
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Physiol., 18 August 2021
Sec. Integrative Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.703069
This article is part of the Research TopicPhysiological and Pathological Responses to Hypoxia and High Altitude, Volume IIView all 22 articles
 Juliane Hannemann1,2
Juliane Hannemann1,2 Kathrin Cordts1†
Kathrin Cordts1† Anika Seniuk3,4
Anika Seniuk3,4 Chi-un Choe5Lena Schmidt-Hutten1Jorge Duque Escobar3,6‡Florian Weinberger7
Chi-un Choe5Lena Schmidt-Hutten1Jorge Duque Escobar3,6‡Florian Weinberger7 Rainer Böger1,2,3
Rainer Böger1,2,3 Edzard Schwedhelm1,3*
Edzard Schwedhelm1,3*Objective: Chronic hypoxia induces pulmonary and cardiovascular pathologies, including pulmonary hypertension (PH). L-arginine:glycine amidinotransferase (AGAT) is essential for homoarginine (hArg) and guanidinoacetate synthesis, the latter being converted to creatine by guanidinoacetate methyltransferase. Low hArg concentrations are associated with cardiovascular morbidity and predict mortality in patients with PH. We therefore aimed to investigate the survival and cardiac outcome of AGAT knockout (Agat−/−) mice under hypoxia and a possible rescue of the phenotype.
Methods: Agat−/− mice and wild-type (WT) littermates were subjected to normoxia or normobaric hypoxia (10% oxygen) for 4 weeks. A subgroup of Agat−/− mice was supplemented with 1% creatine from weaning. Survival, hematocrit, blood lactate and glucose, heart weight-to-tibia length (HW/TL) ratio, hArg plasma concentration, and Agat and Gamt expression in lung, liver, and kidneys were evaluated.
Results: After 6 h of hypoxia, blood lactate was lower in Agat−/−-mice as compared to normoxia (p < 0.001). Agat−/− mice died within 2 days of hypoxia, whereas Agat−/− mice supplemented with creatine and WT mice survived until the end of the study. In WT mice, hematocrit (74 ± 4 vs. 55 ± 2%, mean ± SD, p < 0.001) and HW/TL (9.9 ± 1.3 vs. 7.3 ± 0.7 mg/mm, p < 0.01) were higher in hypoxia, while hArg plasma concentration (0.25 ± 0.06 vs. 0.38 ± 0.12 μmol/L, p < 0.01) was lower. Agat and Gamt expressions were differentially downregulated by hypoxia in lung, liver, and kidneys.
Conclusion: Agat and Gamt are downregulated in hypoxia. Agat−/− mice are nonviable in hypoxia. Creatine rescues the lethal phenotype, but it does not reduce right ventricular hypertrophy of Agat−/− mice in hypoxia.
In patients with newly diagnosed, treatment-naïve pulmonary hypertension (PH), low homoarginine (hArg) plasma concentration was identified as an independent predictor of morbidity and mortality (Atzler et al., 2016). PH is a rare disease with a poor prognosis; it is based on an increase in vascular resistance, which leads to elevated blood pressure in the pulmonary circulation. The increased afterload leads to an increased stress on the right ventricle, causing right ventricular hypertrophy. Eventually, heart failure evolves, which is a major cause of death in PH patients (Giordano, 2005).
The detailed pathophysiological mechanisms leading to idiopathic pulmonary hypertension are not yet fully understood. Pulmonary vasoconstriction is one of the compensatory mechanisms that help to maintain sufficient oxygen supply in focal pulmonary hypoxia; however, excessive and global pulmonary vasoconstriction in global hypoxia may lead to pulmonary hypertension and fatal outcome (Hannemann et al., 2020b). Increased vasoconstriction of the pulmonary vessels is assumed to play an important role, due to an increased release of vasoconstrictor prostaglandins and endothelin-1, and a reduced release of the vasodilator nitric oxide (NO; Lourenco et al., 2012; Giannakoulas et al., 2014). Accumulation of endogenous inhibitors of NO synthesis contributed to this dysregulation, and we and others have shown that chronic hypoxia induces pulmonary and cardiovascular phenotypes that are similar to PH (Hannemann et al., 2020a; Smith et al., 2020); therefore, chronic hypoxia was used as an experimental model in this study.
hArg is a non-proteinogenic amino acid that is synthesized by L-arginine:glycine amidinotransferase (AGAT, Kayacelebi et al., 2015). The same enzyme catalyzes the formation of guanidinoacetic acid, which is further metabolized to creatine by guanidinoacetate methyltransferase (GAMT, Kan et al., 2004). Accordingly, Agat-deficient mice (Agat−/− mice) are characterized by low circulating hArg and creatine concentrations (Choe et al., 2013a; Nabuurs et al., 2013). hArg is a weak substrate of NO synthase (NOS) and an inhibitor of arginase (Hrabák et al., 1994; Moali et al., 1998). Phosphorylated creatine, on the other hand, plays a key role in energy metabolism and acts as a rapidly available energy buffer due to its capacity to recycle adenosine triphosphate (ATP, Lygate et al., 2013; Stockebrand et al., 2018).
Our group has previously shown that Agat−/− mice develop significantly larger stroke areas in a temporary middle cerebral artery occlusion model as compared to WT littermates. Stroke size was significantly reduced to WT size in Agat−/− mice supplemented with hArg, but only slightly in creatine-supplemented animals (Choe et al., 2013a). Likewise, Faller and co-workers have shown that impaired cardiac contractile function in Agat−/− mice is rescued by hArg, but not creatine (Faller et al., 2018). By contrast, Laasmaa et al. showed that impaired calcium handling by isolated cardiomyocytes from Agat−/− mice is normalized by creatine supplementation when creatine is supplemented from weaning (Laasmaa et al., 2021). Taken together, these data suggest that hArg and creatine play differential roles in mediating the pathophysiological phenotypes of Agat−/− mice, which are not yet fully understood.
Chronic hypoxia is a condition that is associated with a global dysbalance of energy metabolism. We therefore hypothesized that Agat−/− mice may be specifically vulnerable in chronic hypoxia and that creatine or hArg supplementation may help to maintain a healthy phenotype.
Agat−/− mice used in the present study were described previously and have a life expectancy similar to WT littermates (Choe et al., 2013a,b; Nabuurs et al., 2013). Female mice were kept at 20 to 24°C and a 12:12 light–dark cycle with free access to chow and water. Agat−/− mice and WT littermates aged 3–4 months were placed in normobaric hypoxia (10% O2) or normoxia (21% O2) for 4 weeks (8–9 animals per group, Supplementary Figure 1). A separate group of Agat−/− mice was supplemented with 1% creatine from weaning in standard chow (Ssniff, Soest, Germany) and placed in normoxia or hypoxia, respectively, together with WT littermates in hypoxia (8–9 animals per group). For each group separately, mice from the same litter were kept in one cage. Throughout the study period, all animals were monitored daily for survival and chow was changed from dry to moistened starting 4 to 7 days prior to hypoxia for Agat−/− mice. For the first hours of hypoxia, mice were housed on external heating mates. After 6 h of hypoxia, a blood sample was collected by puncture; surviving animals were killed in anesthesia after 4 weeks of hypoxia or normoxia. Blood and tissues were collected and processed for analysis as previously described (Hannemann et al., 2020a). This study was approved by the Animal Care and Use Committee of the Freie und Hansestadt Hamburg (Approval no. 40/16). The use of animals was consistent with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996).
Hematocrit, glucose, and lactate were determined after puncture of the Vena facialis and subsequent blood analysis with an ABL 90 FLEX (Radiometer, Krefeld, Germany) after 6 h of hypoxia and after 4 weeks of hypoxia or normoxia. Cardiac troponin I (cTnI) was determined by ARCHITECT STAT high-sensitive cTnI immunoassay (i2000SR, Abbott Diagnostics, IL, United States) in serum samples diluted 1:5 with 0.9% NaCl. The epitope of the antigen showed 96% similarity between mice and human. Body weight and wet weights of the right ventricle (RV) and left ventricle (LV), septum, and the tibia length were determined. The Fulton index was calculated by dividing the RV to the sum of LV and septum. Cardiac echography was performed using the Vevo 3100 System (VisualSonics, Toronto, Canada) using standardized procedures (Zacchigna et al., 2021). Mice were anaesthetized with isoflurane (1–2%) and taped to a warming platform in a supine position. B-mode images were obtained using a MX400 transducer. Images were obtained in a parasternal short- and long-axis view, and dimensions of the left ventricle were measured in a short-axis view in diastole and systole. Fractional area shortening (FAS) was calculated as:
hArg plasma concentrations were determined from EDTA plasma obtained from cardiac puncture; blood samples were centrifuged at 3,000 × g for 3 min at 4°C and stored at −80°C until analysis. hArg concentration was determined by liquid chromatography–tandem mass spectrometry (LC–MS/MS) as previously described (Atzler et al., 2011). Briefly, 25 μl of plasma was diluted with 100 μl of stable isotope-labeled hArg dissolved in methanol. After protein precipitation, hArg was converted to its butyl ester derivative and analyzed by LC–MS/MS (Varian 1200, Agilent Technologies, Santa Clara, CA, United States). Quantification was performed by calculation of peak area ratios and calibration with known concentrations of hArg in dialyzed EDTA plasma (Atzler et al., 2011).
RNA preparation, reverse transcription, and quantitative real-time PCR (qRT-PCR) were performed as previously described (Hannemann et al., 2020a). Briefly, total RNA was isolated from snap-frozen, homogenized lung, liver, and renal tissues using Trizol (Thermo Fisher Scientific, Waltham, MA, United States). Genomic DNA was removed by an on-column clean-up procedure using PureLink™ RNA Mini Kit in combination with PureLink™ DNase (both Thermo Fisher). RNA was reverse transcribed with SuperScript™ IV VILO™ (Thermo Fisher). qRT-PCR was performed with gene-specific, FAM-labeled Taqman™ assays for Agat (Mm00491882_m1) and Gamt (Mm00487473_m1) and a VIC-labeled control assay (Tbp, Mm01277042_m1) for normalization (all Thermo Fisher). For all experiment, technical triplicates were analyzed. Relative gene expression was calculated using the ΔΔCt method (Schmittgen and Livak, 2008).
Preparation of tissue lysates was performed as previously described (Hannemann et al., 2020a). Samples were prepared for SDS-PAGE (kidney, 30 μg total protein; liver, 50 μg total protein per lane) by boiling in beta-mercaptoethanol containing sample buffer; samples were subjected to 10% SDS-PAGE. Subsequently, proteins were transferred onto a 0.2 μm nitrocellulose membrane (GE Healthcare, Munich, Germany). Membranes were incubated with the primary antibody (anti-AGAT, 1:300 in liver, 1:500 in kidney; 12801, Proteintech Chicago, Illinois, United States; anti-β-tubulin: 1:500, Abcam ab6046, Cambridge, United Kingdom) for 1 h at room temperature. Incubation with the secondary antibody (1:10,000; A0545, Merck, Darmstadt, Germany) was done for 1 h at room temperature. Protein was detected using enhanced chemiluminescence detection according to the manufacturer’s protocol (GE Healthcare, Munich, Germany).
Data are expressed as mean ± standard deviation (SD). A two-sided independent Student’s t-test was used to compare two groups. One-way ANOVA with Bonferroni post-hoc test was used to compare more than two groups. Chi-square test was used to compare dichotomized variables; Kaplan–Meier curves were generated for survival analysis. p < 0.05 was considered statistically significant in all analyses. Statistical analyses were performed by GraphPad Prism 6.0 (La Jolla, CA, United States).
We analyzed the acute effects of hypoxia after 6 h of exposure to 10% oxygen. By comparison with normoxia, there was a significant reduction of blood lactate concentration in Agat−/− mice (7.5 ± 1.8 vs. 2.6 ± 0.3 mmol/L; p < 0.001), while no significant reduction was observed in WT littermates (Figure 1A). No significant difference was observed in hematocrit or glucose after 6 h of hypoxia in Agat−/− or WT animals (Figures 1B,C).
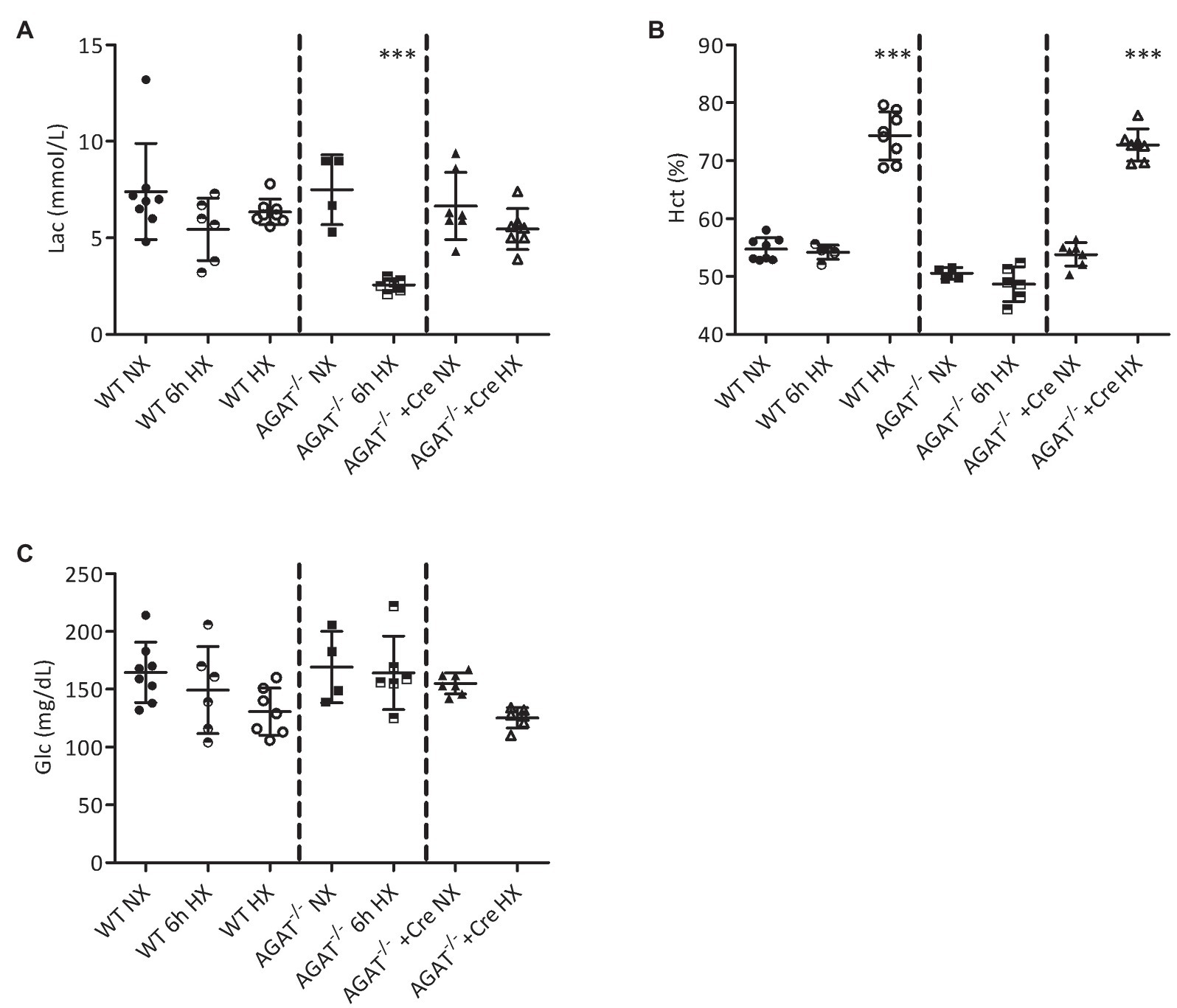
Figure 1. Blood analyses of Agat−/− mice and wild-type (WT) littermates. (A–C) Mean values ± SD from blood gas analyses under normoxia (NX), after 6 h of hypoxia (6 h HX) and after 4 weeks of hypoxia (HX) in unsupplemented and Agat−/− mice supplemented with 1% creatine from weaning (Cre). (A) Lactate (Lac). (B) Hematocrit (Hct %). (C) Glucose (Glc). Agat, arginine:glycine amidinotransferase; Cre, supplemented with 1% creatine. ***p < 0.001 vs. NX (ANOVA with Bonferroni post-hoc test).
All Agat−/− mice died within 48 h of hypoxia, whereas all WT littermates survived until the end of the study after 4 weeks of hypoxia (p < 0.001 Chi-square test; Figure 2A). The lethal phenotype in hypoxia was completely reversed in Agat−/− mice supplemented with creatine (Figure 2A). Agat−/− mice showed a trend of cTnI increase as compared with WT littermates and Agat−/−-mice supplemented with creatine in hypoxia (Figure 2B).
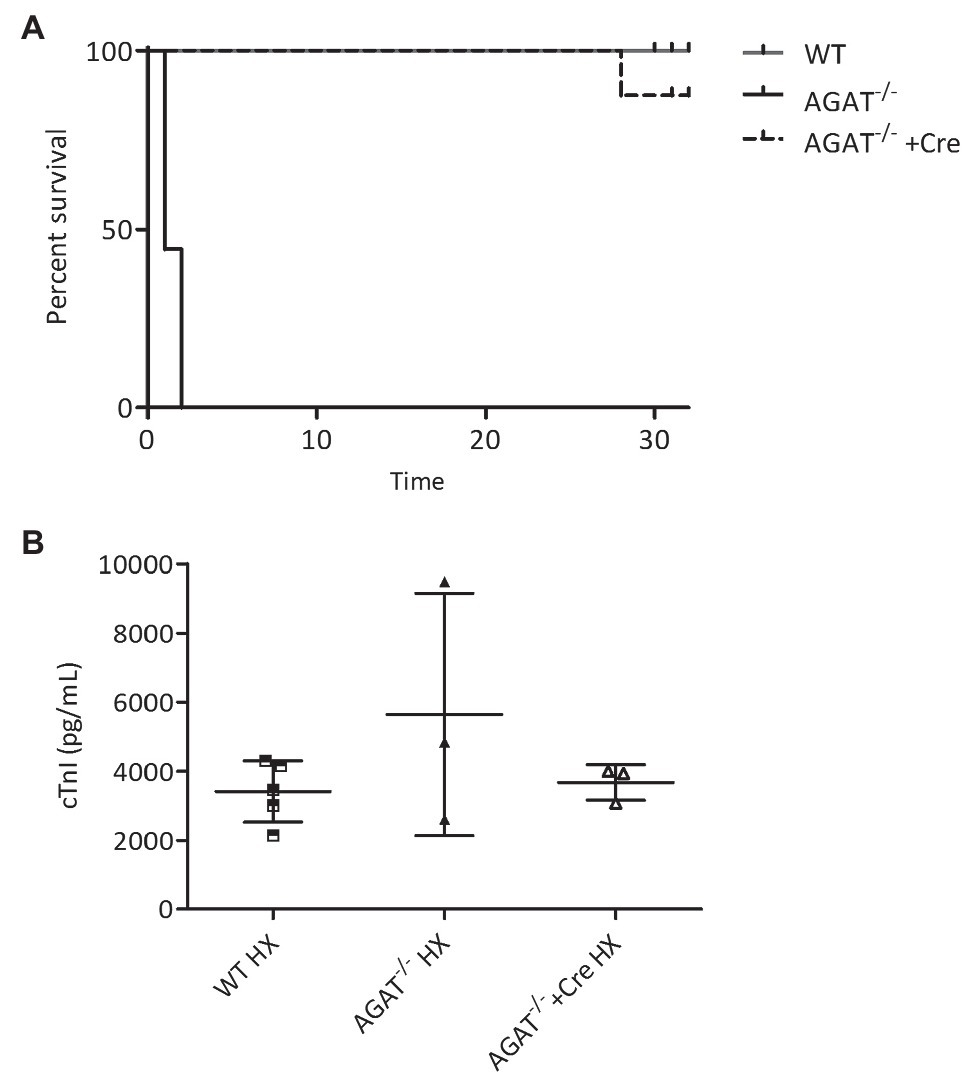
Figure 2. Kaplan–Meier survival analysis and cardiac troponin I (cTnI). (A,B) Outcome of Agat−/− mice with and w/o creatine (Cre) supplementation and WT littermates in hypoxia (HX). (A) Kaplan–Meier survival analysis of Agat−/− mice (n = 9), WT littermates (WT, n = 17) and Agat−/− mice supplemented with 1% creatine from weaning (Agat−/−+Cre, n = 8). All mice were kept in hypoxia and followed for up to 32 days. (B) cTnI in Agat−/− mice (n = 3, 1–2 days of HX), WT littermates (WT, n = 5, 4 weeks of HX) and Agat−/− mice supplemented with 1% creatine from weaning (Agat−/−+Cre, n = 3, 4 weeks of HX) Agat, arginine:glycine amidinotransferase; Cre, supplemented with 1% creatine.
Agat mRNA was highly expressed in kidneys and to a lower extent in liver, heart, and lungs, while Gamt showed highest gene expression in liver and low gene expression levels in kidneys, heart, and lungs (Figure 3). As compared to normoxia, chronic hypoxia caused a significant reduction of Agat mRNA expression in kidneys (p = 0.001) and lungs (p = 0.005), but not in heart and liver (Figure 3A). Likewise, Gamt gene expression in liver was lower in chronic hypoxia (p = 0.001), but remained unchanged in kidneys, heart, and lungs (Figure 3B). In line with the gene expression results, AGAT protein is downregulated in the hypoxic kidney. In the liver, where Agat expression is rather low, no effect of hypoxia was obvious (Figure 3C). Plasma hArg concentrations were significantly lower in hypoxia as compared to normoxia (0.25 ± 0.06 vs. 0.38 ± 0.12 μmol/L, p < 0.01).
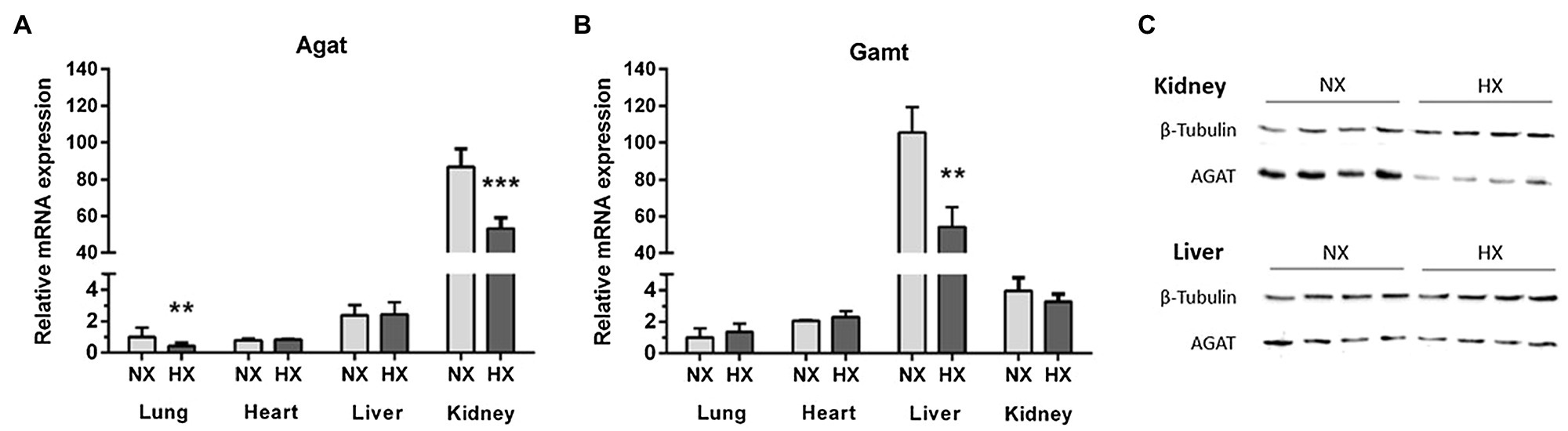
Figure 3. Agat and Gamt mRNA expression in lungs, liver, heart, and kidneys, and L-arginine:glycine amidinotransferase (AGAT) protein expression in liver and kidneys. mRNA expression of Agat (A) and Gamt (B) in normoxia (NX) and hypoxia (HX) as determined by quantitative real-time PCR normalized to Tbp as housekeeping gene. (C) Western Blot of AGAT in liver and kidneys as compared to β-tubulin as loading control. **p < 0.01 and ***p < 0.001 vs. NX (ANOVA with Bonferroni post-hoc test).
At the beginning of the intervention, Agat−/− mice had a lower mean body weight than their WT littermates (13.3 ± 1.3 vs. 19.0 ± 1.7 g, respectively, p < 0.001). Agat−/− mice supplemented with creatine from weaning had a body weight similar to WT littermates (18.0 ± 1.4 g, p = n.s. vs. WT).
After 4 weeks of hypoxia, hematocrit was significantly elevated in Agat−/− mice supplemented with creatine (73 ± 3% vs. 54 ± 2%, p < 0.001) and their unsupplemented WT littermates (74 ± 4% vs. 55 ± 2%, p < 0.001; Figure 1B) as compared with normoxic littermates. No significant differences in blood lactate and glucose concentrations were observed between mice of both groups exposed to either normoxia or hypoxia (Figures 1A,C).
Heart weight-to-tibia length (HW/TL) ratio was significantly elevated in WT mice exposed to chronic hypoxia; however, it was not significantly different between normoxia and hypoxia in Agat−/− mice supplemented with creatine (Figure 4A). Of note, there was a significant increase in Fulton index in both, Agat−/− mice supplemented with creatine (0.43 ± 0.04 vs. 0.27 ± 0.05, p < 0.001) and unsupplemented WT mice after 4 weeks of hypoxia (0.43 ± 0.06 vs. 0.29 ± 0.04, p < 0.001; Figure 4B) as compared with normoxic littermates. Hypoxia did not change tibia length or the sum of LV and septum (Supplementary Figure 2).
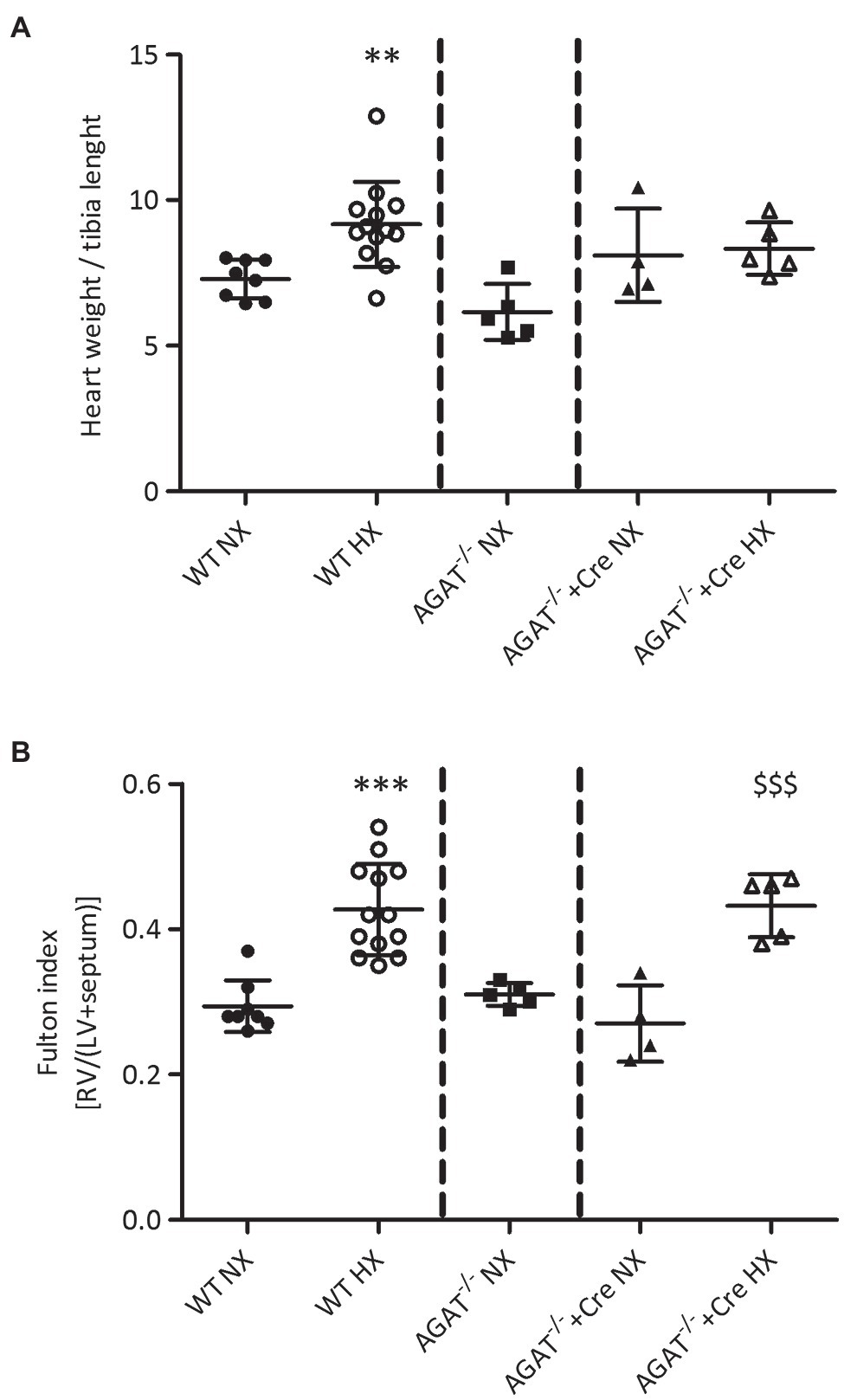
Figure 4. Phenotypes of cardiac hypertrophy of Agat−/− mice and WT littermates. (A,B) Mean values ± SD from hearts from Agat−/− mice supplemented with 1% creatine and WT littermates (WT) under normoxia (NX) and after 4 weeks of hypoxia (HX). (A) Heart weight to tibia length. (B) Fulton index {right ventricle (RV) weight/[left ventricle (LV) + septum weight]}. Agat, arginine:glycine amidinotransferase; Cre, supplemented with 1% creatine. **p < 0.01 vs. WT NX, ***p < 0.001 vs. WT NX, $$$p < 0.001 vs. Agat−/−+Cre NX (ANOVA with Bonferroni post-hoc test).
Echocardiography revealed higher FAS and ejection fraction in Agat−/− mice as compared with WT littermates and Agat−/− mice supplemented with creatine (Figures 5A,B). Hypoxia did not change FAS and ejection fraction in WT mice or AGAT−/− mice supplemented with creatine.
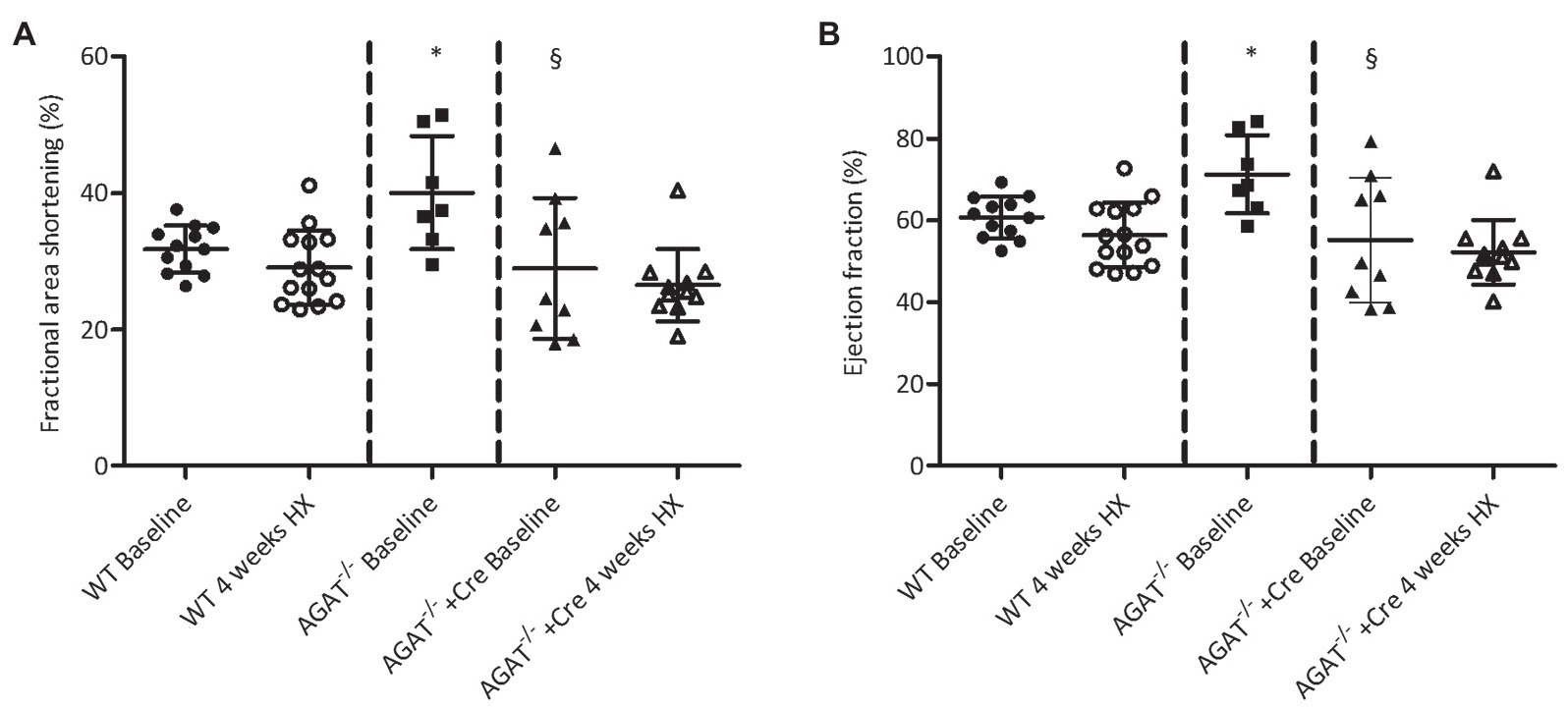
Figure 5. Echocardiography of Agat−/− mice and WT littermates. (A,B) B-mode images were obtained in a parasternal short- and long-axis view, and dimensions of the left ventricle were measured in a short-axis view in diastole and systole from Agat−/− mice supplemented with 1% creatine and WT littermates (WT) at baseline and after 4 weeks of hypoxia (HX). Mean values ± SD are presented. (A) Fractional area shortening. (B) Ejection fraction. Agat, arginine:glycine amidinotransferase; Cre, supplemented with 1% creatine. *p < 0.05 vs. WT baseline, §p < 0.05 vs. Agat−/− baseline (ANOVA with Bonferroni post-hoc test).
The main findings of our study are threefold: (1) Chronic hypoxia downregulates Agat expression and reduces hArg plasma concentrations. (2) Agat-deficient mice are not viable in hypoxia. (3) Supplementation with creatine rescues the lethal phenotype and cardiac hypertrophy.
The enzyme AGAT catalyzes the formation of guanidinoacetate from L-glycine and L-arginine; guanidinoacetate is subsequently methylated to creatine by GAMT activity (Kan et al., 2004; Barsunova et al., 2020). AGAT is involved not only in creatine synthesis but also in the formation of hArg from L-arginine and L-lysine (Kayacelebi et al., 2015). Relative mRNA expression of Agat and Gamt was highest in kidney and liver, respectively (Figure 3). Under hypoxia, Agat and Gamt were found downregulated in kidney and liver, respectively. Downregulation of Agat mRNA was paralleled by decreased AGAT protein detected in the kidney. Moreover, we found impaired hArg in the circulation of hypoxic WT mice. We thus conclude that renal AGAT is essential for hArg synthesis and whole body supply via the circulation.
hArg is a non-essential amino acid that differs from arginine by an additional methylene group in the carboxylic acid chain (Bell, 1962). Both L-arginine and hArg serve as substrates for NOS, with a 5- to 15-fold higher catalytic efficiency for L-arginine (Moali et al., 1998). Furthermore, hArg has been shown to be a weak inhibitor of the L-arginine-degrading enzyme arginase (Hrabák et al., 1994). In various clinical studies, low hArg concentrations have been associated with an increased risk of cardiovascular disease and mortality, as recently reviewed by our group (Atzler et al., 2015). Moreover, hArg plasma concentrations in treatment-naive PH patients are low and indicate a risk for mortality (Atzler et al., 2016). In line with these observations in PH patients, chronic hypoxia decreased hArg plasma concentration as well as Agat mRNA expression in mice. While this decrease in AGAT expression and function might be maladaptive, the absence of AGAT under hypoxia was deleterious. Agat−/− mice were not viable in hypoxia for more than 2 days. This phenotype was reversed by supplementation with 1% creatine from weaning and enabled Agat−/− mice to survive under hypoxic conditions for the complete study period of 4 weeks.
In neuronal and in muscle cells, creatine is phosphorylated to phosphocreatine by creatine kinase, thus serving as a quickly available energy buffer (Lygate et al., 2013; Nabuurs et al., 2013). Adenosine monophosphate-activated protein kinase (AMPK) is a key enzyme regulating energy metabolism; it downregulates energy-consuming metabolic processes in the presence of ATP deficiency. AMPK has been shown to be involved in hypoxic pulmonary vasoconstriction (Evans et al., 2015). In the event of a lack of energy in the cells, AMPK upregulates voltage-dependent potassium channels (Kv5.1), which contributes to cellular repolarization in pulmonary artery smooth muscle cells (Firth et al., 2011). In line with this, we previously observed chronic activation of AMPK in skeletal muscle of AGAT-deficient mice (Choe et al., 2013b). Even though we do not show AMPK activation in tissues of Agat−/− mice in hypoxia in the current study, we observed a dramatic decrease of blood lactate in Agat−/− mice as early as 6 h after the initiation of hypoxia. The heart can utilize glucose, lactate, fatty acids, and creatine for maintaining energy supply (Neubauer, 2007; Branovets et al., 2021). The decrease of blood lactate may thus indicate an increased anaerobic energy demand triggered by hypoxia, which cannot be buffered by cellular stores of creatine/phosphocreatine in the heart of Agat−/− mice as previously shown in hypoxic muscle tissue of these mice (Nabuurs et al., 2013).
Four weeks of hypoxia caused a significant increase of hematocrit as well as cardiac and – more specifically – right ventricular hypertrophy, as assessed by the HW/TL ratio for global cardiac hypertrophy and by the Fulton index for right ventricular hypertrophy. Clearly, the same extent of right ventricular hypertrophy was also found in Agat−/− mice supplemented with creatine, suggesting that right ventricular hypertrophy is not prevented by creatine but hArg supplementation might have an impact. Global cardiac hypertrophy seemed less pronounced in Agat−/− mice supplemented with creatine than in unsupplemented WT mice; however, this finding may have been influenced by the slightly reduced total body growth of the Agat−/− mice supplemented with creatine.
Our study has several limitations. Firstly, Agat−/− mice were supplemented with creatine from weaning; therefore, we cannot extend our findings to a possible effect of creatine supplementation if initiated after the induction of hypoxia. Secondly, animal welfare prevented us from studying the effects of hArg supplementation on the phenotype of Agat−/− mice. Supplementation of WT animals with hArg in hypoxia might be an alternative strategy; however, this was beyond the focus of our study. Although hArg has been implicated with heart failure and cardiomyocyte contractility (Faller et al., 2018), we have no definite proof if the PH-like phenotype, i.e., increase in Fulton index, may be caused, at least in part, by lack of hArg.
We show here that creatine rescues lethality of Agat−/− mice in hypoxia, showing a pathophysiological significance of creatine-related metabolic pathways. Creatine may therefore be a potentially valuable supplement in individuals having impaired AGAT activity, e.g., caused by genetic missense polymorphisms in the Agat gene, during conditions of impaired oxygen availability, e.g., at high altitude. However, creatine supplementation did not affect the PH-like phenotype in hypoxia, suggesting that mechanisms other than creatine-related ones may be involved in its pathogenesis. Further studies in animals and humans are needed to investigate the potentially differential roles of creatine and hArg in PH.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Animal Care and Use Committee of the Freie und Hansestadt Hamburg (Approval no. 40/16), BGV, Billestrasse 80, 20539 Hamburg, Germany. Written informed consent was obtained from the owners for the participation of their animals in this study.
JH, KC, RB, and ES contributed to the conception and design of the study. KC organized the database. JH, KC, FW, and ES performed the statistical analysis. JH and ES wrote the first draft of the manuscript. KC, AS, JDE, and FW wrote the sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors gratefully acknowledge support by grants from the Georg & Jürgen Rickertsen Foundation, Hamburg, Germany (to JH), by the German Federal Ministry of Education and Research (BMBF grant 01DN17046, DECIPHER, to JH and RB, DZHK grant 81Z1710102 to ES), an Else Kröner Exzellenzstipendiu by the Else Kröner-Fresenius-Stiftung (to C-UC) and the Werner Otto Foundation, Hamburg, Germany (02/96, to JH). JDE was supported by the German Center for Cardiovascular Research (DZHK e.V.) under grant number 81Z0710102.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Mariola Kastner, Fiona Franke, and Birgit Geertz for excellent technical assistance.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fphys.2021.703069/full#supplementary-material
Atzler, D., Cracowski, J. L., Cordts, K., Böger, R. H., Humbert, M., and Schwedhelm, E. (2016). Homoarginine predicts mortality in treatment-naive patients with pulmonary arterial hypertension. Int. J. Cardiol. 217, 12–15. doi: 10.1016/j.ijcard.2016.04.161
Atzler, D., Mieth, M., Maas, R., Böger, R. H., and Schwedhelm, E. (2011). Stable isotope dilution assay for liquid chromatography-tandem mass spectrometric determination of L-homoarginine in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 2294–2298. doi: 10.1016/j.jchromb.2011.06.016
Atzler, D., Schwedhelm, E., and Choe, C. U. (2015). L-homoarginine and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 18, 83–88. doi: 10.1097/MCO.0000000000000123
Barsunova, K., Vendelin, M., and Birkedal, R. (2020). Marker enzyme activities in hindleg from creatine-deficient AGAT and GAMT KO mice - differences between models, muscles, and sexes. Sci. Rep. 10:7956. doi: 10.1038/s41598-020-64740-8
Bell, E. A. (1962). α,γ-Diaminobutyric acid in seeds of twelve species of Lathyrus and identification of a new natural amino-acid, L-homoarginine, in seeds of other species toxic to man and domestic animals. Nature 193, 1078–1079. doi: 10.1038/1931078b0
Branovets, J., Karro, N., Barsunova, K., Laasmaa, M., Lygate, C. A., Vendelin, M., et al. (2021). Cardiac expression and location of hexokinase changes in a mouse model of pure creatine deficiency. Am. J. Physiol. Heart Circ. Physiol. 320, H613–H629. doi: 10.1152/ajpheart.00188.2020
Choe, C. U., Atzler, D., Wild, P. S., Carter, A. M., Böger, R. H., Ojeda, F., et al. (2013a). Homoarginine levels are regulated by L-arginine:glycine amidinotransferase and affect stroke outcome: results from human and murine studies. Circulation 128, 1451–1461. doi: 10.1161/CIRCULATIONAHA.112.000580
Choe, C. U., Nabuurs, C., Stockebrand, M. C., Neu, A., Nunes, P., Morellini, F., et al. (2013b). L-arginine:glycine amidinotransferase deficiency protects from metabolic syndrome. Hum. Mol. Genet. 22, 110–123. doi: 10.1093/hmg/dds407
Evans, A. M., Lewis, S. A., Ogunbayo, O. A., and Moral-Sanz, J. (2015). Modulation of the LKB1-AMPK signalling pathway underpins hypoxic pulmonary vasoconstriction and pulmonary hypertension. Adv. Exp. Med. Biol. 860, 89–99. doi: 10.1007/978-3-319-18440-1_11
Faller, K. M. E., Atzler, D., McAndrew, D. J., Zervou, S., Whittington, H. J., Simon, J. N., et al. (2018). Impaired cardiac contractile function in arginine:glycine amidinotransferase knockout mice devoid of creatine is rescued by homoarginine but not creatine. Cardiovasc. Res. 114, 417–430. doi: 10.1093/cvr/cvx242
Firth, A. L., Remillard, C. V., Platoshyn, O., Fantozzi, I., Ko, E. A., and Yuan, J. X. (2011). Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm. Circ. 1, 48–71. doi: 10.4103/2045-8932.78103
Giannakoulas, G., Mouratoglou, S. A., Gatzoulis, M. A., and Karvounis, H. (2014). Blood biomarkers and their potential role in pulmonary arterial hypertension associated with congenital heart disease. A systematic review. Int. J. Cardiol. 174, 618–623. doi: 10.1016/j.ijcard.2014.04.156
Giordano, F. (2005). Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508. doi: 10.1172/JCI200524408
Hannemann, J., Glatzel, A., Hillig, J., Zummack, J., Schumacher, U., Lüneburg, N., et al. (2020a). Upregulation of DDAH2 llimits pulmonary hypertension and right ventricular hypertrophy during chronic hypoxia in Ddah1 knockout mice. Front. Physiol. 11:597559. doi: 10.3389/fphys.2020.597559
Hannemann, J., Zummack, J., Hillig, J., and Böger, R. (2020b). Metabolism of asymmetric dimethylarginine in hypoxia: from bench to bedside. Pulm. Circ. 10:2045894020918846. doi: 10.1177/2045894020918846
Hrabák, A., Bajor, T., and Temesi, A. (1994). Comparison of substrate and inhibitor specificity of arginase and nitric oxide (NO) synthase for arginine analogues and related compounds in murine and rat macrophages. Biochem. Biophys. Res. Commun. 198, 206–212. doi: 10.1006/bbrc.1994.1029
Kan, H. E., Renema, W. K. J., Isbrandt, D., and Heerschap, A. (2004). Phosphorylated guanidinoacetate partly compensates for the lack of phosphocreatine in skeletal muscle of mice lacking guanidinoacetate methyltransferase. J. Physiol. 560, 219–229. doi: 10.1113/jphysiol.2004.067926
Kayacelebi, A. A., Langen, J., Weigt-Usinger, K., Chobanyan-Jürgens, K., Mariotti, F., Schneider, J. Y., et al. (2015). Biosynthesis of homoarginine (hArg) and asymmetric dimethylarginine (ADMA) from acutely and chronically administered free L-arginine in humans. Amino Acids 47, 1893–1908. doi: 10.1007/s00726-015-2012-3
Laasmaa, M., Branovets, J., Barsunova, K., Karro, N., Lygate, C. A., Birkedal, R., et al. (2021). Altered calcium handling in cardiomyocytes from arginine-glycine amidinotransferase-knockout mice is rescued by creatine. Am. J. Physiol. Heart Circ. Physiol. 320, H805–H825. doi: 10.1152/ajpheart.00300.2020
Lourenco, A. P., Fontoura, D., Henriques-Coelho, T., and Leite-Moreira, A. F. (2012). Current pathophysiological concepts and management of pulmonary hypertension. Int. J. Cardiol. 155, 350–361. doi: 10.1016/j.ijcard.2011.05.066
Lygate, C. A., Aksentijevic, D., Dawson, D., Hove, M. T., Phillips, D., Bono, J. D., et al. (2013). Living without creatine unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ. Res. 112, 945–955. doi: 10.1161/CIRCRESAHA.112.300725
Moali, C., Boucher, J. L., Sari, M. A., Stuehr, D. J., and Mansuy, D. (1998). Substrate specificity of NO synthases: detailed comparison of L-arginine, homo-L-arginine, their N omega-hydroxy derivatives, and N omega-hydroxynor-L-arginine. Biochemistry 37, 10453–10460. doi: 10.1021/bi980742t
Nabuurs, C. I., Choe, C. U., Veltien, A., Kan, H. E., van Loon, L. J., Rodenburg, R. J., et al. (2013). Disturbed energy metabolism and muscular dystrophy caused by pure creatine deficiency are reversible by creatine intake. J. Physiol. 591, 571–592. doi: 10.1113/jphysiol.2012.241760
Neubauer, S. (2007). The failing heart–an engine out of fuel. N. Engl. J. Med. 356, 1140–1151. doi: 10.1056/NEJMra063052
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Smith, K. A., Waypa, G. B., Dudley, V. J., Budinger, G. R. S., Abdala-Valencia, H., Bartom, E., et al. (2020). Role of hypoxia-inducible factors in regulating right ventricular function and remodeling during chronic hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 63, 652–664. doi: 10.1165/rcmb.2020-0023OC
Stockebrand, M., Sasani, A., Das, D., Hornig, S., Hermans-Borgmeyer, I., Lake, H. A., et al. (2018). A mouse model of creatine transporter deficiency reveals impaired motor function and muscle energy metabolism. Front. Physiol. 9:773. doi: 10.3389/fphys.2018.00773
Zacchigna, S., Paldino, A., Falcão-Pires, I., Daskalopoulos, E. P., Dal Ferro, M., Vodret, S., et al. (2021). Towards standardization of echocardiography for the evaluation of left ventricular function in adult rodents: a position paper of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 117, 43–59. doi: 10.1093/cvr/cvaa110
Keywords: creatine, homoarginine, hypoxia, L-arginine:glycine amidinotransferase, pulmonary hypertension
Citation: Hannemann J, Cordts K, Seniuk A, Choe C-u, Schmidt-Hutten L, Duque Escobar J, Weinberger F, Böger R and Schwedhelm E (2021) Arginine:Glycine Amidinotransferase Is Essential for Creatine Supply in Mice During Chronic Hypoxia. Front. Physiol. 12:703069. doi: 10.3389/fphys.2021.703069
Edited by:
Rodrigo Iturriaga, Pontificia Universidad Católica de Chile, ChileReviewed by:
Roberto V. Reyes, University of Chile, ChileCopyright © 2021 Hannemann, Cordts, Seniuk, Choe, Schmidt-Hutten, Duque Escobar, Weinberger, Böger and Schwedhelm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edzard Schwedhelm, c2Nod2VkaGVsbUB1a2UuZGU=
†Present address: Kathrin Cordts, Institute of Forensic Medicine, University Medical Center Schleswig-Holstein, Kiel, Germany
‡ORCID: Jorge Duque Escobar, orcid.org/0000-0002-6215-1555
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.