
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 30 August 2021
Sec. Metabolic Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.710420
This article is part of the Research Topic Novel Insights into Obesity-Related Diseases View all 14 articles
 Qianrang Lu1,2Xinyao Tian1,2Hao Wu1,2Jiacheng Huang1,2Mengxia Li1,2
Qianrang Lu1,2Xinyao Tian1,2Hao Wu1,2Jiacheng Huang1,2Mengxia Li1,2 Zhibin Mei1,2Lin Zhou1,2
Zhibin Mei1,2Lin Zhou1,2 Haiyang Xie1,2
Haiyang Xie1,2 Shusen Zheng1,2,3*
Shusen Zheng1,2,3*Nonalcoholic fatty liver disease (NAFLD) is often accompanied by systemic metabolic disorders such as hyperglycemia, insulin resistance, and obesity. The relationship between NAFLD and systemic metabolic disorders has been well reviewed before, however, the metabolic changes that occur in hepatocyte itself have not been discussed. In NAFLD, many metabolic pathways have undergone significant changes in hepatocyte, such as enhanced glycolysis, gluconeogenesis, lactate production, tricarboxylic acid (TCA) cycle, and decreased ketone body production, mitochondrial respiration, and adenosine triphosphate (ATP) synthesis, which play a role in compensating or exacerbating disease progression, and there is close and complex interaction existed between these metabolic pathways. Among them, some metabolic pathways can be the potential therapeutic targets for NAFLD. A detailed summary of the metabolic characteristics of hepatocytes in the context of NAFLD helps us better understand the pathogenesis and outcomes of the disease.
The liver is the hub of the metabolism of glucose, fat, protein, vitamin, hormone, etc., and play a major part in metabolic homeostasis. All kinds of metabolic processes such as glycolysis, gluconeogenesis, lipogenesis, glycogen production can be conducted in liver. Therefore, nonalcoholic fatty liver disease (NAFLD) is often accompanied by systemic metabolic disorders (Byrne and Targher, 2015; Tilg et al., 2017; Khan et al., 2019). In view of the importance and complexity of liver metabolism, it is meaningful to study the metabolic characteristics of the liver under healthy as well as pathological conditions.
Nonalcoholic fatty liver disease, a continuum of liver abnormalities including nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH), and can progress to liver cirrhosis and hepatocellular carcinoma (Friedman et al., 2018). NAFL, also named hepatic steatosis, is the benign stage of NAFLD which is characterized by excessive lipid stored within the cytosol of hepatocytes. Patients in the state of NAFL take a lower risk of deleterious outcomes, while NASH can progress to cirrhosis, liver failure and even hepatic carcinoma (Singh et al., 2015). The presence of NASH means the existence of liver inflammation and injury, accompanied with elevated aminotransferases. Biopsy is the main method of diagnosis which is characterized by various degrees of fat accumulation, hepatocellular ballooning, lobular inflammation and fibrosis (Brunt et al., 2020).
Nowadays NAFLD accounts for the major part of liver disease worldwide. With the decreased incidence of hepatitis and increased obesity (Fan et al., 2017; Wu et al., 2020), the primary cause of end-stage liver disease will be replaced by NAFLD in the next few decades (Younossi et al., 2018). Currently, the worldwide prevalence of NAFLD is nearly 24% (2015) (Younossi et al., 2016). Therefore there is an increasing interest in studying NAFLD. To understand the pathogenesis of NAFLD, a useful conceptual framework is that the excess energy metabolic substrates, fatty acids (FAs), and carbohydrates, exceed the liver’s processing capacity, resulting in the exacerbated formation of reactive oxygen species (ROS), by-products and lipid peroxidation, which will induce hepatocellular injury, inflammation and death (Friedman et al., 2018). In this review, we will discuss the hepatic metabolism changes and their effect in NAFLD.
Glycolysis refers to the process that one molecule of glucose is metabolized through a ten-step process in the cytoplasm to finally generate two molecules of pyruvate, providing with two molecules of NADH and two adenosine triphosphate (ATP). The glycolysis rate is mainly controlled by several rate-limiting enzymes, hexokinase (HK), phosphofructokinase (PFKM), and pyruvate kinase (PK), which are controlled by allosteric and hormonal regulation. In liver, the major isoform of hexokinase is hexokinase IV, glucokinase, which is primarily regulated by insulin but not inhibited by its product. Therefore, it is of great importance in the modulation of postprandial blood glucose. Insulin is the main hormone that regulates blood glucose. It bound to the insulin receptor (INSR) of target cells on the plasma membrane, then act through IRS(insulin receptor substrate)/PI3K(phosphoinositide 3-kinase)/AKT(protein kinase B) axis to regulate glucose metabolism (Haeusler et al., 2018; Petersen and Shulman, 2018). Phosphatase and tensin homolog (PTEN) catalyzed the reverse reaction of P13K and antagonize insulin function, but its activity is inhibited by insulin (Hodakoski et al., 2014).
Changes in glycolysis are found in many diseases. The WARBURG effect, known widely in tumor cells, is characterized by enhanced aerobic glycolysis and lactate production (Vander Heiden et al., 2009; Koppenol et al., 2011). It used to be misread that the damaged respiration is accounted for increased glucose fermentation in cancers. Actually, the tumor cells exhibit the WARBURG effect and meanwhile retain normal or even enhanced mitochondrial respiration (Koppenol et al., 2011; Lu et al., 2015). Recently, the similar metabolic change has also been found in many benign diseases, such as pulmonary hypertension, idiopathic pulmonary fibrosis (IPF), tuberculosis, failing heart (Chen et al., 2018), liver cirrhosis (Trivedi et al., 2021), etc., which indicate that WARBURG effect may not be unique to malignant tumor.
In the hepatocytes of fatty liver, similar metabolic changes also exist. Compared with regular chow (RC)-fed mice, the mRNA levels of key glycolysis related enzymes (HK2, PFKm, and PKm) were increased in the liver of high-fat diet (HFD)-fed mice (Liu et al., 2018). In the PTEN-null mice, HK2, PKM2, and other glycolytic enzymes are overexpressed via P13K/AKT2/PPARγ axis, which partially account for the susceptibility to fatty liver and hepatocellular carcinoma of PTEN-null mice (Horie et al., 2004). Moreover adenoviral-mediated overexpression of HK2 and PKM2 promotes liver growth and liver steatosis (Panasyuk et al., 2012), which may bridge the enhanced glycolysis with carcinogenesis in fatty liver. Using PET/CT to monitor the phosphorylation rate of liver glucose, which represents the activity of hexokinase, proved that the rate of phosphorylation of liver hexose is higher in patients with steatohepatitis (Keramida et al., 2016). Seahorse analysis also showed that the rate of extracellular acidification in HFD-fed mice increased suggesting enhanced glycolysis (Liu et al., 2018). Since NAFLD always accomplished by insulin resistance, hyperinsulinemia may be one of the reasons for enhanced glycolysis.
Recent studies have found that knocking down NOD-like receptor (NLR)X1 NLRX1 can inhibit glycolysis and enhance fat oxidation to reduce liver steatosis (Kors et al., 2018). Liver specific knockdown of SIRT6 will lead to the deacetylation of histone H3 lysine 9 (H3K9) in the promoter of glycolytic genes and elevated the expression of these genes, which consequently cause fatty liver dependent on enhancing glycolysis (Kim et al., 2010). Knockdown of Geranylgeranyl diphosphate synthase (GGPPS) can induce strengthened glycolysis through the LKB1–AMPK pathway, and lead to an increase in the inflammatory factors secreted in mouse primary hepatocytes. Furthermore, using 2-dexoy-D-glucose to inhibit glycolysis dramatically alleviates liver inflammation, injury and fibrosis due to the deletion of GGPPS in HFD mice (Liu et al., 2018). However, the pathway that glycolysis contributes to liver injury is unclear and deserves further study.
The product of glycolysis, pyruvate, will be dehydrogenated by lactate dehydrogenase to lactate or decarboxylated to acetyl-coenzyme A by pyruvate dehydrogenase complex (PDC) in mitochondria which thereby connects the glycolysis with the tricarboxylic acid (TCA) cycle (Zhou et al., 2001). The activity of the complex is regulated by pyruvate dehydrogenase phosphatase (PDP) and pyruvate dehydrogenase kinase (PDK1-4) through reversible dephosphorylation and phosphorylation. Phosphorylation is the inactive state of PDC (Bowker-Kinley et al., 1998).
The activity of PDC in hepatocytes of fatty liver is still controversial. Bajotto et al. (2006) and Go et al. (2016) found that the activity of pyruvate dehydrogenase complex in the liver of OLETF mice and HFD C57BL/6J mice was significantly lower than that of control group, which will consequently reduce the TCA cycle flux of pyruvate. Moreover, the activity, abundance and mRNA level of PDK2 and PDK4 are consistently increased, and positively related to plasma free fatty acid (FFA) concentrations (Bajotto et al., 2006; Go et al., 2016). Consistently, in the NAFLD patients and obese mice, the serum pyruvate concentration and liver pyruvate content are significantly higher than those of the control group (Fletcher et al., 2019; Wang et al., 2020; Shannon et al., 2021), though there is no evidence that it is caused by inactivity of PDC. A current study by Shannon et al. (2021) revealed a contradictory result that in obese mice, the activity of PDC is enhanced accompanied by the reduced activity of PDK4 and enhanced activity of PDP2. The insulin sensitizer Metformin can normalize the activity of PDC and mitigated the flow of TCA derived from pyruvate (Shannon et al., 2021). However, given that the acetyl-CoA produced by pyruvate only accounts for 5% of the liver TCA cycle (Cappel et al., 2019), the effect of pyruvate oxidative decarboxylation by PDC may not directly participate the pathological process of fatty liver.
The alternative metabolic pathway of pyruvate is lactate production. Under physiological conditions, lactate is mainly used by the heart, skeletal muscle and brain as an energy source (Gladden, 2004). The hepatocyte can convert excessive plasma lactate into glucose then bring back to plasma (Brooks, 2018). Lactate dehydrogenase(LDH) is composed of two subunits, H and M, with five isoenzymes: LDH1, LDH2, LHD3, LDH4, and LDH5 (Adeva-Andany et al., 2014). LDH-M, also known as LDHA, is more inclined to convert pyruvate into lactate; while LDH-H, also known as LDHB, is more inclined to convert lactate to pyruvate to dispose lactate (Dawson et al., 1964).
The production of lactate in fatty liver is significantly higher than normal (Liu et al., 2018; Wang et al., 2020), which is another feature of WARBURG effect (de la Cruz-López et al., 2019). P300/CBP-associated factor (PCAF)-mediated high acetylation level of LDH-B is one of the main causes in the accumulation of lactate in NAFLD. The activity of acetylated LDH-B is reduced, which hinders the ability of hepatocyte to dispose of lactate and results in the lactate accumulation (Wang et al., 2020). The increased lactate can not only worsen the hepatic steatosis, but also increase the acetylation of histone H3K9 by reducing the activity of nuclear histone deacetylase (HDAC), and therefore increase the expression of genes involved in lipogenesis and FA uptake (Wang et al., 2020).
Gluconeogenesis refers to the conversion of non-glucose compounds (glycerol, glycogen-producing amino acids, lactic acid, etc.) into glucose (Hers and Hue, 1983). Hepatic gluconeogenesis occurs during prolonged fasting episodes after depletion of hepatic glycogen, and is primarily in response to the insulin and glucagon. After an overnight fasting, about the half of the total hepatic glucose production is attributed to gluconeogenesis (Rothman et al., 1991). In order to achieve gluconeogenesis, the key is to bypass the three irreversible reactions in glycolysis that the conversion of glucose to glucose-6-phosphate by HK, fructose-6-phosphate to fructose-1,6-diphosphate by PFK and phosphoenolpyruvate to pyruvate via PK (Petersen et al., 2017). Conversion of pyruvate to phosphoenolpyruvate is the first reverse reaction in gluconeogenesis catalyzed by pyruvate carboxylase (PC) and phosphoenolpyruvate carboxykinase (PEPCK). The production of pyruvate carboxylation catalyzed by PC, oxaloacetate, is the intermediates of TCA cycle, and such flux is also named anaplerosis. Then oxaloacetate is converted to phosphoenolpyruvate, which is called cataplerosis (Owen et al., 2002). Thus, a close link exists between gluconeogenesis and TCA cycle. The enzymes that catalyze the other two reverse reactions are fructose diphosphatase-1 and glucose-6-phosphatase.
In the individuals with high intrahepatic triglycerides (IHTG), the endogenous glucose is higher than that in normal people, among which derived from gluconeogenesis is increased by 25%, consistent with the flow of anaplerosis increased by 50% (Sunny et al., 2011). The same trend also appears on mice supplied with excessive lipid, either by HFD or by exogenously perfused. Moreover, the increasement of anaplerosis/cataplerosis is linked to enhanced oxidative metabolism and subsequently oxidative stress and inflammation (Satapati et al., 2016). Inhibition of anaplerotic/cataplerotic flux can attenuates gluconeogenesis and TCA cycle and consequently alleviates oxidative stress and hepatic steatosis (Go et al., 2016; Satapati et al., 2016; Yuan et al., 2016). The presumable mechanism could be that, first, the raise of allosteric activation of PK, acetyl-COA, due to enhanced FA oxidation may partially contribute to the enhanced gluconeogenesis. Second, as mentioned before, the increased content of pyruvate in the liver increases the substrate for gluconeogenesis. Third the increased TCA cycle flux that we will discuss later facilitates gluconeogenesis from TCA cycle cataplerosis.
Hepatic de novo lipogenesis (DNL) includes FAs synthesized from acetyl-CoA and then esterified with 3-phosphoglycerol to generate triglycerides (TG). Acetyl-CoA carboxylase (ACC) is the rate-limiting enzyme of FA synthesis, which carboxylate the acetyl-CoA to produce malonyl-CoA. Acetyl-CoA and citric acid are main allosteric activators of ACC. The expression and activity of the enzymes involved in DNL also modulated by various transcription factors like sterol regulatory element-binding protein 1C (SREBP1C) and carbohydrate-responsive element-binding protein (ChREBP; Shimomura et al., 1999; Dentin et al., 2005; Wang et al., 2015). Under physiological conditions, the TG synthesized in liver will not be stored in it, and will be exported in the form of very low density lipoproteins (VLDL; Kawano and Cohen, 2013).
The major feature of NAFLD is the lipid accumulation in hepatocytes. Under fasting conditions, the proportion of TG stored in liver of NAFLD patients are 26.1 ± 6.7% from DNL, 59.0 ± 9.9% from plasma free FAs and 14.9 ± 7.0% from dietary FAs, which is similar to the composition of VLDL-TG (Donnelly et al., 2005), and secretion rate of VLDL-TG is linearly related to the content of IHTG when IHTG content was ≤10% but reached a plateau in subjects with high IHTG (Fabbrini et al., 2008). Since NAFLD is often accompanied by insulin resistance (Watt et al., 2019), the lipolysis of peripheral adipose tissue increased, leading to an elevated in plasma FFA (Fabbrini et al., 2008; Samuel and Shulman, 2016). However, when compared to the individuals with low IHTG, no significant difference has been found in VLDL-TG derived from adipose tissue or diet in patients with high IHTG. Only the VLDL-TG derived from DNL was significantly increased to the three times of low IHTG group. The percentage of newly-synthesized palmitate in VLDL-TG was two times higher in high IHTG group, indicating that it was the increased DNL that contributed to the accumulation of liver fat (Lambert et al., 2014). Fabbrini et al. (2008) also proved that the increase in VLDL-TG secretion was primarily attributed to the FAs derived from DNL and lipolysis of abdominal and hepatic lipid, other than lipolysis of subcutaneous fat.
Moreover, the expression of lipogenic regulatory factors and enzymes, such as SREBP1c, fatty acid synthase (FAS), liver X receptor α (LXRα), ChREBP, and ACC1, is higher in fatty liver (Kohjima et al., 2007; Higuchi et al., 2008). LXRs are classified as the nuclear hormone receptor superfamily, which can be activated by oxysterols due to elevated intracellular cholesterol concentration. LXR is widely involved in the regulation of many metabolic processes including cholesterol, glucose and lipid metabolism (Hiebl et al., 2018). LXRα can promote lipogenic gene transcription by activating SREBP1c (Repa et al., 2000). The expression level of LXR in NAFLD patients was four times that of the control group, and it was significantly related with the expression of SREBP-1c (Higuchi et al., 2008). Moreover The expression of SREBP1c can also be induced by tumor necrosis factor-alpha (TNF-α) (Endo et al., 2007) and the mRNA level of TNF-α is related to the severity of NASH (Crespo et al., 2001). ChREBP is also found to overexpress in hepatic steatosis patient and indicate the severity of hepatic steatosis but is negatively correlated with insulin resistance in NASH patients. Overexpression of ChREBP induce the hepatic steatosis but increase the insulin sensitivity (Benhamed et al., 2012). In addition, due to the systemic insulin resistance, the high levels of insulin will contribute to DNL (Mansouri et al., 2018).
Fatty acids oxidation (FAO) is conducted in mitochondria, endoplasmic reticulum (ER) and peroxisomes which can produce abundant acetyl-CoA. The first step is activation of FAs to fatty acyl-CoA by acyl-CoA synthase in the outer mitochondrial membrane or ER. Mitochondria are the main site of FAs β-oxidation. The long-chain FAs rely on the assistance of carnitine palmitoyltransferase-1 (CPT1) to shuttled across the membrane while medium - and short-chain FAs do not. CPT1 is the key enzyme for β-oxidation, and malonyl-CoA, the intermediates of DNL, is a potent allosteric inhibitor of CPT (Bonnefont et al., 2004). Some transcription factors such as peroxisome proliferator-activated receptor α (PPARα) and its coactivator peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC1α), can enhance the expression of CPT1 and other genes related to FAO (Bougarne et al., 2018). In the context of diabetes or FA overload, a part of FAs will be disposed through ω-oxidation in the endoplasmic reticulum which deteriorate ROS production and lipid peroxidation (Reddy, 2001; Wanders et al., 2011).
Theoretically increased DNL should inhibit the FA oxidation for the intermediate malonyl-CoA. However, in the patient with NASH or obesity, mitochondrial β-oxidation is still enhanced to coordinate with the excess FFA load, which has been proved by 13C-octanoate breath test and positron emission tomography and is correlated with systemic insulin resistance (Miele et al., 2003; Iozzo et al., 2010). In 12-week and 32-week HFD mice, the respiratory exchange ratio (ERE) value of the mice decreased and the oxygen consumption rate increased, suggesting increased FAO (Liu et al., 2018; Pittala et al., 2019). As the upregulation of fatty acid translocase (CD36/FAT), the uptake of FFA from plasma to hepatocytes also increased (Bechmann et al., 2010). CD36/FAT is one of the FA transport proteins which facilitates the uptake and utilization of FFAs in hepatocytes (Silverstein and Febbraio, 2009). Its expression is associated with serum FFAs and apoptosis mediators (Bechmann et al., 2010). The increased FFAs in hepatocytes could enhance the FAO consequently (Mansouri et al., 2018).
In fatty liver mouse model and NAFLD patients, the expression of genes involved in hepatic mitochondrial beta-oxidation such as PPARα, PGC1α and CPT1a is elevated as expected (Kohjima et al., 2007; Liu et al., 2018; Pittala et al., 2019). However, some research has shown that compared high-trans-fat high-fructose diet (TFD)-8 weeks-induced simple steatosis mice with NASH mice by 24 weeks, the expression of genes related beta-oxidation was decreased in liver with onset of NASH (Patterson et al., 2016). That may indicate that the ability of FAO may alter in different stages of NAFLD progression.
In addition, ω-oxidation conducted in the ER and β-oxidation in peroxisomes are upregulated in NASH as compensation (Robertson et al., 2001; Kohjima et al., 2007). However, it may lead to an increasement of ROS (Wanders et al., 2011; Del Río and López-Huertas, 2016). In addition, the increasement of the by-products of fat metabolism such as Cer and DAGs indicated that fat metabolism is insufficient during NSAH (Patterson et al., 2016), and these by-products will hinder insulin signaling pathway and result in insulin resistance (Samuel et al., 2010).
There are three major flux for acetyl-CoA pool produced from FAO and glycolysis that ketogenesis, TCA cycle and DNL in liver. Ketone bodies are mainly synthesized by hepatocyte using acetyl-CoA derived from FAO. However, they cannot be used in liver for lack of enzymes that can utilize ketone bodies, and have to be exported to extrahepatic tissues for terminal oxidation (Puchalska and Crawford, 2017). This physiological process provides an alternative energy source which is enhanced when glucose is unavailable (Robinson and Williamson, 1980). Primary regulators of ketogenesis include the rate of mitochondrial β-oxidation, CPT1 activity, TCA cycle flux and its intermediate concentrations, and the hormones such as glucagon and insulin. When content of acetyl-CoA was elevated due to increased β-oxidation or impaired glycolysis reduced the oxaloacetate derived from anaplerosis, ketogenesis will strengthen as consequence.
Under physiological conditions, in terms of the ratio of TCA cycle and ketogenesis, 86 ± 2% of acetyl-CoA is used for ketone body production, while in patients with NAFLD, only 77 ± 2% of acetyl-CoA with P = 0.003 enters ketogenesis after a 24-h fast, and the serum β-hydroxybutyric acid concentration was 30% lower than that of the control group (Fletcher et al., 2019), even though there was no significant difference in the production of ketone bodies after overnight fasting (Sunny et al., 2011). In addition, the serum β-hydroxybutyric acid concentration was negatively correlated with the fat content in the liver (Fletcher et al., 2019). Compared with HFD-fed mice, ketogenic insufficiency aggravates the hepatic inflammation and injury in HDF-fed 3-hydroxymethylglutaryl CoA synthase (HMGCS2) antisense oligonucleotide (ASO)-treated mice. In addition, ketogenic insufficiency will increase DNL since more the acetyl-CoA will be catalyzed by citrate synthase to citrate (Cotter et al., 2014).
Another metabolic pathway of acetyl-CoA is TCA cycle. The TCA cycle is a series of closed loop reactions. One molecule of acetyl-CoA that go through TCA cycle will produce 3 molecules of NADH, 1 molecule of FADH2, 2 molecules of C02 and 1 molecule of guanosine triphosphate GTP but the carbon does not directly drive from acetyl-CoA. Then NADH and FADH2 will be further oxidized by mitochondrial respiratory chain (MRC) and provide abundant ATP. The TCA cycle serves as the center of cell catabolism because no matter FA, protein or glucose, the terminal decomposition products are all acetyl-CoA. The regulation of TCA cycle mainly depends on allosteric regulation of the substrates, ATP, NADH, etc., but hardly by hormones (Martínez-Reyes and Chandel, 2020).
Under the fasting condition for 12 and 24 h, the acetyl-CoA flow into the TCA cycle in the liver of NAFLD patients was 2 and 1.4 times of normal people, respectively. which is also positively correlated with the liver triglyceride content, the rate of gluconeogenesis, and the blood glucose concentration (Sunny et al., 2011; Fletcher et al., 2019), although there is mitochondrial damage in term of morphology (Sanyal et al., 2001). Combined with attenuated ketogenesis we discussed before, we can draw the conclusion that the excess acetyl-CoA in hepatocytes of NAFLD patients was prone to be catabolized through the TCA cycle rather than ketogenesis, which reduced the fat consumption. Given that activity of TCA cycle is seldom controlled by hormones, and expression of related enzymes is constitutive. Therefore, the increased fat oxidation should be the main reason for TCA enhancement. The enhanced gluconeogenesis accompanied by enrichment of oxaloacetate that is able to activate pyruvate carboxylase may also contribute to overactive TCA cycle. Moreover the mitochondrial TCA cycle flux parallels to the ROS production and consequently cause oxidative stress (Satapati et al., 2012), thereby participate in the progression from liver steatosis to steatohepatitis (Browning and Horton, 2004). By inhibiting anaplerotic/cataplerotic flow, mitochondrial TCA flow and oxidative metabolism was reduced, and thereby mitigating inflammation and oxidative stress (Satapati et al., 2016). The enhancement of the TCA cycle increases the intermediates, citrate. It not only allosterically activates acetyl-CoA carboxylase, but also provides acetyl-CoA for DNL through the citrate pyruvate cycle.
Embedded in the inner mitochondrial membrane, four kinds of protein complexes that complex I, II, III, IV make up the MRC which can transfer proton and electrons from NADH and FADH2. Then the oxygen molecules eventually accept electrons and hydrogen ions to produce water. During the delivery of electrons, protons are transported from the mitochondrial matrix at CI, CIII, and CIV and create proton-motive force (PMF). Then PMF is used to drive ATP generation by CV, coupling the MRC with ATP synthesis (Rich and Maréchal, 2010). As a consequence of normal mitochondrial respiratory, mitochondria generate ROS in a physiological range. Most of the electrons delivered to the MRC are correctly accepted by oxygen and then form water with protons catalyzed by cytochrome c oxidase (CIV). However, a small part of the electrons directly combined with oxygen to form superoxide anion radical (O2–) and other ROS, which usually happens in complex I and complex III (Willems et al., 2015; Mansouri et al., 2018). ROS in physiological level can activate various signaling pathways involved in cell proliferation, inflammation and etc. However, excessive ROS will attack intracellular macromolecular compound such as proteins, nucleic acids, lipids and lead to cell death or induce apoptosis (Zorov et al., 2014).
The efficiency of MRC also alters in NAFLD. In patients with obesity or simple fatty liver, hepatic mitochondrial respiration increased compared with healthy people. However, in patients with steatohepatitis, 31–40% of patients have lower hepatic mitochondrial respiratory levels than obese patients. It indicates that in the early stage of fatty liver, the respiration rate of mitochondria increases as compensation. When it progresses to the stage of steatohepatitis, the respiration of mitochondria begins to decrease, and this adaptive change disappears (Koliaki et al., 2015), and the activity of various complexes in the MRC of NASH patients is decreased consistently (Pérez-Carreras et al., 2003). As attenuated mitochondrial respiration is inadequate to the increased TCA cycle, which means the excess electrons cannot be captured by oxygen appropriately, NASH is always accompanied with more ROS production, DNA damage, inflammation and liver injury (Serviddio et al., 2008; Koliaki et al., 2015).
Moreover, in NASH patients and ob/ob mice, the expression of the uncoupling protein-2 (UCP-2) elevated, which aggravated the mitochondrial uncoupling and proton leakage of liver mitochondria, and reduced the efficiency of liver ATP synthesis (Chavin et al., 1999; Serviddio et al., 2008), All this leads to impaired ATP synthesis which has been validated in obesity and NAFLD patients. by nuclear magnetic resonance spectroscopy (Nair et al., 2003). Interestingly, uncoupling of liver mitochondria seems to play a protective role in the progress of fatty liver working by accelerating energy expenditure. Using adenine nucleotide translocase (ANT) inhibitor, carboxyatractyloside, can alleviate fatty liver and insulin resistance in mice by enhancing uncoupling (Cho et al., 2017).
The treatment for NAFLD targeted on hepatocyte metabolism has demonstrated positive efficacy in some clinical trials. PPARα is one of the PPAR isotypes that mainly expressed in the liver, which can regulate liver FA uptake and the expression of FAO-related enzymes. Fibrates, PPARAα agonist, is mainly used to treat hyperlipidemia (Dubois et al., 2017). However, no obvious effect has been achieved in several clinical trials for the treatment of NAFLD. Nevertheless, several dual PPAR agonists have shown promising efficacy in recent clinical trials on the treatment of NAFLD. Saroglitazar, a dual PPAR α/γ agonist, has achieved a promising effect in the treatment of NAFLD in a phase 2 study(NCT03061721) (Gawrieh et al., 2021). Elafibranor, a dual PPARα/δ agonist, demonstrated the improvement of liver function tests and NASH resolution without worsening of fibrosis in a phase II study based on a post-hoc analysis(NCT01694849) (Ratziu et al., 2016). However, a phase III trial about it was terminated for not meeting the predefined primary surrogate efficacy endpoint (NCT02704403). FGF21 (fibroblast growth factor 21) is one of the target genes of PPARα, which mainly plays a role in the liver, such as enhancing FAO and reducing FA production (Kharitonenkov et al., 2005). Accordingly, Pegbelfermin, a FGF21 analogue, significantly reduced the hepatic fat fraction in patients with NASH determined by MRI-PDFF (magnetic resonance imaging-proton density fat fraction) in a phase 2a trial (NCT02413372) (Sanyal et al., 2019).
ACC is a key enzyme for lipogenesis. Therefore, it is reasonable to speculate that ACC inhibitors are able to reduce fatty liver. In a Phase 2 trial, GS-0976 (firsocostat), an ACC inhibitor, can significantly reduce DNL and ameliorate hepatic steatosis and markers of liver injury (NCT02856555) (Lawitz et al., 2018; Loomba et al., 2018). When combined with cilofexor (Farnesoid X Receptor agonist), firsocostat displayed greater improvement in hepatic steatosis and fibrosis of patients with advanced fibrosis caused by NASH defined by histology and clinically relevant biomarkers (NCT03449446) (Loomba et al., 2021).
Lifestyle intervention and weight loss are also effective methods to treat NAFLD. Among them, the ketogenic diet that increases the hepatic ketone production is popular in weight loss and is also helpful for NAFLD (Thoma et al., 2012; Saeed et al., 2019). In a clinical trial, 10 overweight/obese participants, who were restricted in carbohydrate intake while the intake of fat and protein remained unchanged, demonstrated decreased IHTG content and improved insulin sensitivity, despite increased circulating FFA (Luukkonen et al., 2020).
There is no FDA-approved drug for NAFLD treatment so far. Therefore, it is of great interest to explore novel targets for NAFLD treatment. In animal models, changes in glycolysis, lactate production, and mitochondrial uncoupling can affect the progression of NAFLD, which can be the potential pharmacological targets for NAFLD.
Although the metabolism of liver is generally regulated by insulin and glucagon, NALFD is often accompanied by systemic insulin resistance and the insulin resistance is selective (Brown and Goldstein, 2008), which makes the actual metabolic state hard to predict. In this review we found that the changes in DNL, glycolysis, and ketogenesis is in accord with insulin effect, whereas gluconeogenesis and FAO is less inhibited by insulin.
Therefore, it is meaningful to study the specific changes in metabolism and their role in the pathogenesis of NAFLD. In this review, we mainly discussed changes in lipid and glucose metabolism. Glycolysis is significantly enhanced in the hepatocytes of fatty liver, resulting in the increased content of pyruvate in plasma and liver. Enhanced glycolysis can cause liver inflammation, but the specific pathway is still unclear. PDC is the main enzyme that controls the flux of pyruvate into the TCA cycle. The alteration of its activity in fatty liver is still controversial. Pyruvate can also be converted to oxaloacetate through anaplerosis that provides intermediates for TCA, or to lactate, both of which are enhanced in NAFLD. Increased lactate will elevate the expression of lipogenic enzymes dependent on acetylation of H3K9, and aggravate NAFLD.
Excessive accumulation of fat in hepatocyte is the fundamental feature of NAFLD, accompanied by an increase in VLDL secretion, which is mainly derived from strengthened DNL. Meanwhile, the FAs oxidation is also enhanced in the mitochondria as well as the peroxisomes and ER. The oxidation that occurs in the latter can lead to more production of ROS that causes inflammation and liver damage.
Increased glycolysis and FAO will elevate acetyl-CoA and enhance the TCA cycle, which will increase liver mitochondrial burden. However, the production of ketone bodies is reduced, indicating that the acetyl-CoA is more inclined to enter the flux of TCA cycle. Nevertheless, the activity of MRC is reduced, which cannot match the enhanced TCA cycle, resulting in more ROS generation. Moreover, the expression of UCP2 also increased in fatty liver, which impaired efficiency of ATP synthesis. The global metabolic changes are shown in Figure 1.
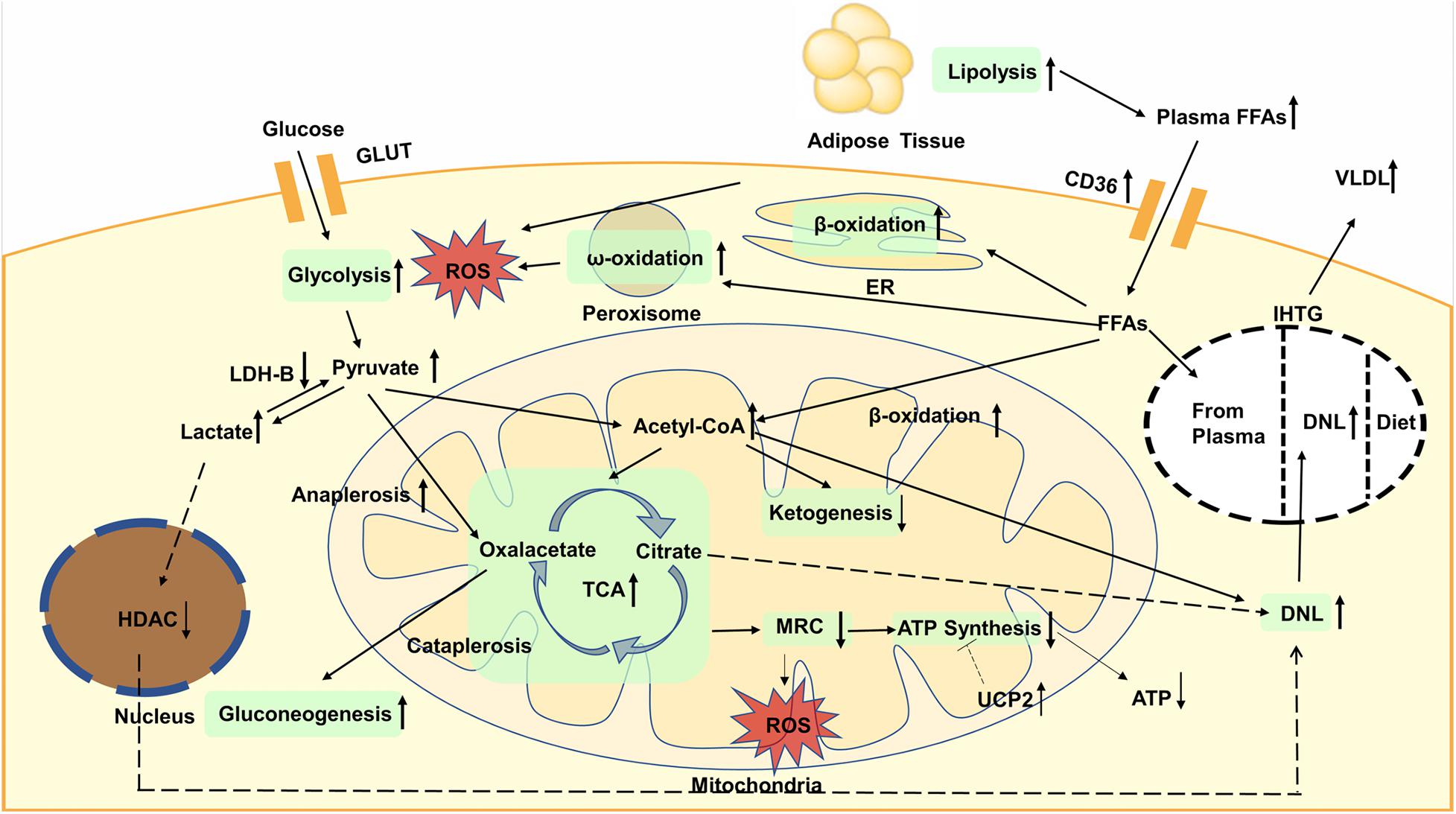
Figure 1. Glycolysis is enhanced, resulting in the increased content of pyruvate in plasma and liver. Then pyruvate is converted to oxaloacetate through anaplerosis or to lactate, both of which are enhanced in NAFLD. Increased lactate will elevate the expression of lipogenic enzymes dependent on decreased activity of nuclear HDAC. Gluconeogenesis is increased and contribute to TCA cycle. Lipolysis is increased, leading to elevated plasma FFAs. accumulation of TAGs is mainly caused by increased DNL. The oxidation of fatty acids is also enhanced, not only in the mitochondria, but also in the endoplasmic reticulum and peroxisomes. The oxidation that occurs in the latter can lead to more production of ROS that causes inflammation and liver damage. Increased glycolysis and fatty acid oxidation will elevate acetyl-CoA and enhance the TCA cycle whereas ketogenesis is reduced. The activity of MRC is reduced, resulting in more ROS generation. The expression of UCP2 increased in fatty liver, which impaired efficiency of ATP synthesis and decreased ATP content. GLUT, glucose transporter; FFAs, plasma free fatty acids; CD36, cluster of designation 36; ROS, reactive oxygen species; ER, endoplasmic reticulum; VLDL, very low density lipoproteins; IHTG, intrahepatic triglycerides; DNL, de novo lipogenesis; LDH, lactate dehydrogenase; HDAC, histone deacetylase; TCA, tricarboxylic acid; MRC, mitochondrial respiratory chain; ATP, adenosine triphosphate; UCP, uncoupling protein.
QL, XT, LZ, and SZ: conception. QL, XT, HW, JH, ML, and ZM: drafting manuscript. XT, HX, LZ, and SZ: revising manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Innovative Research Groups of National Natural Science Foundation of China (No. 81721091).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thanks to Innovative Research Groups of National Natural Science Foundation of China for financial support.
Adeva-Andany, M., López-Ojén, M., Funcasta-Calderón, R., Ameneiros-Rodríguez, E., Donapetry-García, C., Vila-Altesor, M., et al. (2014). Comprehensive review on lactate metabolism in human health. Mitochondrion 17, 76–100. doi: 10.1016/j.mito.2014.05.007
Bajotto, G., Murakami, T., Nagasaki, M., Qin, B., Matsuo, Y., Maeda, K., et al. (2006). Increased expression of hepatic pyruvate dehydrogenase kinases 2 and 4 in young and middle-aged Otsuka Long-Evans Tokushima Fatty rats: induction by elevated levels of free fatty acids. Metabolism 55, 317–323. doi: 10.1016/j.metabol.2005.09.014
Bechmann, L. P., Gieseler, R. K., Sowa, J. P., Kahraman, A., Erhard, J., Wedemeyer, I., et al. (2010). Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 30, 850–859. doi: 10.1111/j.1478-3231.2010.02248.x
Benhamed, F., Denechaud, P. D., Lemoine, M., Robichon, C., Moldes, M., Bertrand-Michel, J., et al. (2012). The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest. 122, 2176–2194. doi: 10.1172/jci41636
Bonnefont, J. P., Djouadi, F., Prip-Buus, C., Gobin, S., Munnich, A., and Bastin, J. (2004). Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol. Aspects Med. 25, 495–520. doi: 10.1016/j.mam.2004.06.004
Bougarne, N., Weyers, B., Desmet, S. J., Deckers, J., Ray, D. W., Staels, B., et al. (2018). Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 39, 760–802. doi: 10.1210/er.2018-00064
Bowker-Kinley, M. M., Davis, W. I., Wu, P., Harris, R. A., and Popov, K. M. (1998). Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329(Pt 1), 191–196. doi: 10.1042/bj3290191
Brooks, G. A. (2018). The science and translation of lactate shuttle theory. Cell Metab. 27, 757–785. doi: 10.1016/j.cmet.2018.03.008
Brown, M. S., and Goldstein, J. L. (2008). Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96. doi: 10.1016/j.cmet.2007.12.009
Browning, J. D., and Horton, J. D. (2004). Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152. doi: 10.1172/jci22422
Brunt, E. M., Kleiner, D. E., Carpenter, D. H., Rinella, M., Harrison, S. A., Loomba, R., et al. (2020). Nonalcoholic fatty liver disease: reporting histologic findings in clinical practice. Hepatology 73, 2028–2038. doi: 10.1002/hep.31599
Byrne, C. D., and Targher, G. (2015). NAFLD: a multisystem disease. J. Hepatol. 62(Suppl_1), S47–S64. doi: 10.1016/j.jhep.2014.12.012
Cappel, D. A., Deja, S., Duarte, J. A. G., Kucejova, B., Iñigo, M., Fletcher, J. A., et al. (2019). Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 29, 1291–1305.e8. doi: 10.1016/j.cmet.2019.03.014
Chavin, K. D., Yang, S., Lin, H. Z., Chatham, J., Chacko, V. P., Hoek, J. B., et al. (1999). Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J. Biol. Chem. 274, 5692–5700. doi: 10.1074/jbc.274.9.5692
Chen, Z., Liu, M., Li, L., and Chen, L. (2018). Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 233, 2839–2849. doi: 10.1002/jcp.25998
Cho, J., Zhang, Y., Park, S. Y., Joseph, A. M., Han, C., Park, H. J., et al. (2017). Mitochondrial ATP transporter depletion protects mice against liver steatosis and insulin resistance. Nat. Commun. 8:14477. doi: 10.1038/ncomms14477
Cotter, D. G., Ercal, B., Huang, X., Leid, J. M., D’Avignon, D. A., Graham, M. J., et al. (2014). Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Invest. 124, 5175–5190. doi: 10.1172/jci76388
Crespo, J., Cayón, A., Fernández-Gil, P., Hernández-Guerra, M., Mayorga, M., Domínguez-Díez, A., et al. (2001). Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 34, 1158–1163. doi: 10.1053/jhep.2001.29628
Dawson, D. M., Goodfriend, T. L., and Kaplan, N. O. (1964). Lactic dehydrogenases: functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science 143, 929–933. doi: 10.1126/science.143.3609.929
de la Cruz-López, K. G., Castro-Muñoz, L. J., Reyes-Hernández, D. O., García-Carrancá, A., and Manzo-Merino, J. (2019). Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 9:1143. doi: 10.3389/fonc.2019.01143
Del Río, L. A., and López-Huertas, E. (2016). ROS generation in peroxisomes and its role in cell signaling. Plant Cell Physiol. 57, 1364–1376. doi: 10.1093/pcp/pcw076
Dentin, R., Girard, J., and Postic, C. (2005). Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 87, 81–86. doi: 10.1016/j.biochi.2004.11.008
Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J., Boldt, M. D., and Parks, E. J. (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351. doi: 10.1172/jci23621
Dubois, V., Eeckhoute, J., Lefebvre, P., and Staels, B. (2017). Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Invest. 127, 1202–1214. doi: 10.1172/jci88894
Endo, M., Masaki, T., Seike, M., and Yoshimatsu, H. (2007). TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c). Exp. Biol. Med. 232, 614–621.
Fabbrini, E., Mohammed, B. S., Magkos, F., Korenblat, K. M., Patterson, B. W., and Klein, S. (2008). Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134, 424–431. doi: 10.1053/j.gastro.2007.11.038
Fan, J. G., Kim, S. U., and Wong, V. W. (2017). New trends on obesity and NAFLD in Asia. J. Hepatol. 67, 862–873. doi: 10.1016/j.jhep.2017.06.003
Fletcher, J. A., Deja, S., Satapati, S., Fu, X., Burgess, S. C., and Browning, J. D. (2019). Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 5:e127737. doi: 10.1172/jci.insight.127737
Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M., and Sanyal, A. J. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922. doi: 10.1038/s41591-018-0104-9
Gawrieh, S., Noureddin, M., Loo, N., Mohseni, R., Awasty, V., Cusi, K., et al. (2021). Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology. doi: 10.1002/hep.31843 [Epub ahead of print].
Gladden, L. B. (2004). Lactate metabolism: a new paradigm for the third millennium. J. Physiol. 558(Pt 1), 5–30. doi: 10.1113/jphysiol.2003.058701
Go, Y., Jeong, J. Y., Jeoung, N. H., Jeon, J. H., Park, B. Y., Kang, H. J., et al. (2016). Inhibition of pyruvate dehydrogenase kinase 2 protects against hepatic steatosis through modulation of tricarboxylic acid cycle anaplerosis and ketogenesis. Diabetes 65, 2876–2887. doi: 10.2337/db16-0223
Haeusler, R. A., McGraw, T. E., and Accili, D. (2018). Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 19, 31–44. doi: 10.1038/nrm.2017.89
Hers, H. G., and Hue, L. (1983). Gluconeogenesis and related aspects of glycolysis. Annu. Rev. Biochem. 52, 617–653. doi: 10.1146/annurev.bi.52.070183.003153
Hiebl, V., Ladurner, A., Latkolik, S., and Dirsch, V. M. (2018). Natural products as modulators of the nuclear receptors and metabolic sensors LXR, FXR and RXR. Biotechnol. Adv. 36, 1657–1698. doi: 10.1016/j.biotechadv.2018.03.003
Higuchi, N., Kato, M., Shundo, Y., Tajiri, H., Tanaka, M., Yamashita, N., et al. (2008). Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. 38, 1122–1129. doi: 10.1111/j.1872-034X.2008.00382.x
Hodakoski, C., Hopkins, B. D., Barrows, D., Mense, S. M., Keniry, M., Anderson, K. E., et al. (2014). Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 111, 155–160. doi: 10.1073/pnas.1213773111
Horie, Y., Suzuki, A., Kataoka, E., Sasaki, T., Hamada, K., Sasaki, J., et al. (2004). Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 113, 1774–1783. doi: 10.1172/jci20513
Iozzo, P., Bucci, M., Roivainen, A., Någren, K., Järvisalo, M. J., Kiss, J., et al. (2010). Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139, 846–856.e6. doi: 10.1053/j.gastro.2010.05.039
Kawano, Y., and Cohen, D. E. (2013). Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 48, 434–441. doi: 10.1007/s00535-013-0758-5
Keramida, G., Hunter, J., and Peters, A. M. (2016). Hepatic glucose utilization in hepatic steatosis and obesity. Biosci. Rep. 36:e00402. doi: 10.1042/bsr20160381
Khan, R. S., Bril, F., Cusi, K., and Newsome, P. N. (2019). Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology 70, 711–724. doi: 10.1002/hep.30429
Kharitonenkov, A., Shiyanova, T. L., Koester, A., Ford, A. M., Micanovic, R., Galbreath, E. J., et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635. doi: 10.1172/jci23606
Kim, H. S., Xiao, C., Wang, R. H., Lahusen, T., Xu, X., Vassilopoulos, A., et al. (2010). Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12, 224–236. doi: 10.1016/j.cmet.2010.06.009
Kohjima, M., Enjoji, M., Higuchi, N., Kato, M., Kotoh, K., Yoshimoto, T., et al. (2007). Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 20, 351–358.
Koliaki, C., Szendroedi, J., Kaul, K., Jelenik, T., Nowotny, P., Jankowiak, F., et al. (2015). Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21, 739–746. doi: 10.1016/j.cmet.2015.04.004
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. doi: 10.1038/nrc3038
Kors, L., Rampanelli, E., Stokman, G., Butter, L. M., Held, N. M., Claessen, N., et al. (2018). Deletion of NLRX1 increases fatty acid metabolism and prevents diet-induced hepatic steatosis and metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 1864(5 Pt A), 1883–1895. doi: 10.1016/j.bbadis.2018.03.003
Lambert, J. E., Ramos-Roman, M. A., Browning, J. D., and Parks, E. J. (2014). Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735. doi: 10.1053/j.gastro.2013.11.049
Lawitz, E. J., Coste, A., Poordad, F., Alkhouri, N., Loo, N., McColgan, B. J., et al. (2018). Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 16, 1983–1991.e3. doi: 10.1016/j.cgh.2018.04.042
Liu, J., Jiang, S., Zhao, Y., Sun, Q., Zhang, J., Shen, D., et al. (2018). Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J. Pathol. 246, 277–288. doi: 10.1002/path.5131
Loomba, R., Kayali, Z., Noureddin, M., Ruane, P., Lawitz, E. J., Bennett, M., et al. (2018). GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 155, 1463–1473.e6. doi: 10.1053/j.gastro.2018.07.027
Loomba, R., Noureddin, M., Kowdley, K. V., Kohli, A., Sheikh, A., Neff, G., et al. (2021). Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 73, 625–643. doi: 10.1002/hep.31622
Lu, J., Tan, M., and Cai, Q. (2015). The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 356(2 Pt A), 156–164. doi: 10.1016/j.canlet.2014.04.001
Luukkonen, P. K., Dufour, S., Lyu, K., Zhang, X. M., Hakkarainen, A., Lehtimäki, T. E., et al. (2020). Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. U.S.A. 117, 7347–7354. doi: 10.1073/pnas.1922344117
Mansouri, A., Gattolliat, C. H., and Asselah, T. (2018). Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 155, 629–647. doi: 10.1053/j.gastro.2018.06.083
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11:102. doi: 10.1038/s41467-019-13668-3
Miele, L., Grieco, A., Armuzzi, A., Candelli, M., Forgione, A., Gasbarrini, A., et al. (2003). Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 98, 2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x
Nair, S., Pc, V., Arnold, C., and Diehl, A. M. (2003). Hepatic ATP reserve and efficiency of replenishing: comparison between obese and nonobese normal individuals. Am. J. Gastroenterol. 98, 466–470. doi: 10.1111/j.1572-0241.2003.07221.x
Owen, O. E., Kalhan, S. C., and Hanson, R. W. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412. doi: 10.1074/jbc.R200006200
Panasyuk, G., Espeillac, C., Chauvin, C., Pradelli, L. A., Horie, Y., Suzuki, A., et al. (2012). PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 3:672. doi: 10.1038/ncomms1667
Patterson, R. E., Kalavalapalli, S., Williams, C. M., Nautiyal, M., Mathew, J. T., Martinez, J., et al. (2016). Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 310, E484–E494. doi: 10.1152/ajpendo.00492.2015
Pérez-Carreras, M., Del Hoyo, P., Martín, M. A., Rubio, J. C., Martín, A., Castellano, G., et al. (2003). Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38, 999–1007. doi: 10.1053/jhep.2003.50398
Petersen, M. C., and Shulman, G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223. doi: 10.1152/physrev.00063.2017
Petersen, M. C., Vatner, D. F., and Shulman, G. I. (2017). Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 13, 572–587. doi: 10.1038/nrendo.2017.80
Pittala, S., Krelin, Y., Kuperman, Y., and Shoshan-Barmatz, V. (2019). A mitochondrial VDAC1-based peptide greatly suppresses steatosis and NASH-associated pathologies in a mouse model. Mol. Ther. 27, 1848–1862. doi: 10.1016/j.ymthe.2019.06.017
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. doi: 10.1016/j.cmet.2016.12.022
Ratziu, V., Harrison, S. A., Francque, S., Bedossa, P., Lehert, P., Serfaty, L., et al. (2016). Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e5. doi: 10.1053/j.gastro.2016.01.038
Reddy, J. K. (2001). Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1333–G1339. doi: 10.1152/ajpgi.2001.281.6.G1333
Repa, J. J., Liang, G., Ou, J., Bashmakov, Y., Lobaccaro, J. M., Shimomura, I., et al. (2000). Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 14, 2819–2830. doi: 10.1101/gad.844900
Rich, P. R., and Maréchal, A. (2010). The mitochondrial respiratory chain. Essays Biochem. 47, 1–23. doi: 10.1042/bse0470001
Robertson, G., Leclercq, I., and Farrell, G. C. (2001). Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol Gastrointest. Liver Physiol. 281, G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135
Robinson, A. M., and Williamson, D. H. (1980). Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 60, 143–187. doi: 10.1152/physrev.1980.60.1.143
Rothman, D. L., Magnusson, I., Katz, L. D., Shulman, R. G., and Shulman, G. I. (1991). Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 254, 573–576. doi: 10.1126/science.1948033
Saeed, N., Nadeau, B., Shannon, C., and Tincopa, M. (2019). Evaluation of dietary approaches for the treatment of non-alcoholic fatty liver disease: a systematic review. Nutrients 11:3064. doi: 10.3390/nu11123064
Samuel, V. T., Petersen, K. F., and Shulman, G. I. (2010). Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277. doi: 10.1016/s0140-6736(10)60408-4
Samuel, V. T., and Shulman, G. I. (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest. 126, 12–22. doi: 10.1172/jci77812
Sanyal, A., Charles, E. D., Neuschwander-Tetri, B. A., Loomba, R., Harrison, S. A., Abdelmalek, M. F., et al. (2019). Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392, 2705–2717. doi: 10.1016/s0140-6736(18)31785-9
Sanyal, A. J., Campbell-Sargent, C., Mirshahi, F., Rizzo, W. B., Contos, M. J., Sterling, R. K., et al. (2001). Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192. doi: 10.1053/gast.2001.23256
Satapati, S., Kucejova, B., Duarte, J. A., Fletcher, J. A., Reynolds, L., Sunny, N. E., et al. (2016). Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Invest. 126:1605. doi: 10.1172/jci86695
Satapati, S., Sunny, N. E., Kucejova, B., Fu, X., He, T. T., Méndez-Lucas, A., et al. (2012). Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 53, 1080–1092. doi: 10.1194/jlr.M023382
Serviddio, G., Bellanti, F., Tamborra, R., Rollo, T., Capitanio, N., Romano, A. D., et al. (2008). Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 57, 957–965. doi: 10.1136/gut.2007.147496
Shannon, C. E., Ragavan, M., Palavicini, J. P., Fourcaudot, M., Bakewell, T. M., Valdez, I. A., et al. (2021). Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 45:101154. doi: 10.1016/j.molmet.2020.101154
Shimomura, I., Bashmakov, Y., and Horton, J. D. (1999). Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 274, 30028–30032. doi: 10.1074/jbc.274.42.30028
Silverstein, R. L., and Febbraio, M. (2009). CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2:re3. doi: 10.1126/scisignal.272re3
Singh, S., Allen, A. M., Wang, Z., Prokop, L. J., Murad, M. H., and Loomba, R. (2015). Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 13, 643–654.e1-9; quiz e39–e40. doi: 10.1016/j.cgh.2014.04.014
Sunny, N. E., Parks, E. J., Browning, J. D., and Burgess, S. C. (2011). Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14, 804–810. doi: 10.1016/j.cmet.2011.11.004
Thoma, C., Day, C. P., and Trenell, M. I. (2012). Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J. Hepatol. 56, 255–266. doi: 10.1016/j.jhep.2011.06.010
Tilg, H., Moschen, A. R., and Roden, M. (2017). NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 14, 32–42. doi: 10.1038/nrgastro.2016.147
Trivedi, P., Wang, S., and Friedman, S. L. (2021). The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 33, 242–257. doi: 10.1016/j.cmet.2020.10.026
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. doi: 10.1126/science.1160809
Wanders, R. J., Komen, J., and Kemp, S. (2011). Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 278, 182–194. doi: 10.1111/j.1742-4658.2010.07947.x
Wang, T., Chen, K., Yao, W., Zheng, R., He, Q., Xia, J., et al. (2020). Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J. Hepatol. 74, 1038–1052. doi: 10.1016/j.jhep.2020.11.028
Wang, Y., Viscarra, J., Kim, S. J., and Sul, H. S. (2015). Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 16, 678–689. doi: 10.1038/nrm4074
Watt, M. J., Miotto, P. M., De Nardo, W., and Montgomery, M. K. (2019). The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr. Rev. 40, 1367–1393. doi: 10.1210/er.2019-00034
Willems, P. H., Rossignol, R., Dieteren, C. E., Murphy, M. P., and Koopman, W. J. (2015). Redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. doi: 10.1016/j.cmet.2015.06.006
Wu, L. M., He, H., Chen, G., Kuang, Y., Lin, B. Y., Chen, X. H., et al. (2020). Associations between obesity and metabolic health with nonalcoholic fatty liver disease in elderly Chinese. Hepatobiliary Pancreat. Dis. Int. 19, 252–257. doi: 10.1016/j.hbpd.2020.02.010
Younossi, Z., Anstee, Q. M., Marietti, M., Hardy, T., Henry, L., Eslam, M., et al. (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15, 11–20. doi: 10.1038/nrgastro.2017.109
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. doi: 10.1002/hep.28431
Yuan, X., Li, H., Bai, H., Zhao, X., Zhang, C., Liu, H., et al. (2016). The 11β-hydroxysteroid dehydrogenase type 1 inhibitor protects against the insulin resistance and hepatic steatosis in db/db mice. Eur. J. Pharmacol. 788, 140–151. doi: 10.1016/j.ejphar.2016.05.034
Zhou, Z. H., McCarthy, D. B., O’Connor, C. M., Reed, L. J., and Stoops, J. K. (2001). The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U.S.A. 98, 14802–14807. doi: 10.1073/pnas.011597698
Keywords: WARBURG effect, NAFLD, Mitochondrial dysfunction, Insulin resistance, Metabolism
Citation: Lu Q, Tian X, Wu H, Huang J, Li M, Mei Z, Zhou L, Xie H and Zheng S (2021) Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 12:710420. doi: 10.3389/fphys.2021.710420
Received: 16 May 2021; Accepted: 04 August 2021;
Published: 30 August 2021.
Edited by:
Xiang Zhang, Qilu Hospital, Shandong University, ChinaReviewed by:
Sonia Michael Najjar, Heritage College of Osteopathic Medicine, Ohio University, United StatesCopyright © 2021 Lu, Tian, Wu, Huang, Li, Mei, Zhou, Xie and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shusen Zheng, c2h1c2VuemhlbmdAemp1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.