 Kyuwon Lee
Kyuwon Lee Hyeonju Yu1
Hyeonju Yu1 Stephanie Shouse
Stephanie Shouse Byungwhi Kong
Byungwhi Kong Seong-Ho Lee
Seong-Ho Lee Kwang Suk Ko
Kwang Suk Ko- 1Department of Nutritional Science and Food Management, College of Science and Industry Convergence, Ewha Womans University, Seoul, South Korea
- 2Center of Excellence for Poultry Science, University of Arkansas System Division of Agriculture, Fayetteville, AR, United States
- 3Department of Nutrition and Food Science, College of Agriculture and Natural Resources, University of Maryland, College Park, MD, United States
- 4Karsh Division of Gastroenterology and Hepatology, Department of Medicine, Cedars-Sinai Medical Center, Beverly Hills, CA, United States
Prohibitin 1 (PHB1) is an evolutionarily conserved and ubiquitously expressed protein that stabilizes mitochondrial chaperone. Our previous studies showed that liver-specific Phb1 deficiency induced liver injuries and aggravated lipopolysaccharide (LPS)-induced innate immune responses. In this study, we performed RNA-sequencing (RNA-seq) analysis with liver tissues to investigate global gene expression among liver-specific Phb1−/−, Phb1+/−, and WT mice, focusing on the differentially expressed (DE) genes between Phb1+/− and WT. When 78 DE genes were analyzed for biological functions, using ingenuity pathway analysis (IPA) tool, lipid metabolism-related genes, including insulin receptor (Insr), sterol regulatory element-binding transcription factor 1 (Srebf1), Srebf2, and SREBP cleavage-activating protein (Scap) appeared to be downregulated in liver-specific Phb1+/− compared with WT. Diseases and biofunctions analyses conducted by IPA verified that hepatic system diseases, including liver fibrosis, liver hyperplasia/hyperproliferation, and liver necrosis/cell death, which may be caused by hepatotoxicity, were highly associated with liver-specific Phb1 deficiency in mice. Interestingly, of liver disease-related 5 DE genes between Phb1+/− and WT, the mRNA expressions of forkhead box M1 (Foxm1) and TIMP inhibitor of metalloproteinase (Timp1) were matched with validation for RNA-seq in liver tissues and AML12 cells transfected with Phb1 siRNA. The results in this study provide additional insights into molecular mechanisms responsible for increasing susceptibility of liver injuries associated with hepatic Phb1.
Introduction
Liver disease is a comprehensive term, including diverse stages of the disorder related to liver injury, and is one of the main causes of illness and death worldwide. According to Globocan 2018 by the WHO, the incidence and mortality of liver cancer are the sixth and third highest in both sexes and all ages, respectively (Bray et al., 2018). Although the risk factors include hepatitis B and C virus, alcohol, obesity, etc., the cellular regulatory mechanisms for liver disease and accountable evidence for physiological responses are yet to be explored (Smalling et al., 2013). It may be crucial to identify specific genes and pathway changes by liver injuries through whole transcriptome analyses of a relevant animal model. RNA-sequencing (RNA-seq) can proceed with a whole-genome survey of gene expression and provide a digital measure of the presence and prevalence of transcripts from known and previously unknown genes (Wang et al., 2010; Benjamin et al., 2014). The objective of the present study was to conduct global gene expression analyses of liver-specific Prohibitin 1 (Phb1) knockout (KO) mice as a liver disease model. PHB1 is an evolutionarily conserved and ubiquitously expressed protein that stabilizes mitochondrial chaperone (Yang et al., 2018). The protein is located to inner mitochondrial membrane and nucleus, so it regulates several important cellular processes, including apoptosis, cell proliferation, and transcriptional regulation by interacting with retinoblastoma protein (Rb), p53, and E2F transcription factors (Nijtmans et al., 2000; Ramani et al., 2016).
Liver-specific Phb1 homozygous KO (referred to as Phb1−/−) mouse showed positive staining for OV-6, an oval cell marker, and glutathione S-transferase Pi (GST Pi), a preneoplastic marker, at 3 weeks old and exhibited spontaneous liver injury, fibrosis, and hepatocellular carcinoma (HCC) from 8 months of age (Ko et al., 2010). A follow-up research study demonstrated that lipopolysaccharide (LPS)-innate immune responses were exacerbated by Phb1 deficiency, which mimics Phb1−/−, in murine macrophages (Jung et al., 2020). Furthermore, another research showed that there were no significant differences in body weight between liver-specific Phb1 heterozygote (referred to as Phb1+/−) and wild-type (WT, referred to as Phb1+/+) mice, but Phb1+/− showed more severe liver damage when feeding a methionine or choline-deficient diet, compared with WT (Heo and Ko, 2019).
Collectively, these results suggested that liver-specific Phb1 deficient mice may be a suitable model to investigate molecular mechanisms related to hepatotoxicity. In an earlier study, we observed phenotypic differences and differential gene expression, using microarray assay between Phb1−/− and WT (Ko et al., 2010). On the basis of the result, we have also investigated the vulnerability of hepatotoxicity in deletion of Phb1 (Heo and Ko, 2019; Jung et al., 2020). As part of research elucidating the susceptibility, it is necessary to observe the genetic change in liver-specific Phb1+/−, compared with WT to determine which genetic factors increases the susceptibility of liver injuries. In the current study, we aimed to investigate genome-wide gene expression among liver-specific Phb1−/−, Phb1+/−, and WT mice by using the RNA-seq method, focusing on the differentially expressed (DE) genes between Phb1+/− and WT. Also, we verified the expression level of hepatic disease-related genes in normal murine hepatocytes transfected with Phb1 siRNA to compare RNA-seq data. Through this comparison, DE genes showing greater differences between Phb1+/− and WT may provide sharper perceptions of alteration of physiological responses in a liver-specific Phb1-deficient mouse as a liver disease model.
Materials and Methods
Animals, Liver Tissue, and Total RNA Isolation
This study about sample collection was approved by Cedars-Sinai Medical Centers (Los Angeles, CA, USA) (IACUC 9370). Liver-specific Phb1 deficient mice were generated as previously described (Ko et al., 2010). Briefly, liver-specific Phb1 KO [Phb1loxp/loxp, Albumin-Cre+/+ (Alb-Cre+/+)] mice on a C57BL/6 background, and Alb-Cre+/+ mice were maintained for the generation of liver-specific Phb1-deficient mice. Selected three males and three females per Phb1−/−, Phb1+/−, and WT (4–5 weeks old, n = 6/genotype) were sacrificed for the harvest of liver tissue. Obtained samples were stored at −80°C until analysis. Total RNA was isolated from liver tissue, using TRIzol reagent (ThermoFisher, Waltham, MA, USA) according to the guideline of the manufacturer.
RNA-seq and Data Analysis
The RNA quality based on the RNA integrity number (RIN) was assessed using the RNA R6K assay for the Agilent 2200 TapeStation (Agilent Technology, Santa Clara, CA, USA). RIN scores were ranged between 8.5 and 9.5. For the RNA-seq analysis, library preparation and sequencing analyses were carried out at the Research Technology Support Facility of Michigan State University (East Lansing, MI, USA). Transcriptome (n = 6 for each WT, Phb1+/−, and Phb1−/− mice, including three males and three females per group) was analyzed, using a 1 × 50 bp single-end-read method of Illumina HiSeq system (Illumina Inc., San Diego, CA, USA) as described in earlier reports (Kong et al., 2017; Lee et al., 2020). The raw reads were aligned with the mouse reference genome (GRCm38.p6) downloaded from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.26) using ArrayStar program in Lagergene software package (DNAStar Inc., Madison, WI, USA). Total mapped counts were transformed into log2 values of the number of reads per million (RPM) to stabilize the variance, and then further quantile normalization was performed. Normalized values were subjected to further statistical analyses performed by JMP Genomics 9 (SAS Institute Inc., Cary, NC). The one-way ANOVA statistics was used to compare among WT, Phb1+/−, and Phb1−/− mice. Genes showing >2-fold changes (1 in log2) differences and <0.05 false discovery rate (FDR) in the comparisons were considered DE genes. The raw data were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under accession numbers: SRR14876823, SRR14876822, SRR14876815, SRR14876812, SRR14876818, SRR14876817, SRR14876813, SRR14876821, SRR14876820, SRR14876819, SRR14876816, SRR14876814, SRR14876807, SRR14876811, SRR14876810, SRR14876809, SRR14876808, and SRR14876806.
Bioinformatics Pathway Analyses
For pathway analyses, ingenuity pathway analysis (IPA; Qiagen, Valencia, CA; http://www.ingenuity.com) software was used for functional annotation, canonical pathway analysis, upstream analysis, and network discovery. The IPA core analyses are based on previous knowledge of the associations of upstream regulators and their downstream target genes archived in the ingenuity knowledge base. The p-values were calculated by Fisher's exact test for the upstream regulator analysis.
Validation for RNA-seq Using RT-PCR in Liver Tissues
For cDNA synthesis and PCR, a Verso cDNA synthesis kit (ThermoFisher, Waltham, MA, USA) and Promega PCR master mix (Promega Corp., Madison, WI, USA) were used. PCR reactions were carried out at 95°C for 2 min, followed by 22 or 32 cycles with denaturation at 95°C for 30 s and annealing and elongation at 55°C for 45 s and 72°C for 1 min. Band density was determined, using Image J software (National Institutes of Health, Bethesda, MD, USA). The gene expression levels were normalized to the expression levels of the GAPDH housekeeping gene. Relative quantification was reported as fold changes compared to control samples. The primer sequence of RT-PCR used in the experiment is shown in Supplementary Material 1.
Cell Culture
The normal murine hepatocyte cell line, AML12 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). AML12 cells were cultured in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium (Cytiva/Hyclone, Marlborough, MA, USA), supplemented with 10% fetal bovine serum (FBS) (ThermoFisher, Waltham, MA, USA), 1% penicillin and streptomycin (ThermoFisher, Waltham, MA, USA), 0.5 mM sodium pyruvate (Sigma-Aldrich, St. Louis, MO, USA), 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenium (ITS) (Sigma-Aldrich, St. Louis, MO, USA), 40 ng/ml dexamethasone (Sigma-Aldrich, St. Louis, MO, USA), and 0.6 g of sodium bicarbonate (Daejung Co., Siheung, Korea). The cells were maintained at 37°C and in a 5% CO2 humidified incubator.
Knockdown of Prohibitin 1 by Small Interfering RNAs
Predesigned small interfering RNA (siRNA) targeting mouse Phb1 (ThermoFisher, Waltham, MA, USA) with different knockdown (KD) efficiency and nonspecific scrambled siRNA (ThermoFisher, Waltham, MA, USA) were purchased. Briefly, AML12 cells were seeded and transfected in a six-well plates at density of 0.2 × 106 cells/well with 13 nM siPhb1 (sense: AGAGCGAGCGGCAACAUUTT, antisense: AAAUGUUGCCGCUCGCUCUGT), which mimics Phb1−/−, 50-nM siPhb1 (sense: GCCGCUGUCAAUAAAUCACTT, antisense: GUCAUUUAUUGACAGCGGCTT), which mimics Phb1+/−, or scramble siRNA supplemented with Lipofectacmine RNAi MAX (ThermoFisher, Waltham, MA, USA), following the guideline of the manufacturer.
qRT-PCR for Expression of Target Genes
Total RNA was isolated from AML12 cells transfected with siRNAs by using TRIzol reagent (ThermoFisher, Waltham, MA, USA) according to the guideline of the manufacturer. First-strand complementary DNA (cDNA) was prepared from 2 μg of total RNA, using RevertAid First Strand cDNA Synthesis Kit (ThermoFisher, Waltham, MA, USA) according to manufacturer's protocol. Quantitative PCR (qPCR) was performed with the Maxima SYBR Green/ROX qPCR Master Mix (2X) (ThermoFisher, Waltham, MA, USA). The reaction was performed in a volume of 10 μl, containing 50 ng of cDNA, 0.3 μM of each primer (Supplementary Material 2) and 5μl maxima SYBR Green/ROX qPCR Master Mix (2X), which were run in duplicate, using a QuantStudio3 thermocycler (ThermoFisher, Waltham, MA, USA). Amplification was carried out with a template denaturation at 95°C for 10 min, followed at 95°C for 15 s and 60°C for 1 min for 40 cycles. The gene expression levels were normalized to the expression levels of the β-actin housekeeping gene and calculated using the 2−ΔΔCt method.
Statistical Analysis
Data on validation using RT-PCR and qRT-PCR were presented as the mean ± standard deviation. Differences among groups were determined by one-way ANOVA analysis with Duncan post-hoc test, using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). Values of p < 0.05 were considered statistically significant.
Results
RNA-seq Results
A total of 18 RNA-seq libraries were constructed using RNA samples extracted from the liver obtained from WT, Phb1+/−, and Phb1−/−. After filtering low read counts and normalization, a total of 8,982 transcripts remained. Of those, only mRNAs showing FDR <0.05 and log2 fold change >1 or <−1 (linear fold change >2 or <−2) were considered differentially expressed (DE) genes for further bioinformatics analyses and PCR validation. Using these criteria, this study was able to select 78 (15 up- and 63 downregulation) DE genes in Phb1+/− mice, compared with WT. The complete gene list for 78DE genes is provided in Supplementary Material 3. Several genes, including optic atrophy 1 (Opa1), mitofusin 1 (Mfn1), and mitofusin 2 (Mfn2), which are involved in mitochondrial fusion and fission, contributing to mitochondrial morphology, were not differentially expressed between Phb1+/− and WT. Expression of mitochondrial cytochrome c oxidase (Cox) responsible for aerobic energy generation was also no difference in Phb1+/−, compared with WT (data not shown). But these mitochondrial-related genes are downregulated in Phb1−/− (data not shown). This result can indicate that half of the reduction in Phb1 expression may not affect mitochondrial dysfunction.
The 10 Most DE Genes in Hetero Compared With WT
The full name of the 10 most DE genes in the Phb1+/− group compared WT (plus FC values and FDR in KO compared with WT) and their functional characteristics are shown in Tables 1, 2. Of those, upregulated genes were involved in nucleolar functions (Nol3, Hist1h2b, and Snora17), xenobiotic enzyme (Sult1e1), ubiquitin peptidase (Usp2), and circadian functions (Ciart, Per2). The 10 most downregulated genes appeared to be involved in potential transcription factors (Mybl1), telomere length regulation (Rad51ap1), mitochondrial enzyme (Pycr1), DNA synthesis (Kifc1), receptors (Fzd3), coagulation factor (F13a1), TIMP inhibitor of metalloproteinase (Timp1), adapter protein in receptor tyrosine kinase (Shc4), membrane transporter (Abcc12), and structural protein (Tubb2b).
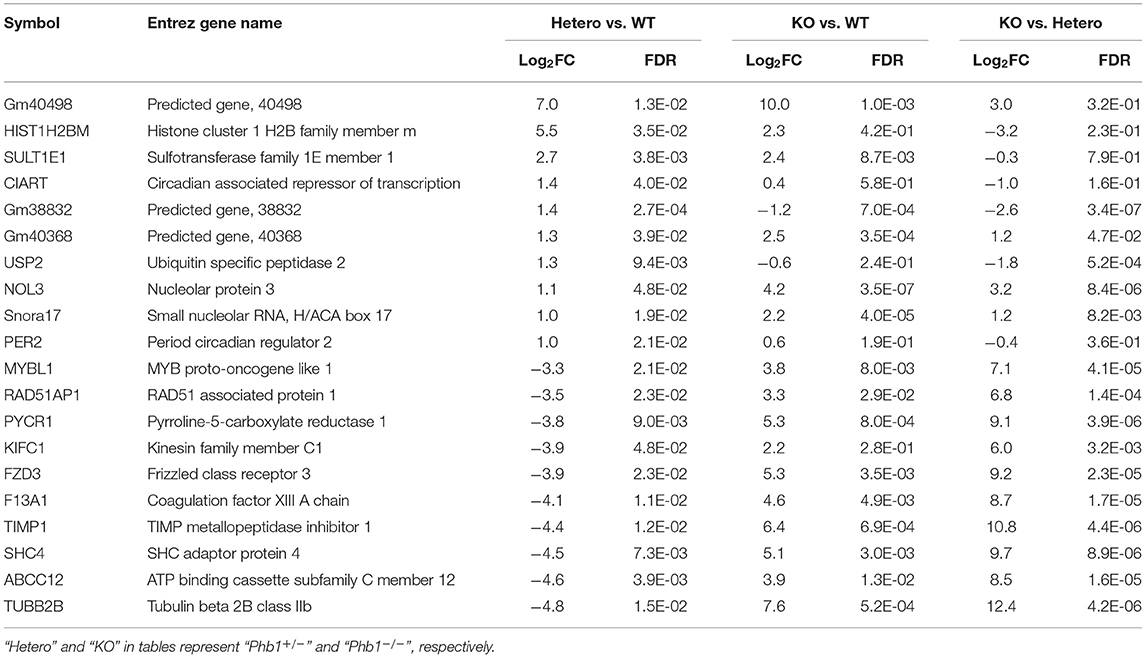
Table 1. The 20 most differentially expressed genes, comparing between Phb1+/− vs. WT, Phb1−/− vs. WT, and Phb1−/− vs. Phb1+/−.

Table 2. Biological functions of the 10 most up- and down-regulated genes between Phb1+/− vs. WT.
Validation of Five DE Genes
Of 78 DE genes in the liver-specific Phb1+/− group, forkhead box M1 (Foxm1), Timp1, Usp2, stearoyl-CoA-desaturase 1 (Scd1), and Sult1e1 are known to be associated with hepatic system disease (Haimerl et al., 2009; Wang et al., 2011; Hu et al., 2014; Lounis et al., 2016; Matsushita et al., 2017). We verified the five DE genes obtained RNA-seq data, using RT-PCR in the same liver tissues, in addition to qRT-PCR in normal murine hepatocytes transfected with siPhb1. Phb1 was silenced by siRNAs, which mimic Phb1−/− and Phb1+/− (Supplementary Materials 4, 5). The mRNA expression levels of all 5 DE genes were well-matched between Phb1+/− and WT in terms of the direction (Table 3; Figures 1A,B). But, in in vitro validation, the mRNA expression levels of Foxm1 and Timp1 were only matched with RNA-seq and RT-PCR validation in liver tissues (Table 3; Figure 1C), which may be due to differences between animal tissues and cell culture. These results confirmed both validity of RNA-seq data for further pathway analyses and the reliability of our liver disease model, indicating that half deficiency of Phb1 in the liver can elucidate the altered molecular mechanisms related to liver injuries.
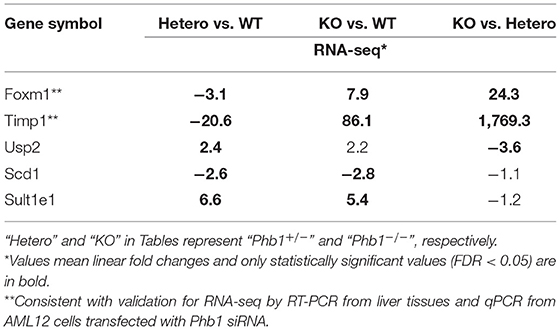
Table 3. The selected 5 DE genes in comparison between Phb1−/−, Phb1+/−, and WT.
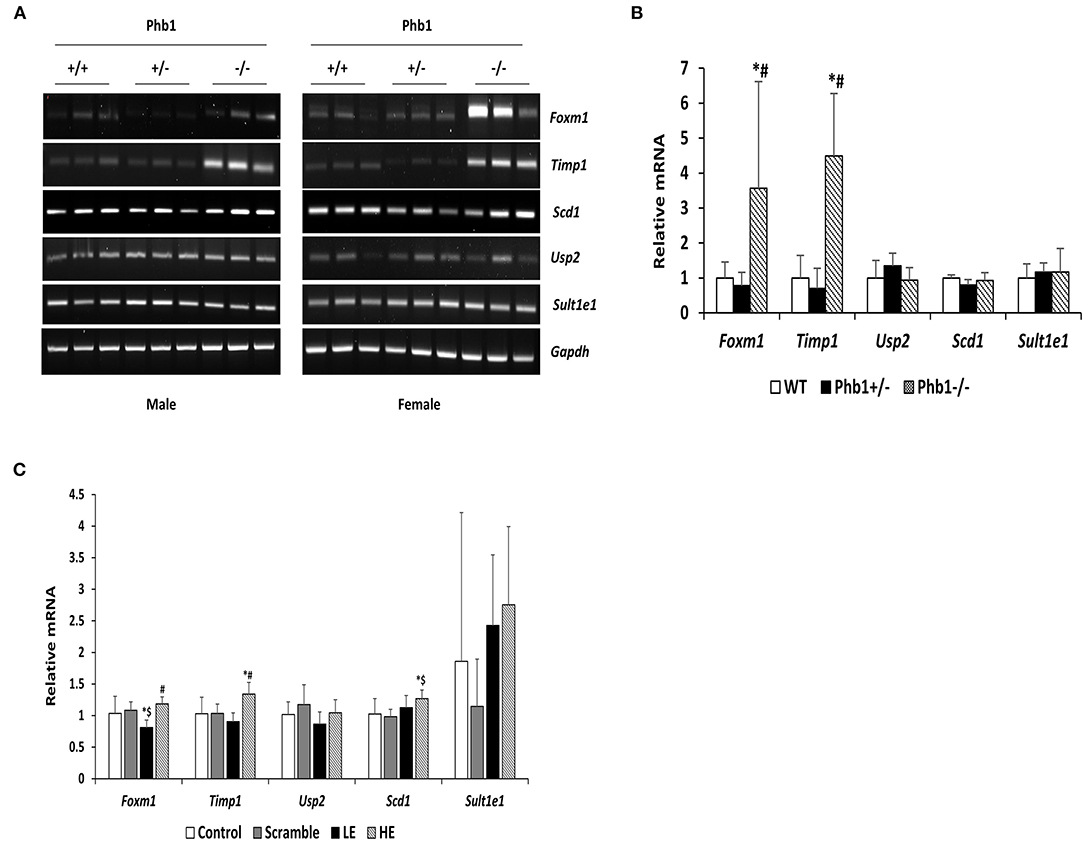
Figure 1. The mRNA expression levels of selected five DE genes in liver tissues and AML12 cells transfected with Phb1 siRNA. (A) The mRNA expression of selected five DE genes in each group of three males and three females (n = 6/ genotype) was detected by RT-PCR. (B) Band density from RT-PCR was quantified with Image J software and represented as the fold change of control and normalized Gapdh. (C) The mRNA expression levels of selected five DE genes in AML12 cells transfected siPhb1 (n = 9) were detected by qRT-PCR and normalized b-actin. “LE” means “low efficiency”, represented Phb1+/−, and “HE” means “high efficiency”, represented Phb1−/−. All data were expressed as means ± standard deviation. One-way ANOVA followed by Duncan's post hoc test was performed, and differences were considered statistically significant. *p < 0.05 vs. wild type (WT) and control, respectively. #p < 0.05 vs. Phb1+/− and low efficiency (LE). $p < 0.05 vs. scramble.
Bioinformatic Pathway Analyses: Upstream Regulators and Molecular Networks
The 78 DE genes in Phb1+/− compared with WT were subjected to core pathway analyses, using ingenuity pathway analysis (IPA) software. Upstream regulator analysis was conducted with enriched pathways and p-value computed by IPA literature review algorithm. The resulting causal networks were scored for known physiological relationships with diseases, functions, genes, or chemicals in the liver tissue and liver cell lines. Causal networks derived by participating the regulator molecule, which controls the expression effector molecules in the given dataset. Thus, causal networks are small hierarchical networks of regulators that control the expression of the given data set. Upstream regulator analyses containing networks provide positive and negative z-scores (≥2 and ≤−2, respectively), which are considered significant predictions of activation and inhibition, respectively. Results showed that sterol regulatory element-binding transcription factor 2 (Srebf2) (z-score = −3.12), Srebf1 (z-score = −3.08), SREBP cleavage-activating protein (Scap) (z-score = −2.98), insulin receptor (Insr) (z-score = −2.62), ATPase copper transporting beta (Atp7b) (z-score = −2.65), and nuclear receptor coactivator 2 (Ncoa2) (z-score = −2.00) were predicted to be inhibited, while peroxisome proliferator-activated receptor alpha (Pparα) (z-score = 2.16) was predicted to be activated in liver of Phb1+/− mice (Table 4). Figure 2 showed these three potential interactions were centered with: (1) Scap, Srebf1, Srebf2; (2) Atp7b, Ncoa2; (3) Pparα, Ncoa2. Similarly, causal network regulator analysis showed that Srebf2 (z-score = −3.16), Srebf1 (z-score = −3.16), Scap (z-score = −3), Insr (z-score = −2.65), Atp7b (z-score = −2.65), insulin-like growth factor 1 (Igf1) (z-score = −2.53) were predicted to be inhibited, while arachidonic acid (z-score = 2.31) was predicted to be activated (Table 5). Srebf1, Srebf2, and Scap, which are involved in regulating cellular lipid metabolism, were listed as the highest ranks of causal network regulators. The predicted downstream target molecules of arachidonic acid were mitogen-activated protein kinase 14 (Mapk14) and Srebf1, followed by downregulating molecules involved in cholesterol and triglycerides (TG) synthesis such as farnesyl diphosphate synthase (Fdps), cytochrome P450 family 51 subfamily A member 1 (Cyp51a1), and Scd1 (Figure 3A). Igf1 was predicted to be inhibited and downregulated similar genes associated with cholesterol and triglycerides via Insr (Figure 3B). Also, Timp1 is one of the target molecules of Igf1 via signal transducer and activator of transcription 3 (Stat3) (Figure 3B).
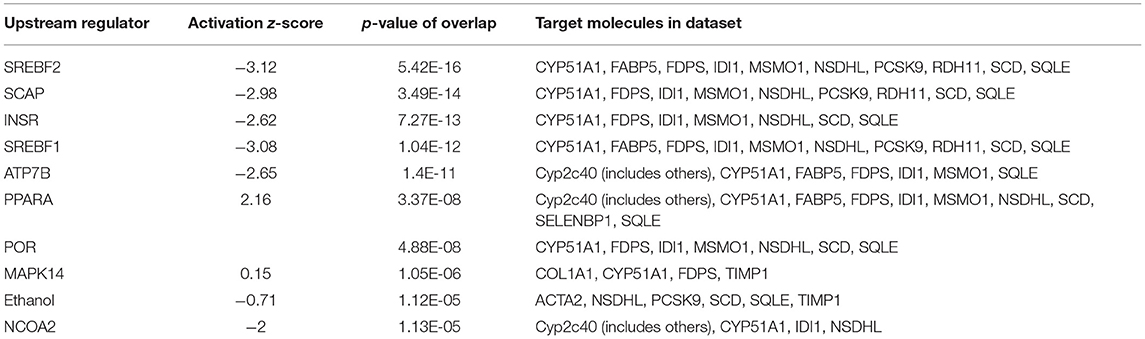
Table 4. Upstream regulators between Phb1+/− and WT.
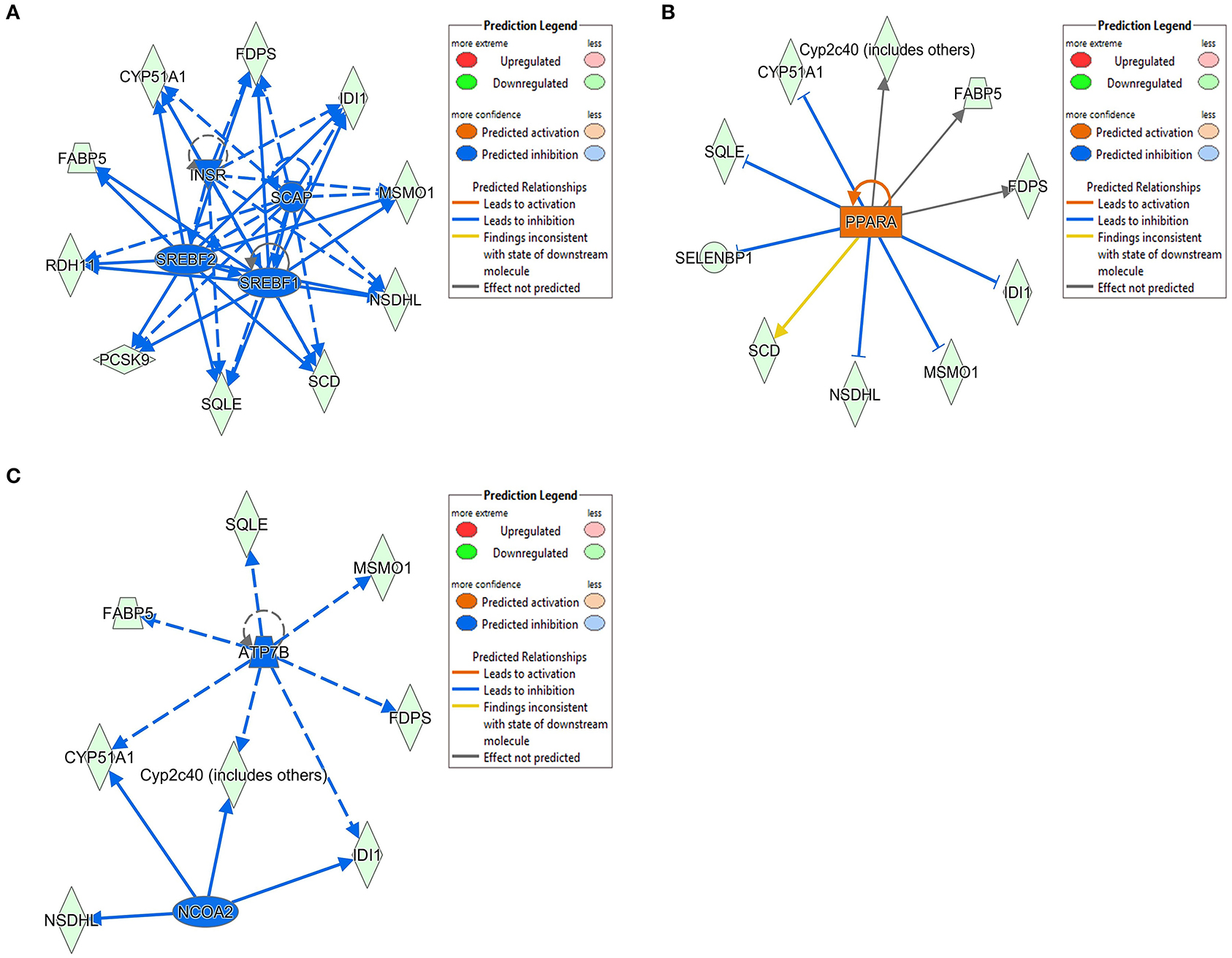
Figure 2. DE genes between Phb1+/− and WT, regulated by (A) Insr, Scap, Srebf1, and Srebf2; (B) Pparα; (C) Atp7b and Ncoa2. Red, green, orange, and blue represent upregulation, downregulation, predicted upregulation, and predicted downregulation, respectively.
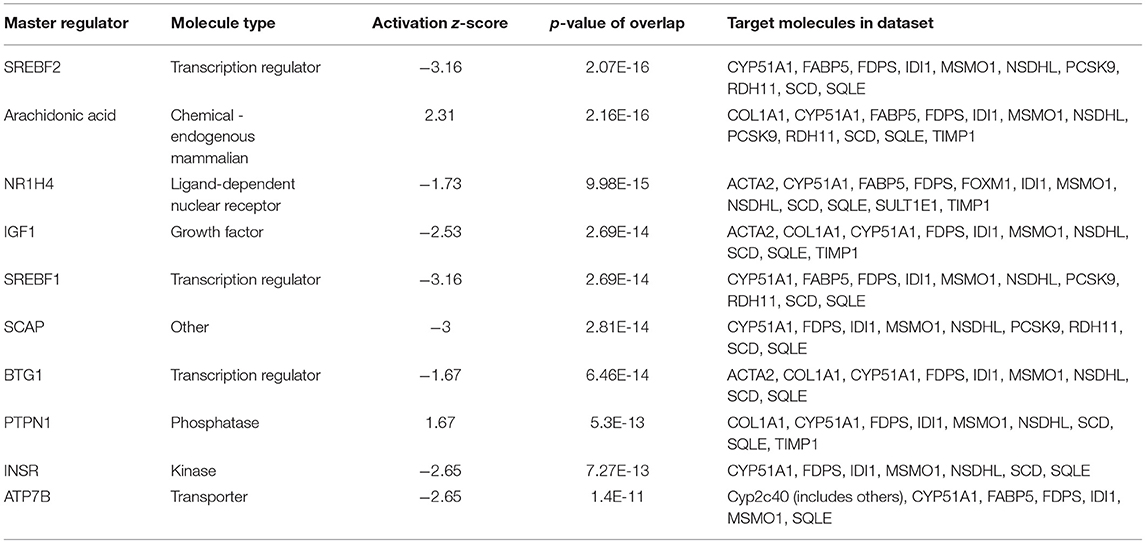
Table 5. Causal network regulators between Phb1+/− and WT.
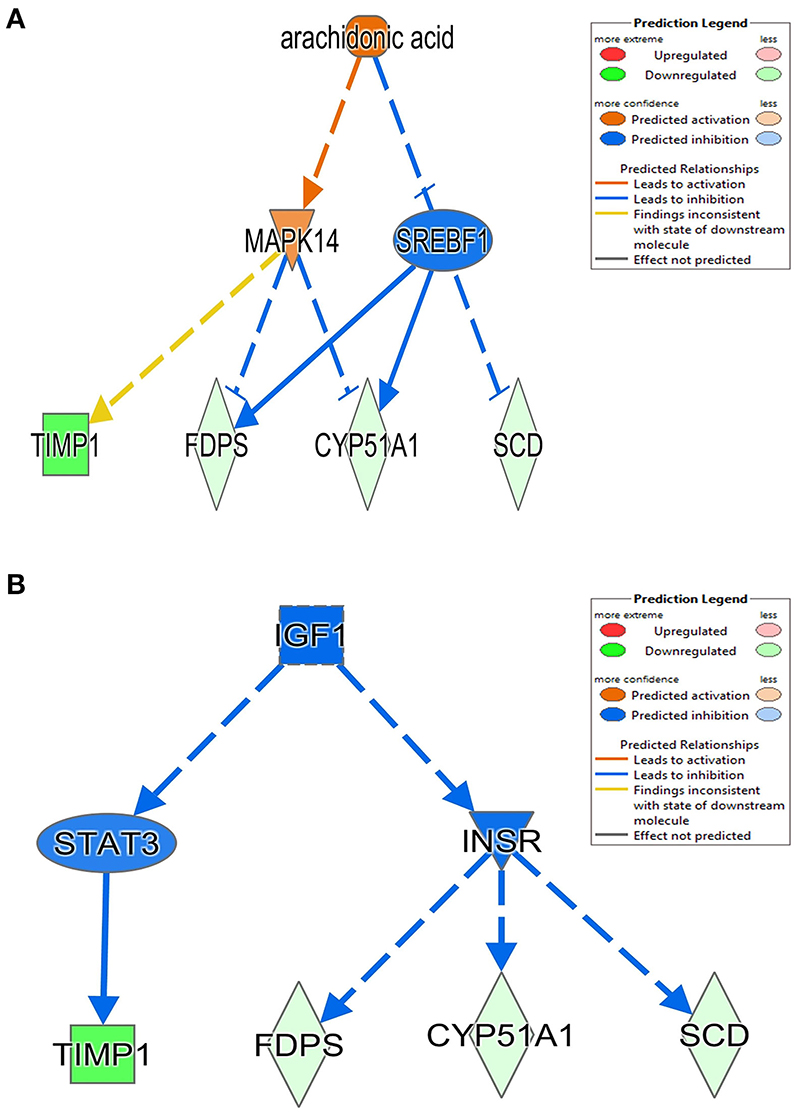
Figure 3. Causal network between Phb1+/− and WT, regulated by (A) arachidonic acid and (B) Igf1. Red, green, orange, and blue represent upregulation, downregulation, predicted upregulation, and predicted downregulation, respectively.
The most critical diseases and biofunctions (Table 6), molecular and cellular functions (Table 7), and physiological system development and functions (Table 8) were identified with influences of 78 DE genes, and specific genes involved in all diseases and functions were listed in Supplementary Material 6. Of diseases and biofunctions listed in Table 6, hepatic system diseases were ranked as the second. The molecules, including Scd1, Acta2, collagen type I alpha 1 chain (Col1a1), Timp1, Foxm1, Sult1e1, and plasminogen activator (Plat), are also involved in hepatotoxicity pathways, such as liver fibrosis, liver hyperplasia/hyperproliferation, liver proliferation, and liver necrosis/cell death (Table 9). Molecules associated with hepatotoxicity were shown in Figure 4. Hepatic DE genes in Phb1+/− mice showed increased risks of hyperplasia of the liver, apoptosis of liver cells, hepatic injury, and hepatoma and decreased functions in the proliferation of liver cells and fibrosis. The hepatotoxicity analysis indicates that Phb1+/− mice may become more susceptible with liver damages/injuries.
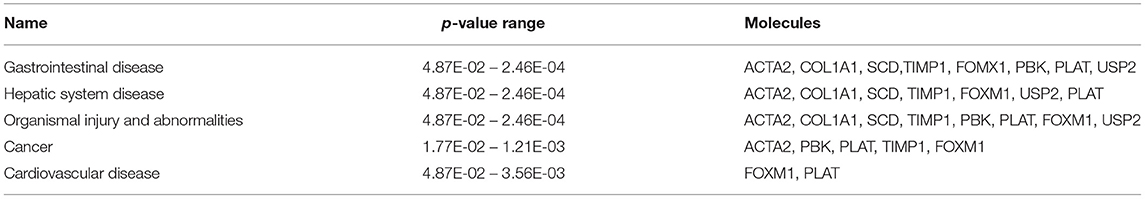
Table 6. Top diseases and biofunctions between Phb1+/− and WT.
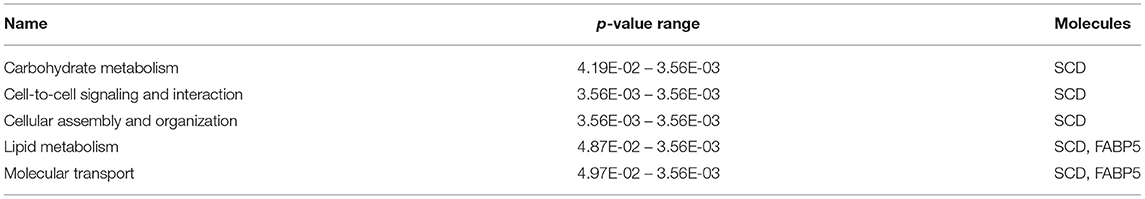
Table 7. Molecular and cellular functions between Phb1+/− and WT.

Table 8. Physiological system development and function between Phb1+/− and WT.
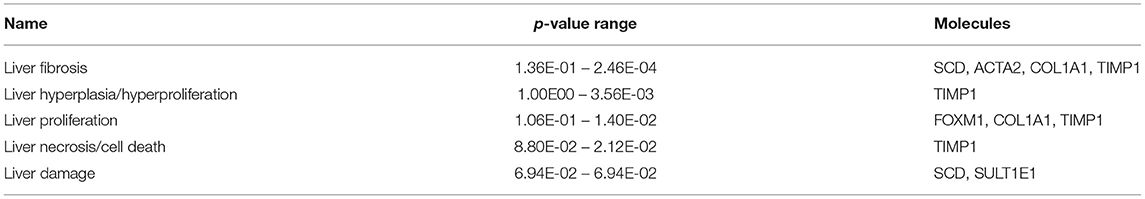
Table 9. Hepatotoxicity between Phb1+/− and WT.
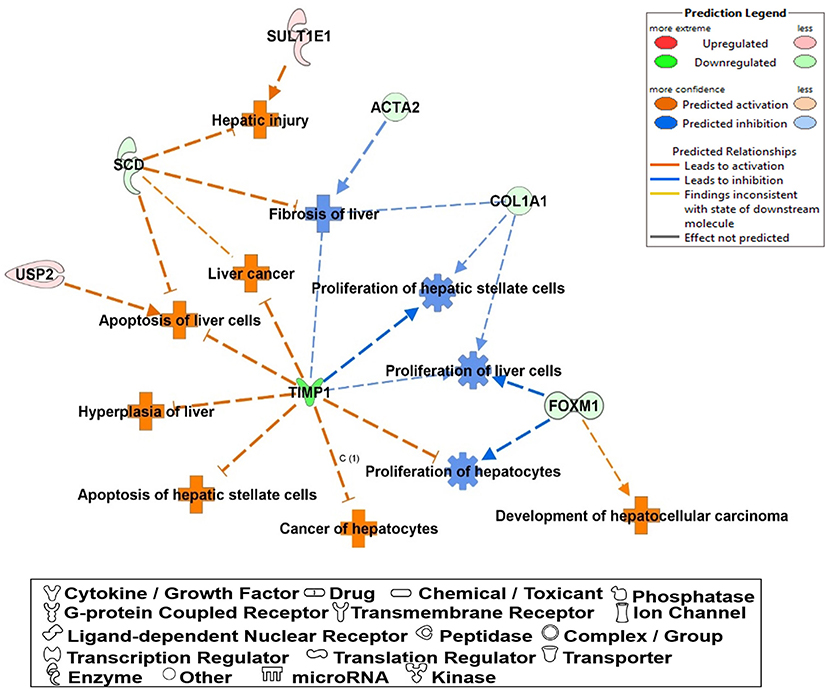
Figure 4. A network of DE genes affecting functions related to “hepatotoxicity” between Phb1+/− and WT. Red, green, orange, and blue represent upregulation and downregulation, predicted upregulation, and predicted downregulation, respectively.
Molecular network analysis produced five potential molecular interactions among 78 DE genes (Table 10). The most interesting network #1 centered with regulatory molecules, including Pparα and Srebf1, which were discussed in upstream regulators above, in addition to tumor necrosis factor (TNF), which is predicted to be inhibited. Molecules interacting network #1 are connected to functions of lipid metabolism, small molecule biochemistry, and vitamin and mineral metabolism (Table 10; Figure 5).
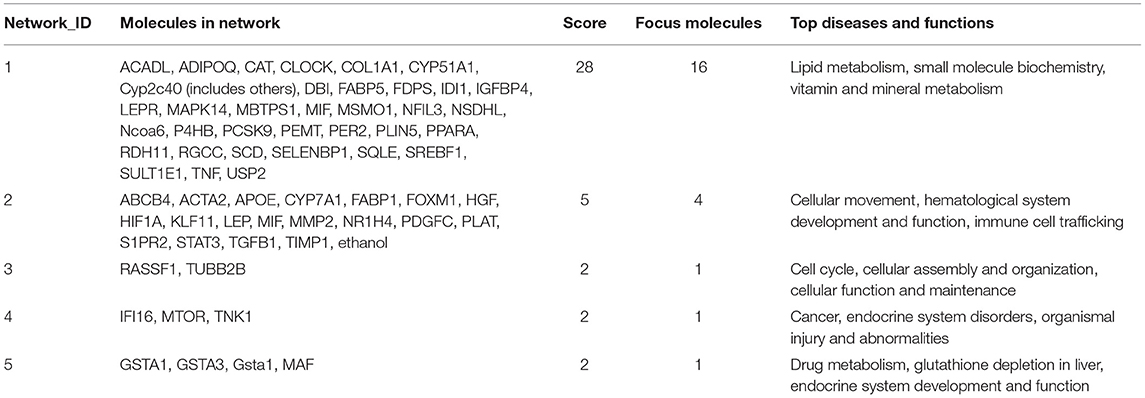
Table 10. Molecular networks between Phb1+/− and WT.
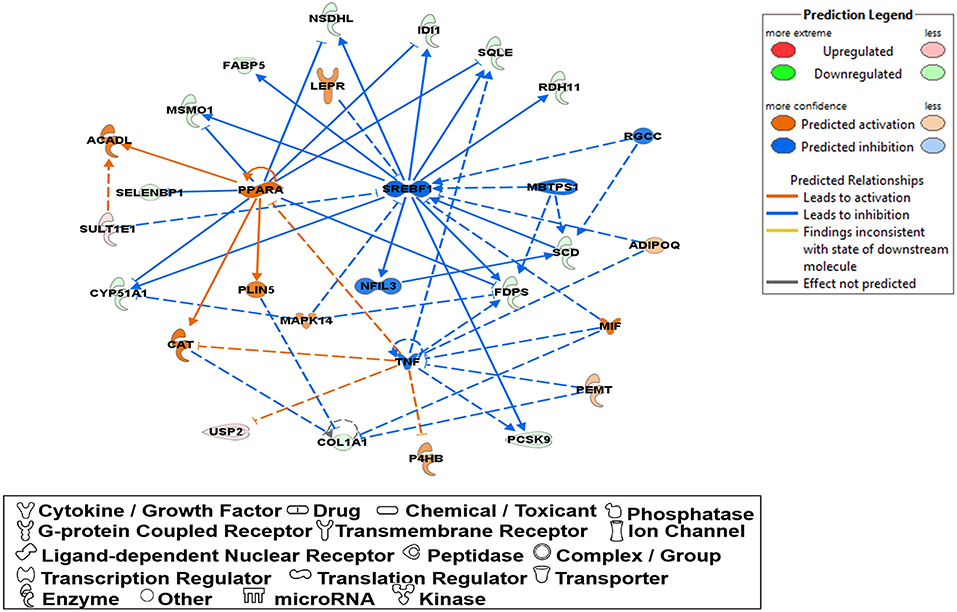
Figure 5. A network relevant to “lipid metabolism”, “small molecule biochemistry”, “vitamin and mineral represented in a gene network (network ID 1). Red, green, orange, and blue represent upregulation and downregulation, and predicted upregulation and downregulation, respectively.
Discussion
In this study, we examined the hepatic transcriptome of liver-specific Phb1 deficient mice and identified biological functions and genes associated with hepatotoxicity, lipid metabolism, and metabolic disorders. IPA suggested a network related to hepatotoxicity, which is composed of Foxm1, Timp1, Usp2, Scd1, and Sult1e1. These data exhibited that decreased Phb1 in hepatocytes contributed to abnormal proliferation of various liver cell types and cancer cell transformation by upregulated or downregulated genes. Also, IPA analysis with DE genes suggested increasing arachidonic acid and suppressing Igf1-mediated signaling as causal networks in liver-specific Phb1+/−, compared with WT. They had in common with downstream targets as Timp1, Scd1, Fdps, and Cyp51a1 (Figure 3), which are mainly associated with the extracellular matrix, fatty acid metabolism, and steroid biosynthesis (Ge et al., 2020; Yan et al., 2020) and downregulated via Mapk14, Srebf1, Stat3, and Insr. This finding is in line with the blocking of activation of phosphoinositide 3-kinase by insulin via p38 Mapk in hepatocytes with arachidonic acid (Talukdar et al., 2005) and evidence that arachidonic acid has the potential to decrease insulin-mediated activation of Srebp-1c by inhibiting liver X receptor (Lxr) activation in rat hepatocytes (Chen et al., 2004). Also, Igf1 and Insr-mediated Cyp51a1 were predicted as a downregulated enzyme, and related research reported that hepatocyte Cyp51KO mice showed inflammation and mild-to-moderate portal fibrosis and abnormal hepatic sterol metabolism (Urlep et al., 2017). Collectively, these results may elucidate physiological changes by Phb1 and provide a comprehensive understanding of prognosis and prevention of liver injuries, containing liver cancer.
Liver Disease-Related Genes
Of 78 DE genes, which were filtered out from liver-specific Phb1+/− vs. WT, Foxm1, Timp1, Usp2, Scd1, Sult1e1, are known to be associated with hepatotoxicity (Figure 4). Foxm1, a transcription regulator, is located to the nucleus and highly expressed in proliferating normal cells and various cancer cells (Teh, 2012). The knockdown of FOXM1 in human hepatocellular carcinoma (HCC) cell lines significantly alleviated cell cycle arrest and cell growth suppression (Hu et al., 2014). In addition, overexpression of FOXM1 is associated with an aggressive tumor feature and poor prognosis of HCC (Sun et al., 2011). Unlike earlier results of positive correlation between Foxm1 and HCC, our transcriptomic profile demonstrated that fold change of Foxm1 was rather reduced in Phb1+/− compared with WT (Phb1+/− vs. WT, log2 FC = −1.6) (Supplementary Material 3) and then increased in Phb1−/− compared with Phb1+/− (Phb1−/− vs. Phb1+/−, log2 FC = 4.6) (data not shown). This finding was consistent with validation using the PCR method in liver tissues and AML12 cells transfected with siPhb1 (Figure 1). Deletion of Foxm1 is correlated with Ras-induced HCC with stem cell features by accumulating reactive oxygen species (ROS) (Kopanja et al., 2015). Another study revealed a negative association with Foxm1/nuclear factor kappa B (NF-kB) and methionine adenosyltransferase 1A (Mat1a) that is responsible for catalyzes in the conversion of methionine to homocysteine (Li et al., 2020). It was reported that Foxm1 directly interacts with Mat1a, which is a tumor suppressor in the liver. Collectively, it can be interpreted that Phb1 leads to a unique change of the expression pattern of Foxm1 and may be a key regulator, modulating the transcriptional activity of Foxm1 in terms of tumorigenesis.
Likewise, Timp1 expression in the transcriptomic profile and PCR validation was similar to the results of Foxm1 expression. Previous studies explained increasing Timp1 expression as a marker of extrahepatic and intrahepatic tumors from liver tissues and serum (Ylisirniö et al., 2000; Yoshiji et al., 2000; Yukawa et al., 2007). Also, Thiele et al. (2017) suggested that hepatic Timp1 mRNA expression from WT mice was upregulated in HCC tissue compared with adjacent paired normal tissue when co-treated with diethylnitrosamine and CCL4. In agreement with these studies, our transcriptomic profile showed increased Timp1 expression in Phb1−/−, compared with Phb1+/− (log2 FC = 10.8) (Table 1). On the contrary, Timp1 expression was rather decreased in Phb1+/−, compared with WT, which was similar to Foxm1 (log2 FC = −4.4) (Table 1). Wang et al. (2011) demonstrated the newly hepatoprotective role of Timp1 during acute and chronic liver injuries, which is positively regulated by the interleukin-6 (IL-6)/STAT3 signaling pathway. Our liver-specific Phb1-deficient mouse showed different Timp1 expression patterns according to Phb1 depletion, which encompassed both protumor- and antitumor activity of Timp1. It was previously reported that Timp1 was increased in the liver from adipocyte-specific nuclear form of SREBP-1c (nSREBP-1c) transgenic mice, showing human nonalcoholic steatohepatitis (NASH) (Kakino et al., 2018). However, Upregulation of Timp1 was reduced in the liver from the Tnf−/− nSREBP-1c transgenic mice (Kakino et al., 2018). Tomita et al. explained that control mice-fed-MCD diet rapidly increased mRNA expression of Timp1 in the whole liver, compared with both TNF receptors 1 (Tnfr1) and 2 (Tnfr2) knockout mice, representing Tnfr-double KO mice (Tomita et al., 2006). We can interpret that decreased Timp1 may act as an initial signal of liver injuries by Phb1+/−, and then chronic liver injuries by Phb1−/− may represent a rapid increase of Timp1. Since we recognize the importance to uncover a direct correlation between Phb1 and Timp1, further study is underway to assess the association. Nevertheless, both RNA-seq and PCR validation in siPhb1-transfected hepatocytes showed indirect inverse relevance between Phb1 and Timp1 in this research. Especially, Ko et al. already explained the relation using microarray analyses (Ko et al., 2010), showing an increase in the expression of Timp1 in liver-specific Phb1−/− mice. Therefore, Timp1 may be an important enzyme that predicts the extent of liver injuries by lack of the Phb1 gene. Further investigations are needed to characterize functional connections between Phb1 and Timp1 in the liver disease model. Despite increased expression of Foxm1 and Timp1 in an environment with liver injuries, another possibility to interpret the differential gene expression in Phb1+/− vs. Phb1−/− may be deciphered by genetic compensation by the gene knockout (El-Brolosy and Stainier, 2017). Genetic compensation means that another gene takes over the role of the knocked-out gene (De Souza et al., 2006). This concept is used for explaining the discrepancy between knockdown and knockout phenotypes. For example, it was reported that upregulated Emilin 3, which shares the functional domain with egfl7 represented for endothelial extracellular matrix genes, was observed only in the zebrafish egfl7 knockout, not in the knockdown, demonstrating that Emilin 3 may compensate for the egfl7 knockout (Rossi et al., 2015). Besides, both Prnp and Sprn proteins share biological functions in early embryogenesis according to the need for either Prnp or Sprn expression, showing that one can compensate for the absence of the other in PrP-knockout mammals (Young et al., 2009). According to these cases in which gene expression is reversely regulated in the knockout and knockdown comparison of the unique expression pattern of Foxm1 and Timp1 in Phb1+/− and Phb1−/− may be explained by genetic compensation. To specify the concept in our liver disease model, further intensive research is necessary in the environment with regulating the expression of Phb1.
Ubiquitin carboxyl-terminal hydrolase 2 (Usp2), which is upregulated in Phb1+/− compared with WT (log2 FC = 1.3) (Table 1), is a deubiquitinating enzyme and involved in controlling activity and levels of protein under certain physiological conditions. Usp2 has been reported to stabilize mouse double minute 2 (Mdm2) and Mdm4, which are proto-oncogenes with both p53-dependent or independent activities, to degrade p53 (Benassi et al., 2012; Young et al., 2019), maintaining the expression of p53 at a low level. Specifically, protein-protein interaction assays using the bacterial two-hybrid system showed that USP2 can deubiquitinate MDM2 and promotes p53 degradation, showing the association between USP2 and MDM2 (Stevenson et al., 2007). Especially, it was reported that transfection of tumor-derived cell lines with siUsp2 resulted in increasing p53 protein expression and its target gene, p21 (Stevenson et al., 2007). PHB1 has an ability to physically interact with p53 and enhance p53-mediated transcriptional activation by promoting its recruitment to promoters in two breast cancer cell lines where it co-localizes with p53 (Fusaro et al., 2003). Our transcriptome analysis showed that the expression level of Usp2 was upregulated in Phb1+/− compared with WT (log2 FC = 1.3) and then decreased in Phb1−/− compared with Phb1+/− (log2 FC = −1.8) (Table 1). We can infer that Phb1 may participate in enhancing Usp2-mediated proteosome activity, and Phb1 deficiency may result in abnormal p53 stabilization and activation (Haupt et al., 1997; Honda and Yasuda, 2000). Thus, we can explain that there is no defense response against a high-stress environment, such as an increasing susceptibility of liver injuries by the drastic shortage of Phb1, represented to Phb1−/−.
Stearoyl-CoA desaturase-1 (SCD1) plays an important role in the conversion of saturated fatty acids to monosaturated fatty acids and further synthesis of TG (Lounis et al., 2016). We expected that Scd1 expression in Phb1+/−, compared with WT, also increased in transcriptome analysis, since our previous research on the effect of Phb1 deficiency with siPhb1 knockdown on impaired lipid metabolism demonstrated that palmitic acid promoted Scd1 mRNA expression levels (unpublished data, under review). In the current study, Scd1 expression was decreased in Phb1+/− compared with WT (log2 FC = −1.4) (Supplementary Material 3), and there was no statistically significant difference between Phb1+/− and Phb1−/− (Table 3). Wang et al. (2002a) reported that PHB1-mediated transcriptional repression required histone deacetylase (HDAC), and additional corepressors like N-CoR are involved. Also, E2F transcription factors, which play a key role in regulating mammalian cell cycle progression, were controlled by PHB1, interacting with retinoblastoma protein (Rb) (Wang et al., 2002b; Mishra et al., 2006). Recently, combining research using lipidomics and transcriptome analyses in Rb depletion in mouse embryonic fibroblast has shown that Rb deficiency increases the concentration of fatty acid, acylcarnitine, phosphatidylcholine, and arachidonoyl ethanolamine (Muranaka et al., 2017). Also, Scd1 is most strongly controlled by Rb, possibly through E2F and SREBP transcription factors (Muranaka et al., 2017). Collectively, a decrease in Scd1 expression in Phb1+/− compared with WT may be due to the interaction between PHB1 and SCD1 at the protein level, maintaining a balance between lipid catabolism and anabolism.
Sulfotransferase family 1E member 1 (SULT1E1) is a cytosolic enzyme, which is called as an estrogen sulfotransferase and is responsible for the inactivation of β-estradiol (E2) involved in changing estrogen metabolism and liver function (Li and Falany, 2007; Li et al., 2009; Xu et al., 2018). According to a study on cystic fibrosis, which is an inherited disorder and affects the lung, intestine, pancreas, and livers, hepatic Sult1e1 activity was increased in cystic fibrosis transmembrane conductance regulator (CFTR) −/− mice compared with CFTR+/+ (Li and Falany, 2007). Also, the estrogen receptor α protein level was reduced in CFTR−/−mice with high Sult1e1 activity, showing high affinity between E2 and substrate of SULT1E1 and thereby altering the level of estrogen-regulated proteins (Song, 2001; Li and Falany, 2007). Wang et al. (2004) demonstrated the association of PHB1 with E2F transcription factor 1 (E2F1) and the repressive function of estrogen antagonists in human breast cancer cells (Wang et al., 2004). Moreover, the PHB1 gene and protein expression were both markedly enhanced by estrogen antagonists (Wang et al., 2004). In agreement with these studies, Sult1e1 expression in the current transcriptomic profile was increased by depletion of Phb1 in between Phb1+/− and Phb1−/− compared with WT (log2 FC = 2.7 and 2.4, respectively) (Supplementary Material 3). Both transcriptome analyses and validation, using AML12 cells transfected with siPhb1, showed a negative relationship between Phb1 and Sult1e1 (Figure 1C). We can infer that Phb1 deficiency is likely to have relevance to increasing expression of Sult1e1. Also, Phb1 depletion may be a crucial status to elevate hepatic Sult1e1 activity responsible for repressing estrogen-related genes in terms of its transcriptional activities. Thus, the association of Phb1 depletion with its effect on estrogen metabolism in livers needs to be investigated further.
Lipid Metabolism and Metabolic Diseases
In addition to the hepatotoxicity of 5 DE genes between liver-specific Phb1+/− and WT, there are some associations with lipotoxicity. The role of fatty acid metabolism in cancer initiation, progression, and drug resistance through the FOXO3-FOXM1 axis had been mentioned (Saavedra-García et al., 2018). Besides, compared with adipose tissue from controls, toll-like receptor 2 (Tlr2) KO mice fed a high-fat diet that decreased levels of Timp1, collagen 1, and transforming growth factor-β1 (TGFβ1) (Song et al., 2018). Usp2 enhances the stability of fatty acid synthase (FASN) by impeding proteasome-dependent degradation in human HCC (Calvisi et al., 2011; Kitamura and Hashimoto, 2021). The high-fat diet-induced NAFLD rat model showed increased protein levels of SULT1E1, compared with mice-fed normal standard diet through proteomics analysis and western blots analysis (Cong et al., 2021). From upstream regulator analysis and causal network regulators, Insr, Scap, Srebf1, and Srebf2 were predicted to be inhibited in liver-specific Phb1+/− compared with WT (Tables 4, 5). SREBPs regulate the biosynthesis of triglyceride (TG), fatty acids (FAs), and cholesterol, which can increase the expression of genes related to lipid synthesis and lipid uptake (Brown and Goldstein, 1997). SREBF1 and SREBF2 encode three SREBP isoforms: SREBP-1a, SREBP-1c, and SREBP-2, respectively. SREBP-1a and−1c are usually expressed in the liver, adipose tissue, and adrenal gland of mice and humans, whereas SREBP-2 is ubiquitously expressed in cell lines, spleen, and intestinal tissues (Jeon and Osborne, 2012; Moslehi and Hamidi-Zad, 2018). When cellular sterol levels are high, SCAP, which is called ER membrane protein, interacts with SREBPs in the endoplasmic reticulum (ER) membrane, and the SREBP/SCAP complex moves to Golgi to translocate to the nucleus and bind to the target gene promoters (Xiao and Song, 2013; Moslehi and Hamidi-Zad, 2018). SREBP-1a and−1c mostly activate fatty acid and TG synthesis, and SREPB-2 increases transcription of genes related to cholesterol synthesis and uptake. Remarkably, SREBP-1c is known to be controlled by insulin treatment. Hegarty et al. (2005) showed that full induction of mature and transcriptionally active form of SREBP-1 in rat hepatocyte resulted from insulin treatment (Hegarty et al., 2005). Many studies reported that insulin is one of the most potent activators of SREBP-1c (Shimomura et al., 1999; Azzout-Marniche et al., 2000; Zeng et al., 2017). We found that Insr, SREBPs, and Scap were predicted to be inhibited in liver-specific Phb1+/−, compared with WT (Tables 4, 5). Potential decrease of Insr may interpret Insr-related insulin resistance in non-adipose tissues like the liver, contributing to metabolic diseases. The liver regulates various metabolism by insulin, which is triggered by INSR (Samuel and Shulman, 2012; Knebel et al., 2018). Liver-specific Insr-KO mice showed dramatic insulin resistance, abnormal glucose homeostasis, and hyperinsulinemia (Michael et al., 2000; Miao et al., 2014). In addition, according to a study on patients with simple steatosis and NASH, downregulation of SREBP-1c may be associated with the development of burned-out NASH through decreasing quantification of SREBP-1c positive hepatocyte nuclei and increasing mature SREBP-1c levels by immunoblot analysis (Nagaya et al., 2010). The study demonstrated that hepatic expression of SREBP-1c is increased in simple steatosis but gradually decreases with fibrosis progression. Based on these findings, we can speculate that Phb1 is relevant to the downregulation of hepatic expression levels of SREBPs and Insr, which control lipid metabolism and metabolic homeostasis by insulin. However, SREBPs were not predicted as upstream regulators between Phb1−/− and Phb1+/− (data not shown); further study is necessary to investigate the hepatic expression level of Insr and SREBPs in our liver disease model for elucidating contribution by gradual depletion of Phb1 on Insr-related insulin resistance and pathogenesis of liver injuries.
Peroxisome proliferator-activated receptor alpha (Pparα) was predicted as an increased upstream regulator in liver-specific Phb1+/− compared with WT (z-score = 2.16) (Table 4). PPARα is a key regulator of lipid oxidation, which regulates genes involved in lipid and glucose metabolism and inflammation in the liver (Li and Palinski, 2006). PPARα agonists, such as fenofibrate and bezafibrate, are widely used to treat dyslipidemia, preventing hepatic steatosis and improving insulin sensitivity (Chou et al., 2002; Harano et al., 2006). We expected that if Pparα is an upstream regulator, it will decrease its function by Phb1 deficiency. Because our previous study showed that the Pparγ mRNA expression level, one of the markers of hepatic steatosis, was increased in normal murine hepatocytes transfected with siPhb1, which mimics liver-specific Phb1−/− (unpublished data, under review). On the other hand, our current study depicted that a slight decrease in Phb1 may attribute to increase Pparα. This trend was consistent with SREBPs as other upstream regulators. In a previous study on a patient with liver diseases, hepatic expression of SREBP-1c was increased in simple steatosis (SS) but gradually decreased in mild NASH and advanced NASH. Likewise, the hepatic mRNA level of PPARα was significantly increased in SS and decreased in the mild NASH and advanced NASH (Nagaya et al., 2010). Taken together, we can infer that Phb1 deficiency at the early stage reflects a defective mechanism via Pparα against abnormal lipogenesis and/or lipid catabolism. But dramatic depletion of Phb1 is no capable of preventing impaired lipid metabolism and rapidly exacerbates liver injuries.
Conclusions
In summary, our study demonstrated that the degree of depletion in Phb1 reflects different physiological responses. We characterized that Foxm1 and Timp1 can provide a basis for investigating a molecular mechanism on the increased susceptibility of liver injuries by Phb1 deficiency. Additionally, SREBPs and Pparα may be key genes to elucidate the pathogenesis of liver diseases relevant to Phb1 deficiency in terms of lipid metabolism. Taken together, these insights may lead to the establishment of novel therapeutic strategies against liver diseases containing liver cancer.
Data Availability Statement
The data generated in this study can be accessed from NCBI using the accession numbers of the project and samples PRJNA738620, SAMN19735929.
Ethics Statement
The animal study was reviewed and approved by Cedars-Sinai Medical Center IACUC (The IACUC protocol number is 9370).
Author Contributions
KL performed pathway analysis and experiments and wrote the manuscript. HY performed the experiments. SS analyzed the data. BK performed RNA-seq analysis, data analysis, manuscript review, and pathway analysis. JL and S-HL performed RNA expression test. KK was responsible for mouse experiments, tissue collection, funding, designing and supervising the study, and revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (Nos. 2020R1A2C1102451 and 2016R1D1A1B04935653).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors appreciate Dr. Shelly Lu and Dr. Tony Li at Cedars-Sinai Medical Center for providing liver-specific prohibitin 1 knockout mice and for their permission to use the lab space and the facility for this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.717911/full#supplementary-material
References
Ahmed, S. B., and Prigent, S. A. (2014). A nuclear export signal and oxidative stress regulate ShcD subcellular localisation: a potential role for ShcD in the nucleus. Cell. Signal. 26, 32–40. doi: 10.1016/j.cellsig.2013.09.003
Aoki, D., Oda, Y., Hattori, S., Taguchi, K.-i., Ohishi, Y., Basaki, Y., et al. (2009). Overexpression of class III β-tubulin predicts good response to taxane-based chemotherapy in ovarian clear cell adenocarcinoma. Clin. Cancer Res. 15, 1473–1480. doi: 10.1158/1078-0432.CCR-08-1274
Azzout-Marniche, D., Bécard, D., Guichard, C., Foretz, M., Ferré, P., Foufelle, et al. (2000). Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem. J. 350, 389–393. doi: 10.1042/bj3500389
Barroso-González, J., García-Expósito, L., Hoang, S. M., Lynskey, M. L., Roncaioli, J. L., Ghosh, A., et al. (2019). RAD51AP1 is an essential mediator of alternative lengthening of telomeres. Mol. Cell 76, 11–26.e17. doi: 10.1016/j.molcel.2019.06.043
Benassi, B., Flavin, R., Marchionni, L., Zanata, S., Pan, Y., Chowdhury, D., et al. (2012). MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov. 2, 236–247. doi: 10.1158/2159-8290.CD-11-0219
Benjamin, A. M., Nichols, M., Burke, T. W., Ginsburg, G. S., and Lucas, J. E. (2014). Comparing reference-based RNA-Seq mapping methods for non-human primate data. BMC Genom. 15, 1–14. doi: 10.1186/1471-2164-15-570
Bolcun-Filas, E., Bannister, L. A., Barash, A., Schimenti, K. J., Hartford, S. A., Eppig, J. J., et al. (2011). A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 138, 3319–3330. doi: 10.1242/dev.067645
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. doi: 10.1016/S0092-8674(00)80213-5
Calvisi, D. F., Wang, C., Ho, C., Ladu, S., Lee, S. A., Mattu, S., et al. (2011). Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071–1083.e1075. doi: 10.1053/j.gastro.2010.12.006
Chen, G., Liang, G., Ou, J., Goldstein, J. L., and Brown, M. S. (2004). Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. 101, 11245–11250. doi: 10.1073/pnas.0404297101
Chou, C. J., Haluzik, M., Gregory, C., Dietz, K. R., Vinson, C., Gavrilova, O., et al. (2002). WY14, 643, a peroxisome proliferator-activated receptor α (PPARα) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 mice. J. Biol. Chem. 277, 24484–24489. doi: 10.1074/jbc.M202449200
Cong, S., Li, Z., Yu, L., Liu, Y., Hu, Y., Bi, Y., et al. (2021). Integrative proteomic and lipidomic analysis of Kaili Sour Soup-mediated attenuation of high-fat diet-induced nonalcoholic fatty liver disease in a rat model. Nutr. Metabol. 18, 1–12. doi: 10.1186/s12986-021-00553-4
De Ingeniis, J., Ratnikov, B., Richardson, A. D., Scott, D. A., Aza-Blanc, P., De, S. K., et al. (2012). Functional specialization in proline biosynthesis of melanoma. PLoS ONE 7:e45190. doi: 10.1371/journal.pone.0045190
De Souza, A. T., Dai, X., Spencer, A. G., Reppen, T., Menzie, A., Roesch, P. L., et al. (2006). Transcriptional and phenotypic comparisons of Ppara knockout and siRNA knockdown mice. Nucl. Acids Res. 34, 4486–4494. doi: 10.1093/nar/gkl609
El-Brolosy, M. A., and Stainier, D. Y. (2017). Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13:e1006780. doi: 10.1371/journal.pgen.1006780
Esmaeili, M., Mirzaahmadi, S., and Asaadi Tehrani, G. (2018). Evaluation of additional ABCC12 gene expression character in breast cancer samples using formal diagnostic profile. Gene Cell Tissue 5:e84392. doi: 10.5812/gct.84392
Fusaro, G., Dasgupta, P., Rastogi, S., Joshi, B., and Chellappan, S. (2003). Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 278, 47853–47861. doi: 10.1074/jbc.M305171200
Ge, Q., Feng, F., Liu, L., Chen, L., Lv, P., Ma, S., et al. (2020). RNA-Seq analysis of the pathogenesis of STZ-induced male diabetic mouse liver. J. Diabet. Complications 34:107444. doi: 10.1016/j.jdiacomp.2019.107444
Guo, Y., Hu, B., Huang, H., Tsung, A., Gaikwad, N. W., Xu, M., et al. (2015). Estrogen sulfotransferase is an oxidative stress-responsive gene that gender-specifically affects liver ischemia/reperfusion injury. J. Biol. Chem. 290, 14754–14764. doi: 10.1074/jbc.M115.642124
Gustafsson, Å. B., Tsai, J. G., Logue, S. E., Crow, M. T., and Gottlieb, R. A. (2004). Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J. Biol. Chem. 279, 21233–21238. doi: 10.1074/jbc.M400695200
Haimerl, F., Erhardt, A., Sass, G., and Tiegs, G. (2009). Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific protease 2 contributes to tumor necrosis factor-α-induced hepatocyte survival. J. Biol. Chem. 284, 495–504. doi: 10.1074/jbc.M803533200
Han, J., Wang, F., Lan, Y., Wang, J., Nie, C., Liang, Y., et al. (2019). KIFC1 regulated by miR-532-3p promotes epithelial-to-mesenchymal transition and metastasis of hepatocellular carcinoma via gankyrin/AKT signaling. Oncogene 38, 406–420. doi: 10.1038/s41388-018-0440-8
Harano, Y., Yasui, K., Toyama, T., Nakajima, T., Mitsuyoshi, H., Mimani, M., et al. (2006). Fenofibrate, a peroxisome proliferator-activated receptor α agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 26, 613–620. doi: 10.1111/j.1478-3231.2006.01265.x
Hatanaka, F., and Takumi, T. (2017). CHRONO integrates behavioral stress and epigenetic control of metabolism. Ann. Med. 49, 352–356.
Haupt, Y., Maya, R., Kazaz, A., and Oren, M. (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299. doi: 10.1038/387296a0
Hegarty, B. D., Bobard, A., Hainault, I., Ferré, P., Bossard, P., and Foufelle, F. (2005). Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory elementbinding protein-1c. Proc. Natl. Acad. Sci. U. S. A. 102, 791–796. doi: 10.1073/pnas.0405067102
Heo, G., and Ko, K. S. (2019). Long-term feeding of soy protein attenuates choline deficient-induced adverse effects in wild type mice and prohibitin 1 deficient mice response more sensitively. Prev. Nutr. Food Sci. 24:32. doi: 10.3746/pnf.2019.24.1.32
Honda, R., and Yasuda, H. (2000). Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19, 1473–1476. doi: 10.1038/sj.onc.1203464
Hu, C., Liu, D., Zhang, Y., Lou, G., Huang, G., Chen, B., et al. (2014). LXRα-mediated downregulation of FOXM1 suppresses the proliferation of hepatocellular carcinoma cells. Oncogene 33, 2888–2897. doi: 10.1038/onc.2013.250
Jeon, T.-I., and Osborne, T. F. (2012). SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol. Metabol. 23, 65–72. doi: 10.1016/j.tem.2011.10.004
Jung, S., Park, J., and Ko, K. S. (2020). Lipopolysaccharide-induced innate immune responses are exacerbated by Prohibitin 1 deficiency and mitigated by S-adenosylmethionine in murine macrophages. PLoS ONE 15:e0241224. doi: 10.1371/journal.pone.0241224
Kakino, S., Ohki, T., Nakayama, H., Yuan, X., Otabe, S., Hashinaga, T., et al. (2018). Pivotal role of TNF-α in the development and progression of nonalcoholic fatty liver disease in a murine model. Hormone Metabolic Res. 50, 80–87. doi: 10.1055/s-0043-118666
Kitamura, H., and Hashimoto, M. (2021). USP2-related cellular signaling and consequent pathophysiological outcomes. Int. J. Mol. Sci. 22:1209. doi: 10.3390/ijms22031209
Knebel, B., Hartwig, S., Jacob, S., Kettel, U., Schiller, M., Passlack, W., et al. (2018). Inactivation of SREBP-1a phosphorylation prevents fatty liver disease in mice: identification of related signaling pathways by gene expression profiles in liver and proteomes of peroxisomes. Int. J. Mol. Sci. 19:980. doi: 10.3390/ijms19040980
Ko, K. S., Tomasi, M. L., Iglesias-Ara, A., French, B. A., French, S. W., Ramani, K., et al. (2010). Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology 52, 2096–2108. doi: 10.1002/hep.23919
Kong, B.-W., Hudson, N., Seo, D., Lee, S., Khatri, B., Lassiter, K., et al. (2017). RNA sequencing for global gene expression associated with muscle growth in a single male modern broiler line compared to a foundational Barred Plymouth Rock chicken line. BMC Genom. 18, 1–19. doi: 10.1186/s12864-016-3471-y
Kopanja, D., Pandey, A., Kiefer, M., Wang, Z., Chandan, N., Carr, J. R., et al. (2015). Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J. Hepatol. 63, 429–436. doi: 10.1016/j.jhep.2015.03.023
Lee, J., Kong, B., and Lee, S.-H. (2020). Patchouli alcohol, a compound from Pogostemoncablin, inhibits obesity. J. Med. Food 23, 326–334. doi: 10.1089/jmf.2019.0182
Lee, J.-K., Wang, J., Sa, J. K., Ladewig, E., Lee, H.-O., Lee, I.-H., et al. (2017). Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 49, 594–599. doi: 10.1038/ng.3806
Li, A. C., and Palinski, W. (2006). Peroxisome proliferator-activated receptors: how their effects on macrophages can lead to the development of a new drug therapy against atherosclerosis. Annu. Rev. Pharmacol. Toxicol. 46, 1–39. doi: 10.1146/annurev.pharmtox.46.120604.141247
Li, L., and Falany, C. N. (2007). Elevated hepatic SULT1E1 activity in mouse models of cystic fibrosis alters the regulation of estrogen responsive proteins. J. Cystic Fibr. 6, 23–30. doi: 10.1016/j.jcf.2006.05.001
Li, L., He, D., Wilborn, T. W., Falany, J. L., and Falany, C. N. (2009). Increased SULT1E1 activity in HepG2 hepatocytes decreases growth hormone stimulation of STAT5b phosphorylation. Steroids 74, 20–29. doi: 10.1016/j.steroids.2008.09.002
Li, Y., Lu, L., Tu, J., Zhang, J., Xiong, T., Fan, W., et al. (2020). Reciprocal regulation between forkhead box M1/NF-κB and methionine adenosyltransferase 1A drives liver cancer. Hepatology 72, 1682–1700. doi: 10.1002/hep.31196
Loayza-Puch, F., Rooijers, K., Buil, L. C., Zijlstra, J., Vrielink, J. F. O., Lopes, R., et al. (2016). Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494. doi: 10.1038/nature16982
Lounis, M. A., Escoula, Q., Veillette, C., Bergeron, K.-F., Ntambi, J. M., and Mounier, C. (2016). SCD1 deficiency protects mice against ethanol-induced liver injury. Biochim. Biophys. Acta 1861, 1662–1670. doi: 10.1016/j.bbalip.2016.07.012
Matsushita, N., Hassanein, M. T., Martinez-Clemente, M., Lazaro, R., French, S. W., Xie, W., et al. (2017). Gender difference in NASH susceptibility: roles of hepatocyte Ikkβ and Sult1e1. PLoS ONE 12:e0181052. doi: 10.1371/journal.pone.0181052
Miao, J., Haas, J. T., Manthena, P., Wang, Y., Zhao, E., Vaitheesvaran, B., et al. (2014). Hepatic insulin receptor deficiency impairs the SREBP-2 response to feeding and statins. J. Lip. Res. 55, 659–667. doi: 10.1194/jlr.M043711
Michael, M. D., Kulkarni, R. N., Postic, C., Previs, S. F., Shulman, G. I., Magnuson, M. A., et al. (2000). Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6, 87–97. doi: 10.1016/S1097-2765(05)00015-8
Mishra, S., Murphy, L. C., and Murphy, L. J. (2006). The Prohibitins: emerging roles in diverse functions. J. Cell. Mol. Med. 10, 353–363. doi: 10.1111/j.1582-4934.2006.tb00404.x
Moslehi, A., and Hamidi-Zad, Z. (2018). Role of SREBPs in liver diseases: a mini-review. J. Clin. Transl. Hepatol. 6:332. doi: 10.14218/JCTH.2017.00061
Muranaka, H., Hayashi, A., Minami, K., Kitajima, S., Kohno, S., Nishimoto, Y., et al. (2017). A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 6:e350. doi: 10.1038/oncsis.2017.51
Nagaya, T., Tanaka, N., Suzuki, T., Sano, K., Horiuchi, A., Komatsu, M., et al. (2010). Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 53, 724–731. doi: 10.1016/j.jhep.2010.04.033
Nijtmans, L. G., de Jong, L., Sanz, M. A., Coates, P. J., Berden, J. A., Back, J. W., et al. (2000). Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 19, 2444–2451. doi: 10.1093/emboj/19.11.2444
Ramani, K., Mavila, N., Ko, K. S., Mato, J. M., and Lu, S. C. (2016). Prohibitin 1 regulates the H19-Igf2 axis and proliferation in hepatocytes. J. Biol. Chem. 291, 24148–24159. doi: 10.1074/jbc.M116.744045
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Saavedra-García, P., Nichols, K., Mahmud, Z., Fan, L. Y.-N., and Lam, E. W. (2018). Unravelling the role of fatty acid metabolism in cancer through the FOXO3-FOXM1 axis. Mol. Cell. Endocrinol. 462, 82–92. doi: 10.1016/j.mce.2017.01.012
Samuel, V. T., and Shulman, G. I. (2012). Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871. doi: 10.1016/j.cell.2012.02.017
Savarin, C., Bergmann, C. C., Hinton, D. R., and Stohlman, S. A. (2013). MMP-independent role of TIMP-1 at the blood brain barrier during viral encephalomyelitis. ASN Neuro 5:AN20130033. doi: 10.1042/AN20130033
Shimomura, I., Bashmakov, Y., Ikemoto, S., Horton, J. D., Brown, M. S., and Goldstein, J. L. (1999). Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U. S. A. 96, 13656–13661. doi: 10.1073/pnas.96.24.13656
Smalling, R. L., Delker, D. A., Zhang, Y., Nieto, N., Mcguiness, M. S., Liu, S., et al. (2013). Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am. J. Physiol. Gastrointestinal Liver Physiol. 305, G364–G374. doi: 10.1152/ajpgi.00077.2013
Song, B., Zhang, H., and Zhang, S. (2018). Toll-like receptor 2 mediates deposition of collagen I in adipose tissue of high fat diet-induced obese mice. Mol. Med. Rep. 17, 5958–5963. doi: 10.3892/mmr.2018.8590
Song, W. C. (2001). Biochemistry and reproductive endocrinology of estrogen sulfotransferase. Ann. New York Acad. Sci. 948, 43–50.
Souri, M., Osaki, T., and Ichinose, A. (2015). The non-catalytic B subunit of coagulation factor XIII accelerates fibrin cross-linking. J. Biol. Chem. 290, 12027–12039. doi: 10.1074/jbc.M114.608570
Stevenson, L. F., Sparks, A., Allende-Vega, N., Xirodimas, D. P., Lane, D. P., and Saville, M. K. (2007). The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 26, 976–986. doi: 10.1038/sj.emboj.7601567
Stuebner, S., Faus-Kessler, T., Fischer, T., Wurst, W., and Prakash, N. (2010). Fzd3 and Fzd6 deficiency results in a severe midbrain morphogenesis defect. Dev. Dyn. 239, 246–260. doi: 10.1002/dvdy.22127
Sun, H.-C., Li, M., Lu, J.-L., Yan, D.-W., Zhou, C.-Z., Fan, J.-W., et al. (2011). Overexpression of Forkhead box M1 protein associates with aggressive tumor features and poor prognosis of hepatocellular carcinoma. Oncol. Rep. 25, 1533–1539. doi: 10.3892/or.2011.1230
Sun, Q., Zhao, Y., Yang, Y., Yang, X., Li, M., Xu, X., et al. (2017). Loss of the clock protein PER2 shortens the erythrocyte life span in mice. J. Biol. Chem. 292, 12679–12690. doi: 10.1074/jbc.M117.783985
Talukdar, I., Szeszel-Fedorowicz, W., and Salati, L. M. (2005). Arachidonic acid inhibits the insulin induction of glucose-6-phosphate dehydrogenase via p38 MAP kinase. J. Biol. Chem. 280, 40660–40667. doi: 10.1074/jbc.M505531200
Tang, H., and Goldberg, E. (2012). A-MYB (MYBL1) stimulates murine testis-specific Ldhc expression via the cAMP-responsive element (CRE) site. Biol. Reprod. 86, 30, 31–38. doi: 10.1095/biolreprod.111.095661
Teh, M.-T. (2012). FOXM1 coming of age: time for translation into clinical benefits? Front. Oncol. 2:146. doi: 10.3389/fonc.2012.00146
Thiele, N. D., Wirth, J. W., Steins, D., Koop, A. C., Ittrich, H., Lohse, A. W., et al. (2017). TIMP-1 is upregulated, but not essential in hepatic fibrogenesis and carcinogenesis in mice. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-00671-1
Tomita, K., Tamiya, G., Ando, S., Ohsumi, K., Chiyo, T., Mizutani, A., et al. (2006). Tumour necrosis factor α signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424. doi: 10.1136/gut.2005.071118
Urlep, Ž., Lorbek, G., Perše, M., Jeruc, J., Juvan, P., Matz-Soja, M., et al. (2017). Disrupting hepatocyte Cyp51 from cholesterol synthesis leads to progressive liver injury in the developing mouse and decreases RORC signalling. Sci. Rep. 7, 1–13. doi: 10.1038/srep40775
Wang, H., Lafdil, F., Wang, L., Yin, S., Feng, D., and Gao, B. (2011). Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 1, 1–10. doi: 10.1186/2045-3701-1-14
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wang, L., Zhang, C.-g., Jia, Y.-l., and Hu, L. (2020). Tissue Inhibitor of Metalloprotease-1 (TIMP-1) Regulates Adipogenesis of Adipose-derived Stem Cells (ASCs) via the Wnt Signaling Pathway in an MMP-independent Manner. Curr. Med. Sci. 40, 989–996. doi: 10.1007/s11596-020-2265-2
Wang, S., Fusaro, G., Padmanabhan, J., and Chellappan, S. P. (2002a). Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 21, 8388–8396. doi: 10.1038/sj.onc.1205944
Wang, S., Zhang, B., and Faller, D. V. (2002b). Prohibitin requires Brg-1 and Brm for the repression of E2F and cell growth. EMBO J. 21, 3019–3028. doi: 10.1093/emboj/cdf302
Wang, S., Zhang, B., and Faller, D. V. (2004). BRG1/BRM and prohibitin are required for growth suppression by estrogen antagonists. EMBO J. 23, 2293–2303. doi: 10.1038/sj.emboj.7600231
Wei, Y.-L., and Yang, W.-X. (2019). Kinesin-14 motor protein KIFC1 participates in DNA synthesis and chromatin maintenance. Cell Death Dis. 10, 1–14. doi: 10.1038/s41419-019-1619-9
Wills, M. K., Tong, J., Tremblay, S. L., Moran, M. F., and Jones, N. (2014). The ShcD signaling adaptor facilitates ligand-independent phosphorylation of the EGF receptor. Mol. Biol. Cell 25, 739–752. doi: 10.1091/mbc.e13-08-0434
Wu, L., Nam, Y.-J., Kung, G., Crow, M. T., and Kitsis, R. N. (2010). Induction of the apoptosis inhibitor ARC by Ras in human cancers. J. Biol. Chem. 285, 19235–19245. doi: 10.1074/jbc.M110.114892
Wu, Y., Wang, H., Qiao, L., Jin, X., Dong, H., and Wang, Y. (2019). Silencing of RAD51AP1 suppresses epithelial–mesenchymal transition and metastasis in non-small cell lung cancer. Thoracic Cancer 10, 1748–1763. doi: 10.1111/1759-7714.13124
Xiao, X., and Song, B.-L. (2013). SREBP: a novel therapeutic target. Actabiochim. BiophysicaSinica 45, 2–10. doi: 10.1093/abbs/gms112
Xu, Y., Lin, X., Xu, J., Jing, H., Qin, Y., and Li, Y. (2018). SULT1E1 inhibits cell proliferation and invasion by activating PPARγ in breast cancer. J. Cancer 9:1078. doi: 10.7150/jca.23596
Yan, W., Jiang, M., and Zheng, J. (2020). Identification of key pathways and differentially expressed genes in bronchopulmonary dysplasia using bioinformatics analysis. Biotechnol. Lett. 42, 2569–2580. doi: 10.1007/s10529-020-02986-y
Yang, J., Li, B., and He, Q.-Y. (2018). Significance of prohibitin domain family in tumorigenesis and its implication in cancer diagnosis and treatment. Cell Death Dis. 9, 1–10. doi: 10.1038/s41419-018-0661-3
Ylisirniö, S., Höyhtyä, M., and Turpeenniemi-Hujanen, T. (2000). Serum matrix metalloproteinases-2,-9 and tissue inhibitors of metalloproteinases-1,-2 in lung cancer–TIMP-1 as a prognostic marker. Anticancer Res. 20, 1311–1316.
Yoshiji, H., Kuriyama, S., Miyamoto, Y., Thorgeirsson, U. P., Gomez, D. E., Kawata, M., et al. (2000). Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 32, 1248–1254. doi: 10.1053/jhep.2000.20521
Young, M.-J., Hsu, K.-C., Lin, T. E., Chang, W.-C., and Hung, J.-J. (2019). The role of ubiquitin-specific peptidases in cancer progression. J. Biomed. Sci. 26, 1–14. doi: 10.1186/s12929-019-0522-0
Young, R., Passet, B., Vilotte, M., Cribiu, E.P., Béringue, V., Le Provost, F., et al. (2009). The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS letters 583, 3296–3300.
Yukawa, N., Yoshikawa, T., Akaike, M., Sugimasa, Y., Rino, Y., Masuda, M., et al. (2007). Impact of plasma tissue inhibitor of matrix metalloproteinase-1 on long-term survival in patients with colorectal cancer. Oncology 72, 205–208. doi: 10.1159/000112827
Keywords: prohibitin 1, RNA-seq, gene expression, liver disease, hepatotoxicity
Citation: Lee K, Yu H, Shouse S, Kong B, Lee J, Lee S-H and Ko KS (2021) RNA-Seq Reveals Different Gene Expression in Liver-Specific Prohibitin 1 Knock-Out Mice. Front. Physiol. 12:717911. doi: 10.3389/fphys.2021.717911
Received: 31 May 2021; Accepted: 27 July 2021;
Published: 03 September 2021.
Edited by:
Atsushi Masamune, Tohoku University, JapanReviewed by:
Derick Han, Keck Graduate Institute of Applied Life Sciences, United StatesEiji Kakazu, National Center for Global Health and Medicine, Japan
Copyright © 2021 Lee, Yu, Shouse, Kong, Lee, Lee and Ko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwang Suk Ko, a2tvQGV3aGEuYWMua3I=