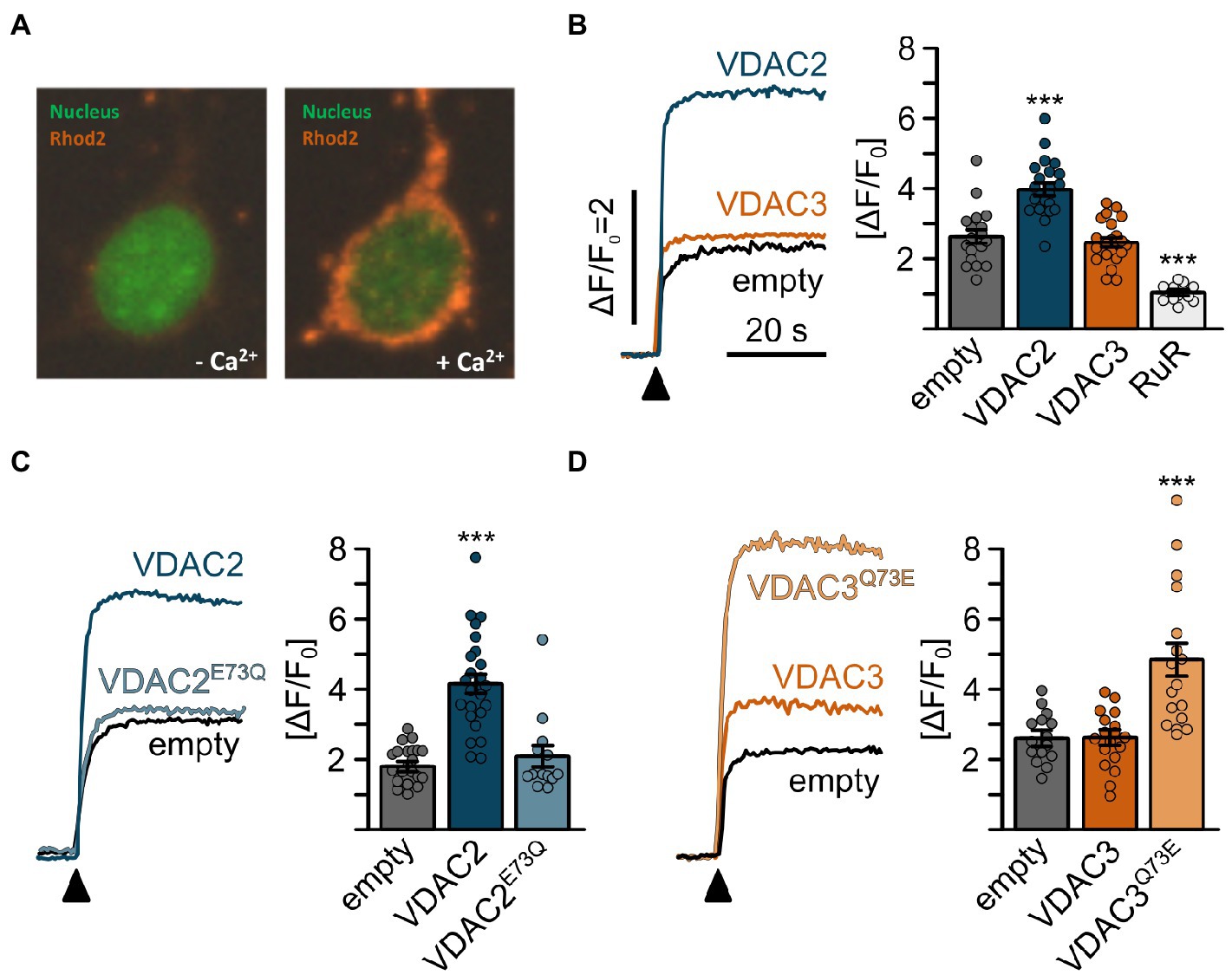
Figure 7. Mitochondrial Ca2+ uptake in HeLa cells is promoted by E73. (A) Representative confocal images of permeabilized HeLa cells loaded with Rhod2 after transiently transfection with VDAC expression constructs. Mitochondrial Rhod2 fluorescence is minimal at the basal state (left). Upon addition of Ca2+, the Rhod2 fluorescence rapidly concentrates in mitochondria (right). (B-D) Mitochondrial Ca2+ uptake assays of HeLa cells transiently transfected with VDAC constructs; (B) mitochondrial Ca2+ uptake was observed in empty vector transfected control cells (empty, ∆F/F0 = 2.64 ± 0.82, n = 18), which was significantly enhanced by overexpression of VDAC2 (∆F/F0 = 3.99 ± 0.80, n = 22) but not VDAC3 (∆F/F0 = 2.47 ± 0.61, n = 22) and significantly inhibited by ruthenium red (∆F/F0 = 1.06 ± 0.23, n = 12). (C) While wild-type VDAC2 enhanced mitochondrial Ca2+ uptake (∆F/F0 = 1.83 ± 0.52 for empty cells vs. ∆F/F0 = 4.18 ± 1.4 for VDAC2, n = 22 and 24, respectively), VDAC2E73Q failed to induce this effect (∆F/F0 = 2.09 ± 1.15, n = 13). (D) Conversely, wild-type VDAC3 did not induce a significant increase in mitochondrial Ca2+ uptake (∆F/F0 = 2.63 ± 0.70 for empty cells vs. ∆F/F0 = 2.59 ± 0.81 for VDAC3, n = 15 and 18, respectively); however, VDAC3Q73E significantly increased mitochondrial Ca2+ uptake to ∆F/F0 = 4.85 ± 1.99 (n = 18).
E73 Critically Regulates VDAC2-Mediated Transfer of Ca2+ From the SR Into Mitochondria
Considerable differences in mitochondrial Ca2+ handling have been observed between excitable and non-excitable cells. In particular, in cardiomyocytes, VDAC2 was suggested to interact with RyR2, the major Ca2+ release channel on the SR, and thereby facilitate Ca2+ transfer from the SR to mitochondria (Subedi et al., 2011; Min et al., 2012). We therefore examined whether E73 regulates VDAC2’s ability to mediate the transfer of Ca2+ from the SR into mitochondria in cultured HL-1 cardiomyocytes (Claycomb et al., 1998; Schweitzer et al., 2017; Wilting et al., 2020). Stable knockdown of the endogenously expressed murine VDAC2 by siRNA (Subedi et al., 2011; Min et al., 2012; Wilting et al., 2020) significantly reduced uptake of Ca2+ released from the SR by a caffeine pulse into mitochondria, while this uptake was comparable to native HL-1 cells when a scrambled control shRNA was used (Figure 8). We subsequently overexpressed our VDAC constructs, VDAC2, VDAC2E73Q, VDAC3, and VDAC3Q73E in mVDAC2 knockdown cells and evaluated their abilities to restore SR-mitochondria Ca2+ transfer. VDAC2 restored mitochondrial Ca2+ uptake to levels above baseline consistent with the idea of VDAC2 as the main mediator of SR-mitochondria Ca2+ transfer. Strikingly, replacing E73 of VDAC2 with a glutamine (VDAC2E73Q) abrogated its ability to promote SR-mitochondria Ca2+ transfer (Figure 8B). On the other hand, while VDAC3 failed to restore SR-mitochondria Ca2+ transfer, introduction of E73 in VDAC3Q73E restored SR-mitochondria Ca2+ transfer to control levels (Figure 8C). These data indicate that VDAC-mediated SR-mitochondria Ca2+ transfer is dependent on the presence of E73 in cardiomyocytes.
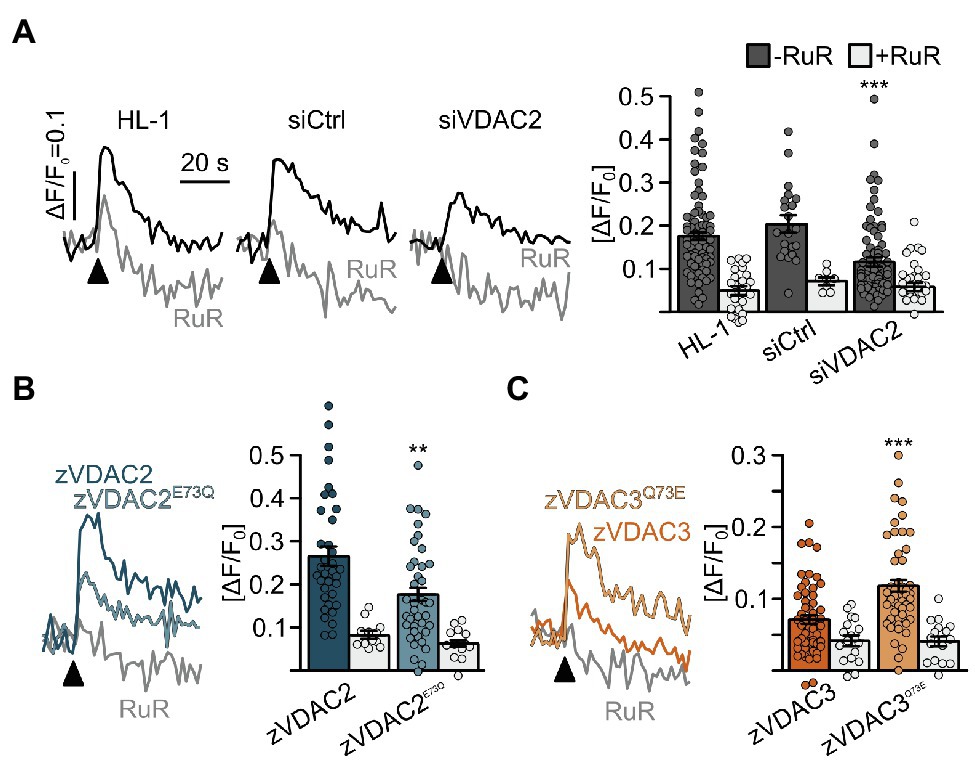
Figure 8. Restoration of SR-mitochondria Ca2+ transfer in permeabilized HL-1 cardiomyocytes. (A) Knockdown of the endogenous mVDAC2 in HL-1 cells significantly reduced SR-mitochondria Ca2+ transfer after addition of caffeine (arrowhead) from ΔF/F0 = 0.18 ± 0.01 (n = 84) to 0.12 ± 0.01 (n = 83) while a scrambled siRNA control did not affect Ca2+ transfer (0.20 ± 0.02, n = 21). (B) Wild-type zVDAC2 restored mitochondrial Ca2+ uptake to 0.27 ± 0.02 (n = 36), well above mVDAC2 knockdown cells. Overexpression of zVDAC2E73Q restored mitochondrial Ca2+ uptake to only 0.18 ± 0.02 (n = 39), which is significantly lower compared to wild-type zVDAC2. (C) Conversely, zVDAC3 did not restore mitochondrial Ca2+ uptake in mVDAC2 knockdown cells (0.12 ± 0.01, n = 54), while VDAC3Q73E restored mitochondrial Ca2+ uptake to ΔF/F0 = 0.20 ± 0.02, (n = 50). Arrowheads indicate injection of 10 mM caffeine. Statistical analysis was performed using Kruskal-Wallis test with Dunn’s post-hoc test.
Discussion
Despite structural and functional similarities, divergent biological functions have been noted among VDAC isoforms. Accumulating evidence indicates that VDAC2 is a critical gatekeeper for mitochondrial Ca2+ entry in physiology and in disease within the heart (Min et al., 2012; Shimizu et al., 2015, 2017; Schweitzer et al., 2017; Wilting et al., 2020; Sander et al., 2021). In this study, we used zebrafish tremblor/ncx1h mutants as an in vivo platform to assess the ability of VDAC1 and VDAC3 to regulate cardiac rhythmicity through mitochondrial Ca2+ uptake. We demonstrate that like VDAC2, VDAC1 is capable of transporting Ca2+ across the outer mitochondrial membrane. When overexpressed, VDAC1 effectively restores rhythmic cardiac contractions in ncx1 deficient hearts. VDAC3, on the contrary, cannot facilitate efficient Ca2+ transport across the mitochondrial membrane, demonstrating divergent roles for these isoforms in Ca2+ trafficking. Structure-function analysis further revealed that the evolutionarily conserved E73/Q73 confers this divergent VDAC isoform-dependent calcium trafficking function.
Across all vertebrate species, a glutamate occupies position 73 in VDAC1 and VDAC2 (E73), while a glutamine is present at this position in VDAC3 (Q73). Using multiple model systems, we demonstrate that only isoforms with E73 are able to promote mitochondrial Ca2+ uptake and to regulate cardiac rhythmicity. While VDAC3 did not mediate mitochondrial Ca2+ uptake in its wild-type form, replacing Q73 with a glutamate allows VDAC3 to facilitate SR-mitochondrial Ca2+ transfer, enhance mitochondrial Ca2+ uptake, and suppress cardiac arrhythmia. These data are consistent with the established notion that E73 is an important regulator of VDAC function. However, the precise mechanism by which E73 influences Ca2+ trafficking remains elusive. Our data suggest that E73 confers cardiac protection by facilitating mitochondrial Ca2+ uptake. In line with these findings, E73 has been suggested to be involved in the Ca2+ transport activity of VDAC (Gincel et al., 2001; Ge et al., 2016; Peng et al., 2020) and is also the binding site for VDAC blockers like ruthenium red and its derivatives, which compete with Ca2+ for binding to this site (Israelson et al., 2007). Also supporting the notion that E73 can facilitate the inter-organellar transport of Ca2+ is the recent finding that it influences mitochondrial uptake of Ca2+ from lysosomes (Peng et al., 2020). Given the location of E73 on the outside of the fourth repeat of the VDAC barrel facing the lipid membrane (Ujwal et al., 2008; Schredelseker et al., 2014), it is unlikely that E73 serves as a direct binding site for Ca2+ ions, but instead may indirectly promote the uptake of Ca2+ by VDAC. Indeed, E73 alone was unable to modify voltage gating of purified VDAC1 channels (Queralt-Martín et al., 2019). An indirect effect is further supported by the findings that the positive charge of E73 can induce a thinning of the local plasma membrane (Villinger et al., 2010) and that E73 promotes binding of VDAC protein partners like hexokinase (Abu-Hamad et al., 2008) and mediates VDAC dimerization (Bergdoll et al., 2018). Though our data demonstrate an essential role for E73 in mitochondrial Ca2+ uptake, these reports clearly highlight the need for further biochemical and structural studies to fully elucidate the molecular mechanisms of E73’s involvement in VDAC-mediated Ca2+ transit.
In cardiomyocytes, VDAC2 interacts with the ryanodine receptor to shape intracellular Ca2+ signals (Subedi et al., 2011; Min et al., 2012). Knockout of VDAC2 alone is sufficient to attenuate SR-mitochondria Ca2+ transfer in cardiomyocytes and cardiac-specific elimination of VDAC2 was reported to result in early cardiac dysfunction (Raghavan et al., 2012). While our study suggests that both VDAC1 and VDAC2 are capable of mediating mitochondrial Ca2+ uptake in cardiomyocytes, VDAC2 appears to be more relevant to mammalian cardiac biology. VDAC1 is known to couple with the IP3-receptor to mediate mitochondrial Ca2+ uptake in non-excitable cells. However, inactivation of glycogen synthase kinase-3β was shown to reduce coupling of the IP3-receptor to VDAC1 in cardiomyocytes and to reduce mitochondrial Ca2+ uptake during cardiac ischemia-reperfusion (Gomez et al., 2016). It would thus be interesting to explore differential interaction of VDAC1 and VDAC2 with the ryanodine receptor or other Ca2+ release channels on various organelles to determine if VDAC2 plays a more acute and VDAC1 plays a more subtle role in healthy cardiomyocytes.
The presence of the highly conserved VDAC3 family, which lacks E73, in vertebrates suggests that this difference may impart crucial functional properties to VDAC3. Whether the VDAC proteins in vertebrates have evolved subtype-specific distinct functions, with each one assuming part of the role played by the single VDAC protein in basal metazoans, or if they have acquired novel functions are still an open question that could be examined to better understand the role of mitochondria in the evolution of the highly demanding vertebrate heart muscle. Whether the E73/Q73 residues identified in fungi by sequence homology are functionally and structurally homologous to zebrafish VDAC, E73/Q73 has not been studied. It would be interesting to examine whether E73 containing and/or Q73 containing fungal VDACs could substitute for the activity of VDAC2 in zebrafish embryos or mammalian cells, and whether these proteins exhibit differing calcium permeability properties. Most species of fungi examined have only a single VDAC protein, with the notable exception of S. cerevisiae. Q73 is present in Dikarya, a subkingdom of Fungi, while E73 is present in all other fungal species surveyed. It is intriguing to note that similar to the evolution of vertebrate VDACs, fungal E73 and Q73 are conserved within their respective clades, suggesting that differences at this residue may be functionally relevant. If indeed Q73 reduces the calcium permeability of fungal VDACs, this would suggest that a reduced requirement for mitochondrial calcium uptake or alternative routes of mitochondrial calcium uptake may have evolved in Dikarya. Alternatively, structural differences between animal and fungal VDACs may render fungal E73 less crucial for calcium uptake.
Together, our data demonstrate that the evolutionarily conserved E73 residue mediates the divergent anti-arrhythmic effects of VDAC isoforms by modulating mitochondrial Ca2+ uptake in cardiomyocytes. These findings underscore the important role of mitochondria in cellular Ca2+ dynamics and suggest that further exploring VDAC1- and VDAC2-mediated mitochondrial Ca2+ uptake may reveal novel regulatory mechanisms for normal cardiac physiology and heart disease.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by University of California Los Angeles Animal Care and Use Committee.
Author Contributions
HS, SH, AL, LC, JH, KW, FW, and JS performed the experiments. JS and J-NC conceptualized the study. HS, JS, TG, and J-NC acquired the funding. HS, AL, LC, JS, and J-NC wrote the manuscript. All authors commented on the manuscript.
Funding
This work was supported by the Heiwa Nakajima foundation (to HS), the Austrian Science Fund FWF (J3065-B11 to JS), the German research foundation (SCHR 1471/1-1 to JS, TRR 152 to TG), Deutsche Forschungsgemeinschaft, and the National Heart, Lung and Blood Institute (HL096980 and HL140472 to J-NC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to acknowledge Brigitte Mayerhofer for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fphys.2021.724828/full#supplementary-material
Footnotes
References
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490. doi: 10.1074/jbc.M708216200
Anflous, K., Armstrong, D. D., and Craigen, W. J. (2001). Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J. Biol. Chem. 276, 1954–1960. doi: 10.1074/jbc.M006587200
Bergdoll, L. A., Lerch, M. T., Patrick, J. W., Belardo, K., Altenbach, C., Bisignano, P., et al. (2018). Protonation state of glutamate 73 regulates the formation of a specific dimeric association of mVDAC1. Proc. Natl. Acad. Sci. U. S. A. 115, E172–E179. doi: 10.1073/pnas.1715464115
Caterino, M., Ruoppolo, M., Mandola, A., Costanzo, M., Orrù, S., and Imperlini, E. (2017). Protein-protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol. BioSyst. 13, 2466–2476. doi: 10.1039/C7MB00434F
Chen, J. N., and Fishman, M. C. (1996). Zebrafish tinman homolog demarcates the heart field and initiates myocardial differentiation. Development 122, 3809–3816. doi: 10.1242/dev.122.12.3809
Cheng, E. H. Y., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003). VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517. doi: 10.1126/science.1083995
Claycomb, W. C., Lanson, N. A., Stallworth, B. S., Egeland, D. B., Delcarpio, J. B., Bahinski, A., et al. (1998). HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U. S. A. 95, 2979–2984. doi: 10.1073/pnas.95.6.2979
Dia, M., Gomez, L., Thibault, H., Tessier, N., Leon, C., Chouabe, C., et al. (2020). Reduced reticulum–mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 115:74. doi: 10.1007/s00395-020-00835-7
Ebert, A. M., Hume, G. L., Warren, K. S., Cook, N. P., Burns, C. G., Mohideen, M. A., et al. (2005). Calcium extrusion is critical for cardiac morphogenesis and rhythm in embryonic zebrafish hearts. Proc. Natl. Acad. Sci. U. S. A. 102, 17705–17710. doi: 10.1073/pnas.0502683102
Feng, Y., Madungwe, N. B., Imam Aliagan, A. D., Tombo, N., and Bopassa, J.-C. (2019). Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 520, 606–611. doi: 10.1016/j.bbrc.2019.10.006
Ge, L., Villinger, S., Mari, S. A., Giller, K., Griesinger, C., Becker, S., et al. (2016). Molecular plasticity of the human voltage-dependent anion channel embedded into a membrane. Structure 24, 585–594. doi: 10.1016/j.str.2016.02.012
Gincel, D., Zaid, H., and Shoshan-Barmatz, V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155. doi: 10.1042/bj3580147
Gomez, L., Thiebaut, P.-A., Paillard, M., Ducreux, S., Abrial, M., Crola Da Silva, C., et al. (2016). The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 23, 313–322. doi: 10.1038/cdd.2015.101
Israelson, A., Abu-Hamad, S., Zaid, H., Nahon, E., and Shoshan-Barmatz, V. (2007). Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium 41, 235–244. doi: 10.1016/j.ceca.2006.06.005
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kwan, K. M., Fujimoto, E., Grabher, C., Mangum, B. D., Hardy, M. E., Campbell, D. S., et al. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099. doi: 10.1002/dvdy.21343
Langenbacher, A. D., Dong, Y., Shu, X., Choi, J., Nicoll, D. A., Goldhaber, J. I., et al. (2005). Mutation in sodium-calcium exchanger 1 (NCX1) causes cardiac fibrillation in zebrafish. Proc. Natl. Acad. Sci. U. S. A. 102, 17699–17704. doi: 10.1073/pnas.0502679102
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Lu, F., Langenbacher, A., and Chen, J.-N. (2017). Tbx20 drives cardiac progenitor formation and cardiomyocyte proliferation in zebrafish. Dev. Biol. 421, 139–148. doi: 10.1016/j.ydbio.2016.12.009
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees.” in 2010 Gateway Computing Environments Workshop (GCE) November 14, 2010; (IEEE), 1–8.
Min, C. K., Yeom, D. R., Lee, K.-E., Kwon, H.-K., Kang, M., Kim, Y.-S., et al. (2012). Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 447, 371–379. doi: 10.1042/BJ20120705
Naghdi, S., and Hajnóczky, G. (2016). VDAC2-specific cellular functions and the underlying structure. Biochim. Biophys. Acta, Mol. Cell Res. 1863, 2503–2514. doi: 10.1016/j.bbamcr.2016.04.020
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H. W., Mueller, S., Miller, T., et al. (2016). There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157. doi: 10.1016/j.tins.2016.01.008
Peng, W., Wong, Y. C., and Krainc, D. (2020). Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. U. S. A. 117, 19266–19275. doi: 10.1073/pnas.2003236117
Queralt-Martín, M., Bergdoll, L., Jacobs, D., Bezrukov, S. M., Abramson, J., and Rostovtseva, T. K. (2019). Assessing the role of residue E73 and lipid headgroup charge in VDAC1 voltage gating. Biochim. Biophys. Acta Bioenerg. 1860, 22–29. doi: 10.1016/j.bbabio.2018.11.001
Raghavan, A., Sheiko, T. V., Graham, B. H., and Craigen, W. J. (2012). Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim. Biophys. Acta Biomembr. 1818, 1477–1485. doi: 10.1016/j.bbamem.2011.10.019
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Rosencrans, W. M., Rajendran, M., Bezrukov, S. M., and Rostovtseva, T. K. (2021). VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 94:102356. doi: 10.1016/j.ceca.2021.102356
Sampson, M. J., Decker, W. K., Beaudet, A. L., Ruitenbeek, W., Armstrong, D. D., Hicks, M. J., et al. (2001). Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 276, 39206–39212. doi: 10.1074/jbc.M104724200
Sander, P., Gudermann, T., and Schredelseker, J. (2021). A calcium guard in the outer membrane: is VDAC a regulated gatekeeper of mitochondrial calcium uptake? Int. J. Mol. Sci. 22:946. doi: 10.3390/ijms22020946
Santin, Y., Fazal, L., Sainte-Marie, Y., Sicard, P., Maggiorani, D., Tortosa, F., et al. (2020). Mitochondrial 4-HNE derived from MAO-A promotes mitoCa2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 27, 1907–1923. doi: 10.1038/s41418-019-0470-y
Schredelseker, J., Paz, A., López, C. J., Altenbach, C., Leung, C. S., Drexler, M. K., et al. (2014). High-resolution structure and double electron-electron resonance of the zebrafish voltage dependent anion channel 2 reveal an oligomeric population. J. Biol. Chem. 289, 12566–12577. doi: 10.1074/jbc.M113.497438
Schweitzer, M. K., Wilting, F., Sedej, S., Dreizehnter, L., Dupper, N. J., Tian, Q., et al. (2017). Suppression of arrhythmia by enhancing mitochondrial Ca2+ uptake in Catecholaminergic ventricular tachycardia models. JACC Basic Transl. Sci. 2, 737–746. doi: 10.1016/j.jacbts.2017.06.008
Shimizu, H., Langenbacher, A. D., Huang, J., Wang, K., Otto, G., Geisler, R., et al. (2017). The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. eLife 6:e27955. doi: 10.7554/eLife.27955
Shimizu, H., Schredelseker, J., Huang, J., Lu, K., Naghdi, S., Lu, F., et al. (2015). Mitochondrial Ca2+ uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity. eLife 4:e04801. doi: 10.7554/eLife.04801
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Subedi, K. P., Kim, J.-C., Kang, M., Son, M.-J., Kim, Y.-S., and Woo, S.-H. (2011). Voltage-dependent anion channel 2 modulates resting Ca2+ sparks, but not action potential-induced Ca2+ signaling in cardiac myocytes. Cell Calcium 49, 136–143. doi: 10.1016/j.ceca.2010.12.004
Ujwal, R., Cascio, D., Colletier, J.-P., Faham, S., Zhang, J., Toro, L., et al. (2008). The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U. S. A. 105, 17742–17747. doi: 10.1073/pnas.0809634105
Villinger, S., Briones, R., Giller, K., Zachariae, U., Lange, A., de Groot, B. L., et al. (2010). Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. U. S. A. 107, 22546–22551. doi: 10.1073/pnas.1012310108
Wilting, F., Kopp, R., Gurnev, P. A., Schedel, A., Dupper, N. J., Kwon, O., et al. (2020). The antiarrhythmic compound efsevin directly modulates voltage-dependent anion channel 2 by binding to its inner wall and enhancing mitochondrial Ca2+ uptake. Br. J. Pharmacol. 177, 2947–2958. doi: 10.1111/bph.15022
Wojtkowska, M., Jąkalski, M., Pieńkowska, J. R., Stobienia, O., Karachitos, A., Przytycka, T. M., et al. (2012). Phylogenetic analysis of mitochondrial outer membrane β-barrel channels. Genome Biol. Evol. 4, 110–125. doi: 10.1093/gbe/evr130
Keywords: mitochondria, voltage-dependent anion channel, calcium, cardiac rhythmicity, zebrafish
Citation: Shimizu H, Huber S, Langenbacher AD, Crisman L, Huang J, Wang K, Wilting F, Gudermann T, Schredelseker J and Chen J-N (2021) Glutamate 73 Promotes Anti-arrhythmic Effects of Voltage-Dependent Anion Channel Through Regulation of Mitochondrial Ca2+ Uptake. Front. Physiol. 12:724828. doi: 10.3389/fphys.2021.724828
Edited by:
Vito De Pinto, University of Catania, ItalyReviewed by:
Radhakrishnan Mahalakshmi, Indian Institute of Science Education and Research, IndiaCarmen A. Mannella, University of Maryland Medical Center, United States
Copyright © 2021 Shimizu, Huber, Langenbacher, Crisman, Huang, Wang, Wilting, Gudermann, Schredelseker and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johann Schredelseker, johann.schredelseker@lmu.de; Jau-Nian Chen, chenjn@mcdb.ucla.edu
†These authors share first authorship
‡These authors share senior authorship