
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 09 September 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.718404
This article is part of the Research Topic Calcium and Heart Failure: From Bench to Bedside View all 11 articles
 Ingunn E. Setterberg1,2
Ingunn E. Setterberg1,2 Christopher Le1,2†
Christopher Le1,2† Michael Frisk1,2†
Michael Frisk1,2† Harmonie Perdreau-Dahl1,2Jia Li1,2
Harmonie Perdreau-Dahl1,2Jia Li1,2 William E. Louch1,2*
William E. Louch1,2*In cardiomyocytes, invaginations of the sarcolemmal membrane called t-tubules are critically important for triggering contraction by excitation-contraction (EC) coupling. These structures form functional junctions with the sarcoplasmic reticulum (SR), and thereby enable close contact between L-type Ca2+ channels (LTCCs) and Ryanodine Receptors (RyRs). This arrangement in turn ensures efficient triggering of Ca2+ release, and contraction. While new data indicate that t-tubules are capable of exhibiting compensatory remodeling, they are also widely reported to be structurally and functionally compromised during disease, resulting in disrupted Ca2+ homeostasis, impaired systolic and/or diastolic function, and arrhythmogenesis. This review summarizes these findings, while highlighting an emerging appreciation of the distinct roles of t-tubules in the pathophysiology of heart failure with reduced and preserved ejection fraction (HFrEF and HFpEF). In this context, we review current understanding of the processes underlying t-tubule growth, maintenance, and degradation, underscoring the involvement of a variety of regulatory proteins, including junctophilin-2 (JPH2), amphiphysin-2 (BIN1), caveolin-3 (Cav3), and newer candidate proteins. Upstream regulation of t-tubule structure/function by cardiac workload and specifically ventricular wall stress is also discussed, alongside perspectives for novel strategies which may therapeutically target these mechanisms.
Forceful contraction of the heart requires coordinated contraction of cardiac muscle cells, called cardiomyocytes. Within these cells, electrical excitation during the action potential is linked to cell shortening by a process known as excitation-contraction (EC) coupling. In mammalian heart, this process is made possible by membrane invaginations called t-tubules which propagate the action potential into the cell interior. Critical in normal cardiac physiology and importantly disrupted during disease, t-tubules have been the focus of considerable research focus in the past decades. This review will summarize this progress and outline future perspectives, with particular attention given to potential roles of t-tubules as therapeutic targets.
Retzius (1881) first suggested t-tubules’ existence in 1881 after he noted that muscle cells exhibit a quick inward spread of electrical depolarization (“negative variation”). Thereafter, the first visual evidence of t-tubules was provided by Nyström (1897), who found that India ink entered rabbit heart muscle in a transverse pattern. In 1957, with the help of electron microscopy, Lindner (1957) clearly described t-tubule structures in dog ventricular cardiomyocytes. Subsequent studies from both skeletal and cardiac muscle revealed that t-tubules serve to rapidly conduct electrical excitation and facilitate communication with the sarcoplasmic reticulum (SR) (Huxley and Taylor, 1955; Cheng et al., 1996), thereby playing a central role in EC coupling. More recently, the advent of newer techniques, such as three-dimensional (3D) electron microscopy, super-resolution microscopy, and ion-conductance microscopy have provided ever greater detail in current understanding of t-tubule biology (Gu et al., 2002; Hayashi et al., 2009; Pinali et al., 2013; Crossman et al., 2017; Frisk et al., 2021).
A schematic overview of cardiomyocyte t-tubule structure is provided in Figure 1. Originally named transverse tubules, the majority of these structures are indeed oriented transversely across the cell, in a well-organized network along the Z-lines (Lindner, 1957; McNutt, 1975; Kostin et al., 1998). However, a fraction of tubules is positioned along the cardiomyocyte’s longitudinal axis, extending into the A-band of the sarcomere (Simpson, 1965; Forssmann and Girardier, 1970; Sperelakis and Rubio, 1971; Forbes and Sperelakis, 1976; Soeller and Cannell, 1999; Pinali et al., 2013). In an effort to accurately describe this arrangement, and include both populations of t-tubules, some have referred to the overall network as the transverse-axial tubule system (TATS) (Sperelakis and Rubio, 1971; Forbes and Sperelakis, 1976). In rat cardiomyocytes, the proportions of transverse and longitudinal t-tubules are estimated at 60 and 40%, respectively (Soeller and Cannell, 1999), although as noted below there are large species differences. In addition to variation in overall orientation, t-tubules also exhibit non-uniform branching, tortuosity, bulges, and folds (Jayasinghe et al., 2012; Hong et al., 2014), and the lumen diameter of the t-tubules varies from 20 to 450 nm (Soeller and Cannell, 1999). These narrow dimensions of the t-tubules are believed to restrict solute movement compared to the extracellular environment, with important physiological consequences (Swift et al., 2006; Kong et al., 2018b; Uchida and Lopatin, 2018). However, a recent publication demonstrated that there is significant deformation of t-tubules throughout the cardiomyocyte contractile cycle. During contraction, extracellular solution is thus actively pumped in and out of the t-tubular compartment in a rate-dependent manner, counteracting geometrical diffusion constraints, and representing a new form of t-tubule functional autoregulation which has been hitherto underappreciated (Rog-Zielinska et al., 2021b).
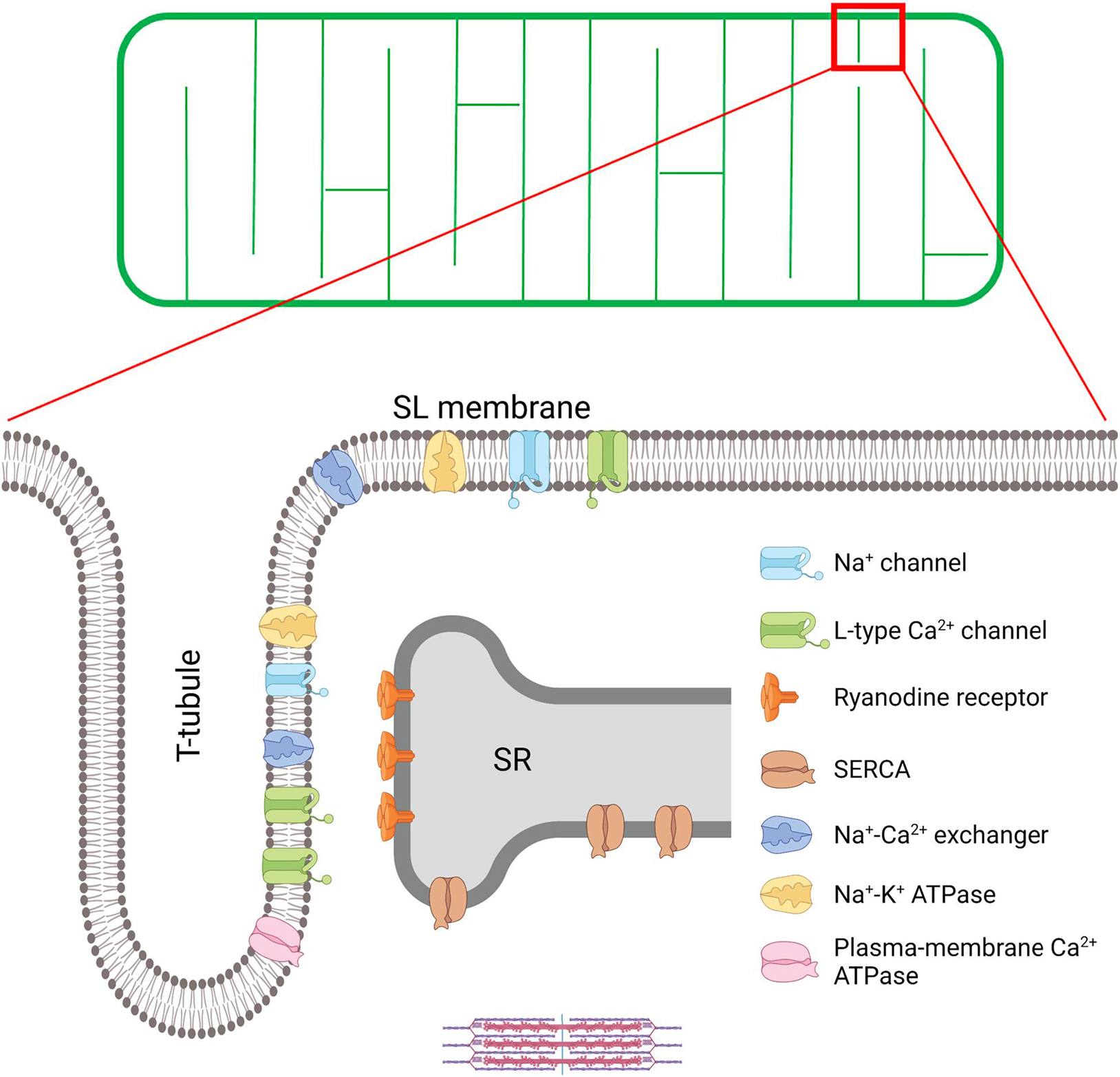
Figure 1. T-tubule structure and key proteins involved in excitation-contraction (EC) coupling in the cardiomyocyte. A schematic overview of the t-tubule network is provided in the upper panel, while an enlargement of the indicated region is provided below to illustrate positioning of EC coupling proteins. EC coupling is initiated as Na+ channels are opened, and the cell membrane depolarizes during the action potential. This depolarization triggers the opening of voltage-gated L-type Ca2+-channels (LTCCs) in the t-tubules, and subsequent Ca2+-induced Ca2+ release from the SR through the opening of Ryanodine Receptors (RyRs). This process occurs at specialized junctions called dyads, where LTCCs and RyRs are in close proximity. After released Ca2+ binds to the myofilaments to trigger contraction, Ca2+ is recycled into the SR by the sarco-endoplasmic reticulum ATPase (SERCA), and removed from the cell by the Na+-Ca2+ exchanger (NCX) and the plasma-membrane Ca2+ ATPase. NCX activity is critically regulated by Na+ levels, set by the Na+ channel and the Na+-K+ ATPase within t-tubules. Created with BioRender.com.
There are considerable species differences in t-tubule density and organization (Soeller and Cannell, 1999). Indeed, depending on species, t-tubules constitute 0.8–3.6% of the total cell volume, and 21–64% of the total sarcolemma (Stewart and Page, 1978; Severs et al., 1985; Bers, 2002; Hayashi et al., 2009). In smaller species with higher heart rates (mouse and rat), t-tubule arrangement is the most complex, with denser, more branched and thinner structures than large mammals (rabbit, pig, and human) (Soeller and Cannell, 1999; Cannell et al., 2006; Savio-Galimberti et al., 2008; Jayasinghe et al., 2012; Kong et al., 2018b). Mouse cardiomyocytes also exhibit smaller t-tubule openings, and thus it has been proposed that t-tubular solute movement is more restricted in these cells than in larger species (Kong et al., 2018b). T-tubule topology also varies across the different heart chambers. While t-tubule structure is reported to be similarly well-organized in both left and right ventricle (Tidball et al., 1991; Chen et al., 2013; Guo et al., 2013), there is greater variance reported in the atria. Although t-tubules have been identified in atrial myocytes in both small (rat) (Frisk et al., 2014) and larger mammals (pig, dog, sheep, cow, horse, and human) (Dibb et al., 2009; Lenaerts et al., 2009; Richards et al., 2011; Frisk et al., 2014; Arora et al., 2017), studies in rat and pig indicated that only a minority of atrial cells contain tubular structures (Frisk et al., 2014; Gadeberg et al., 2016). When they are present in atrial cells, t-tubules are considerably less developed than those present in ventricular myocytes, with generally lower density and often a more longitudinal orientation (Dibb et al., 2009; Lenaerts et al., 2009; Smyrnias et al., 2010; Richards et al., 2011; Frisk et al., 2014; Glukhov et al., 2015; Gadeberg et al., 2016; Arora et al., 2017). Notably, t-tubule density is reported to be higher in the atria’s epicardium than in the endocardium (Frisk et al., 2014). Although not yet closely examined, there may also be intra-chamber variability in t-tubule structure, as lower t-tubule density has been reported in the apex than other regions of the left ventricle (Wright et al., 2018; Frisk et al., 2021).
As central players in EC-coupling, t-tubule structure is closely linked to contractile function. T-tubules carry the action potential (AP) deep into the cell interior, and the resulting depolarization of the t-tubular membrane leads to opening of voltage-gated L-type Ca2+ channels (LTCCs, Cav1.2; see Figure 1). This influx of Ca2+ from the extracellular space triggers a larger amount of Ca2+ release from the SR through ryanodine receptors (RyRs); a process called Ca2+-induced Ca2+ release (CICR) (Fabiato, 1983; Bers, 2002). Binding of the released Ca2+ to the myofilaments results in the contraction of the cell, and thus the whole heart. Thereafter, relaxation occurs as Ca2+ is recycled into the SR by the SR Ca2+-ATPase (SERCA), or extruded from the cell by the Na+/Ca2+ exchanger (NCX), and to a lesser extent, the sarcolemmal Ca2+ ATPase (Louch et al., 2012).
The presence of a dense and well-organized t-tubule network ensures synchronous Ca2+ release across the cell. This is particularly apparent in mouse and rat ventricular cardiomyocytes, due to the robust presence of these structures (Heinzel et al., 2002; Louch et al., 2006; Song et al., 2006). However, in larger animals, such as pig, less synchronous Ca2+ release has been linked to the lower cardiomyocyte t-tubule density (Heinzel et al., 2002, 2008). Similarly, in atrial cells with a less developed t-tubule network, the Ca2+ transient is less synchronous, with a wave-like propagation of released Ca2+ from the periphery toward the cell interior (Frisk et al., 2014). This dyssynchronous pattern of Ca2+ release can be reproduced by experimentally detubulating cardiomyocytes (Louch et al., 2004; Brette et al., 2005).
Triggering of Ca2+ release from the SR requires its close alignment with the t-tubule membrane at “dyads” (Figure 1), and close proximity between LTCCs and RyRs (Sun et al., 1995). While EM data have historically indicated that the dyadic cleft measures only ≈12 nm across (Takeshima et al., 2000), recent data have suggested that the true dimensions may be even more narrow (<10 nm), and that artifactual expansion of the cleft could have occurred in earlier work as a result of sample fixation procedures (Rog-Zielinska et al., 2021a). Consistent dyadic dimensions are ensured by junctophilin-2 (JPH2) which stabilizes the membranes, but also functionally interacts with both RyRs and LTCCs (Jiang et al., 2016; Munro et al., 2016; Reynolds et al., 2016; Gross et al., 2021). Insight into the precise positioning of these dyadic proteins has been made possible by new advances in super-resolution microscopy, including STED (Wagner et al., 2012), 3d STORM (Shen et al., 2019), and DNA-PAINT techniques (Jayasinghe et al., 2018; Sheard et al., 2019). For example, nanoscale visualization of RyRs has shown that these proteins are organized into clusters containing an average of 9–14 channels (Baddeley et al., 2009; Jayasinghe et al., 2018; Shen et al., 2019). Tight packing of RyRs within these clusters is thought to synchronize their gating (Marx et al., 2001; Sobie et al., 2006). At the larger scale, RyR clusters that are in close proximity have been predicted to cooperate as a Ca2+ release unit (CRU), or a “super-cluster,” with released Ca2+ jumping between nearby clusters to produce a Ca2+ spark (Sobie et al., 2006; Baddeley et al., 2009; Louch et al., 2013; Kolstad et al., 2018). CRUs are estimated to contain an average of between 18 and 23 RyRs (Shen et al., 2019), and this composition is a key factor in determining Ca2+ spark frequency and amplitude (Xie et al., 2019).
On the t-tubule side of the dyad, LTCCs are localized opposite RyR clusters. LTCCs also form clusters, with sizes estimated to be about 67% of those reported for RyRs (Scriven et al., 2010). Exciting new data indicate that the interaction between LTCCs facilitates Ca2+ influx via coupled gating of the channels (Dixon et al., 2015). Recently, Ito and colleagues found that β-adrenergic receptor activation augments LTCC abundance and promotes enhanced channel interaction, thereby amplifying Ca2+ influx via a protein kinase A-dependent pathway (Ito et al., 2019). NCX localization in dyads remains a controversial topic (Scriven et al., 2000; Thomas et al., 2003), but it is reported that at least a fraction of NCX is colocalized with RyRs (Mohler et al., 2005; Jayasinghe et al., 2009; Schulson et al., 2011; Wang et al., 2014; Figure 1). While the main function of NCX is extrusion of Ca2+ during relaxation (Louch et al., 2012), numerous reports have suggested that Ca2+ entry via NCX can also act in “reverse mode” to trigger SR Ca2+ release (Sipido et al., 1997; Henderson et al., 2004; Lines et al., 2006; Larbig et al., 2010). This mechanism is proposed to follow Na+ influx during the rising phase of the action potential, which creates driving force for NCX-mediated Ca2+ entry.
Besides ion channels, such as the LTCC and NCX, the t-tubular membrane is also enriched in molecules which critically regulate their structure and function. Notable examples are amphiphysin-2 (BIN1), junctophilin-2 (JPH2), and caveolin-3 (Cav-3) (Figure 1), which are involved in functions spanning t-tubule growth, t-tubule microdomain formation, and regulation of LTCC and RyR localization and activity. A detailed discussion of these important t-tubule regulators will be made in the following sections.
The process of t-tubule development varies between species (Ziman et al., 2010; Louch et al., 2015; Jones et al., 2018). In species such as cow, guinea pig, sheep and human, t-tubules have been found in the fetal stages of development (Forbes and Sperelakis, 1976; Sheldon et al., 1976; Forsgren and Thornell, 1981; Brook et al., 1983; Kim et al., 1992). A recent study in sheep specified that t-tubule development started at a late fetal stage (108 days out of 145 days), with further maturation occurring after birth (Munro and Soeller, 2016). In contrast, t-tubules only appear postnatally in mouse, rat, rabbit, and cat cardiomyocytes (Hirakow et al., 1980; Hoerter et al., 1981; Gotoh, 1983; Ziman et al., 2010; Hamaguchi et al., 2013; Lipsett et al., 2019). In rats, t-tubules appear gradually and become visible around 4–9 days after birth (Ziman et al., 2010; Mackova et al., 2017; Lipsett et al., 2019), with an initial appearance at the cell surface as sparse openings along Z-spines, with a rudimentary internal structure that is largely longitudinal in orientation (Figure 2). During further development, t-tubule density strikingly increases, with a re-orientation into a clear striated pattern at roughly 20 days of age, and a progressively more dominant presence of transverse over longitudinal elements (Ziman et al., 2010; Louch et al., 2015; Mackova et al., 2017; Figure 2). This process continues until surprisingly late stages of postnatal development (Oyehaug et al., 2013).
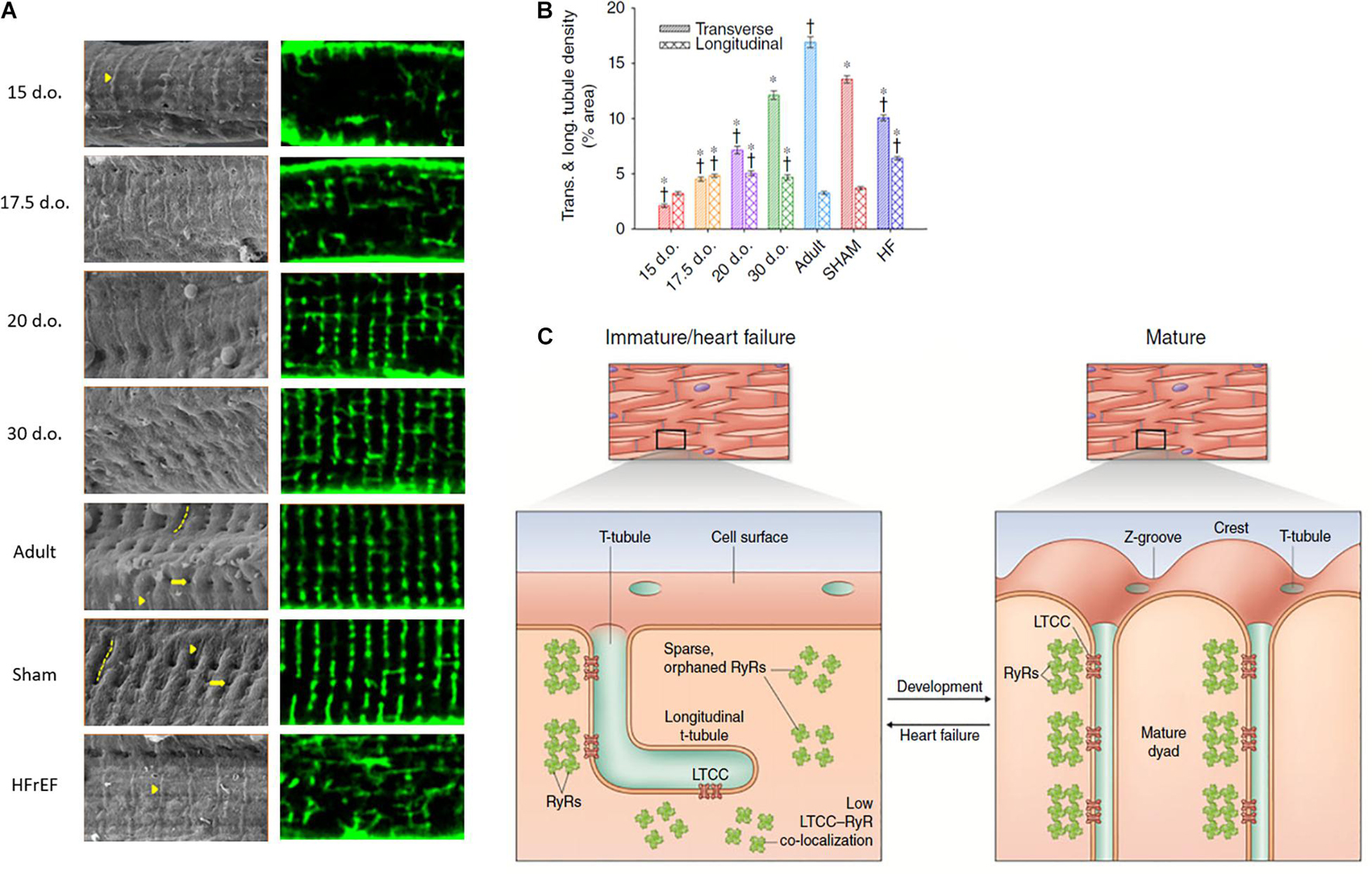
Figure 2. Changes in cardiomyocyte surface topography and dyadic organization in the developing and HFrEF rat heart. (A) Left panels: Scanning electron micrographs show the appearance of Z-spines (arrowheads) from an early time point during development and their re-appearance during post-infarction HFrEF. Z-grooves (dashed lines) and t-tubule openings (arrows) appear gradually during development and are lost in HFrEF. Right panels: temporally matched confocal captures of internal t-tubules (di-8-ANEPPS staining). (B) Quantification of these t-tubule signals illustrates an organizational shift from a predominantly longitudinal to largely transverse orientation during maturation, but reversion to an immature phenotype during HFrEF. (C) Structural parallels between developing and failing myocytes include not only t-tubule structure, but also similarly sparse arrangements of RyRs that exhibit low co-localization with LTCCs. d.o., days old. *P < 0.05 vs. adult in same category, †P < 0.05 vs. SHAM in same category. Reproduced from Lipsett et al. (2019) with permission.
It is noteworthy that the heart exhibits robust contractile function at early embryonic stages of development in the absence of t-tubules (Brand, 2003). Since these embryonic myocytes are quite thin, EC coupling is sufficiently supported by U-shaped propagation of the Ca2+ transient from the surface of the cells to the interior (Rapila et al., 2008; Korhonen et al., 2010), reminiscent of Ca2+ release patterns observed in detubulated myocytes described above. This pattern of Ca2+ release is initiated at the cell surface, where LTCCs and RyRs are co-localized before birth (Snopko et al., 2008), and propagated by RyRs assembled on SR extensions positioned at intervals of 2 μm (de Diego et al., 2008; Rapila et al., 2008). These internal RyR clusters continue to be assembled along Z-lines as t-tubules arrive, and dyadic pairings with LTCCs are quickly formed (Ziman et al., 2010; Lipsett et al., 2019). Thus, dyadic functionality is rapidly attained once these structures are formed, allowing progressive increases in Ca2+ release efficiency with further maturation (Ziman et al., 2010; Lipsett et al., 2019).
Recent data suggest that the aging heart may exhibit reversal of the processes of dyadic assembly that take place during development. Aging is associated with reduced cardiomyocyte contractility, especially in males (Grandy and Howlett, 2006; Feridooni et al., 2015), and these changes are linked to reduction in cardiomyocyte t-tubule density (Kong et al., 2018a; Lyu et al., 2021). An accompanying reduction of t-tubule Ca2+ current density during aging has also been linked to loss of a cav-3-dependent mechanism that augments t-tubular Ca2+ current density (Kong et al., 2018a).
Heart failure (HF) is a leading cause of death worldwide. It is therefore of great importance to understand the different mechanisms underlying this condition, to facilitate discovery of novel treatment targets. HF can be divided into two main entities; HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). HFrEF describes a state where the cardiac muscle is unable to contract adequately, leaving the heart unable to meet the body’s oxygen demand. This state is associated with ventricular dilation, thinning of ventricular walls and high wall stress. HFpEF, on the contrary, is predominantly linked to cardiac hypertrophy, wall thickening, and maintained wall stress (Pieske et al., 2019). Although, ejection fraction is preserved in this condition, compromised cardiac chamber relaxation and filling yields impaired cardiac output. Hence, both HF entities are severe diseases with 2-year mortality rates between 14 and 19% (Lam et al., 2018).
HFrEF mechanisms have been extensively investigated, and numerous studies in a range of species and disease etiologies have linked disease progression to alterations in t-tubule structure and function (Jones et al., 2018; Figure 3). Typically this remodeling includes reduced t-tubule density (Maron et al., 1975; Heinzel et al., 2008; Swift et al., 2008; Wei et al., 2010; Wu et al., 2011, 2012; Xie et al., 2012; Ibrahim et al., 2013; Frisk et al., 2021; Yamakawa et al., 2021), including a lower density of transversely oriented tubules (Louch et al., 2006, 2013; Song et al., 2006; Swift et al., 2008; Ibrahim et al., 2010, 2013; Wei et al., 2010; van Oort et al., 2011; Wu et al., 2011; Lyon et al., 2012; Wagner et al., 2012; Xie et al., 2012; Oyehaug et al., 2013; Frisk et al., 2021). Based on this finding, it has often been claimed that t-tubules are “lost” in HFrEF. However, recent work has indicated that the t-tubule frame may in fact be maintained in this condition, although cell size increases (Frisk et al., 2021). Thus, the observed decrease in t-tubule density is suggested to reflect a lack of adaptive remodeling to meet the developing cellular hypertrophy, rather than a degradation of t-tubule structure per se. Further complicating the picture is the observation that t-tubule remodeling in HFrEF frequently includes an increased fraction of longitudinally oriented tubules (Kaprielian et al., 2000; Louch et al., 2006, 2013; Song et al., 2006; Sachse et al., 2012; Swift et al., 2012; Wagner et al., 2012; Oyehaug et al., 2013; Frisk et al., 2016, 2021), t-tubule dilation (Maron et al., 1975; Schaper et al., 1991; Kostin et al., 1998; Kaprielian et al., 2000; Lyon et al., 2012; Pinali et al., 2017), loss of tubule openings at the cell surface (Lyon et al., 2009; Lipsett et al., 2019), and the appearance of broad t-tubule “sheets” (Pinali et al., 2017; Seidel et al., 2017a; Fiegle et al., 2020). The animal models employed in these examinations have included myocardial infarction (Louch et al., 2006; Swift et al., 2008; Lyon et al., 2009; Wagner et al., 2012; Oyehaug et al., 2013; Crocini et al., 2014; Frisk et al., 2016, 2021; Sanchez-Alonso et al., 2016; Schobesberger et al., 2017; Lipsett et al., 2019), aortic stenosis (Wei et al., 2010; Ibrahim and Terracciano, 2013; Pinali et al., 2013; Caldwell et al., 2014), hypertension (Song et al., 2006; Xie et al., 2012; Shah et al., 2014; Singh et al., 2017), tachycardia (He et al., 2001; Balijepalli et al., 2003; Caldwell et al., 2014), and diabetes (Stolen et al., 2009; Ward and Crossman, 2014). Similar disruption of t-tubule structure has also been reported during chronic ischemia (Heinzel et al., 2008) and atrial fibrillation (Lenaerts et al., 2009). Notably, t-tubule remodeling is not limited to the left ventricle, as comparable alterations have been reported in the right ventricle (Wei et al., 2010; Xie et al., 2012) and atrial cells as well (Melnyk et al., 2002; Dibb et al., 2009; Lenaerts et al., 2009; Wakili et al., 2010; Richards et al., 2011). In disease models with a non-uniform myocardial affliction, such as myocardial infarction, there are reports that t-tubule remodeling exhibits a spatial gradient, with the most marked changes occurring proximal to the infarcted myocardium (Frisk et al., 2016; Pinali et al., 2017; Wang et al., 2018). Perhaps most importantly, examinations in human tissue (Crossman et al., 2015; Frisk et al., 2021) broadly concur with the findings described in animal disease models, as reviewed in Louch et al. (2010b), Ibrahim et al. (2011), Guo et al. (2013), and Hong and Shaw (2017).
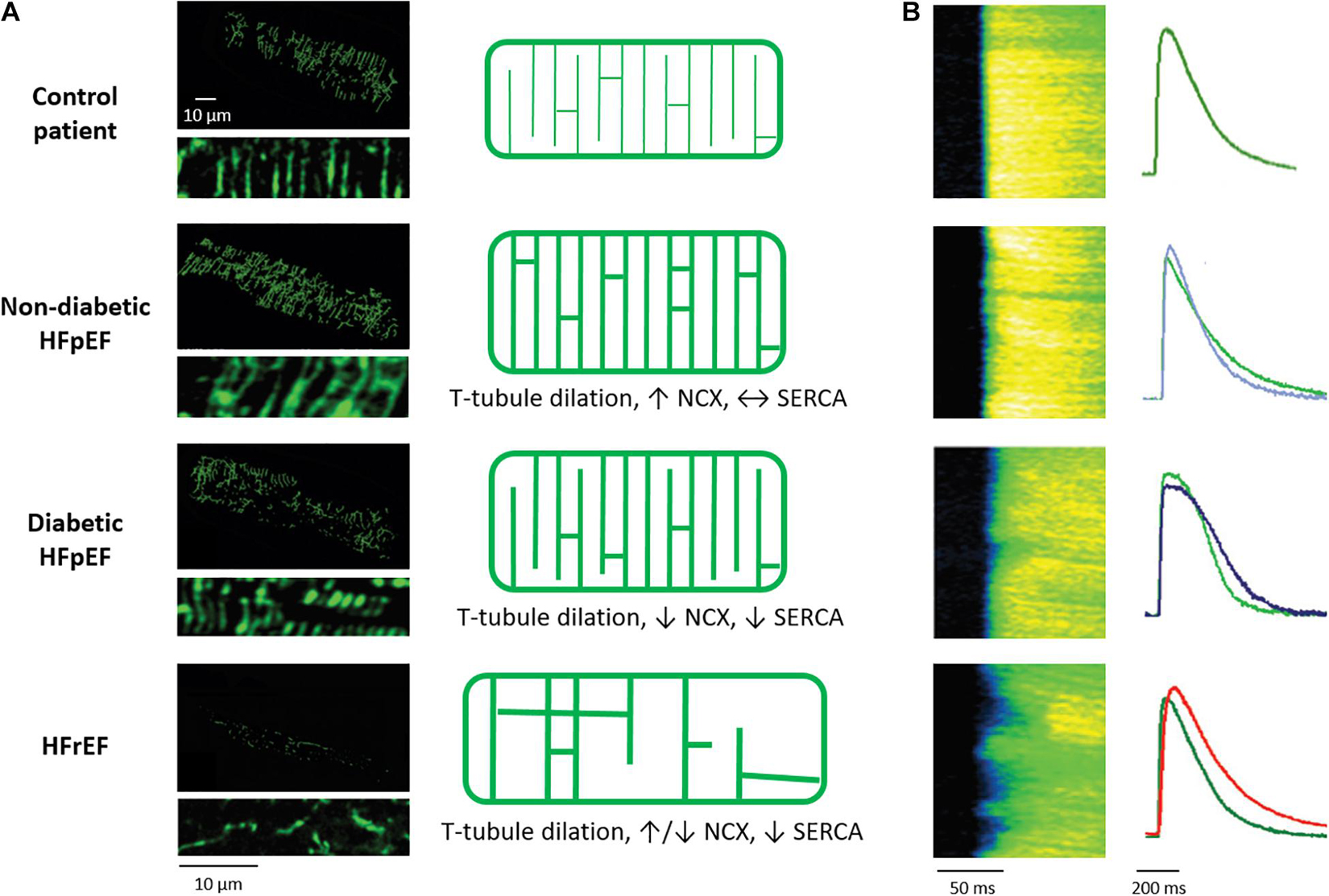
Figure 3. Alterations in cardiomyocyte t-tubule structure and Ca2+ release in different HF entities. (A) Left panels: 3D reconstructions and 2D zoom-ins of t-tubules imaged in human cardiac biopsies (wheat germ agglutinin staining). Healthy individuals are compared with HFpEF patients, with and without diabetes mellitus, and HFrEF patients. As illustrated in the schematics (right panels), t-tubule density is unchanged in diabetic HFpEF, increased in non-diabetic HFpEF, and decreased in HFrEF. In all HF entities, t-tubules dilate. However, whereas t-tubule remodeling is accompanied by increased NCX and SERCA function in non-diabetic HFpEF, Ca2+ removal by these proteins is decreased in diabetic HFpEF and HFrEF. (B) T-tubule disruption promotes dyssynchronous and slowed Ca2+ release in rats with HFrEF but not HFpEF, while slowed Ca2+ removal promotes diastolic dysfunction in diabetic HFpEF and HFrEF. For each disease state, representative Ca2+ transients (right panels) are presented with comparison to a control cardiomyocyte (green) for the individual rat models. Micrographs and Ca2+ transients are reproduced from Frisk et al. (2021) with permission.
T-tubule disorganization during HFrEF causes spatial dissociation between key players in EC coupling, most notably LTCCs and RyRs. This rearrangement leads to the formation of “orphaned” RyRs which are no longer co-localized with t-tubules (Louch et al., 2006; Song et al., 2006; Figure 2C). A resulting de-synchronization of the Ca2+ transient occurs, as Ca2+ release from orphaned RyRs can only be triggered after diffusion of Ca2+ from intact dyads (He et al., 2001; Balijepalli et al., 2003; Cannell et al., 2006; Kemi et al., 2011; Xie et al., 2012; Louch et al., 2013). Ca2+ release dyssynchrony is further augmented in failing cells by reduced SR Ca2+ content (Oyehaug et al., 2013). The net result of this de-synchronized Ca2+ transient is slowed and reduced magnitude of contraction; a hallmark of HFrEF (Louch et al., 2010b).
Impaired efficiency of CICR is not only linked to reorganized t-tubules and orphaned RyRs, but also a reduced ability of t-tubules to trigger Ca2+ release where they are present. This deficit is at least partly attributable to reduced t-tubular LTCC current (Bryant et al., 2015; Sanchez-Alonso et al., 2016; Lipsett et al., 2019). Reduced L-type current has, in turn, been linked to altered localization of LTCCs, but also changes in AP configuration, that prevent optimal channel recruitment (Sah et al., 2002; Louch et al., 2010a). Interesting data have suggested that there is also impairment of AP propagation into some t-tubules in failing cardiomyocytes, resulting in desynchronization of Ca2+ release (Crocini et al., 2014). Continuing work has suggested that this loss of electrical activation may be caused by constriction of t-tubule geometry in this condition (Scardigli et al., 2017; Uchida and Lopatin, 2018).
Recent work has indicated that loss of t-tubule functionality during HFrEF additionally stems from alterations on the SR side of the dyad. We observed “dispersion” of RyRs in MI-induced HFrEF, characterized by break-up of RyR clusters into smaller sub-clusters (Kolstad et al., 2018; Figure 2C). Functionally, we observed that this dispersion was associated with increased “silent” Ca2+ leak, not visible as sparks. Furthermore, we found that larger multi-cluster CRUs exhibited low fidelity Ca2+ spark generation. When successfully triggered, sparks in failing cells displayed slow kinetics as Ca2+ spread across dispersed CRUs. Previous work performed by us and others demonstrated that sparks occur almost exclusively at t-tubules (Brette et al., 2005; Meethal et al., 2007; Louch et al., 2013), suggesting that CRU dispersion and slow sparks solely occur at intact dyads. Thus, t-tubule and CRU remodeling may occur independently. Ultimately, it seems likely that t-tubule and CRU disruption have additive effects, yielding even more marked de-synchronization and slower SR Ca2+ release (Kolstad et al., 2018).
Importantly, not all dyadic changes during HFrEF are detrimental. For example, at early stages of disease, longitudinal tubules appear before transverse elements have disappeared (Louch et al., 2006). LTCCs are co-localized with RyRs at longitudinal dyads, allowing these structures to actively release Ca2+ (Lipsett et al., 2019). Thus, their early growth following the initiating insult (for example myocardial infarction or induction of aortic banding) is thought to be compensatory (Mork et al., 2007). At later time points, contractile function declines as transverse elements are lost, supporting that this latter type of remodeling is a direct cause of HFrEF (Wei et al., 2010; Shah et al., 2014).
The above discussion has highlighted convincing evidence that disrupted t-tubule structure and Ca2+ homeostasis are a root cause of HFrEF. However, approximately 50% of HF patients exhibit HFpEF (Lekavich et al., 2015), and the mechanisms underlying this disease entity are merely beginning to be unraveled. Recent evidence from our group indicates that a distinct form of subcellular remodeling occurs in this condition. Using patient biopsies we observed that, conversely to HFrEF, cardiomyocytes from HFpEF individuals exhibited increased t-tubule density (Figure 3A), and a positive correlation between t-tubule density and the severity of in vivo diastolic dysfunction (Frisk et al., 2021). Higher t-tubule densities resulted from a combination of t-tubule dilation and proliferation, consistent with adaptive remodeling. These data contribute to growing evidence that t-tubules have compensatory capacity, as similar increases in t-tubule density have been reported during physiological hypertrophy following exercise training (Kemi et al., 2011). In this sense, t-tubule growth during concentric, non-dilatory hypertrophy may be viewed as a continuation of processes set in motion during development. Importantly, functional data support such a compensatory role, as cardiomyocytes obtained from hypertensive and ischemic rat models of HFpEF revealed maintained Ca2+ release and reuptake despite decreased SERCA protein levels (Figure 3B). Marked phosphorylation of the SERCA inhibitory protein phospholamban was identified as the key motif for unchanged diastolic Ca2+ homeostasis (Frisk et al., 2021). Other studies examining Dahl salt sensitive rats and aorta banded rats with impaired cardiac relaxation have reported similar maintenance of t-tubule structure and Ca2+ homeostasis (Roe et al., 2017; Curl et al., 2018; Kilfoil et al., 2020).
HFpEF includes a set of patients with diverse etiologies, including those suffering from cardiac ischemia or hypertension, as noted above, but also diabetes (Lekavich et al., 2015). In contrast to other disease etiologies, diabetic HFpEF appears to negatively affect both t-tubule integrity and Ca2+ handling. Indeed, we observed that HFpEF patients and animal models with diabetes exhibited less compensatory t-tubule growth than their non-diabetic counterparts, as t-tubule density was merely maintained (Frisk et al., 2021; Figure 3A). We hypothesize that this reduced capacity for adaptive remodeling may be related to abnormal caveolin-3 and/or phospoinositol-3 kinase expression and activity, since cholesterol, fatty acid and phosphoinositide composition of the sarcolemmal membrane are altered during diabetes (Russell et al., 2017). As discussed later, decreased autophagy might also play a role (Hsu et al., 2016; Seidel et al., 2019). Despite less adaptive t-tubule remodeling, systolic Ca2+ release was observed to be quite well-maintained in diabetic HFpEF. However, diastolic dysfunction in this condition was linked to impairment of diastolic Ca2+ homeostasis, caused by reduced activity of both SERCA and NCX (Frisk et al., 2021; Figure 3B). Accumulating evidence has indicated that the reduced NCX activity in diabetic HFpEF may be related to elevated cytosolic Na+ levels, which reduce the driving force for t-tubular Ca2+ removal. This Na+ accumulation has in turn been linked to increased activity of the Na+-glucose co-transporter 1 (Lambert et al., 2015; Frisk et al., 2021) and/or the Na+-H+-exchanger (Jaquenod De Giusti et al., 2019). Thus, there appears to be impairment of diastolic Ca2+ homeostasis in diabetic HFpEF, which includes detrimental alterations in t-tubule function.
While investigations of HFpEF mechanisms are ongoing, the above studies present an emerging view that t-tubule structure remains adequate to maintain near-normal SR Ca2+ release in this condition. This striking difference from the cardiomyocyte phenotype linked to HFrEF is perhaps not surprising. In addition to the aforementioned differences in chamber remodeling, HFrEF is primarily associated with activation of the renin-angiotensin- aldosterone system and stretch-mediated signaling pathways, while inflammation, endothelial dysfunction, and alteration of the extracellular matrix are key components of HFpEF (Paulus and Tschope, 2013). Perhaps most importantly, established therapies for HFrEF, such as neurohumoral blockage, have proven ineffective for treatment of HFpEF (Paulus and Tschope, 2013). Thus, there appears to be a growing consensus that management of HFpEF will require novel strategies, and that these therapies may be best directed at non-cardiomyocyte alterations occurring in the endothelium and/or extracellular matrix.
Alterations in t-tubule structure and function during HF not only impact systolic and diastolic function, but also have complex consequences for arrhythmia generation (Orchard et al., 2013). HFrEF is frequently associated with increased RyR “leak,” which can be exacerbated by SR Ca2+ overload during β-adrenergic stimulation (Dridi et al., 2020). Extrusion of this spontaneously released Ca2+ by NCX triggers an inward, depolarizing current. If this current occurs during the downstroke of the action potential, an early afterdepolarization (EAD) results, while delayed afterdepolarizations (DADs) are triggered by spontaneous Ca2+ release between beats (Orchard et al., 2013). How would t-tubule remodeling during HFrEF affect these processes? As noted above, t-tubule reorganization in this disease results in the formation of orphaned RyRs. However, Ca2+ sparks occur mostly at intact dyads where t-tubules are present, and only to a much lesser degree at orphaned RyRs (Brette et al., 2005; Meethal et al., 2007; Louch et al., 2013). In the event that spontaneous release does occur at an orphaned RyR, an EAD or DAD would be less likely to occur because NCX is more distally located (Biesmans et al., 2011; Edwards and Louch, 2017). On the other hand, propagation of spontaneously released Ca2+ as a wave is more likely to occur, since distally localized NCX does not draw Ca2+ away from the wavefront. Recent data have painted a more complex picture that considers also the relationship between Ca2+ release and LTCC activity. Less Ca2+-dependent inactivation of LTCCs, due to displaced (orphaned) RyRs, and resulting increased Ca2+ influx has been predicted to contribute to SR Ca2+ overload (Shiferaw et al., 2020). Thus, t-tubule remodeling may make spontaneous Ca2+ release more likely in HFrEF, while at the same time attenuating the link between these events and EADs/DADs.
T-tubule remodeling also has complex implications for EADs triggered by channel reopening. Phase-2 EADs initiated by LTCCs may be favored in HFrEF, as L-type current is redistributed to the cell surface (Sanchez-Alonso et al., 2016; Loucks et al., 2018). This increase in LTCC open probability has been linked to calcium-calmodulin kinase II-dependent phosphorylation of the channel, which augments window current (Sanchez-Alonso et al., 2016), but also phosphorylation by protein kinase A, based on the presence of β2 adrenergic receptors and phosphodiesterases (Loucks et al., 2018). Conversely, loss of LTCCs and NCX in t-tubules, and their respective currents, may shorten the AP making phase-2 EADs less likely, but Na+ channel re-activation and phase-3 EADs more likely (Orchard et al., 2013; Edwards and Louch, 2017). A shorter AP, and accompanying reduction in refractory period, also increases the chance of re-entrant arrhythmia (Herring et al., 2019). In addition to changes in ion channel expression and regulation, Hong and colleagues have provided evidence that alterations in the geometry of the t-tubules themselves can promote the occurrence of EADs (Hong et al., 2014). They observed that intricate folding of the t-tubule lumen creates a microenvironment with slowed ion diffusion. Upon loss of these membrane folds, more rapid exchange of ions between the t-tubule lumen and extracellular space was linked to prolonged action potential duration, and the generation of EADs and arrhythmia (Hong et al., 2014). This interesting observation raises the possibility that t-tubule dilation observed during HFrEF and HFpEF (Figure 3A) may also have arrhythmogenic consequences.
Finally, growing evidence supports that t-tubule remodeling during HFrEF can promote pro-arrhythmic alternans. Two mathematical modeling studies have linked spatially discordant alternans to the increased fraction of orphaned RyRs that yield a phase of secondary release following Ca2+ diffusion from intact dyads (Li et al., 2012; Song et al., 2018). Jiang et al. (2014) suggested that there is, however, an intermediate range of t-tubule remodeling where this phenomenon occurs. Thus, failing ventricular myocytes and healthy atrial myocytes are likely susceptible to this mechanism of alternans, while failing atrial myocytes may be less prone due to their very low t-tubule densities (Jiang et al., 2014). Importantly, since Ca2+-voltage coupling is an additional determinant of alternans generation (Gaeta et al., 2009), the susceptibility of failing myocytes to alternans is expected to be complicated by changes in AP configuration. Interrogating and integrating these mechanisms will be an important topic of future work.
The above sections have highlighted a growing consensus that t-tubule remodeling is a key trigger for reduced contractility and arrhythmia in HFrEF. This understanding has led to a concerted effort to reveal the processes that control t-tubule structure in both health and disease. We review the emerging findings from this work in the following sections.
Accumulating evidence has indicated that ventricular workload plays a key role in regulating t-tubule structure. Pioneering work by the Terraciano group was the first to indicate that the loss of t-tubules during HFrEF could be directly triggered by the elevated ventricular workload (reviewed in Ibrahim and Terracciano, 2013). In their work, failing hearts were unloaded by heterotopic transplantation into healthy animals, resulting in rescue of t-tubular structure (Ibrahim et al., 2012). Similar approaches to unloading of the failing heart either pharmacologically (Chen et al., 2012; Xie et al., 2012; Huang et al., 2016) or via resynchronization therapy (Sachse et al., 2012; Lichter et al., 2014) have similarly shown improved cardiac function linked to restoration of t-tubules. Interestingly, unloading of healthy hearts promoted loss of t-tubules (Ibrahim et al., 2012). These findings support the notion that there is an optimal range of loads necessary to maintain t-tubule integrity (Ibrahim et al., 2015).
Workload is a broad term, and ongoing efforts are aimed at revealing the precise critical mechanical stimuli which control t-tubule structure. The dilated, thin-walled ventricle leads to elevated ventricular wall stress in HFrEF, and this appears to be a critical mechanical signal. Indeed, when we examined regional differences across the post-infarction rat heart we observed a correlation between wall stress and t-tubule disruption. This remodeling included significant t-tubule disruption near the infarct, where thinning of the myocardium markedly increases wall stress, together with locally impaired Ca2+ homeostasis and in vivo systolic and diastolic dysfunction (Frisk et al., 2016; Roe et al., 2019). Ex vivo studies supported this direct role of wall stress in the regulation of t-tubular structure. A likely implication of this wall stress-t-tubule relationship is in differentiating the pathophysiology of HFrEF and HFpEF, since as noted above, HFpEF is associated with concentric remodeling and maintained wall stress, and maintained t-tubule density (Frisk et al., 2021).
How does mechanical overload lead to the t-tubule loss? What are the associated mechanosensors and signaling pathways? While the precise mechanisms continue to be investigated, titin may be of importance, as it plays a key role in cardiomyocyte mechanotransduction by regulating interactions between the extracellular matrix and sarcomeres (Linke, 2008). Telethonin, or titin cap (TCap), a stretch-sensitive protein located in the Z-disc of cardiomyocytes, also integrates mechanical signals (Ibrahim and Terracciano, 2013; Ibrahim et al., 2013). Indeed, TCap knockout mice exhibited progressive disruption of the t-tubules during development (Ibrahim et al., 2013), and TCap downregulation is reported in HFrEF (Lyon et al., 2012). Furthermore, increased Tcap expression was associated with recovery of t-tubules during reverse remodeling induced by SERCA2a gene therapy (Lyon et al., 2012). While further studies are needed to examine the interplay between myocardial load, expression of Tcap, and t-tubule organization, this protein is viewed as a promising therapeutic target (Roe et al., 2015).
Other studies have linked membrane-mediated mechanosensation by stretch-activated channels, integrins, proteoglycans and angiotensin II type I receptors to activation of a variety of pathways, including MAPK, AKT, calcineurin-NFAT, and microRNA-24 (miR-24) (Lammerding et al., 2004; Dostal et al., 2014). Of these, it should be noted that miR-24 is a member of the miR23a-27a-24-2 cluster, which is upregulated in HF (Xu et al., 2012). Xu et al. (2012) observed that overexpression of miR-24 disrupted dyadic structure, and reduced CICR efficiency. This group also found that miR-24 suppression protected against HFrEF progression (Li et al., 2013).
As Jones et al. (2018) discussed in their recent review article, elevated workload and wall stress regulate not only cardiomyocyte remodeling, but also the extracellular matrix. Interestingly, recent evidence indicates that fibrosis occurs within t-tubules in HFrEF, but not in HFpEF where wall stress is maintained (Crossman et al., 2017; Frisk et al., 2021). Crossman and colleagues in fact detected fibroblast filopodia within the t-tubules of HFrEF hearts, suggesting a mechanism for local collagen production (Crossman et al., 2017). Although the consequences of this collagen deposition are unclear, it is proposed that accompanying stiffening of the membrane might mark the t-tubule for degradation (Louch and Nattel, 2017). In apparent support of this hypothesis, it should be noted that in the post-infarcted heart, the most marked t-tubule loss occurs in regions proximal to the infarct, where the fibrosis is most pronounced (Frisk et al., 2016; Seidel et al., 2017b). Fibrosis is expected to impair mechanosignaling, and new data indicate that this might occur within t-tubules themselves during stretch and contraction (Dyachenko et al., 2009; McNary et al., 2011). Such a role is suggestive of t-tubules having a self-regulating feature.
In summary of the above discussion, it appears to be no coincidence that the vast majority of present HFrEF therapies relieve symptoms and delay disease progression by reducing the workload of the heart, and specifically the physical stress placed on the ventricular wall (Cohn, 1996; Tarzia et al., 2016; Berliner and Bauersachs, 2017; Schmitto et al., 2018; Vaidya and Dhamoon, 2019). However, there is great potential for improvement. For example, wall stress and t-tubule structure might be longitudinally tracked as biomarkers, aimed at optimizing ventricular load. Future treatment strategies could also be envisioned which directly inhibit the mechanosensing that signals t-tubule remodeling.
In the quest to unravel signaling pathways involved in triggering subcellular remodeling in HFrEF, newfound attention has turned to the importance of the fetal gene program. Indeed, our group has recently observed striking similarities between developing and diseased cardiomyocytes (Figure 2). Structurally, these similarities include a disorganized and predominantly longitudinal t-tubule configuration in both types of cells. There also appear to be similarities in dyadic configuration, as these junctions are progressively “packed” with LTCCs and RyRs in developing cardiomyocytes and “unpacked” in HFrEF, consistent with a “last in, first out” paradigm (Lipsett et al., 2019). However, even though immature and failing cardiomyocytes share rather similar subcellular structure, functional differences were observed. For example, while dyads were observed to effectively trigger Ca2+ release from early developmental states, impaired release Ca2+ release was noted along t-tubules in failing cells (Lipsett et al., 2019). Thus, it is unlikely that there is a complete reversion of subcellular structure/function to an immature state in this disease.
The above observations suggest that subcellular remodeling during HFrEF progression likely shares signaling mechanisms with the developing heart, which may include fetal genes that are reactivated in disease, and/or adult genes that are suppressed (Rajabi et al., 2007; Louch et al., 2015; Figure 4). Until recently, however, these signals and cardiomyocyte developmental biology in general have been rather under-investigated. This has changed as interest in the generation of stem cell-derived cardiomyocytes and myocardium has come to the forefront, and included an ever-expanding use of human induced pluripotent stem cells (iPSCs). Most early work on iPSC-derived cardiomyocytes generated cells with quite immature features, including a round instead of rod shape, lack of t tubules, poor cooperation of LTCCs and RyRs, and dyssynchronous Ca2+ transients with delayed Ca2+ release occurring in the cell center. More recently, however, improved differentiation has been achieved by treating iPSCs with hormones thought critical for cardiac maturation. Parikh et al. (2017) observed that supplementing the culture medium with thyroid and glucocorticoid hormones, followed by single-cell culture on a Matrigel mattress, stimulated rudimentary t-tubule development and functional coupling of LTCCs and RyRs. Similarly, Huang et al. (2020) documented t-tubule formation close to z-lines in iPSC-derived cardiomyocytes treated with thyroid hormone, dexamethasone, and insulin-like growth factor-1 in both 2D and 3D culture conditions. It is noteworthy that both of these studies included a cellular environment which provided mechanical cues, reinforcing the view expressed above that t-tubule growth and maintenance are highly workload-sensitive. Indeed, even without hormone treatment, Silbernagel et al. (2020) found that reshaping single iPSC-CMs in rectangular 3D-micro-scaffolds triggered t tubule formation and improved Ca2+ handling. Ronaldson-Bouchard et al. (2018) on the other hand, co-cultured hiPSC-CMs with human cardiac fibroblasts, exposed the developing tissue to mechanical load, and gradually increased electrical stimulation. With this method, the authors observed an impressive degree of cellular maturation, including transversely oriented t-tubules, functional Ca2+ handling, and adult-like gene expression profiles. This progress has given credence to the application of iPSC-derived cardiomyocytes for human cardiac disease modeling, drug development, and eventually, engineered cardiac tissue which may be suitable for in vivo implantation.
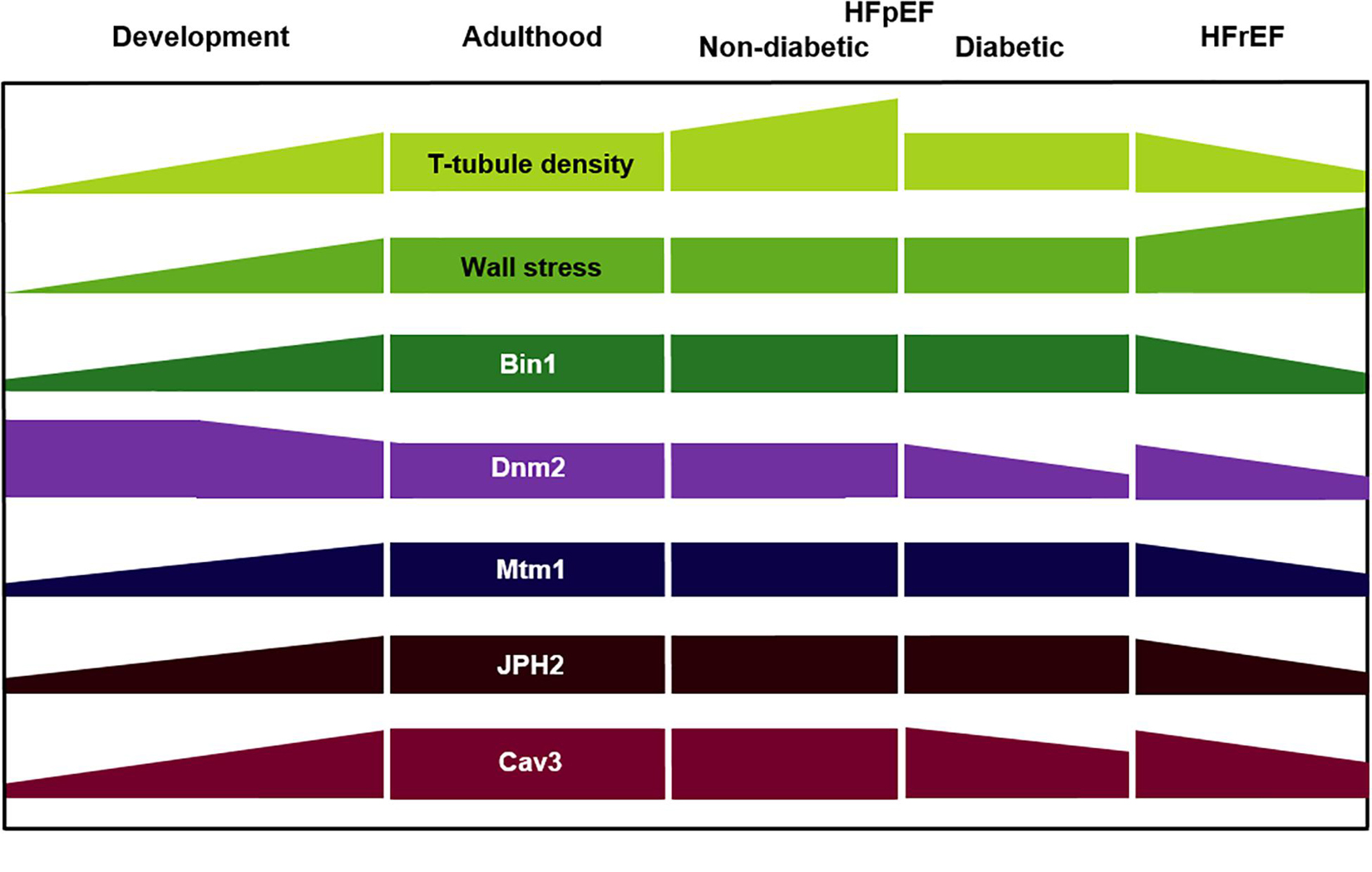
Figure 4. Key regulators of t-tubule structure during development, adulthood, and heart failure of various etiologies. HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; Dnm2, dynamin-2; Mtm1, myotubularin-1; JPH2, junctophilin-2; Cav3, caveolin-3.
In addition to the broader t-tubule regulatory processes described above, several specific proteins have been attributed roles in dyadic structure and function. Of these, BIN1 has received particular attention. This membrane sensing and bending protein has been identified as a key regulator of t-tubule structure assembly and maintenance in both developing and diseased hearts (Lee et al., 2002; Muller et al., 2003; Hong et al., 2010, 2014; see Figure 4). However, understanding of these roles has been complicated by the fact that BIN1 is expressed in several tissue- and species-specific isoforms. Early work suggested that the presence of exon 11 is required to induce tubulogenesis (Lee et al., 2002; Kojima et al., 2004), and exon 11-containing BIN1 isoforms expressed in rat, sheep, and human myocardium have indeed been observed to induce t-tubule formation (Caldwell et al., 2014; De La Mata et al., 2019; Lawless et al., 2019; Li L. L. et al., 2020). However, other studies have reported that exon 11 is dispensable for t-tubule development (Prokic et al., 2020), and shown that the mouse heart expresses four BIN1 splice variants which do not contain this motif (BIN1, BIN1+13, BIN1+17, and BIN1+13+17) (Forbes and Sperelakis, 1976; Hong et al., 2014). In addition to gross t-tubule biogenesis, there may also be isoform-specific roles in determining their fine structure and function. Hong and colleagues reported that isoform BIN1+13+17 creates microdomains by folding the tubular inner membrane (Hong et al., 2014), and attracts phosphorylated RyRs on the SR membrane (Fu et al., 2016). Since BIN1 also anchors microtubules transporting LTCCs, in a process known as targeted delivery (Hong et al., 2012b; De La Mata et al., 2019), an emerging view is that this protein serves as a master regulator of dyadic structure and function. Consistent with this paradigm, genetic knockout of BIN1 has been shown to be embryonically lethal (Muller et al., 2003).
Accumulating data have identified BIN1 as a culprit in cardiac pathology, with its downregulation linked to decreased t-tubule density in HFrEF (Caldwell et al., 2014; Hong et al., 2014; Figure 4). An associated reduction in t-tubule folding has also been predicted to augment diffusion of ions within individual t-tubules which, as noted above, may predispose for cardiac arrhythmias (Hong et al., 2014). Furthermore, given BIN1’s proposed role in trafficking of LTCCs and RyRs to the dyad, it seems highly plausible that “unpacking” of these proteins during HFrEF progression may be linked to declining BIN1 levels (Manfra et al., 2017). These findings indicate that BIN1 may serve as a therapeutic target in HFrEF, and preclinical data support this view. Treating mice with isoproterenol-induced HFrEF with adenoviral BIN1 overexpression was shown to attenuate hypertrophy, increase t-tubular microfolding, normalize SERCA2a distribution, and decrease the LTCC-RyR nearest neighbor distance (Liu Y. et al., 2020). This group additionally showed that BIN1 transduction rescued pre-existing global cardiac global dysfunction following aortic banding (Li J. et al., 2020). Beyond BIN1 overexpression as a therapeutic alternative, it appears that this protein may even serve as a biomarker. With normal, continuous turnover of BIN1 from dyads in healthy human patients, high levels of BIN1 are maintained in the blood. Thus, lowering of BIN1 levels has been linked to HF and arrhythmia (Hong et al., 2012a), including HFpEF disease severity and hospitalization risk (Nikolova et al., 2018), and HFrEF-associated risk of cardiovascular events (Hitzeman et al., 2020).
Given the exciting basic science and clinical data described above, more thorough investigation of BIN1’s protein partners seems warranted. Evidence from skeletal muscle has indicated that BIN1’s interaction with dynamin-2 (Dnm2) critically regulates tubulogenesis (Lee et al., 2002; Picas et al., 2014). This membrane-bound GTPase mediates membrane fission of clathrin-coated pits and plays a central role in membrane and vesicle trafficking (Gonzalez-Jamett et al., 2013). Dnm2 knockdown was found to rescue perinatal death in Bin1 knockout mice, and to normalize t-tubule formation and muscle function in animals with myopathies caused by Bin1 mutations (Tasfaout et al., 2017). An inhibitory role of Dnm2 in tubulogenesis was further supported by the observation that increasing its expression disrupts Bin1-induced tubulation in skeletal muscle (Gibbs et al., 2014).
A key aspect of Bin1’s function appears to be its ability to cluster phosphoinositides (PIs), in particular PI4, 5P2 (Lee et al., 2002). The dominant precursors of this phosphoinositide are PI and PI5P, which are created through the actions of the PI3P phosphatase myotubularin-1 (MTM1) (Ketel et al., 2016). This protein is expressed in most tissues, where it regulates endolysosomal sorting and trafficking by controlling PI expression patterns (Ketel et al., 2016; Figure 5, yellow arrows). Exciting data from the skeletal muscle field indicate that Bin1-mediated tubulogenesis is dependent on MTM1’s phosphatase activity, as MTM1 expression levels predict the extent of t-tubule biogenesis (Al-Qusairi and Laporte, 2011; Royer et al., 2013). Although MTM1’s role in t-tubule formation in cardiomyocytes is not yet resolved, PIs are indeed thought to be important for maintaining t-tubule integrity (Wu et al., 2011) and MTM1 dysregulation can induce dilated cardiomyopathy (Agrawal et al., 2014). Collectively, an emerging view is that balanced expression and activity of Bin1, Dnm2 and MTM1 are crucial for controlling tubule growth and maintenance (Figures 4, 5).
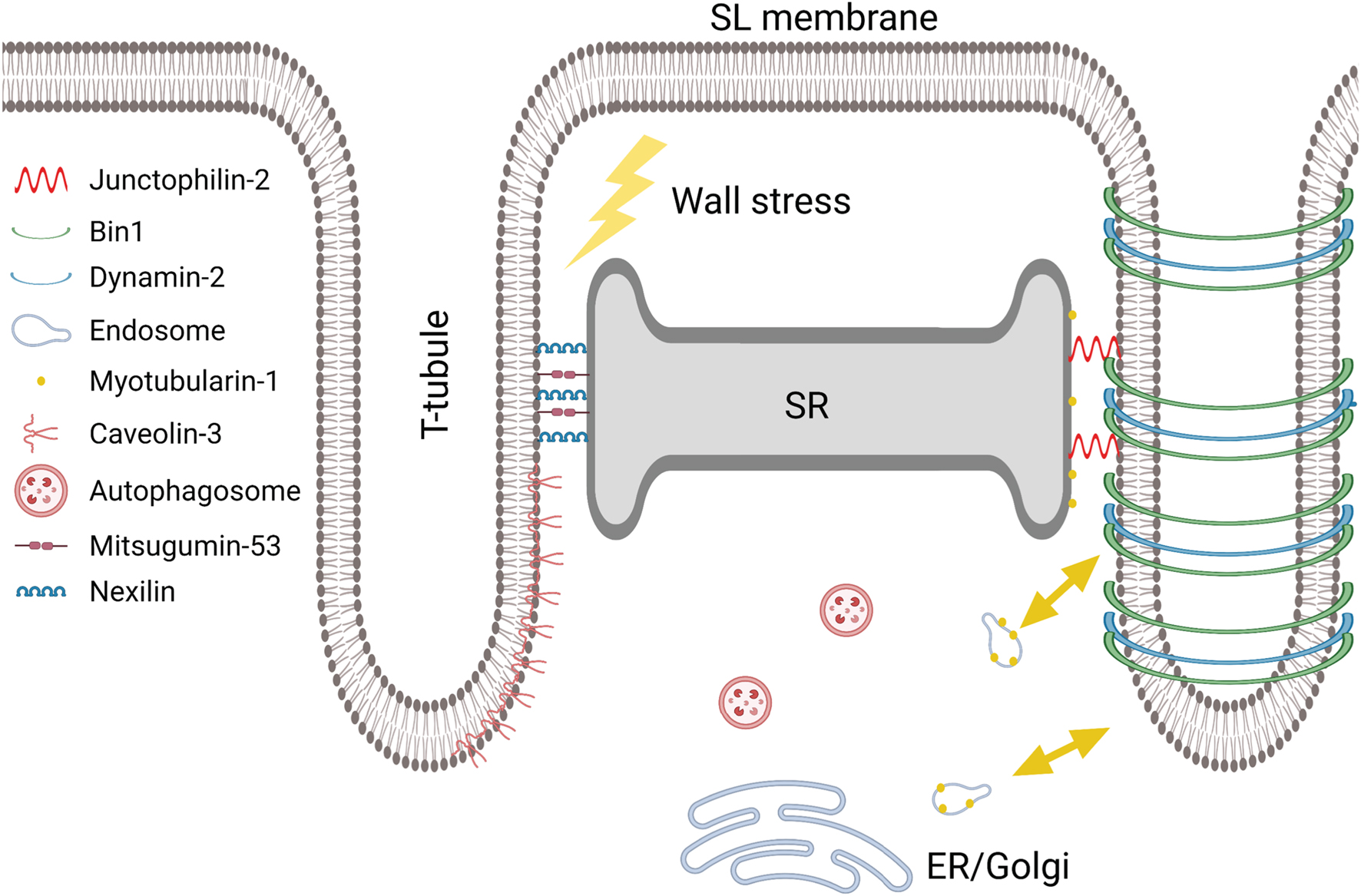
Figure 5. Proposed proteins and pathways involved in dyadic biogenesis, maintenance, and disruption. Created with BioRender.com.
As noted in the first chapter, JPH2 is a membrane anchoring protein, important for connecting the sarcolemma and its t-tubules to the SR in the dyad (Takeshima et al., 2000; Minamisawa et al., 2004; Han et al., 2013; Beavers et al., 2014; Figure 5). In this capacity, JPH2 maintains dyadic dimensions, and efficient crosstalk between LTCCs and RyRs. Importantly, JPH2 also interacts with both LTCCs and RyRs (Jiang et al., 2016; Reynolds et al., 2016), although the precise nature of these connections is not completely understood.
As with BIN1, JPH2 has been shown to play a key role in the development of t-tubules and dyads, both pre- and postnatally (Ziman et al., 2010; Munro and Soeller, 2016; Figure 4). This function is enabled by the membrane on receptor nexus (MORN) motif of JPH2, which allows anchoring to the t-tubule membrane, while the carboxy terminal is secured within the lumen of the SR (Nishi et al., 2000; Takeshima et al., 2000). The role of JPH2 in forming dyads is supported by the parallel appearance of JPH2 and t-tubules along z-lines in advance of developing t-tubules (Ziman et al., 2010). Furthermore, in mouse studies, reduced expression of JPH2 has been found to prevent t-tubule growth or result in an immature longitudinal configuration (Chen et al., 2013; Reynolds et al., 2013), consistent with a role of JPH2 in anchoring transverse, but not longitudinal elements (Chen et al., 2013). When JPH2 is fully knocked out in mice, it results in embryonic mortality, suggesting that it is required for dyad formation at the surface of the cell, even before the development of t-tubules starts (Takeshima et al., 2000; Franzini-Armstrong et al., 2005; Jones et al., 2018).
In adult failing cardiomyocytes, declining expression of JPH2 has been linked to t-tubule remodeling and disrupted dyads (Minamisawa et al., 2004; Xu et al., 2007; Wei et al., 2010; Landstrom et al., 2011; Chen et al., 2012; Xie et al., 2012; Zhang et al., 2014; Frisk et al., 2016; Figure 4). A causative role of JPH2 as a promoter of this remodeling is supported by studies showing that overexpression of JPH2 protects against t-tubule degradation, abnormal SR Ca2+ release, and HFrEF (Guo et al., 2014; Reynolds et al., 2016).
Emerging data support the role of JPH2 as a regulator of dyadic proteins. JPH2 overexpression was reported to result in the formation of larger RyR clusters within CRUs (Munro et al., 2016), while Wang et al. (2014) observed a reduction in RyR and NCX co-localization following JPH2 knockdown. JPH2 binding to RyRs may directly stabilize the channel’s function, as JPH2 knockdown induced RyR hyperactivity (van Oort et al., 2011), while overexpression inhibited Ca2+ sparks (Munro et al., 2016). JPH2 is also reported to modulate LTCC activity, which has important implications for L-type current in HFrEF, where JPH2 expression is lowered (Jiang et al., 2016). Interestingly, recent data from Gross et al. (2021) indicate that the “Joining Region” of JPH2 may exert direct effects on the localization of LTCC in t-tubules, indicating that not only the presence of JPH2 but specifically JPH2-LTCC binding is necessary for maintenance of dyadic integrity and Ca2+ homeostasis.
What causes JPH2 changes during disease? A suggested mechanism for suppression of JPH2 expression is via upregulation of microRNA-24 (miR-24), as Xu et al. (2012) showed that a miR-24 antagomir protected against JPH2 downregulation and associated changes in t-tubule architecture. Mislocalization of JPH2 in the failing heart has also been reported, and linked to reorganization of the microtubules necessary for its delivery to dyads (Zhang et al., 2014; Prins et al., 2016).
Calpain cleavage of JPH2 is believed to be one of the mechanisms behind its downregulation, as first reported in mouse models of reversible heart failure (Guo et al., 2015), and ischemia-reperfusion injury (Wu et al., 2014). Furthermore, Wang et al. (2018) demonstrated increased calpain-mediated JPH2 cleavage in several mouse HFrEF models (myocardial infarction, transaortic banding, and chronic isoproterenol infusion), and found that inhibition of calpain partially restored both JPH2 expression levels and t-tubular density. These authors also observed that in a dual overexpression model of calpain and JPH2, JPH2 expression was only transiently maintained, and that subsequent deterioration in t-tubules and higher risk of cardiac death temporally correlated with declining JPH2 levels. Interestingly, contrasting reports also indicate that two different cleavage products of JPH2 may relocate to the nucleus, and attenuate (Guo et al., 2018b), or exacerbate cardiomyocyte stress responses (Lahiri et al., 2020).
Finally, the function of JPH2 has been shown to be dependent on its phosphorylation status (Guo et al., 2015), which is in turn regulated by “striated muscle preferentially expressed protein kinase” (SPEG). SPEG is downregulated in HFrEF, and loss of SPEG-dependent phosphorylation of JPH2 is suggested to promote t-tubule disruption (Quick et al., 2017).
Taken together, there is an abundance of evidence to support that JPH2 is a critical regulator of dyadic assembly and maintenance, and that this function extends far beyond a passive role in anchoring membrane positions. Thus, JPH2 is a promising target for future therapies.
Cav-3 is known to play a key role in the formation of t-tubules and caveolae (Parton et al., 1997; Figures 4, 5). However, as with other dyadic regulators, emerging data suggest that decreased expression of Cav-3 has important functional consequences in the failing heart. Data from the Orchard group have shown that Cav-3 knockout mice exhibit cardiac dysfunction, associated with t-tubule reorganization and decreased LTCC current density (Bryant et al., 2018b). In another study, this group found that pressure overload in mice triggered loss of t-tubular Ca2+ current density and impairment of Ca2+ release, linked to lowered levels at Cav-3 (Bryant et al., 2018a). Interestingly, follow-up work with Cav-3 overexpressing mice exposed to pressure overload showed cardioprotective effects linked to maintained t-tubular Ca2+ current (Kong et al., 2019). Of note, these protective effects may also be linked to associations between Cav-3 and JPH2, as Cav-3 overexpression reportedly stabilizes JPH2, and thus t-tubules (Minamisawa et al., 2004).
Recent work has identified a role of autophagy in regulating the assembly and disassembly of cardiac t-tubules (Figure 5). First, dexamethasone, a synthetic glucocorticoid, was shown to aid t-tubule growth in stem cell-derived cardiomyocytes (Parikh et al., 2017), and knockout of cardiac glucocorticoid receptors was found to induce heart failure and disturbances in Ca2+ handling (Oakley et al., 2013). However, it wasn’t until Seidel and colleagues (Seidel et al., 2019) recently examined the t-tubule network in glucocorticoid receptor knockout mice that the former results were linked to autophagy. Here they demonstrated that t-tubule loss caused by glucocorticoid receptor knockout can be rescued by treatment with dexamethasone, and that this is associated with upregulation of the autophagy markers LC3BII and p62. Furthermore, treatment with rapamycin, an autophagy enhancer, reproduced the findings with dexamethasone treatment. Conversely, treatment with chloroquine and bafilomycin A1 (autophagy blockers) exacerbated detrimental effects on t-tubules (Seidel et al., 2019). In line with these findings, it has also been shown that a high-fat diet, and diabetes, induce apoptosis and cardiac alterations through inhibition of autophagy (Hsu et al., 2016).
Very recently, several studies from the Chen group have identified an exciting new dyadic regulator, called Nexilin (NEXN) (Figure 5). Previously known as an actin-binding and Z-disk protein, NEXN was shown to be critical to the formation of dyadic membranes, as myocytes from knockout animals did not develop t-tubules and exhibited early postnatal cardiomyopathy and lethality (Liu et al., 2019). Furthermore, conditional knockout of NEXN in adult cardiomyocytes resulted in a remodeling of t-tubule structure that was highly reminiscent of HFrEF, with loss of transverse t-tubules and an increased proportion of longitudinal elements (Spinozzi et al., 2020). The authors additionally reported that NEXN interacts with both JPH2 and RyRs, and that its loss results in decreased expression of these and other dyadic proteins, and accompanying impairment of Ca2+ homeostasis (Liu et al., 2019; Spinozzi et al., 2020). These findings are of considerable interest, as nexilin mutations are linked to cardiopathy in mice and humans (Hassel et al., 2009; Wang H. et al., 2010; Haas et al., 2015; Liu C. et al., 2020). Further work is required to determine the exact mechanism by which NEXN grows and maintains dyads, and whether NEXN alterations occur in acquired HFrEF. In this regard, it is noteworthy that NEXN expression levels were found to be maintained in HFrEF patients (Chen et al., 2018).
The muscle-specific membrane repair protein mitsugumin 53 (MG53) is an up and coming t-tubule regulator (Kitmitto et al., 2019; Figure 5). This “wound-healing” protein is part of the tripartite motif family (TRIM), and is often referred to as TRIM72. Wang X. et al. (2010) were the first to identify MG53 as critical for maintaining cardiomyocyte sarcolemmal stability. The sarcolemmal membrane of the cardiomyocyte is the first line of defense against external stresses, such as oxygen and nutrient deprivation, inflammation and oxidative stress, and indeed, loss of sarcolemmal viability is a key step in cell death via necrosis (Kitmitto et al., 2019). Regulatory injury-repair proteins, such as MG53, are thus important for the integrity of both the sarcolemma and the cell as a whole. A new study suggests that while MG53 is not necessary for the development or maintenance of t-tubules during health, it may crucially preserve their integrity and function when the heart is placed under pathological stress (Zhang et al., 2017). When an injury or defect appears in the cell membrane, MG53 travels to the injury site and “plugs the hole,” by re-sealing the membrane (Cooper and McNeil, 2015). MG53 coordinates this role through interplay with caveolaer proteins (Kitmitto et al., 2019). He et al. (2012) found that elevated expression of MG53 improved heart function and augmented membrane repair capacity. While these authors did not specifically link these findings to changes in t-tubule structure/function, such effects seem likely based on the well-established role of declining t-tubule integrity in disease.
Protein kinase C (PKC) activation has been implicated in t-tubule remodeling in a recent study by Guo et al. (2018a), where the authors found a transient elevation in PKC activity in the days following transaortic banding. Inhibition of PKC during this period ameliorated subsequent t-tubule remodeling and heart failure development. It was shown that the increase in PKC coincided with a transient biphasic mode of actin depolymerization and repolarization, and that PKC inhibition abolished this response. Furthermore, the use of various inhibitors or stabilizers of F-actin polymers seemed to protect the t-tubule system after aortic banding, indicating that the sudden biphasic response is detrimental to t-tubule integrity. Blocking stretch-activated channels diminished the PKC-mediated effect on t-tubules, implicating these channels in mechanosensitive regulation of t-tubule structure downstream of PKC. These exciting findings are consistent with a growing appreciation for the importance of workload in regulating t-tubule structure, but are the first to implicate a role of PKC and actin filament dynamics in these processes.
The above discussion has highlighted recent work implicating a plethora of proteins involved in the growth and maintenance of t-tubules and their remodeling during disease. How should we make sense of this increasingly complex array of proteins? Are there shared, overarching signaling pathways which coordinate changing protein expression patterns? We believe that changing physical stress experienced by the myocardium is at least one such signal. Indeed, when viewed from a mechanosensatory viewpoint we can see that many of the proposed t-tubule regulators can be functionally clustered together. For example, several of these proteins have proposed roles in mechanosensing at the cell membrane (PKC via stretch-activated channels) or Z-disks (T-Cap), or are linked to the mechanosensitive process of actin polymerization (BIN1, Nexilin, and PKC). A perhaps somewhat distinct group of t-tubule regulatory proteins appears to be focused on maintenance of the membrane itself, and notably includes Mtm1, BIN1, and the general autophagic and endocytic processes. It is less clear that these processes are mechanosensitive in cardiomyocytes, and we might rather speculate that the changing metabolic environment of the diseased heart is an overarching signal that regulates membrane integrity.
Despite promising results from rodent studies, it seems unlikely that targeting of only a single t-tubule regulator would be sufficient to therapeutically protect t-tubule structure in humans. Rather, we believe that new interventions should instead be aimed at the overarching signals and functional groups of proteins described above. For example, upregulating expression of JPH2 has been suggested as an approach to maintain dyadic structure during HFrEF (Reynolds et al., 2016). However, we expect that this approach would yield only temporary benefits since JPH2 downregulation is itself driven by elevated workload (Frisk et al., 2016), and thus the persistence of these mechanical cues would continue to signal detrimental changes in JPH2 and other mechanosensitive proteins. Indeed, it is notable that existing therapeutics for HFrEF act largely to alleviate cardiac workload, and thus are expected to normalize expression of a spectrum of key mechanosensitive t-tubule regulators. As an alternative approach, we speculate that more precisely interrupting the processes of mechanosensation at the levels of the cardiomyocyte membrane and/or cytoskeleton could be broadly beneficial in the treatment of HFrEF patients.
This review has summarized our growing appreciation for the role of t-tubules and dyads as critical regulators of cardiomyocyte Ca2+ homeostasis, and thus systolic and diastolic function of the heart as a working organ. The discussion has highlighted a consensus view that t-tubule remodeling is a key mechanism contributing to disrupted Ca2+ handling and contractility in HFrEF, and a likely contributor to arrythmogenesis, but that distinct forms of remodeling occur during HFpEF. We have described a wealth of evidence indicating key roles of dyadic proteins JPH2, BIN1, and Cav-3, but also newer players and signaling pathways which hold promise. We believe that several current HFrEF therapies preserve t-tubule structure and cardiac function by normalizing expression of these proteins, and that this involves relief of the high workload that drives dyadic disruption. We anticipate that improved safeguarding of t-tubule integrity will serve as a basis for future HFrEF therapy.
All authors contributed to the writing of the article and development of the figures.
This work was financially supported by the European Union’s Horizon 2020 Research and Innovation Programme (Consolidator grant, WL) under grant agreement No. 647714, the Norwegian Research Council (WL and MF), the South-Eastern Norway Regional Health Authority (WL and MF), the KG Jebsen Centre for Cardiac Research (WL and CL), the Norwegian Institute for Public Health (MF and JL), and the University of Oslo (IS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CICR, Ca2+-induced Ca2+ release; Cav-3, Caveolin-3; CRU, Ca2+ release unit; DAD, Delayed afterdepolarization; EAD, Early afterdepolarization; EC coupling, Excitation-contraction coupling; HF, Heart failure; HFpEF, Heart failure with preserved ejection fraction; HFrEF, Heart failure with reduced ejection fraction; JPH2, Junctophilin-2; LTCC, L-type Ca2+ channel; miR-24, microRNA-24; MG53, Mitsugumin 53; NEXN, Nexilin; NCX, Sodium-calcium exchanger 1; PKC, Protein kinase C; RyR, Ryanodine Receptor; SR, Sarcoplasmic Reticulum; Tcap, Titin cap/Telethonin.
Agrawal, P. B., Pierson, C. R., Joshi, M., Liu, X., Ravenscroft, G., Moghadaszadeh, B., et al. (2014). SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am. J. Hum. Genet. 95, 218–226. doi: 10.1016/j.ajhg.2014.07.004
Al-Qusairi, L., and Laporte, J. (2011). T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 1:26. doi: 10.1186/2044-5040-1-26
Arora, R., Aistrup, G. L., Supple, S., Frank, C., Singh, J., Tai, S., et al. (2017). Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm 14, 273–281. doi: 10.1016/j.hrthm.2016.09.022
Baddeley, D., Jayasinghe, I. D., Lam, L., Rossberger, S., Cannell, M. B., and Soeller, C. (2009). Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 22275–22280. doi: 10.1073/pnas.0908971106
Balijepalli, R. C., Lokuta, A. J., Maertz, N. A., Buck, J. M., Haworth, R. A., Valdivia, H. H., et al. (2003). Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc. Res. 59, 67–77.
Beavers, D. L., Landstrom, A. P., Chiang, D. Y., and Wehrens, X. H. (2014). Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc. Res. 103, 198–205. doi: 10.1093/cvr/cvu151
Berliner, D., and Bauersachs, J. (2017). Current drug therapy in chronic heart failure: the new guidelines of the European society of cardiology (ESC). Korean Circ. J. 47, 543–554. doi: 10.4070/kcj.2017.0030
Biesmans, L., Macquaide, N., Heinzel, F. R., Bito, V., Smith, G. L., and Sipido, K. R. (2011). Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One 6:e25100. doi: 10.1371/journal.pone.0025100
Brand, T. (2003). Heart development: molecular insights into cardiac specification and early morphogenesis. Dev. Biol. 258, 1–19. doi: 10.1016/s0012-1606(03)00112-x
Brette, F., Despa, S., Bers, D. M., and Orchard, C. H. (2005). Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J. Mol. Cell. Cardiol. 39, 804–812. doi: 10.1016/j.yjmcc.2005.08.005
Brook, W. H., Connell, S., Cannata, J., Maloney, J. E., and Walker, A. M. (1983). Ultrastructure of the myocardium during development from early fetal life to adult life in sheep. J. Anat. 137(Pt 4), 729–741.
Bryant, S. M., Kong, C. H. T., Watson, J. J., Gadeberg, H. C., James, A. F., Cannell, M. B., et al. (2018a). Caveolin 3-dependent loss of t-tubular ICa during hypertrophy and heart failure in mice. Exp. Physiol. 103, 652–665. doi: 10.1113/ep086731
Bryant, S. M., Kong, C. H. T., Watson, J. J., Gadeberg, H. C., Roth, D. M., Patel, H. H., et al. (2018b). Caveolin-3 KO disrupts t-tubule structure and decreases t-tubular ICa density in mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 315, H1101–H1111.
Bryant, S. M., Kong, C. H., Watson, J., Cannell, M. B., James, A. F., and Orchard, C. H. (2015). Altered distribution of ICa impairs Ca release at the t-tubules of ventricular myocytes from failing hearts. J. Mol. Cell. Cardiol. 86, 23–31. doi: 10.1016/j.yjmcc.2015.06.012
Caldwell, J. L., Smith, C. E., Taylor, R. F., Kitmitto, A., Eisner, D. A., Dibb, K. M., et al. (2014). Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ. Res. 115, 986–996. doi: 10.1161/circresaha.116.303448
Cannell, M. B., Crossman, D. J., and Soeller, C. (2006). Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J. Muscle Res. Cell Motil. 27, 297–306. doi: 10.1007/s10974-006-9089-y
Chen, B., Guo, A., Zhang, C., Chen, R., Zhu, Y., and Hong, J. (2013). Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc. Res. 100, 54–62. doi: 10.1093/cvr/cvt180
Chen, B., Li, Y., Jiang, S., Xie, Y. P., Guo, A., Kutschke, W., et al. (2012). beta-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. 26, 2531–2537. doi: 10.1096/fj.11-199505
Chen, C. Y., Caporizzo, M. A., Bedi, K., Vite, A., Bogush, A. I., Robison, P., et al. (2018). Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 24, 1225–1233. doi: 10.1038/s41591-018-0046-2
Cheng, H., Lederer, M. R., Lederer, W. J., and Cannell, M. B. (1996). Calcium sparks and [Ca2+ ]i waves in cardiac myocytes. Am. J. Physiol. 270, C148–C159.
Cooper, S. T., and McNeil, P. L. (2015). Membrane repair: mechanisms and pathophysiology. Physiol. Rev. 95, 1205–1240. doi: 10.1152/physrev.00037.2014
Crocini, C., Coppini, R., Ferrantini, C., Yan, P., Loew, L. M., Tesi, C., et al. (2014). Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure. Proc. Natl. Acad. Sci. U.S.A. 111, 15196–15201. doi: 10.1073/pnas.1411557111
Crossman, D. J., Shen, X., Jullig, M., Munro, M., Hou, Y., Middleditch, M., et al. (2017). Increased collagen within the transverse tubules in human heart failure. Cardiovasc. Res. 113, 879–891. doi: 10.1093/cvr/cvx055
Crossman, D. J., Young, A. A., Ruygrok, P. N., Nason, G. P., Baddelely, D., Soeller, C., et al. (2015). T-tubule disease: relationship between t-tubule organization and regional contractile performance in human dilated cardiomyopathy. J. Mol. Cell. Cardiol. 84, 170–178. doi: 10.1016/j.yjmcc.2015.04.022
Curl, C. L., Danes, V. R., Bell, J. R., Raaijmakers, A. J. A., Ip, W. T. K., Chandramouli, C., et al. (2018). Cardiomyocyte functional etiology in heart failure with preserved ejection fraction is distinctive-a new preclinical model. J. Am. Heart Assoc. 7:e007451.
de Diego, C., Chen, F., Xie, L. H., Dave, A. S., Thu, M., Rongey, C., et al. (2008). Cardiac alternans in embryonic mouse ventricles. Am. J. Physiol. Heart Circ. Physiol. 294, H433–H440.
De La Mata, A., Tajada, S., O’Dwyer, S., Matsumoto, C., Dixon, R. E., Hariharan, N., et al. (2019). BIN1 induces the formation of T-Tubules and adult-like Ca2+ release units in developing cardiomyocytes. Stem Cells 37, 54–64. doi: 10.1002/stem.2927
Dibb, K. M., Clarke, J. D., Horn, M. A., Richards, M. A., Graham, H. K., Eisner, D. A., and Trafford, A. W. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482–489. doi: 10.1161/circheartfailure.109.852228
Dixon, R. E., Moreno, C. M., Yuan, C., Opitz-Araya, X., Binder, M. D., Navedo, M. F., et al. (2015). Graded Ca2+ /calmodulin-dependent coupling of voltage-gated CaV1.2 channels. eLife 4:e05608.
Dostal, D. E., Feng, H., Nizamutdinov, D., Golden, H. B., Afroze, S. H., Dostal, J. D., et al. (2014). Mechanosensing and regulation of cardiac function. J. Clin. Exp. Cardiol. 5:314.
Dridi, H., Kushnir, A., Zalk, R., Yuan, Q., Melville, Z., and Marks, A. R. (2020). Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 17, 732–747. doi: 10.1038/s41569-020-0394-8
Dyachenko, V., Husse, B., Rueckschloss, U., and Isenberg, G. (2009). Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 45, 38–54. doi: 10.1016/j.ceca.2008.06.003
Edwards, A. G., and Louch, W. E. (2017). Species-dependent mechanisms of cardiac arrhythmia: a cellular focus. Clin. Med. Insights Cardiol. 1:1179546816686061.
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–C14.
Feridooni, H. A., Dibb, K. M., and Howlett, S. E. (2015). How cardiomyocyte excitation, calcium release and contraction become altered with age. J. Mol. Cell. Cardiol. 83, 62–72. doi: 10.1016/j.yjmcc.2014.12.004
Fiegle, D. J., Schober, M., Dittrich, S., Cesnjevar, R., Klingel, K., Volk, T., et al. (2020). Severe T-System remodeling in pediatric viral myocarditis. Front. Cardiovasc. Med. 7:624776. doi: 10.3389/fcvm.2020.624776
Forbes, M. S., and Sperelakis, N. (1976). The presence of transverse and axial tubules in the ventricular myocardium of embryonic and neonatal guinea pigs. Cell Tissue Res. 166, 83–90. doi: 10.1007/bf00215127
Forsgren, S., and Thornell, L. E. (1981). The development of Purkinje fibres and ordinary myocytes in the bovine fetal heart. An ultrastructural study. Anat. Embryol. 162, 127–136. doi: 10.1007/bf00306485
Forssmann, W. G., and Girardier, L. (1970). A study of the T system in rat heart. J. Cell Biol. 44, 1–19. doi: 10.1083/jcb.44.1.1
Franzini-Armstrong, C., Protasi, F., and Tijskens, P. (2005). The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 1047, 76–85. doi: 10.1196/annals.1341.007
Frisk, M., Koivumaki, J. T., Norseng, P. A., Maleckar, M. M., Sejersted, O. M., and Louch, W. E. (2014). Variable t-tubule organization and Ca2+ homeostasis across the atria. Am. J. Physiol. Heart Circ. Physiol. 307, H609–H620.
Frisk, M., Le, C., Shen, X., Roe, A. T., Hou, Y., and Manfra, O. (2021). Etiology-dependent impairment of diastolic cardiomyocyte calcium homeostasis in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 77, 405–419.
Frisk, M., Ruud, M., Espe, E. K., Aronsen, J. M., Roe, A. T., Zhang, L., et al. (2016). Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 112, 443–451. doi: 10.1093/cvr/cvw111
Fu, Y., Shaw, S. A., Naami, R., Vuong, C. L., Basheer, W. A., Guo, X., et al. (2016). Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation 133, 388–397. doi: 10.1161/circulationaha.115.018535
Gadeberg, H. C., Bond, R. C., Kong, C. H., Chanoit, G. P., Ascione, R., Cannell, M. B., et al. (2016). Heterogeneity of T-Tubules in pig hearts. PLoS One 11:e0156862. doi: 10.1371/journal.pone.0156862
Gaeta, S. A., Bub, G., Abbott, G. W., and Christini, D. J. (2009). Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ. Res. 105, 335–342. doi: 10.1161/circresaha.109.197590
Gibbs, E. M., Davidson, A. E., Telfer, W. R., Feldman, E. L., and Dowling, J. J. (2014). The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis. Model. Mech. 7, 157–161.
Glukhov, A. V., Balycheva, M., Sanchez-Alonso, J. L., Ilkan, Z., Alvarez-Laviada, A., and Bhogal, N. (2015). Direct evidence for microdomain-specific localization and remodeling of functional L-Type calcium channels in rat and human atrial myocytes. Circulation 132, 2372–2384. doi: 10.1161/circulationaha.115.018131
Gonzalez-Jamett, A. M., Momboisse, F., Haro-Acuna, V., Bevilacqua, J. A., Caviedes, P., and Cardenas, A. M. (2013). Dynamin-2 function and dysfunction along the secretory pathway. Front. Endocrinol. (Lausanne) 4:126. doi: 10.3389/fendo.2013.00126
Gotoh, T. (1983). Quantitative studies on the ultrastructural differentiation and growth of mammalian cardiac muscle cells. The atria and ventricles of the cat. Acta Anat. 115, 168–177. doi: 10.1159/000145687
Grandy, S. A., and Howlett, S. E. (2006). Cardiac excitation-contraction coupling is altered in myocytes from aged male mice but not in cells from aged female mice. Am. J. Physiol. Heart Circ. Physiol. 291, H2362–H2370.
Gross, P., Johnson, J., Romero, C. M., Eaton, D. M., Poulet, C., Sanchez-Alonso, J., et al. (2021). Houser, interaction of the joining region in junctophilin-2 with the L-Type Ca2+ channel is pivotal for cardiac dyad assembly and intracellular Ca2+ dynamics. Circ. Res. 128, 92–114.
Gu, Y., Gorelik, J., Spohr, H. A., Shevchuk, A., Lab, M. J., and Harding, S. E. (2002). High-resolution scanning patch-clamp: new insights into cell function. FASEB J. 16, 748–750. doi: 10.1096/fj.01-1024fje
Guo, A., Chen, R., Wang, Y., Huang, C. K., Chen, B., Kutschke, W., et al. (2018a). Transient activation of PKC results in long-lasting detrimental effects on systolic [Ca2+ ]i in cardiomyocytes by altering actin cytoskeletal dynamics and T-tubule integrity. J. Mol. Cell. Cardiol. 115, 104–114. doi: 10.1016/j.yjmcc.2018.01.003
Guo, A., Hall, D., Zhang, C., Peng, T., Miller, J. D., Kutschke, W., et al. (2015). Molecular determinants of calpain-dependent cleavage of Junctophilin-2 protein in cardiomyocytes. J. Biol. Chem. 290, 17946–17955. doi: 10.1074/jbc.m115.652396
Guo, A., Wang, Y., Chen, B., Wang, Y., Yuan, J., Zhang, L., et al. (2018b). E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 362:eaan3303. doi: 10.1126/science.aan3303
Guo, A., Zhang, C., Wei, S., Chen, B., and Song, L. S. (2013). Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc. Res. 98, 204–215. doi: 10.1093/cvr/cvt020
Guo, A., Zhang, X., Iyer, V. R., Chen, B., Zhang, C., Kutschke, W. J., et al. (2014). Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc. Natl. Acad. Sci. U.S.A. 111, 12240–12245. doi: 10.1073/pnas.1412729111
Haas, J., Frese, K. S., Peil, B., Kloos, W., Keller, A., Nietsch, R., et al. (2015). Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 36, 1123–1135.
Hamaguchi, S., Kawakami, Y., Honda, Y., Nemoto, K., Sano, A., Namekata, I., et al. (2013). Developmental changes in excitation-contraction mechanisms of the mouse ventricular myocardium as revealed by functional and confocal imaging analyses. J. Pharmacol. Sci. 123, 167–175. doi: 10.1254/jphs.13099fp
Han, J., Wu, H., Wang, Q., and Wang, S. (2013). Morphogenesis of T-tubules in heart cells: the role of junctophilin-2. Sci. China Life Sci. 56, 647–652. doi: 10.1007/s11427-013-4490-4
Hassel, D., Dahme, T., Erdmann, J., Meder, B., Huge, A., Stoll, M., et al. (2009). Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 15, 1281–1288. doi: 10.1038/nm.2037
Hayashi, T., Martone, M. E., Yu, Z., Thor, A., Doi, M., Holst, M. J., et al. (2009). Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 122, 1005–1013. doi: 10.1242/jcs.028175
He, B., Tang, R. H., Weisleder, N., Xiao, B., Yuan, Z., Cai, C., et al. (2012). Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol. Ther. 20, 727–735. doi: 10.1038/mt.2012.5
He, J., Conklin, M. W., Foell, J. D., Wolff, M. R., Haworth, R. A., Coronado, R., et al. (2001). Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc. Res. 49, 298–307. doi: 10.1016/s0008-6363(00)00256-x
Heinzel, F. R., Bito, V., Biesmans, L., Wu, M., Detre, E., von Wegner, F., et al. (2008). Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ. Res. 102, 338–346. doi: 10.1161/circresaha.107.160085
Heinzel, F. R., Bito, V., Volders, P. G., Antoons, G., Mubagwa, K., and Sipido, K. R. (2002). Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ. Res. 91, 1023–1030. doi: 10.1161/01.res.0000045940.67060.dd
Henderson, S. A., Goldhaber, J. I., So, J. M., Han, T., Motter, C., Ngo, A., et al. (2004). Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ. Res. 95, 604–611. doi: 10.1161/01.res.0000142316.08250.68
Herring, N., Kalla, M., and Paterson, D. J. (2019). The autonomic nervous system and cardiac arrhythmias: current concepts and emerging therapies. Nat. Rev. Cardiol. 16, 707–726. doi: 10.1038/s41569-019-0221-2
Hirakow, R., Gotoh, T., and Watanabe, T. (1980). Quantitative studies on the ultrastructural differentiation and growth of mammalian cardiac muscle cells. I. The atria and ventricles of the rat. Acta Anat. 108, 144–152. doi: 10.1159/000145293
Hitzeman, T. C., Xie, Y., Zadikany, R. H., Nikolova, A. P., Baum, R., Caldaruse, A. M., et al. (2020). cBIN1 score (CS) identifies ambulatory HFrEF patients and predicts cardiovascular events. Front. Physiol. 11:503. doi: 10.3389/fphys.2020.00503
Hoerter, J., Mazet, F., and Vassort, G. (1981). Perinatal growth of the rabbit cardiac cell: possible implications for the mechanism of relaxation. J. Mol. Cell. Cardiol. 13, 725–740. doi: 10.1016/0022-2828(81)90255-8
Hong, T. T., Cogswell, R., James, C. A., Kang, G., Pullinger, C. R., Malloy, M. J., et al. (2012a). Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 9, 961–967. doi: 10.1016/j.hrthm.2012.01.024
Hong, T. T., Smyth, J. W., Chu, K. Y., Vogan, J. M., Fong, T. S., Jensen, B. C., et al. (2012b). BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm 9, 812–820. doi: 10.1016/j.hrthm.2011.11.055
Hong, T. T., Smyth, J. W., Gao, D., Chu, K. Y., Vogan, J. M., Fong, T. S., et al. (2010). BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 8:e1000312. doi: 10.1371/journal.pbio.1000312
Hong, T., and Shaw, R. M. (2017). Cardiac T-Tubule microanatomy and function. Physiol. Rev. 97, 227–252. doi: 10.1152/physrev.00037.2015
Hong, T., Yang, H., Zhang, S. S., Cho, H. C., Kalashnikova, M., Sun, B., et al. (2014). Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 20, 624–632. doi: 10.1038/nm.3543
Hsu, H. C., Chen, C. Y., Lee, B. C., and Chen, M. F. (2016). High-fat diet induces cardiomyocyte apoptosis via the inhibition of autophagy. Eur. J. Nutr. 55, 2245–2254. doi: 10.1007/s00394-015-1034-7
Huang, C. K., Chen, B. Y., Guo, A., Chen, R., Zhu, Y. Q., Kutschke, W., et al. (2016). Sildenafil ameliorates left ventricular T-tubule remodeling in a pressure overload-induced murine heart failure model. Acta Pharmacol. Sin. 37, 473–482. doi: 10.1038/aps.2016.13
Huang, C. Y., Peres Moreno Maia-Joca, R., Ong, C. S., Wilson, I., DiSilvestre, D., Tomaselli, G. F., et al. (2020). Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J. Mol Cell. Cardiol. 138, 1–11. doi: 10.1016/j.yjmcc.2019.10.001
Ibrahim, M., Al Masri, A., Navaratnarajah, M., Siedlecka, U., Soppa, G. K., Moshkov, A., et al. (2010). Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 24, 3321–3329. doi: 10.1096/fj.10-156638
Ibrahim, M., and Terracciano, C. M. (2013). Reversibility of T-tubule remodelling in heart failure: mechanical load as a dynamic regulator of the T-tubules. Cardiovasc. Res. 98, 225–232. doi: 10.1093/cvr/cvt016
Ibrahim, M., Gorelik, J., Yacoub, M. H., and Terracciano, C. M. (2011). The structure and function of cardiac t-tubules in health and disease. Proc. Biol. Sci. 278, 2714–2723. doi: 10.1098/rspb.2011.0624
Ibrahim, M., Kukadia, P., Siedlecka, U., Cartledge, J. E., Navaratnarajah, M., Tokar, S., et al. (2012). Cardiomyocyte Ca2+ handling and structure is regulated by degree and duration of mechanical load variation. J. Cell. Mol. Med. 16, 2910–2918. doi: 10.1111/j.1582-4934.2012.01611.x
Ibrahim, M., Nader, A., Yacoub, M. H., and Terracciano, C. (2015). Manipulation of sarcoplasmic reticulum Ca2+ release in heart failure through mechanical intervention. J. Physiol. 593, 3253–3259. doi: 10.1113/jp270446
Ibrahim, M., Siedlecka, U., Buyandelger, B., Harada, M., Rao, C., Moshkov, A., et al. (2013). A critical role for Telethonin in regulating t-tubule structure and function in the mammalian heart. Hum. Mol. Genet. 22, 372–383.
Ito, D. W., Hannigan, K. I., Ghosh, D., Xu, B., Del Villar, S. G., and Xiang, Y. K. (2019). beta-adrenergic-mediated dynamic augmentation of sarcolemmal CaV 1.2 clustering and co-operativity in ventricular myocytes. J. Physiol. 597, 2139–2162. doi: 10.1113/jp277283
Jaquenod De Giusti, C., Blanco, P. G., Lamas, P. A., Carrizo Velasquez, F., Lofeudo, J. M., Portiansky, E. L., et al. (2019). Carbonic anhydrase II/sodium-proton exchanger 1 metabolon complex in cardiomyopathy of ob(-/-) type 2 diabetic mice. J. Mol. Cell. Cardiol. 136, 53–63. doi: 10.1016/j.yjmcc.2019.09.005
Jayasinghe, I. D., Cannell, M. B., and Soeller, C. (2009). Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys. J. 97, 2664–2673. doi: 10.1016/j.bpj.2009.08.036
Jayasinghe, I., Clowsley, A. H., Lin, R., Lutz, T., Harrison, C., Green, E., et al. (2018). True molecular scale visualization of variable clustering properties of ryanodine receptors. Cell Rep. 22, 557–567. doi: 10.1016/j.celrep.2017.12.045
Jayasinghe, I., Crossman, D., Soeller, C., and Cannell, M. (2012). Comparison of the organization of T-tubules, sarcoplasmic reticulum and ryanodine receptors in rat and human ventricular myocardium. Clin. Exp. Pharmacol. Physiol. 39, 469–476. doi: 10.1111/j.1440-1681.2011.05578.x
Jiang, M., Zhang, M., Howren, M., Wang, Y., Tan, A., Balijepalli, R. C., et al. (2016). JPH-2 interacts with Cai-handling proteins and ion channels in dyads: contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 13, 743–752. doi: 10.1016/j.hrthm.2015.10.037
Jiang, Y., Tanaka, H., Matsuyama, T. A., Yamaoka, Y., and Takamatsu, T. (2014). Pacing-induced non-uniform Ca2+ dynamics in rat atria revealed by rapid-scanning confocal microscopy. Acta Histochem. Cytochem. 47, 59–65. doi: 10.1267/ahc.14014
Jones, P. P., MacQuaide, N., and Louch, W. E. (2018). Dyadic plasticity in cardiomyocytes. Front. Physiol. 9:1773. doi: 10.3389/fphys.2018.01773
Kaprielian, R. R., Stevenson, S., Rothery, S. M., Cullen, M. J., and Severs, N. J. (2000). Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 101, 2586–2594. doi: 10.1161/01.cir.101.22.2586
Kemi, O. J., Hoydal, M. A., Macquaide, N., Haram, P. M., Koch, L. G., Britton, S. L., et al. (2011). The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J. Cell Physiol. 226, 2235–2243. doi: 10.1002/jcp.22559
Ketel, K., Krauss, M., Nicot, A. S., Puchkov, D., Wieffer, M., Muller, R., et al. (2016). A phosphoinositide conversion mechanism for exit from endosomes. Nature 529, 408–412. doi: 10.1038/nature16516
Kilfoil, P. J., Lotteau, S., Zhang, R., Yue, X., Aynaszyan, S., Solymani, R. E., et al. (2020). Distinct features of calcium handling and beta-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 598, 5091–5108. doi: 10.1113/jp280425
Kim, H. D., Kim, D. J. I, Lee, J., Rah, B. J., Sawa, Y., and Schaper, J. (1992). Human fetal heart development after mid-term: morphometry and ultrastructural study. J. Mol. Cell. Cardiol. 24, 949–965. doi: 10.1016/0022-2828(92)91862-y
Kitmitto, A., Baudoin, F., and Cartwright, E. J. (2019). Cardiomyocyte damage control in heart failure and the role of the sarcolemma. J. Muscle Res. Cell Motil. 40, 319–333. doi: 10.1007/s10974-019-09539-5
Kojima, C., Hashimoto, A., Yabuta, I., Hirose, M., Hashimoto, S., Kanaho, Y., et al. (2004). Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J. 23, 4413–4422. doi: 10.1038/sj.emboj.7600442
Kolstad, T. R., van den Brink, J., MacQuaide, N., Lunde, P. K., Frisk, M., Aronsen, J. M., et al. (2018). Ryanodine receptor dispersion disrupts Ca2+ release in failing cardiac myocytes. eLife 7:e39427.
Kong, C. H. T., Bryant, S. M., Watson, J. J., Gadeberg, H. C., Roth, D. M., Patel, H. H., et al. (2018a). The effects of aging on the regulation of T-Tubular ICa by caveolin in mouse ventricular myocytes. J. Gerontol. A Biol. Sci. Med. Sci. 73, 711–719. doi: 10.1093/gerona/glx242
Kong, C. H. T., Bryant, S. M., Watson, J. J., Roth, D. M., Patel, H. H., Cannell, M. B., et al. (2019). Cardiac-specific overexpression of caveolin-3 preserves t-tubular ICa during heart failure in mice. Exp. Physiol. 104, 654–666. doi: 10.1113/ep087304
Kong, C. H. T., Rog-Zielinska, E. A., Kohl, P., Orchard, C. H., and Cannell, M. B. (2018b). Solute movement in the t-tubule system of rabbit and mouse cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 115, E7073–E7080.
Korhonen, T., Rapila, R., Ronkainen, V. P., Koivumaki, J. T., and Tavi, P. (2010). Local Ca2+ releases enable rapid heart rates in developing cardiomyocytes. J. Physiol. 588, 1407–1417. doi: 10.1113/jphysiol.2009.185173
Kostin, S., Scholz, D., Shimada, T., Maeno, Y., Mollnau, H., Hein, S., et al. (1998). The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 294, 449–460. doi: 10.1007/s004410051196
Lahiri, S. K., Quick, A. P., Samson-Couterie, B., Hulsurkar, M., Elzenaar, I., van Oort, R. J., et al. (2020). Nuclear localization of a novel calpain-2 mediated junctophilin-2 C-terminal cleavage peptide promotes cardiomyocyte remodeling. Basic Res. Cardiol. 115:49.
Lam, C. S. P., Gamble, G. D., Ling, L. H., Sim, D., Leong, K. T. G., Yeo, P. S. D., et al. (2018). Mortality associated with heart failure with preserved vs. reduced ejection fraction in a prospective international multi-ethnic cohort study. Eur. Heart J. 39, 1770–1780. doi: 10.1093/eurheartj/ehy005
Lambert, R., Srodulski, S., Peng, X., Margulies, K. B., Despa, F., and Despa, S. (2015). Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-Glucose cotransport. J. Am. Heart Assoc. 4:e002183.
Lammerding, J., Kamm, R. D., and Lee, R. T. (2004). Mechanotransduction in cardiac myocytes. Ann. N. Y. Acad. Sci. 1015, 53–70. doi: 10.1196/annals.1302.005
Landstrom, A. P., Kellen, C. A., Dixit, S. S., van Oort, R. J., Garbino, A., Weisleder, N., et al. (2011). Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ. Heart Fail. 4, 214–223. doi: 10.1161/circheartfailure.110.958694
Larbig, R., Torres, N., Bridge, J. H., Goldhaber, J. I., and Philipson, K. D. (2010). Activation of reverse Na+-Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: evidence from NCX knockout mice. J. Physiol. 588, 3267–3276. doi: 10.1113/jphysiol.2010.187708
Lawless, M., Caldwell, J. L., Radcliffe, E. J., Smith, C. E. R., Madders, G. W. P., Hutchings, D. C., et al. (2019). Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci. Rep. 9:6801.
Lee, E., Marcucci, M., Daniell, L., Pypaert, M., Weisz, O. A., Ochoa, G. C., et al. (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196. doi: 10.1126/science.1071362
Lekavich, C. L., Barksdale, D. J., Neelon, V., and Wu, J. R. (2015). Heart failure preserved ejection fraction (HFpEF): an integrated and strategic review. Heart Fail. Rev. 20, 643–653. doi: 10.1007/s10741-015-9506-7
Lenaerts, I., Bito, V., Heinzel, F. R., Driesen, R. B., Holemans, P., D’Hooge, J., et al. (2009). Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ. Res. 105, 876–885. doi: 10.1161/circresaha.109.206276
Li, J., Agvanian, S., Zhou, K., Shaw, R. M., and Hong, T. (2020). Exogenous cardiac bridging Integrator 1 benefits mouse hearts with pre-existing pressure overload-induced heart failure. Front. Physiol. 11:708. doi: 10.3389/fphys.2020.00708
Li, L. L., Guo, Q. J., Lou, H. Y., Liang, J. H., Yang, Y., Xing, X., et al. (2020). Nanobar array assay revealed complementary roles of BIN1 splice isoforms in cardiac T-Tubule morphogenesis. Nano Lett. 20, 6387–6395. doi: 10.1021/acs.nanolett.0c01957
Li, Q., O’Neill, S. C., Tao, T., Li, Y., Eisner, D., and Zhang, H. (2012). Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys. J. 102, 1471–1482. doi: 10.1016/j.bpj.2012.03.007
Li, R. C., Tao, J., Guo, Y. B., Wu, H. D., Liu, R. F., Bai, Y., et al. (2013). In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ. Res. 112, 601–605. doi: 10.1161/circresaha.112.300806
Lichter, J. G., Carruth, E., Mitchell, C., Barth, A. S., Aiba, T., Kass, D. A., et al. (2014). Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. J. Mol. Cell. Cardiol. 72, 186–195. doi: 10.1016/j.yjmcc.2014.03.012
Lindner, E. (1957). [Submicroscopic morphology of the cardiac muscle]. Z. Zellforsch. Mikrosk. Anat. (Vienna, Austria: 1948) 45, 702–746.
Lines, G. T., Sande, J. B., Louch, W. E., Mork, H. K., Grottum, P., and Sejersted, O. M. (2006). Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys. J. 91, 779–792. doi: 10.1529/biophysj.105.072447
Linke, W. A. (2008). Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 77, 637–648.
Lipsett, D. B., Frisk, M., Aronsen, J. M., Norden, E. S., Buonarati, O. R., Cataliotti, A., et al. (2019). Cardiomyocyte substructure reverts to an immature phenotype during heart failure. J. Physiol. 59, 1833–1853. doi: 10.1113/jp277273
Liu, C., Spinozzi, S., Chen, J. Y., Fang, X., Feng, W., Perkins, G., et al. (2019). Nexilin is a new component of junctional membrane complexes required for cardiac T-Tubule formation. Circulation 140, 55–66. doi: 10.1161/circulationaha.119.039751
Liu, C., Spinozzi, S., Feng, W., Chen, Z., Zhang, L., Zhu, S., et al. (2020). Homozygous G650del nexilin variant causes cardiomyopathy in mice. JCI Insight 5:e138780.
Liu, Y., Zhou, K., Li, J., Agvanian, S., Caldaruse, A. M., Shaw, S., et al. (2020). In mice subjected to chronic stress, exogenous cBIN1 preserves calcium-handling machinery and cardiac function. JACC Basic Transl. Sci. 5, 561–578. doi: 10.1016/j.jacbts.2020.03.006
Louch, W. E., and Nattel, S. (2017). T-tubular collagen: a new player in mechanosensing and disease? Cardiovasc. Res. 113, 839–840. doi: 10.1093/cvr/cvx091
Louch, W. E., Bito, V., Heinzel, F. R., Macianskiene, R., Vanhaecke, J., Flameng, W., et al. (2004). Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc. Res. 62, 63–73. doi: 10.1016/j.cardiores.2003.12.031
Louch, W. E., Hake, J., Jolle, G. F., Mork, H. K., Sjaastad, I., Lines, G. T., et al. (2010a). Control of Ca2+ release by action potential configuration in normal and failing murine cardiomyocytes. Biophys. J. 99, 1377–1386. doi: 10.1016/j.bpj.2010.06.055
Louch, W. E., Hake, J., Mork, H. K., Hougen, K., Skrbic, B., and Ursu, D. (2013). Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes: evidence for altered configuration of Ca2+ release units? J. Mol. Cell. Cardiol. 58, 41–52. doi: 10.1016/j.yjmcc.2013.01.014
Louch, W. E., Koivumaki, J. T., and Tavi, P. (2015). Calcium signalling in developing cardiomyocytes: implications for model systems and disease. J. Physiol. 593, 1047–1063. doi: 10.1113/jphysiol.2014.274712
Louch, W. E., Mork, H. K., Sexton, J., Stromme, T. A., Laake, P., Sjaastad, I., et al. (2006). T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J. Physiol. 574, 519–533. doi: 10.1113/jphysiol.2006.107227
Louch, W. E., Sejersted, O. M., and Swift, F. (2010b). There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J. Biomed. Biotechnol. 2010:503906.
Louch, W. E., Stokke, M. K., Sjaastad, I., Christensen, G., and Sejersted, O. M. (2012). No rest for the weary: diastolic calcium homeostasis in the normal and failing myocardium. Physiology (Bethesda) 27, 308–323. doi: 10.1152/physiol.00021.2012
Loucks, A. D., O’Hara, T., and Trayanova, N. A. (2018). Degradation of T-Tubular microdomains and altered cAMP compartmentation lead to emergence of arrhythmogenic triggers in heart failure myocytes: an in silico study. Front. Physiol. 9:1737. doi: 10.3389/fphys.2018.01737
Lyon, A. R., MacLeod, K. T., Zhang, Y., Garcia, E., Kanda, G. K., Lab, M. J., et al. (2009). Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. U.S.A. 106, 6854–6859. doi: 10.1073/pnas.0809777106
Lyon, A. R., Nikolaev, V. O., Miragoli, M., Sikkel, M. B., Paur, H., Benard, L., et al. (2012). Plasticity of surface structures and beta(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ. Heart Fail. 5, 357–365. doi: 10.1161/circheartfailure.111.964692
Lyu, Y., Verma, V. K., Lee, Y., Taleb, I., Badolia, R., Shankar, T. S., et al. (2021). Remodeling of t-system and proteins underlying excitation-contraction coupling in aging versus failing human heart. NPJ Aging Mech. Dis. 7:16.
Mackova, K., Zahradnikova, A. Jr., Hotka, M., Hoffmannova, B., Zahradnik, I., and Zahradnikova, A. (2017). Calcium release-dependent inactivation precedes formation of the tubular system in developing rat cardiac myocytes. Eur. Biophys. J. 46, 691–703. doi: 10.1007/s00249-017-1249-z
Manfra, O., Frisk, M., and Louch, W. E. (2017). Regulation of cardiomyocyte t-tubular structure: opportunities for therapy. Curr. Heart Fail. Rep. 14, 167–178. doi: 10.1007/s11897-017-0329-9
Maron, B. J., Ferrans, V. J., and Roberts, W. C. (1975). Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am. J. Pathol. 79, 387–434.
Marx, S. O., Gaburjakova, J., Gaburjakova, M., Henrikson, C., Ondrias, K., and Marks, A. R. (2001). Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. 88, 1151–1158. doi: 10.1161/hh1101.091268
McNary, T. G., Bridge, J. H., and Sachse, F. B. (2011). Strain transfer in ventricular cardiomyocytes to their transverse tubular system revealed by scanning confocal microscopy. Biophys. J. 100, L53–L55.
McNutt, N. S. (1975). Ultrastructure of the myocardial sarcolemma. Circ. Res. 37, 1–13. doi: 10.1161/01.res.37.1.1
Meethal, S. V., Potter, K. T., Redon, D., Munoz-del-Rio, A., Kamp, T. J., Valdivia, H. H., et al. (2007). Structure-function relationships of Ca spark activity in normal and failing cardiac myocytes as revealed by flash photography. Cell Calcium 41, 123–134. doi: 10.1016/j.ceca.2006.05.006
Melnyk, P., Zhang, L., Shrier, A., and Nattel, S. (2002). Differential distribution of Kir2.1 and Kir2.3 subunits in canine atrium and ventricle. Am. J. Physiol. Heart Circ. Physiol. 283, H1123–H1133.
Minamisawa, S., Oshikawa, J., Takeshima, H., Hoshijima, M., Wang, Y., Chien, K. R., et al. (2004). Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem. Biophys. Res. Commun. 325, 852–856. doi: 10.1016/j.bbrc.2004.10.107
Mohler, P. J., Davis, J. Q., and Bennett, V. (2005). Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3:e423. doi: 10.1371/journal.pbio.0030423
Mork, H. K., Sjaastad, I., Sande, J. B., Periasamy, M., Sejersted, O. M., and Louch, W. E. (2007). Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J. Mol. Cell. Cardiol. 43, 177–186. doi: 10.1016/j.yjmcc.2007.05.004
Muller, A. J., Baker, J. F., DuHadaway, J. B., Ge, K., Farmer, G., Donover, P. S., et al. (2003). Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol. Cell. Biol. 23, 4295–4306. doi: 10.1128/mcb.23.12.4295-4306.2003
Munro, M. L. I, Jayasinghe, D., Wang, Q., Quick, A., Wang, W., Baddeley, D., et al. (2016). Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J. Cell Sci. 129, 4388–4398.
Munro, M. L., and Soeller, C. (2016). Early transverse tubule development begins in utero in the sheep heart. J. Muscle Res. Cell. Motil. 37, 195–202. doi: 10.1007/s10974-016-9462-4
Nikolova, A. P., Hitzeman, T. C., Baum, R., Caldaruse, A. M., Agvanian, S., Xie, Y., et al. (2018). Association of a novel diagnostic biomarker, the plasma cardiac bridging Integrator 1 score, with heart failure with preserved ejection fraction and cardiovascular hospitalization. JAMA Cardiol. 3, 1206–1210. doi: 10.1001/jamacardio.2018.3539
Nishi, M., Mizushima, A., Nakagawara, K., and Takeshima, H. (2000). Characterization of human junctophilin subtype genes. Biochem. Biophys. Res. Commun. 273, 920–927. doi: 10.1006/bbrc.2000.3011
Nyström, G. (1897). Über die lymphbahnen des Herzens. Arch. Anat. Physiol. (Anat. Abt.) 12, 361–378.
Oakley, R. H., Ren, R., Cruz-Topete, D., Bird, G. S., Myers, P. H., Boyle, M. C., et al. (2013). Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. U.S.A. 110, 17035–17040. doi: 10.1073/pnas.1302546110
Orchard, C. H., Bryant, S. M., and James, A. F. (2013). Do t-tubules play a role in arrhythmogenesis in cardiac ventricular myocytes? J. Physiol. 591, 4141–4147. doi: 10.1113/jphysiol.2013.254540
Oyehaug, L., Loose, K. O., Jolle, G. F., Roe, A. T., Sjaastad, I., Christensen, G., et al. (2013). Synchrony of cardiomyocyte Ca2+ release is controlled by T-tubule organization, SR Ca2+ content, and ryanodine receptor Ca2+ sensitivity. Biophys. J. 104, 1685–1697. doi: 10.1016/j.bpj.2013.03.022
Parikh, S. S., Blackwell, D. J., Gomez-Hurtado, N., Frisk, M., Wang, L., Kim, K., et al. (2017). Thyroid and glucocorticoid hormones promote functional t-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 121, 1323–1330. doi: 10.1161/circresaha.117.311920
Parton, R. G., Way, M., Zorzi, N., and Stang, E. (1997). Caveolin-3 associates with developing T-tubules during muscle differentiation. J. Cell Biol. 136, 137–154. doi: 10.1083/jcb.136.1.137
Paulus, W. J., and Tschope, C. (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271.
Picas, L., Viaud, J., Schauer, K., Vanni, S., Hnia, K., Fraisier, V., et al. (2014). BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun. 5:5647.
Pieske, B., Tschope, C., de Boer, R. A., Fraser, A. G., Anker, S. D., Donal, E., et al. (2019). How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European society of cardiology (ESC). Eur. Heart J. 40, 3297–3317. doi: 10.1093/eurheartj/ehz641
Pinali, C., Bennett, H., Davenport, J. B., Trafford, A. W., and Kitmitto, A. (2013). Three-dimensional reconstruction of cardiac sarcoplasmic reticulum reveals a continuous network linking transverse-tubules: this organization is perturbed in heart failure. Circ. Res. 113, 1219–1230. doi: 10.1161/circresaha.113.301348
Pinali, C., Malik, N., Davenport, J. B., Allan, L. J., Murfitt, L., Iqbal, M. M., et al. (2017). Post-myocardial infarction T-tubules form enlarged branched structures with dysregulation of Junctophilin-2 and Bridging Integrator 1 (BIN-1). J. Am. Heart Assoc. 6:e004834.
Prins, K. W., Asp, M. L., Zhang, H., Wang, W., and Metzger, J. M. (2016). Microtubule-mediated misregulation of Junctophilin-2 underlies T-Tubule disruptions and calcium mishandling in mdx Mice. JACC Basic Transl. Sci. 1, 122–130. doi: 10.1016/j.jacbts.2016.02.002
Prokic, I., Cowling, B. S., Kutchukian, C., Kretz, C., Tasfaout, H., Gache, V., et al. (2020). Differential physiological roles for BIN1 isoforms in skeletal muscle development, function and regeneration. Dis. Model. Mech. 13:dmm044354.
Quick, A. P., Wang, Q., Philippen, L. E., Barreto-Torres, G., Chiang, D. Y., Beavers, D., et al. (2017). SPEG (Striated Muscle Preferentially Expressed Protein Kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ. Res. 120, 110–119. doi: 10.1161/circresaha.116.309977
Rajabi, M., Kassiotis, C., Razeghi, P., and Taegtmeyer, H. (2007). Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail. Rev. 12, 331–343. doi: 10.1007/s10741-007-9034-1
Rapila, R., Korhonen, T., and Tavi, P. (2008). Excitation-contraction coupling of the mouse embryonic cardiomyocyte. J. Gen. Physiol. 132, 397–405. doi: 10.1085/jgp.200809960
Reynolds, J. O., Chiang, D. Y., Wang, W., Beavers, D. L., Dixit, S. S., Skapura, D. G., et al. (2013). Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc. Res. 100, 44–53. doi: 10.1093/cvr/cvt133
Reynolds, J. O., Quick, A. P., Wang, Q., Beavers, D. L., Philippen, L. E., and Showell, J. (2016). Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int. J. Cardiol. 225, 371–380. doi: 10.1016/j.ijcard.2016.10.021
Richards, M. A., Clarke, J. D., Saravanan, P., Voigt, N., Dobrev, D., Eisner, D. A., et al. (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 301, H1996–H2005.
Roe, A. T., Aronsen, J. M., Skardal, K., Hamdani, N., Linke, W. A., Danielsen, H. E., et al. (2017). Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc. Res. 113, 1161–1172. doi: 10.1093/cvr/cvx087
Roe, A. T., Frisk, M., and Louch, W. E. (2015). Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 21, 431–448. doi: 10.2174/138161282104141204124129
Roe, A. T., Ruud, M., Espe, E. K., Manfra, O., Longobardi, S., Aronsen, J. M., et al. (2019). Regional diastolic dysfunction in post-infarction heart failure: role of local mechanical load and SERCA expression. Cardiovasc. Res. 115, 752–764. doi: 10.1093/cvr/cvy257
Rog-Zielinska, E. A., Moss, R., Kaltenbacher, W., Greiner, J., Verkade, P., Seemann, G., et al. (2021a). Nano-scale morphology of cardiomyocyte t-tubule/sarcoplasmic reticulum junctions revealed by ultra-rapid high-pressure freezing and electron tomography. J. Mol. Cell. Cardiol. 153, 86–92. doi: 10.1016/j.yjmcc.2020.12.006
Rog-Zielinska, E. A., Scardigli, M., Peyronnet, R., Zgierski-Johnston, C. M., Greiner, J., Madl, J., et al. (2021b). Beat-by-Beat cardiomyocyte t-tubule deformation drives tubular content exchange. Circ. Res. 128, 203–215. doi: 10.1161/circresaha.120.317266
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi: 10.1038/s41586-018-0016-3
Royer, B., Hnia, K., Gavriilidis, C., Tronchere, H., Tosch, V., and Laporte, J. (2013). The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. EMBO Rep. 14, 907–915. doi: 10.1038/embor.2013.119
Russell, J., Du Toit, E. F., Peart, J. N., Patel, H. H., and Headrick, J. P. (2017). Myocyte membrane and microdomain modifications in diabetes: determinants of ischemic tolerance and cardioprotection. Cardiovasc. Diabetol. 16:155.
Sachse, F. B., Torres, N. S., Savio-Galimberti, E., Aiba, T., Kass, D. A., Tomaselli, G. F., et al. (2012). Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ. Res. 110, 588–597. doi: 10.1161/circresaha.111.257428
Sah, R., Ramirez, R. J., and Backx, P. H. (2002). Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ. Res. 90, 165–173. doi: 10.1161/hh0202.103315
Sanchez-Alonso, J. L., Bhargava, A., O’Hara, T., Glukhov, A. V., Schobesberger, S., Bhogal, N., et al. (2016). Microdomain-specific modulation of L-Type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ. Res. 119, 944–955. doi: 10.1161/circresaha.116.308698
Savio-Galimberti, E., Frank, J., Inoue, M., Goldhaber, J. I., Cannell, M. B., Bridge, J. H., et al. (2008). Novel features of the rabbit transverse tubular system revealed by quantitative analysis of three-dimensional reconstructions from confocal images. Biophys. J. 95, 2053–2062. doi: 10.1529/biophysj.108.130617
Scardigli, M., Crocini, C., Ferrantini, C., Gabbrielli, T., Silvestri, L., Coppini, R., et al. (2017). Quantitative assessment of passive electrical properties of the cardiac T-tubular system by FRAP microscopy. Proc. Natl. Acad. Sci. U.S.A. 114, 5737–5742. doi: 10.1073/pnas.1702188114
Schaper, J., Froede, R., Hein, S., Buck, A., Hashizume, H., Speiser, B., et al. (1991). Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 83, 504–514. doi: 10.1161/01.cir.83.2.504
Schmitto, J. D., Krabatsch, T., Damme, L., and Netuka, I. (2018). Less invasive HeartMate 3 left ventricular assist device implantation. J. Thorac. Dis. 10, S1692–S1695.
Schobesberger, S., Wright, P., Tokar, S., Bhargava, A., Mansfield, C., Glukhov, A. V., et al. (2017). T-tubule remodelling disturbs localized beta2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 113, 770–782. doi: 10.1093/cvr/cvx074
Schulson, M. N., Scriven, D. R., Fletcher, P., and Moore, E. D. (2011). Couplons in rat atria form distinct subgroups defined by their molecular partners. J. Cell Sci. 124, 1167–1174. doi: 10.1242/jcs.080929
Scriven, D. R., Asghari, P., Schulson, M. N., and Moore, E. D. (2010). Analysis of Cav1.2 and ryanodine receptor clusters in rat ventricular myocytes. Biophys. J. 99, 3923–3929. doi: 10.1016/j.bpj.2010.11.008
Scriven, D. R., Dan, P., and Moore, E. D. (2000). Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 79, 2682–2691. doi: 10.1016/s0006-3495(00)76506-4
Seidel, T., Fiegle, D. J., Baur, T. J., Ritzer, A., Nay, S., Heim, C., et al. (2019). Glucocorticoids preserve the t-tubular system in ventricular cardiomyocytes by upregulation of autophagic flux. Basic Res. Cardiol. 114:47. doi: 10.3390/cells9010047
Seidel, T., Navankasattusas, S., Ahmad, A., Diakos, N. A., Xu, W. D., Tristani-Firouzi, M., et al. (2017a). Sheet-like remodeling of the transverse tubular system in human heart failure impairs excitation-contraction coupling and functional recovery by mechanical unloading. Circulation 135, 1632–1645. doi: 10.1161/circulationaha.116.024470
Seidel, T., Sankarankutty, A. C., and Sachse, F. B. (2017b). Remodeling of the transverse tubular system after myocardial infarction in rabbit correlates with local fibrosis: a potential role of biomechanics. Prog. Biophys. Mol. Biol. 130, 302–314. doi: 10.1016/j.pbiomolbio.2017.07.006
Severs, N. J., Slade, A. M., Powell, T., Twist, V. W., and Jones, G. E. (1985). Morphometric analysis of the isolated calcium-tolerant cardiac myocyte. Organelle volumes, sarcomere length, plasma membrane surface folds, and intramembrane particle density and distribution. Cell Tissue Res. 240, 159–168.
Shah, S. J., Aistrup, G. L., Gupta, D. K., O’Toole, M. J., Nahhas, A. F., Schuster, D., et al. (2014). Ultrastructural and cellular basis for the development of abnormal myocardial mechanics during the transition from hypertension to heart failure. Am. J. Physiol. Heart Circ. Physiol. 306, H88–H100.
Sheard, T. M. D., Hurley, M. E., Colyer, J., White, E., Norman, R., Pervolaraki, E., et al. (2019). Three-dimensional and chemical mapping of intracellular signaling nanodomains in health and disease with enhanced expansion microscopy. ACS Nano 13, 2143–2157.
Sheldon, C. A., Friedman, W. F., and Sybers, H. D. (1976). Scanning electron microscopy of fetal and neonatal lamb cardiac cells. J. Mol. Cell. Cardiol. 8, 853–862. doi: 10.1016/0022-2828(76)90068-7
Shen, X., van den Brink, J., Hou, Y., Colli, D., Le, C., Kolstad, T. R., et al. (2019). 3D dSTORM imaging reveals novel detail of ryanodine receptor localization in rat cardiac myocytes. J. Physiol. 597, 399–418. doi: 10.1113/jp277360
Shiferaw, Y., Aistrup, G. L., Louch, W. E., and Wasserstrom, J. A. (2020). Remodeling promotes proarrhythmic disruption of calcium homeostasis in failing atrial myocytes. Biophys. J. 118, 476–491. doi: 10.1016/j.bpj.2019.12.012
Silbernagel, N., Korner, A., Balitzki, J., Jaggy, M., Bertels, S., Richter, B., et al. (2020). Shaping the heart: structural and functional maturation of iPSC-cardiomyocytes in 3D-micro-scaffolds. Biomaterials 227:119551. doi: 10.1016/j.biomaterials.2019.119551
Simpson, F. O. (1965). The transverse tubular system in mammalian myocardial cells. Am. J. Anat. 117, 1–17. doi: 10.1002/aja.1001170102
Singh, J. K., Barsegyan, V., Bassi, N., Marszalec, W., Tai, S., Mothkur, S., et al. (2017). T-tubule remodeling and increased heterogeneity of calcium release during the progression to heart failure in intact rat ventricle. Physiol. Rep. 5:e13540. doi: 10.14814/phy2.13540
Sipido, K. R., Maes, M., and Van de Werf, F. (1997). Low efficiency of Ca2+ entry through the Na(+)-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na(+)-Ca2+ exchange. Circ. Res. 81, 1034–1044. doi: 10.1161/01.res.81.6.1034
Smyrnias, I., Mair, W., Harzheim, D., Walker, S. A., Roderick, H. L., and Bootman, M. D. (2010). Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium 47, 210–223. doi: 10.1016/j.ceca.2009.10.001
Snopko, R. M., Ramos-Franco, J., Di Maio, A., Karko, K. L., Manley, C., Piedras-Renteria, E., et al. (2008). Ca2+ sparks and cellular distribution of ryanodine receptors in developing cardiomyocytes from rat. J. Mol. Cell. Cardiol. 44, 1032–1044. doi: 10.1016/j.yjmcc.2008.03.015
Sobie, E. A., Guatimosim, S., Gomez-Viquez, L., Song, L. S., Hartmann, H., Saleet Jafri, M., et al. (2006). The Ca2+ leak paradox and rogue ryanodine receptors: SR Ca 2+ efflux theory and practice. Prog. Biophys. Mol. Biol. 90, 172–185. doi: 10.1016/j.pbiomolbio.2005.06.010
Soeller, C., and Cannell, M. B. (1999). Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ. Res. 84, 266–275. doi: 10.1161/01.res.84.3.266
Song, L. S., Sobie, E. A., McCulle, S., Lederer, W. J., Balke, C. W., and Cheng, H. (2006). Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 103, 4305–4310. doi: 10.1073/pnas.0509324103
Song, Z., Liu, M. B., and Qu, Z. (2018). Transverse tubular network structures in the genesis of intracellular calcium alternans and triggered activity in cardiac cells. J. Mol. Cell. Cardiol. 114, 288–299. doi: 10.1016/j.yjmcc.2017.12.003
Sperelakis, N., and Rubio, R. (1971). An orderly lattice of axial tubules which interconnect adjacent transverse tubules in guinea-pig ventricular myocardium. J. Mol. Cell. Cardiol. 2, 211–220. doi: 10.1016/0022-2828(71)90054-x
Spinozzi, S., Liu, C., Chen, Z., Feng, W., Zhang, L., Ouyang, K., et al. (2020). Nexilin is necessary for maintaining the transverse-axial tubular system in adult cardiomyocytes. Circ. Heart Fail. 13:e006935.
Stewart, J. M., and Page, E. (1978). Improved stereological techniques for studying myocardial cell growth: application to external sarcolemma, T system, and intercalated disks of rabbit and rat hearts. J. Ultrastruct. Res. 65, 119–134. doi: 10.1016/s0022-5320(78)90050-3
Stolen, T. O., Hoydal, M. A., Kemi, O. J., Catalucci, D., Ceci, M., Aasum, E., et al. (2009). Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 105, 527–536. doi: 10.1161/circresaha.109.199810
Sun, X. H., Protasi, F., Takahashi, M., Takeshima, H., Ferguson, D. G., and Franzini-Armstrong, C. (1995). Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J. Cell Biol. 129, 659–671. doi: 10.1083/jcb.129.3.659
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na+/Ca2+ -exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Swift, F., Franzini-Armstrong, C., Oyehaug, L., Enger, U. H., Andersson, K. B., Christensen, G., et al. (2012). Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc. Natl. Acad. Sci. U.S.A. 109, 3997–4001. doi: 10.1073/pnas.1120172109
Swift, F., Stromme, T. A., Amundsen, B., Sejersted, O. M., and Sjaastad, I. (2006). Slow diffusion of K+ in the T tubules of rat cardiomyocytes. J. Appl. Physiol. (1985) 101, 1170–1176. doi: 10.1152/japplphysiol.00297.2006
Takeshima, H., Komazaki, S., Nishi, M., Iino, M., and Kangawa, K. (2000). Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22. doi: 10.1016/s1097-2765(00)00003-4
Tarzia, V., Di Giammarco, G., Di Mauro, M., Bortolussi, G., Maccherini, M., Tursi, V., et al. (2016). From bench to bedside: can the improvements in left ventricular assist device design mitigate adverse events and increase survival? J. Thorac. Cardiovasc. Surg. 151, 213–217. doi: 10.1016/j.jtcvs.2015.09.107
Tasfaout, H., Buono, S., Guo, S., Kretz, C., Messaddeq, N., Booten, S., et al. (2017). Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 8:15661.
Thomas, M. J., Sjaastad, I., Andersen, K., Helm, P. J., Wasserstrom, J. A., Sejersted, O. M., et al. (2003). Localization and function of the Na+/Ca2+ -exchanger in normal and detubulated rat cardiomyocytes. J. Mol. Cell. Cardiol. 35, 1325–1337. doi: 10.1016/j.yjmcc.2003.08.005
Tidball, J. G., Cederdahl, J. E., and Bers, D. M. (1991). Quantitative analysis of regional variability in the distribution of transverse tubules in rabbit myocardium. Cell Tissue Res. 264, 293–298. doi: 10.1007/bf00313966
Uchida, K., and Lopatin, A. N. (2018). Diffusional and electrical properties of T-Tubules are governed by their constrictions and dilations. Biophys. J. 114, 437–449. doi: 10.1016/j.bpj.2017.11.3742
Vaidya, Y., and Dhamoon, A. S. (2019). StatPearls. Treasure Island (FL): StatPearls Publishing StatPearls Publishing LLC.
van Oort, R. J., Garbino, A., Wang, W., Dixit, S. S., Landstrom, A. P., Gaur, N., et al. (2011). Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123, 979–988. doi: 10.1161/circulationaha.110.006437
Wagner, E., Lauterbach, M. A., Kohl, T., Westphal, V., Williams, G. S., and Steinbrecher, J. H. (2012). Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 111, 402–414.
Wakili, R., Yeh, Y. H., Yan Qi, X., Greiser, M., Chartier, D., Nishida, K., et al. (2010). Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ. Arrhythm. Electrophysiol. 3, 530–541.
Wang, H., Li, Z., Wang, J., Sun, K., Cui, Q., Song, L., et al. (2010). Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 87, 687–693.
Wang, W., Landstrom, A. P., Wang, Q., Munro, M. L., Beavers, D., Ackerman, M. J., et al. (2014). Reduced junctional Na+/Ca2+ -exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin-2-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 307, H1317–H1326.
Wang, X., Xie, W., Zhang, Y., Lin, P., Han, L., Han, P., et al. (2010). Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83.
Wang, Y., Chen, B., Huang, C. K., Guo, A., Wu, J., Zhang, X., et al. (2018). Targeting calpain for heart failure therapy: implications from multiple murine models. JACC Basic Transl. Sci. 3, 503–517.
Ward, M. L., and Crossman, D. J. (2014). Mechanisms underlying the impaired contractility of diabetic cardiomyopathy. World J. Cardiol. 6, 577–584. doi: 10.4330/wjc.v6.i7.577
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531. doi: 10.1161/circresaha.109.212324
Wright, P. T., Bhogal, N. K., Diakonov, I., Pannell, L. M. K., Perera, R. K., and Bork, N. I. (2018). Cardiomyocyte membrane structure and cAMP compartmentation produce anatomical variation in beta2AR-cAMP responsiveness in murine hearts. Cell Rep. 23, 459–469. doi: 10.1016/j.celrep.2018.03.053
Wu, C. Y., Chen, B., Jiang, Y. P., Jia, Z., Martin, D. W., Liu, S., et al. (2014). Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J. Am. Heart Assoc. 3: e000527.
Wu, C. Y., Jia, Z., Wang, W., Ballou, L. M., Jiang, Y. P., Chen, B., et al. (2011). PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS One 6:e24404. doi: 10.1371/journal.pone.0024404
Wu, H. D., Xu, M., Li, R. C., Guo, L., Lai, Y. S., Xu, S. M., et al. (2012). Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc. Res. 95, 430–438. doi: 10.1093/cvr/cvs195
Xie, Y. P., Chen, B., Sanders, P., Guo, A., Li, Y., Zimmerman, K., et al. (2012). Sildenafil prevents and reverses transverse-tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension 59, 355–362. doi: 10.1161/hypertensionaha.111.180968
Xie, Y., Yang, Y., Galice, S., Bers, D. M., and Sato, D. (2019). Size matters: ryanodine receptor cluster size heterogeneity potentiates calcium waves. Biophys. J. 116, 530–539. doi: 10.1016/j.bpj.2018.12.017
Xu, M., Wu, H. D., Li, R. C., Zhang, H. B., Wang, M., Tao, J., et al. (2012). Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ. Res. 111, 837–841. doi: 10.1161/circresaha.112.277418
Xu, M., Zhou, P., Xu, S. M., Liu, Y., Feng, X., Bai, S. H., et al. (2007). Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol 5:e21. doi: 10.1371/journal.pbio.0050021
Yamakawa, S., Wu, D., Dasgupta, M., Pedamallu, H., Gupta, B., Modi, R., et al. (2021). Role of t-tubule remodeling on mechanisms of abnormal calcium release during heart failure development in canine ventricle. Am. J. Physiol. Heart Circ. Physiol. 320, H1658–H1669.
Zhang, C., Chen, B., Guo, A., Zhu, Y., Miller, J. D., Gao, S., et al. (2014). Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129, 1742–1750. doi: 10.1161/circulationaha.113.008452
Zhang, C., Chen, B., Wang, Y., Guo, A., Tang, Y., Khataei, T., et al. (2017). MG53 is dispensable for T-tubule maturation but critical for maintaining T-tubule integrity following cardiac stress. J. Mol. Cell. Cardiol. 112, 123–130. doi: 10.1016/j.yjmcc.2017.08.007
Keywords: t-tubules, dyad, cardiomyocyte, calcium homeostasis, heart failure
Citation: Setterberg IE, Le C, Frisk M, Perdreau-Dahl H, Li J and Louch WE (2021) The Physiology and Pathophysiology of T-Tubules in the Heart. Front. Physiol. 12:718404. doi: 10.3389/fphys.2021.718404
Received: 31 May 2021; Accepted: 07 July 2021;
Published: 09 September 2021.
Edited by:
Daniel M. Johnson, The Open University, United KingdomReviewed by:
Jessica Caldwell, University of California, Davis, United StatesCopyright © 2021 Setterberg, Le, Frisk, Perdreau-Dahl, Li and Louch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William E. Louch, dy5lLmxvdWNoQG1lZGlzaW4udWlvLm5v
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.