
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 11 August 2021
Sec. Craniofacial Biology and Dental Research
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.715687
This article is part of the Research TopicAlveolar Bone: a Pivotal Role in Periodontal Disease Pathobiology and Treatment, Volume IView all 11 articles
 Satoru Yamada1,2*
Satoru Yamada1,2* Kenichiro Tsushima1Masaki Kinoshita1Hiromi Sakashita1
Kenichiro Tsushima1Masaki Kinoshita1Hiromi Sakashita1 Tetsuhiro Kajikawa1†
Tetsuhiro Kajikawa1† Chiharu Fujihara1Hang Yuan2Shigeki Suzuki2Takayuki Morisaki3
Chiharu Fujihara1Hang Yuan2Shigeki Suzuki2Takayuki Morisaki3 Shinya Murakami1
Shinya Murakami1Loeys–Dietz syndrome (LDS) is a syndromic connective tissue disorder caused by a heterozygous missense mutation in genes that encode transforming growth factor (TGF)-β receptor (TGFBR) 1 and 2. We encountered a patient with LDS, who had severe periodontal tissue destruction indicative of aggressive periodontitis. The patient had a missense mutation in the glycine and serine-rich domain of TGFBR1 exon 3. This G-to-T mutation at base 563 converted glycine to valine. We established an LDS model knock-in mouse that recapitulated the LDS phenotype. Homozygosity of the mutation caused embryonic lethality and heterozygous knock-in mice showed distorted and ruptured elastic fibers in the aorta at 24 weeks of age and died earlier than wildtype (WT) mice. We stimulated mouse embryonic fibroblasts (MEFs) from the knock-in mouse with TGF-β and examined their responses. The knock-in MEFs showed downregulated Serpine 1 mRNA expression and phosphorylation of Smad2 to TGF-β compared with WT MEFs. To clarify the influence of TGF-β signaling abnormalities on the pathogenesis or progression of periodontitis, we performed pathomolecular analysis of the knock-in mouse. There were no structural differences in periodontal tissues between WT and LDS model mice at 6 or 24 weeks of age. Micro-computed tomography revealed no significant difference in alveolar bone resorption between WT and knock-in mice at 6 or 24 weeks of age. However, TGF-β-related gene expression was increased significantly in periodontal tissues of the knock-in mouse compared with WT mice. Next, we assessed a mouse periodontitis model in which periodontal bone loss was induced by oral inoculation with the bacterial strain Porphyromonas gingivalis W83. After inoculation, we collected alveolar bone and carried out morphometric analysis. P. gingivalis-induced alveolar bone loss was significantly greater in LDS model mice than in WT mice. Peritoneal macrophages isolated from Tgfbr1G188V/+ mice showed upregulation of inflammatory cytokine mRNA expression induced by P. gingivalis lipopolysaccharide compared with WT macrophages. In this study, we established an LDS mouse model and demonstrated that LDS model mice had elevated susceptibility to P. gingivalis-induced periodontitis, probably through TGF-β signal dysfunction. This suggests that TGF-β signaling abnormalities accelerate the pathogenesis or progression of periodontitis.
Marfan syndrome (MFS) is an autosomal dominant connective tissue disease caused by a mutation in fibrillin-1 (Dietz et al., 1991), which leads to systemic diseases with various phenotypes. It affects approximately 1 in 5,000 people and includes patients with mild disease (Dietz et al., 1991). In particular, MFS causes characteristic signs in the skeletal system (e.g., bone hyperplasia, joint relaxation, height, arachnoid finger, and spinal scoliosis), cardiovascular system (e.g., aortic aneurysm and mitral valve deviation), and ocular system (e.g., lens subluxation) (Schorr et al., 1951). Fibrillin-1 is a component of microfibrils in the extracellular matrix (Godfrey et al., 1990) and is involved in the control of transforming growth factor (TGF)-β expression and function (Neptune et al., 2003). TGF-β is a cytokine involved in the regulation of cell proliferation, differentiation, and death (Huang and Chen, 2012). In particular, it promotes collagen production and extracellular matrix remodeling (Dietz, 2007). Various MFS symptoms result from excessive TGF-β in serum caused by fibrillin-1 mutations (Matt et al., 2009). Additionally, various MFS-related diseases are caused by abnormalities in TGF-β signaling, which include Beals syndrome caused by mutations in fibrillin-2 (Putnam et al., 1995), Loeys–Dietz syndrome (LDS) caused by mutations in TGF-β receptor (TGFBR) 1 or TGFBR2 (Mizuguchi et al., 2004; Loeys et al., 2005, 2006), juvenile polyposis syndrome caused by mutations in Smad4, a TGF-β signaling factor (Howe et al., 1998), and Shprintzen–Goldberg syndrome caused by mutations in SKI, a gene that suppresses Smad signaling (Doyle et al., 2012).
There have been reports of patients with MFS and severe periodontitis (De Coster et al., 2002; Straub et al., 2002; Jain and Pandey, 2013; Staufenbiel et al., 2013). Patients with MFS have greater morbidity and severity of periodontitis than otherwise healthy individuals (Suzuki et al., 2015). In patients who exhibit MFS, severe chronic periodontitis has been reported with periodontitis substantial palatal and dental irregularities. To our knowledge, details of the relationships between genetic mutations and periodontitis in MFS patients and its related diseases are unclear. Thus, in this study, we first investigated the relationships between MFS, related diseases, and periodontitis. We identified a patient with LDS, who had an aggressive periodontitis-like pathology. Periodontitis progresses under a range of conditions that include environmental and genetic factors. Periodontitis is broadly divided into chronic and aggressive periodontitis, with aggressive periodontitis assumed to be more closely related to genetic factors (Meng et al., 2007). Recently, a new periodontitis classification scheme has been adopted, in which forms of the disease recognized as “chronic” or “aggressive” are now characterized by a multi-dimensional staging and grading system (Papapanou et al., 2018).
LDS is caused by mutations in TGFBR1 or TGFBR2 (Loeys et al., 2005). LDS and MFS share many common clinical symptoms that include aortic lesions (e.g., basal dilatation, aortic aneurysm, and aortic dissection) and skeletal system lesions (e.g., scoliosis, joint laxity, and spider finger). However, features not evident in patients with MFS (i.e., characteristic facial features such as cleft palate, dichotomy uvula, interocular dissociation, systemic blood vessel meandering, craniosynostosis, congenital heart disease, and intellectual disability) are observed at high rates in patients with LDS (Loeys et al., 2006). In vascular smooth muscle cells collected from patients with LDS, TGF-β levels are low (Gallo et al., 2014). However, a compensatory change comprises TGF-β overexpression in aortic tissues. This likely results in the onset of cardiovascular symptoms such as aortic aneurysms and aortic dissection (Loeys et al., 2005; Gallo et al., 2014). On the basis of the data collected from the LDS patient who had aggressive periodontitis, we developed genetically modified mice with the candidate genetic mutation to analyze the disease state and the effects of a mutation in a TGF-β-related gene on periodontitis.
All human experiments were approved by the Institutional Ethics Committee of Osaka University Graduate School of Dentistry (No. H22-E10) and the National Cerebral and Cardiovascular Center (No. M22-34). The epidemiological study included 120 patients who had visited the National Cerebral and Cardiovascular Center Hospital in Osaka, Japan, and were diagnosed with Marfan syndrome or Marfan-related syndrome by the revised Ghent nosology (Loeys et al., 2010) to survey the prevalence rate of periodontal disease in Marfan syndrome and Marfan-related syndrome patients. Informed consent was obtained from all patients involved in the study. Genomic DNA isolated from peripheral white blood cells was amplified by polymerase chain reaction (PCR) using primers in the flanking introns of TGFBR1 and TGFBR2. Sequence analyses were performed using Applied Biosystems automated DNA sequencer (ABI3770, Waltham, MA, United States) in accordance with the manufacturer’s protocol.
All animal experiments were approved by the Institutional Animal Care and Use Committee of Osaka University Graduate School of Dentistry and complied with the guidelines for the care and use of laboratory animals at Osaka University. This study was carried out in compliance with the ARRIVE guidelines, where applicable. To generate Tgfbr1G188V/+ mice, site-directed mutagenesis was performed to replace glycine with valine at codon 188 of Tgfbr1 (guanine to thymine at nucleotide 563). This mutated Tgfbr1 cDNA was cloned into pBSIISK + with a floxed neomycin resistance cassette (NeoR). Bac-based long-range PCR was used to amplify murine genomic fragments of Tgfbr1. The long arm (6 kb, exon 3 with G188V) and short arm (2.8 kb, exon 4) were cloned into pBS-DTA and pBS-LNL, respectively. The final targeting vector was constructed and then linearized. The targeting vector DNA was electroporated into C57BL/6 ES cells. Homologous recombination-positive ES cells were identified by Southern blot analysis. Positive clones were injected into BALB/c blastocysts and transferred into pseudopregnant female mice. Chimeric offspring were mated with C57BL/6 mice and germline transmission was confirmed by RT-PCR. The loxP-flanked NeoR was removed by mating Tgfbr1G188V/+ founder mice with CAG-Cre recombinase transgenic mice (Sakai and Miyazaki, 1997). Experimental mice were backcrossed with C57BL/6 mice to remove the CAG-Cre transgene. Genotype analysis of Tgfbr1G188V/+ mice was performed by genomic PCR using flanking loxP site-specific primers (5′CTAAGAGAAGTGTGCCTCCTTTACA-3′ and 5′-CCAAAGTCATAGAGCATGTGTTAGA-3′).
Wildtype (WT) and Tgfbr1G188V/+ mouse embryonic fibroblasts (MEFs) were isolated from embryos on day 13.5 by a previously described method (Awata et al., 2015). MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 60 μg/mL kanamycin. MEFs from passages 3–5 were used in this study. Each genotype of MEFs (WT, Tgfbr1G188V/+, and Tgfbr1G188V/G188V) was cultured in a 12-well plate until confluency. The next day, the medium was replaced with serum-free DMEM. After serum deprivation for 24 h, the cells were stimulated with TGF-β (R&D Systems, Minneapolis, MN, United States) in serum-free DMEM for 30 min for western blot analysis and 12 h for quantitative PCR analysis.
cDNA of the mouse Tgfbr1 gene was cloned into the p3XFLAG-CMV-14 expression vector (Sigma-Aldrich, St. Louis, MO, United States). cDNA for the Tgfbr1 mutation (G188V: Tgfbr1G188V) was ligated to Tgfbr1 cDNA by the Quick Change Site-Directed Mutagenesis kit (Stratagene, LA Jolla, CA, United States) in accordance with the manufacturer’s protocol. The sequence was verified by DNA sequencing. For luciferase assays, human embryonic kidney (HEK) 293 cells were seeded in a 12-well plate. After 24 h, the cells were transfected with the Tgfbr1G188V/3XFLAG-CMV-14 expression vector or wildtype Tgfbr1/3XFLAG-CMV-14 expression vector mixed with Transcription Factor Reporter using a Signal SMAD Reporter Assay kit (Polyscience, Inc., Warrington, PA, United States) in accordance with the manufacturer’s protocol. At 48 h after transfection, the medium was replaced with serum-free DMEM. After serum deprivation for 24 h, the cells were stimulated with 0–5 ng/ml TGF-β in serum-free DMEM for 8 h. Luciferase activity was measured by a GloMax 96 Microplate Luminometer (Promega, Madison, WI, United States).
Thoracic aortae were collected from WT and Tgfbr1G188V/+ mice at 24 weeks of age and fixed overnight in 4% paraformaldehyde (PFA)/phosphate buffer (Wako Pure Chemical Industries, Osaka, Japan). Samples were embedded in paraffin and sectioned at 5 μm thicknesses using a LEICA RM2245 microtome (Leica Microsystems, Wetzlar, Germany). Sections were stained with Elastica van Gieson (EVG) for elastin staining. Maxillae from WT and Tgfbr1G188V/+ mice at 6 and 24 weeks of age were fixed in 4% PFA/phosphate-buffered saline (PBS) (Wako Pure Chemical Industries) overnight at 4°C and decalcified in 0.5 M EDTA (Wako) for 1 week. After decalcification, periodontal tissues were dehydrated using 15, 20, and 25% sucrose in PBS. Then, periodontal tissues were embedded in O.C.T. Compound (Sakura Finetek, Tokyo, Japan). They were frozen and sectioned at 5 μm thicknesses in a mesiodistal orientation using the LEICA RM2245 microtome. Sections were stained with hematoxylin-eosin (HE) or using a Tartrate-Resistance Acid Phosphatase (TRAP) Staining Kit (Wako) in accordance with the manufacturer’s protocol.
Thoracic aortae, maxillae, and periodontal tissues were extracted from 6-week-old male WT and Tgfbr1G188V/+ mice. Total RNA from tissues or MEFs was extracted using a PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA, United States). Total RNA was reversed transcribed to cDNA using a High-Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, United States). Quantitative PCR was performed with the StepOnePlus Real-time PCR system (Applied Biosystems) using Fast SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA, United States) and gene-specific primers (Table 1).
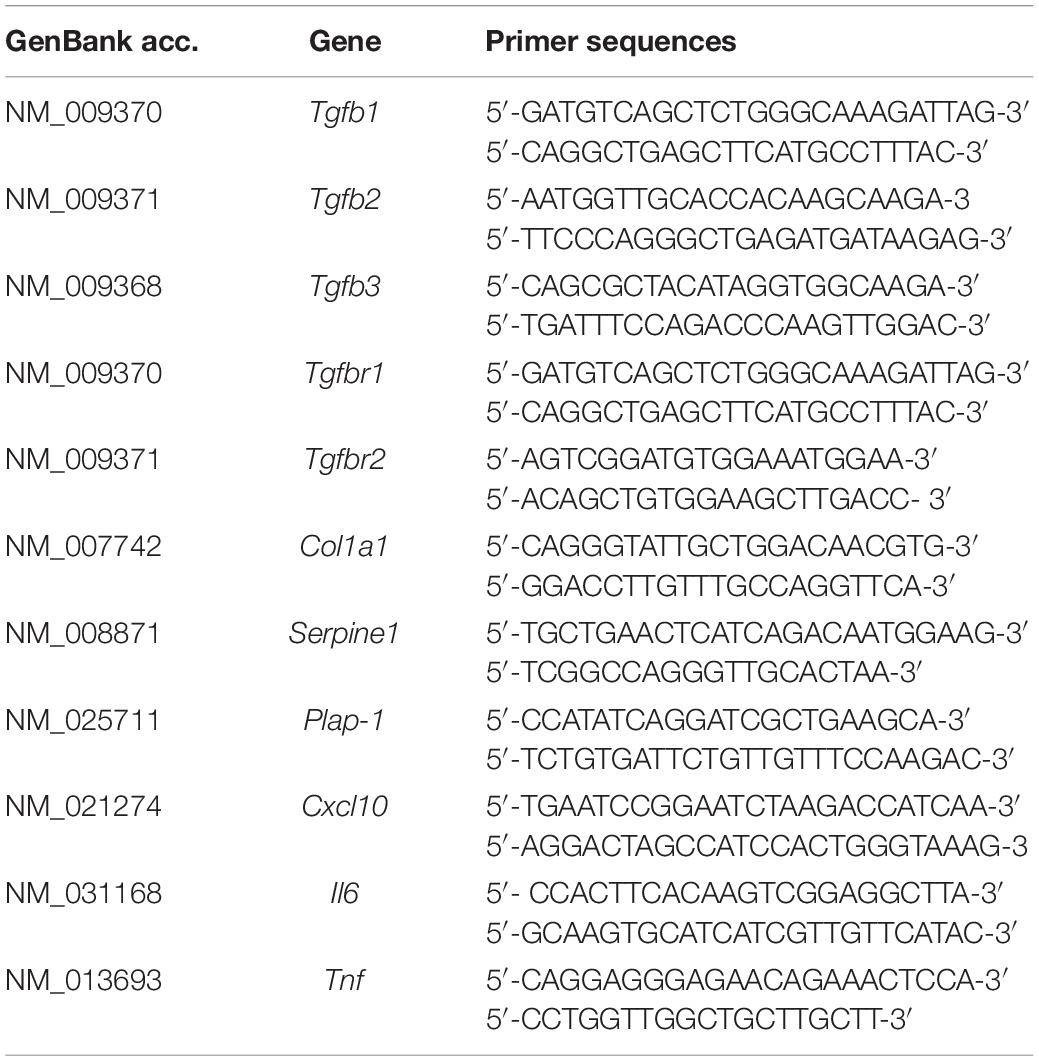
Table 1. Primer sequences.
Cells were lysed with RIPA buffer (Millipore, Billerica, MA, United States) that contained phosphatase and proteinase inhibitors. The protein concentration of the lysates was measured by the Bradford assay (Bio-Rad, Hercules, CA, United States). Aliquots of lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subjected to western blot analysis. Primary antibodies included a rabbit anti-TGFBR1 antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, United States), mouse anti-beta actin antibody (1:10,000, Sigma-Aldrich), rabbit anti-phospho-Smad2 antibody (1:1,000, Millipore), and rabbit anti-Smad2 antibody (1:1,000, Cell Signaling Technology, Danvers, MA, United States). Secondary antibodies were a horseradish peroxidase (HRP)-linked anti-mouse IgG antibody (1:10,000, GE Healthcare, Piscataway, NJ, United States) and HRP-linked anti-rabbit IgG antibody (1:10,000, GE Healthcare). Immunoreactive proteins were detected by SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, United States) with an ImageQuant LAS4,000 (GE Healthcare).
Maxillae were collected from WT and Tgfbr1G188V/+ mice at 6 and 24 weeks of age and imaged by a 3D micro X-ray CT R_mCT2 (Rigaku, Tokyo, Japan). The images were analyzed using TRI/3D-BON software (RATOC System Engineering, Tokyo, Japan). The root surface area between the alveolar bone crest and cementoenamel junction was measured using WinROOF software (Mitani, Fukui, Japan). The total value of these three distances was regarded as alveolar bone resorption. Alveolar bone resorption was measured in the root surface area between the alveolar apex from the cementoenamel junction.
Porphyromonas gingivalis strain W83 was cultured in modified Gifu anaerobic medium broth (Nissui, Tokyo, Japan) in an anaerobic jar (Becton Dickinson Microbiology System, Cockeysville, MD, United States) in the presence of an AnaeroPack (Mitsubishi Gas Chemical Co., Inc., Tokyo, Japan) for 48 h at 37°C. Bacterial suspensions were prepared in PBS without Mg2+/Ca2+ using established growth curves and spectrophotometric analysis. The number of CFUs was standardized by measuring optical density at 600 nm. The murine experimental periodontal infection model was established in accordance with a previously described method (Aoki-Nonaka et al., 2013). The 8-week-old male mice received sulfamethoxazole and trimethoprim at final concentrations of 700 and 400 μg/mL, respectively, in water bottles administered ad libitum for 10 days. This treatment was followed by 3 days without antibiotics. The experimental group was then infected as follows. In total, 1 × 109 CFUs of live P. gingivalis were suspended in 100 μL PBS with 2% carboxymethyl cellulose (Sigma−Aldrich) and administered to each mouse through a feeding needle 10 times at 3-day intervals. The control group received the same pretreatment and was sham infected without P. gingivalis. At 1 day after the final treatment, the mice were sacrificed with CO2 affixation and their maxillae were collected for micro-CT and histological analyses.
Macrophages were harvested from the peritoneal cavity of 8-week-old male WT and Tgfbr1G188V/+ mice at 3 days after injection of thioglycollate (Yamaba et al., 2015) and seeded on 6-well plates. After 2 h of incubation, non-adherent cells were washed out and the remaining cells were subjected to further analysis. The peritoneal macrophages were stimulated with 1 Uμg/ml P. gingivalis lipopolysaccharide (LPS) (InvivoGen, Inc., San Diego, CA, United States) for the indicated times and then total RNA was isolated for quantitative PCR analysis.
Data are represented as the mean ± SD. Statistical analyses were performed using the Student’s t-test for paired comparisons and one-way analysis of variance for multiple comparisons using Bonferroni’s post hoc test with Excel statistics software (Bellcurve, Tokyo, Japan). A value of P < 0.05 was considered statistically significant.
We surveyed 120 patients with Marfan syndrome or Marfan-related syndrome to determine the prevalence rate of periodontal disease in these syndromes. We found that the prevalence of chronic periodontitis in Marfan syndrome and Marfan-related syndrome patients was significantly higher compared with that in healthy controls (manuscript in preparation). In this clinical study, we encountered a 44-year-old Japanese female with LDS, who had localized vertical bone loss around molars in spite of good oral hygiene (O’Leary’s plaque control record: 13.0%) as shown in Figure 1. She was only one patient diagnosed with aggressive periodontitis (new classification: stage III and grade B). She was diagnosed with familial thoracic aortic aneurysms and dissections. She was treated by total arch aortic and descending aorta replacement. Systemic features of the patient were a tall height (−), down slanting palpebral fissures (+), retrognathia (+), pectus carinatum deformity (−), pneumothorax (−), myopia (−), hypertelorism (+), and bifid uvula (−). Genomic sequencing of the patient revealed a mutation in the glycine and serine-rich domain of TGFBR1 exon 3 (referred to as the GS domain). This G-to-T mutation at base 563 converted glycine to valine at residue 188 (G188V) (Figure 2A). To assess the effects of the G188V mutation on TGF-β signaling, we introduced mouse Tgfbr1WT and Tgfbr1G188V expression vectors into HEK293 cells. We confirmed that the TGF-β type I receptor was sufficiently expressed in both groups (Figure 2B). Next, we performed luciferase assays and found that TGF-β-induced luciferase activity was significantly elevated in the group with high levels of Tgfbr1WT and significantly suppressed in the group with Tgfbr1G188V (Figure 2C). These results suggested that the G188V mutation resulted in loss of TGF-β signaling.
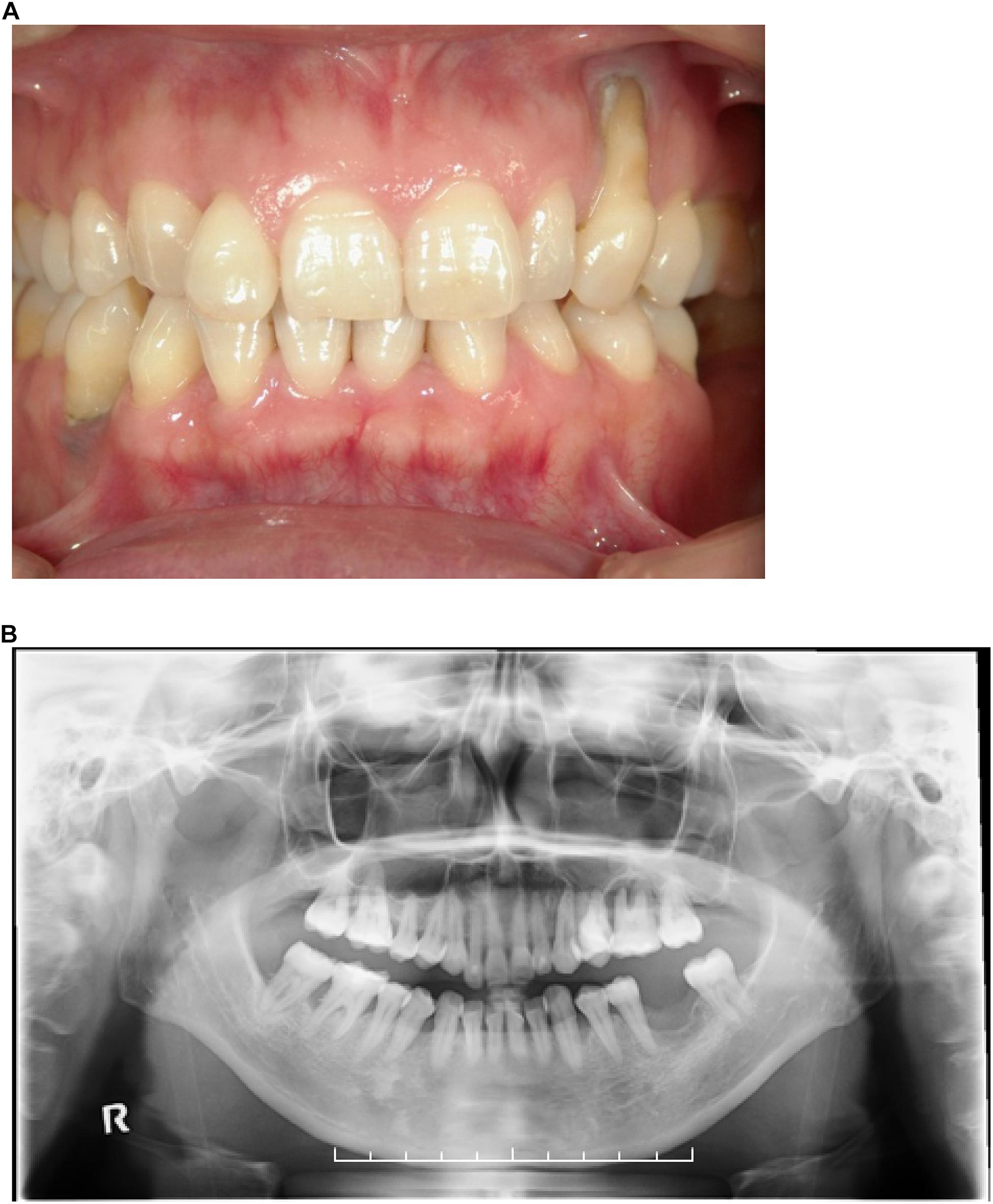
Figure 1. (A) Front view of the dentition of a patient with LDS and aggressive periodontitis. (B) Orthopantomograph of the patient. Note the presence of localized vertical bone loss around molars.
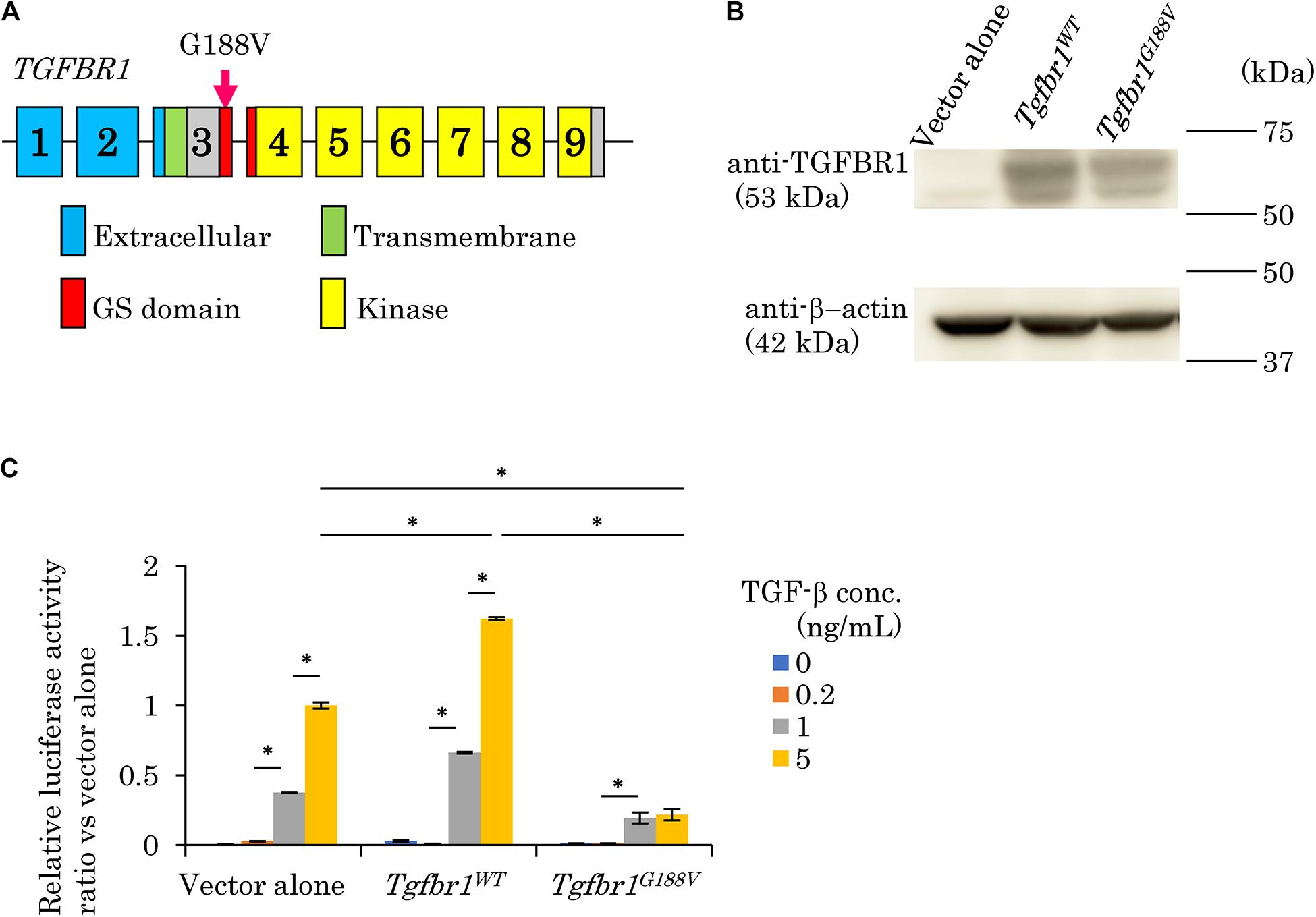
Figure 2. Mutation in exon 3 of TGFBR1 isolated from a patient with LDS and aggressive periodontitis diminishes the response to TGF-β (G188V mutation). (A) Location of the mutation in exon 3 of TGFBR1. (B) HEK293 cells were transfected with a vector alone, wildtype mouse Tgfbr1 (Tgfbr1WT), or mouse Tgfbr1 with the G188V mutation (Tgfbr1G188V). Lysates of transfected cells were subjected to western blot analysis using an anti-TGFBR1 antibody. (C) TGF-β-induced promoter activity was assessed by the dual luciferase reporter assay system. Values represent the mean ± SD in triplicate assays. ∗P < 0.05, compared with the indicated TGF-β concentration.
To introduce the G188V mutation into mice, we generated a knock-in construct (Figure 3A). Tgfbr1G188V/+ mice were delivered spontaneously, able to breed, and were fertile with no apparent abnormalities at 6 weeks of age (Figure 3B). Mouse genotyping was performed using genomic DNA by PCR with a primer to identify the Tgfbr1 allele and mutant Tgfbr1 allele that contained the loxP sequence (Figure 3C). After generation of Tgfbr1G188V/+ mice, genotyping was performed using embryos on days 13.5 and 14.5, and after birth. WT, Tgfbr1G188V/+, and Tgfbr1G188V/G188V embryos were obtained at day 14.5 (Figure 3D). We observed no conspicuous morphological or patterning defects, although some Tgfbr1G188V/G188V embryos at day 14.5 showed fewer bloodstreams in craniofacial, trunk, and spinal cord regions. Among embryos on days 13.5 and 14.5, WT, Tgfbr1G188V/+, and Tgfbr1G188V/G188V mice had survived. However, with respect to the number of births, only one Tgfbr1G188V/G188V mouse was obtained. Moreover, the Tgfbr1G188V/G188V mutation caused embryonic lethality after day 14.5 (Table 2). Therefore, the Tgfbr1G188V/+ mouse was analyzed as an LDS model.
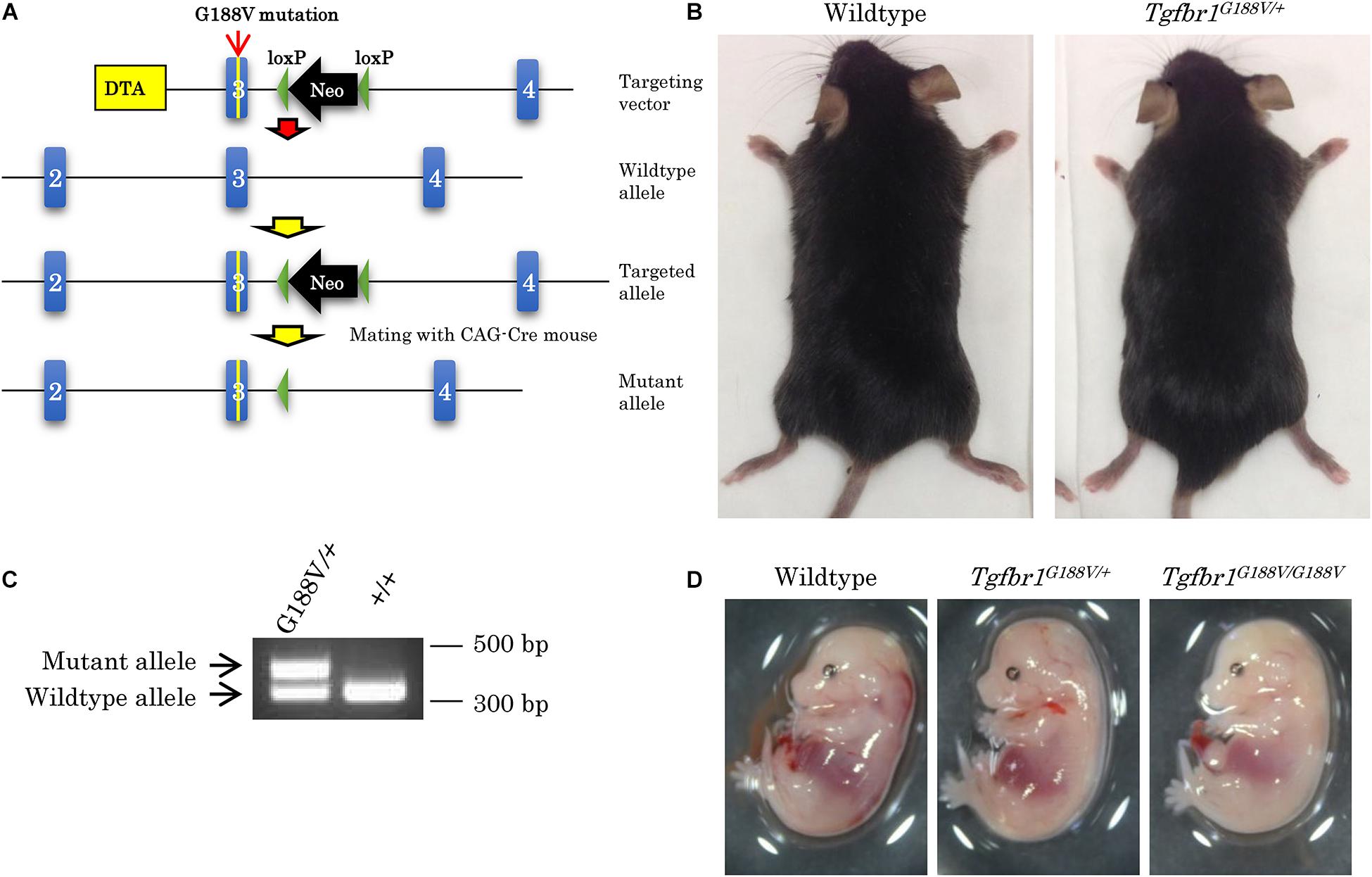
Figure 3. Generation of knock-in mice with the Tgfbr1 G188V mutation. (A) Structure of the Tgfbr1 G188V mutant allele. (B) No clear differences in 6-week-old Tgfbr1G188V/+ mice. (C) PCR genotyping of genomic DNA using tail specimens collected from Tgfbr1G188V/+ and WT mice. (D) Embryos of WT, Tgfbr1G188V/+, and Tgfbr1G188V/G188V at embryonic day 14.5.
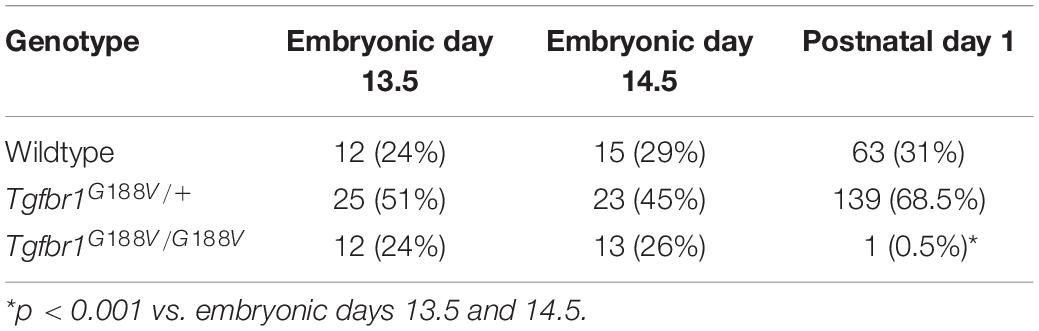
Table 2. Number of mice with each genotype.
The survival rate of Tgfbr1G188V/+ mice was observed over 180 days by a Kaplan–Meier survival curve. Mutant mice had died substantially earlier than WT mice (Figure 4A). To analyze the cause of premature death in Tgfbr1G188V/+ mice, tissue sections of the aorta were prepared, and elastic fibers were observed. Aortae were collected from 24-week-old male WT and Tgfbr1G188V/+ mice. Thinly sectioned axial specimens were prepared for EVG staining. In aortic samples from Tgfbr1G188V/+ mice, elastic fibers appeared to be distorted (Figure 4B, arrow) and fine rupture of elastic fibers was observed (Figure 4B, arrowhead). Next, TGF-β-related gene expression was analyzed in aortae from Tgfbr1G188V/+ mice. The thoracic aortae of 24-week-old male WT and Tgfbr1G188V/+ mice were collected. mRNA expression levels of Tgfb1, Tgfb2, and Tgfb3, TGF-β receptor genes Tgfbr1 and Tgfbr2, and TGF-β targets Col1a1 and Serpine1 were analyzed by real-time PCR. Although the differences were not statistically significant, the mRNA levels of Tgfb1, Tgfb3, Tgfbr1, Tgfbr2, Col1a1, and Serpine1 tended to be higher in Tgfbr1G188V/+ mice than in WT mice (Figure 4C). Using MEFs, we analyzed changes in TGF-β activity caused by the Tgfbr1 G188V mutation. MEFs of each genotype were cultured for 24 h in the absence of FBS and then stimulated with TGF-β (0–5 ng/mL). The expression levels of TGF-β-induced Serpine1 were evaluated after 12 h (Figure 4D). In WT MEFs, Serpine1 expression was increased in a TGF-β concentration-dependent manner. In Tgfbr1G188V/+ and Tgfbr1G188V/G188V MEFs, the increase in Serpine1 expression induced by TGF-β was reduced significantly. Next, we performed intracellular signal transduction analysis of TGF-β in MEFs. MEFs were cultured for 24 h in the absence of FBS and then stimulated with TGF-β (0–10 ng/mL). Cells were recovered after 30 min and Smad2 phosphorylation was examined by western blotting. Although phosphorylation of Smad2 in WT MEFs was increased in a TGF-β concentration-dependent manner, the levels of Smad2 phosphorylation were lower in Tgfbr1G188V/+ and Tgfbr1G188V/G188V MEFs than in WT MEFs (Figure 4E). These findings demonstrated that cellular TGF-β responses had decreased because of the Tgfbr1 G188V mutation.
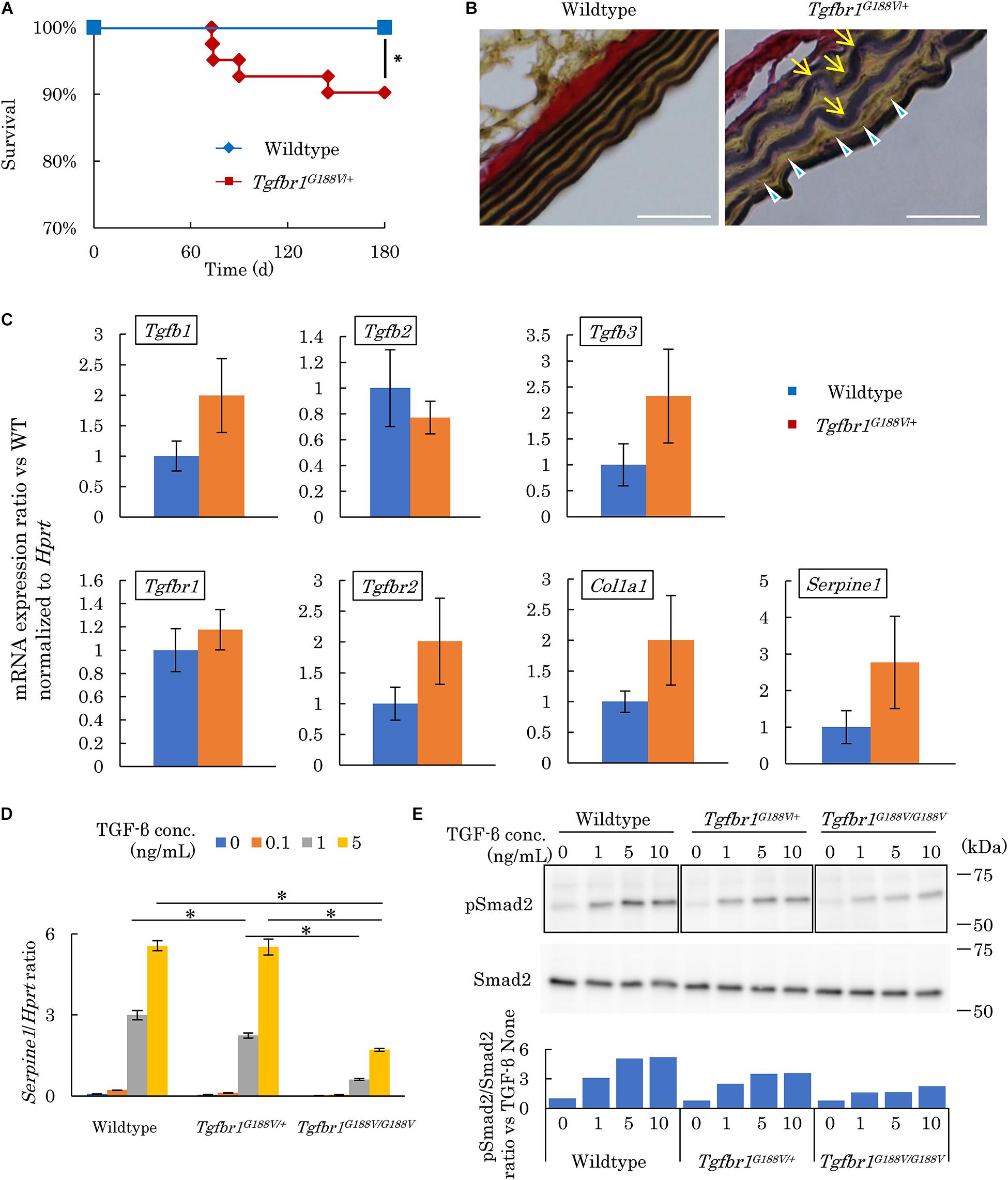
Figure 4. Tgfbr1G188V/+ mice recapitulate vascular LDS phenotypes. (A) Kaplan–Meier survival curve showing a diminishing lifespan of Tgfbr1G188V/+ mice. ∗P < 0.05. (B) Representative aortic wall sections of Tgfbr1G188V/+ mice at 24 weeks of age stained with EVG to identify elastin fibers. Scale bar: 25 μm. Arrows show elastic fiber distortion. Arrowheads show elastic fiber fragmentation. (C) Analysis of TGF-β-related genes in aortic tissues derived from wildtype and Tgfbr1G188V/+ mice (n = 4). Data represent means ± SD in triplicate assays. (D) Serpine1 gene expression in MEFs derived from wildtype, Tgfbr1G188V/+, and Tgfbr1G188V/G188V embryos at embryonic day 13.5 stimulated with TGF-β (0–5 ng/mL). Data represent means ± SD in triplicate assays. ∗P < 0.05, compared with the indicated TGF-β concentration. (E) Phospho-Smad2 (pSmad2) in MEFs derived from wildtype, Tgfbr1G188V/+, and Tgfbr1G188V/G188V embryos at embryonic day 13.5 stimulated with TGF-β (0–10 ng/mL). Quantitative western blot analysis is shown as the ratios of the intensities of phospho-Smad2 to Smad2.
Micro-CT contrast imaging was performed to evaluate maxillary alveolar bones collected from WT and Tgfbr1G188V/+ mice. Alveolar bone resorption was measured in the area between the alveolar apex from the cementoenamel boundary (Figure 5A, red line). The sums of values obtained for the first, second, and third molars were calculated. WT and Tgfbr1G188V/+ mice showed significant increases in bone resorption with aging. However, there was no significant difference in alveolar bone resorption between WT and Tgfbr1G188V/+ mice (Figure 5B). HE staining of periodontal tissues derived from WT and Tgfbr1G188V/+ mice at 6 and 24 weeks of age revealed no structural differences between WT and Tgfbr1G188V/+ mice (Figure 5C). Next, TGF-β-related gene expression was analyzed in periodontal tissues from Tgfbr1G188V/+ mice. Periodontal tissues were collected from 6-week-old male WT and Tgfbr1G188V/+ mice, and mRNA expression levels of TGF-β-related genes were analyzed by real-time PCR. Expression levels of Tgfb1, Tgfb2, Tgfb3, Col1a1, Serpine1, and Plap-1 were significantly higher in Tgfbr1G188V/+ mice than in WT mice (Figure 5D).
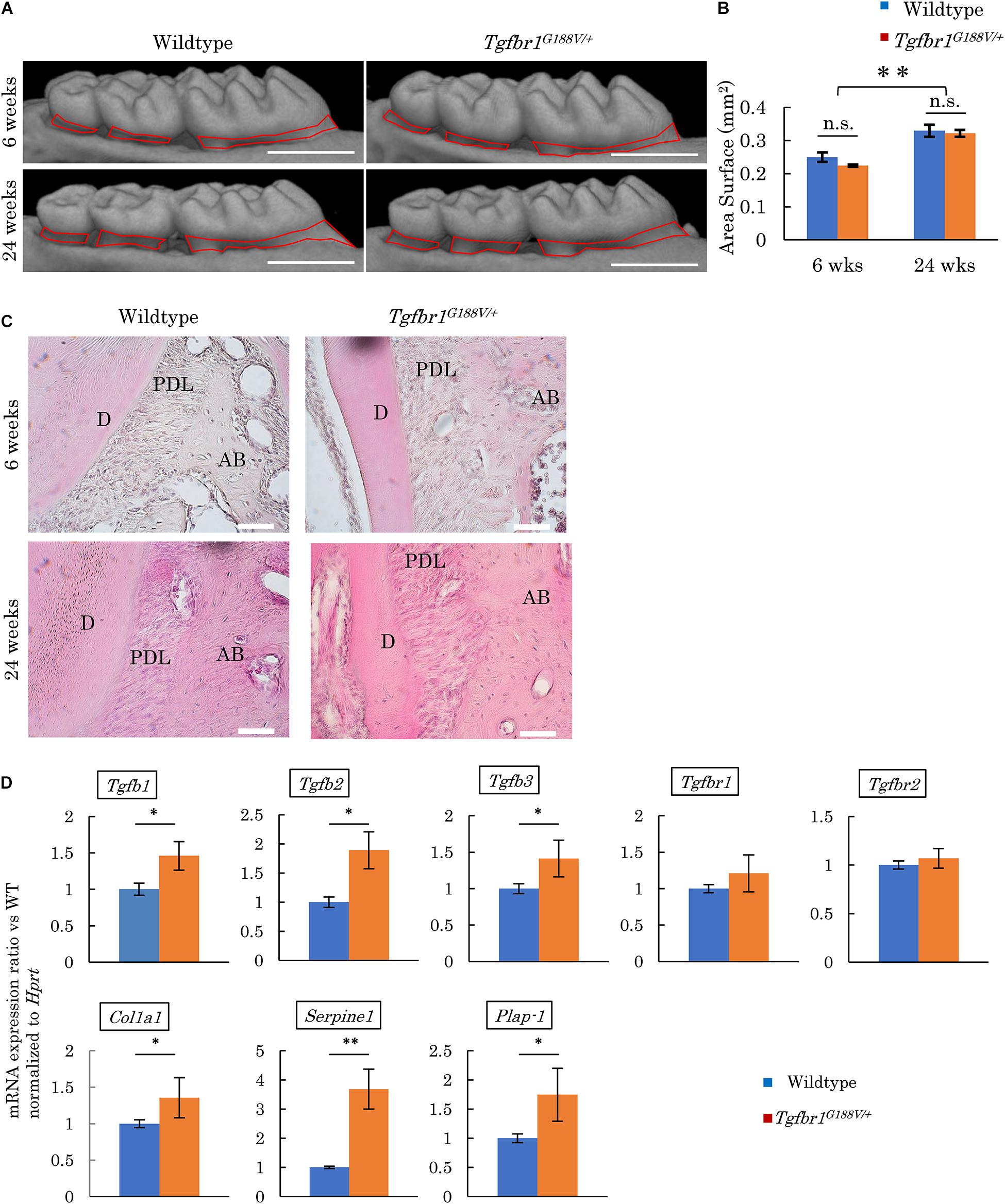
Figure 5. Analysis of phenotypes in periodontal tissues of Tgfbr1G188V/+ mice. (A) Representative images from micro-CT analysis of maxillary alveolar bones in Tgfbr1G188V/+ mice. Scale bar: 1 mm. (B) Alveolar bone resorption was measured in the root surface area between the alveolar apex from the cementoenamel junction (red line in Figure 4A). Data represent means ± SD. Six-week-old wildtype: n = 6, 6-week-old Tgfbr1G188V/+: n = 8, 24-week-old wildtype: n = 20, and 24-week-old Tgfbr1G188V/+: n = 14. (C) Representative images of HE staining of the periodontium from WT and Tgfbr1G188V/+ mice at 6 and 24 weeks of age. AB, alveolar bone; PDL, periodontal ligament; D, dentin. Scale bar: 50 μm. (D) Analysis of TGF-β-related gene expression in periodontal tissues derived from wildtype and Tgfbr1G188V/+ mice (n = 4). Data represent means ± SD in triplicate assays. ∗P < 0.05, ∗∗P < 0.01, compared with wildtype.
To analyze changes in periodontal tissue caused by bacterial invasion in WT and Tgfbr1G188V/+ mice, P. gingivalis was orally administered to generate an experimental periodontitis model. After P. gingivalis administration, the maxillary bones of the mice were recovered, micro-CT contrast imaging of periodontal tissues was performed (Figure 6A), and sections were prepared. Bone resorption after P. gingivalis administration was significantly greater in Tgfbr1G188V/+ mice than in control mice (Figure 6B). Periodontal tissue sections were prepared and subjected to HE staining. Multinucleated cells near the alveolar bone were observed in WT and Tgfbr1G188V/+ mice in the P. gingivalis treatment group (Figure 6C). To observe osteoclasts in close proximity to the alveolar bone, sections of each periodontal tissue were prepared, and TRAP staining was performed. In the P. gingivalis treatment group, osteoclasts were found near the alveolar bone (Figure 6D, arrowhead). For quantitative analysis, the numbers of osteoclasts in close proximity to the alveolar bone were counted between the first and second molars and between the second and third molars. These values were divided by the length of the alveolar bone surface for comparison (Figure 6D, red line). In Tgfbr1G188V/+ mice, the number of osteoclasts was increased significantly after P. gingivalis administration (Figure 6E).
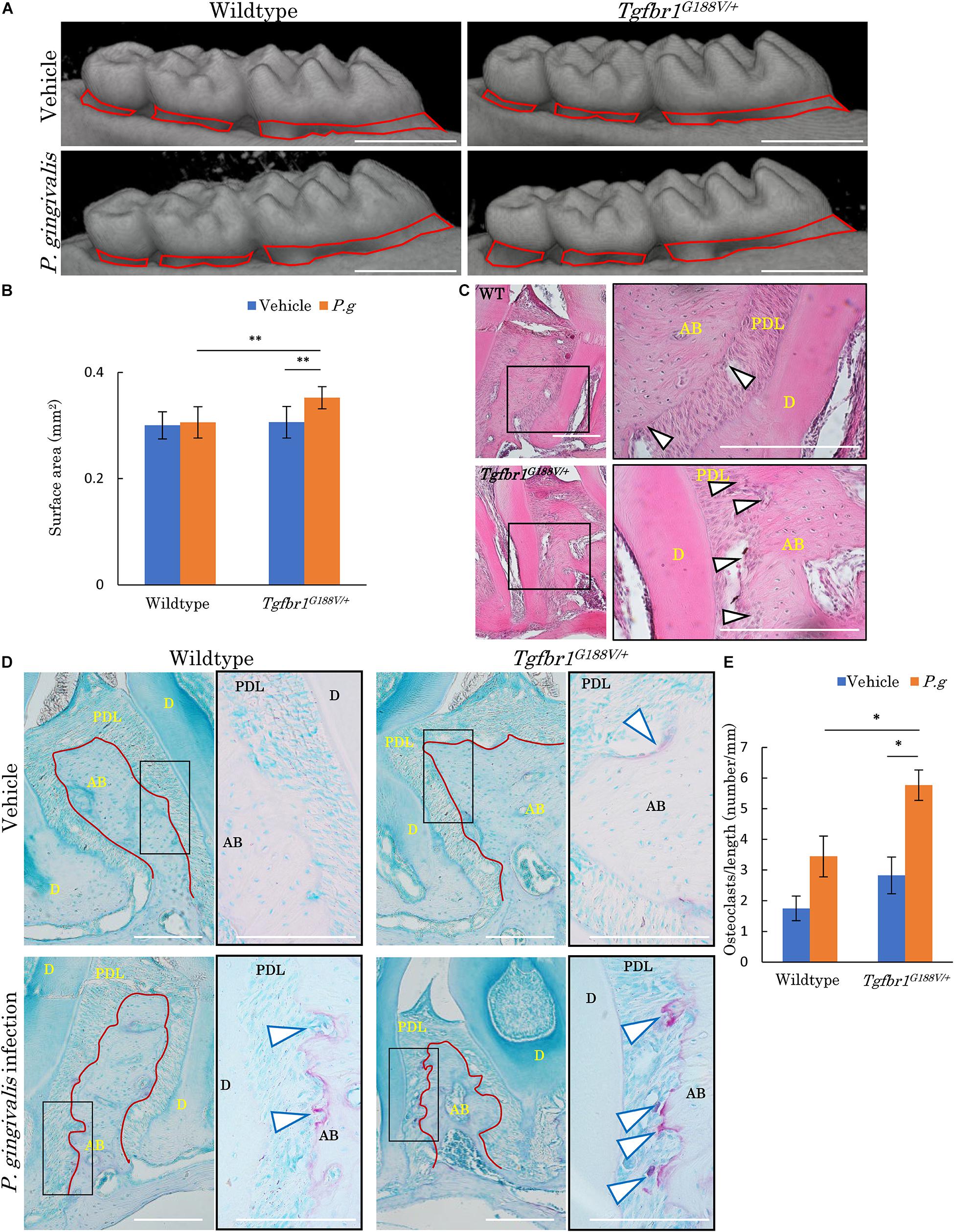
Figure 6. Oral infection with P. gingivalis induces alveolar bone resorption in Tgfbr1G188V/+ mice. (A) Representative images from micro-CT analysis of maxillary alveolar bones in P. gingivalis-infected WT and Tgfbr1G188V/+ mice. Scale bar: 1 mm. (B) Alveolar bone resorption was measured in the root surface area between the alveolar apex from the cementoenamel junction (red line in A). Data represent means ± SD in triplicate assays. ∗∗P < 0.01. Wildtype: n = 11 in vehicle, n = 6 in P. gingivalis infection. (C) Representative images of HE staining of the periodontium from P. gingivalis-infected wildtype and Tgfbr1G188V/+ mice. Right panel shows a high magnification images of the rectangular area indicated in the left panel. Scale bar: 200 μm. Arrowheads indicate multinucleated cells. AB, alveolar bone; PDL, periodontal ligament; D, dentin. (D) Representative images of TRAP-stained periodontium from P. gingivalis-infected wildtype and Tgfbr1G188V/+ mice. Right panel shows a high magnification image of the rectangular area indicated in the left panel. Scale bar: 200 μm. Arrowheads indicate TRAP-positive multinucleated cells. (E) Quantification of TRAP-positive cell numbers in each group. TRAP-positive cell numbers on the surface of alveolar bone were counted and divided by the length of the alveolar bone (red line in D). Data represent means ± SD in wildtype: n = 3 in vehicle, n = 5 in P. gingivalis infection. Tgfbr1G188V/+: n = 4 in vehicle, n = 6 in P. gingivalis infection. Vehicle: sham control, P.g, P. gingivalis infection, AB, alveolar bone; D, dentin; PDL, periodontal ligament. ∗P < 0.05, compared with Vehicle.
To assess the innate immunological response of Tgfbr1G188V/+ mice to P. gingivalis, we isolated peritoneal macrophages from WT and Tgfbr1G188V/+ mice, stimulated these cells with P. gingivalis LPS, and then analyzed inflammatory cytokine expression by quantitative PCR (Figure 7). We found that mRNA expression of Cxcl10, Il6 and Tnf was significantly upregulated in Tgfbr1G188V/+ macrophages compared with WT macrophages.
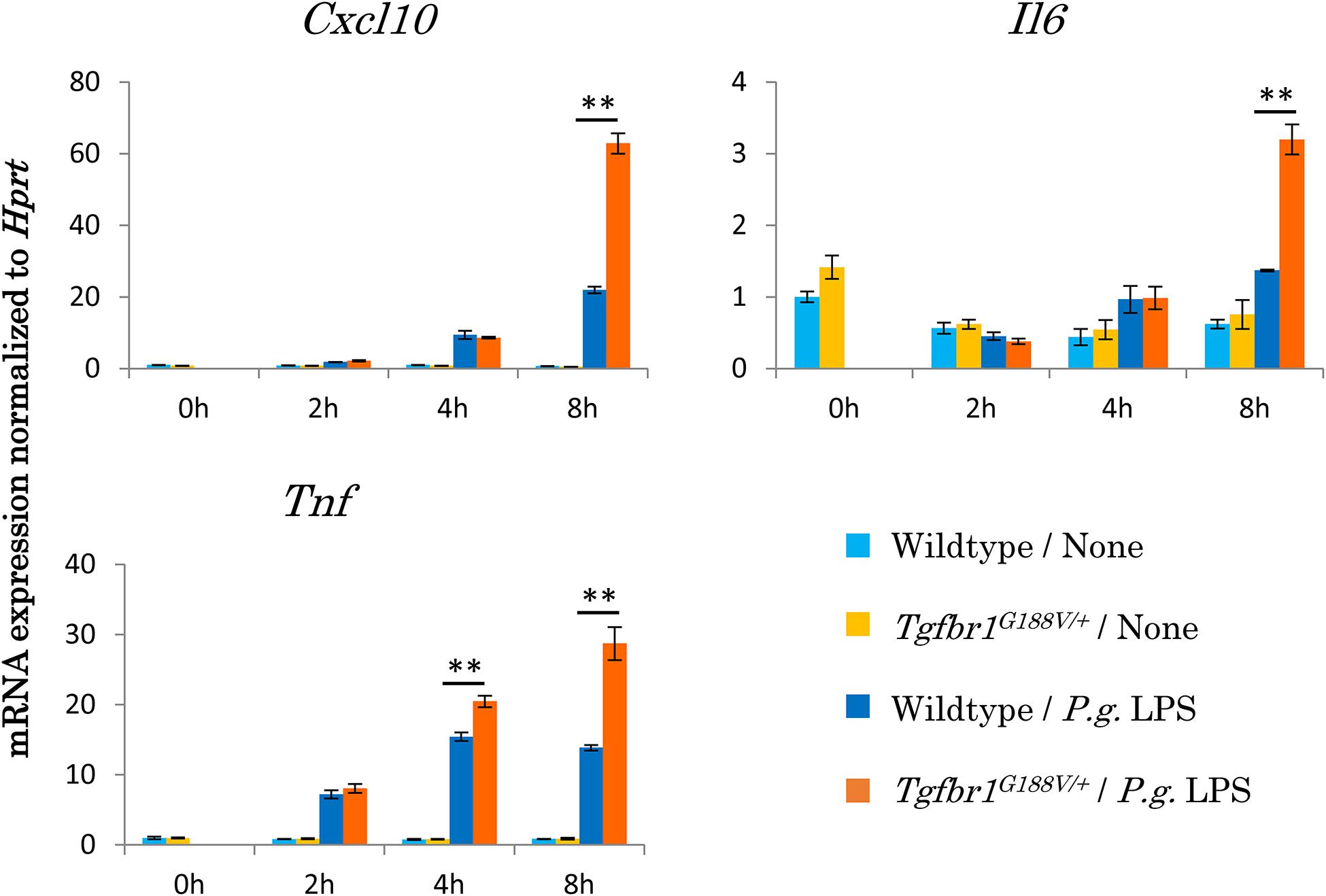
Figure 7. Upregulation of inflammatory cytokine mRNA expression in peritoneal macrophages from Tgfbr1G188V/+ mice. Macrophages were harvested from the peritoneal cavity of 8-week-old male WT and Tgfbr1G188V/+ mice at 3 days after injection of thioglycolate. The peritoneal macrophages were stimulated with 1 μg/ml P. gingivalis LPS for the indicated times and then total RNA was isolated for quantitative PCR analysis. Data represent means ± SD in triplicate assays. ∗∗P < 0.01, compared with wildtype.
In this study, we generated a knock-in mouse that reproduced the TGFBR1 gene mutation in an LDS patient who exhibited localized and remarkable vertical bone resorption, despite generally good plaque control. We then analyzed the phenotypes of this mouse model. Aggressive periodontitis is strongly affected by genetic factors and the morbidity rate in Japan is 0.05–1% (Takahashi et al., 2011). More than 40 genetic variants associated with LDS have been reported thus far (Loeys et al., 2006). However, the patient in this study had a previously unreported mutation, namely a novel G188V mutation in the GS domain of TGFBR1. TGF-β binds to type 2 receptors on cell membranes, thereby forming a complex with type 1 receptors. Type 2 receptors have serine–threonine kinase activity in the intracellular region and phosphorylate the GS domain of bound type 1 receptors. The GS domain of type 1 receptors is phosphorylated and kinase activation results in signal transmission to cells via R-Smad, an intracellular signal transduction molecule (Doyle et al., 2012). The genetic mutation identified in the LDS patient in this study attenuated Smad signaling. Structural changes in the GS domain of TGFBR1 may prevent phosphorylation of the GS domain and inhibit signal transduction. However, distinct point mutations in TGFBR1, which spontaneously activate TGF-β signaling, have been reported as the cause of multiple self-healing squamous epithelioma, also known as Ferguson–Smith disease (Goudie et al., 2011). The detailed changes in receptor locality and the three-dimensional structure are unclear and should be examined in future studies.
In the mouse model established in this study, the Tgfbr1 G188V/G188V mutation was lethal in embryos. Similarly, this mutation has not been identified in human homozygous patients with LDS. Therefore, Tgfbr1 is considered to be essential for survival. Some Tgfbr1G188V/G188V embryos collected at day 14.5 showed fewer bloodstreams in craniofacial, trunk, and spinal cord regions. It is important to investigate the effects of the Tgfbr1 G188V/G188V mutation on cardiovascular development during embryogenesis to reveal the mechanisms of embryonic lethality. In Tgfbr1G188V/+ mice, distortion and fine tearing of elastic fibers in aortic tissue were confirmed and a tendency toward premature death was observed. Structural abnormalities of the elastic fibers of the aorta trigger pathological diseases (e.g., aortic aneurysm, aortic dissection, hemorrhage, and fibrosis) and premature death. However, because the relationship between aortic structural abnormalities and aortic aneurysm was not examined, additional studies are necessary.
In this study, Tgfbr1G188V/+ MEFs showed attenuation of TGF-β signaling, but had elevated TGF-β-related gene expression in aortic tissues. In studies of aortic tissues, overexpression of TGF-β has been reported in patients with LDS and an LDS mouse model (Loeys et al., 2005, 2006; Gallo et al., 2014). TGF-β signaling involves canonical pathways via Smad and non-canonical pathways via ERK, JNK, and other molecules. Their expression patterns are regulated by a feedback mechanism that involves Smad (Lindsay and Dietz, 2011). In LDS, mutations in TGF-β receptors selectively attenuate the canonical pathway, thereby inhibiting the negative feedback mechanism that underlies TGF-β expression. Accordingly, increased levels of TGF-β in tissues contribute to excessive canonical pathway signaling. Non-canonical pathways are also presumed to cause aortic lesions by excessive activation (Lindsay and Dietz, 2011). In Tgfbr1G188V/+ mice, aortic elastic fiber abnormalities likely occurred through the same mechanism.
Periodontal tissues showed significantly higher expression levels of TGF-β-related genes in Tgfbr1G188V/+ mice than in WT mice, with no obvious differences in alveolar bone resorption or periodontal tissue status. TGF-β overexpression in tissues was presumed to compensate for the decrease in TGF-β signaling at the cellular level similar to the findings in aortic tissues. However, no apparent periodontal tissue-specific structural abnormalities were observed by HE staining. Therefore, abnormalities related to excessive TGF-β signaling were not triggered in periodontal tissues without pathological stimulation. In the future, detailed analyses at cellular and molecular levels are needed to evaluate structural abnormalities in periodontal tissues.
In the P. gingivalis-induced periodontitis model, significantly more osteoclasts were present in Tgfbr1G188V/+ mice than in WT mice. Moreover, alveolar bone resorption was significantly increased in Tgfbr1G188V/+ mice. P. gingivalis is commonly used to induce periodontitis (Baker et al., 1994; Suda et al., 2013; Arimatsu et al., 2014; Papathanasiou et al., 2016). In this experimental periodontitis model, the oral administration of P. gingivalis causes changes in the intestinal flora, which leads to systemic inflammation and subsequent bone resorption through immune cell responses in the oral cavity (Baker et al., 1994; Arimatsu et al., 2014). Notably, the oral administration of P. gingivalis may have triggered systemic inflammation in Tgfbr1G188V/+ mice. A previous report has demonstrated that TGF-β signaling in macrophages suppresses Toll-like receptor (TLR) signaling through myeloid differentiation factor 88 (Naiki et al., 2005). In Tgfbr1G188V/+ mouse macrophages, the mutation in Tgfbr1 may promote TLR signaling by attenuation of TGF-β signaling, thereby increasing the macrophage response to P. gingivalis LPS intrinsically. In the future, more details of the mechanisms that underlie osteoclastogenesis induced by P. gingivalis in Tgfbr1G188V/+ mice should be analyzed to understand the pathophysiology of periodontal disease caused by the Tgfbr1G188V/+ mutation.
Smad-mediated TGF-β signaling inhibits osteoblast differentiation (Borton et al., 2001). In periodontal tissues of Tgfbr1G188V/+ mice, excessive TGF-β signaling may suppress osteoblast differentiation. In LDS model mice with mutations in TGF-β II receptors, the femur is thinner and bone mass decreases, thereby increasing fragility (Dewan et al., 2015). In Tgfbr1G188V/+ mice, cell differentiation into osteoblasts and osteocytes may be suppressed, which suggests that the alveolar bone tends to be susceptible to resorption.
In conclusion we established an LDS mouse model that showed elevated susceptibility to P. gingivalis-induced periodontitis, probably through TGF-β signal dysfunction. This suggests that TGF-β signaling abnormalities accelerate the pathogenesis or progression of periodontitis.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of Osaka University Graduate School of Dentistry, and National Cerebral and Cardiovascular Center. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Osaka University Graduate School of Dentistry.
SY, KT, TM, and SM conceived, designed the experiments, and wrote the manuscript. SY, KT, MK, HS, and TK performed the experiments. SY, KT, CF, TM, HY, SS, and SM analyzed the data. All authors contributed to the article and approved the submitted version.
This study was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Nos. 15H02579, 17H04417, 19H01069, and 20H03862).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Ryan Chastain-Gross, Ph.D., and Mitchell Arico from Edanz Group (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Aoki-Nonaka, Y., Nakajima, T., Miyauchi, S., Miyazawa, H., Yamada, H., Domon, H., et al. (2013). Natural killer T cells mediate alveolar bone resorption and a systemic inflammatory response in response to oral infection of mice with Porphyromonas gingivalis. J. Periodontal. Res. 49, 69–76. doi: 10.1111/jre.12080
Arimatsu, K., Yamada, H., Miyazawa, H., Minagawa, T., Nakajima, M., Ryder, M. I., et al. (2014). Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 4:4828. doi: 10.1038/srep04828
Awata, T., Yamada, S., Tsushima, K., Sakashita, H., Yamaba, S., Kajikawa, T., et al. (2015). PLAP-1/Asporin positively regulates FGF-2 activity. J. Dent. Res. 94, 1417–1424. doi: 10.1177/0022034515598507
Baker, P. J., Evans, R. T., and Roopenian, D. C. (1994). Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch. Oral Biol. 39, 1035–1040. doi: 10.1016/0003-9969(94)90055-8
Borton, A. J., Frederick, J. P., Datto, M. B., Wang, X. F., and Weinstein, R. S. (2001). The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 16, 1754–1764. doi: 10.1359/jbmr.2001.16.10.1754
De Coster, P. J., Martens, L. C., and De Paepe, A. (2002). Oral manifestations of patients with Marfan syndrome: a case-control study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 93, 564–572. doi: 10.1067/moe.2002.121430
Dewan, A. K., Tomlinson, R. E., Mitchell, S., Goh, B. C., Yung, R. M., Kumar, S., et al. (2015). Dysregulated TGF-beta signaling alters bone microstructure in a mouse model of Loeys-Dietz syndrome. J. Orthop. Res. 33, 1447–1454. doi: 10.1002/jor.22920
Dietz, H. C., Cutting, G. R., Pyeritz, R. E., Maslen, C. L., Sakai, L. Y., Corson, G. M., et al. (1991). Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–346. doi: 10.1038/352337a0
Dietz, H. (2007). Marfan syndrome: from molecules to medicines. Am. J. Hum. Genet. 81, 662–667. doi: 10.1086/521409
Doyle, A. J., Doyle, J. J., Bessling, S. L., Maragh, S., Lindsay, M. E., Schepers, D., et al. (2012). Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254. doi: 10.1038/ng.2421
Gallo, E. M., Loch, D. C., Habashi, J. P., Calderon, J. F., Chen, Y., Bedja, D., et al. (2014). Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Invest. 124, 448–460. doi: 10.1172/JCI69666
Godfrey, M., Menashe, V., Weleber, R. G., Koler, R. D., Bigley, R. H., Lovrien, E., et al. (1990). Cosegregation of elastin-associated microfibrillar abnormalities with the Marfan phenotype in families. Am. J. Hum. Genet. 46, 652–660.
Goudie, D. R., D’Alessandro, M., Merriman, B., Lee, H., Szeverényi, I., Avery, S., et al. (2011). Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat. Genet. 43, 365–371. doi: 10.1038/ng.780
Howe, J. R., Roth, S., Ringold, J. C., Summers, R. W., Jarvinen, H. J., Sistonen, P., et al. (1998). Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 280, 1086–1088. doi: 10.1126/science.280.5366.1086
Jain, E., and Pandey, R. K. (2013). Marfan syndrome. BMJ Case Rep. 2013:bcr2013201632. doi: 10.1136/bcr-2013-201632
Lindsay, M. E., and Dietz, H. C. (2011). Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473, 308–316. doi: 10.1038/nature10145
Loeys, B. L., Chen, J., Neptune, E. R., Judge, D. P., Podowski, M., Holm, T., et al. (2005). A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281. doi: 10.1038/ng1511
Loeys, B. L., Schwarze, U., Holm, T., Callewaert, B. L., Thomas, G. H., Pannu, H., et al. (2006). Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 355, 788–798. doi: 10.1056/NEJMoa055695
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., Backer, J. D., Devereux, R. B., et al. (2010). The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 47, 476–485. doi: 10.1136/jmg.2009.072785
Matt, P., Schoenhoff, F., Habashi, J., Holm, T., Van Erp, C., Loch, D., et al. (2009). Circulating transforming growth factor-beta in Marfan syndrome. Circulation 120, 526–532. doi: 10.1161/CIRCULATIONAHA.108.841981
Meng, H., Xu, L., Li, Q., Han, J., and Zhao, Y. (2007). Determinants of host susceptibility in aggressive periodontitis. Periodontol. 2000 43, 133–159. doi: 10.1111/j.1600-0757.2006.00204.x
Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T., et al. (2004). Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 36, 855–860. doi: 10.1038/ng1392
Naiki, Y., Michelsen, K. S., Zhang, W., Chen, S., Doherty, T. M., and Arditi, M. (2005). Transforming growth factor-beta differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J. Biol. Chem. 280, 5491–5495. doi: 10.1074/jbc.C400503200
Neptune, E. R., Frischmeyer, P. A., Arking, D. E., Myers, L., Bunton, T. E., Gayraud, B., et al. (2003). Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 33, 407–411. doi: 10.1038/ng1116
Papapanou, P. N., Sanz, M., Buduneli, N., Dietrich, T., Feres, M., Fine, D. H., et al. (2018). Periodontitis: consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Clin. Periodontol. 45, S162–S170. doi: 10.1111/jcpe.12946
Papathanasiou, E., Kantarci, A., Konstantinidis, A., Gao, H., and Van Dyke, T. E. (2016). SOCS-3 Regulates Alveolar Bone Loss in Experimental Periodontitis. J. Dent. Res. 95, 1018–1025. doi: 10.1177/0022034516645332
Putnam, E. A., Zhang, H., Ramirez, F., and Milewicz, D. M. (1995). Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 11, 456–464. doi: 10.1038/ng1295-456
Sakai, K., and Miyazaki, J. (1997). A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun. 237, 318–324. doi: 10.1006/bbrc.1997.7111
Schorr, S., Braun, K., and Wildman, J. (1951). Congenital aneurysmal dilatation of the ascending aorta associated with arachnodactyly. Am. Heart J. 42, 610–616. doi: 10.1016/0002-8703(51)90157-3
Straub, A. M., Grahame, R., Scully, C., and Tonetti, M. S. (2002). Severe periodontitis in Marfan’s syndrome: a case report. J. Periodontol. 73, 823–826. doi: 10.1902/jop.2002.73.7.823
Staufenbiel, I., Hauschild, C., Kahl-Nieke, B., Vahle-Hinz, E., von Kodolitsch, Y., Berner, M., et al. (2013). Periodontal conditions in patients with Marfan syndrome - a multicenter case control study. BMC Oral Health 13:59. doi: 10.1186/1472-6831-13-59
Suda, N., Moriyama, K., and Ganburged, G. (2013). Effect of angiotensin II receptor blocker on experimental periodontitis in a mouse model of Marfan syndrome. Infect. Immun. 81, 182–188. doi: 10.1128/IAI.00886-12
Suzuki, J., Imai, Y., Aoki, M., Fujita, D., Aoyama, N., Tada, Y., et al. (2015). High incidence and severity of periodontitis in patients with Marfan syndrome in Japan. Heart Vessels 30, 692–695. doi: 10.1007/s00380-013-0434-y
Takahashi, M., Chen, Z., Watanabe, K., Kobayashi, H., Nakajima, T., Kimura, A., et al. (2011). Toll-like receptor 2 gene polymorphisms associated with aggressive periodontitis in Japanese. Open Dent. J. 5, 190–194. doi: 10.2174/1874210601105010190
Keywords: TGF-beta, periodontal ligament, extracelluar matix, periodontitis, knock-in mice
Citation: Yamada S, Tsushima K, Kinoshita M, Sakashita H, Kajikawa T, Fujihara C, Yuan H, Suzuki S, Morisaki T and Murakami S (2021) Mouse Model of Loeys–Dietz Syndrome Shows Elevated Susceptibility to Periodontitis via Alterations in Transforming Growth Factor-Beta Signaling. Front. Physiol. 12:715687. doi: 10.3389/fphys.2021.715687
Received: 27 May 2021; Accepted: 23 July 2021;
Published: 11 August 2021.
Edited by:
Frédéric Lézot, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Olivier Huck, Université de Strasbourg, FranceCopyright © 2021 Yamada, Tsushima, Kinoshita, Sakashita, Kajikawa, Fujihara, Yuan, Suzuki, Morisaki and Murakami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoru Yamada, c2F0b3J1eUB0b2hva3UuYWMuanA=
†Present address: Tetsuhiro Kajikawa, Laboratory of Innate Immunity and Inflammation, Department of Basic and Translational Sciences, Penn Dental Medicine, University of Pennsylvania, Philadelphia, PA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.