 Gustavo Jose Justo da Silva
Gustavo Jose Justo da Silva Raffaele Altara
Raffaele Altara George W. Booz
George W. Booz Alessandro Cataliotti
Alessandro Cataliotti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 08 July 2021
Sec. Clinical and Translational Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.691407
This article is part of the Research Topic Natriuretic Peptides in Cardiovascular Pathophysiology View all 12 articles
The characterization of the cardiac hormone atrial natriuretic peptide (ANP99–126), synthesized and secreted predominantly by atrial myocytes under stimulation by mechanical stretch, has established the heart as an endocrine organ with potent natriuretic, diuretic, and vasodilating actions. Three additional distinct polypeptides resulting from proteolytic cleavage of proANP have been identified in the circulation in humans. The mid-sequence proANP fragment 31–67 (also known as proANP31–67) has unique potent and prolonged diuretic and natriuretic properties. In this review, we report the main effects of this circulating hormone in different tissues and organs, and its mechanisms of actions. We further highlight recent evidence on the cardiorenal protective actions of chronic supplementation of synthetic proANP31–67 in preclinical models of cardiorenal disease. Finally, we evaluate the use of proANP31–67 as a new therapeutic strategy to repair end-organ damage secondary to hypertension, diabetes mellitus, renal diseases, obesity, heart failure, and other morbidities that can lead to impaired cardiac function and structure.
The human natriuretic peptides (NPs) consist of a family of three known peptides encoded in the human genome, with each being a distinct gene product with similar structure (Figure 1). The atrial natriuretic peptide (ANP99–126), a hormone synthesized and secreted predominantly by cardiac cells, was the first member of the NP family to be discovered in de Bold et al. (1981), and established the heart as an endocrine organ. Wall stretch, due to increased intravascular volume and/or cardiac transmural pressure, is the major stimulus for cardiac ANP release (Goetze et al., 2020). ANP is encoded by the NPPA gene located on chromosome 1 in the human genome, and is primarily expressed by atrial myocytes. The NPPA gene translates a 151-amino acid polypeptide known as preproANP. A post-translational modification process cleaves the 25 amino acid signal sequence to produce proANP, a 126 amino acid peptide that is stored in intracellular granules of atrial myocytes (Yan et al., 2000). Under stimulation, atrial cells release proANP that is rapidly converted to the 28-amino-acid C-terminal mature ANP by the transmembrane serine protease corin (Yan et al., 2000), a transmembrane cardiac serine protease, to form the biologically active carboxyl-terminal 28-amino-acid peptide called ANP99–126 (Forssmann et al., 1998). The 28-amino acid peptide contains a 17-amino acid ring in the center of the molecule (Figure 1), formed by a disulfide bond between two cysteine residues at positions 7 and 23. The highly biologically active ANP99–126 is formed at equimolar amounts as the biologically inactive amino-terminal portion (98 amino acid) of proANP (termed NT-proANP, or proANP1–98) (Yan et al., 2000). However, as ANP99–126 has a very short half-life (less than 5 min) compared with NT-proANP (60–120 min), NT-proANP is considered a more reliable biomarker than ANP99–126 (Buckley et al., 1999). Originally, the ring structure was thought to be essential for the ANP99–126 biological actions (Currie et al., 1984), but linear forms of the N-terminal ANP prohormone containing internal sequences believed to account for activity without the ring structure were shown also to have biological activity, albeit significantly reduced (Brenner et al., 1990; Vesely, 2007). The B-type natriuretic peptide (BNP), also known as brain natriuretic peptide, is a hormone secreted primarily by cardiomyocytes in the heart atria and ventricles (Sudoh et al., 1988) in response to stretching caused by increased ventricular blood volume and increased filling pressure (de Lemos et al., 2003). While the main source of BNP in normal conditions is the atrium, the production of BNP from the ventricles increases under pathological conditions such as cardiac remodeling (Luchner et al., 1998). Under stimulation, a 32-amino acid polypeptide is secreted attached to a 76-amino acid N-terminal fragment in the prohormone called NT-proBNP. A specific convertase (furin or corin) subsequently cleaves proBNP between arginine-102 and serine-103 into NT-proBNP and the biologically active 32-amino acid polypeptide BNP 1–32. Last, the C-type natriuretic peptide (CNP), encoded by the gene NPPC located on human chromosome 2, is synthesized and secreted from the central nervous system (e.g., cerebellum, hypothalamus, and anterior pituitary), kidney, and vascular endothelial cells (Mukoyama et al., 1991; Heublein et al., 1992; Stingo et al., 1992; Suga et al., 1992), and by the heart (Vollmar et al., 1993; Del Ry et al., 2011; Sangaralingham et al., 2020), in response to shear stress and certain proinflammatory cytokines. CNP is structurally related to ANP and BNP molecules, but has less intensive natriuretic and diuretic effects (Goetze et al., 2020). A recent study revealed that CNP regulates distal arteriolar and capillary blood flow via NPR-B-induced cGMP signaling in microvascular smooth muscle cells and pericytes (Spiranec et al., 2018), controlling microvascular resistance and blood pressure through vasodilating actions (Sangaralingham and Burnett, 2018). Other NPs have been identified in nature. The Dendroaspis natriuretic peptide (DNP), is structurally similar to ANP, BNP, and CNP, and possesses comparable biologic properties to other NPs. Additionally, the NP urodilatin (URO or CDD/ANP 95–126), known as renal ANP among the NPs, is secreted by cells of the distal tubule and collecting duct in the kidney in response to increased blood pressure and blood volume. Urodilatin is transcribed by the NPPA gene, but differentially processed in the kidney, and only detected in urine (Meyer et al., 1998). Both DNP and Urodilatin bind to NPR-A resulting in a cGMP-dependent signal transduction (Pandey, 2014).
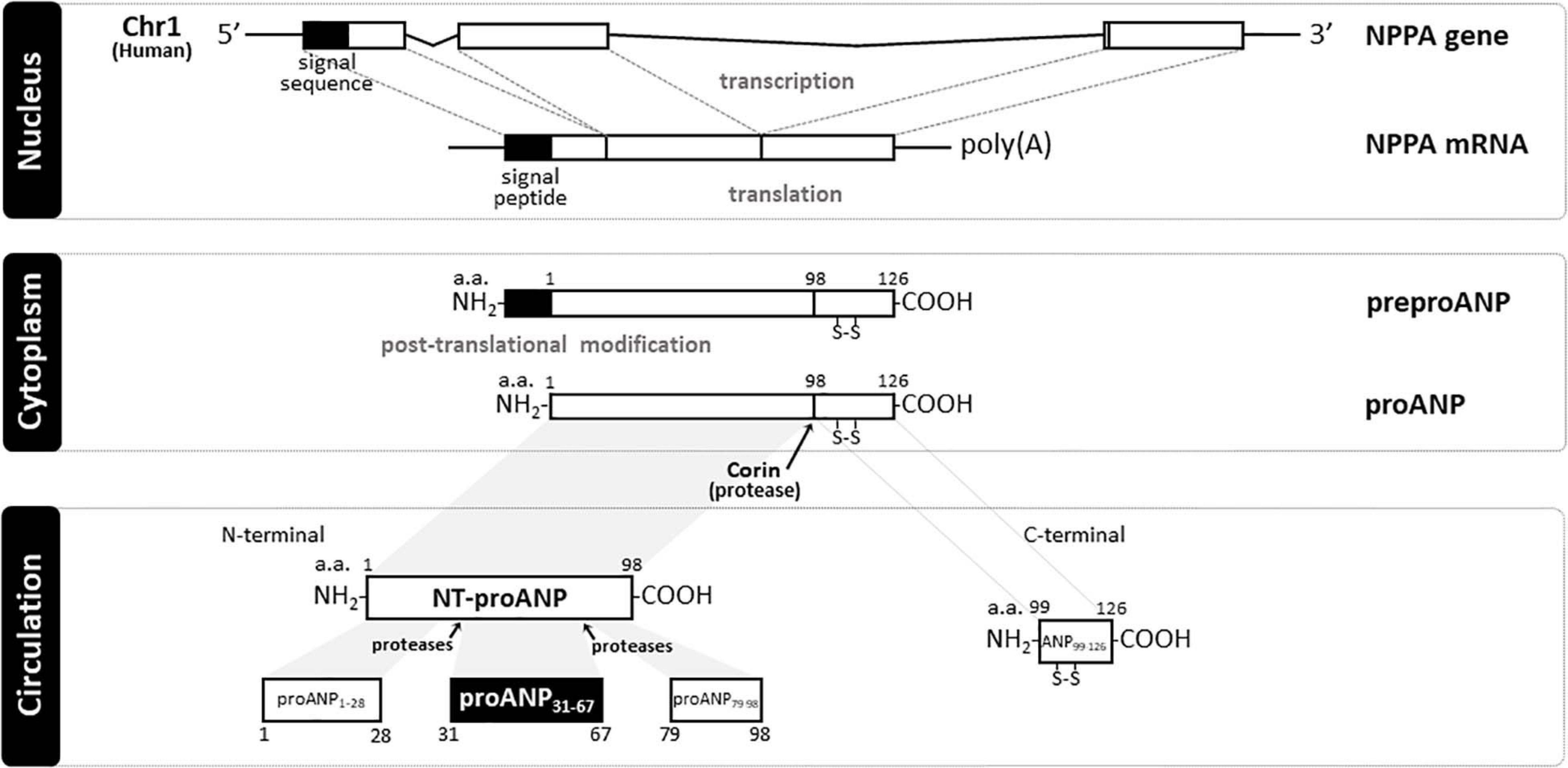
Figure 1. Schematic representation of natriuretic peptide gene product processing and generated peptide fragments. In humans, atrial natriuretic peptide (ANP) is encoded by NPPA gene (Chr1:11,845,709-11,848,345:-) that translates a 151 a.a. polypeptide (preproANP). Post-translational modification process cleavages the 25 a.a. signal sequence to produce proANP, a 126 a.a. peptide that is stored in intracellular granules of atrial myocytes. Under stimulation, atrial cells release proANP that is rapidly converted to the 28-a.a. C-terminal mature ANP by the transmembrane serine protease Corin, and a 98 a.a. N-terminal mature ANP (NT-proANP or ANP1–98). Further cleavage by proteases of NT-proANP generates different fragments, i.e., amino acids 1–28 (proANPl–28), amino acids 31–67 (proANP31–67), and amino acids 79–98 (proANP78–98). NH2, free amine group located at the N-terminal end of a polypeptide; COOH, free carboxyl group located at the C-terminal end of a polypeptide; poly(A), multiple adenosine monophosphates mRNA tail; a.a., amino acid; Chr 1, Chromosome 1; NPPA, human atrial natriuretic peptide gene; ANP, Atrial natriuretic peptide; NT-proANP, N-terminal proatrial natriuretic peptide; preproANP, precursors to prohormone of atrial natriuretic peptide; proANP, prohormone of atrial natriuretic peptide.
Biological actions of NPs are mediated by membrane-bound guanylyl cyclase receptors, which are expressed in a variety of cells. Three NP receptors are known: NPR-A (or NPR1), NPR-B (or NPR2), and NPR-C (or NPR3). ANP and BNP bind primarily to NPR-A and CNP binds to NPR-B (Charles et al., 1996; Kuhn, 2004). When activated, NPR-A and NPR-B receptors generate the second messenger cyclic guanosine monophosphate (cGMP), while the activation of NPR-C does not generate cGMP. In cardiac myocytes, cGMP-mediated signaling is regulated in a spatial and temporal manner by specific phosphodiesterases, which act to localize and temper levels of this signaling second messenger (Dunkerly-Eyring and Kass, 2020). All three NPs are ligands with similar affinity (Anand-Srivastava and Trachte, 1993) to the receptor NPR-C, which is not a guanylyl cyclase-linked receptor (Kuhn, 2004). It is known that NPR-C couples to inhibitory G proteins (Gi) and causes inhibition of adenylyl cyclase and activation of phospholipase-C (Anand-Srivastava, 2005).
A comprehensive biological understanding of NPs emerged following studies in cultured cells, rodent models of altered NPs production or receptor function, and integrative physiologic studies in disease models and in humans. The biological properties of the NPs, which include natriuresis, vasodilatation, inhibition of the renin-angiotensin-aldosterone system (RAAS), positive lusitropy, and inhibition of fibrosis, have led to the unique concept of cardiorenal protection by activation of cGMP (de Bold et al., 1981; Burnett et al., 1984; Wada et al., 1994; Kishimoto et al., 1996; Stevens et al., 1996; Wright et al., 1996; Lainchbury et al., 2000). An accumulating body of evidence demonstrated the tissue-specific distribution of NPs (Saito et al., 1989; Kojima et al., 1990; Ogawa et al., 1990; Mukoyama et al., 1991) and their receptors (Fuller et al., 1988; Martin et al., 1989; Schulz et al., 1989). Additionally, proANP1–98 can break down into multiple peptides (Figure 1), i.e., amino acids 1–28 (proANPl–28), amino acids 31–67 (proANP31–67), and amino acids 79–98 (proANP78–98), which also have potent vasodilatory properties (Vesely et al., 1987). Interestingly, these proANP forms have been identified in the circulation in humans (Vesely, 1995). For instance, by using high performance-gel permeation chromatography (HPGPC) and radioimmunoassay (RAI), Vesely (1995) was able to demonstrate that proANP1–98 is further cleaved by proteases to generate these proANP fragments in the circulation. Among these forms, proANP31–67 has unique potent and prolonged diuretic and natriuretic properties (Gunning et al., 1992) and will be the main form described in the current review.
Here, we review in detail, the actions of the linear fragment proANP31–67, in particular on the heart, kidneys, and metabolism, which are independent of cGMP production. Originally, Gower et al. (1994) demonstrated the presence of different circulating molecular forms of the N-terminal and the C-terminal ANP prohormone peptides in plasma and their metabolites excreted in urine. These authors subjected plasma and urine samples from humans to high performance gel permeation chromatography (HP-GPC), followed by radioimmunoassay assessment of all ANP fragments, to reveal that proANP31–67 and ANP circulate as distinct peptides (Gower et al., 1994). Interestingly, the proANP31–67 levels in the circulation were found to be 10–20-fold higher than ANP99–126 in normal humans (Winters et al., 1989; Hartter et al., 2000) and dogs (Habibullah et al., 1995). This is explained by differences in the clearance rates of both peptides, i.e., it takes 45 min for proANP31–67 to be removed from the body compared to a half-life of 3–5 min for ANP (Greenwald et al., 1992). Additionally, proANP31–67 appears to be resistant to degradation by endopeptidases, such as neutral endopeptidase (NEP), being excreted in the urine largely intact (less terminal 2–3 a.a.) (Gower et al., 1994; Hartter et al., 2000). This unique characteristic of this polypeptide, contributes to the prolonged renal actions, and the potential therapeutic effects of proANP31–67.
It has been shown that the upstream and C-terminus fragments of the ANP prohormone are released by central hypervolemia induced by head-out water immersion (Vesely et al., 1989) or cardiac pacing or tachycardia (Ngo et al., 1989). These ANP fragments have physiologic actions similar to the ring structured ANP form, producing vasodilation (Vesely, 1995), natriuresis (Martin et al., 1990; Villarreal et al., 1999b), diuresis (Martin et al., 1990), and affecting metabolic phenotypes (Moro, 2013). In rodents, intravenous infusion of proANP31–67 (at doses of 0, 10, 30, and 100 ng/kg/min) in anesthetized normotensive and spontaneously hypertensive rats elicited natriuresis and diuresis (Villarreal et al., 1999b). Moreover, an increase in sodium excretion was also observed in intravenously infused anesthetized Munich-Wistar (Martin et al., 1990) and Sprague-Dawley (Dietz et al., 1994) rats. Both ANP and proANP3l–67 also inhibit sodium transport in suspensions of inner medullary collecting duct cells (Zeidel et al., 1986; Gunning et al., 1992). Because both peptides inhibit sodium transport in the collecting duct, it is possible that they act in an additive fashion on these cells in the intact animal. In conscious non-human primates (Macaca fascicularis), Benjamin and Peterson showed that infusion of proANP31–67 (15 pmol. kg–l. min–l i.v.) increases renal sodium excretion, due to tubular and hemodynamic components (Benjamin and Peterson, 1995). Similarly, Vesely et al. (1994) demonstrated that intravenous infusion of proANP31–67 (100 ng/kg body weight/min, for 60 min) produced blood pressure-lowering, and diuretic and natriuretic properties in healthy individuals. Interestingly, these authors additionally showed that proANP31–67 has natriuretic properties that are significantly prolonged compared with ANP (Vesely et al., 1994).
NPs play also a key role in human metabolism (Cannone et al., 2019), thus connecting the heart with insulin−sensitive organs like adipose tissue, skeletal muscle, and liver. In fact, accumulation of NPs is associated with protein energy wasting and activation of browning in white adipose tissue (Luce et al., 2020). Importantly, ANP increases mitochondrial uncoupling and thermogenic gene expression in human adipocytes, induces thermogenic programs in brown (BAT) and white (WAT) adipose tissue and so increases energy expenditure (Bordicchia et al., 2012). These actions are considered favorable effects. However, in pathological conditions, these favorable actions are blunted or abolished. Accumulating evidence suggests that impaired cardiac endocrine function contributes to the development of obesity, type 2 diabetes, and other cardiometabolic complications (Verboven et al., 2017). The ring-structured form of ANP was reported to induce lipid mobilization and oxidation and to enhance insulin sensitivity (Coue and Moro, 2016). ANP infusion in humans increases energy expenditure and leads to lipolysis, with an increase in plasma levels of glycerol and non-esterified fatty acids regardless of body mass index (Birkenfeld et al., 2005, 2008). Also, intravenous administration of ANP increases plasma levels of adiponectin (Tsukamoto et al., 2009), an adipocyte-derived cytokine, which protects against atherosclerosis and insulin resistance or diabetes. However, the potential clinical utility of ANP might be limited by its inherent, sustained blood pressure lowering effects that can cause hypotension. Regarding the mid-sequence of the ANP, it has been shown that proANP31–67 might play an important role in the acute and chronic physiological responses to physical exercise. For instance, Cappellin et al. (2004) showed that measured proANP31–67 levels before and at the end of dynamic exercise in 28 trained cyclists and found that a single bout of exercise induce an increase in the urinary proANP31–67 levels. This could be, at least in part, explained by the increase in the venous return to the heart, and perhaps the higher heart rate levels, during a single exercise session. Interestingly, Freund et al. (1988) have demonstrated that the increase in the ANP levels occurs in a dose- and time-dependent manner. Additionally, proANP31–67 plasma concentration was also found higher in endurance trained athletes than in sedentary subjects (De Palo et al., 2000). Although protection of the vasculature, heart, and kidneys are favorable effects in the setting of metabolic diseases, the role of proANP31–67 in metabolism is unknown.
One of the hallmarks of heart failure (HF) is the marked increase in plasma levels of NPs (Burnett et al., 1986; Sugawara et al., 1988; Yamamoto et al., 1996; Cataliotti et al., 2001; Maisel et al., 2003; Richards and Troughton, 2004). It is established that elevated cardiac filling pressure is accompanied by increased circulating levels of ANP, and that congestive HF is not characterized by a deficiency in ANP, but with its elevation (Burnett et al., 1986). The increased circulating NP levels during HF are a compensatory response to volume overload and to hyperactivation of the adrenergic system and renin-angiotensin-aldosterone system (RAAS). However, not all HF patients seem to increase the circulating levels of NPs. In a recent clinical study, approximately 26% of acutely decompensated heart failure patients presented a lack of increase of circulating levels of ANP (Reginauld et al., 2019), which might suggest the existence of a relative state of ANP deficiency in a subgroup of patients, possibly due to reduced production, altered release, or enhanced enzymatic degradation by neprilysin. It should also be underscored, however, that the role of potential confounders responsible for this apparent ANP deficiency status remains yet to be fully elucidated (Richards and Januzzi, 2019). Nevertheless, as discussed herein, the increased cardiac production and circulation of NPs can be differently processed in the periphery in chronic HF patients, resulting in inactive forms with no efficient benefit, thus supporting the rational for using NPs or their analogs as anti-HF therapy (Belluardo et al., 2006; Macheret et al., 2012). Others and we have previously demonstrated the existence of a deficiency state of the endogenous biologically active NPs system in HF patients starting with the early stage of HF (Hawkridge et al., 2005; Belluardo et al., 2006; Niederkofler et al., 2008; Macheret et al., 2012). Additionally, a blunted natriuretic response has been observed after treatment with different pharmacological agents (e.g., angiotensin-converting enzyme inhibitors, angiotensin-II blockers, β-blockers, and spironolactone) in experimental models and in patients with chronic heart failure, suggesting a resistance to the biological effects of NPs (Cody et al., 1986; Saito et al., 1987; Komeichi et al., 1995; Charloux et al., 2003). This resistance to biological effects of ANP is probably mainly due to up-regulation of clearance receptors in patients with chronic heart failure (Andreassi et al., 2001; Clerico et al., 2006).
Winters et al. (1989) have evaluated the N-terminus and C-terminus ANP fragments in the circulation of thirty patients with varying severity degrees of congestive HF using high-pressure liquid chromatography. Compared to the other ANP peptides, proANP31–67 was the only one that accurately discriminated the severity of congestive HF (Winters et al., 1989). In light of these findings, the impaired production and release of mature forms of the NPs and of their linear precursors seems to play a fundamental role in the evolution and progression of HF, and thus the exogenous supplementation of such cardiac hormones may prove to be of therapeutic importance in HF. In fact, the biologic properties of the NPs have supported the development of as therapeutic agents for cardiovascular diseases (Marcus et al., 1996; Yamamoto et al., 1997; Colucci et al., 2000; Hobbs et al., 2001; Boerrigter and Burnett, 2004; Rubattu et al., 2019; Rubattu and Volpe, 2019). Here we will further discuss the development of novel therapeutic strategies based on exogenous supplementation of a linear fragment of the ANP, the proANP31–67, in HF.
The effort to develop novel therapeutic strategies to prevent the progression of cardiovascular disease is also focused on restoring the impaired NP system, for instance, by augmenting the circulating levels of NPs through exogenous supplementation. For instance, the NP drugs carperitide and nesiritide have been approved for use in patients in Japan and United States, respectively, as intravenous agents for the treatment of acute decompensated HF. These forms stimulate the production of cGMP, and frequently result in inadequate cardioprotective effects due to significant reductions in blood pressure levels, leading to reduced renal perfusion and further deterioration of kidney function.
ProANP31–67 was shown to enhance renal function acutely in persons with congestive heart failure (Vesely et al., 2001) and to protect against ischemia-induced acute tubular necrosis and renal failure in a rat model of ischemic non-oliguric acute renal failure (Clark et al., 2000). Overall, proANP31–67 induced renal vasodilation and diuresis with enhanced sodium excretion, but with no associated increase in oxygen consumption. Of note, as mentioned above, the biological actions of proANP31–67 were independent of cGMP activation and therefore, are characterized by a less intense vasodilatory effect (Vesely et al., 1987).
Potential clinical indications for proANP31–67 include reducing symptoms in patients with worsening HF or those diagnosed with stable congestive HF and compromised renal function, or cardiorenal syndrome. Clinical trials showing safety and efficacy of synthetic proANP31–67 peptide were conducted on stable congestive HF and renal impairment patients (ACTRN12612000576820 and ACTRN12611000806965), and on acute decompensated congestive HF patients (ACTRN12609000998246). Intravenous and subcutaneous delivery of proANP31–67 was shown to preserve renal function in both chronic and acute heart failure with reduced ejection fraction (HFrEF) (Delacroix et al., 2016). Additionally, the infusion of proANP31–67 (100 ng/kg/min, i.v. for 1 h) has been shown to possess several cardiac enhancing effects in congestive HF patients (NYHA III), including augmenting cardiac output, cardiac index, and stroke volume index, while reducing pulmonary capillary wedge pressure (Vesely et al., 1998). Of note, proANP31–67 has similar effects to those observed with the ring forms of the NPs, which are currently in use for the treatment of acute decompensated overt HF, but has shown a less intense blood pressure lowering effect.
Based on these observations and on the known unique renal protective effects of proANP31–67, we investigated the therapeutic value of proANP31–67 for maladaptive cardiac and renal remodeling in a rat experimental model of salt-induced hypertension (Altara et al., 2020). This is a preclinical model for heart failure with preserved ejection fraction (HFpEF), as evidenced by concentric remodeling/hypertrophy and diastolic dysfunction, i.e., increased cardiac stiffness. We also sought to extend current knowledge on the protective actions of chronic exogenous supplementation of proANP31–67 on the kidney, knowing that it stimulates natriuresis and diuresis, but has moderate effect on blood pressure. With hypertension in this preclinical model, we observed that proANP31–67 increased urine output, natriuresis, and glomerular filtration rate (GFR), while preventing detrimental perivascular collagen deposition in the renal cortex. Remarkably, proANP31–67 was shown to be beneficial to the heart. Characteristic signs of adverse cardiac remodeling and function that manifested as diastolic dysfunction were attenuated with chronic administration of proANP31–67. These beneficial actions on the heart, included attenuated cardiac hypertrophy, as indicated by decreased heart weight to body weight ratio and left atrial diameter, as well as reduced fibrosis (both interstitial and perivascular left ventricular fibrosis) and normalized ratio of the diastolic mitral inflow E wave to A wave, a measure of cardiac stiffness (Altara et al., 2020). Of note, the beneficial effects on the heart were retained absent of a marked lowering of blood pressure and when animals were treated with a renal sub-therapeutic dose of proANP31–67, suggesting a unique mode of action directly on the heart, beyond its renal actions, which warrants further investigation.
The mechanisms of action of proANP31–67 associated with the cardiorenal protective and diuretic effects were not attributed to any effects on blood pressure. Competitive binding experiments revealed that proANP31–67 does not activate the canonical NP receptors (e.g., NPR-A and NPR-B), resulting in the activation of the cGMP pathway but rather has its own separate and distinct receptor (Vesely et al., 1990, 1992). However, the nature of the proANP31–67 receptor is still unknown.
With regard to its mechanism of action in the kidney, several studies have shown that proANP31–67 endogenously (i.e., paracrine like) induces prostaglandin E2 (PGE2) formation (Gunning et al., 1992; Vesely et al., 2000). PGE2, a product of the cyclooxygenase 2 (COX-2) pathway (Figure 2), is an important homeostatic regulator of nephropathy, as well as hypertension, adipogenesis, dyslipidemia, diabetes, neuropathy, atherogenesis and retinopathy, contributing to global cardiovascular risk (Nasrallah et al., 2016). PGE2 was also reported to modulate growth, fibrosis, and apoptosis phenotypes by influencing inflammatory, immune, and oxidative stress responses (Makino et al., 2002; Zahner et al., 2009; Nasrallah et al., 2015). Felder et al. (2017) reported that proANP31–67 directly stimulated PGE2 release in the renal medullary tissue, more precisely by cells located in the collecting tubules/ducts at the cortical-medullary interface. Through PGE2, proANP31–67 is also a potent inhibitor of the Na+-K+-ATPase (or sodium-potassium pump) of inner medullary collecting duct cells, resulting in Na+ transport inhibition and natriuretic actions. Furthermore, proANP31–67 was shown to increase GFR, both in preclinical models as well as in humans with congestive HF, and to attenuate tubular necrosis in a rat model of acute renal failure (Afsar et al., 2017). Intrarenal administration of proANP31–67 also increases creatinine clearance and inhibits renin secretion in a Na+ depleted canine model of renin system activation induced by unilateral nephrectomy (Villarreal et al., 1999a). These data suggest that inhibition of renin secretion is, at least in part, in response to a proANP31–67-induced increase in the sodium load delivered to the macula densa.
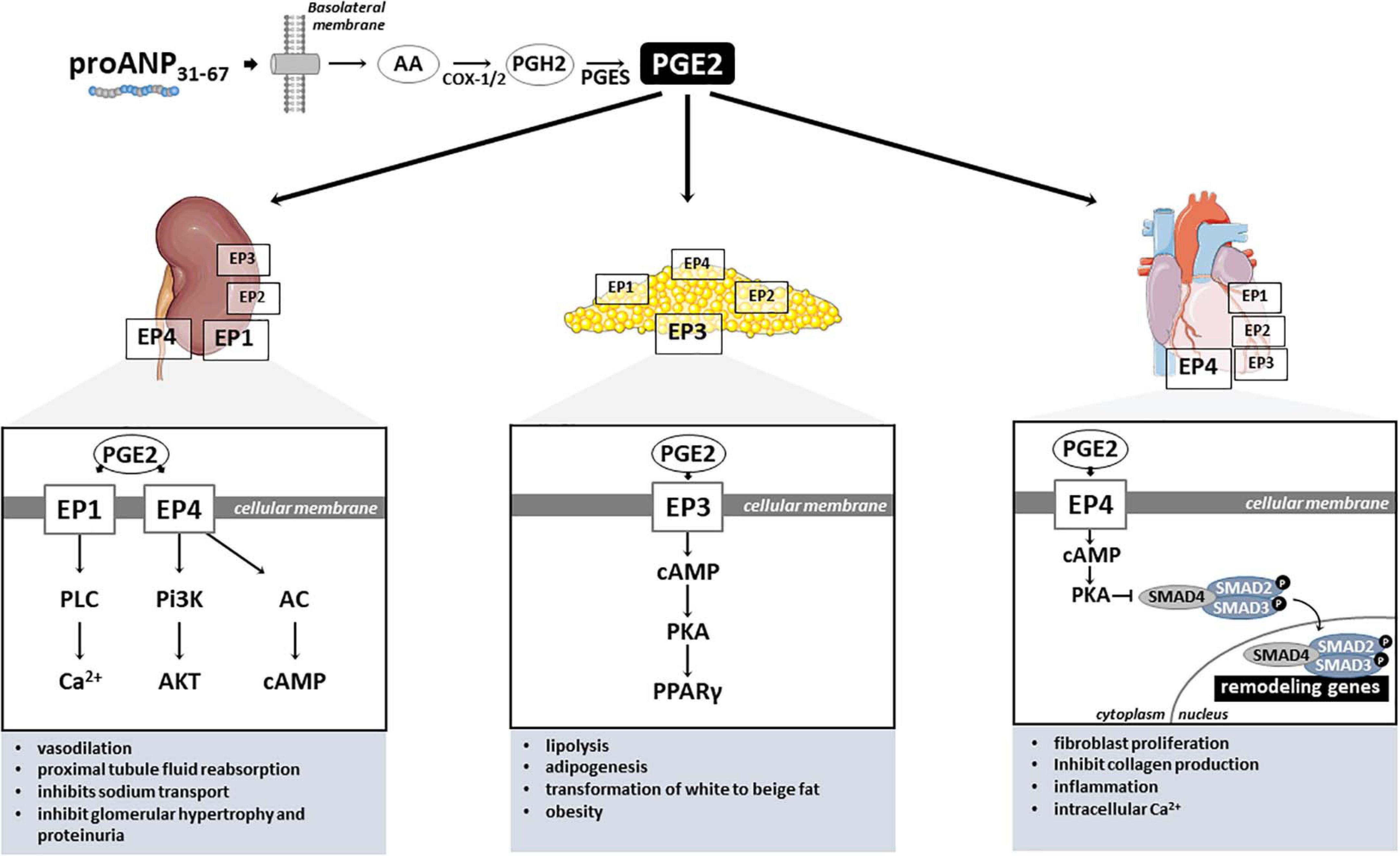
Figure 2. PGE2-mediated proANP31–67 cardiac, renal and metabolic mechanisms of action. PGE2, Prostaglandin E2; EP1–4, PGE2 receptors (coupled to Gq); AA, Arachidonic acid; COX-1/2, Cyclooxygenase-1 and –2; PGH2, Prostaglandin H2; PGES, Prostaglandin E synthase; PLC, Phospholipase C; Pi3K, Phosphoinositide 3-kinases; AC, Adenylate cyclase; Ca2+, Calcium ion; AKT, protein kinase B; cAMP, Adenosine 3′,5′-cyclic monophosphate; PKA, protein kinase A; SMAD2–4, SMAD (Mothers against decapentaplegic) family member 2, 3, and 4; PPARγ, Peroxisome proliferator-activated receptor gamma.
In mammals, PGE2 exerts its signals through four G protein-coupled receptors, designated EP1, EP2, EP3, and EP4 (Figure 2; Sugimoto and Narumiya, 2007). Although highly conserved among mammals, the PGE2 receptors have distinct signal transduction pathways, and tissue and cellular distribution, reflecting their diverse properties (Nasrallah et al., 2016). In the kidney, EP1 and EP4 receptors seem to mediate PGE2 microcirculation actions (Figure 2). Purdy and Arendshorst (2000) identified by RT-PCR the expression of EP1 (Ptger1) and EP4 (Ptger4) receptors in freshly isolated preglomerular arterioles of Sprague-Dawley rats. These authors also demonstrate that the EP4 receptor is the major receptor located in preglomerular vascular smooth muscle cells, mediating PGE2-induced vasodilation through cAMP formation and reduction of cytosolic Ca2+ levels (Purdy and Arendshorst, 2000). Curiously, the renal vascular tone response induced by PGE2 stimulation seems to vary depending on the type of the receptor (Schweda et al., 2004). For instance, EP2–/– and EP4–/– mice presented an augmented vasoconstriction in response to higher PGE2 concentrations, contrasting with the markedly blunted response observed in EP1 and EP3 knockout mice (Schweda et al., 2004). Furthermore, EP1 and EP4 were detected in transformed murine proximal tubular cells (MCTs), mediating PGE2-induce fluid reabsorption (Nasrallah et al., 2015). Loss-of-function in vivo experiments in mice have shown that PGE2 stimulates the renin-angiotensin-aldosterone system by activation of EP4 receptor (Poschke et al., 2012). Similarly, Schweda et al. (2004) demonstrated that PGE2 stimulates renin release in juxtaglomerular cells via activation of both EP2 and EP4 receptors. Interestingly, the EP1 receptor attenuates vasopressin-dependent water reabsorption and inhibits sodium transport in the collecting duct (Nasrallah et al., 2018). Activation of PGE2-EP4 signaling with proANP31–67 also can exert multiple biochemical effects on the kidney and other organs, suggesting the potential wide-ranging use of EP4 in both cardiovascular and metabolic disorders. For instance, by inhibiting Na+ transport in the inner medullary collecting duct cells, proANP31–67 is known to reduce renal oxygen consumption (Gunning et al., 1992).
Under different pathological conditions, the PGE2 receptors seem to be involved in the development of renal disease. For instance, the oral administration of PGE2 receptor EP1-selective antagonist prevented the progression of nephropathy, evidenced by improved glomerular hypertrophy, decreased mesangial expansion, and suppression of proteinuria in streptozotocin-induced diabetic rats (Makino et al., 2002). Mechanistically, the authors demonstrated that mesangial cells cultured under high-glucose conditions and treated with this selective agonist for EP1 receptor exhibit inhibited transforming growth factor-beta (TGF-β) and fibronectin upregulation, key regulators of the extracellular matrix. Similarly, an EP4-specific agonist significantly attenuated the development of tubulointerstitial fibrosis induced by unilateral ureteral obstruction in mice by suppressing inflammatory responses (Nakagawa et al., 2012). On the other hand, knockout mice for EP4 showed exacerbated tubulointerstitial fibrosis response after ureteral obstruction. Additionally, cultured renal fibroblasts treated with EP4 agonist significantly inhibited the platelet-derived growth factor (PDGF)-induced proliferation and profibrotic connective tissue growth factor production (Nakagawa et al., 2012). Hence, these data indicate that both PGE2 receptors EP1 and EP4 play critical roles in the development of renal injury (Figure 2), and might explain the renal protective benefits of proANP31–67 observed by our group in hypertensive rats. For instance, chronic administration of proANP31–67 prevented perivascular collagen deposition in the rat experimental model of salt-induced hypertension, accompanied by improvements in renal function (Altara et al., 2020).
Cardiac phenotypes are equally affected by the different PGE2 receptors (Figure 2). Although all four PGE2 receptors are detected in the cardiac tissue, EP4 is highly expressed in the heart (Muraoka et al., 2019), and seems to have protective effects against adverse remodeling. In fact, EP4 agonist administration to mice subjected to pressure overload (Wang et al., 2017) and cardiac injury (Hishikari et al., 2009; Pang et al., 2016) exhibited antifibrotic effects and prevented the progression to systolic dysfunction. In a mouse model of cardiac hypertrophy generated by transverse aortic constriction (TAC) surgical procedure, EP4 agonist ONO-0260164 treatment significantly prevented myocardial fibrosis and progression of systolic dysfunction 5 weeks after pressure overload (Wang et al., 2017). Hishikari et al. (2009) used another EP4 selective agonist (EP4RAG) to treat rats submitted to myocardial ischemia-reperfusion injury and demonstrated that EP4RAG significantly reduced ischemic myocardium, attenuated interstitial fibrosis, and ameliorated cardiac contractility and dilatation compared with vehicle.
The generation of genetically engineered animals has contributed with the understanding of the role of PGE2 receptors in the cardiac tissue. Qian et al. (2008) generated cardiac specific EP4 deficiency, using site-specific recombination by the Cre recombinase method (Cre-loxP) to inactivate EP4 only in cardiomyocytes (CM- EP4 knockout [KO]), and showed that CM-EP4 KO mice are defective in their ability to activate Stat-3, presenting a worsening of systolic function after myocardial infarction injury. These studies are interpreted as indicating that EP4 plays both protective and damaging roles in the heart with the protective effects of EP4 due at least in part to its ability to suppress inflammation.
We cannot exclude, however, the role of PGE2 stimulation of its receptors in cells other than cardiomyocytes, for instance cardiac fibroblasts, endothelial cells, and smooth muscle cells. In fact, it has been demonstrated recently that EP4 signal also regulates fibrotic phenotypes in cardiac fibroblasts. In this regards, Umemura et al. (2019) showed that cardiac fibroblasts isolated from adult rats treated with EP4 agonist (ONO-AE1-437) decreased the expression of transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF) and ACTA2 (a-smooth muscle actin) mRNA, suggesting that that EP4 signaling suppresses fibroblasts to myofibroblast transdifferentiation. Consistently, Wang et al. (2017) demonstrated in cultured neonatal rat cardiac fibroblasts that treatment with EP4 agonist ONO-0260164 inhibited the TGF-β1 induced upregulation of collagen type 1 (Col1a1) and type 3 (Col3a1) gene expression.
Mechanistically, EP4 is a G protein-coupled receptor with seven transmembrane domains that when bound to PGE2 or another agonists, mobilizes G proteins containing the Gs alpha subunit (i.e., Gαs) and G beta-gamma (i.e., Gβγ) (Tuteja, 2009). In particular, Gsα stimulates adenylyl cyclase to raise the production of cyclic adenosine monophosphate (cAMP) (Yokoyama et al., 2013), that subsequently activates protein kinase A (PKA), which in turn phosphorylates downstream proteins, such as the transcription factor cAMP response element binding protein (CREB). Of note, CREB regulates the expression of genes that control cellular proliferation, cellular differentiation, cellular survival, and angiogenesis. The activated CREB(p) binds to specific sites and regulates the expression of genes, such as B-cell lymphoma 2 and tumor necrosis factor α (TNFα), which are involved in development of ischemic heart disease (Ichiki, 2006). EP4 activation of G proteins also triggers PI3K/AKT/mTOR, ERK, and p38 MAPK pathways (Xu et al., 2018). Regarding the other PGE2 receptors expressed in the cardiac tissue, it is known that PGE2 stimulates cardiac fibroblast proliferation via both EP1 and EP3, p42/44 MAPK and Akt-regulation of cyclin D3, possibly modulating cardiac fibrosis (Harding and LaPointe, 2011).
As mentioned, in addition to its renoprotective effects, proANP31–67 inhibited cardiac hypertrophy and early onset of diastolic dysfunction in our salt-induced hypertension model of HFpEF, as indicated by reduced cardiac fibrosis (Altara et al., 2020). The cardioprotective actions of proANP31–67 may have resulted from a local increase in PGE2 and activation of the EP4, which recently has been demonstrated to have antifibrotic and antihypertrophic actions in the heart (Yamagami et al., 2015; Harada et al., 2017; Wang et al., 2017; Bryson et al., 2018; Lai et al., 2018; Zhu et al., 2019; Jin et al., 2020). ProANP31–67 seems to activate the PGE2-EP4-SMAD signaling pathway, reducing the phosphorylation of SMAD2 (Altara et al., 2020), possibly inhibiting the activation in TGF-β1 mediated collagen deposition. Evidence indicates that EP4 attenuates cardiac fibrosis by inhibiting SMAD signaling through activation of protein kinase A (PKA) (Harada et al., 2017; Wang et al., 2017). In our study, urine levels of PGE2 were elevated by proANP31–67, although we did not observe a significant increase in plasma PGE2. However, local PGE2 production in the heart, where levels tended to be increased by treatment with proANP31–67, may have been responsible for the inhibition of cardiac remodeling process observed in our study (Altara et al., 2020), Therefore, the cardioprotective actions of proANP31–67 observed in our study (e.g., improved diastolic function, attenuated cardiac fibrosis and hypertrophy, and anti-remodeling effect on cardiomyocytes) (Altara et al., 2020) may have resulted from the activation of the EP4.
However, this response might be dependent on the cardiac cell type involved. In fact, proANP31–67 may have direct effects on the ultrastructure of cardiomyocytes. We have demonstrated that chronic administration of proANP31–67 reduced t-tubule density in our rat model of hypertensive heart disease and renal damage (Altara et al., 2020). Normal ultrastructure of cardiac t-tubules is important in electrical-mechanical coupling and Ca2+ handling in cardiomyocytes as any abnormalities may predispose toward heart failure (Manfra et al., 2017). In HF patients, we have demonstrated etiology-dependent differences in mechanisms for diastolic dysfunction (Frisk et al., 2021). For instance, myocardial biopsies from HFrEF hearts under high ventricular wall stress were linked to disruption of t-tubules, local collagen deposition and of systolic calcium homeostasis impairment. In contrast, maintained wall stress in HFpEF patients was associated with compensatory t-tubule proliferation and largely maintained calcium release (Frisk et al., 2021). In keeping with this, we observed that proANP31–67 treatment protects t-tubular structure and density, and also preserves intracellular distances between t-tubules and the sarcolemmal membrane. In our study, we did not examine the effect of proANP31–67 on Ca2+ dynamics in cardiomyocytes, which remains an area for future investigation.
Taken together, our study previously discussed supports the hypothesis that proANP31–67 cardioprotective benefits might directly affect both cardiac fibroblasts and cardiomyocytes, via the activation of PGE2/EP4 signaling. We cannot exclude the possibility that proANP31–67 mediates the secretion of growth factors by myofibroblasts indirectly induces hypertrophy of cardiomyocytes via a paracrine like-manner, which is a landmark of heart failure. Therefore, the detailed investigation of the proANP31–67 molecular mechanisms involving anti-fibrotic signaling pathways and cellular processes, including inflammation, signaling kinases, apoptosis, fibroblast-to-myofibroblast differentiation, cardiomyocytes ultrastructure is absolutely crucial to understand the cardiorenal protective actions of this compound in a heart failure scenario.
With regard to the role of PGE2 mediating metabolic phenotypes, these effects seem to be mainly mediated by EP3 and EP4 receptors in the adipose tissue. Of those, the EP3 is the most widely abundant receptor in adipose tissue (Tang et al., 2015; Xu et al., 2016), and is involved in various pathophysiological processes (Cai et al., 2015). Accordingly, PGE2 receptor EP3 seems to regulate both lipolysis and adipogenesis in white adipose tissue (Strong et al., 1992; Fain et al., 2000; Xu et al., 2016), as well as adipocyte transformation of white to beige fat, protecting against obesity and metabolic disease (Garcia-Alonso et al., 2013). In fact, it has been shown that loss-of-function of EP3 in mice resulted in obese and insulin resistant phenotypes (Sanchez-Alavez et al., 2007; Ceddia et al., 2016). Mechanistically, using both pharmacological blockade and genetic disruption, Xu et al. (2016) elegantly showed that PGE2 EP3 receptor inhibits adipogenesis via the cAMP/PKA/PPARγ pathway, and blocks lipolysis mainly through the cAMP/PKA/HSL pathway in white adipose tissue. These data demonstrates that PGE2/EP3 axis is critical for lipid and glucose metabolism.
Activation of PGE2-EP4 signaling seems also to exert important role in adipose tissue and metabolic disorders. Loss-of-function mice model for EP4 submitted to high fat diet exhibited reduced body weight gain and adiposity, and shorter life span when compared with wild type (Cai et al., 2015). Additionally, EP4 deficiency induced disruption in lipid metabolism due to impaired triglyceride clearance (Cai et al., 2015). Nevertheless, it is still unknown any direct or indirect metabolic properties of proANP31–67 and future investigation is fundamental. However, there are some evidence of the possible connection of proANP31–67 and metabolic phenotypes. For instance, in inner medullary collecting duct (IMCD) cells it has been previous shown that proANP31–67 reduces O2 not by direct inhibition of mitochondrial O2 consumption, but by reducing the demand for metabolic energy of the Na+-K+-ATPase (Gunning et al., 1992). Accumulation of NPs is, in fact, associated with protein energy wasting and activation of browning in white adipose tissue (Luce et al., 2020). The incubation of primary adipose cells exposed to ANP led to a significant increase of uncoupling protein 1 content. Therefore, it is reasonable to believe that proANP31–67 might also has potential in metabolic disorders associated or not with cardiovascular diseases.
Notwithstanding the substantial advance achieved in treatment, the incidence of heart failure has not been reduced and remains the major cause of morbidity and mortality in developing and developed countries (Roger, 2013). Nevertheless, current medical procedures aim to increase survival of cardiac tissue and limit cardiac damage, whereas an effective treatment to improve and/or protect renal function, which often deteriorates after cardiac injury, is still lacking and urgently needed. More recently, the combination of NEP inhibitor (Sacubitril) and angiotensin II receptor blocker (Valsartan) (sold as Entresto) became a first-choice treatment for HFrEF patients (Volpe et al., 2015; Seferovic et al., 2019; Volpe et al., 2019), based on its superior benefits to reduce cardiovascular death, and HF symptoms and hospitalizations compared to angiotensin-converting enzyme inhibitor (ACEi) (McMurray et al., 2014). Inhibition of NEP, by decreasing NPs degradation (Zile et al., 2016), elicits hypotensive actions. Of note, Entresto presented higher proportions of hypotension and non-serious angioedema cases, but lower proportions of patients with renal impairment, hyperkalemia, and cough than ACEi (Enalapril). In this regards, proANP31–67 may provide an ideal complementary therapeutic strategy by directly targeting end-organ remodeling in the setting of HFpEF. Given the fact that proANP31–67 does not appreciably lower blood pressure, this peptide may be especially efficacious as an add-on-therapy to target end organ damage, and could be tested in other heart disease settings, including coronary heart disease, as well as aortic valve stenosis, cardiac dysfunction in the presence of metabolic disease, along with both forms of HF (e.g., HFpEF or HFrEF), and cardiorenal syndrome, for which it has been first developed. An additional beneficial action of proANP31–67 is increased urinary excretion of potassium, suggesting that this peptide might be combined with potassium sparing drugs or medications frequently used in the treatment of HF (Altara et al., 2020). An interesting area of future investigation would be the impact of proANP31–67 on the metabolic syndrome, a collection of conditions that contribute to the development of heart disease, diabetes and stroke. We anticipate a synergistic beneficial action of proANP31–67 in preventing end organ damage and attenuating metabolic dysfunction when used in combination with current therapies to ameliorate lipid and glucose metabolism. Of note, proANP31–67 pharmacokinetics and high stability also render it an interesting candidate as therapeutic agent for chronic administration. Equally important, no adverse effects, gross or microscopic pathology changes were observed when proANP31–67 was tested in various pre-clinical models (Benjamin and Peterson, 1995; Villarreal et al., 1999b; Clark et al., 2000; Vesely, 2007; Altara et al., 2020), ranging from mice to non-human primates (monkeys or Macaca fascicularis). With respect to the administration to patients, proANP31–67 proved to be well tolerated at all doses via both intravenous (IV) and subcutaneous (SQ) infusions, without the profound vasodilatory and hypotensive complications evident with CT ring agents (blood pressure was maintained with no adverse hemodynamic effects noted) (Vesely et al., 1994, 1998, 2000). Additionally, pharmacokinetic analysis with pharmacodynamic parameters showed no adverse effects on any parameters measured, including cardiac and renal performance.
Considering that unique mechanism of action, its intrinsic resistance to enzymatic degradation, and its complementary actions to other members of the NPs system, make it compelling to evaluate the effects of proANP31–67, as single therapy as well as in combination with current medications, in the treatment of cardiac diseases and metabolic syndrome. The long half-life, and its safe pharmacological profile, make proANP31–67 a promising therapeutic option for currently difficult to treat clinical conditions. Therefore, further studies aimed to demonstrate its protective effects and exploit its clinical potential are clearly warranted.
AC was the lead author and conceived the concept of the manuscript. All authors contributed to the literature review and writing of the manuscript and approved the final version.
GS was supported by the South-Eastern Norway Regional Health Authority (HSØ-RHF, Project No. 25674). RA was supported by the Olav Raagholt og Gerd Meidel Raagholts, and internal grant funding. AC was funded by the Nasjonalforeningen for folkehelsen (Project No. 13173) and the HSØ-RHF (Project Nos. 2016089 and 2019009).
AC serves as consultant for the advisory board of Madeleine Pharmaceuticals. RA and AC have applied for a patent related to the cardiac protective properties of proANP31–67 (UK # 2000945.2).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Lawrence Mahan for his valuable comments in the preparation of this article.
Afsar, B., Rossignol, P., van Heerebeek, L., Paulus, W. J., Damman, K., Heymans, S., et al. (2017). Heart failure with preserved ejection fraction: a nephrologist-directed primer. Heart Fail. Rev. 22, 765–773. doi: 10.1007/s10741-017-9619-2
Altara, R., da Silva, G. J. J., Frisk, M., Spelta, F., Zouein, F. A., Louch, W. E., et al. (2020). Cardioprotective effects of the novel compound vastiras in a preclinical model of end-organ damage. Hypertension 75, 1195–1204. doi: 10.1161/HYPERTENSIONAHA.120.14704
Anand-Srivastava, M. B. (2005). Natriuretic peptide receptor-C signaling and regulation. Peptides 26, 1044–1059. doi: 10.1016/j.peptides.2004.09.023
Anand-Srivastava, M. B., and Trachte, G. J. (1993). Atrial natriuretic factor receptors and signal transduction mechanisms. Pharmacol. Rev. 45, 455–497.
Andreassi, M. G., Del Ry, S., Palmieri, C., Clerico, A., Biagini, A., and Giannessi, D. (2001). Up-regulation of ‘clearance’ receptors in patients with chronic heart failure: a possible explanation for the resistance to biological effects of cardiac natriuretic hormones. Eur. J. Heart Fail. 3, 407–414. doi: 10.1016/s1388-9842(01)00161-1
Belluardo, P., Cataliotti, A., Bonaiuto, L., Giuffre, E., Maugeri, E., Noto, P., et al. (2006). Lack of activation of molecular forms of the BNP system in human grade 1 hypertension and relationship to cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 291, H1529–H1535. doi: 10.1152/ajpheart.00107.2006
Benjamin, B. A., and Peterson, T. V. (1995). Effect of proANF-(31–67) on sodium excretion in conscious monkeys. Am. J. Physiol. 269(6 Pt 2), R1351–R1355. doi: 10.1152/ajpregu.1995.269.6.R1351
Birkenfeld, A. L., Boschmann, M., Moro, C., Adams, F., Heusser, K., Franke, G., et al. (2005). Lipid mobilization with physiological atrial natriuretic peptide concentrations in humans. J. Clin. Endocrinol. Metab. 90, 3622–3628. doi: 10.1210/jc.2004-1953
Birkenfeld, A. L., Budziarek, P., Boschmann, M., Moro, C., Adams, F., Franke, G., et al. (2008). Atrial natriuretic peptide induces postprandial lipid oxidation in humans. Diabetes Metab. Res. Rev. 57, 3199–3204. doi: 10.2337/db08-0649
Boerrigter, G., and Burnett, J. C. Jr. (2004). Recent advances in natriuretic peptides in congestive heart failure. Expert Opin. Investig. Drugs 13, 643–652. doi: 10.1517/13543784.13.6.643
Bordicchia, M., Liu, D., Amri, E. Z., Ailhaud, G., Dessi-Fulgheri, P., Zhang, C., et al. (2012). Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 122, 1022–1036. doi: 10.1172/JCI59701
Brenner, B. M., Ballermann, B. J., Gunning, M. E., and Zeidel, M. L. (1990). Diverse biological actions of atrial natriuretic peptide. Physiol. Rev. 70, 665–699. doi: 10.1152/physrev.1990.70.3.665
Bryson, T. D., Gu, X., Khalil, R. M., Khan, S., Zhu, L., Xu, J., et al. (2018). Overexpression of prostaglandin E2 EP4 receptor improves cardiac function after myocardial infarction. J. Mol. Cell Cardiol. 118, 1–12. doi: 10.1016/j.yjmcc.2018.03.005
Buckley, M. G., Marcus, N. J., and Yacoub, M. H. (1999). Cardiac peptide stability, aprotinin and room temperature: importance for assessing cardiac function in clinical practice. Clin. Sci. 97, 689–695.
Burnett, J. C. Jr., Granger, J. P., and Opgenorth, T. J. (1984). Effects of synthetic atrial natriuretic factor on renal function and renin release. Am. J. Physiol. 247(5 Pt 2), F863–F866. doi: 10.1152/ajprenal.1984.247.5.F863
Burnett, J. C. Jr., Kao, P. C., Hu, D. C., Heser, D. W., and Heublein, D. (1986). Atrial natriuretic peptide elevation in congestive heart failure in the human. Science 231, 1145–1147. doi: 10.1126/science.2935937
Cai, Y., Ying, F., Song, E., Wang, Y., Xu, A., Vanhoutte, P. M., et al. (2015). Mice lacking prostaglandin E receptor subtype 4 manifest disrupted lipid metabolism attributable to impaired triglyceride clearance. FASEB J. 29, 4924–4936. doi: 10.1096/fj.15-274597
Cannone, V., Cabassi, A., Volpi, R., and Burnett, J. C. Jr. (2019). Atrial natriuretic peptide: a molecular target of novel therapeutic approaches to cardio-metabolic disease. Int. J. Mol. Sci. 20:3265. doi: 10.3390/ijms20133265
Cappellin, E., De Palo, E. F., Gatti, R., Solda, G., Woloszczuk, W., and Spinella, P. (2004). Effect of prolonged physical exercise on urinary proANP1-30 and proANP31-67. Clin. Chem. Lab. Med. 42, 1058–1062. doi: 10.1515/CCLM.2004.212
Cataliotti, A., Malatino, L. S., Jougasaki, M., Zoccali, C., Castellino, P., Giacone, G., et al. (2001). Circulating natriuretic peptide concentrations in patients with end-stage renal disease: role of brain natriuretic peptide as a biomarker for ventricular remodeling. Mayo Clin. Proc. 76, 1111–1119. doi: 10.4065/76.11.1111
Ceddia, R. P., Lee, D., Maulis, M. F., Carboneau, B. A., Threadgill, D. W., Poffenberger, G., et al. (2016). The PGE2 EP3 receptor regulates diet-induced adiposity in male mice. Endocrinology 157, 220–232. doi: 10.1210/en.2015-1693
Charles, C. J., Espiner, E. A., Nicholls, M. G., Richards, A. M., Yandle, T. G., Protter, A., et al. (1996). Clearance receptors and endopeptidase 24.11: equal role in natriuretic peptide metabolism in conscious sheep. Am. J. Physiol. 271(2 Pt 2), R373–R380. doi: 10.1152/ajpregu.1996.271.2.R373
Charloux, A., Piquard, F., Doutreleau, S., Brandenberger, G., and Geny, B. (2003). Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur. J. Clin. Invest. 33, 769–778. doi: 10.1046/j.1365-2362.2003.01222.x
Clark, L. C., Farghaly, H., Saba, S. R., and Vesely, D. L. (2000). Amelioration with vessel dilator of acute tubular necrosis and renal failure established for 2 days. Am. J. Physiol. Heart Circ. Physiol. 278, H1555–H1564. doi: 10.1152/ajpheart.2000.278.5.H1555
Clerico, A., Recchia, F. A., Passino, C., and Emdin, M. (2006). Cardiac endocrine function is an essential component of the homeostatic regulation network: physiological and clinical implications. Am. J. Physiol. Heart Circ. Physiol. 290, H17–H29. doi: 10.1152/ajpheart.00684.2005
Cody, R. J., Atlas, S. A., Laragh, J. H., Kubo, S. H., Covit, A. B., Ryman, K. S., et al. (1986). Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J. Clin. Invest. 78, 1362–1374. doi: 10.1172/JCI112723
Colucci, W. S., Elkayam, U., Horton, D. P., Abraham, W. T., Bourge, R. C., Johnson, A. D., et al. (2000). Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N. Engl. J. Med. 343, 246–253. doi: 10.1056/NEJM200007273430403
Coue, M., and Moro, C. (2016). Natriuretic peptide control of energy balance and glucose homeostasis. Biochimie 124, 84–91. doi: 10.1016/j.biochi.2015.05.017
Currie, M. G., Geller, D. M., Cole, B. R., Siegel, N. R., Fok, K. F., Adams, S. P., et al. (1984). Purification and sequence analysis of bioactive atrial peptides (atriopeptins). Science 223, 67–69. doi: 10.1126/science.6419347
de Bold, A. J., Borenstein, H. B., Veress, A. T., and Sonnenberg, H. (1981). A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 28, 89–94. doi: 10.1016/0024-3205(81)90370-2
de Lemos, J. A., McGuire, D. K., and Drazner, M. H. (2003). B-type natriuretic peptide in cardiovascular disease. Lancet 362, 316–322. doi: 10.1016/S0140-6736(03)13976-1
De Palo, E. F., Woloszczuk, W., Meneghetti, M., De Palo, C. B., Nielsen, H. B., and Secher, N. H. (2000). Circulating immunoreactive proANP(1-30) and proANP(31-67) in sedentary subjects and athletes. Clin. Chem. 46(6 Pt 1), 843–847.
Del Ry, S., Cabiati, M., Vozzi, F., Battolla, B., Caselli, C., Forini, F., et al. (2011). Expression of C-type natriuretic peptide and its receptor NPR-B in cardiomyocytes. Peptides 32, 1713–1718. doi: 10.1016/j.peptides.2011.06.014
Delacroix, S., Chokka, R., Nelson, A., Sidharta, S., Teo, K., Tuke, J., et al. (2016). Safety and efficacy of pro ANP 31-67 in cardiorenal syndrome: a first-in-human trial. J. Am. Coll. Cardiol. 67:1351.
Dietz, J. R., Vesely, D. L., and Nazian, S. J. (1994). Possible mechanisms involved in the natriuretic response to atrial natriuretic factor (ANF) and proANF 31-67 in the rat. Clin. Exp. Pharmacol. Physiol 21, 599–606. doi: 10.1111/j.1440-1681.1994.tb02560.x
Dunkerly-Eyring, B., and Kass, D. A. (2020). Myocardial phosphodiesterases and their role in cGMP regulation. J. Cardiovasc. Pharmacol. 75, 483–493. doi: 10.1097/FJC.0000000000000773
Fain, J. N., Leffler, C. W., and Bahouth, S. W. (2000). Eicosanoids as endogenous regulators of leptin release and lipolysis by mouse adipose tissue in primary culture. J. Lipid Res. 41, 1689–1694.
Felder, R., Schiermeyer, K., Xu, P., Gildea, J. J., Geimer, T. R., and Mahan, L. (2017). ProANP 31-67 and renal function in heart failure. Hypertension 70:27.
Forssmann, W. G., Richter, R., and Meyer, M. (1998). The endocrine heart and natriuretic peptides: histochemistry, cell biology, and functional aspects of the renal urodilatin system. Histochem. Cell Biol. 110, 335–357.
Freund, B. J., Wade, C. E., and Claybaugh, J. R. (1988). Effects of exercise on atrial natriuretic factor. Release mechanisms and implications for fluid homeostasis. Sports Med. 6, 364–377. doi: 10.2165/00007256-198806060-00003
Frisk, M., Le, C., Shen, X., Røe, ÅT., Hou, Y., Manfra, O., et al. (2021). HFpEF is associated with preserved t-tubule integrity but etiology-dependent impairment of diastolic Ca2+ homeostasis. J. Am. Coll. Cardiol. 77, 405–419.
Fuller, F., Porter, J. G., Arfsten, A. E., Miller, J., Schilling, J. W., Scarborough, R. M., et al. (1988). Atrial natriuretic peptide clearance receptor. Complete sequence and functional expression of cDNA clones. J. Biol. Chem. 263, 9395–9401.
Garcia-Alonso, V., Lopez-Vicario, C., Titos, E., Moran-Salvador, E., Gonzalez-Periz, A., Rius, B., et al. (2013). Coordinate functional regulation between microsomal prostaglandin E synthase-1 (mPGES-1) and peroxisome proliferator-activated receptor gamma (PPARgamma) in the conversion of white-to-brown adipocytes. J. Biol. Chem. 288, 28230–28242. doi: 10.1074/jbc.M113.468603
Goetze, J. P., Bruneau, B. G., Ramos, H. R., Ogawa, T., de Bold, M. K., and de Bold, A. J. (2020). Cardiac natriuretic peptides. Nat. Rev. Cardiol. 17, 698–717. doi: 10.1038/s41569-020-0381-0
Gower, W. R. Jr., Chiou, S., Skolnick, K. A., and Vesely, D. L. (1994). Molecular forms of circulating atrial natriuretic peptides in human plasma and their metabolites. Peptides 15, 861–867. doi: 10.1016/0196-9781(94)90043-4
Greenwald, J. E., Needleman, P., Siegel, N., Tetens, E., Biel, B., and Ritter, D. (1992). Processing of atriopeptin prohormone by nonmyocytic atrial cells. Biochem. Biophys. Res. Commun. 188, 644–654. doi: 10.1016/0006-291x(92)91105-y
Gunning, M. E., Brady, H. R., Otuechere, G., Brenner, B. M., and Zeidel, M. L. (1992). Atrial natriuretic peptide(31-67) inhibits Na+ transport in rabbit inner medullary collecting duct cells. Role of prostaglandin E2. J. Clin. Invest. 89, 1411–1417. doi: 10.1172/JCI115730
Habibullah, A. A., Villarreal, D., Freeman, R. H., Dietz, J. R., Vesley, D. L., and Simmons, J. C. (1995). Atrial natriuretic peptide fragments in dogs with experimental heart failure. Clin. Exp. Pharmacol. Physiol. 22, 130–135. doi: 10.1111/j.1440-1681.1995.tb01969.x
Harada, M., Takeda, N., and Toko, H. (2017). Prostaglandin E2 signaling as a target of anti-cardiac fibrosis in heart failure treatment. Int. Heart J. 58, 3–4. doi: 10.1536/ihj.16-614
Harding, P., and LaPointe, M. C. (2011). Prostaglandin E2 increases cardiac fibroblast proliferation and increases cyclin D expression via EP1 receptor. Prostaglandins Leukot. Essent. Fatty Acids 84, 147–152. doi: 10.1016/j.plefa.2011.01.003
Hartter, E., Khalafpour, S., Missbichler, A., Hawa, G., and Woloszczuk, W. (2000). Enzyme immunoassays for fragments (epitopes) of human proatrial natriuretic peptides. Clin. Chem. Lab. Med 38, 27–32. doi: 10.1515/CCLM.2000.005
Hawkridge, A. M., Heublein, D. M., Bergen, H. R. III, Cataliotti, A., and Burnett, J. C. Jr. (2005). Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc. Natl. Acad. Sci. U.S.A. 102, 17442–17447. doi: 10.1073/pnas.0508782102
Heublein, D. M., Clavell, A. L., Stingo, A. J., Lerman, A., Wold, L., and Burnett, J. C. Jr. (1992). C-type natriuretic peptide immunoreactivity in human breast vascular endothelial cells. Peptides 13, 1017–1019. doi: 10.1016/0196-9781(92)90065-b
Hishikari, K., Suzuki, J., Ogawa, M., Isobe, K., Takahashi, T., Onishi, M., et al. (2009). Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 81, 123–132. doi: 10.1093/cvr/cvn254
Hobbs, R. E., Mills, R. M., and Young, J. B. (2001). An update on nesiritide for treatment of decompensated heart failure. Expert Opin. Investig. Drugs 10, 935–942. doi: 10.1517/13543784.10.5.935
Ichiki, T. (2006). Role of cAMP response element binding protein in cardiovascular remodeling: good, bad, or both? Arterioscler. Thromb. Vasc. Biol. 26, 449–455. doi: 10.1161/01.ATV.0000196747.79349.d1
Jin, L., Zhang, J., Deng, Z., Liu, J., Han, W., Chen, G., et al. (2020). Mesenchymal stem cells ameliorate myocardial fibrosis in diabetic cardiomyopathy via the secretion of prostaglandin E2. Stem Cell Res. Ther. 11:122. doi: 10.1186/s13287-020-01633-7
Kishimoto, I., Dubois, S. K., and Garbers, D. L. (1996). The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: acute handling of sodium and water in response to volume expansion. Proc. Natl. Acad. Sci. U.S.A. 93, 6215–6219. doi: 10.1073/pnas.93.12.6215
Kojima, M., Minamino, N., Kangawa, K., and Matsuo, H. (1990). Cloning and sequence analysis of a cDNA encoding a precursor for rat C-type natriuretic peptide (CNP). FEBS Lett. 276, 209–213. doi: 10.1016/0014-5793(90)80544-s
Komeichi, H., Moreau, R., Cailmail, S., Gaudin, C., and Lebrec, D. (1995). Blunted natriuresis and abnormal systemic hemodynamic responses to C-type and brain natriuretic peptides in rats with cirrhosis. J. Hepatol. 22, 319–325. doi: 10.1016/0168-8278(95)80285-1
Kuhn, M. (2004). Molecular physiology of natriuretic peptide signalling. Basic Res. Cardiol. 99, 76–82. doi: 10.1007/s00395-004-0460-0
Lai, Y. J., Chen, I. C., Li, H. H., and Huang, C. C. (2018). EP4 Agonist L-902,688 Suppresses EndMT and attenuates right ventricular cardiac fibrosis in experimental pulmonary arterial hypertension. Int. J. Mol. Sci. 19:727. doi: 10.3390/ijms19030727
Lainchbury, J. G., Burnett, J. C. Jr., Meyer, D., and Redfield, M. M. (2000). Effects of natriuretic peptides on load and myocardial function in normal and heart failure dogs. Am. J. Physiol. Heart Circ. Physiol. 278, H33–H40. doi: 10.1152/ajpheart.2000.278.1.H33
Luce, M., Barba, C., Yi, D., Mey, A., Roussel, D., Bres, E., et al. (2020). Accumulation of natriuretic peptides is associated with protein energy wasting and activation of browning in white adipose tissue in chronic kidney disease. Kidney Int. 98, 663–672. doi: 10.1016/j.kint.2020.03.027
Luchner, A., Stevens, T. L., Borgeson, D. D., Redfield, M., Wei, C. M., Porter, J. G., et al. (1998). Differential atrial and ventricular expression of myocardial BNP during evolution of heart failure. Am. J. Physiol. 274, H1684–H1689. doi: 10.1152/ajpheart.1998.274.5.H1684
Macheret, F., Heublein, D., Costello-Boerrigter, L. C., Boerrigter, G., McKie, P., Bellavia, D., et al. (2012). Human hypertension is characterized by a lack of activation of the antihypertensive cardiac hormones ANP and BNP. J. Am. Coll. Cardiol. 60, 1558–1565. doi: 10.1016/j.jacc.2012.05.049
Maisel, A. S., McCord, J., Nowak, R. M., Hollander, J. E., Wu, A. H., Duc, P., et al. (2003). Bedside B-Type natriuretic peptide in the emergency diagnosis of heart failure with reduced or preserved ejection fraction. Results from the Breathing Not Properly Multinational Study. J. Am. Coll. Cardiol. 41, 2010–2017. doi: 10.1016/s0735-1097(03)00405-4
Makino, H., Tanaka, I., Mukoyama, M., Sugawara, A., Mori, K., Muro, S., et al. (2002). Prevention of diabetic nephropathy in rats by prostaglandin E receptor EP1-selective antagonist. J. Am. Soc. Nephrol. 13, 1757–1765. doi: 10.1097/01.asn.0000019782.37851.bf
Manfra, O., Frisk, M., and Louch, W. E. (2017). Regulation of cardiomyocyte T-tubular structure: opportunities for therapy. Curr. Heart Fail. Rep. 14, 167–178. doi: 10.1007/s11897-017-0329-9
Marcus, L. S., Hart, D., Packer, M., Yushak, M., Medina, N., Danziger, R. S., et al. (1996). Hemodynamic and renal excretory effects of human brain natriuretic peptide infusion in patients with congestive heart failure. A double-blind, placebo-controlled, randomized crossover trial. Circulation 94, 3184–3189. doi: 10.1161/01.cir.94.12.3184
Martin, D. R., Pevahouse, J. B., Trigg, D. J., Vesely, D. L., and Buerkert, J. E. (1990). Three peptides from the ANF prohormone NH(2)-terminus are natriuretic and/or kaliuretic. Am. J. Physiol. 258(5 Pt 2), F1401–F1408. doi: 10.1152/ajprenal.1990.258.5.F1401
Martin, E. R., Lewicki, J. A., Scarborough, R. M., and Ballermann, B. J. (1989). Expression and regulation of ANP receptor subtypes in rat renal glomeruli and papillae. Am. J. Physiol. 257(4 Pt 2), F649–F657. doi: 10.1152/ajprenal.1989.257.4.F649
McMurray, J. J., Packer, M., Desai, A. S., Gong, J., Lefkowitz, M. P., Rizkala, A. R., et al. (2014). Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 371, 993–1004. doi: 10.1056/NEJMoa1409077
Meyer, M., Richter, R., and Forssmann, W. G. (1998). Urodilatin, a natriuretic peptide with clinical implications. Eur. J. Med. Res. 3, 103–110.
Moro, C. (2013). Natriuretic peptides and fat metabolism. Curr. Opin. Clin. Nutr. Metab. Care 16, 645–649. doi: 10.1097/MCO.0b013e32836510ed
Mukoyama, M., Nakao, K., Hosoda, K., Suga, S., Saito, Y., Ogawa, Y., et al. (1991). Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Invest. 87, 1402–1412. doi: 10.1172/JCI115146
Muraoka, N., Nara, K., Tamura, F., Kojima, H., Yamakawa, H., Sadahiro, T., et al. (2019). Role of cyclooxygenase-2-mediated prostaglandin E2-prostaglandin E receptor 4 signaling in cardiac reprogramming. Nat. Commun. 10:674. doi: 10.1038/s41467-019-08626-y
Nakagawa, N., Yuhki, K., Kawabe, J., Fujino, T., Takahata, O., Kabara, M., et al. (2012). The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 82, 158–171. doi: 10.1038/ki.2012.115
Nasrallah, R., Hassouneh, R., and Hebert, R. L. (2016). PGE2, kidney disease, and cardiovascular risk: beyond hypertension and diabetes. J. Am. Soc. Nephrol. 27, 666–676. doi: 10.1681/ASN.2015050528
Nasrallah, R., Hassouneh, R., Zimpelmann, J., Karam, A. J., Thibodeau, J. F., Burger, D., et al. (2015). Prostaglandin E2 increases proximal tubule fluid reabsorption, and modulates cultured proximal tubule cell responses via EP1 and EP4 receptors. Lab. Invest. 95, 1044–1055. doi: 10.1038/labinvest.2015.79
Nasrallah, R., Zimpelmann, J., Eckert, D., Ghossein, J., Geddes, S., Beique, J. C., et al. (2018). PGE2 EP1 receptor inhibits vasopressin-dependent water reabsorption and sodium transport in mouse collecting duct. Lab. Invest. 98, 360–370. doi: 10.1038/labinvest.2017.133
Ngo, L., Wyeth, R. P., Bissett, J. K., Hester, W. L., Newton, M. T., Sallman, A. L., et al. (1989). Prohormone atrial natriuretic peptides 1-30, 31-67, and 99-126 increase in proportion to right ventricular pacing rate. Am. Heart J. 117, 385–390. doi: 10.1016/0002-8703(89)90784-9
Niederkofler, E. E., Kiernan, U. A., O’Rear, J., Menon, S., Saghir, S., Protter, A. A., et al. (2008). Detection of endogenous B-type natriuretic peptide at very low concentrations in patients with heart failure. Circ. Heart Fail. 1, 258–264. doi: 10.1161/CIRCHEARTFAILURE.108.790774
Ogawa, Y., Nakao, K., Mukoyama, M., Shirakami, G., Itoh, H., Hosoda, K., et al. (1990). Rat brain natriuretic peptide–tissue distribution and molecular form. Endocrinology 126, 2225–2227. doi: 10.1210/endo-126-4-2225
Pandey, K. N. (2014). Guanylyl cyclase/natriuretic peptide receptor-A signaling antagonizes phosphoinositide hydrolysis, Ca(2+) release, and activation of protein kinase C. Front. Mol. Neurosci. 7:75. doi: 10.3389/fnmol.2014.00075
Pang, L., Cai, Y., Tang, E. H., Irwin, M. G., Ma, H., and Xia, Z. (2016). Prostaglandin E receptor subtype 4 signaling in the heart: role in ischemia/reperfusion injury and cardiac hypertrophy. J. Diabetes Res. 2016:1324347. doi: 10.1155/2016/1324347
Poschke, A., Kern, N., Maruyama, T., Pavenstadt, H., Narumiya, S., Jensen, B. L., et al. (2012). The PGE(2)-EP4 receptor is necessary for stimulation of the renin-angiotensin-aldosterone system in response to low dietary salt intake in vivo. Am. J. Physiol. Renal Physiol. 303, F1435–F1442. doi: 10.1152/ajprenal.00512.2011
Purdy, K. E., and Arendshorst, W. J. (2000). EP(1) and EP(4) receptors mediate prostaglandin E(2) actions in the microcirculation of rat kidney. Am. J. Physiol. Renal Physiol. 279, F755–F764. doi: 10.1152/ajprenal.2000.279.4.F755
Qian, J. Y., Harding, P., Liu, Y., Shesely, E., Yang, X. P., and LaPointe, M. C. (2008). Reduced cardiac remodeling and function in cardiac-specific EP4 receptor knockout mice with myocardial infarction. Hypertension 51, 560–566. doi: 10.1161/HYPERTENSIONAHA.107.102590
Reginauld, S. H., Cannone, V., Iyer, S., Scott, C., Bailey, K., Schaefer, J., et al. (2019). Differential regulation of ANP and BNP in Acute Decompensated Heart Failure: deficiency of ANP. JACC Heart Fail. 7, 891–898. doi: 10.1016/j.jchf.2019.05.012
Richards, A. M., and Januzzi, J. L. Jr. (2019). Are some patients with acute heart failure ANP-deficient? JACC Heart Fail. 7, 899–901. doi: 10.1016/j.jchf.2019.07.003
Richards, M., and Troughton, R. W. (2004). NT-proBNP in heart failure: therapy decisions and monitoring. Eur. J. Heart Fail. 6, 351–354. doi: 10.1016/j.ejheart.2004.01.003
Roger, V. L. (2013). Epidemiology of heart failure. Circ. Res. 113, 646–659. doi: 10.1161/CIRCRESAHA.113.300268
Rubattu, S., Forte, M., Marchitti, S., and Volpe, M. (2019). Molecular implications of natriuretic peptides in the protection from hypertension and target organ damage development. Int. J. Mol. Sci. 20:798. doi: 10.3390/ijms20040798
Rubattu, S., and Volpe, M. (2019). Natriuretic peptides in the cardiovascular system: multifaceted roles in physiology, pathology and therapeutics. Int. J. Mol. Sci. 20:3991. doi: 10.3390/ijms20163991
Saito, Y., Nakao, K., Itoh, H., Yamada, T., Mukoyama, M., Arai, H., et al. (1989). Brain natriuretic peptide is a novel cardiac hormone. Biochem. Biophys. Res. Commun. 158, 360–368. doi: 10.1016/s0006-291x(89)80056-7
Saito, Y., Nakao, K., Nishimura, K., Sugawara, A., Okumura, K., Obata, K., et al. (1987). Clinical application of atrial natriuretic polypeptide in patients with congestive heart failure: beneficial effects on left ventricular function. Circulation 76, 115–124. doi: 10.1161/01.cir.76.1.115
Sanchez-Alavez, M., Klein, I., Brownell, S. E., Tabarean, I. V., Davis, C. N., Conti, B., et al. (2007). Night eating and obesity in the EP3R-deficient mouse. Proc. Natl. Acad. Sci. U.S.A. 104, 3009–3014. doi: 10.1073/pnas.0611209104
Sangaralingham, S. J., and Burnett, J. C. Jr. (2018). Relaxing with C-type natriuretic peptide, the guanylyl cyclase B receptor, and pericytes. Circulation 138, 509–512. doi: 10.1161/CIRCULATIONAHA.118.035132
Sangaralingham, S. J., Chen, Y., and Burnett, J. C. (2020). C-type natriuretic peptide: the heart’s guardian angel. Eur. Heart J. 41, 1021–1023. doi: 10.1093/eurheartj/ehz142
Schulz, S., Singh, S., Bellet, R. A., Singh, G., Tubb, D. J., Chin, H., et al. (1989). The primary structure of a plasma membrane guanylate cyclase demonstrates diversity within this new receptor family. Cell 58, 1155–1162. doi: 10.1016/0092-8674(89)90513-8
Schweda, F., Klar, J., Narumiya, S., Nusing, R. M., and Kurtz, A. (2004). Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am. J. Physiol. Renal Physiol. 287, F427–F433. doi: 10.1152/ajprenal.00072.2004
Seferovic, P. M., Ponikowski, P., Anker, S. D., Bauersachs, J., Chioncel, O., Cleland, J. G. F., et al. (2019). Clinical practice update on heart failure 2019: pharmacotherapy, procedures, devices and patient management. An expert consensus meeting report of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 21, 1169–1186. doi: 10.1002/ejhf.1531
Spiranec, K., Chen, W., Werner, F., Nikolaev, V. O., Naruke, T., Koch, F., et al. (2018). Endothelial C-type natriuretic peptide acts on pericytes to regulate microcirculatory flow and blood pressure. Circulation 138, 494–508. doi: 10.1161/CIRCULATIONAHA.117.033383
Stevens, T. L., Rasmussen, T. E., Wei, C. M., Kinoshita, M., Matsuda, Y., and Burnett, J. C. Jr. (1996). Renal role of the endogenous natriuretic peptide system in acute congestive heart failure. J. Card. Fail. 2, 119–125. doi: 10.1016/s1071-9164(96)80030-3
Stingo, A. J., Clavell, A. L., Heublein, D. M., Wei, C. M., Pittelkow, M. R., and Burnett, J. C. Jr. (1992). Presence of C-type natriuretic peptide in cultured human endothelial cells and plasma. Am. J. Physiol. 263(4 Pt 2), H1318–H1321. doi: 10.1152/ajpheart.1992.263.4.H1318
Strong, P., Coleman, R. A., and Humphrey, P. P. (1992). Prostanoid-induced inhibition of lipolysis in rat isolated adipocytes: probable involvement of EP3 receptors. Prostaglandins 43, 559–566. doi: 10.1016/0090-6980(92)90115-a
Sudoh, T., Kangawa, K., Minamino, N., and Matsuo, H. (1988). A new natriuretic peptide in porcine brain. Nature 332, 78–81. doi: 10.1038/332078a0
Suga, S., Nakao, K., Itoh, H., Komatsu, Y., Ogawa, Y., Hama, N., et al. (1992). Endothelial production of C-type natriuretic peptide and its marked augmentation by transforming growth factor-beta. Possible existence of “vascular natriuretic peptide system”. J. Clin. Invest. 90, 1145–1149. doi: 10.1172/JCI115933
Sugawara, A., Nakao, K., Morii, N., Yamada, T., Itoh, H., Shiono, S., et al. (1988). Synthesis of atrial natriuretic polypeptide in human failing hearts. Evidence for altered processing of atrial natriuretic polypeptide precursor and augmented synthesis of beta-human ANP. J. Clin. Invest. 81, 1962–1970. doi: 10.1172/JCI113544
Sugimoto, Y., and Narumiya, S. (2007). Prostaglandin E receptors. J. Biol. Chem. 282, 11613–11617. doi: 10.1074/jbc.R600038200
Tang, E. H., Cai, Y., Wong, C. K., Rocha, V. Z., Sukhova, G. K., Shimizu, K., et al. (2015). Activation of prostaglandin E2-EP4 signaling reduces chemokine production in adipose tissue. J. Lipid Res. 56, 358–368. doi: 10.1194/jlr.M054817
Tsukamoto, O., Fujita, M., Kato, M., Yamazaki, S., Asano, Y., Ogai, A., et al. (2009). Natriuretic peptides enhance the production of adiponectin in human adipocytes and in patients with chronic heart failure. J. Am. Coll. Cardiol. 53, 2070–2077. doi: 10.1016/j.jacc.2009.02.038
Tuteja, N. (2009). Signaling through G protein coupled receptors. Plant Signal. Behav. 4, 942–947. doi: 10.4161/psb.4.10.9530
Umemura, M., Osawa, K., Tanaka, R., Hikichi, M., Nakakaji, R., Rafikul, I., et al. (2019). Prostaglandin E2 receptor EP4 regulates fibrotic changes via intracellular calcium in cardiac fibroblast cells. Circulation 140:A13193.
Verboven, K., Hansen, D., Jocken, J. W. E., and Blaak, E. E. (2017). Natriuretic peptides in the control of lipid metabolism and insulin sensitivity. Obes. Rev. 18, 1243–1259. doi: 10.1111/obr.12598
Vesely, D. L. (1995). Atrial natriuretic hormones originating from the N-terminus of the atrial natriuretic factor prohormone. Clin. Exp. Pharmacol. Physiol. 22, 108–114. doi: 10.1111/j.1440-1681.1995.tb01965.x
Vesely, D. L. (2007). Discovery of new cardiovascular hormones for the treatment of congestive heart failure. Cardiovasc. Hematol. Disord. Drug Targets 7, 47–62. doi: 10.2174/187152907780059128
Vesely, D. L., Cornett, L. E., MacLeod, S. L., Nash, A. A., and Norris, J. S. (1990). Specific binding sites for prohormone atrial natriuretic peptides 1-30, 31-67 and 99-126. Peptides 11, 193–197. doi: 10.1016/0196-9781(90)90070-l
Vesely, D. L., Dietz, J. R., Parks, J. R., Baig, M., McCormick, M. T., Cintron, G., et al. (1998). Vessel dilator enhances sodium and water excretion and has beneficial hemodynamic effects in persons with congestive heart failure. Circulation 98, 323–329. doi: 10.1161/01.cir.98.4.323
Vesely, D. L., Douglass, M. A., Dietz, J. R., Gower, W. R. Jr., and McCormick, M. T. (1994). Three peptides from the atrial natriuretic factor prohormone amino terminus lower blood pressure and produce diuresis, natriuresis, and/or kaliuresis in humans. Circulation 90, 1129–1140. doi: 10.1161/01.cir.90.3.1129
Vesely, D. L., Norris, J. S., Walters, J. M., Jespersen, R. R., and Baeyens, D. A. (1987). Atrial natriuretic prohormone peptides 1-30, 31-67, and 79-98 vasodilate the aorta. Biochem. Biophys. Res. Commun. 148, 1540–1548. doi: 10.1016/s0006-291x(87)80307-8
Vesely, D. L., Norsk, P., Winters, C. J., Rico, D. M., Sallman, A. L., and Epstein, M. (1989). Increased release of the N-terminal and C-terminal portions of the prohormone of atrial natriuretic factor during immersion-induced central hypervolemia in normal humans. Proc. Soc. Exp. Biol. Med. 192, 230–235. doi: 10.3181/00379727-192-42990
Vesely, D. L., Perez-Lamboy, G. I., and Schocken, D. D. (2000). Vessel dilator, long acting natriuretic peptide, and kaliuretic peptide increase circulating prostaglandin E2. Life Sci. 66, 905–913. doi: 10.1016/s0024-3205(99)00674-8
Vesely, D. L., Perez-Lamboy, G. I., and Schocken, D. D. (2001). Long-acting natriuretic peptide, vessel dilator, and kaliuretic peptide enhance urinary excretion rate of albumin, total protein, and beta(2)-microglobulin in patients with congestive heart failure. J. Card. Fail. 7, 55–63. doi: 10.1054/jcaf.2001.23109
Vesely, D. L., Sallman, A. L., and Bayliss, J. M. (1992). Specific binding site for pro atrial natriuretic factors 1-30, 31-67, and 99-126 on distal nephrons, proximal tubules, renal cortical and medullary membranes. Ren. Physiol. Biochem. 15, 23–32. doi: 10.1159/000173438
Villarreal, D., Freeman, R. H., Taraben, A., and Reams, G. P. (1999a). Modulation of renin secretion by atrial natriuretic factor prohormone fragment 31-67. Am. J. Med. Sci. 318, 330–335. doi: 10.1097/00000441-199911000-00008
Villarreal, D., Reams, G. P., Taraben, A., and Freeman, R. H. (1999b). Hemodynamic and renal effects of ProANF31-67 in hypertensive rats. Proc. Soc. Exp. Biol. Med. 221, 166–170. doi: 10.1046/j.1525-1373.1999.d01-71.x
Vollmar, A. M., Gerbes, A. L., Nemer, M., and Schulz, R. (1993). Detection of C-type natriuretic peptide (CNP) transcript in the rat heart and immune organs. Endocrinology 132, 1872–1874. doi: 10.1210/endo.132.4.8462485
Volpe, M., Rubattu, S., and Battistoni, A. (2019). ARNi: a novel approach to counteract cardiovascular diseases. Int. J. Mol. Sci. 20:2092. doi: 10.3390/ijms20092092
Volpe, M., Tocci, G., Battistoni, A., and Rubattu, S. (2015). Angiotensin II receptor blocker neprilysin inhibitor (ARNI): new avenues in cardiovascular therapy. High Blood Press. Cardiovasc. Prev. 22, 241–246. doi: 10.1007/s40292-015-0112-5
Wada, A., Tsutamoto, T., Matsuda, Y., and Kinoshita, M. (1994). Cardiorenal and neurohumoral effects of endogenous atrial natriuretic peptide in dogs with severe congestive heart failure using a specific antagonist for guanylate cyclase-coupled receptors. Circulation 89, 2232–2240. doi: 10.1161/01.cir.89.5.2232
Wang, Q., Oka, T., Yamagami, K., Lee, J. K., Akazawa, H., Naito, A. T., et al. (2017). An EP4 receptor agonist inhibits cardiac fibrosis through activation of PKA signaling in hypertrophied heart. Int. Heart J. 58, 107–114. doi: 10.1536/ihj.16-200
Winters, C. J., Sallman, A. L., Baker, B. J., Meadows, J., Rico, D. M., and Vesely, D. L. (1989). The N-terminus and a 4,000-MW peptide from the midportion of the N-terminus of the atrial natriuretic factor prohormone each circulate in humans and increase in congestive heart failure. Circulation 80, 438–449. doi: 10.1161/01.cir.80.3.438
Wright, R. S., Wei, C. M., Kim, C. H., Kinoshita, M., Matsuda, Y., Aarhus, L. L., et al. (1996). C-type natriuretic peptide-mediated coronary vasodilation: role of the coronary nitric oxide and particulate guanylate cyclase systems. J. Am. Coll. Cardiol. 28, 1031–1038. doi: 10.1016/s0735-1097(96)00241-0
Xu, H., Fu, J. L., Miao, Y. F., Wang, C. J., Han, Q. F., Li, S., et al. (2016). Prostaglandin E2 receptor EP3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. J. Mol. Cell Biol. 8, 518–529. doi: 10.1093/jmcb/mjw035
Xu, S., Zhou, W., Ge, J., and Zhang, Z. (2018). Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMPPKA/PI3KAkt signaling pathway. Mol. Med. Rep. 17, 4702–4712. doi: 10.3892/mmr.2018.8415
Yamagami, K., Oka, T., Wang, Q., Ishizu, T., Lee, J. K., Miwa, K., et al. (2015). Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am. J. Physiol. Heart Circ. Physiol. 309, H512–H522. doi: 10.1152/ajpheart.00137.2015
Yamamoto, K., Burnett, J. C. Jr., Jougasaki, M., Nishimura, R. A., and Bailey, K. R. (1996). Superiority of brain natriuretic peptide as a hormonal marker of ventricular systolic and diastolic dysfunction and ventricular hypertrophy. Hypertension 28, 988–994. doi: 10.1161/01.hyp.28.6.988
Yamamoto, K., Burnett, J. C. Jr., and Redfield, M. M. (1997). Effect of endogenous natriuretic peptide system on ventricular and coronary function in failing heart. Am. J. Physiol. 273, H2406–H2414. doi: 10.1152/ajpheart.1997.273.5.H2406
Yan, W., Wu, F., Morser, J., and Wu, Q. (2000). Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc. Natl. Acad. Sci. U.S.A. 97, 8525–8529. doi: 10.1073/pnas.150149097
Yokoyama, U., Iwatsubo, K., Umemura, M., Fujita, T., and Ishikawa, Y. (2013). The prostanoid EP4 receptor and its signaling pathway. Pharmacol. Rev. 65, 1010–1052. doi: 10.1124/pr.112.007195
Zahner, G., Schaper, M., Panzer, U., Kluger, M., Stahl, R. A., Thaiss, F., et al. (2009). Prostaglandin EP2 and EP4 receptors modulate expression of the chemokine CCL2 (MCP-1) in response to LPS-induced renal glomerular inflammation. Biochem. J. 422, 563–570. doi: 10.1042/BJ20090420
Zeidel, M. L., Seifter, J. L., Lear, S., Brenner, B. M., and Silva, P. (1986). Atrial peptides inhibit oxygen consumption in kidney medullary collecting duct cells. Am. J. Physiol. 251(2 Pt 2), F379–F383. doi: 10.1152/ajprenal.1986.251.2.F379
Zhu, L., Xu, C., Huo, X., Hao, H., Wan, Q., Chen, H., et al. (2019). The cyclooxygenase-1/mPGES-1/endothelial prostaglandin EP4 receptor pathway constrains myocardial ischemia-reperfusion injury. Nat. Commun. 10:1888. doi: 10.1038/s41467-019-09492-4
Zile, M. R., Jhund, P. S., Baicu, C. F., Claggett, B. L., Pieske, B., Voors, A. A., et al. (2016). Plasma biomarkers reflecting profibrotic processes in heart failure with a preserved ejection fraction: data from the prospective comparison of ARNI with ARB on management of heart failure with preserved ejection fraction study. Circ. Heart Fail. 9:e002551. doi: 10.1161/CIRCHEARTFAILURE.115.002551
Keywords: natriuretic peptides, atrial natriuretic peptide, proANP31–67, cardiovascular disease, heart Failure
Citation: da Silva GJJ, Altara R, Booz GW and Cataliotti A (2021) Atrial Natriuretic Peptide31–67: A Novel Therapeutic Factor for Cardiovascular Diseases. Front. Physiol. 12:691407. doi: 10.3389/fphys.2021.691407
Received: 06 April 2021; Accepted: 14 June 2021;
Published: 08 July 2021.
Edited by:
Speranza Rubattu, Istituto Neurologico Mediterraneo Neuromed (IRCCS), ItalyReviewed by:
Valentina Cannone, University Hospital of Parma, ItalyCopyright © 2021 da Silva, Altara, Booz and Cataliotti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Cataliotti, YWxlc3NhbmRyby5jYXRhbGlvdHRpQG1lZGlzaW4udWlvLm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.