
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 27 July 2021
Sec. Respiratory Physiology and Pathophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.690531
This article is part of the Research Topic Respiratory Barrier Versus Exogenous and Endogenous Noxious Stimuli: at the Crossroad Between Integrity and Dysfunction View all 9 articles
 Virinchi N. S. Kuchibhotla1,2,3,4*†
Virinchi N. S. Kuchibhotla1,2,3,4*† Malcolm R. Starkey2,5,6†
Malcolm R. Starkey2,5,6† Andrew T. Reid1,7
Andrew T. Reid1,7 Irene H. Heijink3,4,8
Irene H. Heijink3,4,8 Martijn C. Nawijn3,4
Martijn C. Nawijn3,4 Philip M. Hansbro1,2,9,10‡Darryl A. Knight1,2,11,12‡
Philip M. Hansbro1,2,9,10‡Darryl A. Knight1,2,11,12‡Excessive mucus production is a major feature of allergic asthma. Disruption of epithelial junctions by allergens such as house dust mite (HDM) results in the activation of β-catenin signaling, which has been reported to stimulate goblet cell differentiation. β-catenin interacts with various co-activators including CREB binding protein (CBP) and p300, thereby regulating the expression of genes involved in cell proliferation and differentiation, respectively. We specifically investigated the role of the β-catenin/CBP signaling pathway in goblet cell metaplasia in a HDM-induced allergic airway disease model in mice using ICG-001, a small molecule inhibitor that blocks the binding of CBP to β-catenin. Female 6- 8-week-old BALB/c mice were sensitized to HDM/saline on days 0, 1, and 2, followed by intranasal challenge with HDM/saline with or without subcutaneous ICG-001/vehicle treatment from days 14 to 17, and samples harvested 24 h after the last challenge/treatment. Differential inflammatory cells in bronchoalveolar lavage (BAL) fluid were enumerated. Alcian blue (AB)/Periodic acid–Schiff (PAS) staining was used to identify goblet cells/mucus production, and airway hyperresponsiveness (AHR) was assessed using invasive plethysmography. Exposure to HDM induced airway inflammation, goblet cell metaplasia and increased AHR, with increased airway resistance in response to the non-specific spasmogen methacholine. Inhibition of the β-catenin/CBP pathway using treatment with ICG-001 significantly attenuated the HDM-induced goblet cell metaplasia and infiltration of macrophages, but had no effect on eosinophils, neutrophils, lymphocytes or AHR. Increased β-catenin/CBP signaling may promote HDM-induced goblet cell metaplasia in mice.
Asthma is broadly characterized by chronic inflammation and remodeling of the airways, excessive airway mucus production, reversible airflow obstruction with loss of lung function (Fehrenbach et al., 2017). The airway epithelium is the first line of defense against inhaled allergens and is more susceptible to damage by allergens such as house dust mite (HDM) in asthma. This results in the deterioration of cellular junctions (Heijink et al., 2014), and release of pro-inflammatory cytokines like Chemokine (C-C motif) ligand 20 (CCL20), CCL17, interleukin (IL)-25, IL-33 and thymic stromal lymphopoietin (TSLP), which activate and attract immune cells like dendritic cells (DCs) and macrophages, promoting the differentiation of type-2 cells (Heijink et al., 2007; Hansbro et al., 2013; Hallstrand et al., 2014). T helper 2 (Th2) cells and innate lymphoid cells (ILCs) release various cytokines including IL-4, IL-5, and IL-13, which activate B-lymphocytes, induce the infiltration of eosinophils, airway hyperresponsiveness (AHR) (Thorburn et al., 2012; Liu et al., 2017), and enhance the differentiation of goblet cells (Hammad and Lambrecht, 2015; Reid et al., 2018; Starkey et al., 2019). Goblet cells are secretory cells that produce mucus, which consists of polypeptides, water, DNA, enzymes and high molecular weight glycoproteins called mucins (Williams et al., 2006). MUC5AC and MUC5B are the major mucins secreted by goblet cells, and their expression is increased in asthma and is associated with goblet cell metaplasia (Bonser and Erle, 2017; Vieira Braga et al., 2019). MUC5AC production is regulated by the transcription factors SAM pointed domain containing ETS transcription factor (SPDEF) and forkhead box a2 (FOXA2), which activate and repress the expression of MUC5AC, respectively (Evans et al., 2009; Bonser and Erle, 2017).
Adherens junctions, which mainly constitute the transmembrane protein E-cadherin, help in maintaining the integrity and function of the airway epithelium. Reduced expression of E-cadherin has been observed in airway epithelial cells from asthmatic donors along with decreased epithelial barrier function (Post et al., 2013). We have previously shown that allergens like HDM disrupt E-cadherin at the cell junctions and induce goblet cell metaplasia in mice (Post et al., 2012). In mice, embryonic knock-out of E-cadherin in airway epithelial cells induced club cell hyperplasia (Ceteci et al., 2012) and excessive mucus production (Post et al., 2018). The loss of E-cadherin leads to the delocalization of its intracellular binding partner β-catenin into the cytoplasm, where its levels are regulated by a destruction complex consisting of axin, adenomatous polyposis coli APC and GSK-3β. Activation of Wnt and/or growth factor signaling prevents phosphorylation and degradation of β-catenin by the destruction complex in the cytoplasm. The non-phosphorylated β-catenin, which is the transcriptionally active form of β-catenin, translocates to the nucleus and acts as a transcription factor to regulate the expression of various genes involved in cellular development and regulate cell fate decisions (Valenta et al., 2012; Figure 1). Increased transcriptional activity of β-catenin has been shown to induce goblet cell metaplasia in mice (Mucenski et al., 2005). The regulation of a variety of genes by β-catenin is possible because of its ability to bind to various transcriptional coactivators including CREB binding protein (CBP) and p300 leading to divergent cellular processes of cell migration and proliferation versus differentiation, respectively (Ma et al., 2005). Specifically, β-catenin/CBP signaling has been shown to regulate epithelial-to-mesenchymal transition of primary airway epithelial cells, leading to loss of epithelial markers such as E-cadherin (Nawijn et al., 2011; Moheimani et al., 2015). The small molecule inhibitor ICG-001 has been previously shown to inhibit β-catenin/CBP signaling by specifically binding to CBP, thereby preventing β-catenin from interacting with CBP (Emami et al., 2004). In addition, inhibition of β-catenin/CBP pathway by the small molecule inhibitor ICG-001 improved airway epithelial barrier function in vitro upon Ca+2 signaling-induced damage by stabilizing E-cadherin at cell junctions (Kuchibhotla et al., 2020). We hypothesized that inhibition of the β-catenin/CBP pathway upon HDM-induced E-cadherin loss using ICG-001 would attenuate HDM-induced goblet cell metaplasia and AHR in a mouse model of allergic asthma.
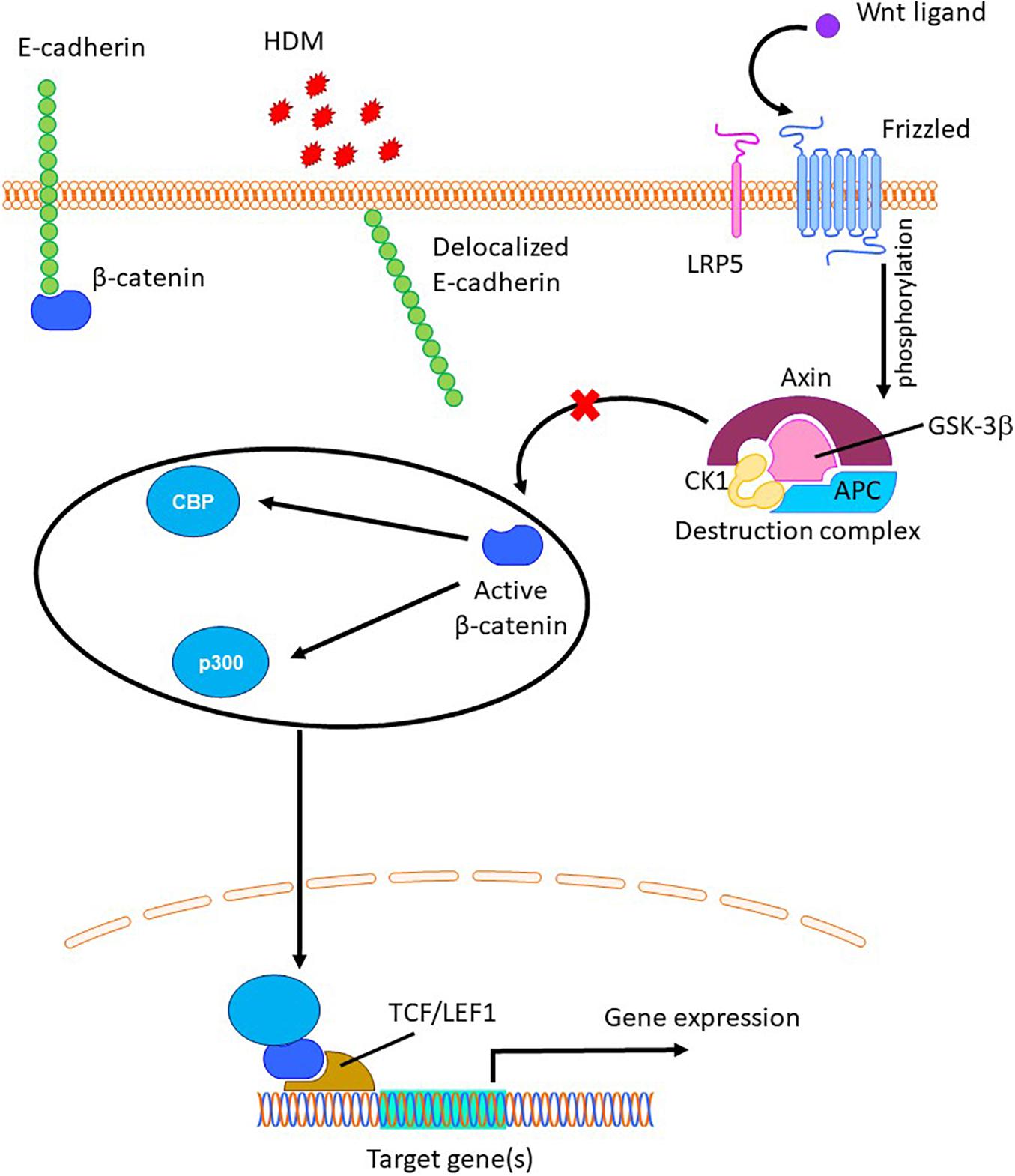
Figure 1. The role of β-catenin in regulation of gene expression. Allergens like house dust mite (HDM) can delocalize junctional E-cadherin (Post et al., 2012), resulting in the release of β-catenin into cytoplasm, where is targeted for proteolytic degradation by the destruction complex (Valenta et al., 2012). Upon Wnt ligand mediated phosphorylation and inactivation of GSK-3β, cytosolic β-catenin escapes from the destruction complex, translocates to the nucleus and binds to TCF/LEF group of transcriptional factors and other co-activators such as CBP and p300, resulting transcription of target genes (Valenta et al., 2012).
6–8 week old female BALB/c mice were obtained from the University of Newcastle’s central animal house and housed at the Hunter Medical Research Institute animal facility in individually ventilated cages in an SPF PC2 facility. Mice were provided standard rodent chow ad libitum. All protocols were approved by the University of Newcastle Animal Care and Ethics Committee.
Mice (n = 6−8/group) were sensitized to HDM (Dermatophagoides pteronyssinus) extract [intranasal (i.n.): days 0, 1, and 2; 50 μg; Greer Labs, NC, United States] in sterile saline (50 μl) and challenged with HDM (i.n.: day 14–17; 5 μg in 50 μl saline) with/without ICG-001 (subcutaneous: day 14–17; 5 mg/Kg) under isoflurane anesthesia and euthanized on day 18 (Figure 2A) as previously described (Thorburn et al., 2012, 2015).
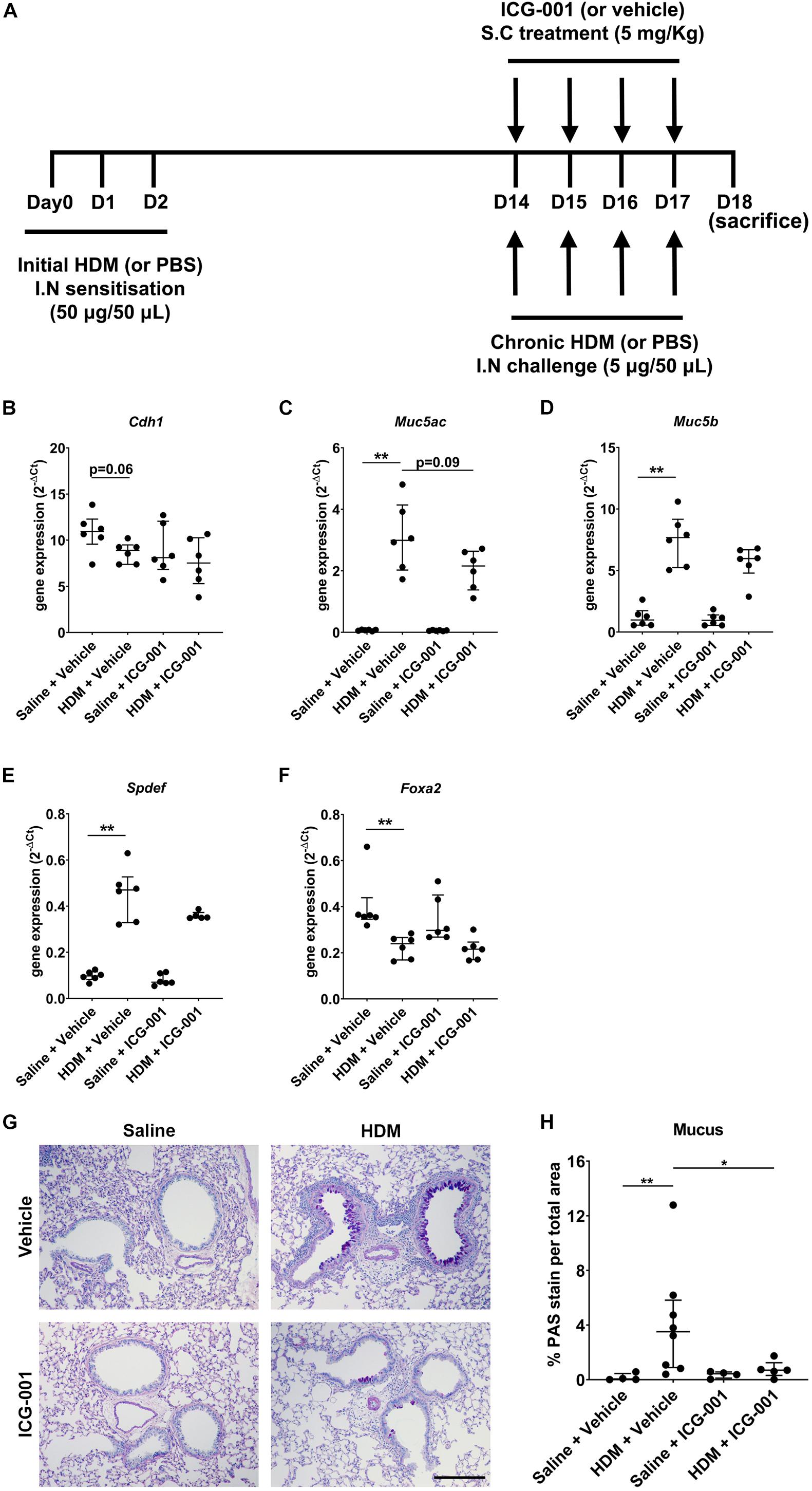
Figure 2. ICG-001 decreases HDM-induced goblet cell metaplasia. (A) HDM-induced allergic airway disease model in mice treated with the ICG-001 inhibitor. (B–F) Gene expression of Cdh1 (n = 6), Muc5ac (n = 6), Muc5b (n = 6), Spdef (n = 5–6) and Foxa2 (n = 6) normalized to Hprt (n = 6) (G) Alcian Blue (AB)/Periodic acid–Schiff (PAS) staining of lung sections from mice for mucus (n = 4–8), scale = 100 μm. (H) Semi-quantification of AB/PAS staining for mucus using color deconvolution (n = 4–8). Data is presented as median ± IQR; *p < 0.05, **p < 0.01, Mann–Whitney U test.
Total RNA was extracted from lung homogenates using TRIZOL (Sigma, MO, United States) as previously described (Horvat et al., 2007; Essilfie et al., 2011; Kim et al., 2017b). Synthesis of cDNA was performed using high-capacity cDNA reverse transcription kit with RNase inhibitor (Thermo Fisher Scientific, Waltham, MA, United States) and T100 Thermal Cycler (Bio-Rad Laboratories, CA, United States). Quantitative real time PCR (qRT-PCR) targeting Cdh1 (Mm01247357_m1, Thermo Fisher Scientific), Muc5ac (Mm01276718_m1, Thermo Fisher Scientific), Muc5b (Mm00466391_m1, Thermo Fisher Scientific), Spdef (Hs00171942_m1, Thermo Fisher Scientific), and Foxa2 (Mm01976556_s1, Thermo Fisher Scientific) was performed on individual biological replicates, which were normalized to Hprt (Mm03024075_m1, Thermo Fisher Scientific) and presented as gene expression (2–ΔCt) as previously described (Schmittgen and Livak, 2008).
Lung sections of 5 μm thickness were obtained for all the experimental mice and stained for goblet cells/mucins using standard Alcian blue (AB) pH 2.5 followed by periodic acid and Schiff’s reagent (PAS) as previously described (Horvat et al., 2007; Starkey et al., 2013; Reid et al., 2020). A minimum of eight different airways per section were imaged at 10× magnification and quantified using color deconvolution algorithm in ImageJ (National institute of Health) by one person as previously described (Horvat et al., 2007; Starkey et al., 2013; Reid et al., 2020). All the AB/PAS staining images were blinded and imported into ImageJ. The airway epithelial cells were selected by drawing a region of interest (ROI) and the outside area was deleted. The “Threshold color” was adjusted to select all airway epithelial cells and the total area of the airway epithelial cells was measured. Next, by selecting the “color deconvolution” function with H-PAS vector, the image was split into three different color channels, of which, the blue/magenta color channel was selected. The AB/PAS staining intensity was quantified using the “Threshold” function. The same threshold value is used all the images for consistency and eliminate any human bias. All the values of AB/PAS staining intensities and area of airway epithelial cells from different airways of a single image section were separately added to get the total AB/PAS staining intensity and total area of airway epithelial cells, respectively. Finally, the % AB/PAS staining was calculated using the following equation: (total AB/PAS staining intensity/total area of airway epithelial cells) × 100. Each data point represents the % AB/PAS staining of mucins per total area of the airway epithelial cells in a section from a single mouse.
Bronchoalveolar lavage fluid (2 ml) was prepared, and total cell numbers were determined with a hemocytometer. Cells prepared by cytocentrifugation (Shandon Cytospin; Thermo Fisher Scientific, Waltham, MA, United States) were stained with May-Grünwald-Giemsa and leukocytes were enumerated on the basis of morphologic criteria [200 cells by light microscopy (×40)] as previously described (Horvat et al., 2007; Kaiko et al., 2008; Thorburn et al., 2010). Further, eosinophils, neutrophils, lymphocytes, and macrophages were calculated and represented as the percentage of the total leukocytes in BAL fluid.
Airway hyperresponsiveness was measured using plethysmography, which provides similar results as Flexivent in HDM models of experimental asthma (Horvat et al., 2007; Kim et al., 2017b,c). Mice were anesthetized [ketamine and xylazine (80–100 and 10 mg/kg, respectively); Troy Laboratories, Smithfield, NSW, Australia] and the tracheas were cannulated. Each cannula was connected to an inline aerosol administrator and ventilator, which were attached to a preamplifier and computer (Buxco, Wilmington, NC, United States) to analyze pressure and flow waveforms and to determine airway resistance and dynamic compliance. Mice were nebulized with saline followed by increasing doses of methacholine (Sigma) as previously described (Horvat et al., 2007; Kim et al., 2017b,c).
All statistical analyses were performed using GraphPad Prism (Graphpad software, San Diego, CA, United States). Non-parametric Mann–Whitney U test was performed to assess for significant differences in gene expression, staining quantification, and the infiltration of immune cells between different groups. For AHR, two-way analysis of variance (ANOVA) was used to compare different groups and multiple comparisons were done by uncorrected Dunn’s test. Outliers were identified by Grubb’s test. We detected one outlier in the HDM + ICG-001 group for the Spdef gene expression analysis (Figure 2F) and one outlier in the HDM + ICG-001 group for the AB/PAS staining quantification (Figure 2H) and removed these from the specific analyses. In addition, we also detected and removed one outlier each in the Saline + ICG-001 group for the measurement of total leukocytes, eosinophils, neutrophils, lymphocytes, and macrophages (Figures 3A–E and Supplementary Figures 1A–E), which are from a single mouse. P < 0.05 was considered statistically significant.
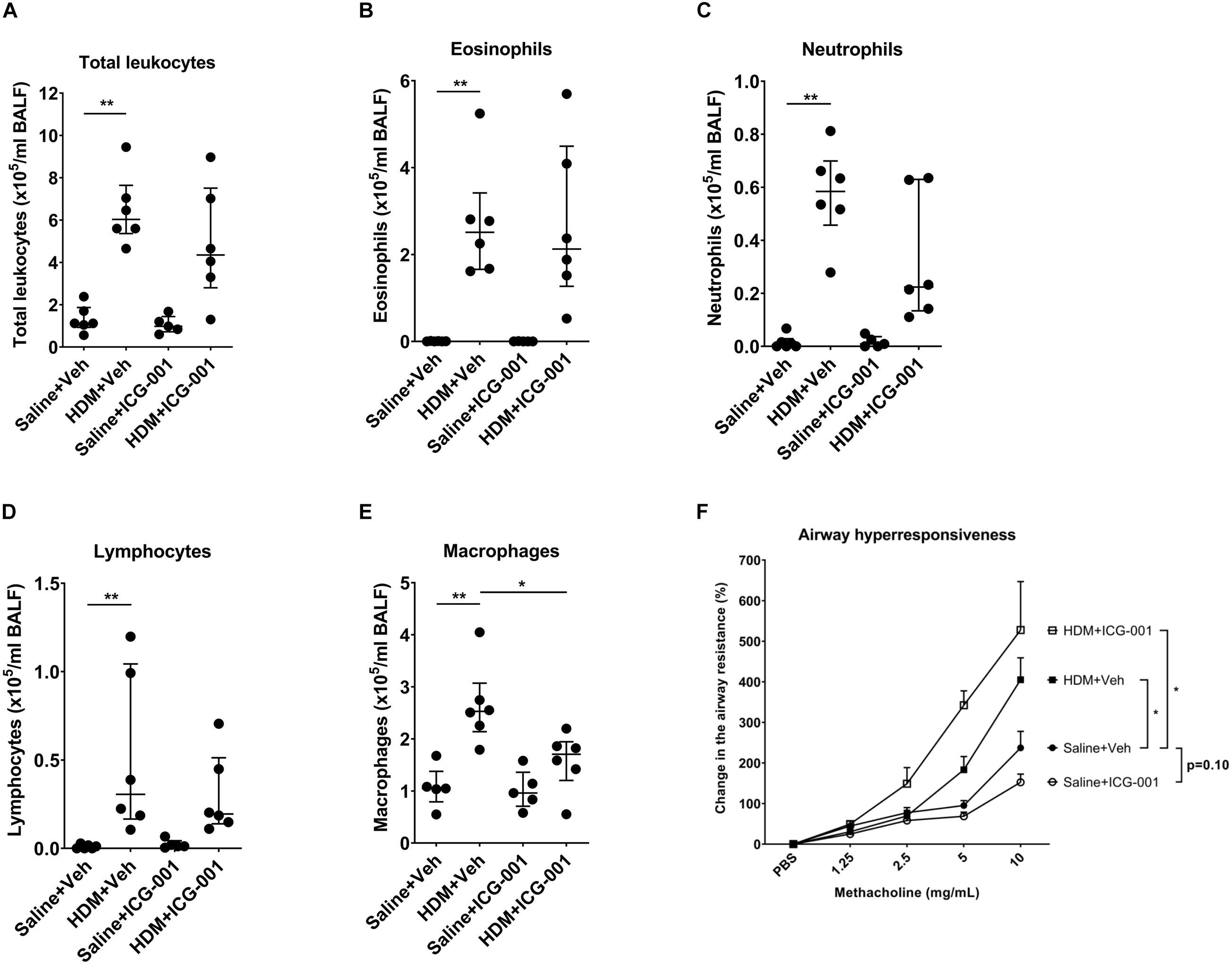
Figure 3. ICG-001 inhibits HDM-induced infiltration of macrophages but does not influence HDM-induced AHR. Numbers of (A) total leukocytes (n = 5–6), (B) eosinophils (n = 5–6), (C) neutrophils (n = 5–6), (D) lymphocytes (n = 5–6), and (E) macrophages (n = 5–6) present in BAL fluid of mice. Data is presented as median ± IQR; n = 5–6, *p < 0.05, **p < 0.01, Mann–Whitney U test. (F) Change in the methacholine-induced airway resistance (%) in mice following exposure to HDM and treated with ICG-001 or vehicle-treated controls. Data is presented as median ± IQR; n = 6, *p < 0.05, two-way ANOVA.
Sensitization of mice for 3 days from day 0 to 3 followed by exposure to HDM for 4 days from day 14 to 17 showed a strong trend (p = 0.06) toward a decrease in the mRNA expression of E-cadherin (Cdh1) compared to PBS-sensitized and PBS-challenged mice. In mice treated with ICG-001, no differences were observed in the Cdh1 mRNA expression levels after HDM exposure compared to the PBS-exposed controls (Figure 2B). HDM significantly increased the mRNA expression of secretory mucins Muc5ac and Muc5b (Figures 2C,D). Treatment of mice with ICG-001 resulted in a trend (p = 0.09) toward the suppression of HDM-induced mRNA expression of Muc5ac but had no effect on Muc5b expression (Figures 2C,D). The Muc5ac transcriptional activator Spdef was significantly increased (Figure 2E) and the Muc5ac transcriptional repressor Foxa2 was significantly decreased (Figure 2F) following HDM exposure, which was not affected by ICG-001 treatment (Figures 2E,F). The increase in Muc5ac and Muc5b expression was validated at the protein level by AB/PAS staining, revealing that HDM exposure promoted goblet cell metaplasia and mucus production compared to the PBS + Vehicle treated controls. Notably, HDM-induced goblet cell metaplasia and mucus production was significantly repressed by ICG-001 treatment (Figures 2G,H).
Airway inflammation with increases in leukocytes and notably eosinophils is a hallmark of the HDM-induced asthma model in mice. HDM significantly increased the total leukocyte counts. The majority of leukocytes in the BAL fluid of PBS exposed mice were macrophages (98.3 ± 1.6%) and the remaining fraction included eosinophils, neutrophils and lymphocytes (Supplementary Figure 1). Exposure to HDM significantly increased the number of eosinophils, neutrophils, lymphocytes, and to a lesser extent, macrophages in BAL fluid (Figures 3A–E), resulting in a decrease in the percentage of macrophages (Supplementary Figure 1E). ICG-001 did not have any effect on HDM-induced increases in the absolute numbers of total leukocytes including eosinophils, neutrophils, and lymphocytes (Figures 3A–D). ICG-001 treatment did result in a significant reduction in HDM-induced macrophages (Figure 3E). Methacholine induced a dose-dependent increase in airway resistance, which was significantly elevated by HDM sensitization and challenge (Figure 3F). In line with the lack of effect on inflammation, ICG-001 treatment did not suppress HDM-induced increase in the airway resistance (Figure 3F).
In this study, we investigated the role of β-catenin/CBP signaling in allergen-induced manifestations of asthma. HDM exposure reduced the expression of E-cadherin, which was accompanied by increases in Muc5ac, Muc5b, and Spdef expression, decreased Foxa2 expression, the induction of goblet cell metaplasia and the infiltration of inflammatory cells, thereby representing the phenotype of asthmatic airway epithelium. Specific inhibition of β-catenin/CBP signaling using ICG-001 significantly decreased goblet cell metaplasia and mucus production. The observed effect of ICG-001 on mucus production is in line with a trend toward a decrease in HDM-induced Muc5ac expression with ICG-001 treatment, which may have failed to reach statistical significance due to the high variation between mice. The RNA analysis of lung tissue exhibits greater within-group variance than AB/PAS staining, as the former is dependent on the relative proportion of RNA coming from the airway epithelial cells in the total lung tissue RNA pool. Furthermore, the design of our study may explain the discrepancy between the effect of ICG-001 on HDM-induced Muc5ac expression and production. We treated mice with ICG-001 after the initial HDM sensitization and we cannot be sure that the changes in Muc5ac gene expression were already induced during the initial sensitization phase. A sham-sensitized and HDM-treated control may be used in the future studies to obtain clarity on the potential of ICG-001 to suppress earlier HDM-induced changes during the sensitization phase, e.g., in Muc5ac gene expression. Furthermore, mRNA expression is usually transient and depends on the time point of measurement and the protein levels of mucins quantified by the staining can be considered as the most important measurement for goblet cell metaplasia. Our findings are in line with a previous study in which ICG-001 was shown to inhibit goblet cell metaplasia in a toluene diisocyanate treated mouse model of asthma (Yao et al., 2017).
It has been recently shown that ICG-001 prevented the differentiation of human airway epithelial progenitor cells into mucous-secreting cells and decreased MUC5B gene expression during culture at the air-liquid interface (Malleske et al., 2018). However, in addition to the lack of effect on Muc5ac, ICG-001 treatment did not affect Muc5b expression in HDM-treated mice. Further investigations into the molecular mechanisms responsible for the inhibition mucus production and goblet cell hyperplasia by ICG-01 will be addressed in future studies. The discrepancy in the effects of ICG-001 on MUC5B expression between our current in vivo observations and the findings in vitro (Malleske et al., 2018), where ICG-001 inhibited MUC5B expression could be due to differences in the cellular composition of airway epithelium between human and mice. Lineage tracing studies revealed that IL-13-induced goblet cells were derived from FOXJ1-expressing ciliated cells in human primary airway epithelial cells in ALI (Turner et al., 2011), but this was not demonstrated in vivo in an Ovalbumin-induced mouse model of allergic airway disease (Pardo-Saganta et al., 2013). Newly differentiated goblet cells generated in response to allergen in mice may originate predominantly from club cells present in the pseudostratified layer of airway epithelium (Park et al., 2007). Different types of progenitors for goblet cells may not be equally sensitive to the inhibitor. In addition to Muc5ac, ICG-001 treatment also had no effect on the gene expression of Foxa2, which was previously identified to be regulated by the β-catenin pathway (Mucenski et al., 2005). Of note, Foxa2 is a transcriptional activator of E-cadherin (Song et al., 2010; Zhang et al., 2015), and the HDM-induced downregulation of Foxa2 may have resulted in reduced expression of E-cadherin. In line with the lack of effect on Foxa2, we did not observe an effect of ICG-001 on E-cadherin expression. ICG-001 was able to significantly decrease the HDM-induced infiltration of macrophages, suggesting the important role of β-catenin/CBP signaling in HDM-induced pro-inflammatory responses in mice. Indeed, we have previously shown that ICG-001 inhibits the HDM-induced granulocyte macrophage-colony stimulating factor (GM-CSF) (Kuchibhotla et al., 2020), a cytokine that stimulates the production of macrophages. Alternatively, ICG-001 was not able to prevent the infiltration of eosinophils, neutrophils, and lymphocytes. This is in contrast to our in vitro data where we showed that ICG-001 reduced HDM-induced CCL20 levels in primary bronchial epithelial cells (Kuchibhotla et al., 2020), as neutrophils and T cells (predominantly Th17) are known to express CCL20 receptor CRR6. Moreover, ICG-001 was previously shown to be able to significantly decrease eosinophil and neutrophil influx into the airways induced by toluene diisocyanate in mice (Yao et al., 2017). These contrasting findings could be due to the difference in the mechanism of action of HDM and toluene diisocyanate. HDM activates pathogen recognition receptors (PRRs) like dectin-1 and Toll-like receptor (TLR)-4 resulting in the release of pro-inflammatory cytokines like CCL17, CCL20, IL-5, and IL-13 (Gregory and Lloyd, 2011; Post et al., 2014), whereas toluene diisocyanate acts on transient receptor potential melastatin 8 (TRPM8) resulting in the release of IL-25, IL-4, and IL-13 (Kim et al., 2017a). This suggests that TRPM8, but not PRR activation is regulated by β-catenin/CBP signaling. As HDM is a major allergen responsible for the airway inflammation in Type-2 driven, atopic asthma, our model may be more relevant for better understanding of the role of β-catenin/CBP signaling in allergic airway inflammation.
Here, we show that ICG-001 attenuates HDM-induced goblet cell metaplasia independent of HDM-induced airway inflammation. Inhibition of β-catenin/CBP pathway could be an alternative strategy to regulate mucus hypersecretion in asthma. Future studies should be directed toward more targeted delivery of the ICG-001 to specific airway epithelial cell types for increased efficiency of the drug.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by University of Newcastle Animal Care and Ethics Committee.
VK was involved in formal analysis, project administration, validation, and writing the original draft of the manuscript. MS and AR were involved in conceptualization, methodology, investigation, formal analysis, project administration, and validation of the project. IH and MN were involved in supervision of the project. PH and DK were involved in conceptualization, funding acquisition, and supervision of the project. All authors have contributed significantly to the manuscript, read the manuscript, agreed with its content, and approve the submission of this manuscript.
DK reports grant from National Health and Medical Research Council (NHMRC #1064405) of Australia. PH was funded by a Fellowship and grants from the NHMRC of Australia (#1079187 and #1175134). MS was funded by fellowships from the NMHRC (#1072000) and Australian Research Council (#170100226).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.690531/full#supplementary-material
Bonser, L. R., and Erle, D. J. (2017). Airway mucus and asthma: the role of MUC5AC and MUC5B. J. Clin. Med. 6:112. doi: 10.3390/jcm6120112
Ceteci, F., Ceteci, S., Zanucco, E., Thakur, C., Becker, M., El-Nikhely, N., et al. (2012). E-cadherin controls bronchiolar progenitor cells and onset of preneoplastic lesions in mice. Neoplasia (United States) 14, 1164–1177. doi: 10.1593/neo.121088
Emami, K. H., Nguyen, C., Ma, H., Kim, D. H., Jeong, K. W., Eguchi, M., et al. (2004). A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. U.S.A. 101, 12682–12687. doi: 10.1073/pnas.0404875101
Essilfie, A. T., Simpson, J. L., Horvat, J. C., Preston, J. A., Dunkley, M. L., Foster, P. S., et al. (2011). Haemophilus influenzae infection drives IL-17-mediated neutrophilic Allergic airways disease. PLoS Pathog. 7:1002244. doi: 10.1371/journal.ppat.1002244
Evans, C. M., Kim, K., Tuvim, M. J., and Dickey, B. F. (2009). Mucus hypersecretion in asthma: causes and effects. Curr. Opin. Pulm. Med. 15, 4–11. doi: 10.1097/mcp.0b013e32831da8d3
Fehrenbach, H., Wagner, C., and Wegmann, M. (2017). Airway remodeling in asthma: what really matters. Cell Tissue Res. 367, 551–569. doi: 10.1007/s00441-016-2566-8
Gregory, L. G., and Lloyd, C. M. (2011). Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 32, 402–411. doi: 10.1016/j.it.2011.06.006
Hallstrand, T. S., Hackett, T. L., Altemeier, W. A., Matute-Bello, G., Hansbro, P. M., and Knight, D. A. (2014). Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin. Immunol. 151, 1–15. doi: 10.1016/j.clim.2013.12.003
Hammad, H., and Lambrecht, B. N. (2015). Barrier epithelial cells and the control of Type 2 immunity. Immunity 43, 29–40. doi: 10.1016/j.immuni.2015.07.007
Hansbro, P. M., Scott, G. V., Essilfie, A.-T., Kim, R. Y., Starkey, M. R., Nguyen, D. H., et al. (2013). Th2 cytokine antagonists: potential treatments for severe asthma. Expert Opin. Investig. Drugs 22, 49–69. doi: 10.1517/13543784.2013.732997
Heijink, I. H., Kies, P. M., Kauffman, H. F., Postma, D. S., van Oosterhout, A. J. M., and Vellenga, E. (2007). Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J. Immunol. 178, 7678–7685. doi: 10.4049/jimmunol.178.12.7678
Heijink, I. H., Nawijn, M. C., and Hackett, T. L. (2014). Airway epithelial barrier function regulates the pathogenesis of allergic asthma. Clin. Exp. Allergy 44, 620–630. doi: 10.1111/cea.12296
Horvat, J. C., Beagley, K. W., Wade, M. A., Preston, J. A., Hansbro, N. G., Hickey, D. K., et al. (2007). Neonatal chlamydial infection induces mixed T-cell responses that drive allergic airway disease. Am. J. Respir. Crit. Care Med. 176, 556–564. doi: 10.1164/rccm.200607-1005oc
Kaiko, G. E., Phipps, S., Hickey, D. K., Lam, C. E., Hansbro, P. M., Foster, P. S., et al. (2008). Chlamydia muridarum Infection subverts dendritic cell function to promote Th2 immunity and airways hyperreactivity. J. Immunol. 180, 2225–2232. doi: 10.4049/jimmunol.180.4.2225
Kim, J. H., Jang, Y. S., Jang, S. H., Jung, K. S., Kim, S. H., Ye, Y. M., et al. (2017a). Toluene diisocyanate exposure induces airway inflammation of bronchial epithelial cells via the activation of transient receptor potential melastatin 8. Exp. Mol. Med. 49:e299. doi: 10.1038/emm.2016.161
Kim, R. Y., Horvat, J. C., Pinkerton, J. W., Starkey, M. R., Essilfie, A. T., Mayall, J. R., et al. (2017b). MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase–mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 139, 519–532. doi: 10.1016/j.jaci.2016.04.038
Kim, R. Y., Pinkerton, J. W., Essilfie, A. T., Robertson, A. A. B., Baines, K. J., Brown, A. C., et al. (2017c). Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 196, 283–297. doi: 10.1164/rccm.201609-1830oc
Kuchibhotla, V. N. S., Jonker, M. R., Bruin, H. G., Noordhoek, J. A., Knight, D. A., Nawijn, M. C., et al. (2020). Inhibition of β-catenin/CBP signalling improves airway epithelial barrier function and suppresses CCL20 release. Allergy 75, 1786–1789. doi: 10.1111/all.14216
Liu, G., Cooley, M. A., Nair, P. M., Donovan, C., Hsu, A. C., Jarnicki, A. G., et al. (2017). Airway remodelling and inflammation in asthma are dependent on the extracellular matrix protein fibulin-1c. J. Pathol. 243, 510–523. doi: 10.1002/path.4979
Ma, H., Nguyen, C., Lee, K.-S., and Kahn, M. (2005). Differential roles for the coactivators CBP and p300 on TCF/b-catenin-mediated survivin gene expression. Oncogene 24, 3619–3631. doi: 10.1038/sj.onc.1208433
Malleske, D. T. Jr., Hayes, D. Jr., Lallier, S. W., Hill, C. L., and Reynolds, S. D. (2018). Regulation of human airway epithelial tissue stem cell differentiation by β-catenin, P300, and CBP. Stem Cells 36, 1905–1916. doi: 10.1002/stem.2906
Moheimani, F., Roth, H. M., Cross, J., Reid, A. T., Shaheen, F., Warner, S. M., et al. (2015). Disruption of β-catenin/CBP signaling inhibits human airway epithelial-mesenchymal transition and repair. Int. J. Biochem. Cell Biol. 68, 59–69. doi: 10.1016/j.biocel.2015.08.014
Mucenski, M. L., Nation, J. M., Thitoff, A. R., Besnard, V., Xu, Y., Wert, S. E., et al. (2005). β-Catenin regulates differentiation of respiratory epithelial cells in vivo. Am. J. Physiol. Cell. Mol. Physiol. 289, L971–L979.
Nawijn, M. C., Hackett, T. L., Postma, D. S., van Oosterhout, A. J. M., and Heijink, I. H. (2011). E-cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 32, 248–255. doi: 10.1016/j.it.2011.03.004
Pardo-Saganta, A., Law, B. M., Gonzalez-Celeiro, M., Vinarsky, V., and Rajagopal, J. (2013). Ciliated cells of pseudostratified airway epithelium do not become mucous cells after ovalbumin challenge. Am. J. Respir. Cell Mol. Biol. 48, 364–373. doi: 10.1165/rcmb.2012-0146oc
Park, K. S., Korfhagen, T. R., Bruno, M. D., Kitzmiller, J. A., Wan, H., Wert, S. E., et al. (2007). SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Invest. 117, 978–988. doi: 10.1172/jci29176
Post, S., Heijink, I. H., Hesse, L., Koo, H. K., Shaheen, F., Fouadi, M., et al. (2018). Characterization of a lung epithelium specific E-cadherin knock-out model: implications for obstructive lung pathology. Sci. Rep. 8:13275.
Post, S., Heijink, I. H., Petersen, A. H., De Bruin, H. G., Van Oosterhout, A. J. M., and Nawijn, M. C. (2014). Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS One 9:e91206. doi: 10.1371/journal.pone.0091206
Post, S., Nawijn, M. C., Hackett, T. L., Baranowska, M., Gras, R., Van Oosterhout, A. J. M., et al. (2012). The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax 67, 488–495. doi: 10.1136/thoraxjnl-2011-200606
Post, S., Nawijn, M. C., Jonker, M. R., Kliphuis, N., Van Den Berge, M., Van Oosterhout, A. J. M., et al. (2013). House dust mite-induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy 68, 1117–1125.
Reid, A. T., Nichol, K. S., Veerati, P. C., Moheimani, F., Kicic, A., Stick, S. M., et al. (2020). Blocking notch3 signaling abolishes MUC5AC production in airway epithelial cells from individuals with asthma. Am. J. Respir. Cell Mol. Biol. 62, 513–523. doi: 10.1165/rcmb.2019-0069oc
Reid, A. T., Veerati, P. C., Gosens, R., Bartlett, N. W., Wark, P. A., Grainge, C. L., et al. (2018). Persistent induction of goblet cell differentiation in the airways: therapeutic approaches. Pharmacol. Ther. 185, 155–169. doi: 10.1016/j.pharmthera.2017.12.009
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Song, Y., Washington, M. K., and Crawford, H. C. (2010). Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 70, 2115–2125. doi: 10.1158/0008-5472.can-09-2979
Starkey, M. R., Essilfie, A. T., Horvat, J. C., Kim, R. Y., Nguyen, D. H., Beagley, K. W., et al. (2013). Constitutive production of IL-13 promotes early-life Chlamydia respiratory infection and allergic airway disease. Mucosal Immunol. 6, 569–579. doi: 10.1038/mi.2012.99
Starkey, M. R., McKenzie, A. N., Belz, G. T., and Hansbro, P. M. (2019). Pulmonary group 2 innate lymphoid cells: surprises and challenges. Mucosal Immunol. 12, 299–311. doi: 10.1038/s41385-018-0130-4
Thorburn, A. N., Foster, P. S., Gibson, P. G., and Hansbro, P. M. (2012). Components of Streptococcus pneumoniae suppress allergic airways disease and NKT cells by inducing regulatory T cells. J. Immunol. 188, 4611–4620. doi: 10.4049/jimmunol.1101299
Thorburn, A. N., McKenzie, C. I., Shen, S., Stanley, D., MacIa, L., Mason, L. J., et al. (2015). Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 6:7320.
Thorburn, A. N., O’Sullivan, B. J., Thomas, R., Kumar, R. K., Foster, P. S., Gibson, P. G., et al. (2010). Pneumococcal conjugate vaccine-induced regulatory T cells suppress the development of allergic airways disease. Thorax 65, 1053–1060. doi: 10.1136/thx.2009.131508
Turner, J., Roger, J., Fitau, J., Combe, D., Giddings, J., Van Heeke, G., et al. (2011). Goblet cells are derived from a FOXJ1-expressing progenitor in a human airway epithelium. Am. J. Respir. Cell Mol. Biol. 44, 276–284. doi: 10.1165/rcmb.2009-0304oc
Valenta, T., Hausmann, G., and Basler, K. (2012). The many faces and functions of β-catenin. EMBO J. 31, 2714–2736. doi: 10.1038/emboj.2012.150
Vieira Braga, F. A., Kar, G., Berg, M., Carpaij, O. A., Polanski, K., Simon, L. M., et al. (2019). A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163.
Williams, O. W., Sharafkhaneh, A., Kim, V., Dickey, B. F., and Evans, C. M. (2006). Airway mucus: from production to secretion. Am. J. Respir. Cell Mol. Biol. 34, 527–536.
Yao, L., Zhao, H., Tang, H., Xiong, J., Zhao, W., Liu, L., et al. (2017). Blockade of β-catenin signaling attenuates toluene diisocyanate-induced experimental asthma. Allergy 72, 579–589. doi: 10.1111/all.13045
Keywords: asthma, airway inflammation, β-catenin, ICG-001, goblet cell metaplasia
Citation: Kuchibhotla VNS, Starkey MR, Reid AT, Heijink IH, Nawijn MC, Hansbro PM and Knight DA (2021) Inhibition of β-Catenin/CREB Binding Protein Signaling Attenuates House Dust Mite-Induced Goblet Cell Metaplasia in Mice. Front. Physiol. 12:690531. doi: 10.3389/fphys.2021.690531
Received: 03 April 2021; Accepted: 05 July 2021;
Published: 27 July 2021.
Edited by:
Aldo Pinto, University of Salerno, ItalyReviewed by:
Hans Haverkamp, Washington State University Spokane, United StatesCopyright © 2021 Kuchibhotla, Starkey, Reid, Heijink, Nawijn, Hansbro and Knight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virinchi N. S. Kuchibhotla, dmlyaW5jaGkuMTI3QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share first and senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.